

Abstract
Cardiovascular diseases are the leading cause of deaths in the world. Endothelial dysfunction followed by inflammation of the vessel wall leads to atherosclerotic lesion formation that causes ischemic heart and myocardial hypertrophy, which ultimately progress into cardiac dysfunction and failure. Histone deacetylases (HDACs) have been recognized to play crucial roles in cardiovascular disease, particularly in the epigenetic regulation of gene transcription in response to a variety of stresses. The unique nature of HDAC regulation includes that HDACs form a complex co-regulatory network with other transcription factors, deacetylate histones and non-histone proteins to facilitate the regulatory mechanism of the vascular system. The selective HDAC inhibitors are considered as the most promising target in cardiovascular disease, especially for preventing cardiac hypertrophy. In this review, we discuss our present knowledge of the cellular and molecular basis of HDACs in mediating the biological function of vascular cells and related pharmacologic interventions in vascular disease.
Keywords: Histone deacetylase, epigenetics, acetylation, deacetylation, cardiovascular disease, vascular smooth muscle cell, endothelial, atherosclerosis
Graphical Abstract
Introduction
Cardiovascular disease (CVD) is highly prevalent among the general population and represents the leading cause of mortality and morbidity in developed countries. CVD refers to a broad range of diseases that inflict the cardiovascular system, which includes the heart and blood vessels. Atherosclerosis is a progressive process in which the inner layers of artery walls become thick and irregular as a result of deposits of fat, cholesterol and other substances. A comprehensive understanding of the regulatory mechanisms of vascular physiology will prove a foundation for understanding the novel mechanism(s) underlying vascular disease and designing more specific molecular targets for vascular function. Lysine acetylation is reversible and is controlled by the opposing actions of acetyltransferase and deacetylase through HATs and HDACs, which manifest an opposing fashion to control the acetylation state of nucleosomal histones [1–5]. A plenty of evidence in recent years reports the critical function of HDACs in regulating vascular cell homeostasis and atherosclerosis. An increased understanding of the regulatory role of HDAC in vascular physiology may result in new therapeutic approaches for specifically targeting CVD that act at the risk-factor level, as well as directly on the vascular system.
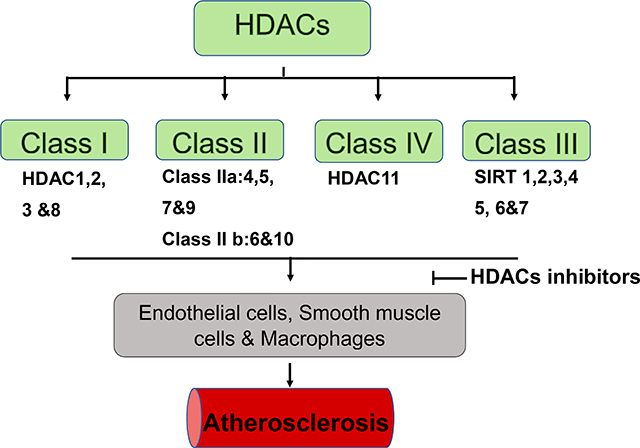
Eighteen mammalian histone deacetylases have been classified into four distinct classes (classes I, IIa, IIb, III and IV) based on their phylogenetic analyses of the sequence homology, enzymatic activity, domain structure, and functional similarity (Table 1). Class I, which is homologous to Rpd3, including HDACs 1, 2, 3, and 8, is found to be ubiquitously expressed in human tissues. Class I HDACs show high enzymatic activity and reside predominantly in the nucleus. Class I HDACs are closely associated with several other subunits such as Sin3 and N- CoR, to modulate histone deacetylation and transcriptional co-repression [2, 6]. Class II is homologous to yeast Hda1, and its N-terminal extension possesses conserved domains that regulates protein-protein interaction. Class II HDACs is further divided into two subclasses: IIa (HDAC 4, 5,7, and 9) and IIb (HDAC 6 and 10). Class IIa HDACs manifest a more restricted pattern of expression, but is traditionally considered to show enzymatic activity under certain condition, and serves as signal transducers that shuttle between the cellular compartments [7, 8]. The enzymatic activity of Class II HDACs depends on the formation of a multiprotein complex [9]. Class III or sirtuin, is homologous to the silent information regulator 2 (Sir2) family, which includes SIRT1–7. Sirtuins require NAD+ for deacetylation and play a critical regulatory function in transcriptional repression [4]. Finally, class IV HDAC consists of a solitary member, HDAC11, which is homologous to class I and class II HDACs. However, because of low sequence similarities, class IV HDACs has not yet been classified into any of the three existing classes [10].
Table 1.
Histone deacetylase (HDAC) classifications.
| Class I HDACs: HDAC1, HDAC2, HDAC3, and HDAC8, |
| Class II HDACs: IIa: HDAC4, HDAC5, HDAC7, HDAC9; IIb: HDAC6 and HDAC10 |
| Class III HDACs: SIRT1, SIRT2, SIRT3, SIRT4, SIRT5, SIRT6, SIRT7 |
| Class IV HDACs: HDAC11 |
1. The function of HDACs in modulating the function of endothelial cell
Endothelial cells control vascular tone, blood coagulation and serve as the most important mediators of inflammatory reaction. HDACs are considered to be critical transcriptional cofactors recruited to promoters by sequence-specific transcription factors to mediate gene expression for vascular homeostasis and vessel development.
1.1. Class I HDACs
It was shown that oscillatory shear stress increased cyclin A, but down-regulated p21 in endothelial cells and induced proliferation, which were mediated by HDACs 1, 2 and 3. Oscillatory shear stress-induced HDAC signaling and responses in EC are highly related to PI3K/Akt [11]. Overexpressed HDAC1 resulted in the downregulation of p53 and von HippelLindau tumor suppressor genes and increased the angiogenesis of human endothelial cells, but inhibition of HDACs led to downregulation of the vascular endothelial growth factor, and also suppressed both angiogenesis in vitro and in vivo [12]. FK228, a macrocyclic polypeptide, was used as a molecular tool to compare its binding conformation in HDAC1 and HDAC6 to study the molecular mechanism for inhibiting HDAC1 [13]. FK228 significantly attenuated the content of vascular endothelial growth factor (VEGF) in response to hypoxia, indicating that FK228 plays a contributable role in modulating angiogenesis. FK228 was also found to block angiogenesis induced by hypoxia in the Lewis lung carcinoma model, suggesting that FK228 downregulates hypoxia-responsive angiogenesis by attenuating HIF-1α activity [14].
Overexpression of HDAC2 suppressed Arg2 expression in human aortic endothelial cells, but knockdown of HDAC2 enhanced the expression of Arg2. HDAC2 overexpression suppressed oxidized low-density lipoprotein-induced vascular dysfunction [15]. HDAC3 plays a critical role in modulating endothelial cell differentiation from embryonic stem cells [16,17]. HDAC3 are also crucial for the maintenance of endothelial integrity associated with the PI3K/Akt- and TGFβ2-dependent mechanism [18]. Nox4, a member of the NADPH oxidase family, represents a major source of generation of reactive oxygen species (ROS) in the vascular wall. siRNA-mediated knockdown of HDAC3 was reported to be associated with down- regulation of Nox4 in human umbilical vein endothelial cells (HUVEC) [19]. The evidence suggests the critical role of class I HDACs involved in mediating the function of endothelial cells.
1.2. Class II HDACs
Class IIa HDACs are signal-responsive regulators of gene expression, involved in modulating vascular homeostasis. Nitric oxide modulates the function of endothelial cells and initiates class II HDACs 4 and 5 nuclear shuttling. MC1568, a class II HDAC selective inhibitor, rescued serum-dependent histone acetylation in nitric oxide-treated HUVECs. Nitric oxide-induced HDAC4 and HDAC5 nuclear shuttling related to the activation of the protein phosphatase 2A (PP2A). In addition, nitric oxide was found to induce the formation of a macromolecular complex with HDAC3, HDAC4, HDAC5, and an active PP2A [20]. HDAC5 acts as a repressor of angiogenesis, which mediates the angiogenic gene expression pattern of endothelial cells. HDAC5 overexpression attenuated sprout formation [21]. Inhibition of HDAC5 demonstrated a unique pro-angiogenic effect by stimulating endothelial cell migration, sprouting, and tube formation. However, HDAC5 overexpression mitigated sprout formation, suggesting that HDAC5 serves as a negative regulator of angiogenesis [22]. Blockade of HDAC5 in the ECs of systemic sclerosis restored normal angiogenesis, which is consistent with the evidence that knockdown of HDAC5 in ECs led to systemic sclerosis, identifying key HDAC5-regulated genes involved in angiogenesis [23]. Impaired MEF2 activity in pulmonary arterial hypertension is mediated by excess nuclear accumulation of HDAC4 and HDAC5. Pharmacological inhibition of class IIa HDACs caused a restoration of MEF2 activity in pulmonary artery endothelial cells, as evident by increases in its transcriptional factors and a reduced proliferative rate[24]. In addition, treatment of mice with a selective HDAC6 inhibitor, tubastatin A, alleviated angiotensin II-induced high blood pressure and vasoconstriction, which is related to the recovered AngII-mediated decreased H2S levels in endothelial cells that contribute to the alleviation of hypertension [25]. It has been noted that new potent and selective hydroxamate-based HDAC6 inhibitors were developed on the phenothiazine system as a cap group scaold. On the basis of a HDAC pharmacophore model containing cap group, linker, and ZBG [26], the introduction of nitrogen atoms into the phenothiazine system has resulted in inhibitors for HDAC6, with azaphenothiazine 7i as the most potent and selective inhibitor, which provides an excellent tool for identifying its physiological role in the regulation of vascular biology.
Silencing of HDAC7 upregulated the key factors platelet-derived growth factor-B (PDGF-B) and its receptor (PDGFR-beta). HDAC7 silencing was also partially contributable for the inhibition of endothelial cell migration [27]. It was also reported that vascular endothelial growth factor treatment resulted in a decrease in HDAC7 via the PLCgamma-IP3K pathway and partially rescued HDAC7-induced attenuation of proliferation. HDAC7 knockout mice died at E11.5 from vascular defects, which is likely resulting from suppressing a matrix metalloproteinase, Mmp10, to maintain the matrix environment and integrity of vessels [28]. In agreement with this, Mottet et al. reported that HDAC7 inhibition altered the morphology, migration, and capacity to form capillary tube-like structures in endothelial cells in vitro. Furthermore, HDAC7 also interacts with beta-catenin to form a complex with 14–3-3 epsilon, zeta, and eta proteins to regulate endothelial cell development [29].
Hypoxia-inducible factor (HIF)-1 is known as a transcription factor to regulate the expression of genes such as VEGF and erythropoietin. HDAC7 enhanced the transcriptional activity of HIF-1α in the nucleus through the formation of a complex between HIF-1α and HDAC7 [30]. Clinical data shows that HDAC7 reduction causes a defect in HIF-1α in response to hypoxia in association with an impaired VEGF gene expression in COPD [31]. In addition, activation of the HDAC7 pathway could stimulate endothelial progenitor cell migration and tube formation through a phospholipase Cg/protein kinase C/protein kinase D1 (PKDl)-dependent mechanism [32]. Infection of ECs with adenoviruses encoding HDAC7 deficient mice blocked VEGF-induced primary aortic endothelium cell migration [33]. Very recently, PCI-34051, which is an indole-based derivative, is currently considered as among the most selective HDAC8 inhibitors, serving as an excellent chemical tool for assessing the functional role of HDAC8 [34]. In an angiotensin II-regulated mouse model, PCI34051 administration could lower high blood pressure by modulating arterial remodeling and vasoconstriction, indicating that HDAC8 inhibitor is a potential therapeutic target for hypertension [35]. Lobera et al. have identified trifluoromethyloxadiazole (TFMO)-based metal binding group inhibitors that target the acetyl- lysine sites of class IIa HDACs, circumventing the selectivity and pharmacologic liabilities of hydroxamates 36]. It was demonstrated that TMP269 mitigated the abnormal endothelial cell permeability changes in a cerebral ischemia/reperfusion model, suggesting that TFMO-based HDAC inhibitors could be developed as a target to treat vascular damage [37]. HDAC9 was found to contribute to for oxygen-glucose deprivation-induced brain microvessel endothelial cell dysfunction and endothelial cell permeability dysfunction in the ischemic cerebral hemisphere[38]. HDAC10 overexpression stimulated tube formation in HUVECs, whereas silencing of HDAC10 in HUVECs attenuated tube formation involving extracellular-regulated kinase 1/2 (ERK1/2) activation [39].
1.3. Class IV HDAC
Class IV HDAC consists of the solitary member HDAC11, homologous to both class I and class II HDACs. However, because the overall sequence similarities are low, it has not yet been grouped into any of the three existing classes [10]. HDAC11 restored angiogenic factor with G-patch and FHA domain 1 (Aggf1) expression and abrogated vascular smooth muscle cell phenotypic modulation in the vessels after carotid artery ligation in mice, but treatment with a HDAC11 inhibitor attenuated vascular injury in mice, indicating that the key role of HDAC11 is related to vascular injury [40].
1.4. SIRTs
SIRT1 is crucially required for neovascularization by controlling the angiogenic activity in response to angiogenic cues. SIRT1 is found to be highly expressed in the growing vascular endothelium and contributes critically to the angiogenic activity [41]. SIRT1 deacetylate to repress the transcription of Foxo transcription factors in vascular endothelial homeostasis [42–44], which are important negative regulators of postnatal angiogenesis and restrain endothelium cell proliferation, migration, and neovessel formation [45,46]. Knockdown of Foxo1 partially rescued the inhibitory effects of SIRT1 gene silencing on the angiogenic activity of endothelial cells, revealing that SIRT1 mediates endothelial angiogenic responses, at least in part, through modulating Foxo1 transcription factor.
Angiotensin II resulted in a significant increase in cell migration, which was mitigated through pretreatment with sirtinol (a cell-permeable inhibitor of sirtuin NAD+-dependent deacetylases) and depletion of SIRT2. Angiotensin II and mechanical stretch induced microtubule redistribution and deacetylation via SIRT2, implying an important role of SIRT in mediating hypertension-induced vascular remodeling in endothelial cells [47].
SIRT6 is demonstrated to prevent endothelial cells from senescence. SIRT6 depletion by RNA interference reduces cell proliferation, increases the fraction of senescence-associated-beta- galactosidase-positive cells in the models of HUVEC and aortic endothelial cells, and suppresses the formation of tubule networks. SIRT6 confers protection from telomeres and genomic DNA damage in endothelial cells, thus preventing a decrease in replicative capacity and the onset of premature senescence[48].
2. HDAC modulates smooth muscle cell proliferation and migration
Vascular smooth muscle cells play a key role in modulating blood pressure, and tissue repair and progression to aging and diseased arteries. The migration of VSMCs is associated with vascular remodeling, which contributes largely to the pathogenesis of vascular diseases, for instance atherosclerosis and restenosis.
2.1. Class I HDAC
It was shown that short interfering RNA-mediated suppression of HDACs 1, 2, or 3 and pharmacological blockade of HDAC attenuated mitogen-induced smooth muscle cell proliferation [49]. HDAC1 and HDAC5 were elevated in lungs from human idiopathic pulmonary arterial hypertension and also in the right ventricle. Inhibition of class I, II, and IV HDACs attenuated the development of hypoxia-induced pulmonary hypertension, which was associated with inhibition of the imprinted highly proliferative phenotypes of fibroblasts and PDGF-induced growth of human VSMCs [50]. In response to PDGF, inhibition of class I HDAC counteracted the hyperproliferative effect, suppressing both proliferation and migration in pulmonary artery smooth muscle cells, while class II was ineffective [51].
HDAC3 mitigates smooth muscle differentiation, which is related to downregulation of the Notch ligand Jagged1, a key driver of smooth muscle differentiation in the aortic arch arteries[52].. Loss of HDAC3 in the neural crest leads to perinatal lethality and vascular abnormalities such as interrupted aortic arch type B, aortic arch hypoplasia, double-outlet right ventricle, and ventricular septal defect. ROS attenuated IGF1R expression and induced apoptosis in VSMCs[53], which is related to recruiting HDAC1 to the IGF1R promoter. HDAC8 is a prominent cytosolic marker of smooth muscle differentiation [54]. HDAC8 shows a striking stress fiber-like pattern of distribution and is coexpressed in vivo with smooth muscle alpha-actin. HDAC8 silencing through RNAi strongly mitigates the capacity of human vascular smooth muscle cells to contract collagen lattices. HDAC8 forms a partnership with the smooth muscle actin cytoskeleton to regulate the contractile capacity of smooth muscle actin [55].
2.2. Class II HDACs
The suppession of activity of class I, II HDACs by apicidin and HDACi VIII attenuated proliferation of newborn pulmonary arterial smooth muscle cells and induced cell cycle arrest in G1 phase [56]. In human aortic SMCs, HDAC inhibition mitigated the effect of Notch to increase smooth muscle a-actin levels, indicating that HDAC activity was required for Notch differentiation signaling [57]. HDAC4 and HDAC5 are regulated in a CaMKII-dependent manner in vascular smooth muscle cells [58–60]. CaMKII-5 mediated MEF2-dependent gene transcription through modulation of HDAC4 and HDAC5 in VSMCs, [59]. Mechanical cyclic strain inhibited migration and upregulated acetylated histone H3 and HDAC7 while mitigating the magnitudes of HDAC3/4 in VSMCs, but the mechanically induced VSMC migration was diminished by treatment with tributyrin, a HDAC inhibitor [61]. In addition, LMK235, a class I and histone deacetylase (HDAC6)-preferential HDAC inhibitor, augmented nitric oxide production in HUVECs and inhibited the increase of aortic wall thickness in hypertensive rats. This may be related to suppressing calcium calmodulin-dependent protein kinase II (CaMKII) a, which is related to vascular smooth muscle cell proliferation [62]. It was shown that HDAC inhibition using butyrate abrogated the activation of Akt, resulting in differential effects on Akt downstream targets, promoting convergence of their cross-talks leading to vascular smooth muscle growth by arresting proliferation [63]. It has been shown that mercaptoacetamide histone deacetylase inhibitors act as neuroprotective agents, without exhibiting the genotoxicity induced by hydroxamate inhibitors [64], revealing chemical differences in the binding of mercaptoacetamides and hydroxamates to HDAC6 and HDAC8 [65] and mediating their selectivity and activity [66]. Moreover, the mercaptoacetamide HDAC inhibitor, ubacin, a compound to selectively inhibit HDAC6, enhanced eNOS content in endothelial cells and protected endothelial cells in mice, respectively [ 67, 68].
2. 3. SIRT
Overexpression of SIRT1 inhibited the expression of angiotensin II type I receptor whereas nicotinamide, an inhibitor of SIRT1, enhanced the expression of angiotensin II type I receptor [69]. SIRT1 inhibited angiotensin II-induced hypertrophy of VSMCs [58]. VSMC-specific SIRT1 transgene alleviated angiotensin II-induced systolic blood pressure, attenuated angiotensin II -induced vascular remodeling, abrogated the associated pathological changes, and TGF-01 contents [70]. Li et al. found that SIRT1 modulated vascular smooth muscle cell proliferation and migration and led to cell cycle arrest at G1/S transition [71]. SIRT3 was stably expressed in mouse VSMCs and angiotensin II can elevate SIRT3 in VSMCs. However, SIRT3 silencing was found to result in an increase in cell proliferation [72].
3. The function of HDACs in regulation of atherosclerosis
Many biological processes are implicated in the development of pathophysiologic events during atherogenesis. These include endothelial denudation, injury, or activation, shear stress; local adherence of platelets; lipoprotein oxidation; lipoprotein aggregation; macrophage-elicited inflammatory responses and foam cell formation [73–75]. Macrophages play a key regulatory role as immune cells in forming atherosclerotic plaques and critically shape atherosclerotic disease development [76–79]. Class I, IIa, IIb, and IV HDAC isoenzymes were reported to be significantly elevated in human atherosclerotic tissue and in the atherosclerotic aorta of ApoE’1’ mice. Treatment of ApoE−/− mice with SAHA attenuated the extent of atherosclerotic lesions and NADPH-stimulated ROS production and expression of pro-inflammatory markers [80]. The expression of HDAC1 was dramatically decreased in atherosclerotic lesions and Ox-LDL treated human aortic endothelial cells. TargetScan predicted that HDAC1 is a potential target of miR-34 and suggested that HDAC1 was directly targeted by miR-34a to mediate the cell viability of human aortic endothelial cells and cell apoptosis [81]. Arginase 2 (Arg2) acts as a critical target in developing atherosclerosis because it controls the production of endothelial nitric oxide, proliferation, fibrosis, and inflammation. HDAC2 over-expression in human aortic endothelial cells suppressed Arg2 expression, and knockdown of HDAC2 by siRNA enhanced Arg2 expression. HDAC2 overexpression attenuates oxidized low-density lipoprotein-mediated activation in human aortic endothelial cells[82]. Zampetaki et al. showed that HDAC3 level was up-regulated in areas in the vicinity to branch opening area in which disturbed flow occurs [83]. In aortic isografts of apolipoprotein E-knockout mice, which are treated with shHDAC3, there was a robust atherosclerotic lesion. These findings indicate that HDAC3 in the artery endothelium is crucial in preventing atherosclerosis. The inhibition of HDACs increased gene expression of the cholesterol efflux regulators: ATP-binding cassette transporters ABCA1 and ABCG1. HDAC3 inhibition phenocopies the atheroprotective effects of pan-HDAC inhibitors, indicating that the inhibition of HDACs, and in particular HDAC3, in macrophages serves as a novel potential target to treat atherosclerosis [84]. Moreover, HDAC3 was the sole HDAC upregulated in human atherosclerotic lesions in association with inflammatory macrophages, identifying HDAC3 as a potential novel therapeutic target in cardiovascular disease [85]. HDAC3 inhibits aspirin-stimulated lysine acetylation, the enzymatic activity of eNOS, NO production, and subsequent binding of eNOS to calmodulin, but downregulation of HDAC3 promotes NO lysine acetylation and generation, addressing that HDAC3 suppressed the effect of aspirin on endothelial NO production [86].
Endothelial cell-specific SIRT1 transgenic/ApoE−/− compound mice showed fewer atherosclerotic lesions relative to ApoE−/− controls, which did not affect peripheral lipids and glucose contents. The endothelium-specific SIRT1 overexpression largely attenuated atherogenesis by improving endothelial cell survival rate as well as its physiological function [87]. In endothelial cells and macrophages, SIRT1 demonstrated anti-inflammatory responses by suppressing various pro-inflammatory cytokines through deacetylation of RelA/p65-NF-kB. Deacetylation of RelA/p65-NF-kB in macrophages could prevent the formation of macrophage foam cells [88].
Macrophage-elicited chronic inflammation and SMC-induced vascular remodeling are considered to be as two major pathophysiologic events during atherogenesis. CIITA (major histocompatibility class II transactivator) is found to be a key mediator of these processes by regulating interferon gamma (IFN-gamma)-induced major histocompatibility class II activation and repressing type I collagen. It was found that HDAC2 antagonized CIITA activation through committing CIITA to protein degradation and decreasing the interaction of CIITA in a deacetylation-dependent manner [89]. HDAC6 protein levels in aortas from high-fat diet-fed ApoE−/− mice were comparable to those in controls, whereas HDAC6 activity was robustly upregulated, indicating that inhibition of HDAC6 activity may improve endothelial function and prevent or reverse the development of atherosclerosis [90].
Treatment of A10 vascular smooth muscle cells with IGF-1 enhanced HDAC5 phosphorylation. Blockade of the IGF-1 receptor tyrosine kinase mitigated IGF-1-induced HDAC5 phosphorylation. Inhibition of the NAD(P)H oxidase also suppressed IGF-1-induced phosphorylation of HDAC5, indicating that IGF-1-induced HDAC5 phosphorylation involves the NAD(P)H oxidase-dependent reactive oxygen species generation, which is related to vascular disorders, such as atherosclerosis [91].
Recently, a genome-wide association analysis showed that a single nucleotide polymorphism of HDAC9 on chromosome 7p21.1 is related to large vessel ischemic stroke [92]. Unlike the Caucasian population, rs11984041 was not polymorphic in the Chinese population but two other SNPs, rs2389995 and rs2240419 on HDAC9 of chromosome 7q21.1, were identified to be largely related to a large-vessel stroke risk. HDAC9 expression was upregulated in human atherosclerotic plaques in different arteries [93]. Cao et al. showed that deletions of HDAC9 in systemic and bone marrow cells attenuated the magnitude of atherosclerosis in LDLr−/−mice in the absence of significant effects on peripheral lipid levels. Deletion of HDAC9 led to upregulation of lipid homeostatic genes and downregulation of inflammation in macrophages. HDAC9 upregulation was associated with atherogenic functions by suppressing cholesterol efflux and generation of alternatively activated macrophages in atherosclerosis [94].
Summary
In this review, we have summarized the recent progress about the regulatory role of histone deacetylase in modulating the development of vascular cells as well as atherosclerosis, which is outlined in Table 2. We provided the latest views and knowledge about the interplay between specific HDACs and related partners in controlling vascular cell development and modulating the progression of vascular pathology, which provides new insights into our understanding of the critical function of histone deacetylases that contribute critically to the modulation of vascular biology and diseases. Fully understanding the critical function of HDACs in vascular biology will provide potential strategies for vascular disease by targeting HDAC as a molecular therapy.
Table 2.
The physiological role of major HDACs in the phenotype on vascular cells.
| Subtypes | Models | Functions and phenotypes | References |
|---|---|---|---|
| HDAC1 | ECs | Stimulates angiogenesis | Kim M S (12) |
| HAECs | Promote cell survival and prevent cell apoptosis | Li Y (81) | |
| HDAC2 | SMC | Inhibit cell proliferation | Findeisen HM (49) |
| HAECs | Inhibit vascular dysfunction | Pandey D 82) | |
| HDAC3 | ECs | Stimulate EC differentiation | Zang L (17) |
| Macrophages | Upregulate in atherosclerosis | Hoeksema MA (85) | |
| HDAC5 | ECs | Repress angiogenesis | Urbich CL (22) |
| HUVECs | Repress angiogenesis | Ha CH (21) | |
| HDAC6 | Mice | Increase blood pressure and vasoconstriction | Chi Z (25) |
| SHR | Increase vascular hyperplasia or vasoconstriction | Choi SY (62) | |
| ApoE-/- mice | Develop atherosclerosis | Leucker TM (90) | |
| HDAC7 | ECs | Stimulate cell migration | Mottet D(27) |
| Mouse | Rupture of blood vessels | Chang S (28) | |
| HDAC8 | SMCs | The marker of smooth muscle differentiation | Waltregny D (54) |
| HDAC9 | Rats | Increase EC permeability | Shi W (38) |
| LDLr-/- mice | Develop atherosclerosis | Cao Q (94) | |
| HDAC10 | ECs | Stimulate tube formation | Duan B (39) |
| HDAC11 | Rats | Induce vessel injury | Zhou B (40) |
| SIRT1 | EC | Modulate homeostasis | Wan, Z (44) |
| Mice | Inhibit vascular remodeling | Gao P (70) | |
| Mice | Inhibit neointima formation | Li L (71) | |
| HUVECs | Enhance EC survival | Zhang QJ (87) | |
| ECs | Anti-inflammation | Stein S (88) | |
| SIRT3 | SMCs | Decrease proliferation | Wu X (72) |
| SIRT6 | ECs | Prevent senescence | Cardus A (48) |
HAECs: human aortic endothelial cells; HUVECs: human umbilical vein endothelial cells. SHR: spontaneously hypertensive rat. ECs: endothelial cells: SMCs: smooth muscle cells.
Acknowledgements
The work is supported by the National Heart, Lung, and Blood Institute Grant (R01 HL089405 and R01 HL115265) to TCZ
List of abbreviations
- CVD
Cardiovascular disease
- HDAC
Histone deacetylase
- HAT
Histone acetyltransferases
- Sir
silent information regulator
- ROS
reactive oxygen species
- HUVEC
human umbilical vein endothelial cells
- PP2A
protein phosphatase 2A
- MEF2
Myocardial enhancing factor 2
- VEGF
Vascular endothelial growth factor
- HIF
Hypoxia- inducible factor
- PDK1
protein kinase D1
- VSMC
Vascular smooth muscle cells
- IGF1R
Insulin-like growth factor 1 receptor
- TBP
TATA-binding protein
- SMC
smooth muscle cell
- PDGF
platelet-derived growth factor
- CaMKII
Ca2+/calmodulin-dependent protein kinase II
- Arg2
Arginase 2
- oxLDL
oxidized low-density lipoproteins
- CIITA
Major histocompatibility class II transactivator
- IFN-gamma
interferon gamma
Footnotes
Conflict of interest
The authors declared they do not have anything to disclose regarding conflict of interest with respect to this manuscript.
The manuscript reports the new progress and novel molecular mechanism(s) for which HDAC regulates vascular cell and atherosclerosis, which provides new insight into understanding of HDAC’s function and could develop a new therapy based on targeting HDAC for vascular disease.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Grozinger CM, Hassig CA and Schreiber SL Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc Natl Acad Sci US A. 1999;96(9): 4868–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003; 370(Pt 3): 737–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang XJ and Seto E The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol. 2008. 9(3); 206–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blander G and Guarente L, The Sir2 family of protein deacetylases. Annu Rev Biochem, 2004;73: 417–35. [DOI] [PubMed] [Google Scholar]
- 5.Haberland M, Montgomery RL and Olson EN The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10(1): 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thiagalingam S, Cheng KH, Lee HJ, Mineva N, Thiagalingam A, Ponte JF. Histone deacetylases: unique players in shaping the epigenetic histone code. Ann N Y Acad Sci. 2003; 983: 84–100. [DOI] [PubMed] [Google Scholar]
- 7.Fischle W, Kiermer V, Dequiedt F, Verdin E. The emerging role of class II histone deacetylases. Biochem Cell Biol. 2001; 79(3): 337–48. [PubMed] [Google Scholar]
- 8.Martin M, Kettmann R and Dequiedt F Class IIa histone deacetylases: regulating the regulators. Oncogene. 2007; 26(37): 5450–67. [DOI] [PubMed] [Google Scholar]
- 9.Fischle W, Dequiedt F, Hendzel MJ, Guenther MG, Lazar MA, Voelter W, Verdin E. Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol Cell. 2002; 9(1): 45–57. [DOI] [PubMed] [Google Scholar]
- 10.Gao L, Cueto MA, Asselbergs F, Atadja P. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J Biol Chem. 2002; 277(28): 25748–55. [DOI] [PubMed] [Google Scholar]
- 11.Lee Ding-Yu, Lee Chih-I, Lin Ting-Er, Seh Hong Lim Jing Zhou, Tseng Ying-Chih, Chien Shu, Jeng-Jiann Chiu Role of Histone Deacetylases in Transcription Factor Regulation and Cell Cycle Modulation in Endothelial Cells in Response to Disturbed Flow. Proc Natl Acad Sci U S A. 2012; 109 (6): 1967–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim MS, Kwon HJ, Lee YM, Baek JH, Jang JE, Lee SW, Moon EJ, Kim HS, Lee SK, Chung HY, Kim CW, Kim KW. Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nat Med. 2001; 7(4): 437–43. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Y, Fu TT, Ren YX, Li FC, Zheng GX, Hong J, Yao XJ, Xue WW, Zhu F Selective Inhibition of HDAC1 by Macrocyclic Polypeptide for the Treatment of Glioblastoma: A Binding Mechanistic Analysis Based on Molecular Dynamics. Front Mol Biosci 2020; 7: 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mie Lee Y, Kim SH, Kim HS, Jin Son M, Nakajima H, Jeong Kwon H, Kim KW. Inhibition of hypoxia-induced angiogenesis by FK228, a specific histone deacetylase inhibitor, via suppression of HIF-1alpha activity. Biochem Biophys Res Commun. 2003;300(1):241–6. [DOI] [PubMed] [Google Scholar]
- 15.Pandey D, Sikka G, Bergman Y, Kim JH, Ryoo S, Römer L, Berkowitz D. Transcriptional regulation of endothelial arginase 2 by histone deacetylase 2. Arterioscler Thromb Vasc Biol. 2014;34(7):1556–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xiao Q, Zeng L, Zhang Z, Margariti A, Ali ZA, Channon KM, Xu Q, Hu Y. Sca-1+ progenitors derived from embryonic stem cells differentiate into endothelial cells capable of vascular repair after arterial injury. Arterioscler Thromb Vasc Biol. 2006; 26(10): 2244–51. [DOI] [PubMed] [Google Scholar]
- 17.Zeng L, Xiao Q, Margariti A, Zhang Z, Zampetaki A, Patel S, Capogrossi MC, Hu Y, Xu Q. HDAC3 is crucial in shear- and VEGF-induced stem cell differentiation toward endothelial cells. J Cell Biol. 2006; 174(7): 1059–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zeng L, Wang G, Ummarino D, Margariti A, Xu Q, Xiao Q, Wang W, Zhang Z, Yin X, Mayr M, Cockerill G, Li JY, Chien S, Hu Y, Xu Q. Histone deacetylase 3 unconventional splicing mediates endothelial-to-mesenchymal transition through transforming growth factor beta2. J Biol Chem. 2013; 288(44): 31853–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Siuda D, Zechner U, El Hajj N, Prawitt D, Langer D, Xia N, Horke S, Pautz A, Kleinert H, Förstermann U, Li H. Transcriptional regulation of Nox4 by histone deacetylases in human endothelial cells. Basic Res Cardiol. 2012; 107(5): p. 283 doi: 10.1007/s00395-012-0283-3. [DOI] [PubMed] [Google Scholar]
- 20.Illi B, Dello Russo C, Colussi C, Rosati J, Pallaoro M, Spallotta F, Rotili D, Valente S, Ragone G, Martelli F, Biglioli P, Steinkuhler C, Gallinari P, Mai A, Capogrossi MC, Gaetano C. Nitric oxide modulates chromatin folding in human endothelial cells via protein phosphatase 2A activation and class II histone deacetylases nuclear shuttling. Circ Res. 2008; 102(1): 51–8. [DOI] [PubMed] [Google Scholar]
- 21.Ha CH, Wang W, Jhun BS, Wong C, Hausser A, Pfizenmaier K, McKinsey TA, Olson EN, Jin ZG. Protein kinase D-dependent phosphorylation and nuclear export of histone deacetylase 5 mediates vascular endothelial growth factor-induced gene expression and angiogenesis. J Biol Chem. 2008; 283(21): 14590–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Urbich C, Rössig L, Kaluza D, Potente M, Boeckel JN, Knau A, Diehl F, Geng JG, Hofmann WK, Zeiher AM, Dimmeler S. HDAC5 is a repressor of angiogenesis and determines the angiogenic gene expression pattern of endothelial cells. Blood. 2009; 113(22): 5669–79. [DOI] [PubMed] [Google Scholar]
- 23.Tsou PS, Wren JD, Amin MA, Schiopu E, Fox DA, Khanna D, Sawalha AH. Histone Deacetylase 5 Is Overexpressed in Scleroderma Endothelial Cells and Impairs Angiogenesis via Repression of Proangiogenic Factors. Arthritis Rheumatol 2016;68(12):2975–2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim J, Hwangbo C, Hu X, Kang Y, Papangeli I, Mehrotra D, Park H, Ju H, McLean DL, Comhair SA, Erzurum SC, Chun HJ. Restoration of impaired endothelial myocyte enhancer factor 2 function rescues pulmonary arterial hypertension. Circulation. 2015;131(2):190–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chi Z, Byeon HE, Seo E, Nguyen QT, Lee W, Jeong Y, Choi J, Pandey D, Berkowitz DE, Kim JH, Lee SY. Histone deacetylase 6 inhibitor tubastatin A attenuates angiotensin II-induced hypertension by preventing cystathionine y-lyase protein degradation. Pharmacol Res. 2019;146:104281. [DOI] [PubMed] [Google Scholar]
- 26.Vögerl K, Ong N, Senger J, Herp D, Schmidtkunz K, Marek M, Müller M, Bartel K, Shaik TB, Porter NJ, Robaa D, Christianson DW, Romier C, Sippl W, Jung M, Bracher F.Synthesis and Biological Investigation of Phenothiazine-Based Benzhydroxamic Acids as Selective Histone Deacetylase 6 Inhibitors. J Med Chem. 2019;62(3):1138–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mottet D, Bellahcène A, Pirotte S, Waltregny D, Deroanne C, Lamour V, Lidereau R, Castronovo V. Histone deacetylase 7 silencing alters endothelial cell migration, a key step in angiogenesis. Circ Res. 2007;101(12):1237–46. [DOI] [PubMed] [Google Scholar]
- 28.Chang S, Young BD, Li S, Qi X, Richardson JA, Olson EN, Histone deacetylase 7 maintains vascular integrity by repressing matrix metalloproteinase 10. Cell. 2006; 126(2): 321–34. [DOI] [PubMed] [Google Scholar]
- 29.Andriana Margariti, Anna Zampetaki, Qingzhong Xiao, Boda Zhou, Eirini Karamariti, Daniel Martin, Xiaoke Yin, Manuel Mayr, Hongling Li, Zhongyi Zhang, Elena De Falco, Yanhua Hu, Gillian Cockerill, Qingbo Xu, Lingfang Zeng.Histone Deacetylase 7 Controls Endothelial Cell Growth Through Modulation of Beta-Catenin. Circ Res. 2010; 106 (7): 1202–11. [DOI] [PubMed] [Google Scholar]
- 30.Kato H, Tamamizu-Kato S and Shibasaki F Histone deacetylase 7 associates with hypoxia-inducible factor 1alpha and increases transcriptional activity. J Biol Chem. 2004; 279(40): 41966–74. [DOI] [PubMed] [Google Scholar]
- 31.To M, Yamamura S, Akashi K, Charron CE, Haruki K, Barnes PJ, Ito K. Defect of adaptation to hypoxia in patients with COPD due to reduction of histone deacetylase 7. Chest. 2012; 141(5): 1233–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu H Chen W, Ren J, Zhang T, Yang K, Wu G, Liu H. VEGF-PKD1-HDAC7 signaling promotes endothelial progenitor cell migration and tube formation. Microvasc Res. 2014; 91: 66–72. [DOI] [PubMed] [Google Scholar]
- 33.Ha CH, Jhun BS, Kao HY, Jin ZG. VEGF stimulates HDAC7 phosphorylation and cytoplasmic accumulation modulating matrix metalloproteinase expression and angiogenesis. Arterioscler Thromb Vasc Biol. 2008; 28(10): 1782–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marek M, Shaik TB, Heimburg T, Chakrabarti A, Lancelot J, Ramos-Morales E, Da Veiga C, Kalinin D, Melesina J, Robaa D, Schmidtkunz K, Suzuki T, Holl R, Ennifar E, Pierce RJ, Jung M, Sippl W, Romier C.Characterization of Histone Deacetylase 8 (HDAC8) Selective Inhibition Reveals Specific Active Site Structural and Functional Determinants. J Med Chem. 2018;61(22):10000–10016. [DOI] [PubMed] [Google Scholar]
- 35.Kee HJ, Ryu Y, Seok YM, Choi SY, Sun S, Kim GR, Jeong MH. Selective inhibition of histone deacetylase 8 improves vascular hypertrophy, relaxation, and inflammation in angiotensin II hypertensive mice. Clin Hypertens. 2019;25:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lobera M, Madauss KP, Pohlhaus DT, Wright QG, Trocha M, Schmidt DR, Baloglu E, Trump RP, Head MS, Hofmann GA, Murray-Thompson M, Schwartz B, Chakravorty S, Wu Z, Mander PK, Kruidenier L, Reid RA, Burkhart W, Turunen BJ, Rong JX, Wagner C, Moyer MB, Wells C, Hong X, Moore JT, Williams JD, Soler D, Ghosh S, Nolan MA. Selective class Ila histone deacetylase inhibition via a nonchelating zincbinding group. Nat Chem Biol. 2013;9(5):319–25. [DOI] [PubMed] [Google Scholar]
- 37.Su L, Liang D, Kuang SY, Dong Q, Han X, Wang Z. Neuroprotective mechanism of TMP269, a selective class IIA histone deacetylase inhibitor, after cerebral ischemia/reperfusion injury. Neural Regen Res. 2020;15(2):277–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shi W, Wei X, Wang Z, Han H, Fu Y, Liu J, Zhang Y, Guo J, Dong C, Zhou D, Zhou Q, Chen Y, Yi F. HDAC9 exacerbates endothelial injury in cerebral ischaemia/reperfusion injury. J Cell Mol Med. 2016;20(6):1139–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Duan B, Ye D, Zhu S, Jia W, Lu C, Wang G, Guo X, Yu Y, Wu C, Kang J. HDAC10 promotes angiogenesis in endothelial cells through the PTPN22/ERK axis. Oncotarget. 2017;8(37):61338–61349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou B, Zeng S, Li N, Yu L, Yang G, Yang Y, Zhang X, Fang M, Xia J, Xu Y. Angiogenic Factor With G Patch and FHA Domains 1 Is a Novel Regulator of Vascular Injury. Arterioscler Thromb Vasc Biol. 2017;37(4):675–684. [DOI] [PubMed] [Google Scholar]
- 41.Potente M, Ghaeni L, Baldessari D, Mostoslavsky R, Rossig L, Dequiedt F, Haendeler J, Mione M, Dejana E, Alt FW, Zeiher AM, Dimmeler S. SIRT1 controls endothelial angiogenic functions during vascular growth. Genes Dev. 2007; 21(20): 2644–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li J, Rinaldo F E, Datta K Upregulation of VEGF-C by androgen depletion: the involvement of IGF-IR-FOXO pathway. Oncogene. 2005; 24(35): 5510–20. [DOI] [PubMed] [Google Scholar]
- 43.Sedding DG, FoxO transcription factors in oxidative stress response and ageing--a new fork on the way to longevity? Biol Chem. 2008; 389(3): 279–83. [DOI] [PubMed] [Google Scholar]
- 44.Wan Z, Yu W, Chen Y, Dai YT. Deacetylase SIRT1 and vascular endothelial function]. Zhonghua Nan Ke Xue. 2012; 18(9): 831–4. [PubMed] [Google Scholar]
- 45.Dharaneeswaran H, Abid MR, Yuan L, Dupuis D, Beeler D, Spokes KC, Janes L, Sciuto T, Kang PM, Jaminet SS, Dvorak A, Grant MA, Regan ER, Aird WC. FOXO1-mediated activation of Akt plays a critical role in vascular homeostasis. Circ Res. 2014;115(2): 238–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yan L, Wei X, Tang QZ, Feng J, Zhang Y, Liu C, Bian ZY, Zhang LF, Chen M, Bai X, Wang AB, Fassett J, Chen Y, He YW, Yang Q, Liu PP, Li H. Cardiac-specific mindin overexpression attenuates cardiac hypertrophy via blocking AKT/GSK3beta and TGF-beta1-Smad signalling. Cardiovasc Res.2011; 92(1): 85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hashimoto-Komatsu A, Hirase T, Asaka M, Node K. Angiotensin II induces microtubule reorganization mediated by a deacetylase SIRT2 in endothelial cells. Hypertens Res 2011; 34(8):949–56. [DOI] [PubMed] [Google Scholar]
- 48.Cardus A, Uryga AK, Walters G, Erusalimsky JD. SIRT6 protects human endothelial cells from DNA damage, telomere dysfunction, and senescence. Cardiovasc Res. 2013; 97(3): 571–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Findeisen HM, Gizard F, Zhao Y, Qing H, Heywood EB, Jones KL, Cohn D, Bruemmer D. Epigenetic regulation of vascular smooth muscle cell proliferation and neointima formation by histone deacetylase inhibition. Arterioscler Thromb Vasc Biol. 2011;31(4):851–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhao L, Chen CN, Hajji N, Oliver E, Cotroneo E, Wharton J, Wang D, Li M, McKinsey TA, Stenmark KR, Wilkins MR Histone deacetylation inhibition in pulmonary hypertension: therapeutic potential of valproic acid and suberoylanilide hydroxamic acid. Circulation. 2012;126(4):455–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Galletti M, Cantoni S, Zambelli F, Valente S, Palazzini M, Manes A, Pasquinelli G, Mai A, Galiè N, Ventura C. Dissecting histone deacetylase role in pulmonary arterial smooth muscle cell proliferation and migration. Biochem Pharmacol. 2014;91(2):181–90. [DOI] [PubMed] [Google Scholar]
- 52.Singh N, Trivedi CM, Lu M, Mullican SE, Lazar MA, Epstein JA. Histone deacetylase 3 regulates smooth muscle differentiation in neural crest cells and development of the cardiac outflow tract. Circ Res. 2011; 109(11): 1240–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kavurma MM, Figg N, Bennett MR, Mercer J, Khachigian LM, Littlewood TD. Oxidative stress regulates IGF1R expression in vascular smooth-muscle cells via p53 and HDAC recruitment. Biochem J. 2007; 407(1): 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Waltregny D, De Leval L, Glénisson W, Ly Tran S, North BJ, Bellahcène A, Weidle U, Verdin E, Castronovo V. Expression of histone deacetylase 8, a class I histone deacetylase, is restricted to cells showing smooth muscle differentiation in normal human tissues. Am J Pathol. 2004; 165(2): 553–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Waltregny D, Glénisson W, Tran SL, North BJ, Verdin E, Colige A, Castronovo V. Histone deacetylase HDAC8 associates with smooth muscle alpha-actin and is essential for smooth muscle cell contractility. FASEB J. 2005; 19(8): 966–8. [DOI] [PubMed] [Google Scholar]
- 56.Yang Q, Dahl MJ, Albertine KH, Ramchandran R, Sun M, Raj JU. Role of histone deacetylases in regulation of phenotype of ovine newborn pulmonary arterial smooth muscle cells. Cell Prolif. 2013;46(6):654–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tang Y, Boucher JM, Liaw L. Histone deacetylase activity selectively regulates notch- mediated smooth muscle differentiation in human vascular cells. J Am Heart Assoc. 2012;1 (3):e000901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ellis JJ, Valencia TG, Zeng H, Roberts LD, Deaton RA, Grant SR CaM kinase IldeltaC phosphorylation of 14–3-3beta in vascular smooth muscle cells: activation of class II HDAC repression. Mol Cell Biochem. 2003; 242(1–2): 153–61. [PubMed] [Google Scholar]
- 59.Ginnan R, Sun LY, Schwarz JJ, Singer HA. MEF2 is regulated by CaMKIIdelta2 and a HDAC4-HDAC5 heterodimer in vascular smooth muscle cells. Biochem J. 2012; 444(1): 105–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li H, Li W, Gupta AK, Mohler PJ, Anderson ME, Grumbach IM. Calmodulin kinase II is required for angiotensin II-mediated vascular smooth muscle hypertrophy. Am J Physiol Heart Circ Physiol. 2010; 298(2): H688–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yan ZQ, Yao QP, Zhang ML, Qi YX, Guo ZY, Shen BR, Jiang ZL. Histone deacetylases modulate vascular smooth muscle cell migration induced by cyclic mechanical strain. J Biomech. 2009; 42(7): 945–8. [DOI] [PubMed] [Google Scholar]
- 62.Choi SY, Kee HJ, Sun S, Seok YM, Ryu Y, Kim GR, Kee SJ, Pflieger M, Kurz T, Kassack MU, Jeong MH. Histone deacetylase inhibitor LMK235 attenuates vascular constriction and aortic remodelling in hypertension. J Cell Mol Med. 2019;23(4):2801–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mathew OP, Ranganna K, Mathew J, Zhu M, Yousefipour Z, Selvam C, Milton SG. Cellular effects of butyrate on vascular smooth muscle cells are mediated through disparate actions on dual targets, histone deacetylase (HDAC) activity and PI3K/Akt signaling network. Int J Mol Sci 2019;20(12). pii: E2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kozikowski AP, Chen Y, Gaysin A, Chen B, D’Annibale MA, Suto CM, Langley BC. Functional differences in epigenetic modulators-superiority of mercaptoacetamide-based histone deacetylase inhibitors relative to hydroxamates in cortical neuron neuroprotection studies. J Med Chem. 2007;50(13):3054–61. [DOI] [PubMed] [Google Scholar]
- 65.Porter NJ, Shen S, Barinka C, Kozikowski AP, Christianson DW.Molecular Basis for the Selective Inhibition of Histone Deacetylase 6 by a Mercaptoacetamide Inhibitor. ACS Med Chem Lett. 2018;9(12):1301–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen J, Zhang J, Shaik NF, Yi B, Wei X, Yang XF, Naik UP, Summer R, Yan G, Xu X, Sun J. The histone deacetylase inhibitor tubacin mitigates endothelial dysfunction by up- regulating the expression of endothelial nitric oxide synthase. J Biol Chem. 2019;294(51):19565–19576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Leucker TM, Nomura Y, Kim JH, Bhatta A, Wang V, Wecker A, Jandu S, Santhanam L, Berkowitz D, Romer L, Pandey D. Cystathionine y-lyase protects vascular endothelium: a role for inhibition of histone deacetylase 6. Am J Physiol Heart Circ Physiol. 2017;312(4):H711–H720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Miyazaki R, Ichiki T, Hashimoto T, Inanaga K, Imayama I, Sadoshima J, Sunagawa K. SIRT1, a longevity gene, downregulates angiotensin II type 1 receptor expression in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2008; 28(7): 1263–9. [DOI] [PubMed] [Google Scholar]
- 69.Li L, Gao P, Zhang H, Chen H, Zheng W, Lv X, Xu T, Wei Y, Liu D, Liang C. SIRT1 inhibits angiotensin II-induced vascular smooth muscle cell hypertrophy. Acta Biochim Biophys Sin (Shanghai). 2011; 43(2): 103–9. [DOI] [PubMed] [Google Scholar]
- 70.Gao P, Xu TT, Lu J, Li L, Xu J, Hao DL, Chen HZ, Liu DP. Overexpression of SIRT1 in vascular smooth muscle cells attenuates angiotensin II-induced vascular remodeling and hypertension in mice. J Mol Med (Berl). 2014; 92(4): 347–57. [DOI] [PubMed] [Google Scholar]
- 71.Li L, Zhang HN, Chen HZ, Gao P, Zhu LH, Li HL, Lv X, Zhang QJ, Zhang R, Wang Z, She ZG, Zhang R, Wei YS, Du GH, Liu DP, Liang CC. SIRT1 acts as a modulator of neointima formation following vascular injury in mice. Circ Res. 2011; 108(10): 1180–9. [DOI] [PubMed] [Google Scholar]
- 72.Wu X, Bu P, Liu J, Zhao L, Wang X, Li N. Inhibitory effect of Sirt3 on proliferation of vascular smooth muscle cells induced by angiotensin II. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. 2013;29(3): 237–41. [PubMed] [Google Scholar]
- 73.Williams KJ Atherosclerosis: cell biology and lipoproteins. Curr Opin Lipidol, 1996; 7(6): U202–8. [DOI] [PubMed] [Google Scholar]
- 74.Williams KJ and Tabas I. The response-to-retention hypothesis of atherogenesis reinforced. Curr Opin Lipidol. 1998; 9(5): 471–4. [DOI] [PubMed] [Google Scholar]
- 75.Zernecke A and Weber C. Inflammatory mediators in atherosclerotic vascular disease. Basic Res Cardiol. 2005;100(2): 93–101. [DOI] [PubMed] [Google Scholar]
- 76.Lucas AD and Greaves DR. Atherosclerosis: role of chemokines and macrophages. Expert Rev Mol Med. 2001; 3(25): 1–18. [DOI] [PubMed] [Google Scholar]
- 77.Boyle JJ. Macrophage activation in atherosclerosis: pathogenesis and pharmacology of plaque rupture. Curr Vasc Pharmacol. 2005; 3(1): 63–8. [DOI] [PubMed] [Google Scholar]
- 78.Carter AM. Inflammation. thrombosis and acute coronary syndromes. Diab Vasc Dis Res. 2005; 2(3): 113–21. [DOI] [PubMed] [Google Scholar]
- 79.Johnson JL and Newby AC. Macrophage heterogeneity in atherosclerotic plaques. Curr Opin Lipidol, 2009;20(5): 370–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Manea SA, Vlad ML, Fenyo IM, Lazar AG, Raicu M, Muresian H, Simionescu M, Manea A. Pharmacological inhibition of histone deacetylase reduces NADPH oxidase expression, oxidative stress and the progression of atherosclerotic lesions in hypercholesterolemic apolipoprotein E-deficient mice; potential implications for human atherosclerosis. Redox Biol. 2020;28:101338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li Y, Zhang K, Mao W. Inhibition of miR-34a prevents endothelial cell apoptosis by directly targeting HDAC1 in the setting of atherosclerosis. Mol Med Rep. 2018;17(3):4645–4650. [DOI] [PubMed] [Google Scholar]
- 82.Pandey D, Sikka G, Bergman Y, Kim JH, Ryoo S, Romer L, Berkowitz D. Transcriptional regulation of endothelial arginase 2 by histone deacetylase 2. Arterioscler Thromb Vasc Biol. 2014;34(7): 1556–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zampetaki A, Zeng L, Margariti A, Xiao Q, Li H, Zhang Z, Pepe AE, Wang G, Habi O, deFalco E, Cockerill G, Mason JC, Hu Y, Xu Q. Histone deacetylase 3 is critical in endothelial survival and atherosclerosis development in response to disturbed flow. Circulation.2010;121(1): 132–42. [DOI] [PubMed] [Google Scholar]
- 84.Van den Bossche J, Neele AE, Hoeksema MA, de Heij F, Boshuizen MC, van der Velden S, de Boer VC, Reedquist KA, de Winther MP.Inhibiting epigenetic enzymes to improve atherogenic macrophage functions. Biochem Biophys Res Commun. 2014;455(3–4):396–402. [DOI] [PubMed] [Google Scholar]
- 85.Hoeksema MA, Gijbels MJ, Van den Bossche J, van der Velden S, Sijm A, Neele AE, Seijkens T, Stöger JL, Meiler S, Boshuizen MC, Dallinga-Thie GM, Levels JH, Boon L, Mullican SE, Spann NJ, Cleutjens JP, Glass CK, Lazar MA, de Vries CJ, Biessen EA, Daemen MJ, Lutgens E, de Winther MP. Targeting macrophage Histone deacetylase 3 stabilizes atherosclerotic lesions. EMBO Mol Med. 2014;6(9):1124–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jung SB, Kim CS, Naqvi A, Yamamori T, Mattagajasingh I, Hoffman TA, Cole MP, Kumar A, Dericco JS, Jeon BH, Irani K. Histone deacetylase 3 antagonizes aspirin- stimulated endothelial nitric oxide production by reversing aspirin-induced lysine acetylation of endothelial nitric oxide synthase. Circ Res. 2010;107(7):877–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang QJ, Wang Z, Chen HZ, Zhou S, Zheng W, Liu G, Wei YS, Cai H, Liu DP, Liang CC. Endothelium-specific overexpression of class III deacetylase SIRT1 decreases atherosclerosis in apolipoprotein E-deficient mice. Cardiovasc Res. 2008; 80(2): 191–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Stein S and Matter CM. Protective roles of SIRT1 in atherosclerosis. Cell Cycle.2011; 10(4): 640–7. [DOI] [PubMed] [Google Scholar]
- 89.Kong X, Fang M, Li P, Fang F, Xu Y. HDAC2 deacetylates class II transactivator and suppresses its activity in macrophages and smooth muscle cells. J Mol Cell Cardiol. 2009;46(3): 292–9 [DOI] [PubMed] [Google Scholar]
- 90.Leucker TM, Nomura Y, Kim JH, Bhatta A, Wang V, Wecker A, Jandu S, Santhanam L, Berkowitz D, Romer L, Pandey D. Cystathionine y-lyase protects vascular endothelium: a role for inhibition of histone deacetylase 6. Am J Physiol Heart Circ Physiol. 2017;312(4):H711–H720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pietruczuk P, Jain A, Simo-Cheyou ER, Anand-Srivastava MB, Srivastava AK. Protein kinase B/AKT mediates insulin-like growth factor 1-induced phosphorylation and nuclear export of histone deacetylase 5 via NADPH oxidase 4 activation in vascular smooth muscle cells. J Cell Physiol. 2019;234(10):17337–17350. [DOI] [PubMed] [Google Scholar]
- 92.International Stroke Genetics Consortium (ISGC); Wellcome Trust Case Control Consortium 2 (WTCCC2),Bellenguez C, Bevan S, Gschwendtner A, Spencer CC, Burgess AI, Pirinen M, Jackson CA, Traylor M, Strange A, Su Z, Band G, Syme PD, Malik R, Pera J, Norrving B, Lemmens R, Freeman C, Schanz R, James T, Poole D, Murphy L, Segal H, Cortellini L, Cheng YC, Woo D, Nalls MA, Müller-Myhsok B, Meisinger C, Seedorf U, Ross-Adams H, Boonen S, Wloch-Kopec D, Valant V, Slark J, Furie K, Delavaran H, Langford C, Deloukas P, Edkins S, Hunt S, Gray E, Dronov S, Peltonen L, Gretarsdottir S, Thorleifsson G, Thorsteinsdottir U, Stefansson K, Boncoraglio GB, Parati EA, Attia J, Holliday E, Levi C, Franzosi MG, Goel A, Helgadottir A, Blackwell JM, Bramon E, Brown MA, Casas JP, Corvin A, Duncanson A, Jankowski J, Mathew CG, Palmer CN, Plomin R, Rautanen A, Sawcer SJ, Trembath RC, Viswanathan AC, Wood NW, Worrall BB, Kittner SJ, Mitchell BD, Kissela B, Meschia JF, Thijs V, Lindgren A, Macleod MJ, Slowik A, Walters M, Rosand J, Sharma P, Farrall M, Sudlow CL, Rothwell PM, Dichgans M, Donnelly P, Markus HS. Genome-wide association study identifies a variant in HDAC9 associated with large vessel ischemic stroke. Nat Genet. 2012; 44(3): 328–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Markus HS, Mäkelä KM, Bevan S, Raitoharju E, Oksala N, Bis JC, O’Donnell Hainsworth A, Lehtimaki T. Evidence HDAC9 genetic variant associated with ischemic stroke increases risk via promoting carotid atherosclerosis. Stroke. 2013; 44(5): 1220–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cao Q, Rong S, Repa JJ, St Clair R, Parks JS, Mishra N. Histone deacetylase 9 represses cholesterol efflux and alternatively activated macrophages in atherosclerosis development. Arterioscler Thromb Vasc Biol. 2014; 34(9): 1871–9. [DOI] [PMC free article] [PubMed] [Google Scholar]