

ABSTRACT
The diagnosis of multiple sclerosis (MS) is through clinical assessment and supported by investigations. There is no single accurate and reliable diagnostic test. MS is a disease of young adults with a female predominance. There are characteristic clinical presentations based on the areas of the central nervous system involved, for example optic nerve, brainstem and spinal cord. The main pattern of MS at onset is relapsing–remitting with clinical attacks of neurological dysfunction lasting at least 24 hours. The differential diagnosis includes other inflammatory central nervous system disorders. Magnetic resonance imaging of the brain and lumbar puncture are the key investigations. New diagnostic criteria have been developed to allow an earlier diagnosis and thus access to effective disease modifying treatments.
KEYWORDS: Multiple sclerosis, MS, neurology
Key points
The diagnosis of multiple sclerosis is a clinical diagnosis supported by investigation findings.
There is no single sensitive and specific diagnostic test for multiple sclerosis.
The principle of dissemination of lesions in time and space underpins the diagnosis.
Eighty-five per cent of people with multiple sclerosis have a relapsing-remitting course at onset.
New diagnostic criteria aim to allow an earlier, accurate diagnosis.
Introduction
Multiple sclerosis (MS) is an inflammatory demyelinating central nervous system (CNS) disease. Its onset is typically in adults with peak age at onset between 20–40 years. There is a female predominance of up to 3:1. The course of MS is relapsing–remitting (RRMS) at onset in 85% with episodes of neurological dysfunction followed by complete or incomplete recovery. Fifteen per cent of people present with a gradually progressive disease course from onset known as primary progressive MS (PPMS). A single episode in isolation with no previous clinical attacks in someone who does not fulfil the diagnostic criteria for MS is known as clinically isolated syndrome (CIS). Over time, people with RRMS can develop gradually progressive disability called secondary progressive MS (SPMS). This usually occurs at least 10–15 years after disease onset. These descriptions of clinical disease course are still used in practice (Fig 1). However, increased understanding of MS and its pathology has led to new definitions focused on disease activity (based on clinical or magnetic resonance imaging (MRI) findings) and disease progression.1
Fig 1.
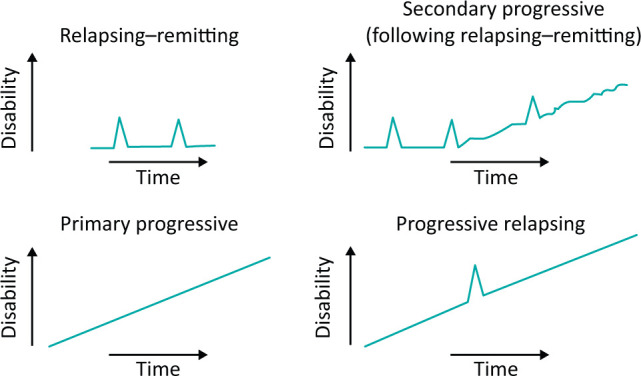
Multiple sclerosis disease course.
Clinical presentation
MS is a CNS disease characterised by demyelinating lesions in regions including the optic nerves, brainstem, cerebellum, periventricular and spinal cord. Histopathology also shows widespread involvement of the cerebral grey matter, although this is not well appreciated on conventional MRI. The clinical features of an MS attack depend on the areas of the brain or spinal cord involved. As this is an inflammatory condition, the onset of symptoms of an attack in RRMS is usually gradual and can evolve over days. Sudden onset with symptoms maximal at onset would be much more suggestive of a vascular event. A clinical attack must last at least 24 hours in the absence of fever or infection. In primary progressive MS, symptoms would be expected to have a gradual and insidious onset over at least 12 months by the time of diagnosis.
A common first presentation of RRMS is with unilateral optic neuritis characterised by gradual onset monocular visual loss, pain on moving the eye and altered colour vision. Visual loss rarely progresses beyond 2 weeks from the onset. Visual recovery usually takes longer than 2 weeks and may not recover to baseline. On examination, visual acuity is typically reduced, there may be a relative afferent pupillary defect, a central scotoma or impaired colour vision. On funduscopy, the optic disc may appear normal (retrobulbar neuritis) or swollen acutely, and may become pale and atrophic over time following the attack.
An inflammatory lesion in the spinal cord causes a myelitis that is usually partial and presents with gradual onset sensory and motor symptoms of the limbs. Evolution is over hours to days. The severity of myelitis can vary from a mild sensory syndrome to a severe disabling attack causing tetraparesis. A lesion in the cervical cord can cause Lhermitte's phenomenon with an electric shock-like sensation down the neck and back on flexing the neck. This can be a useful clue to the diagnosis. Thoracic cord lesions can cause a tight band-like sensation around the trunk or abdomen often described as the ‘MS hug’. In severe cases this has been misinterpreted as being due to a cardiac event. On examination, signs can include sensory signs of reduced fine touch, vibration sense and joint position sense. There may be a sensory level. Motor signs are typical of an upper motor neuron lesion with increased tone or spasticity, pyramidal weakness and hyperreflexia with extensor plantar responses. Myelitis may be partial causing a hemi-cord syndrome or partial Brown-Séquard.
Brainstem syndromes can present with diplopia, oscillopsia, facial sensory loss, vertigo and dysarthria. Typical findings include an isolated sixth nerve palsy, gaze evoked nystagmus or an internuclear ophthalmoplegia. Bilateral internuclear ophthalmoplegia is pathognomonic of MS.
The diagnosis of MS is based on the clinical features of the attacks including the history and examination findings. The guiding principle of the diagnosis is that of dissemination in time (DIT) and dissemination in space (DIS). There is no single diagnostic laboratory test for MS. The diagnosis is based on the clinical findings supported by investigations.
Investigations
Magnetic resonance imaging
MRI has been increasingly used to support the diagnosis of MS and to look for any atypical features suggesting an alternative diagnosis. Brain and spinal cord MRI are used to determine dissemination in space (DIS) and for evidence of dissemination in time (DIT) in patients with a typical CIS. DIS can be demonstrated by one or more MRI T2-hyperintense lesions that are characteristic of MS in two or more of four areas of the CNS: periventricular; cortical or juxtacortical; infratentorial; and the spinal cord. DIT can be demonstrated by the simultaneous presence of gadolinium-enhancing and non-enhancing lesions at any time or a new T2 lesion or gadolinium-enhancing lesion on follow-up MRI.2
Cerebrospinal fluid
Cerebrospinal fluid (CSF) examination remains a valuable diagnostic test, particularly when clinical and MRI evidence is insufficient to confirm the diagnosis of MS. There has been a major change in the most recent MS diagnostic criteria in that oligoclonal bands in the CSF can be used as a surrogate marker of DIT to confirm the diagnosis of RRMS in people with CIS and MRI evidence of DIS.3 CSF findings are also important when there is a progressive course from onset (PPMS) and when there are any atypical clinical or imaging findings. Evidence of intrathecal antibody synthesis (ie oligoclonal bands in the CSF but not in a paired serum sample) supports the diagnosis of MS. An elevated CSF protein >1.0 g/L or significant pleocytosis >50 cells/mm³ or the presence of neutrophils would suggest an alternative diagnosis.
Visually evoked potentials and optical coherence tomography
Visually evoked potentials (VEPs) were historically included in MS diagnostic criteria with an abnormal VEP (delayed but with a well preserved waveform) being used as objective evidence of a second lesion if the clinical presentation did not include the visual pathway.4,5 In the 2017 criteria, it was recommended that further studies are needed to determine the role of VEPs and optical coherence tomography (OCT) in supporting the diagnosis of MS and VEPs are not included in the criteria.3 In clinical practice VEPs can be useful, for example in a patient with a progressive spinal cord syndrome and normal brain MRI.
Differential diagnosis
The differential diagnosis of MS is wide and varies depending on the site of presentation eg optic nerve or spinal cord. It is important for the clinician to be vigilant for atypical clinical findings or investigation results.6,7 Non-specific symptoms with white matter lesions on the MRI can be a common cause of misdiagnosis of common disorders, such as migraine or small vessel vascular disease in the elderly.
Other CNS inflammatory diseases including neuromyelitis optica spectrum disorder (NMOSD), myelin oligodendrocyte glycoprotein (MOG) antibody-associated disease and acute disseminated encephalomyelitis (ADEM) are important differential diagnoses as the treatment approaches are different. Several other rarer inflammatory, infective and metabolic conditions should also be considered (Table 1).
Table 1.
Differential diagnosis of multiple sclerosis
| Autoimmune/inflammatory | CNS infections | Metabolic | Vascular conditions | Other |
|---|---|---|---|---|
| Neuromyelitis optica spectrum disorder (NMOSD) | CNS syphilis | Vitamin B12 deficiency | Small vessel disease | CNS lymphoma |
| Acute disseminated encephalomyelitis (ADEM) | Lyme disease | Copper deficiency | Stroke | Paraneoplastic |
| Myelin oligodendrocyte glycoprotein (MOG) antibody disease | Human T-lymphotropic virus (HTLV) | Mitochondrial disease | CADASIL | |
| Sjögren's syndrome | HIV | Leukodystrophies | Susac's syndrome | |
| CNS lupus | Anti-phospholipid antibody syndrome | |||
| Sarcoidosis | ||||
| Behçet's | ||||
| CNS vasculitis |
CADASIL = cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy; CNS = central nervous system.
NMOSD is an inflammatory CNS syndrome distinct from MS that can be associated with serum aquaporin-4 immunoglobulin G antibodies (AQP4-IgG).8 NMOSD is stratified by serologic testing into NMOSD with or without AQP4-IgG. NMOSD presents with severe episodes of complete transverse myelitis and/or severe episodes of optic neuritis with incomplete recovery or a brainstem syndrome of the area postrema causing nausea and vomiting or hiccups. More stringent clinical criteria with additional neuroimaging findings, are required for diagnosis of NMOSD without AQP4-IgG or when serologic testing is unavailable. The myelitis is usually extensive on MRI with a T2 spinal cord lesion extending over three or more spinal segments (longitudinally extensive transverse myelitis (LETM)). Brain lesions in NMO are located in areas of high expression of aquaporin 4, including the hypothalamus, medulla, and other brainstem areas. Oligoclonal bands in the CSF are detected in 10–20% of patients with NMO.
MOG antibody disease is associated with pathogenic serum antibodies against myelin oligodendrocyte glycoprotein.9 Most are AQP4-IgG-seronegative. The presentation is varied ranging from isolated optic neuritis to classic NMO to ADEM. It affects both children and adults and there is no sex predominance. The disease can relapse, but medium-term immunosuppression is usually effective. Permanent disability is less frequent than in NMOSD associated with positive AQP4 antibodies. Sphincter and sexual dysfunction are common as the transverse myelitis involves the conus.
ADEM tends to present with a subacute encephalopathy with altered level of consciousness, behaviour or cognitive function. It often follows an infectious illness and is most common in children. It was initially thought to be a monophasic condition, but some patients experience recurrence of their initial ADEM symptoms. Around 60% of children with ADEM have MOG antibodies. MRI typically shows symmetrical multifocal or diffuse brain lesions (Table 1).
Diagnostic criteria
Diagnostic criteria for MS have been developed since the first description of MS as ‘La sclérose en plaques disséminées’ by Charcot in 1868. He described a triad of nystagmus, intention tremor and scanning speech. Clinical criteria were supplemented by CSF, MRI and evoked potentials in the Poser criteria in 1983.4 With the more widespread availability of MRI, the McDonald criteria were developed by the International Panel on Diagnosis of Multiple Sclerosis in 2001 giving increasing weight to MRI in the diagnosis of MS.5 There have been subsequent revisions of these criteria in 2005 and 2010 with the most recent revision in 2017 (Table 2).3 The new criteria allow for an earlier diagnosis of MS in patients experiencing a typical clinically isolated syndrome. Earlier diagnosis of MS has become much more important with the availability of highly effective disease modifying treatments (DMTs) for MS. However, the benefits of earlier diagnosis have to be balanced with the risks of misdiagnosis.10 The 2017 position paper addresses concerns about the potential for misdiagnosis, particularly with misinterpretation of non-specific symptoms and non-specific MRI findings. The development of the 2017 McDonald criteria was informed by the new MRI criteria for diagnosing MS proposed by the European Magnetic Resonance Imaging in Multiple Sclerosis (MAGNIMS) network.1,2
Table 2.
2017 McDonald criteria for the diagnosis of multiple sclerosis in patients with an attack at onset3
| Number of attacks at clinical presentation | Number of lesions with objective clinical evidence | Additional data needed for diagnosis of multiple sclerosis |
|---|---|---|
| ≥2 | ≥2 | Nonea |
| ≥2 | 1 (as well as clear-cut historical evidence of a previous attack involving a lesion in a distinct anatomical location) | Nonea |
| ≥2 | 1 | Dissemination in space demonstrated by an additional clinical attack implicating a different CNS site |
| Or by MRI | ||
| 1 | ≥2 | Dissemination in time demonstrated by an additional clinical attack |
| Or by MRI | ||
| Or demonstration of CSF-specific oligoclonal bands | ||
| 1 | 1 | Dissemination in space demonstrated by an additional clinical attack implicating a different CNS site |
| Or by MRI | ||
| And dissemination in time demonstrated by an additional clinical attack | ||
| Or by MRI | ||
| Or demonstration of CSF-specific oligoclonal bands |
a= no additional tests are required to demonstrate dissemination in space and time. However, unless MRI is not possible, brain MRI should be obtained in all patients in whom the diagnosis of multiple sclerosis is being considered. In addition, spinal cord MRI or CSF examination should be considered in patients with insufficient clinical and MRI evidence supporting multiple sclerosis, with a presentation other than a typical clinically isolated syndrome, or with atypical features. If imaging or other tests (eg CSF) are undertaken and are negative, caution needs to be taken before making a diagnosis of multiple sclerosis, and alternative diagnoses should be considered. CNS = central nervous system; CSF = cerebrospinal fluid; MRI = magnetic resonance imaging.
The diagnosis of MS is still based on a combination of clinical, MRI and laboratory (eg CSF) findings. The diagnostic criteria are designed to be used for patients with typical clinical presentations and should not be applied in cases where MRI changes are incidentally identified in asymptomatic individuals. In that situation, cases are referred to as radiologically isolated syndrome (RIS). The risk of misdiagnosis may have harmful consequences if patients are started on DMTs inappropriately, for example, some MS DMTs can worsen outcomes for patients with NMO spectrum disorders.
The aim of the criteria is to make an earlier and accurate diagnosis of MS and lessen the period of uncertainty for the patient and clinician. This enables appropriate management including confirmation of the diagnosis for the patient and access to effective disease modifying treatments.
Conclusion
The clinical diagnosis of MS is based on history and examination providing evidence of typical neurological dysfunction. Dissemination in time and space can be demonstrated clinically but MRI is now routinely used to confirm the diagnosis. With a single episode, or clinically isolated syndrome, MRI evidence can allow an earlier diagnosis using new diagnostic criteria. Over-interpretation of non-specific symptoms and non-specific white matter lesions on MRI can lead to misdiagnosis. The differential diagnosis of MS includes other CNS inflammatory conditions such as NMOSD, ADEM and MOG antibody-related disease. It is important to differentiate these conditions as the treatment approach is different. CNS infections, metabolic conditions and vascular disease also need to be considered. There are now highly effective DMTs for MS and an accurate, timely diagnosis is crucial to ensure appropriate access to treatment.
References
- 1.Lublin FD, Reingold SC, Cohen JA, et al. Defining the clinical course of multiple sclerosis. Neurology 2014;83:278–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Filippi M, Rocca MA, Ciccarelli O, et al. MRI criteria for the diagnosis of multiple sclerosis: MAGNIMS consensus guidelines. Lancet Neurol 2016;15:292–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol 2018;17:162–173. [DOI] [PubMed] [Google Scholar]
- 4.Poser CM, Paty DW, Scheinberg LC, et al. New diagnostic criteria for multiple sclerosis: guidelines for research protocols. Ann Neurol 1983;13:227–31. [DOI] [PubMed] [Google Scholar]
- 5.McDonald WI, Compston DA, Edan G, et al. Recommended diagnostic criteria for multiple sclerosis: guidelines from the International Panel on the Diagnosis of Multiple Sclerosis. Ann Neurol 2001;50:121–7. [DOI] [PubMed] [Google Scholar]
- 6.Miller DH, Weinshenker BG, Filippi M, et al. Differential diagnosis of suspected multiple sclerosis: a consensus approach. Mult Scler 2008;14:1157–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brownlee W. Differential diagnosis of multiple sclerosis: The better explanations in clinical practice. Neurology 2019;92:1037–8. [DOI] [PubMed] [Google Scholar]
- 8.Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015;85:177–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jurynczyk M, Jacob A, Fujihara K, Palace J. Myelin oligodendrocyte glycoprotein (MOG) antibody-associated disease: practical considerations. Pract Neurol 2019;19:187–95. [DOI] [PubMed] [Google Scholar]
- 10.Solomon AJ, Corboy JR. The tension between early diagnosis and misdiagnosis in multiple sclerosis. Nat Rev Neurol 2017;13:567–72. [DOI] [PubMed] [Google Scholar]