

Abstract
The hepatitis C virus (HCV) nonstructural 5A, a phosphorylated zinc metalloprotein, is an essential component of the HCV replication complex. An amphipathic α-helical peptide (HCV peptide [C5A]) derived from nonstructural 5A membrane anchor domain possesses potent anti-HCV and anti-HIV activity in vitro. In this study, we aimed to investigate the potential of HCV peptide (C5A) to regulate host immune responses. The capacity of HCV peptide (C5A) in vitro to induce migration and calcium mobilization of human phagocytes and chemoattractant receptor-transfected cells was investigated. The recruitment of phagocytes in vivo induced by HCV peptide (C5A) and its adjuvant activity were examined. The results revealed that HCV peptide (C5A) was a chemoattractant and activator of human phagocytic leukocytes by using a G-protein coupled receptor, namely formyl peptide receptor. In mice, HCV peptide (C5A) induced massive phagocyte infiltration after injection in the air pouch or the s.c. region. HCV peptide (C5A) also acted as an immune adjuvant by enhancing specific T cell responses to Ag challenge in mice. Our results suggest that HCV peptide (C5A) derived from HCV regulates innate and adaptive immunity in the host by activating the formyl peptide receptor.
Chemoattractants, including chemokines and classic chemotactic factors, are small molecules that play an important role in mediating leukocyte trafficking, hematopoiesis, wound healing, allergy, angiogenesis, HIV infection, and malignancy (1–3). The biologic effects of chemoattractants are mediated via their specific G-protein-coupled receptors. Most chemoattractant receptors can be stimulated by several chemoattractants (4–6). Indeed, the classic chemotactic factor receptor formyl peptide receptor (FPR) binds the bacterial peptide N-formyl-methionyl-leucyl-phenylalanine (fMLF), virus HIV-derived peptide T20 and T21 (7, 8), and endogenous ligands including formyl peptides released by mitochondria of ruptured cells (9,10), annexin I produced by activated epithelia (11), and a neutrophil granule protein, cathepsin G (12). Functional FPR has been detected in phagocytic leukocytes and cells of the non-hematopoietic origin (13–17), such as lung epithelial cells (16) and hepatocytes (17). These findings suggest that FPR may be involved in a broader spectrum of pathophysiologic processes, including viral infection.
Previous studies showed that chemoattractants have an important role in hepatitis C virus (HCV) infection (18–20). The levels of many chemokines in liver tissue or in serum are increased during HCV hepatitis (19, 20). The expression level of the chemokine CXCL10 and its receptor CXCR3 correlates with liver damage (19). Patients with HCV had a lower proportion of peripheral blood CD8 T cells expressing chemokine receptors CCR1 and CCR5; this was associated with significantly reduced migration of CD8 T cells from such patients in response to the CCR1 and CCR5 chemokine agonists in vitro (21). Myeloid dendritic cells (DCs) generated from patients with HCV were less mature. Myeloid and plasmacytoid DCs from these patients express activated receptors for inflammatory chemokines (22), indicating activation of DCs by chemokines produced by the host during HCV infection. It has also been reported that HCV NS2 and NS3/4A proteins bind to chemokine gene promoters and downregulate the expression of genes for chemokines CCL5, CXCL8, and CXCL10 in host cells (23, 24). These observations suggest the capacity of viral components to regulate host responses during HCV infection. However, the involvement of FPR in HCV infection remains unclear.
The HCV nonstructural (NS) protein NS5A is cleaved by caspases 3 and 6 and by Ca2+-dependent calpain proteases resulting in the formation of N-terminally truncated NS5A fragments (25–27). The truncated NS5A fragments accumulate in the nucleus and cytoplasm and impair HCV replication (28). These fragments might be released by damaged cells and interact with phagocytic cells to induce host defense responses during HCV infection. Recently, an amphipathic α-helical peptide (HCV peptide [C5A]) derived from the membrane anchor domain of the NS5A protein has been reported to be virucidal to HCV at submicromolar concentrations in vitro (29). HCV peptide (C5A) prevents de novo HCV infection and suppresses ongoing infection by inactivating both extracellular and intracellular infectious particles. Importantly, the peptide is nontoxic to normal host cells in vitro and in vivo at doses at least 100-fold higher than are required for antiviral activity. In addition to HCV, HCV peptide (C5A) inhibits infection by selected flaviviruses, paramyxoviruses, and HIV (29, 30). In order to understand the potential effects of HCV components on host immune responses, we tested four peptide fragments derived from the membrane anchor domain of NS5A protein (HCV peptide [C5A]) and the putative CD81 binding regions (R1, R2, R3) of transmembrane envelope glycoproteins (E2) of HCV (31) for their capacity to activate human leukocytes. We report that HCV peptide (C5A) is a chemoattractant and activator of human phagocytic leukocytes by using a G-protein coupled formyl peptide receptor FPR.
Materials and Methods
Reagents, cells, and animals
HCV peptide (C5A), an amphipathic α-helical peptide derived from the membrane anchor domain of NS5A protein, a scrambled control of HCV peptide (C5Ascr), and three putative CD81 binding regions of transmembrane envelope glycoproteins (E2) of HCV were synthesized and purified by Pi Proteomics, LLC (Huntsville, AL), according to the published sequences: HCV peptide (C5A): SWLRDIWDWICEVLSDFK-NH2 (aa 3–20 of NS5A) (5); Scrambled control of HCV peptide (C5Ascr): WSRDIWLWIDDVLSCFEK-NH2; R1:YANGSGLDERPYCWHYPPR-NH2 (19 aa, 474–492 of E2); R2: SGAPTYSWGANDTDVFVLNNTRPPLGNWFGG-NH2 (31 aa, 522–551 of E2); R3: PYRLWHYPC-NH2 (9 aa, 612–619 of E2). The purity of the peptides was >95%, and the amino acid composition were verified by mass spectrometry. The endotoxin levels in dissolved peptide were undetectable (<0.125 EU/ml; LAL QCL-1000 Kit; Lonza, Walkersville, MD). The synthetic formyl peptide fMLF was purchased from Sigma (St. Louis, MO). Human PBMCs were isolated from Leukopaks through the courtesy of Transfusion Medicine Department (National Institutes of Health Clinical Center, Bethesda, MD). Monocytes were further purified by elutriation to yield >90% pure preparation. Neutrophils were purified from the same Leukopaks by 3% dextran sedimentation with a purity of 98%. Rat basophilic leukemia cells stably transfected with epitope-tagged FPR (ETFR) were a gift from Dr. R. Snyderman (Duke University, Durham, NC). The cells were designated ETFR and were grown in DMEM, 10% FCS, and 0.8 mg/ml geneticin (G418) to maintain selection pressure. The FPRL1 cDNA was cloned and transfected into human embryonic kidney cells 293 (designated FPRL1/293 cells), as reported previously (32). The cells were maintained in DMEM, 10% FCS, and 2 mg/ml Geneticin. Female C57BL/6 (B6; H-2b) mice, 8–10 wk of age, were purchased from the animal facility at the Sun Yat-Sen University (Guangzhou, China). All mice were treated according to the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research.
Chemotaxis
Leukocyte, ETFR, and FPRL1/293 cell migration was assessed using a 48-well microchemotaxis chamber technique, as described previously (7, 32). Different concentrations of stimulants were placed in wells of the lower compartment of the chamber (Neuro Probe, Cabin John, MA), and 50 μl of the cell suspension (2 × 106 cells/ml) was added in wells of the upper compartment, which was separated from the lower compartment by a polycarbonate filter (pore size: 5 μm for leukocytes, 10 μm for ETFR and FPRL1/293 cells). After incubation at 37°C (90 min for monocytes, 60 min for neutrophils, 180 min for T cells, and 300 min for ETFR or FPRL1/293 cells), migrated cells through the pores (the cells attached to the other side of the membrane and fallen into the bottoms of the lower wells) were collected and counted by FACSCalibur (BD Biosciences, San Jose, CA). The experiments were performed at least five times with each cell type, and the results are presented as the percentage of migrated cells versus the total loading cells.
Calcium mobilization
Calcium mobilization in phagocytes was assayed as described previously (7, 32). Calcium mobilization in ETFR transfectants and parent rat basophilic leukemia cells was measured with the FlexStation II384 system (Molecular Devices) using Fluo-3AM as described previously (33). Florescence in the FlexStation II384 system was measured with the excitation wavelength at 485 nm and the emission wavelength at 525 nm, and it is shown as relative fluorescence units. The assays were performed at least three times, and results from representative experiments are shown.
Flow cytometric analysis
For the analysis of HCV peptide (C5A) binding to monocytes, the cells were incubated with 10 μM biotin-HCV peptide (C5A; with or without 10 μM unlabeled HCV peptide [C5A] or fMLF) for 30 min in PBS containing 0.02% sodium azide at 4°C. The unbound biotin-HCV peptide (C5A) was washed extensively with PBS containing 0.2% BSA and 0.02% sodium azide. Next, the cells were incubated with streptavidin-FITC at 4°C for 30 min in the dark. Subsequently, cells were washed twice in washing solution (0.5% BSA-PBS with 0.02% azide). The cells were analyzed in a single laser for FITC with FACSCalibur (BD Biosciences, San Jose, CA). For the analysis of HCV peptide (C5A) on the regulation of CD11b expression, monocytes were stimulated with fMLF (10−7 M) or HCV peptide (C5A; 10−5 M) at 37°C for 30 min. The cells were collected and stained with FITC-CD11b (Pharmigen, San Diego, CA) and analyzed.
Superoxide production
Human neutrophils were suspended in HBSS containing Ca2+ and Mg2+ at 106 cells/ml; 50 μl (5 × 104 cells) were distributed into wells of a 96-well microtiter chemiluminescence plate and incubated at 37°C for 5 min. Next, a mixture of the superoxide-specific chemiluminescence indicator reagent Diogenes (National Diagnostics, Atlanta, GA) was added to the cells (50% of total reaction volume) with fMLF (10−7 M), HCV peptide (C5A; 10−5 M), or vehicle control. Superoxide dismutase-inhibitable chemiluminescence was measured. To test the effect of HCV peptide (C5A) on LPS-primed cells, neutrophils were incubated with LPS (100 ng/ml) at 37°C for 20 min, and then superoxide was measured. Data are expressed as integrated luminescence (relative light units). A p value <0.05 indicates significant differences.
Degranulation assay
To measure β-glucuronidase release, neutrophils were preincubated with 10 μM cytochalasin B in HBSS containing 20 mM HEPES (pH 7.4) and 0.1% BSA for 15 min on ice, followed by 15 min at 37°C. The cells were then stimulated for 10 min with the indicated amounts of HCV peptide (C5A) or fMLF at 37°C. The reaction was terminated by placing samples on ice, and supernatant was immediately separated from cell pellet by centrifugation. The amount of β-glucuronidase was quantified by incubating 20 μl supernatant with 20 μl of 10 mM 4-methylumbelliferyl β-d-glucuronide hydrate in 0.1 M sodium acetate (pH 4.0) and 0.1% Triton X-100 at 37°C for 15 min. The reaction was terminated by adding 300 μl Stop Solution (pH 10.4) containing 50 mM glycine and 5 mM EDTA. Fluorescence was measured immediately with an excitation wavelength at 365 nm and an emission wavelength at 460 nm. Total cellular β-glucuronidase was determined with cell lysate in 0.1% Triton X-100. Data are presented as a percentage of total β-glucuronidase released.
MAPK assay
Human monocytes (5 × 106 cells/ml) were stimulated with fMLF (10−7 M) or HCV peptide (C5A; 10−5 M) at 37°C for 5 min. The reaction was terminated by brief centrifugation and adding 80 μl of ice-cold SDS-PAGE loading buffer after removing the supernatant. Samples were sonicated twice for 15 s each. After boiling, samples were analyzed by SDS-PAGE and Western blotting using anti-ERK1/2 and anti–phospho-ERK1/2 Abs (Cell Signaling Technology) at a 1/1000 dilution. After washing, the membrane was incubated with an HRP-conjugated anti-rabbit Ab (1/3000 dilution; Bio-Rad). The resulting immunocomplex was visualized using SuperSignal West Pico Chemiluminescence kit (Pierce), according to the manufacturer’s instructions.
Air pouch assay and exudate preparation
To evaluate the effect of HCV peptide (C5A) on in vivo cell migration, an air pouch assay was used. Air pouches were raised on the dorsum by s.c. injection of 3 ml sterile air on days 0 and 3. On day 6, mice with well-formed air pouches were randomized into three different groups, in which each mouse was injected with 1 ml endotoxin-free PBS alone or PBS containing 10 μM HCV peptide (C5A), or 100 nM fMLF into the air pouches. Four hours after the injection, the mice were sacrificed by CO2 asphyxiation, and the air pouches were washed once with 2 ml PBS containing 5 mM EDTA and 20 U/ml heparin. The exudates were centrifuged at 500 × g for 5 min at room temperature. Cells were counted with a hematocytometer following Trypan Blue staining. Characterization of leukocyte subpopulations migrating into the pouch space was performed by eosin-methylene blue staining of cytospin slides.
Measurement of phagocyte accumulation in skin
The backs of mice were shaved and 50 μl HCV peptide (C5A) at 10 μM or PBS was injected s.c. using 27-gauge needles. Ten hours later, the injected sites were removed and fixed in a 10% formaldehyde neutral buffer solution. Paraffin-embedded tissue was cut into 4-μm sections and stained with H&E.
Immunization and detection of splenocyte proliferation and cytokine production
Six- to 8-wk-old C57BL/6 mice (5 mice per group) were injected i.p. on day 0 with 0.2 ml PBS containing 50 μg OVA (Sigma-Aldrich) in the presence of alum (Sigma-Aldrich), 10 μM HCV peptide (C5A), or 1 μM fMLF. On day 14, all mice were booster immunized by i.p. injection of 0.2 ml PBS containing 50 μg OVA. On day 21, immunized mice were sacrificed to remove spleens. OVA-specific splenocyte proliferation, cytokine production, or both were measured as described previously with minor modifications (34). Splenocytes (5 × 105 cells/well) were seeded in triplicate in flat-bottom 96-well plates in complete RPMI 1640 medium (0.2 ml/well) and incubated in the presence or absence of the indicated concentrations of OVA at 37°C in a CO2 incubator for 72 h. MTT solution (10 μl of 5 mg/ml) was added to each well, and the cultures were further incubated for 4 h at 37°C. Cells were then resuspended in 100 μl of 0.04 M HCl/isopropanol solution, and the incubation was continued for 2 h to solubilize formazan violet crystals in the cells. The absorbance in each well was determined by spectrophotometry at the dual wavelengths of 570 and 630 nm on a microplate reader (Pharmacia, Sweden). For cytokine production, pooled splenocytes of each group were cultured in RPMI 1640 in 24-well plates (5 × 106 cells/ml/well) with 20 μg/ml of OVA for 48 h before culture supernatants were harvested for the measurement of cytokines (ELISA kits were obtained from R&D Systems).
Statistical analysis
Data were expressed as the mean ± SD. The unpaired Student t test was used to compare the differences between testing and control groups. Differences resulting in p ≤ 0.05 were considered to be statistically significant.
Results
HCV peptide (C5A) is a chemoattractant and activator of human monocytes and neutrophils
The effects of HCV peptide (C5A) were first investigated for their ability to induce human leukocyte migration, a crucial step for cell homing and accumulation at sites of inflammation or injury. This was measured by testing the migratory response of the cells to concentration gradients of the peptides placed in the bottom wells of a chemotaxis chamber. As shown in Fig. 1A and 1B, human peripheral blood monocytes and neutrophils, but not CD3+ T lymphocytes, migrated in response to HCV peptide (C5A) in a dose-dependent manner. The dose-response curve was bell shaped, a typical pattern shown by well-defined leukocyte chemoattractants, including fMLFs and chemokines (7, 32). However, HCV peptide (C5A) added simultaneously to the cells of the upper wells of the chamber abrogated the cell migration induced by equal concentrations of HCV peptide (C5A) in the lower wells (data not shown). Thus, HCV peptide (C5A)-induced migration of monocytes and neutrophils was based on chemotaxis rather than chemokinesis. The chemotactic activity of scrambled control of HCV peptide (C5Ascr) and other three synthetic peptides (R1, R2, and R3) derived from D81-binding regions of HCV transmembrane envelope glycoproteins (E2) (31) was also tested, but none of them induced the migration of human monocytes (Fig. 1C) and neutrophils (Fig. 1D).
FIGURE 1.

Induction of phagocyte migration by HCV peptide (C5A). Different concentrations of HCV peptide (C5A), scrambled control of HCV peptide (C5Ascr), and R1, R2, and R3 peptides were placed in the lower wells of the chemotaxis chamber; cell suspension was placed in the upper wells. The upper and lower wells were separated by polycarbonate filters. After incubation, the cells that migrated across the filters were stained and counted or collected and counted by FACSCalibur. A, Migration of human monocytes across the filters in response to 10 μM HCV peptide (C5A) or 100 nM fMLF (open arrow, pore; solid arrow, migrated monocyte; original magnification × 200). B, Percentage of leukocyte migration in response to HCV peptide (C5A) over total loading cells. C and D, The migration of monocytes (C) and neutrophils (D) in response to scrambled HCV peptide (C5Ascr), R1, R2, and R3. E, Inhibition of monocyte migration in response to HCV peptide (C5A) (10−5 M) or fMLF (10−7 M) by pretreatment of the cells with PT (100 ng/ml) or cholera toxin (CT, 100 ng/ml) for 30 min at 37°C.
We next examined whether HCV peptide (C5A)-induced monocyte and neutrophil migration was due to activation of a specific receptor. The migration of monocytes to HCV peptide (C5A) was completely inhibited by pretreatment of the cells with pertussis toxin (PT), but not cholera toxin (Fig. 1E). PT also completely inhibited the migration of neutrophils in response to HCV peptide (C5A; Fig. 1E), suggesting that a G-protein of the Gi-type coupled receptor was involved (3). This finding was supported by induction of a dose-dependent and pertussis toxin-sensitive calcium (Ca2+) mobilization in monocytes by HCV peptide (C5A; Fig. 2A, 2B). The capacity of HCV peptide (C5A) to desensitize cell response to subsequent stimulation with other chemoattractants was then tested to determine whether this peptide shared a G-protein–coupled receptor with other chemoattractants. HCV peptide (C5A) did not cross desensitize the Ca2+ flux induced by a number of chemokines, including CXCL8, CCL2, CCL5, CCL7, or CCL3 (Fig. 2C, 2D, and data not shown), suggesting that HCV peptide (C5A) does not share a receptor with any of these chemokines. However, the bacterial chemotactic peptide fMLF markedly desensitized HCV peptide (C5A)-induced Ca2+ mobilization in monocytes (Fig. 2E, 2F). The desensitization of Ca2+ flux between fMLF and HCV peptide (C5A) at equal concentrations (1 μmol/l) is shown in Fig. 2E and 2F. HCV peptide (C5A) had a negligible effect on subsequent cell response to fMLF stimulation. However, fMLF at 10−8 M desensitized the calcium mobilization induced by HCV peptide (C5A) at 10−6 M, whereas HCV peptide (C5A) at 10−6 M desensitized the calcium flux induced by fMLF at 10−8 M (Fig. 2E, 2F). These results suggest that fMLF is a more potent activator of phagocytes than HCV peptide (C5A). In addition, because HCV peptide (C5A) desensitized the effect of low-dose (nM) fMLF in induction of calcium flux in monocytes, we hypothesized that HCV peptide (C5A) might use a cell surface receptor, which interacts also with fMLF in the nanomole range on human phagocytic cells (3, 13).
FIGURE 2.
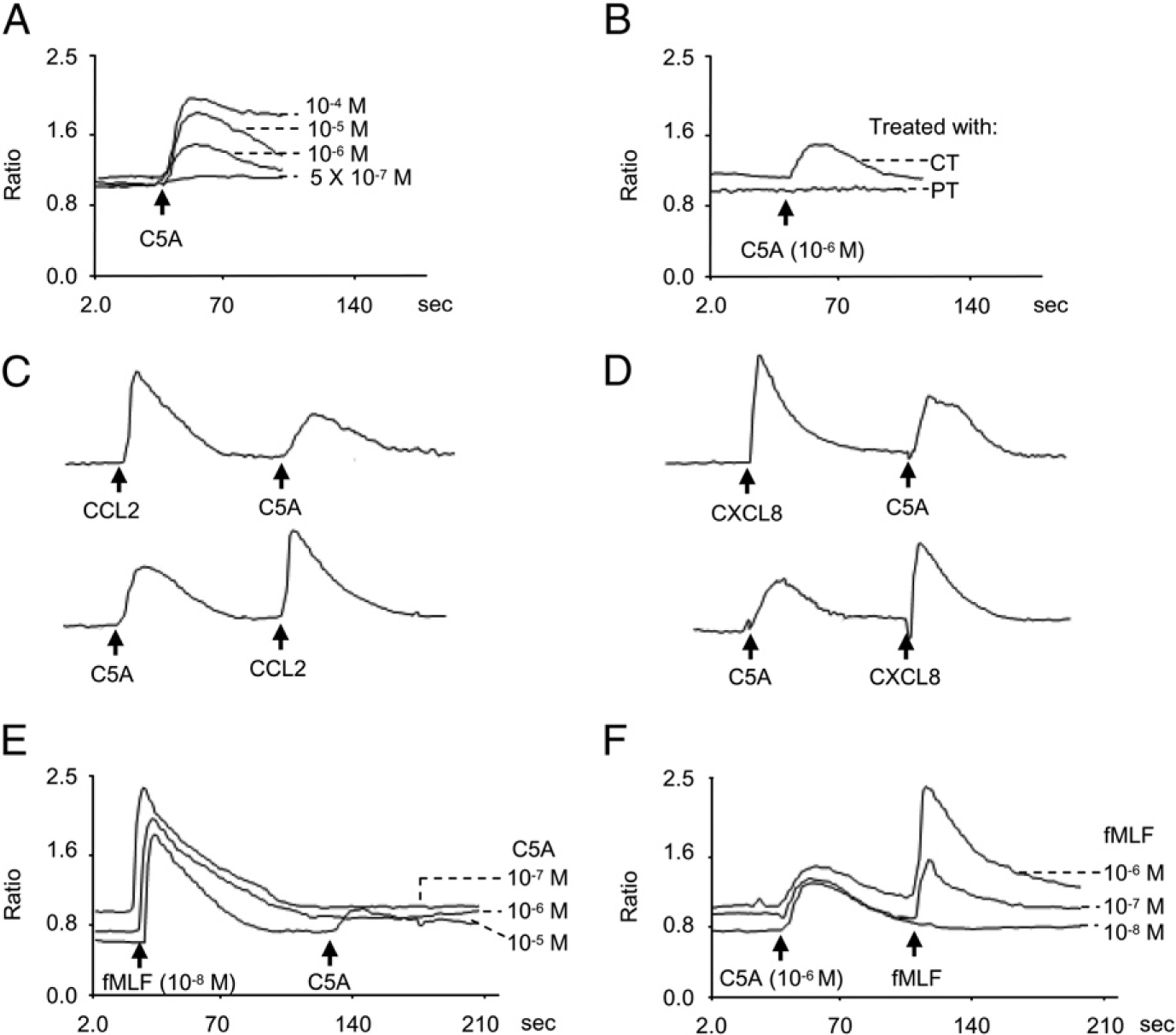
Calcium (Ca2+) mobilization in phagocytes induced by HCV peptide (C5A). Human phagocytes were loaded with Fura-2 and stimulated with HCV peptide (C5A). The ratio of fluorescence at 340 and 380 nm wavelength was recorded and calculated using the FLWinLab program. A, Dose response of HCV peptide (C5A) in monocytes. B, Inhibition of monocyte response to HCV peptide (C5A; 10−6 M) by pretreatment of the cells with PT (100 ng/ml) but not cholera toxin (CT; 100 ng/ml). C, Cross-desensitization of Ca2+ flux between 10−6 M of HCV peptide (C5A) and CCL2 in monocytes. D, Cross-desensitization of Ca2+ flux between 10−6 M HCV peptide (C5A) and CXCL8 in neutrophils. E and F, Desensitization of HCV peptide (C5A)-induced Ca2+ flux by 10−8 M fMLF (E) or desensitization of fMLF-induced Ca2+ flux by 1 μM HCV peptide (C5A) (F) in monocytes.
HCV peptide (C5A) may be a ligand for FPR on phagocytic cells
To determine whether HCV peptide (C5A) interacts with FPR, its variant FPRL1, or both, or the two cognate receptors for fMLF, we performed receptor-binding analyses and cross-desensitization experiments using flow cytometric analyses. Monocytes were stained with biotin-HCV peptide (C5A) and subsequently with streptavidin-FITC. The results showed that HCV peptide (C5A) bound to monocytes (Fig. 3A). The binding was markedly reduced by the addition of unlabeled HCV peptide (C5A) and fMLF. We next evaluated whether the FPR receptor antagonist Boc-PLPLPL could block the effects of HCV peptide (C5A). The results from cell migration assays showed that Boc-PLPLPL blocked the migration of monocytes induced by HCV peptide (C5A; Fig. 3B). These results indicate that HCV peptide (C5A) may bind to FPR on monocytes. To verify whether HCV peptide (C5A) uses FPR, we tested the effect of HCV peptide (C5A) on a rat basophil leukemia cell line transfected to express an epitope-tagged FPR (ETFR cells) (35). HCV peptide (C5A)-induced a dose-dependent Ca2+ mobilization in ETFR cells (Fig. 4A), but not in parental cells or mock-transfected cells (Fig. 4B), indicating that the response was mediated by FPRs in ETFR cells. We also examined the effect of HCV peptide (C5A) on 293 cells transfected with FPRL1 (FPRL1/293) cells, a low-affinity receptor for fMLF. Fig. 4C shows that T21 (8), an FPRL1 receptor cognate ligand, but not HCV peptide (C5A), induced calcium mobilization in FPRL1 transfectants. Thus, FPR was responsible for the unidirectional desensitization between fMLF and HCV peptide (C5A) in calcium mobilization in human phagocytes. Chemotaxis assays were then used as another sensitive biologic parameter to assess the usage of chemoattractant receptors by potential agonists, as reported in our previous studies (7, 32). ETFR cells were induced by fMLF to migrate across polycarbonatefilters coated with collagen type I, whereas HCV peptide (C5A) induced a similar migration of the ETFR cells, although with lower potency than fMLF (Fig. 4D, 4E). In contrast, HCV peptide (C5A) or fMLF did not induce migration of mock-transfected cells (data not shown). The chemotactic activity of HCV peptide (C5A) on cells transfected with FPRL1 was also examined, and the peptide did not induce FPRL1 cell migration at all concentrations tested (data not shown). These results indicate that HCV peptide (C5A) uses FPR as a functional receptor.
FIGURE 3.
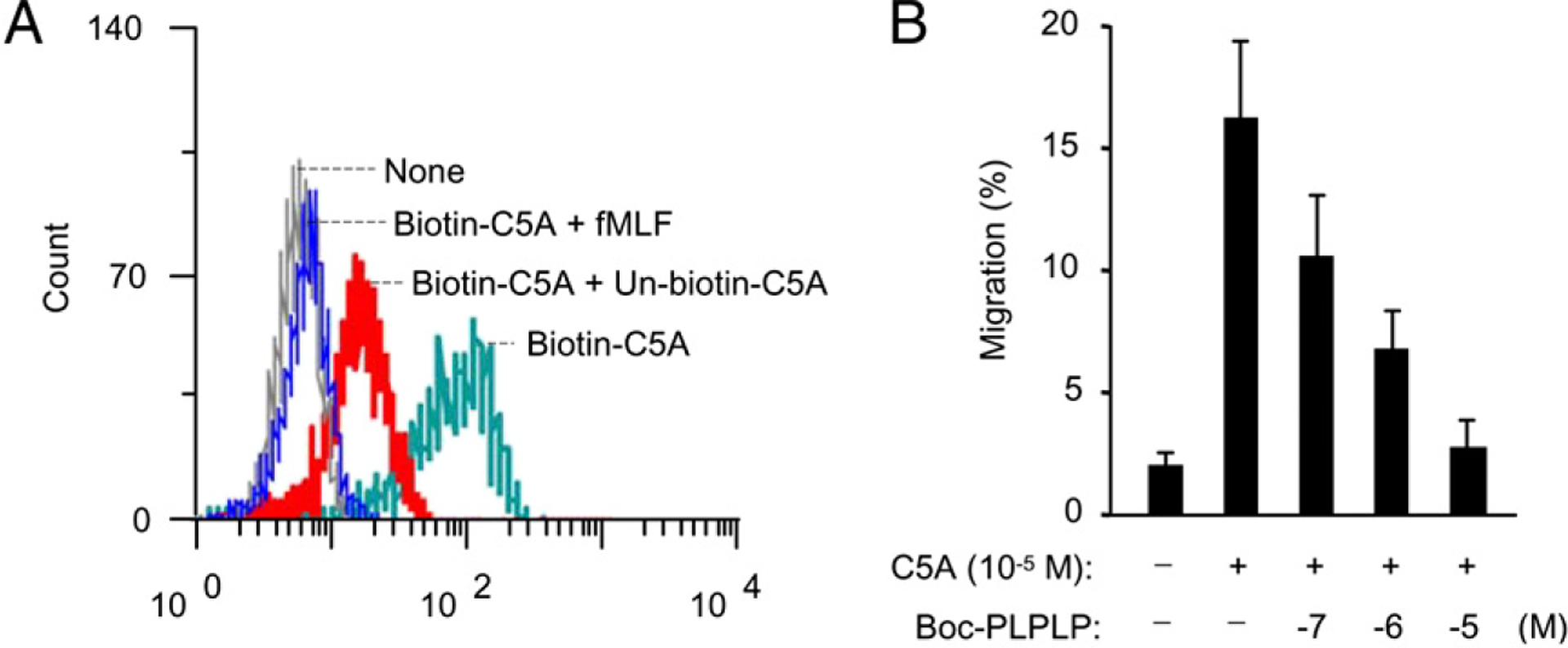
A, Flow cytometric analysis of HCV peptide (C5A) binding to monocytes. Monocytes (1 × 107 cells/ml) were stained with 10 μM biotin-HCV peptide (C5A) in the absence or presence of 10 μM HCV peptide (C5A) or 10 μM fMLF. The samples were further incubated with streptavidin-FITC and analyzed in FACSCalibur as described in Materials and Methods. B, Inhibition of monocyte migration in response to HCV peptide (C5A; 10−5 M) by pretreatment of fMLF antagonist Boc-PLPLPL as described in Materials and Methods.
FIGURE 4.
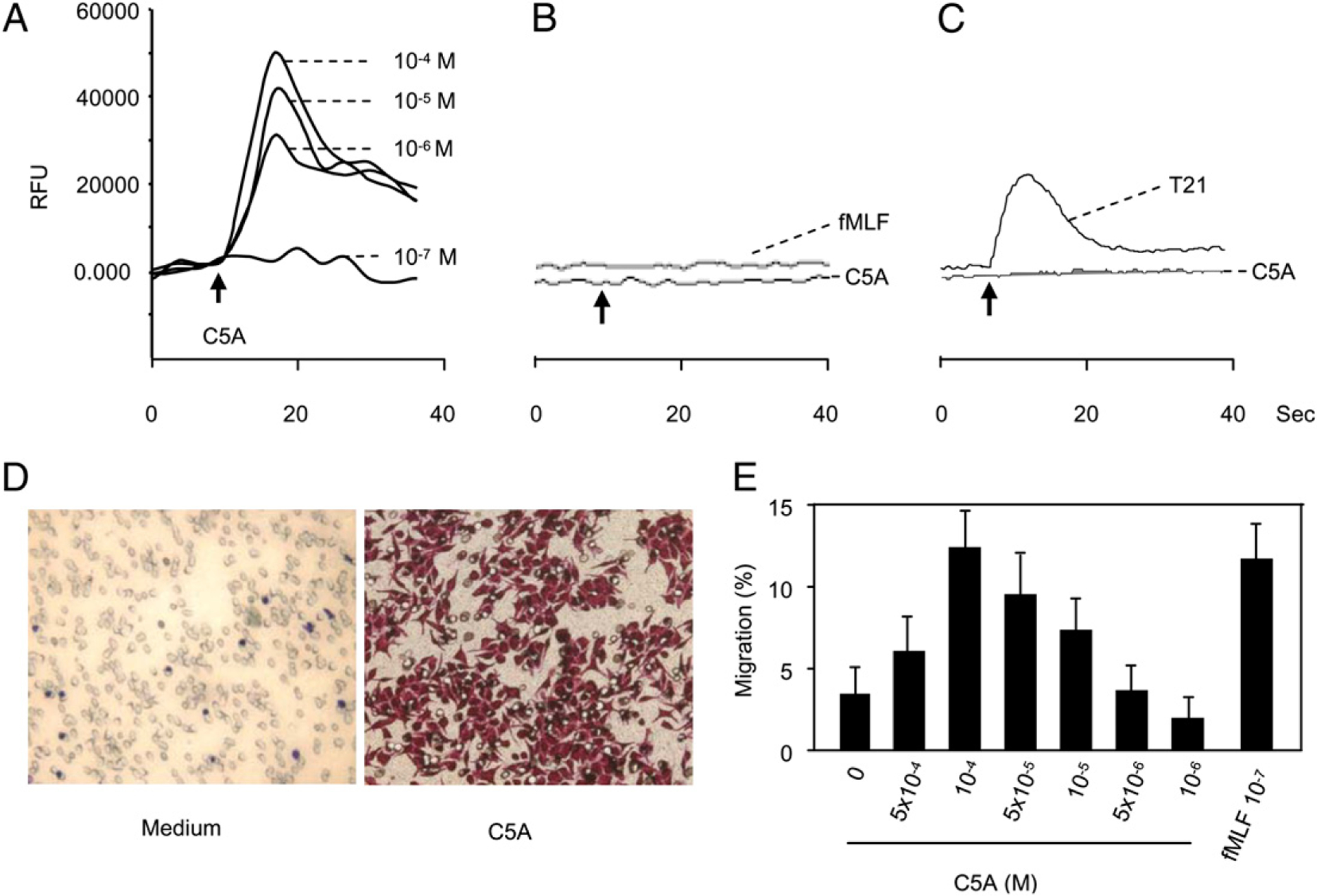
Activation of FPR transfected cells (ETFR cells) by HCV peptide (C5A). Calcium mobilization in ETFR tranfectants (A), rat basophilic leukemia parent cells (B), or FPRL1/293 cells (C) was measured with FlexStation II384 system using Fura-3AM. Changes in intracellular calcium concentration in response to agonists were recorded as relative fluorescence units (RFUs). D and E, Induction of ETFR cell migration by HCV peptide (C5A). Different concentrations of HCV peptide (C5A) were placed in the lower wells of the chemotaxis chamber; cell suspension was placed in the upper wells. The upper and lower wells were separated by polycarbonate filters. After incubation, the cells migrated across the filters were stained and counted or collected and counted by FACSCalibur. D, Migration of ETFR cells across the filters in response to 10−5 M HCV peptide (C5A; original magnification ×200). E, Percentage of ETFR cell migration in response to HCV peptide (C5A) in total loaded cells.
HCV peptide (C5A) induced superoxide production, degranulation, and MAPK activation and enhanced CD11b expression by phagocytes
Superoxide generation is an important bactericidal function of neutrophils. We examined whether HCV peptide (C5A) could induce superoxide production in neutrophils and, if so, whether this activity could be desensitized by prestimulation with fMLF. Fig. 5A shows that both HCV peptide (C5A) and fMLF could induce superoxide production in neutrophils. Furthermore, the effect of HCV peptide (C5A) was cross-desensitized by fMLF or vice versa. The response to HCV peptide (C5A) was markedly attenuated in LPS-primed cells (Fig. 5A). We next investigated whether HCV peptide (C5A) was able to induce the degranulation from neutrophils. Neutrophils were stimulated with fMLF and HCV peptide (C5A), and the release of β-glucuronidase was determined. HCV peptide (C5A) induced significant enzyme release, with an optimum concentration at 10 μM. However, this activity of HCV peptide (C5A) was ~100-fold less than fMLF (Fig. 5B). HCV peptide (C5A) could also induce ERK1/2 phosphorylation at 1 μM (Fig. 5C). In addition, similar to LPS or fMLF, HCV peptide (C5A) upregulated CD11b expression on monocytes (Fig. 5D). Irrelevant peptides R1, R2, and R3 derived from HCV failed to activate phagocytes in all experiments performed (data not shown). These data indicate that HCV peptide (C5A) is an activator of human monocytes.
FIGURE 5.
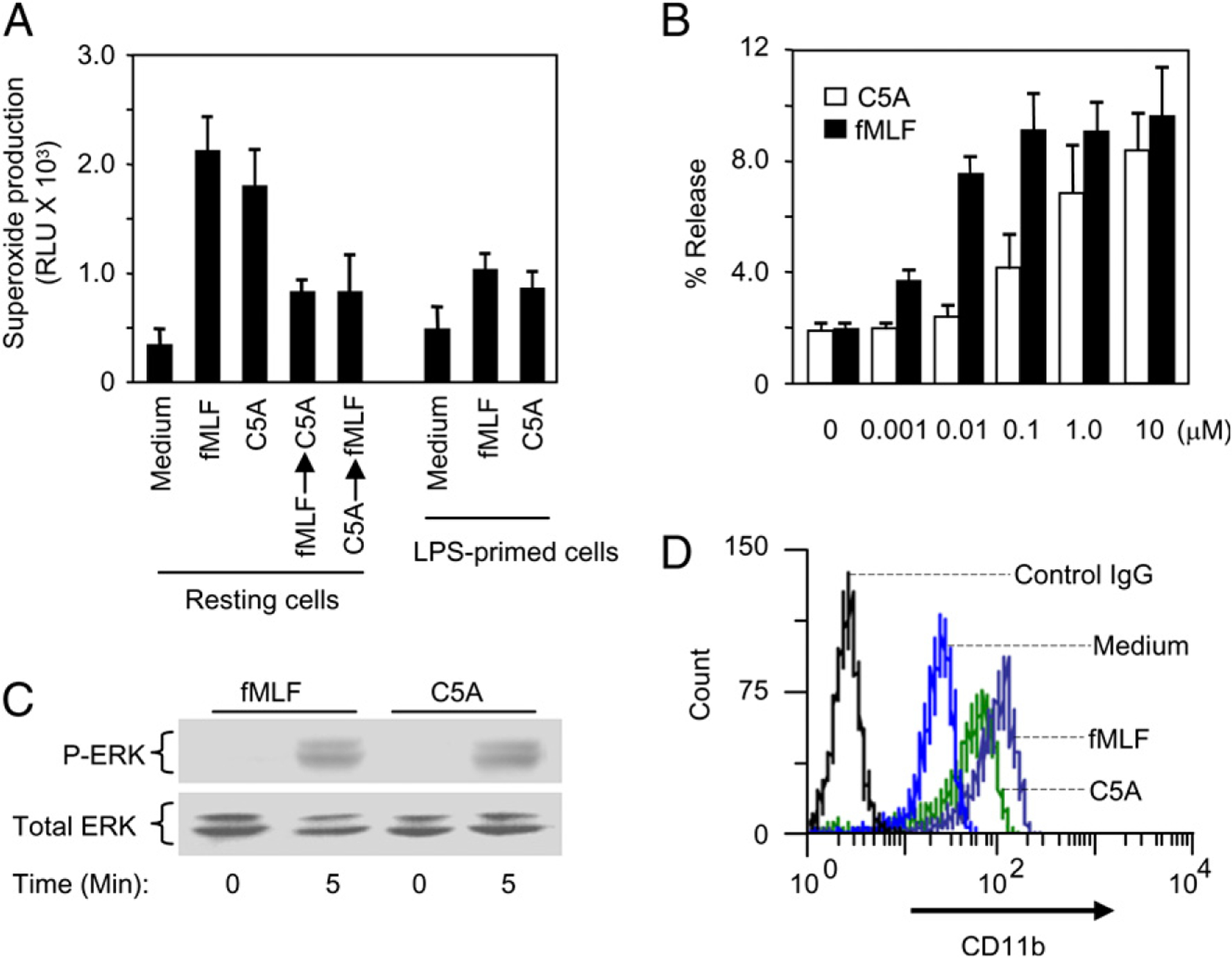
A, Induction of superoxide generation by HCV peptide (C5A) in neutrophils: Resting or LPS-primed neutrophils were stimulated with fMLF, with HCV peptide (C5A), or sequentially with fMLF (10−7 M), HCV peptide (C5A) (10−5 M) in the order shown. The superoxide produced was measured as described in Materials and Methods. B, Release of β-glucuronidase in neutrophils stimulated with HCV peptide (C5A). Neutrophils were stimulated with different concentrations of fMLF or HCV peptide (C5A) as described in Materials and Methods. The results show dose-dependent secretion of β-glucuronidase from the stimulated cells as mean ± SEM, based on triplicate measurements from three independent experiments, using neutrophils from different donors. C, ERK1/2 phosphorylation in monocytes stimulated with HCV peptide (C5A). Monocytes were stimulated with HCV peptide (C5A) 10−5 M or fMLF 10−7 M for 5 min. The cells were harvested and the phosphorylated ERK1/2 were determined by Western blotting using an anti-phospho-ERK Ab. Equal loading of the proteins was determined with an anti-ERK1/2 Ab. Data shown are representative of two independent experiments. D, HCV peptide (C5A) regulated CD11b expression on monocytes. Monocytes were stimulated with fMLF (10−7 M) or HCV peptide (C5A; 10−5 M) at 37°C for 30 min. The cells were collected and stained with FITC-CD11b and analyzed by FACSCalibur.
HCV peptide (C5A) induces the accumulation of phagocytes in vivo
To confirm the chemoattractant activity of HCV peptide (C5A) in vivo, we determined the number and identity of cells accumulating in response to injection of HCV peptide (C5A) into the air pouches of mice. Four hours after injection of PBS, fMLF, or HCV peptide (C5A) into the air pouches, cells infiltrating into the air pouches were washed out and counted. Leukocyte subpopulations were distinguished by eosin-methylene blue staining of cytospin slides. Fig. 6A shows the total number of leukocytes recruited into the air pouches of each mouse group. Injection of HCV peptide (C5A) enhanced the recruitment of total leukocytes into the air pouches. The number of leukocytes recruited by HCV peptide (C5A) increased by 2.5-fold compared with control air pouches injected with PBS. In agreement with the in vitro chemotactic activity, the cells recruited into the air pouches by HCV peptide (C5A) were monocytes and neutrophils rather than lymphocytes (Fig. 6B). We further tested the chemotactic activity of HCV peptide (C5A) in vivo by s.c. injection of the peptide. Ten hours after injection, HCV peptide (C5A) induced typical features of inflammation, consisting of local redness and swelling of the skin with dilated blood vessels and massive numbers of infiltrating leukocytes as observed in a histologic examination (Fig. 6C). These data indicate HCV peptide (C5A) as a chemoattractant for monocytes and neutrophils in vivo.
FIGURE 6.

In vivo recruitment of mouse leukocytes by HCV peptide (C5A). A, One milliliter of endotoxin-free PBS or PBS containing HCV peptide (C5A; 10−5 M) or fMLF (10−7 M) was injected into experimentally formed air pouches in C57BL/6 mice. Four hours later, leukocytes recruited into the pouches were collected, enumerated, and presented as the average cell number (mean ± SD) per pouch. B, The migration of various subpopulations of leukocytes (neutrophils, monocytes, and lymphocytes) into the air pouches of mice in response to HCV peptide (C5A) was determined by differential counting after staining the cytospin slides of leukocytes collected from air pouches. C, Skin redness, swellings, and cell infiltration induced by HCV peptide (C5A) injected s.c. (100 μl/10 μM). The tissues were collected for histologic examination 10 h after challenge (original magnification ×10). In all experiments, three mice per group were used. *p < 0.05 compared with PBS-treated group.
HCV peptide (C5A) acts as an adjuvant to enhance immune responses
Many microbes and their products possess adjuvant activity to enhance immune responses of the host. We determined the potential activity of HCV peptide (C5A) in enhancing Ag-specific immune response in vivo. Naive C57BL/6 mice were immunized with OVA in the presence or absence of HCV peptide (C5A) or fMLF, and measured for the subsequent immune responses. Splenocytes from the mice immunized with OVA in the presence of HCV peptide (C5A) or fMLF showed enhanced OVA-specific T cell proliferative responses (Fig. 7A). Splenocytes of the mice immunized with OVA in the presence of HCV peptide (C5A) or fMLF produced predominantly IFN-γ and IL-17, but not IL-4, suggesting that HCV peptide (C5A) and fMLF polarized OVA-specific Th1 and Th17 T cell responses in mice (Fig. 7B).
FIGURE 7.

HCV peptide (C5A) enhancement of OVA-specific cellular immune responses. C57BL/6 mice were immunized i.p. on day 0 with 0.2 ml PBS containing 50 μg OVA in the presence of alum (3 mg), HCV peptide (C5A; 10 μM) or fMLF (1 μM). On day 14, mice were booster-immunized by i.p. injection of 0.2 ml PBS containing 50 μg OVA. Ag-specific T cell proliferation (A) and cytokine production (B) were measured. The data from one of three representative experiments are shown. *p < 0.05 compared with PBS-treated group.
Discussion
This study demonstrates that HCV peptide (C5A) is able to activate FPR on phagocytic cells. This property of HCV peptide (C5A) might have an important role in the recruitment and activation of phagocytic cells during HCV infection. Phagocytic leukocytes respond to a large number of chemoattractants with activation of integrins, directional cell migration, generation of superoxide anions, and release of granule contents. These functions constitute the first line of host defense against invading microorganisms and tissue injury. Over the past three decades, numerous chemoattractants have been identified and include the classical chemoattractants such as the bacterial chemotactic peptide fMLF, C5a, LTB4, and platelet-activating factor, as well as a superfamily of chemokines (1–3, 7). Both classical chemoattractants and chemokines activate G-protein coupled receptors (GPCRs) expressed on cells of hematopoietic origin and on other cell types. In addition to their role in infection and inflammation, there is growing evidence for the involvement of one or more chemoattractants in leukocyte trafficking, coagulation, hematopoiesis, wound healing, allergy, atherogenesis, angiogenesis, and malignancy (1–3, 7, 36, 37). The scope of chemoattractant receptor research has greatly expanded because of the discovery that several GPCRs, CCR5 and CXCR4 in particular, also serve as coreceptors for HIV type 1 (HIV-1) (7, 38). In addition, some viruses contain components that activate GPCRs on host cells (7, 15, 32, 37). This study revealed an HCV component HCV peptide (C5A) as an activator of host phagocyte by using the GPCR FPR, which was originally identified as a receptor for the gram-negative bacterial product.
The role of FPR in the pathophysiologic processes of HCV infection requires further investigation. HCV peptide (C5A) does not bear any sequence identity with the reported FPR agonistic peptides, including the absence of an N-formyl group, suggesting that it is the conformation rather than the primary sequence that determines its activity. Activation of FPR in leukocytes and glioma cells has been shown to elicit increased production of proinflammatory cytokines, chemokines, and angiogenic factors (36). Interestingly, functional FPR has also been detected in hepatocytes, and it promotes the production of acute phase proteins in response to fMLF (17). Thus, FPR expressed on hepatocytes might participate in the inflammatory courses in liver. Previous studies have shown that the expression levels of chemokines in the liver are positively correlated to the severity of hepatic inflammation in chronic HCV infection (18, 39, 40). The elevated expression of the chemokines in a severely inflamed liver could attract leukocytes more efficiently than express relevant chemokine GPCRs to the liver (40–42). However, it remains to be determined whether HCV peptide (C5A) plays a role in activating hepatocytes to produce increased levels of chemokines.
Although HCV peptide (C5A) could exacerbate liver damage during infection by activating FPR on leukocytes and hepatocytes, it could also promote host immune responses required for the destruction and clearance of infectious agents. Our in vivo experiments showing HCV peptide (C5A) as a phagocyte recruiter and enhancer of OVA-specific T cell responses (Fig. 7A), suggesting that HCV peptide (C5A) has the capacity to promote innate and adaptive immune responses. In this context, HCV peptide (C5A) may be considered as a basis for the development of anti-HCV agents and immune adjuvants.
Acknowledgments
This work was supported by grants from the National Basic Research Program of China (2007CB512206) and the National Natural Science Foundation of China (81072483). W.G., K.C., and J.M.W. were supported by federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. NO1-CO-12400, and by the Intramural Research Program of the National Cancer Institute, National Institutes of Health.
Abbreviations used in this article:
- DC
dendritic cell
- ETFR
epitope-tagged FPR
- fMLF
N-formyl-methionyl-leucyl-phenylalanine
- FPR
formyl peptide receptor
- GPCR
G-protein coupled receptor
- HCV
hepatitis C virus
- NS
nonstructural
- PT
pertussis toxin
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Viola A, and Luster AD. 2008. Chemokines and their receptors: drug targets in immunity and inflammation. Annu. Rev. Pharmacol. Toxicol 48: 171–197. [DOI] [PubMed] [Google Scholar]
- 2.Sahin H, Trautwein C, and Wasmuth HE. 2010. Functional role of chemokines in liver disease models. Nat. Rev. Gastroenterol. Hepatol 7: 682–690. [DOI] [PubMed] [Google Scholar]
- 3.Ye RD, Boulay F, Wang JM, Dahlgren C, Gerard C, Parmentier M, Serhan CN, and Murphy PM. 2009. International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol. Rev 61: 119–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou Y, Bian X, Le Y, Gong W, Hu J, Zhang X, Wang L, Iribarren P, Salcedo R, Howard OM, et al. 2005. Formylpeptide receptor FPR and the rapid growth of malignant human gliomas. J. Natl. Cancer Inst 97: 823–835. [DOI] [PubMed] [Google Scholar]
- 5.Wang JM, Deng X, Gong W, and Su S. 1998. Chemokines and their role in tumor growth and metastasis. J. Immunol. Methods 220: 1–17. [DOI] [PubMed] [Google Scholar]
- 6.Kim SD, Kim JM, Jo SH, Lee HY, Lee SY, Shim JW, Seo SK, Yun J, and Bae YS. 2009. Functional expression of formyl peptide receptor family in human NK cells. J. Immunol 183: 5511–5517. [DOI] [PubMed] [Google Scholar]
- 7.Su SB, Gong WH, Gao JL, Shen WP, Grimm MC, Deng X, Murphy PM, Oppenheim JJ, and Wang JM. 1999. T20/DP178, an ecto-domain peptide of human immunodeficiency virus type 1 gp41, is an activator of human phagocyte N-formyl peptide receptor. Blood 93: 3885–3892. [PubMed] [Google Scholar]
- 8.Su SB, Gao J, Gong W, Dunlop NM, Murphy PM, Oppenheim JJ, and Wang JM. 1999. T21/DP107, A synthetic leucine zipper-like domain of the HIV-1 envelope gp41, attracts and activates human phagocytes by using G-protein-coupled formyl peptide receptors. J. Immunol 162: 5924–5930. [PubMed] [Google Scholar]
- 9.Rabiet MJ, Huet E, and Boulay F. 2005. Human mitochondria-derived N-formylated peptides are novel agonists equally active on FPR and FPRL1, while Listeria monocytogenes-derived peptides preferentially activate FPR. Eur. J. Immunol 35: 2486–2495. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, and Hauser CJ. 2010. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464: 104–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xia SH, Hu LP, Hu H, Ying WT, Xu X, Cai Y, Han YL, Chen BS, Wei F, Qian XH, et al. 2002. Three isoforms of annexin I are preferentially expressed in normal esophageal epithelia but down-regulated in esophageal squamous cell carcinomas. Oncogene 21: 6641–6648. [DOI] [PubMed] [Google Scholar]
- 12.Sun R, Iribarren P, Zhang N, Zhou Y, Gong W, Cho EH, Lockett S, Chertov O, Bednar F, Rogers TJ, et al. 2004. Identification of neutrophil granule protein cathepsin G as a novel chemotactic agonist for the G protein-coupled formyl peptide receptor. J. Immunol 173: 428–436. [DOI] [PubMed] [Google Scholar]
- 13.Le Y, Murphy PM, and Wang JM. 2002. Formyl-peptide receptors revisited. Trends Immunol. 23: 541–548. [DOI] [PubMed] [Google Scholar]
- 14.Le Y, Oppenheim JJ, and Wang JM. 2001. Pleiotropic roles of formyl peptide receptors. Cytokine Growth Factor Rev. 12: 91–105. [DOI] [PubMed] [Google Scholar]
- 15.Le Y, Wang JM, Liu X, Kong Y, Hou X, Ruan L, and Mou H. 2007. Biologically active peptides interacting with the G protein-coupled formylpeptide receptor. Protein Pept. Lett 14: 846–853. [DOI] [PubMed] [Google Scholar]
- 16.Rescher U, Danielczyk A, Markoff A, and Gerke V. 2002. Functional activation of the formyl peptide receptor by a new endogenous ligand in human lung A549 cells. J. Immunol 169: 1500–1504. [DOI] [PubMed] [Google Scholar]
- 17.McCoy R, Haviland DL, Molmenti EP, Ziambaras T, Wetsel RA, and Perlmutter DH. 1995. N-formylpeptide and complement C5a receptors are expressed in liver cells and mediate hepatic acute phase gene regulation. J. Exp. Med 182: 207–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shields PL, Morland CM, Salmon M, Qin S, Hubscher SG, and Adams DH. 1999. Chemokine and chemokine receptor interactions provide a mechanism for selective T cell recruitment to specific liver compartments within hepatitis C-infected liver. J. Immunol 163: 6236–6243. [PubMed] [Google Scholar]
- 19.Larrubia JR, Benito-Martínez S, Calvino M, Sanz-de-Villalobos E, and Parra-Cid T. 2008. Role of chemokines and their receptors in viral persistence and liver damage during chronic hepatitis C virus infection. World J. Gastroenterol 14: 7149–7159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zeremski M, Petrovic LM, and Talal AH. 2007. The role of chemokines as inflammatory mediators in chronic hepatitis C virus infection. J. Viral Hepat 14: 675–687. [DOI] [PubMed] [Google Scholar]
- 21.Lichterfeld M, Leifeld L, Nischalke HD, Rockstroh JK, Hess L, Sauerbruch T, and Spengler U. 2002. Reduced CC chemokine receptor (CCR) 1 and CCR5 surface expression on peripheral blood T lymphocytes from patients with chronic hepatitis C infection. J. Infect. Dis 185: 1803–1807. [DOI] [PubMed] [Google Scholar]
- 22.Cicinnati VR, Kang J, Sotiropoulos GC, Hilgard P, Frilling A, Broelsch CE, Gerken G, and Beckebaum S. 2008. Altered chemotactic response of myeloid and plasmacytoid dendritic cells from patients with chronic hepatitis C: role of alpha interferon. J. Gen. Virol 89: 1243–1253. [DOI] [PubMed] [Google Scholar]
- 23.Sillanpää M, Kaukinen P, Melén K, and Julkunen I. 2008. Hepatitis C virus proteins interfere with the activation of chemokine gene promoters and down-regulate chemokine gene expression. J. Gen. Virol 89: 432–443. [DOI] [PubMed] [Google Scholar]
- 24.Kaukinen P, Sillanpää M, Kotenko S, Lin R, Hiscott J, Melén K, and Julkunen I. 2006. Hepatitis C virus NS2 and NS3/4A proteins are potent inhibitors of host cell cytokine/chemokine gene expression. Virol. J 3: 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hidajat R, Nagano-Fujii M, Deng L, and Hotta H. 2004. Cleavage of the hepatitis C virus NS5A protein by caspase-3 in the interferon sensitivity-determining region in a sequence-dependent manner. Kobe J. Med. Sci 50: 153–166. [PubMed] [Google Scholar]
- 26.Kalamvoki M, Georgopoulou U, and Mavromara P. 2006. The NS5A protein of the hepatitis C virus genotype 1a is cleaved by caspases to produce C-terminal-truncated forms of the protein that reside mainly in the cytosol. J. Biol. Chem 281: 13449–13462. [DOI] [PubMed] [Google Scholar]
- 27.Kalamvoki M, and Mavromara P. 2004. Calcium-dependent calpain proteases are implicated in processing of the hepatitis C virus NS5A protein. J. Virol 78: 11865–11878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sauter D, Himmelsbach K, Kriegs M, Carvajal Yepes M, and Hildt E. 2009. Localization determines function: N-terminally truncated NS5A fragments accumulate in the nucleus and impair HCV replication. J. Hepatol 50: 861–871. [DOI] [PubMed] [Google Scholar]
- 29.Cheng G, Montero A, Gastaminza P, Whitten-Bauer C, Wieland SF, Isogawa M, Fredericksen B, Selvarajah S, Gallay PA, Ghadiri MR, and Chisari FV. 2008. A virocidal amphipathic alpha-helical peptide that inhibits hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. USA 105: 3088–3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bobardt MD, Cheng G, de Witte L, Selvarajah S, Chatterji U, Sanders-Beer BE, Geijtenbeek TB, Chisari FV, and Gallay PA. 2008. Hepatitis C virus NS5A anchor peptide disrupts human immunodeficiency virus. Proc. Natl. Acad. Sci. USA 105: 5525–5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rothwangl KB, Manicassamy B, Uprichard SL, and Rong L. 2008. Dissecting the role of putative CD81 binding regions of E2 in mediating HCV entry: putative CD81 binding region 1 is not involved in CD81 binding. Virol. J 5: 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Su SB, Gong W, Gao JL, Shen W, Murphy PM, Oppenheim JJ, and Wang JM. 1999. A seven-transmembrane, G protein-coupled receptor, FPRL1, mediates the chemotactic activity of serum amyloid A for human phagocytic cells. J. Exp. Med 189: 395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu J, Wang O, Ruan L, Hou X, Cui Y, Wang JM, and Le Y. 2009. The green tea polyphenol (−)-epigallocatechin-3-gallate inhibits leukocyte activation by bacterial formylpeptide through the receptor FPR. Int. Immunopharmacol 9: 1126–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Su SB, Silver PB, Grajewski RS, Agarwal RK, Tang J, Chan CC, and Caspi RR. 2005. Essential role of the MyD88 pathway, but nonessential roles of TLRs 2, 4, and 9, in the adjuvant effect promoting Th1-mediated autoimmunity. J. Immunol 175: 6303–6310. [DOI] [PubMed] [Google Scholar]
- 35.Ali H, Richardson RM, Tomhave ED, Didsbury JR, and Snyderman R. 1993. Differences in phosphorylation of formylpeptide and C5a chemoattractant receptors correlate with differences in desensitization. J. Biol. Chem 268: 24247–24254. [PubMed] [Google Scholar]
- 36.Huang J, Chen K, Gong W, Zhou Y, Le Y, Bian X, and Wang JM. 2008. Receptor “hijacking” by malignant glioma cells: a tactic for tumor progression. Cancer Lett. 267: 254–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang J, Chen K, Huang J, Gong W, Dunlop NM, Howard OM, Bian X, Gao Y, and Wang JM. 2009. Regulation of the leucocyte chemoattractant receptor FPR in glioblastoma cells by cell differentiation. Carcinogenesis 30: 348–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alkhatib G 2009. The biology of CCR5 and CXCR4. Curr. Opin. HIV AIDS 4: 96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kusano F, Tanaka Y, Marumo F, and Sato C. 2000. Expression of C-C chemokines is associated with portal and periportal inflammation in the liver of patients with chronic hepatitis C. Lab. Invest 80: 415–422. [DOI] [PubMed] [Google Scholar]
- 40.Apolinario A, Majano PL, Alvarez-Pérez E, Saez A, Lozano C, Vargas J, and García-Monzón C. 2002. Increased expression of T cell chemokines and their receptors in chronic hepatitis C: relationship with the histological activity of liver disease. Am. J. Gastroenterol 97: 2861–2870. [DOI] [PubMed] [Google Scholar]
- 41.Murdoch C, and Finn A. 2000. Chemokine receptors and their role in inflammation and infectious diseases. Blood 95: 3032–3043. [PubMed] [Google Scholar]
- 42.Moser B, and Loetscher P. 2001. Lymphocyte traffic control by chemokines. Nat. Immunol 2: 123–128. [DOI] [PubMed] [Google Scholar]