

Abstract
Duchenne Muscular Dystrophy is a devastating disease caused by the absence of a functional rod-shaped cytoplasmic protein called dystrophin. Several avenues are being developed aimed to restore dystrophin expression in boys affected by this X-linked disease. However, its complete cure is likely to need combinational approaches which may include regimes aimed at restoring muscle mass. Augmenting muscle growth through the manipulation of the Myostatin/Activin signalling axis has received much attention. However, we have recently shown that while manipulation of this axis in wild type mice using the sActRIIB ligand trap indeed results in muscle growth, it also had a detrimental impact on the testis. Here we examined the impact of administering a powerful Myostatin/Activin antagonist in two mouse models of Duchenne Muscular Dystrophy. We report that whilst the impact on muscle growth was not always positive, both models showed attenuated testis development. Sperm number, motility and ultrastructure were significantly affected by the sActRIIB treatment. Our report suggests that interventions based on Myostatin/Activin should investigate off-target effects on tissues as well as muscle.
Key Words: Duchenne Muscular Dystrophy, Mdx, Muscle hypertrophy, Testis, Activin, Myostatin
Ethical Publication Statement
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Duchenne Muscular Dystrophy (DMD) is a genetic disease caused by the absence of a protein called Dystrophin.1 The dystrophin locus is found on the X-chromosome, hence the disease manifests mainly in boys. It is a lethal disease characterised by progressive muscle weakness and wasting. As well as muscle damage and necrosis, a multitude of other cellular and tissue abnormalities play a role in the development of the pathology including inflammation, fibrosis and abnormal adiposity. Several treatments based on addressing mutations in the dystrophin gene mutations have/are being developed, including exon skipping and viral gene delivery therapies.2 The goal of most of the therapies is to modify DMD into a less severe disease called Becker Muscular Dystrophy. It is more than likely that combinational therapies (comprising gene correction strands and approaches to remedy abnormal tissues) are going to be needed to fully treat DMD/BMD. A major feature of both DMD and BMD is muscle wasting and contraction-induced damage.3 Several approaches are being developed to promote myogenesis for neuromuscular diseases and many of them focus on manipulating Myostatin signalling.4 Myostatin is a member of the TGF-β family of secreted proteins and is a potent inhibitor of muscle development; genetic deletion in the mouse genome leads to massive degree of muscle enlargement mediated by the development of supernumerary muscle fibres (hyperplasia) as well as fibre enlargement (hypertrophy).5 Myostatin and other closely related molecules including Activin activate intracellular signalling cascades by interacting with heterotetrameric receptor complex comprised of two Type I and two Type II receptors located on the cell surface. Myostatin/Activin signalling preferentially utilises the Type I receptors ALK4 or ALK7 and the Type II receptors ACVR2A or ACVR2B (henceforth ActRIIA or ActRIIB, respectively).6 We and others have developed a ligand trap called the sActRIIB (capable of binding Myostatin and Activin) made of a stabilised form of the extracellular portion of the ActRIIB receptor and shown that it can promote considerable muscle growth (20-30%) in rodents in a relatively short period of time (4 weeks).7 However, concerns stemming from the broad ligand binding nature of sActRIIB and the numerous tissue processes controlled by Activin remain, which could lead to unwanted side effects of its use to promote muscle development. Evidence to support such apprehensions have been reported in studies in non-human primates in which changes to visceral organs (pancreas and spleen) developed following sActRIIB treatment.8 More recently we have reported that sActRIIB had a profound effect on the development of the mouse testis (9). We showed that muscle development promoted by sActRIIB was accompanied by a decrease in testis growth and furthermore that the beneficial effects of muscle growth were lost after the cessation of sActRIIB treatment, but the impairment in testis structure persisted.9 As treatments for DMD are being developed using molecules like sActRIIB, we investigated the impact of such an intervention in two mouse models for this disease. Here we show that although sActRIIB had differing degree of effects on muscle enlargement on the two mouse models of DMD, both showed abnormal testis development. Importantly we show that sActRIIB dramatically reduced sperm number as well as inducing numerous ultrastructural abnormalities.
Materials and Methods
Ethical approval
The experiments were performed under a United Kingdom Home Office project license, in agreement with the Animals (Scientific Procedures) Act 1986. The University of Reading Animal Care and Ethical Review Committee approved all procedures. Animals were humanely sacrificed via Schedule 1 killing.
Animal maintenance
All mice used were bred and maintained in accordance to the Animals (Scientific Procedures) Act 1986 (UK) and approved by the Biological Resources Unit of University of Reading. Post-natal Myostatin/Activin block was induced via intraperitoneal (IP) injection with 10mg/kg of sActRIIB-Fc (hereafter sActRIIB) twice weekly. The age of initial injection varied among different studies. All mice were obtained from Jackson laboratories and bred for pups.
Histological analysis and immunohistochemistry
Following dissection, testes were weighed and fixed in 4% PFA prior to 4°C storage. PFA fixed testis were embedded in paraffin following a series of ethanol and histoclear stages. 8μm sections were cut using a Leica RM2155 rotary microtome prior to dewaxing in Xylene. Rehydration with ethanol stages was then performed prior to antigen retrieval in 0.01M citrate buffer pH6. Blocking buffer (10% FBS in TBS-Tx) was incubated on sections for 45 minutes at room temperature prior to incubation with primary antibody overnight at 4°C. Sections were subsequently washed in PBS-Tx three times prior to incubation with secondary antibody. Secondary antibodies attached to a fluorophore were used for detection of all antibodies except PLZF and PCNA which were detected using the Vectastain ABC-HRP kit with a Vector NOVARed HRP substrate. All primary antibodies being detected were subsequently incubated with a secondary antibody for 1hour at room temperature prior to detection. Details of primary and secondary antibodies are given in Table 1.
Table 1.
Antibody details
| Primary antibodies Antigen | Species | Dilution | Supplier |
|---|---|---|---|
| PCNA | Mouse | 1:200 | Cell signalling Technology #25865 |
| PLZF (D-9) | Mouse | 1:250 | Santa Cruz Biotechnology #sc-28319 |
| Stra8 | Rabbit | 1:1000 | Abcam ab49405 |
| Sox9 | Rabbit | 1:1000 | Abcam ab185966 |
| AQP3 | Rabbit | 1:800 | Abcam ab125219 |
| Secondary antibodies | Species | Dilution | Supplier |
| AlexaFluor 488 anti-mouse | Goat | 1:200 | Invitrogen |
| Rabbit anti mouse IgG HRP | Rabbit | 1:200 | DAKO #PO260 |
| Goat anti-rabbit IgG HRP | Goat | 1:5000 | ThermoFisher scientific #65-6120 |
Hematoxylin and eosin
Testes sections were dewaxed in xylene prior to rehydration in ethanol stages. Harris haematoxylin solution (Sigma HHS16) was then incubated on the slides for 2 minutes prior to acidic alcohol and tap water for blueing. Eosin solution (Sigma-Aldrich 318906) was incubated on sections for 2 minutes prior to dehydration in ethanol and clearing with xylene. Slides were mounted with a coverslip and DPX mounting media.
Fig 1.

sActRIIB causes a reduction in testes weight and tubule areas in the Mdx mouse model of Duchenne Muscular Dystrophy. (A) Schematic of experimental design. Black arrows injection of 10mg/kg sActRIIB. Red arrows indicate time of cull. (B) Body weight at P69. (C) Gastrocnemius mass. (D) Teste mass. (E) Average tubule area. (F) Average lumen area. (G) Average differentiation thickness. (H) H&E of seminiferous tubule. Immunostaining for (I) PCNA, (J) PLZF (K) Stra8 (L) Sox9 (M) AQP3. Quantification of (N) PCNA, (O) PLZF (P) Stra8 (Q) Sox9 positive cells per tubule. (R) Quantification of AQP3 positive tubules as a percentage of total tubules. n=3 PBS treated male mdx mice, n=3 sActRIIB treated male Mdx mice. Scale for all images represents 50μm. *p<0.05, ***<0.001.
Sperm motility studies
70μl G-IVF PLUS (Vitrolife) was pipetted in two areas on to 35mm IVF Petri Dish (Thermo Scientific) and covered with mineral oil (Sigma-Aldrich M8410) prior to incubation at 37°C 5% CO2 for a minimum of 30 minutes prior to sperm isolation. Following schedule 1 killing, epididymis was dissected attached to the testes and placed in pre-warmed M2 medium (Sigma Aldrich). The epididymis was placed in to 70μl G-IVF PLUS bubble and cut to release the live spermatozoa. The cut epididymis was incubated at 37°C in 5% CO2 for 40 minutes prior to 50μl being transferred to the second bubble of G-IVF PLUS media. Spermatozoa were imaged using Nikon Eclipse Ti-E Inverted Microscope at 10x magnification with 3050 frames per minute captured. Analyses of spermatozoa speeds were determined using the manual tracking plugin in FIJI. Sperm concentrations were determined by fixing spermatozoa in 4% PFA prior to placing 10μl under a haemocytometer and imaged at 20x magnification.
Transmission electron microscopy
For transmission electron microscopy, sperms were centrifuged and the resulting pellet embedded in 2% agarose. Afterwards, samples were contrasted using 1% osmium tetroxide (in PBS, 45 minutes at RT) and 1% uranyl acetate (45 minutes at RT in 70% ethanol). Agar blocks were dehydrated and embedded in epoxy resin (Durcupan, Sigma, Germany). Ultrathin sections were prepared using a Leica UC6 ultra-microtome and analysed using a Philipps CM100 TEM.
Statistical analysis
Data are presented as mean +/- SE. Significant differences between two groups were performed by the Student’s t-test for independent variables. Statistical analysis was performed on GraphPad Prism 5 with statistically significant differences considered at P<0.05.
Results
Impact of attenuation of Activin Receptor IIB signalling on the development of skeletal muscle and the testis of Mdx and DBA/2-Mdx mice
We injected sActRIIB into Mdx mice from just after the onset of muscle necrosis (p21) until a stabilisation phase (p56) and collected tissues at p69 (Figure 1 A). There was no significant change in body weight induced by sActRIIB treatment (Figure 1 B). However, we found that sActRIIB had a profound impact on muscle growth, epitomized by the 36% increase in gastrocnemius mass (Figure 1 C). During the course of tissue recovery, we noticed that the testes were smaller in the sActRIIB treated group compared to those receiving vehicle (PBS) (Figure 1 D). In the treated Mdx mouse they were 17% lighter than in the untreated group. Thereafter we performed a histological and immunocytochemical analysis to identify the cellular and molecular changes underpinning the change in testis weight. In both the untreated and treated Mdx mice, the seminiferous tubules were readily identifiable with expected peritubular, basal, adluminal cellular distribution and lumen structures. Examination of the H and E images showed that the size of seminiferous tubules was smaller in the treated Mdx cohort which was substantiated following quantitative analysis (Figure 1 E and H). Lumen area was smaller in sActRIIB treated samples than those of controls (Figure 1 F). The area of the cellular component of each tubule was measured and revealed that it was decreased by sActRIIB treatment (Figure 1 G). We next examined if the reduction in seminiferous tubule cross-section was caused by a decrease in cell proliferation. Visualisation of nuclear antigen (PCNA) expression, a marker for cell proliferation, demonstrated that these events were taking place adjacent to the basement membrane in both treated and untreated Mdx samples. Nevertheless, there were significantly fewer proliferating cells in the tubules of sActRIIB treated samples than in Mdx controls (Figure 1 I and N). Next, we quantified the number of spermatogonial stem cells (SSC) using the marker promyelocytic leukemia zinc finger (PLZF) since they give rise to the proliferating A and B type spermatogonial cells located within tubules.10 However, we were unable to detect any significant difference in the number of SSC per tubule between the two cohorts (Figure 1 J and O). Furthermore, we did not detect any difference in the ability of preleptotene spermatocyte to enter meiosis as demonstrated by Stimulated by Retinoic Acid 8 (Stra8) staining (Figure 1 K and P).11 Lastly, we quantified the number of Sertoli cells, identified by Sox9 expression,12 which supports and thus ultimately limits the extent of spermatogenesis in each tubule and found no differences in this parameter between the sActRIIB treated Mdx group and untreated mice(Figure 1 L and Q). Importantly, the number of sperm tails (as detected either through H and E staining or immunostaining with sperm tail specific markers including Aquaporin3 (AQP3)13 was significantly reduced by sActRIIB treatment (Figure 1 M and R). Of note, there was no effect on Leydig cell numbers by sActRIIB treatment (data not shown). Although the mdx mouse has been extensively used for over 30 years since its full molecular characterization,14 its pathology does not fully mirror the human condition. In recent years a more severe mouse model for Duchenne Muscle Dystrophy called the DBA/2-Mdx mouse has been developed as this strain displays a progressive dystrophy with greater degree of fibrosis and less muscle regeneration than the traditional Mdx mouse.15,16 We therefore examined the impact of sActRIIB on muscle and testis mass in the DBA/2-Mdx mouse. Surprisingly we found that an identical regime of sActRIIB treatment in DBA/2-Mdx as the Mdx mouse resulted in a decrease in body mass (Figure 2 A-B). Interestingly, gross muscle mass was not increased by sActRIIB (Figure 2 C) but the mass of the testis was decreased (Figure 2 D). In accordance to our finding in the Mdx mouse, sActRIIB had a profound effect on the seminiferous tubules in DBA/2-Mdx; with decreased in tubule as well as lumen areas and decreased in differentiation thickness (Figure 2 E-G). Examination of key markers impacting on testis structure revealed that proliferation was attenuated as was the development of sperm tail (Figure 2 I and O and M and R). Identical to Mdx mice, treatment of DBA/2-Mdx with sActRIIB had no effect on spermatogonial stem cells, meiosis or Sertoli cells (Figure 2 J-Q). Next, we assessed the impact of sActRIIB treatment on both the quantity and quality of mature sperm in the two genetic models of DMD. Quantification of sperm number revealed a decrease in sperm concentration of 62% in Mdx and 70% in DBA/2-Mdx (Figure 3 A-B). Sperm motility was also reduced by sActRIIB treatment in both mouse models (Figure 3 C-D). Ultra-structural sperm analysis in PBS treated Mdx and DBA/2-Mdx mice displayed normal spermatozoa with regular heads including elongated nuclei and acrosomes (Figure 4 A and F), longitudinal and cross-sections of midpieces (Figure 4 A) including mitochondrial sheaths as well as tails (Figure 4 A) with regular axoneme. sActRIIB treatment in Mdx mice leads to a significant reduction of normal spermatozoa (Figure 4 E). Instead, we found a lot of cell debris (Figure 4 B, C), cytoplasm rich round cells (Figure 4 B-C) and large cells with atypical nuclear shape and doubling of organelles (Figure 4D). DBA/2-Mdx mice sActRIIB treatment resulted in a comparable phenotype. Only very few normal spermatozoa were found. Instead, cytoplasm rich pleomorphic (Figure 4 G, I and J) and round cells with malformed cell organelles (Figure 4 G-H) e.g. nuclei (Figure 4 H, I and J) were detected.
Fig 2.
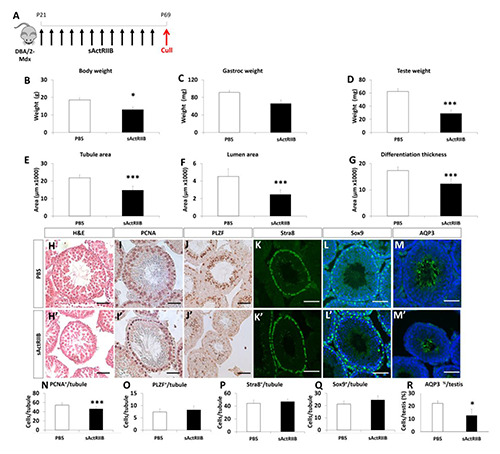
sActRIIB causes a reduction in testes weight and tubule areas in the DBA/2-Mdx mouse model of Duchenne Muscular Dystrophy. (A) Schematic of experimental design. Black arrows injection of 10mg/kg sActRIIB. Red arrows indicate time of cull. (B) Body weight at P69. (C) Gastrocnemius mass. (D) Teste mass. (E) Average tubule area. (F) Average lumen area. (G) Average differentiation thickness. (H) H&E of seminiferous tubule. Immunostaining for (I) PCNA, (J) PLZF (K) Stra8 (L) Sox9 (M) AQP3. Quantification of (N) PCNA, (O) PLZF (P) Stra8 (Q) Sox9 positive cells per tubule. (R) Quantification of AQP3 positive tubules as a percentage of total tubules. n=5 PBS treated male DBA/2-Mdx mice, n=5 sActRIIB treated male DBA/2-Mdx mice. Scale for all images represents 50μm. *p<0.05, ***<0.001.
Fig 3.
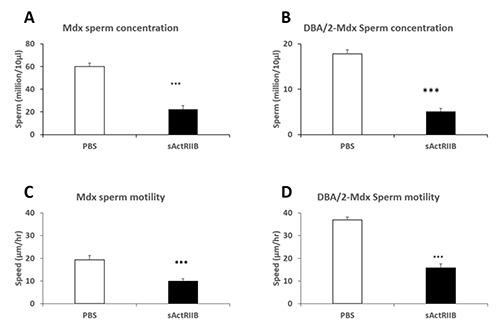
sActRIIB causes a reduction in sperm concentration and speed in the Mdx and DBA/2-Mdx mouse model of Duchenne Muscular Dystrophy. (A) Sperm concentration in the Mdx mouse model. (B) Sperm concentration in the DBA/2-Mdx mouse model. (C) Sperm speed in the Mdx mouse model. (D) Sperm speed in the DBA/2-Mdx mouse model. n=3 PBS treated male DBA/2-Mdx mice, n=3 sActRIIB treated male DBA/2-Mdx mice. *p<0.05, ***<0.001.
Discussion
Regimes to promote muscle growth are likely to feature in combinational therapies that aim to cure patients with neuromuscular diseases. However, it is likely they will need careful optimisation before they can reach their intended milestones due in many cases to factors that cause the disease. One recent example was highlighted in an elegant study which showed that the muscle growth properties of myostatin inhibition could be effectively eliminated by glucocorticoids, drugs that are often used to control inflammation in diseases including DMD. Here we highlight the need to optimise the use of muscle growth regimes due to side effects on the development of the testis. This study adds to a growing body of evidence that Myostatin/Activin neutralisation impacts on non-muscle tissues exemplified by a clinical trial in patients with DMD of the soluble Activin ligand trap ACE-031 which was halted due to telangiectasias and epistaxis.17
Here we show that sActRIIB had a profound impact on testis mass and both quantitatively and qualitatively on sperm in two mouse models of DMD; sActRIIB decreased the number of sperm and those that formed often had abnormal structure, including duplication of mid-piece organelles. Interestingly the sActRIIB did not have the same impact on muscle mass in both models; Mdx showed an increase in this parameter whereas DBA/2-Mdx failed to respond to any significant levels. However, this result may be misleading since the DBA/2-Mdx muscle is very fibrotic and contain a substantial osteogenic component.15. Myostatin as well as Activin are known to promote fibrosis and therefore it is possible that following sActRIIB treatment, there is a concurrent decrease in fibrosis and increase in muscle content.18 Indeed, a detailed examination of the structure of sActRIIB treated DBA/2-Mdx muscle supports this thinking (manuscript in preparation).
Fig 4.

Ultra-structure of sperm from mouse models of DMD. Mdx sperm treated with PBS show normal head structure with elongated nuclei and acrosomes (asterisk), midpieces (longitudinal-section (red arrow) and cross-sections (blue arrows)) mitochondrial sheaths and tail segments (arrowhead). (B-E) Mdx treated with sActRIIB. (E) A few normal sperm with usually elongated nuclei (asterisk) and midpiece (red arrow). (B-C) Presence of cell debris was prominent (arrow heads) and cells with large cytoplasmic component (red arrowheads). (D) Sperm with atypical nuclear shape and doubling of organelles (asterisk and arrows). (F) DBA/2-Mdx sperm treated with PBS show normal head structure with elongated nuclei and acrosomes (asterisk), longitudinal (red arrow) and tails (arrowhead). (G-H) DBA/2-Mdx treated with sActRIIB. Large pleomorphic cells filled with cytoplasm (G, I, J) and (H) round cells with malformed organelle (arrow) and nuclei (H, I, J asterisks).
In some ways, sActRIIB acts as endogenous ligand traps for Myostatin and Activin such as Follistatin (FST) and Follistatin-like 3 (FSTL3, also identified as Follistatin Related Protein or Follistatin Related Gene,19,20 which sequester the ligands and prevent them from interacting with the membrane bound signalling receptors.21 The biochemistry, and associated biology, of these two molecules reveal a large degree of similarity as well as crucial differences. Importantly, in both human and mouse serum, Myostatin was found to be in a complex with FSTL3 and not FST.22 Moreover, the expression pattern of these two ligand traps are different and of the two FSTL3 is more relevant to the testis.23 Importance of FSTL3 in testicular biology is highlighted by in vivo models. Overexpression using a transgenic approach reduces testis size and sperm count,24 mirroring sActRII injection studies. Contrastingly, FSTL3 deletion increases testicular size and prolongs the span spermatogenesis.25 Finally, both FST and FSTL3 mRNA and protein increases in response to Activin,26 setting up a feedback inhibitory loop for activin action. Given that, sActRIIB reduces Myostatin/Activin signalling and therefore FSTL3 expression is not likely to increase in the testis in this model, hence the effect of sActRIIB on inhibiting testicular development and function is also not likely due to the induction or overexpression of FSTL3. Many of the side effects of ligand trap treatments could possibly be attributed to the ability of the molecule to interact with proteins other than Myostatin and Activin and the specific model in which the they are deployed. For example, the incidence of bleeding attributed to ACE-031 could be due to its interaction with BMP9, which has been shown to regulate endothelial cell function.27 It is intriguing that ACE-031 induced bleeding in DMD boys whereas no bleeding issues were noted in our use of sActRIIB in mice. We suggest that each molecule, due to specific (and potentially proprietorial) modifications may harbour unique ligand binding kinetics which implies that each iteration should be experimentally evaluated in individual target organisms rather than relying on anecdotal data. Alternatively, the side-effects could be attributed to interfering with the large number of processes that are regulated by Activin. One would predict that any mechanism that delivers the same outcome of robust dual Myostatin/Activin inhibition could also lead to unwanted side effects. In this respect, one would that regimes used to block the ActRIIB receptor (e.g. Bimagrumab)28 to be equivalent to the sActRIIB molecule used in this study, with the same outcomes in terms of tissue development. However the effects we report on the testis were not observed or reported following the use of Bimagrumab.29 We suggest that in future the side-effects could be limited by either i) using lower doses of the Myostatin/Activin blocker, ii) using a targeted approach that neutralises Myostatin and Activin only in the circulation or iii) targeting either only Myostatin or Activin but not both. We believe for muscle wasting therapies, it may not be necessary to use the high doses of sActRIIB which are used in experimental work since the intended outcomes are different. In the former, the point is to stop muscle mass from decreasing, whereas in the latter proof of principle requires robust muscle growth, typically 30-50% in two weeks. The second point relates to targeting the molecules that need to be neutralised while sparing those performing essential functions. It is worth drawing parallels from the work conducted using Follistatin to promote muscle growth. The group of Mendell has specifically deployed a form of Follistatin (FS315) to neutralise circulating Myostatin/Activin while sparing molecules working in the extracellular matrix (ECM) which results in significant muscle growth but no issues related to the reproductive system.30 Our study induced both a muscle phenotype and one related to the reproductive system. This implies that sActRIIB can neutralise both the circulating ligands and those associated with the ECM. Future efforts should be made to modify sActRIIB’s ability to interact with the ECM. The last point relates to targeting Myostatin alone as opposed to both Myostatin and Activin. This will undoubtedly be less potent in terms of muscle growth. However, it may be adequate when the desired outcome is the prevention of muscle wasting, rather than promotion of growth.
The results presented in this study are supported by likely on-target effect for an intervention that inhibits Activin.31 To our knowledge, no other study has reported a similar undesired side-effect of using a similar molecule. Indeed, many studies fail to mention whether this organ was ever investigated. Given the severity of sActRIIB phenotype on the testis in the two mouse models of DMD, we suggest that it is essential that they should be studied not only in clinical trials but also in other model systems when a similar regime is being deployed.
Our study counsels caution to the use of robust anti-myostatin based therapies developed to promote muscle growth, especially those that simultaneously neutralise the activity of both Activin and Myostatin. Furthermore, we note that other non-skeletal muscle conditions are being targeted using molecules almost identical to that used here. For example, an ActRIIB ligand trap called ACE-536 which is marketed under the tradename Reblozyl for the treatment of anaemia patients with beta thalassemia.32 This molecule has been shown to be a potent binder of both Myostatin and GDF-11(32). However, recent work suggests that the ability of ACE-536 to promote terminal erythropoiesis may be independent of GDF-11 binding.33 We suggest that long term use of this molecule should be tested first in animal models for its impact on testis development.
In summary, we show in two mouse models of DMD that attenuation of Myostatin/Activin signalling using sActRIIB leads to a testicular phenotype which impacts on the structure of the organ as well as the quantity and quality of sperm. A debate needs to be initiated if the benefits in terms of muscle growth outweigh this and possible other side-effects when using a molecule for the treatment of young boys with DMD.
Acknowledgments
Authors are grateful for the Editorial Office of European Journal of Translational Myology guidance during the publication of this paper.
List of acronyms
- ACE-031
Acceleron 031 protein
- ACE-536
Acceleron 536 protein
- Activin Receptor IIB ActRIIA
Actvin receptor IIA
- ActRIIB
Actvin receptor IIB
- ACVR2A
Actvin receptor 2A
- ACVR2B
Actvin receptor 2B
- AQP3
Aquaporin 3
- DMD
Duchenne Muscular Dystrophy
- DPX
Dibutylphthalate Polystyrene Xylene
- ECM
extra cellular matrix
- FSTL3
Follistatin-like 3
- GDF11
Growth and differentiation factor 11
- NOVARed HRP
NOVA Red Horse Radish Peroxidase
- PBS-Tx
Phosphate buffered Salines with Triton X100
- PCNA
Proliferating cell nuscler antigen
- PLZF
Promyelocytic Leukemia Zinc Finger
- RT
room temperature
- sActRIIB-Fc
sActRIIB
- Sigma HHS16
Harris haematoxylin solution 16
- Sox9
SRY Box transcription factor 9
- Stra8
Stimulated By Retinoic Acid 8
- TGFβ
Transforming Growth Factor beta
Funding Statement
Funding: This work was supported by the Biotechnology and Biological Sciences Research Council (Grants BB/J016454/1 to HCH and KP and BB/M014878/1 to DV). AA acknowledges a PhD studentship from the Office of the Assistant Military Attaché for Training at the Embassy of the State of Kuwait in London. TBH was supported by the DFG (CRC 1192, CRC1140, CRC 992), by the BMBF (01GM1518C), by the European Research Council-ERC (grant 616891) and by the H2020-IMI2 consortium BEAt-DKD (Innovative Medicines Initiative 2 Joint Undertaking under grant agreement No 115974).
Contributor Information
Danielle Vaughan, Email: dani_vaughan@hotmail.com.
Oliver Kretz, Email: o.kretz@uke.de.
Ali Alqallaf, Email: a.alqallaf@pgr.reading.ac.uk.
Robert Mitchell, Email: robmitchell1@gmail.com.
Jennie L von der Heide, Email: jennie.vonderheide@gmx.de.
Sakthivel Vaiyapuri, Email: s.vaiyapuri@reading.ac.uk.
Antonios Matsakas, Email: Antonios.Matsakas@hyms.ac.uk.
Arja Pasternack, Email: arja.pasternack@helsinki.fi.
Henry Collins-Hooper, Email: henry_dintle@hotmail.com.
Olli Ritvos, Email: olli.ritvos@gmail.com.
Randy Ballesteros, Email: rballesteros@rvc.ac.uk.
Tobias B. Huber, Email: t.huber@uke.de.
Helge Amthor, Email: helge.amthor@handimedex.onmicrosoft.com.
Abir Mukherjee, Email: amukherjee@rvc.ac.uk.
References
- 1.Hoffman EP, Brown RH, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 1987;51:919-28. [DOI] [PubMed] [Google Scholar]
- 2.Falzarano MS, Scotton C, Passarelli C, Ferlini A. Duchenne Muscular Dystrophy: From Diagnosis to Therapy. Molecules 2015;20:18168-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Petrof BJ, Shrager JB, Stedman HH, et al. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc Natl Acad Sci U S A 1993;90: 3710-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patel K, Amthor H. The function of Myostatin and strategies of Myostatin blockade-new hope for therapies aimed at promoting growth of skeletal muscle. Neuromuscul Disord 2005;15:117-26. [DOI] [PubMed] [Google Scholar]
- 5.McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature 1997;387:83-90. [DOI] [PubMed] [Google Scholar]
- 6.Olsen OE, Wader KF, Hella H, et al. Activin A inhibits BMP-signaling by binding ACVR2A and ACVR2B. Cell Commun Signal 2015;13:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Relizani K, Mouisel E, Giannesini B, et al. Blockade of ActRIIB signaling triggers muscle fatigability and metabolic myopathy. Mol Ther 2014;22:1423-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Latres E, Mastaitis J, Fury W, et al. Activin A more prominently regulates muscle mass in primates than does GDF8. Nat Commun 2017;8:15153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vaughan D, Ritvos O, Mitchell, et al. Inhibition of Activin/Myostatin signalling induces skeletal muscle hypertrophy but impairs mouse testicular development. Eur J Transl Myol 2020;30:8737 DOI https://doi.org/10.4081/ejtm.2019.8737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Costoya JA, Hobbs RM, Barna M, et al. Essential role of Plzf in maintenance of spermatogonial stem cells. Nat Genet 2004;36:653-9. [DOI] [PubMed] [Google Scholar]
- 11.Zhou Q, Nie R, Li Y, et al. Expression of stimulated by retinoic acid gene 8 (Stra8) in spermatogenic cells induced by retinoic acid: an in vivo study in vitamin A-sufficient postnatal murine testes. Biol Reprod 2008;79(1):35-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morais da Silva S, Hacker A, Harley V, et al. Sox9 expression during gonadal development implies a conserved role for the gene in testis differentiation in mammals and birds. Nat Genet 1996;14:62-8. [DOI] [PubMed] [Google Scholar]
- 13.Chen Q, Peng H, Lei L, et al. Aquaporin3 is a sperm water channel essential for postcopulatory sperm osmoadaptation and migration. Cell Res 2011;21:922-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sicinski P, Geng Y, Ryder-Cook AS, et al. The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science 1989;244(4912):1578-80. [DOI] [PubMed] [Google Scholar]
- 15.Coley WD, Bogdanik L, Vila MC, et al. Effect of genetic background on the dystrophic phenotype in mdx mice. Hum Mol Genet 2016;25:130-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fukada S, Morikawa D, Yamamoto Y, et al. Genetic background affects properties of satellite cells and mdx phenotypes. Am J Pathol 2010;176:2414-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Campbell C, McMillan HJ, Mah JK, et al. Myostatin inhibitor ACE-031 treatment of ambulatory boys with Duchenne muscular dystrophy: Results of a randomized, placebo-controlled clinical trial. Muscle Nerve 2017;55:458-64. [DOI] [PubMed] [Google Scholar]
- 18.Li ZB, Kollias HD, Wagner KR. Myostatin directly regulates skeletal muscle fibrosis. J Biol Chem 2008;283:19371-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schneyer A, Tortoriello D, Sidis Y, et al. Follistatin-related protein (FSRP): a new member of the follistatin gene family. Mol Cell Endocrinol 2001;180:33-8. [DOI] [PubMed] [Google Scholar]
- 20.Maguer-Satta V, Bartholin L, Jeanpierre S, et al. Expression of FLRG, a novel activin A ligand, is regulated by TGF-beta and during hematopoiesis [corrected]. Exp Hematol 2001;29:301-8. [DOI] [PubMed] [Google Scholar]
- 21.Sidis Y, Mukherjee A, Keutmann H, et al. Biological activity of follistatin isoforms and follistatin-like-3 is dependent on differential cell surface binding and specificity for activin, myostatin, and bone morphogenetic proteins. Endocrinology 2006;147:3586-97. [DOI] [PubMed] [Google Scholar]
- 22.Hill JJ, Davies MV, Pearson AA, et al. The myostatin propeptide and the follistatin-related gene are inhibitory binding proteins of myostatin in normal serum. J Biol Chem 2002;277:40735-41. [DOI] [PubMed] [Google Scholar]
- 23.Tortoriello DV, Sidis Y, Holtzman DA, Holmes WE, Schneyer AL. Human follistatin-related protein: a structural homologue of follistatin with nuclear localization. Endocrinology 2001;142: 3426-34. [DOI] [PubMed] [Google Scholar]
- 24.Xia Y, Sidis Y, Schneyer A. Overexpression of follistatin-like 3 in gonads causes defects in gonadal development and function in transgenic mice. Mol Endocrinol 2004;18(4):979-94. [DOI] [PubMed] [Google Scholar]
- 25.Oldknow KJ, Seebacher J, Goswami T, et al. Follistatin-like 3 (FSTL3) mediated silencing of transforming growth factor beta (TGFbeta) signaling is essential for testicular aging and regulating testis size. Endocrinology 2013;154(3):1310-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bartholin L, Maguer-Satta V, Hayette S, et al. Transcription activation of FLRG and follistatin by activin A, through Smad proteins, participates in a negative feedback loop to modulate activin A function. Oncogene 2002;21:2227-35. [DOI] [PubMed] [Google Scholar]
- 27.Tillet E, Bailly S. Emerging roles of BMP9 and BMP10 in hereditary hemorrhagic telangiectasia. Front Genet 2014;5:456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lach-Trifilieff E, Minetti GC, Sheppard K, et al. An antibody blocking activin type II receptors induces strong skeletal muscle hypertrophy and protects from atrophy. Mol Cell Biol 2014;34:606-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garito T, Roubenoff R, Hompesch M, et al. Bimagrumab improves body composition and insulin sensitivity in insulin-resistant individuals. Diabetes Obes Metab 2018;20:94-102. [DOI] [PubMed] [Google Scholar]
- 30.Rodino-Klapac LR, Haidet AM, Kota J, et al. Inhibition of myostatin with emphasis on follistatin as a therapy for muscle disease. Muscle Nerve 2009;39:283-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dias V, Meachem S, Rajpert-De Meyts E, et al. Activin receptor subunits in normal and dysfunctional adult human testis. Hum Reprod 2008;23:412-20. [DOI] [PubMed] [Google Scholar]
- 32.Suragani RN, Cadena SM, Cawley SM, et al. Transforming growth factor-beta superfamily ligand trap ACE-536 corrects anemia by promoting late-stage erythropoiesis. Nat Med 2014;20(4):408-14. [DOI] [PubMed] [Google Scholar]
- 33.Guerra A, Oikonomidou PR, Sinha S, et al. Lack of Gdf11 does not improve anemia or prevent the activity of RAP-536 in a mouse model of beta-thalassemia. Blood. 2019;134(6):568-72. [DOI] [PMC free article] [PubMed] [Google Scholar]