

Abstract
Background/Aims
Overwhelming evidence suggests that inflammatory bowel disease (IBD) is caused by a complicated interplay between the multiple genes and abnormal epigenetic regulation in response to environmental factors. It is becoming apparent that epigenetic factors are significantly associated with the development of the disease. DNA methylation remains the most studied epigenetic modification, and hypermethylation of gene promoters is associated with gene silencing.
Methods
DNA methylation alterations may contribute to the many complex diseases development by regulating the interplay between external and internal environmental factors and gene transcriptional expression. In this study, we used 15 tumor suppressor genes (TSGs), originally identified in colon cancer, to detect promoter methylation in patients with Crohn’s disease (CD). Methylation specific polymerase chain reaction and bisulfite sequencing analyses were performed to assess methylation level of TSGs in CD patients.
Results
We found 6 TSGs (sFRP1, sFRP2, sFRP5, TFPI2, Sox17, and GATA4) are robustly hypermethylated in CD patient samples. Bisulfite sequencing analysis confirmed the methylation levels of the sFRP1, sFRP2, sFRP5, TFPI2, Sox17, and GATA4 promoters in the representative CD patient samples.
Conclusions
In this study, the promoter hypermethylation of the TSGs observed indicates that CD exhibits specific DNA methylation signatures with potential clinical applications for the noninvasive diagnosis of IBD and the prognosis for patients with IBD.
Keywords: DNA methylation; Promoter; Crohn disease; Genes, Tumor suppressor
INTRODUCTION
Inflammatory bowel disease (IBD) comprising CD and UC is believed to reflect complex interplay between genetic and environmental factors. CD is one of the major type of IBD. CD and UC exhibit etiologically distinct characters. CD often involves in the ileum and colon mostly but also can involves in any region of the gut. UC mostly common involves the rectum, and it has been known that the inflammation may extend the cecum in a contiguous pattern [1]. Inflammatory diseases such as CD are usually affected due to genetic factors and environmental factors [2]. A recent genome-wide association study successfully identified more than 200 CD-associated loci at the level of genome-wide significance [3,4]. However, these findings could only address a few part of the IBD pathogenesis. Numerous genetic, clinical, and multiple experimental studies should have been proved to our understanding of the etiology of IBD.
DNA methylation, predominantly occurring in the CpG dinucleotide of the genome, is a key epigenetic mechanism that can regulate gene expression and therefore can affect to the development and progression of complex diseases including cancer [5]. Aberrant DNA methylation frequently involves in inactivation of tumor suppressor genes (TSGs) in human cancers [5]. Among others, the transcriptional silencing of tumor suppressor candidate genes is regulated by promoter hypermethylation in colorectal cancer (CRC) by our group and other groups as well [6,7]. High-throughput genome-wide methylation studies provide a brand-new approach to understand the importance of DNA methylation at the whole genome level and its impact on gene regulation [6,8]. Therefore, the role of DNA methylation has been widely studied in many different cancer types and other human diseases, such as IBD [9]. For example, abnormal DNA methylation of the estrogen receptor, p14ARF, and E-cadherin genes has been detected in UC patients. In addition, there is growing evidence that epigenetic changes in the regulation of gene expression are critical factors in the pathogenesis of IBD [10,11]. Many studies have used these wellknown TSG candidates to investigate other cancers, such as lung, breast, and pancreatic cancer [7,12]. We previously have reported that CpG islands in the promoter region of TCERG1L is abnormally hypermethylated at high frequency not only in colon cancer patient tissues [13], but also in patients with UC [14]. However, few studies have been conducted on the DNA methylation profiles of these TSGs in patient with CD.
We tested the promoter hypermethylation of 18 tumor suppressor candidates (TFPI2, GATA4, p16, sFRP1, sFRP4, MGMT, Sox17, HIC1, sFRP2, sFRP5, p14, p15, GATA5, E-cadherin, and Timp3) in a limited number of tissue samples from Korean patients with CD (n = 15). Interestingly, we detected the abnormal promoter hypermethylation of several TSGs in patients with CD: SFRP1, SFRP2, SFRP5, TFPI2, Sox17, and GATA4 were aberrantly hypermethylated with high frequency in most of the CD patient tissues. Our data suggest that the methylation status of 6 genes in patients with CD has the potential to become a marker of risk for the progression of severe disease.
METHODS
1. Patients and Samples
The CD and normal colon biospecimens for this study were provided by the Inje Biobank (Inje University School of Medicine), a member of the National Biobank of Korea, which is supported by the Ministry of Health and Welfare. Tissue samples were collected from the ascending colon at the time of colonoscopy. Of the 15 subjects, 10 were male and 5 female, yielding a male to female ratio of 2:1. The median age at diagnosis of CD was 28.4 years (range, 18–45 years). CD was diagnosed on the basis of conventional clinical, radiologic, endoscopic, and histopathologic criteria. At the time of diagnosis, 3 patients (20%) had disease in the colon alone (L2), and 12 (80%) had disease in the small bowel and colon (L3). Disease behavior at diagnosis was inflammatory (B1) in 10 patients (66.7%), stricturing (B2) in 4 (26.7%), and penetrating (B3) in 1 (6.6%). Clinical characteristics of these patients are shown in Table 1. This study was approved by the respective institutional review board (IRB No. 129792-2015-056) of the participating institutions of the National Biobank of Korea, and written informed consent was obtained for all study participants prior to data collection.
Table 1.
Characteristics of Samples of Patient with CD
| Characteristic | Value (n = 15) |
|---|---|
| Age (yr) | 28.4 (18.0–45.0) |
| Sex | |
| Male | 10 (66.7) |
| Female | 5 (23.3) |
| Disease location at diagnosis | |
| Small bowel alone (L1) | 0 |
| Colon alone (L2) | 3 (20.0) |
| Small bowel and colon (L3) | 12 (80.0) |
| Disease behavior at diagnosis | |
| Inflammatory (B1) | 10 (66.7) |
| Stricturing (B2) | 4 (26.7) |
| Penetrating (B3) | 1 (6.6) |
Values are presented as median (range) or number (%).
2. Genomic DNA Extraction and DNA Methylation Analyses
Genomic DNA was extracted following a standard phenolchloroform extraction protocol. Bisulfite modification of DNA was performed using the EZ DNA Methylation Kit (Zymo Research, Irvine, CA, USA) according to the manufacturer’s instructions. Methylation analysis by methylation-specific PCR (MSP) was carried out in a 25-μL reaction of 10 × PCR buffer, 10 mM dNTPs (deoxyribonucleotide triphosphates), 10 pmol of each of the methylated or unmethylated primers, 1 unit of JumpStartTM REDTaq® DNA polymerase (Sigma, St. Louis, MO, USA) and 4 μL of bisulfite-treated DNA. The detailed information of primer was listed in in Table 2. Cycling condition were: 5 minutes at 95°C followed by 35 repeats of the following cycle: 95°C for 30 seconds, annealing at the appropriate temperature for 30 seconds. In vitro methylated DNA was used as a positive control.
Table 2.
MSP Primers Information in This Study
| Gene | Target | Methylation-specific PCR |
|
|---|---|---|---|
| Forward primer (5’-3’) | Reverse primer (5’-3’) | ||
| TFPI2 | Unmethylation | CCCACATAAAACAAACACCCAAACCA | TGGTTTGTTGGGTAAGGTGTTTG |
| Methylation | CATAAAACGAACACCCGAACCG | GTTCGTTGGGTAAGGCGTTC | |
| BS-Seq | GGTTTATGGTGTAGGGG | CAATCACTAACAAATCATTTCC | |
| GATA4 | Unmethylation | TTTGTATAGTTTTGTAGTTTGTGTTTAGT | CCCAACTCACAACTCAAATCCCCA |
| Methylation | GTATAGTTTCGTAGTTTGCGTTTAGC | AACTCGCGACTCGAATCCCCG | |
| BS-Seq | CTTCCAACCCYACCTTC | GTTTTTAGAAGAAGAGGAGGG | |
| p16 | Unmethylation | TTATTAGAGGGTGGGGTGGATTGT | CAACCCCAAACCACAACCATAA |
| Methylation | TTATTAGAGGGTGGGGCGGATCGC | GACCCCGAACCGCGACCGTAA | |
| sFRP1 | Unmethylation | GTTTTGTAGTTTTTGGAGTTAGTGTTGTGT | CTCAACCTACAATCAAAAACAACACAAACA |
| Methylation | TGTAGTTTTCGGAGTTAGTGTCGCGC | CCTACGATCGAAAACGACGCGAACG | |
| BS-Seq | GTTTTGTTTTTTAAGGGGTGTTGAG | ACACTAACTCCRAAAACTACAAAAC | |
| sFRP4 | Unmethylation | GGGGGTGATGTTATTGTTTTTGTATTGAT | CACCTCCCCTAACATAAACTCAAAACA |
| Methylation | GGGTGATGTTATCGTTTTTGTATCGAC | CCTCCCCTAACGTAAACTCGAAACG | |
| MGMT | Unmethylation | TTTGTGTTTTGATGTTTGTAGGTTTTTGT | AACTCCACACTCTTCCAAAAACAAAACA |
| Methylation | TTTCGACGTTCGTAGGTTTTCGC | GCACTCTTCCGAAAACGAAACG | |
| Sox17 | Unmethylation | CAAACCAAAAACAAATCCCATATCCAACA | GATTTTGTTGTGTTAGTTGTTTGTGTTTG |
| Methylation | CAAAAACCAATCCCGTATCCCACG | TTGCGTTAGTCGTTTGCGTTC | |
| BS-Seq | TAATAAAGTTGATTTTGGGTATTATAG | CCCTACCTACTAAACCTAAAAATTC | |
| HIC1 | Unmethylation | TTGGGTTTGGTTTTTGTGTTTTG | CACCCTAACACCACCCTAAC |
| Methylation | TCGGTTTTCGCGTTTTGTTCGT | AACCGAAAACTATCAACCCTCG | |
| sFRP2 | Unmethylation | GTTTTGTAGTTTTTGGAGTTAGTGTTGTGT | CTCAACCTACAATCAAAAACAACACAAACA |
| Methylation | TGTAGTTTTCGGAGTTAGTGTCGCGC | CCTACGATCGAAAACGACGCGAACG | |
| BS-Seq | TATTTATGTTTGGTAATTTAGTAGAAATT | CCCTAAAATTTCTTTAAACAACAAACA | |
| sFRP5 | Unmethylation | GTAAGATTTGGTGTTGGGTGGGATGTTT | AAAACTCCAACCCAAACCTCACCATACA |
| Methylation | AAGATTTGGCGTTGGGCGGGACGTTC | ACTCCAACCCGAACCTCGCCGTACG | |
| BS-Seq | TTAAATGTTTAGGGAGGTAGGGAGT | AATCGCCCAAATAAATAACAACCTAC | |
| p14 | Unmethylation | TTTTTGGTGTTAAAGGGTGGTGTAGT | CACAAAAACCCTCACTCACAACAA |
| Methylation | GTGTTAAAGGGCGGCGTAGC | AAAACCCTCACTCGCGACGA | |
| p15 | Unmethylation | GGTTGGTTTTTTATTTTGTTAGAGTGAGGT | AACCACTCTAACCACAAAATACAAACACA |
| Methylation | GGTTTTTTATTTTGTTAGAGCGAGGC | TAACCGCAAAATACGAACGCG | |
| GATA5 | Unmethylation | TGGAGTTTGTTTTTAGGTTAGTTTTTGGT | CAAACCAATACAACTAAACAAACAAACCA |
| Methylation | AGTTCGTTTTTAGGTTAGTTTTCGGC | CCAATACAACTAAACGAACGAACCG | |
| E-cadherin | Unmethylation | TGGTTGTAGTTATGTATTTATTTTTAGTGGTGTT | ACACCAAATACAATCAAATCAAACCAAA |
| Methylation | TGTAGTTACGTATTTATTTTTAGTGGCGTC | CGAATACGATCGAATCGAACCG | |
| Timp3 | Unmethylation | TTTTGTTTTGTTATTTTTTGTTTTTGGTTTT | CCCCCAAAAACCCCACCTCA |
| Methylation | CGTTTCGTTATTTTTTGTTTTCGGTTTC | CCGAAAACCCCGCCTCG | |
MSP, methylation-specific PCR; BS-Seq, bisulfite sequencing.
3. Bisulfite Sequencing Analysis
Bisulfite modification of DNA was used for PCR reaction to clone alleles of target amplicons. The PCR amplicons were gelpurified and subcloned into the pCRII-TOPO vector (Invitrogen, Carlsbad CA, USA). At least 7 clones were randomly selected and sequenced on an ABI3730xl DNA analyzer to ascertain the methylation patterns of each locus. All primers for the bisulfite sequencing analysis are listed in Table 2.
4. Statistical Analysis
All statistical analyses were performed using the STATA 9.2 software package (StataCorp., College Station, TX, USA). The data from most analyses were compared using a t-test, while continuous variables were analyzed using the Mann-Whitney U-test. P-values of less than 0.05 were considered significant.
RESULTS
1. Abnormal Hypermethylation of TSGs in CD Patients
In this study, we investigated transcriptional silencing by promoter hypermethylation of 15 well-known TSG candidates (TFPI2, GATA4, p16, sFRP1, sFRP4, MGMT, Sox17, HIC1, sFRP2, sFRP5, p14, p15, GATA5, E-cadherin, and Timp3) in samples from patients with CD. These genes were previously reported on the basis of comprehensive studies of candidate TSGs in colon cancer, and the transcriptional silencing of these suppressors is regulated by promoter hypermethylation in many cancer types [7]. In addition, highly sensitive DNA methylation markers in colon cancer could be used to detect the risk level of patients with inflammatory diseases, such as UC and CD. Motivated by this possibility, we previously analyzed DNA methylation in UC samples [15,16] and serum samples of CD patients [17]. Based on previous studies, we were aware of that unknown TSGs could be abnormally hypermethylated in patients with CD. Here, we extracted genomic DNA from patient tissues with CD (n = 15) (Table 1). First, using MSP analysis, we assessed the methylation level of the promoter regions of 15 genes (TFPI2, GATA4, p16, sFRP1, sFRP4, MGMT, Sox17, HIC1, sFRP2, sFRP5, p14, p15, GATA5, E-cadherin, and Timp3) (Table 2) in tissue samples from patient with CD. Conventional MSP analysis was performed successfully with most samples. Fig. 1 displays the frequency of hypermethylation for the 15 gene promoter regions in patient tissues with CD. For most of the CD samples we tested, sFRP1, sFRP2, sFRP5, and Sox17 showed the highest hypermethylation levels (100% frequencies), and TFPI2, GATA4, sFRP4, GATA5, HIC1, and p15 showed methylation frequencies of 66.7%, 60.0%, 46.7%, 33.3%, 13.3%, and 6.7%, respectively. The other 5 genes (p16, p14, E-cadherin, MGMT, and Timp3) showed no methylation in the CD samples.
Fig. 1.
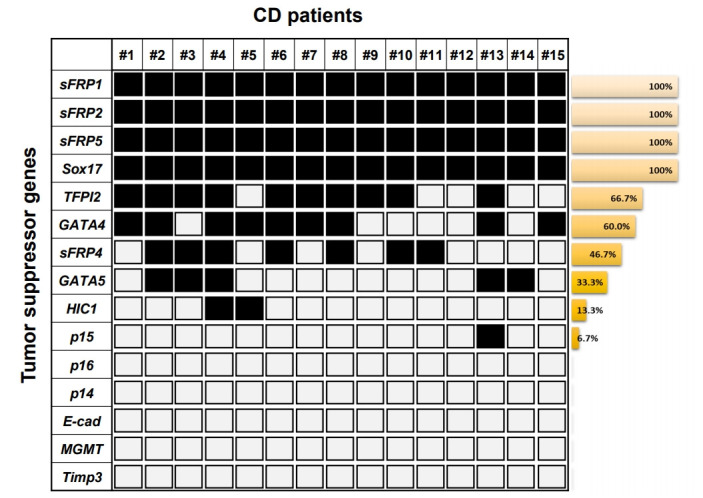
Summarized results from the methylation analyses on the tumor suppressor genes identified from colon cancer and obtained from patients with CD. Each square and number indicates a single CD patient blood sample. The white square indicates the unmethylated CpG sites. The black square indicates the methylated CpG sites. The Bar graph presents methylation frequencies of tested genes. E-cad, Ecadherin.
2. Six TSGs Are Highly Methylated in CD Patients
On the basis of the high frequency of methylation of genes (sFRP1, sFRP2, sFRP5, TFPI2, Sox17, and GATA4; > 60% methylation frequencies) in the samples from patients with CD, we next texted the changes in DNA methylation status in the promoter region of the 6 genes by bisulfite genomic sequencing in representative patient tissues with CD (n = 3). All the genes we tested had previously been shown to be hypermethylated in a cancer-specific manner but are not methylated in normal colon tissues [7]. The location and extent of the methylated promoter regions used for bisulfite sequencing, relative to the transcription start sites of exon 1, are as follows: sFRP1: upstream region from –142 to 59, 26 CG sites; sFRP2: upstream region from –378 to –159, 21 CG sites; sFRP5: upstream region from –296 to –4, 26 CG sites; TFPI2: upstream region from –286 to –76, 30 CG sites; Sox17: region from 339 to 603, 34 CG sites; and GATA4: upstream region from –229 to –161, 46 CG sites. The bisulfite sequencing of the individual clones of 5 PCR products from 3 different individual CD tissue samples revealed densely methylated CG sites within the promoter regions of all the clones (Figs 2, 3). The methylation status in the promoter regions of sFRP1, TFPI2, and sFRP2 in the CD tissues showed 82.5%, 70%, and 63.4% methylation at the CG sites, respectively. The CG sites of the Sox17, sFRP4, and GATA4 genes in the CD tissues were methylated in a range from 55% to 57%. Accordingly, the bisulfite sequencing data confirmed the complete methylation of the 6 gene promoters in patients with CD as had been determined by the MSP analysis (Fig. 1); that is, 6 genes were significantly hypermethylated in patient samples with CD. Overall, these results strongly suggest that we found increased DNA methylation levels of the sFRP1, sFRP2, sFRP5, TFPI2, Sox17, and GATA4 genes in inflammatory diseases, as represented by CD.
Fig. 2.

The methylation level of the promoter region of the sFRP1, sFRP2, and sFRP5 genes in CD patient samples (patient #1, #2, and #3) as determined by bisulfite sequencing analysis. The location of each CpG site related to the transcription start site is shown at the bottom of the panels. Open and filled squares indicate methylation and no methylation, respectively.
Fig. 3.

The methylation levels of the promoter regions of the GATA4, Sox17, and TFPI2 genes in CD patient samples (patient #1, #2, and #3) as determined by bisulfite sequencing analysis. The location of each CpG site related to the transcription start site is shown at the bottom of the panels. Open and filled squares indicate methylation and no methylation, respectively.
DISCUSSION
IBD is identified by chronic uncontrolled inflammation of the intestinal mucosa due to abnormal immune response, leading the disruption of the intestinal epithelium [18]. Patients with IBD can be grouped as lifetime risk (2- to 3-fold greater) of developing CRC, and the most important risk factors for colitis-associated cancer are duration, severity and extent of the IBD [13,14]. There is a growing body of evidence suggesting that inflammation is the critical cause of many cancers, and colitis-associated cancer serves as a great model system to study the general principles understanding for the etiology of inflammation-associated cancer [19]. Prevalent evidence supports the theory that IBD is caused by a complex interplay between the predispositions of multiple genes and abnormal interactions with environmental factors. It is becoming increasingly apparent that epigenetic factors can significantly contribute to the pathogenesis of disease.
We have previously reported that the TCERG1L, sFRP1, FBN2, and TFPI2 genes were hypermethylated at high frequency in patients with UC, which is another type of IBD [16]. In the present study, we expanded a gene panel consisting mostly of tumor suppressor candidates identified from the colon cancer and tested the methylation level of these genes in patients with CD. According to our previous data and present study, these TSGs are transcriptionally regulated by promoter hypermethylation in patients with IBD. We do not have direct evidences the biological role of these TSGs during IBD development but our data suggest that epigenetically regulated TSGs could contribute to the pathogenesis of IBD. Although lack of studies have been reported that the methylation of TSGs in CD, here we found the promoter methylation changes of sFRP families, TFPI2, Sox17, and GATA4 genes in patients with CD.
The tissue factor pathway inhibitor 2 (TFPI2) gene is located on chromosome 7q22 and encodes a Kunitz-type serine proteinase inhibitor [20]. TFPI2 can play as a critical roles involved in protecting the extracellular matrix of cancer cells from degradation as well as regulating tumor invasion [21]. Abnormal promoter hypermethylation of TFPI2 has been observed in CRC [22], gastric cancer [23], hepatocellular carcinoma [24], pancreatic cancer [25], and cervical cancer [26]. Moreover, promoter hypermethylation of TFPI2 has been detected in recurrence and early-stage CRC [27], and in sera from CRC patients with large, poorly differentiated carcinomas, invasion, and several metastasis such as lymph node or distant [28]. Overall, our study provides the evidences that promoter hypermethylation of TFPI2 is also detected in most of the CD patient samples we tested. This data suggests that TFPI2 may be a useful biomarker during colonoscopic surveillance screening. Therefore, further studies using a large series of samples are necessary to confirm the hypothesis on the TFPI2 gene could be a promising methylation biomarker for screening patients with CD.
Sox17 encodes an HMG box transcription factor and its biological roles in many different stage of developmental process such as in vascular development and hematopoiesis [29-32]. The Sox17 promoter is hypermethylated in cholangiocarcinoma tissues, lung cancer [33], gastric cancer [34], liver cancer [35], and breast cancer [36]. In addition, inactivated Sox17 by promoter hypermethylation could contribute to the dysregulaion of the Wnt signaling pathway in human cancer [37]. These accumulating data strongly suggests that epigenetically silenced Sox17 can affect to the aberrant activation of Wnt signaling during tumorigenesis in multiple cancer types [38]. We first report here that Sox17 is hypermethylated in patients with CD, suggesting that epigenetically inactivated Sox17 may contribute to the pathogenesis of IBD.
The transcription factor GATA4 plays an essential role in the development and differentiation of the GI tract and is suggested to be involved in CRC development [39,40]. The lack of GATA4 expression regulated by promoter hypermethylation has been reported in primary CRC, gastric, esophageal, lung, and ovarian cancers [40-43]. Although accumulating studies on the GATA4 gene is epigenetically regulated in many cancer types, there is lack of information whether GATA4 gene is hypermethylated in patients with CD.
SFRPs belong to the family of Wnt pathway antagonists, and the transcriptional expression of SFRPs are regulated by gene promoter hypermethylation in colon, gastric, ovarian, and lung cancers [44-47]. Epigenetic regulation of the Wnt pathway was first characterized in colon cancer, which is known to be related to chronic inflammation. Recently, it was shown that the downregulation of SFRP gene family members in gastric and CRC is commonly regulated by promoter hypermethylation [45,48]. Thus, transcriptional silencing of SFRP gene may affect to leading a mechanism of aberrant Wnt signaling activation [45,48]. It has been reported that the frequent inactivation of SFRP1 and SFRP2 in IBD-associated neoplasia further highlights the importance of the canonical Wnt pathway to understand the pathogenesis of IBD-associated cancer. Our study has some limiting issues. We lacked control samples for CD, such that we could not determine the TSG methylation with specificity in CD patient samples. We also lacked clinical information on the CD samples, such as the disease duration or severity that; therefore, we could not implicate the clinical importance of the promoter methylation of TSGs for the CD patients. Therefore, further study is necessary to define the DNA methylation markers in a large series of clinical data from CD patients, including control samples.
In summary, we investigated the promoter DNA methylation pattern of 15 TSGs in samples from patients with CD. We found that samples from 10 of the 15 patients had TSG promoters that were methylated as determined by conventional MSP analysis. Six TSGs (sFRP1, sFRP2, sFRP5, TFPI2, Sox17, and GATA4) showed a high frequency of hypermethylation in the CD patients, a finding that we confirmed by conducting bisulfite sequencing analysis to determine the methylation levels of the genes in the CD patient tissues. To our knowledge, our study demonstrates for the first time that both the Sox17 and GATA4 genes are epigenetically regulated in CD patients and the methylation levels of these genes in samples from CD patients. Although we found that promoter of TSGs is frequently hypermethylated in CD patients, we could not implicate the clinical relevance between promoter methylation of TSGs and CD progression.
Acknowledgments
We would like to thank Prof. Khadijah A. Mitchell of the Lafayette College for critically reading the manuscript and providing language editing.
Footnotes
FINANCIAL SUPPORT
This study was supported by the 2017 Inje University Research Grant.
CONFLICT OF INTEREST
No potential conflict of interest relevant to this article was reported.
AUTHOR CONTRIBUTION
Conceptualization: Kim TO, Yi JM. Methodology: Han YK, Yi JM. Formal analysis: Han YK. Funding acquisition: Kim TO. Project administration: Yi JM. Visualization: Han YK, Yi JM. Writing - original draft: Yi JM. Writing - review and editing: Kim TO, Yi JM. Approval of final manuscript: all authors.
REFERENCES
- 1.Podolsky DK. Inflammatory bowel disease. N Engl J Med. 2002;347:417–429. doi: 10.1056/NEJMra020831. [DOI] [PubMed] [Google Scholar]
- 2.Rivas MA, Beaudoin M, Gardet A, et al. Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat Genet. 2011;43:1066–1073. doi: 10.1038/ng.952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Franke A, McGovern DP, Barrett JC, et al. Genome-wide metaanalysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 2010;42:1118–1125. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu JZ, van Sommeren S, Huang H, et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet. 2015;47:979–986. doi: 10.1038/ng.3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schuebel KE, Chen W, Cope L, et al. Comparing the DNA hypermethylome with gene mutations in human colorectal cancer. PLoS Genet. 2007;3:1709–1723. doi: 10.1371/journal.pgen.0030157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Esteller M. Epigenetic gene silencing in cancer: the DNA hypermethylome. Hum Mol Genet. 2007:16. doi: 10.1093/hmg/ddm018. Spec No 1:R50-R59. [DOI] [PubMed] [Google Scholar]
- 8.Shen L, Kondo Y, Guo Y, et al. Genome-wide profiling of DNA methylation reveals a class of normally methylated CpG island promoters. PLoS Genet. 2007;3:2023–2036. doi: 10.1371/journal.pgen.0030181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robertson KD. DNA methylation and human disease. Nat Rev Genet. 2005;6:597–610. doi: 10.1038/nrg1655. [DOI] [PubMed] [Google Scholar]
- 10.Maeda O, Ando T, Watanabe O, et al. DNA hypermethylation in colorectal neoplasms and inflammatory bowel disease: a mini review. Inflammopharmacology. 2006;14:204–206. doi: 10.1007/s10787-006-1540-6. [DOI] [PubMed] [Google Scholar]
- 11.Tahara T, Shibata T, Nakamura M, et al. Effect of MDR1 gene promoter methylation in patients with ulcerative colitis. Int J Mol Med. 2009;23:521–527. doi: 10.3892/ijmm_00000160. [DOI] [PubMed] [Google Scholar]
- 12.Heyn H, Esteller M. DNA methylation profiling in the clinic: applications and challenges. Nat Rev Genet. 2012;13:679–692. doi: 10.1038/nrg3270. [DOI] [PubMed] [Google Scholar]
- 13.Kang K, Bae JH, Han K, Kim ES, Kim TO, Yi JM. A genomewide methylation approach identifies a new hypermethylated gene panel in ulcerative colitis. Int J Mol Sci. 2016;17:E1291. doi: 10.3390/ijms17081291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim TO, Park J, Kang MJ, et al. DNA hypermethylation of a selective gene panel as a risk marker for colon cancer in patients with ulcerative colitis. Int J Mol Med. 2013;31:1255–1261. doi: 10.3892/ijmm.2013.1317. [DOI] [PubMed] [Google Scholar]
- 15.Bae JH, Park J, Yang KM, Kim TO, Yi JM; IBD Study Group of Korean Association for Study of Intestinal Diseases (KASID) Detection of DNA hypermethylation in sera of patients with Crohn’s disease. Mol Med Rep. 2014;9:725–729. doi: 10.3892/mmr.2013.1840. [DOI] [PubMed] [Google Scholar]
- 16.Hanauer SB. Inflammatory bowel disease: epidemiology, pathogenesis, and therapeutic opportunities. Inflamm Bowel Dis. 2006:12–Suppl 1:S3-S9. doi: 10.1097/01.mib.0000195385.19268.68. [DOI] [PubMed] [Google Scholar]
- 17.Bernstein CN, Blanchard JF, Kliewer E, Wajda A. Cancer risk in patients with inflammatory bowel disease: a populationbased study. Cancer. 2001;91:854–862. doi: 10.1002/1097-0142(20010215)91:4<854::aid-cncr1073>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 18.Bernstein CN, Blanchard JF, Rawsthorne P, Wajda A. Epidemiology of Crohn’s disease and ulcerative colitis in a central Canadian province: a population-based study. Am J Epidemiol. 1999;149:916–924. doi: 10.1093/oxfordjournals.aje.a009735. [DOI] [PubMed] [Google Scholar]
- 19.Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. 2009;30:1073–1081. doi: 10.1093/carcin/bgp127. [DOI] [PubMed] [Google Scholar]
- 20.Rollin J, Iochmann S, Bléchet C, et al. Expression and methylation status of tissue factor pathway inhibitor-2 gene in nonsmall-cell lung cancer. Br J Cancer. 2005;92:775–783. doi: 10.1038/sj.bjc.6602298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gerecke C, Scholtka B, Löwenstein Y, et al. Hypermethylation of ITGA4, TFPI2 and VIMENTIN promoters is increased in inflamed colon tissue: putative risk markers for colitis-associated cancer. J Cancer Res Clin Oncol. 2015;141:2097–2107. doi: 10.1007/s00432-015-1972-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Glöckner SC, Dhir M, Yi JM, et al. Methylation of TFPI2 in stool DNA: a potential novel biomarker for the detection of colorectal cancer. Cancer Res. 2009;69:4691–4699. doi: 10.1158/0008-5472.CAN-08-0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hibi K, Goto T, Kitamura YH, et al. Methylation of the TFPI2 gene is frequently detected in advanced gastric carcinoma. Anticancer Res. 2010;30:4131–4133. [PubMed] [Google Scholar]
- 24.Sun FK, Fan YC, Zhao J, et al. Detection of TFPI2 methylation in the serum of hepatocellular carcinoma patients. Dig Dis Sci. 2013;58:1010–1015. doi: 10.1007/s10620-012-2462-3. [DOI] [PubMed] [Google Scholar]
- 25.Kisiel JB, Yab TC, Taylor WR, et al. Stool DNA testing for the detection of pancreatic cancer: assessment of methylation marker candidates. Cancer. 2012;118:2623–2631. doi: 10.1002/cncr.26558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dong Y, Tan Q, Tao L, et al. Hypermethylation of TFPI2 correlates with cervical cancer incidence in the Uygur and Han populations of Xinjiang, China. Int J Clin Exp Pathol. 2015;8:1844–1854. [PMC free article] [PubMed] [Google Scholar]
- 27.Rasmussen SL, Krarup HB, Sunesen KG, Pedersen IS, Madsen PH, Thorlacius-Ussing O. Hypermethylated DNA as a biomarker for colorectal cancer: a systematic review. Colorectal Dis. 2016;18:549–561. doi: 10.1111/codi.13336. [DOI] [PubMed] [Google Scholar]
- 28.Hibi K, Goto T, Shirahata A, et al. Detection of TFPI2 methylation in the serum of colorectal cancer patients. Cancer Lett. 2011;311:96–100. doi: 10.1016/j.canlet.2011.07.006. [DOI] [PubMed] [Google Scholar]
- 29.Sohn J, Natale J, Chew LJ, et al. Identification of Sox17 as a transcription factor that regulates oligodendrocyte development. J Neurosci. 2006;26:9722–9735. doi: 10.1523/JNEUROSCI.1716-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matsui T, Kanai-Azuma M, Hara K, et al. Redundant roles of Sox17 and Sox18 in postnatal angiogenesis in mice. J Cell Sci. 2006;119(Pt 17):3513–3526. doi: 10.1242/jcs.03081. [DOI] [PubMed] [Google Scholar]
- 31.Park KS, Wells JM, Zorn AM, Wert SE, Whitsett JA. Sox17 influences the differentiation of respiratory epithelial cells. Dev Biol. 2006;294:192–202. doi: 10.1016/j.ydbio.2006.02.038. [DOI] [PubMed] [Google Scholar]
- 32.Kim I, Saunders TL, Morrison SJ. Sox17 dependence distinguishes the transcriptional regulation of fetal from adult hematopoietic stem cells. Cell. 2007;130:470–483. doi: 10.1016/j.cell.2007.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hulbert A, Jusue-Torres I, Stark A, et al. Early detection of lung cancer using DNA promoter hypermethylation in plasma and sputum. Clin Cancer Res. 2017;23:1998–2005. doi: 10.1158/1078-0432.CCR-16-1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oishi Y, Watanabe Y, Yoshida Y, et al. Hypermethylation of Sox17 gene is useful as a molecular diagnostic application in early gastric cancer. Tumour Biol. 2012;33:383–393. doi: 10.1007/s13277-011-0278-y. [DOI] [PubMed] [Google Scholar]
- 35.Jia Y, Yang Y, Liu S, Herman JG, Lu F, Guo M. SOX17 antagonizes WNT/beta-catenin signaling pathway in hepatocellular carcinoma. Epigenetics. 2010;5:743–749. doi: 10.4161/epi.5.8.13104. [DOI] [PubMed] [Google Scholar]
- 36.Fu DY, Wang ZM, et al. Sox17, the canonical Wnt antagonist, is epigenetically inactivated by promoter methylation in human breast cancer. Breast Cancer Res Treat. 2010;119:601–612. doi: 10.1007/s10549-009-0339-8. [DOI] [PubMed] [Google Scholar]
- 37.Zhang W, Glöckner SC, Guo M, et al. Epigenetic inactivation of the canonical Wnt antagonist SRY-box containing gene 17 in colorectal cancer. Cancer Res. 2008;68:2764–2772. doi: 10.1158/0008-5472.CAN-07-6349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–480. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 39.Gao X, Sedgwick T, Shi YB, Evans T. Distinct functions are implicated for the GATA-4, -5, and -6 transcription factors in the regulation of intestine epithelial cell differentiation. Mol Cell Biol. 1998;18:2901–2911. doi: 10.1128/mcb.18.5.2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Akiyama Y, Watkins N, Suzuki H, et al. GATA-4 and GATA-5 transcription factor genes and potential downstream antitumor target genes are epigenetically silenced in colorectal and gastric cancer. Mol Cell Biol. 2003;23:8429–8439. doi: 10.1128/MCB.23.23.8429-8439.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guo M, Akiyama Y, House MG, et al. Hypermethylation of the GATA genes in lung cancer. Clin Cancer Res. 2004;10:7917–7924. doi: 10.1158/1078-0432.CCR-04-1140. [DOI] [PubMed] [Google Scholar]
- 42.Guo M, House MG, Akiyama Y, et al. Hypermethylation of the GATA gene family in esophageal cancer. Int J Cancer. 2006;119:2078–2083. doi: 10.1002/ijc.22092. [DOI] [PubMed] [Google Scholar]
- 43.Wakana K, Akiyama Y, Aso T, Yuasa Y. Involvement of GATA4/-5 transcription factors in ovarian carcinogenesis. Cancer Lett. 2006;241:281–288. doi: 10.1016/j.canlet.2005.10.039. [DOI] [PubMed] [Google Scholar]
- 44.Suzuki H, Watkins DN, Jair KW, et al. Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nat Genet. 2004;36:417–422. doi: 10.1038/ng1330. [DOI] [PubMed] [Google Scholar]
- 45.Cheng YY, Yu J, Wong YP, et al. Frequent epigenetic inactivation of secreted frizzled-related protein 2 (SFRP2) by promoter methylation in human gastric cancer. Br J Cancer. 2007;97:895–901. doi: 10.1038/sj.bjc.6603968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Su HY, Lai HC, Lin YW, et al. Epigenetic silencing of SFRP5 is related to malignant phenotype and chemoresistance of ovarian cancer through Wnt signaling pathway. Int J Cancer. 2010;127:555–567. doi: 10.1002/ijc.25083. [DOI] [PubMed] [Google Scholar]
- 47.Fukui T, Kondo M, Ito G, et al. Transcriptional silencing of secreted frizzled related protein 1 (SFRP 1) by promoter hypermethylation in non-small-cell lung cancer. Oncogene. 2005;24:6323–6327. doi: 10.1038/sj.onc.1208777. [DOI] [PubMed] [Google Scholar]
- 48.Nojima M, Suzuki H, Toyota M, et al. Frequent epigenetic inactivation of SFRP genes and constitutive activation of Wnt signaling in gastric cancer. Oncogene. 2007;26:4699–4713. doi: 10.1038/sj.onc.1210259. [DOI] [PubMed] [Google Scholar]