

Summary
Abundance of urea cycle enzymes in the liver is regulated by dietary protein intake. Although urea cycle enzyme levels rise in response to a high protein diet, signaling networks that sense dietary protein intake and trigger changes in expression of urea cycle genes have not been identified. The aim of this study was to identify signaling pathway(s) that respond to changes in protein intake and regulate expression of urea cycle genes in mice and human hepatocytes. Mice were adapted to either high (HP) or low (LP) protein diets followed by isolation of liver protein and mRNA and integrated analysis of the proteomic and transcriptomic data. HP diet led to increased expression of mRNA and enzymes in amino acid degradation pathways, and decreased expression of mRNA and enzymes in carbohydrate and fat metabolism, which implicated AMPK as a possible regulator. Primary human hepatocytes, treated with AICAR an activator of AMPK, were used to test whether AMPK regulates expression of urea cycle genes. The abundance of CPS1 and OTC mRNA increased in hepatocytes treated with AICAR, which supports a role for AMPK signaling in regulation of the urea cycle. Because AMPK is either a target of drugs used to treat type-2 diabetes, these drugs might increase the expression of urea cycle enzymes in patients with partial urea cycle disorders, which could be the basis of a new therapeutic approach.
Introduction
The physiological function of the urea cycle is to protect the central nervous system from the neuro-toxic effects of ammonia (Meijer et al 1990; Brusilow and Horwich 2001). Hyperammonemia is the primary pathophysiology of urea cycle disorders (UCD) that results in neuro-cognitive symptoms such as nausea, lethargy, seizures, and, in severe cases, coma and death (Ah Mew et al 1993). While complete block of ureagenesis results in severe hyperammonemia within the first few days of life (Ah Mew et al 1993; Batshaw et al 2014) approximately 70% of patients with UCD have partial defects in urea cycle enzymes (Batshaw et al 2014) with decreased capacity to produce urea. They can develop hyperammonemia at any time, usually due to infections, invasive medical procedures, fasting or lifestyle changes that result in increased protein catabolism (Yeh and Cheng 1997; Caldovic et al 2005; Nurmohamed et al 2007; Marcus et al 2008; Panlaqui et al 2008; Ben-Ari et al 2010; Martinez de Lapiscina et al 2012). Low protein diet and activation of alternative pathways for disposal of ammonia are standard therapies for UCD aimed to reduce ammonia production and increase its elimination, respectively (Adam et al 2012; Haeberle et al 2012; Adam et al 2013; Batshaw et al 2014). Decreased protein intake can be sensed by the body (Nuzum and Snodgrass 1971), leading to decreased expression of urea cycle genes and enzymes, including the defective one in patients with partial UCD. This can further diminish patient’s capacity to produce urea and increases their risk of hyperammonemia upon sudden increase of protein catabolism. However, this also presents an opportunity for development of new treatments for partial UCD with drugs that increase the expression of urea cycle enzymes despite patients’ low protein diet. Development of such drugs requires detailed understanding of the sensory mechanisms and signaling pathways that regulate expression of the urea cycle genes and enzymes is response to dietary protein intake changes.
Expression of urea cycle genes and abundance of urea cycle enzymes in the liver depend on the rates of dietary and cellular protein catabolism (Schimke 1962; Schimke 1962; Nuzum and Snodgrass 1971; Saheki et al 1982). Hormones glucocorticoid and glucagon act via glucocorticoid receptor and cAMP signaling to activate expression of urea cycle genes (Schimke 1963; McLean and Novello 1965; Snodgrass et al 1978; Gebhardt and Mecke 1979; Morris 1987; Nebes and Morris 1988; Ulbright and Snodgrass 1993; Hazra et al 2008) while the role of insulin in the regulation of urea cycle remains unclear (Husson et al 1990). Glucocorticoid receptor regulates expression of the rat carbamoylphosphate synthetase 1 (Cps1) gene (Christoffels et al 1998) and cAMP response element binding (CREB) transcription factor regulates rat Cps1 and human N-acetylglutamate synthase (NAGS) genes (Christoffels et al 1996; Christoffels et al 2000; Heibel et al 2012) but transcription factors that regulate expression of each urea cycle gene (Table S1) do not reveal a common regulatory mechanism that might be responsible for coordinated changes in expression of all urea cycle genes in response to changes in dietary protein intake. Therefore, we carried out an integrated analysis of transcriptional and proteomic profiles of the livers from mice fed either high or low protein diet and determined that activity of AMP-dependent protein kinase (AMPK) correlates with expression of urea cycle enzymes in mice and primary human hepatocytes.
Materials and Methods
The Institutional Animal Care and Use Committee of the Children’s National Medical Center approved all experimental procedures involving mice. All institutional and national guidelines for the care and use of laboratory animals were followed.
Time-Restricted Feeding Study
Two groups of 32 syngeneic adult male C57BL/6 mice were housed individually in the standard mouse cages and fed either low (LP; Harlan Teklad cat# TD.06220) or high (HP; Harlan Teklad cat# TD.91352) protein diet. The HP and LP diets were isocaloric and had 65.3 and 21.3 kcal% derived from protein, and 21.5 and 65.3 kcal%, derived from carbohydrates, respectively, while 13.1 kcal% and 13.4 kcal%, respectively, were derived from fat. Mice were on a fast/feast cycle consisting of 6 hours fasting (10 AM to 4 PM), with unlimited access to water, followed by 18 hours of ad libitum feeding for 11 days. On the 12th day mice were sacrificed immediately before scheduled food reintroduction (T0 or fasting), and at 30 min. (T30), 60 min. (T60) and 120 min. (T120) after reintroduction of food (Figure S1A). Eight mice were sacrificed at each time point; their livers were dissected and immediately frozen in liquid nitrogen. Livers from four mice were used for mRNA and protein profiling. Livers from the remaining four animals were used for validation of the expression data.
Primary Hepatocyte Cultures
Human primary hepatocytes were obtained from the Liver Tissue Cell Distribution System, which provided human liver tissue that was collected with informed consent following ethical and institutional guidelines. Tissue dissociation and subsequent hepatocyte isolation were carried out as described previously (Gramignoli et al 2012). Hepatocytes were allowed to adhere for 2 hours with Hepatocyte Maintenance Media (Lonza, Walkersville, MD) containing 10% FBS, penicillin and streptomycin. This was followed by maintenance culture for 1 day in serum-free Hepatocyte Maintenance Media. Hepatocytes were treated with 5 μM AICAR with or without 40 μM Compound C (Sigma-Alrich, St. Louis, MO) for 24 hours. Cell viability was determined using PrestoBlue Cell Viability Reagent (Life Technologies, Grand Island, NY) according to the manufacturer’s instructions. Cell pellets and supernatants were stored at −80°C until analysis.
Quantitative RT-PCR
RNA was extracted from cell pellets using RNeasy Minikit (Qiagen) according to the manufacturer’s instructions. One microgram of RNA was reverse transcribed to cDNA using a modified MMLV-reverse transcriptase with RNaseH+ activity (iScript, Bio-Rad, Hercules, CA) according to the manufacturer’s instructions. Real-time quantitative PCR reactions were carried out using TaqMan ornithine transcarbamoylase (OTC) and CPS1 probes, and ABI 7500 Fast Real Time PCR System (Applied Biosystems, Foster City, CA).
Description of the LP and HP diets, determination of the food and water consumption, RNA sample preparation, microarray processing, analysis of microarray data, validation of gene expression data, sample preparation for mass spectrometry, database search and stable isotope labeling by amino acids in mammals (SILAM) ratio measurement, validation of proteomic data, and measurements of protein phosphorylation are described in the supplementary materials and methods.
Results
Physiological effects of time-restricted feeding
The body weight of mice in the two experimental groups (HP and LP) were similar at the beginning and at the end of the 12 days of time-restricted feeding (Figure S1B). Daily protein consumption of mice fed HP diet was 88–103 mg protein/g body weight while mice on LP diet consumed 21–31 mg protein/g body weight per day. Mice fed HP diet gained weight during experiment while there was a trend towards higher weight at the end of experiment in mice fed LP (Figure S1B). Food intake was lower in mice fed the high protein diet (Figure S1C), most likely due to the prolonged satiety following consumption of high protein food (Paddon-Jones et al 2008; Veldhorst et al 2008). Mice fed high protein diet drank more water, presumably to allow for increased urea excretion through urination (Figure S1D). While urea excretion was not measured, the high protein diet would result in increased urea production (Das and Waterlow 1974).
Proteomic and Transcriptional Profiling
Livers were harvested from mice fed either HP or LP diet on a time-restricted feeding schedule of 6 hr. fasting and 18 hr. feeding for 11 days. On the 12th day, livers were collected immediately prior to reintroduction of food, and 30, 60, and 120 minutes after reintroduction of food (T0, T30, T60 and T120 time points, respectively); expression microarrays were used to assess mRNA levels at each time point and spike-in differential proteome profiling was used to detect changes in protein abundance in mice fed either LP or HP diets.
Differences in expression levels of urea cycle mRNA and proteins were assessed first. Expression levels of Cps1, Otc, argininosuccinate synthetase (Ass), and argininosuccinate lyase (Asl) mRNA were higher in mice fed HP diet, while the abundance of the Nags and arginase 1 (Arg1) mRNA did not change in response to diet (Figures 1A and 1B). The Cps1, Otc, Ass, Asl and Arg1 proteins were more abundant in the livers of mice fed HP diet (Figures 1C–1E), similar to previously observed changes in their abundance and enzymatic activity in response to HP diet (Schimke 1962; Morris et al 1987). The Nags protein was assayed only by immunoblotting because it could not be detected by mass spectroscopy due to its very low abundance. Unlike other urea cycle proteins, the Nags was more abundant in the livers of mice fed LP diet than in the livers of mice fed HP diet (Figures 1D and 1E).
Figure 1.
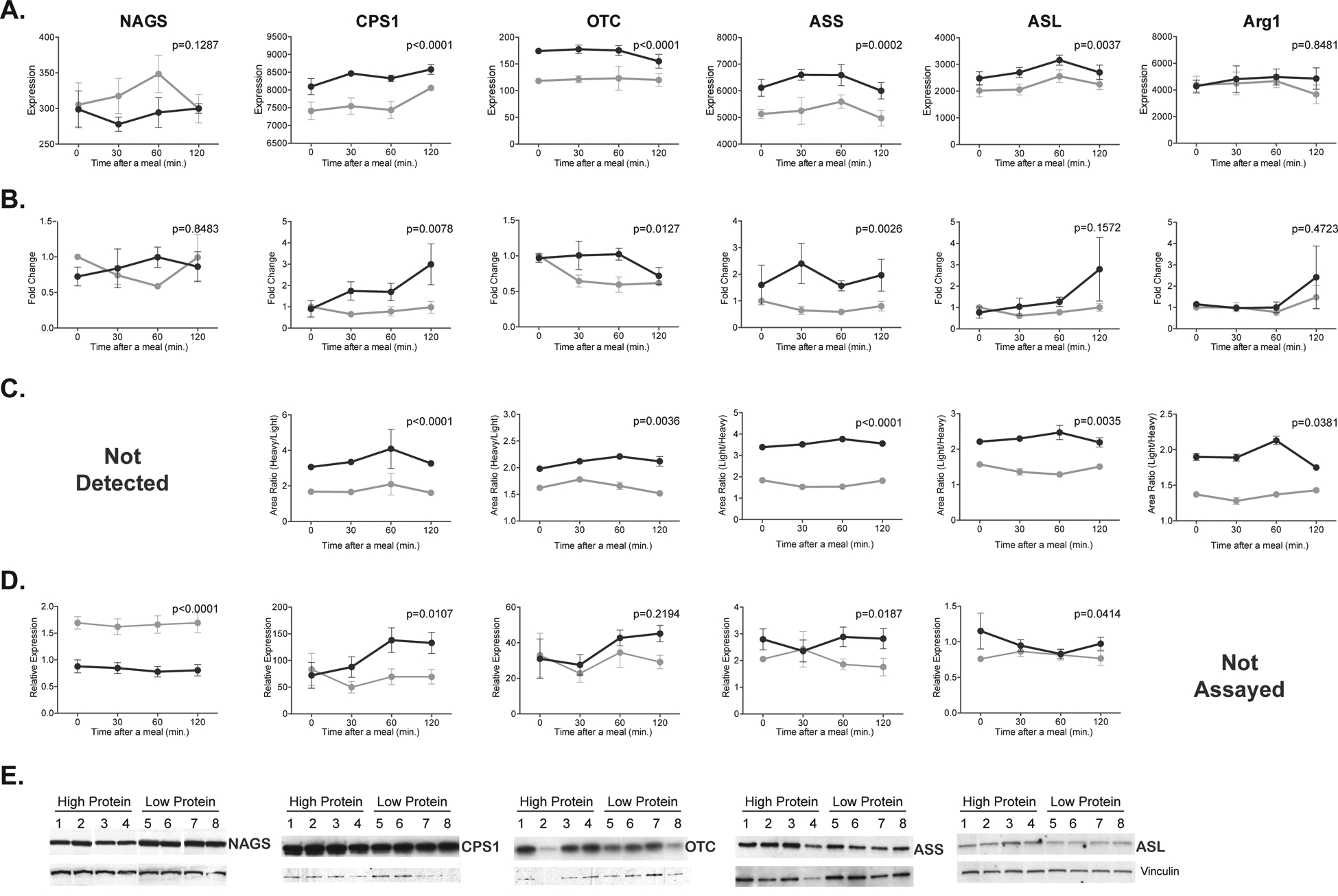
Expression of urea cycle genes and enzymes in mice fed either HP (dark gray) or LP (light gray) diets. The mRNA abundance was measured using Affymetrix microarrays (A) and validated with quantitative RT-PCR in a different cohort of mice(B). The abundance of urea cycle enzymes was measured using spike-in mass spectrometry (C) and validated with immunoblotting in a different cohort of mice (D and E). Abundance of mRNA and proteins was measured at fasting and 30, 60 and 120 min. after introduction of food. Each data point is a mean and associated SEM of 4 measurements.
Of the 45101 probe sets used for analysis 505, 604, 740 and 447 probe sets indicated expression fold change ±1.4 and p<0.05 in mice fed HP and LP at T0, T30, T60 and T120, respectively (Figure S2 and Table S2). Proteomic analysis was initially done on a single mouse fed each diet at each time point. Because none of the detected proteins changed abundance over time we focused on analyzing differences in protein abundance at T0. Of the 1073 proteins that were identified and quantified in mice fed HP and LP at T0, 335 had fold change ±1.2 and p<0.05 (Figure S3 and Table S3). When lists of differentially expressed genes and proteins were analyzed using the Integrated Pathway Analysis software package, there were no obvious signaling or metabolic pathways that responded to dietary protein intake. We then looked simultaneously at ±1.5-fold changes in mRNA and protein abundance in mice fed HP and LP at fasting (Figure S4). Abundance of mRNA and protein for aldehyde dehydrogenase X and glycine dehydrogenase were higher in mice fed HP, while 11 mRNA and proteins were down regulated in these mice (Figure S4 and Table S4). 70 proteins were at least 1.5-fold more abundant in the livers of mice fed HP while 24 proteins were at least 1.5-fold less abundant, while the abundance of their mRNA did not change (Figure S4 and Table S4). Many of the genes and proteins that were up or down-regulated in mice fed HP participate in metabolism of amino acids, carbohydrates, fats and steroids (Figure 2). Defects in many proteins that are up-regulated in mice fed HP cause inborn errors of metabolism. Absence or reduction in activity of PCCA and PCCB cause propionic acidemia (Shchelochkov et al 1993); maple syrup urine disease is caused by defects in BCKDHA and BCKDHB genes and proteins (Strauss et al 1993), methylmalonic acidemia is caused by defects in the MUT gene and protein (Manoli et al 1993). Although mice fed HP and LP had the same fat intake, the acetyl CoA carboxylase α (Accα), fatty acid synthase (Fasn) and 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase (Hmgcr), the key enzymes of the fatty acid and steroid biosynthesis (Barber et al 2005; An et al 2007; DeBose-Boyd 2008) were down regulated in mice fed HP (Figure 2). This prompted us to validate mRNA expression changes of these three genes and to examine expression levels of Ampk mRNA because it regulates activity and expression of the three enzymes (Beg and Brewer 1982; Ha et al 1994; An et al 2007).
Figure 2.

Expression changes of mRNA and enzymes in amino acid catabolism, carbohydrate metabolism, and fatty acid and steroid biosynthesis in mice fed either HP or LP diets. Solid – mRNA expression. Hatched – Protein expression.
Fluorescence intensity data of probe sets that correspond to Ampkα2, Accα, Fasn and Hmgcr were extracted from the data generated by PLIER, and the difference in abundance of these four mRNAs was measured by quantitative RT-PCR (qRT-PCR) in a separate cohort of mice fed HP and LP. Profiling and qRT-PCR data indicated that expression of the Ampkα2 mRNA did not change in response to diet (Figure S5A). The gene expression array indicated lower abundance of the Accα mRNA in the livers of mice fed HP diet while validation of expression data by qRT-PCR revealed only a trend towards lower expression of the Accα mRNA in the livers of mice fed HP diet (Figure S5B). The abundance of the Fasn and Hmgcr mRNA was lower in the livers of mice fed HP (Figures S5C–D).
Activated AMPK signaling in the livers of mice fed HP diet
Because AMPK signaling regulates expression and enzymatic activity of Accα, Fasn and Hmgcr (Beg and Brewer 1982; Ha et al 1994; An et al 2007) we examined whether phosphorylation levels of Ampkα, mammalian target of rapamycin (mTor), and eukaryotic translation initiation factor 4E binding protein 1 (4E-BP1), differ at fasting and at different times after a meal in the livers of mice fed HP and LP; Ampk phosphorylates mTor while 4E-BP1 is a downstream phosphorylation target of mTor. Liver proteins from mice fed HP and LP were resolved using SDS-PAGE and probed with antibodies that detect phospho-Thr172 in Ampkα, phospho-Thr2446 in mTor, and phospho-Thr37 and Thr46 (Thr37/46) in the 4E-BP1, followed by immunoblotting with antibodies that detect the total amount of the three proteins. Beta-actin was used as a loading control (Figure S6). There was a trend (p=0.097) toward different pattern of Thr172 phosphorylation levels of the Ampkα in mice fed HP and LP. The fasting levels of Thr172 phosphorylation levels were lower in mice fed HP and they tended to increase thereafter, while phosphorylation of Thr172 decreased during and after a LP meal (Figure 3A). Patterns of phosphorylation changes of mTor and 4E-BP1 were similar. Phosphorylation of levels Thr2446 and Thr37/46 in the mTor and 4E-BP1, respectively, did not change during and after an LP meal. However, in mice fed HP diet, phosphorylation of the mTor Thr2446 and 4E-BP1 Thr37/46 at fasting was lower than in mice fed LP diet; 30 and 60 min. after introduction of the HP meal, phosphorylation levels of the mTor Thr2446 and 4E-BP1 Thr37/46 increased and then 120 min. after introduction of the HP meal, decreased to levels lower than in mice fed LP diet (Figure 3B–C).
Figure 3.
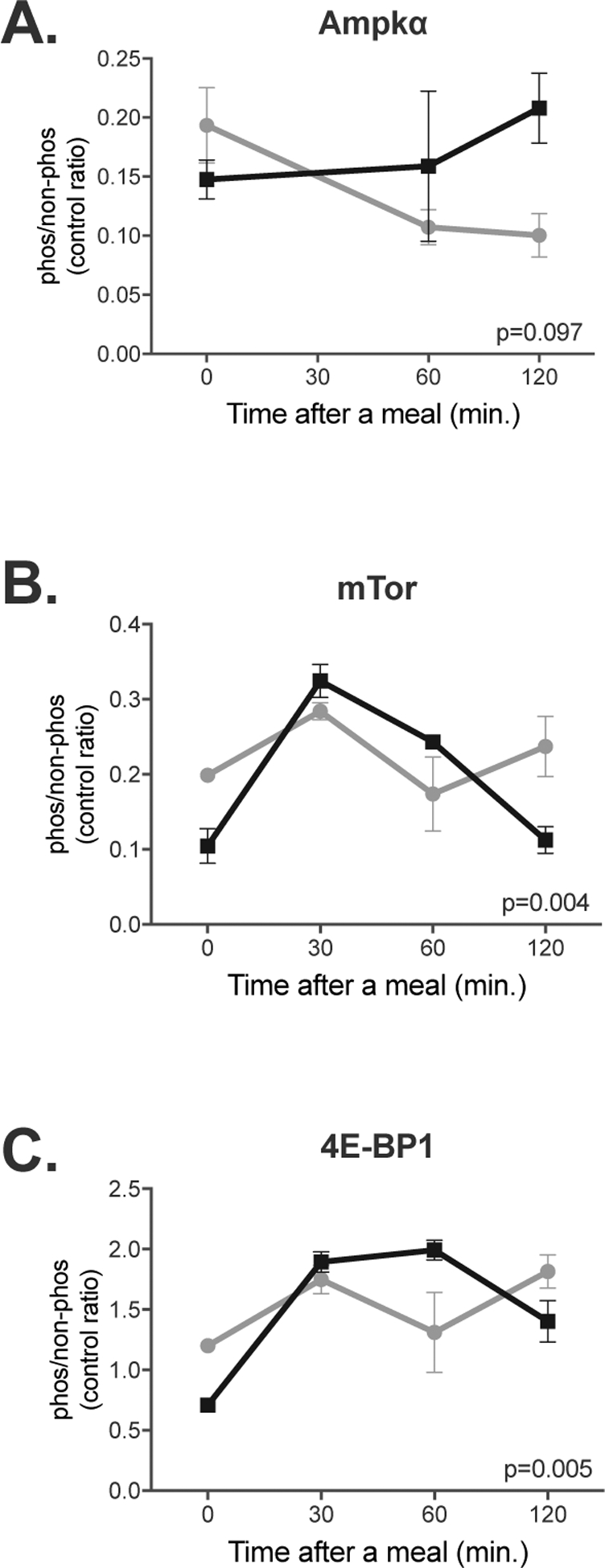
Changes in phosphorylation of Ampk (A), mTor (B) and 4E-BP1 (C) in the livers of mice fed either HP or LP diets. Phosphorylation was measured at fasting and 30, 60 and 120 min. after introduction of food. The ratio of phosphorylated and non-phosphorylated protein was normalized to β-actin abundance. Each data point is a mean and associated SEM of either 4 (Ampk) or 3 (mTor and 4E-BP1) measurements.
Activation of AMPK increases expression of the CPS1 and OTC mRNA
Inverse correlation of expression changes of the urea cycle enzymes and the key enzymes in the fatty acid and steroid synthesis raised the possibility that AMPK signaling may regulate expression of urea cycle enzymes in response to changes in the dietary protein intake. We used human primary hepatocytes to test whether activation of AMPK signaling results in increased expression of the CPS1 and OTC mRNA. Primary human hepatocytes were treated with AICAR, which activates AMPK, or with AICAR and compound C, a reversible inhibitor of AMPK and cell uptake of AICAR (Fryer et al 2002), or with PBS as a control, followed by measurements of cell viability and the CPS1 and OTC mRNA levels. Viability of the primary human hepatocytes was not affected by the treatments with AICAR and compound C (Figure 4A). Expression levels of CPS1 and OTC mRNA increased 1.8- and 2.8-fold, respectively, in primary human hepatocytes treated with AICAR (Figures 4B and 4C). Expression of the CPS1 mRNA decreased two-fold in cells treated with AICAR and compound C either due to inhibition of AMPK activity by compound C or its effects on other signaling pathways (Emerling et al 2007), while expression of the OTC mRNA remained unchanged (Figures 4B–C).
Figure 4.
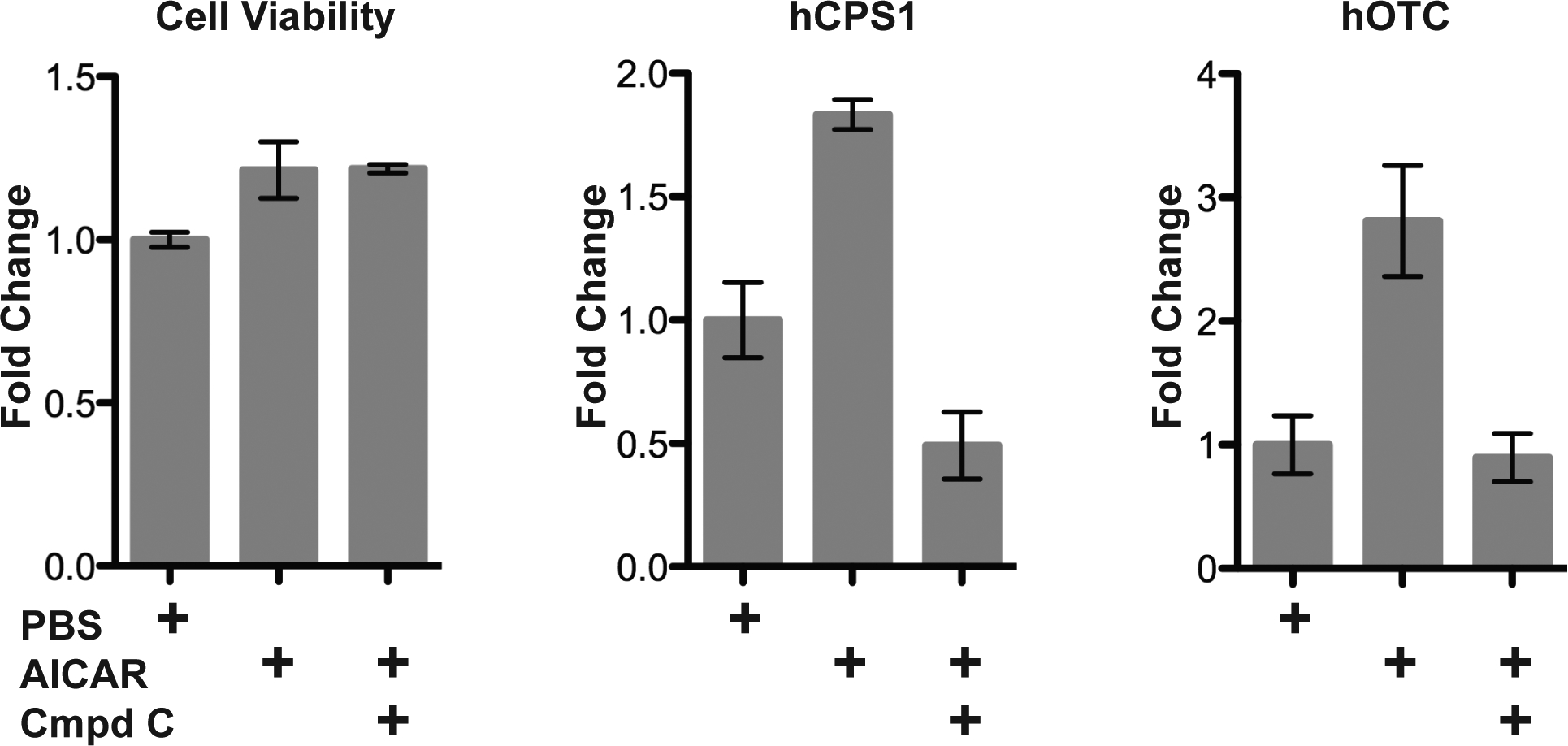
Cell viability (A) and expression of CPS1 (B) and OTC (C) mRNA in primary human hepatocytes treated with either AICAR or AICAR and compound C. Each data point is a mean and associated SEM of 3 measurements.
Discussion
The goal of this study was to identify signaling pathways that regulate expression of urea cycle genes in response to changes in the dietary protein intake. We used integrated transcriptional and proteomic profiling to show that activation of the AMPK signaling correlates with increased expression of urea cycle genes in mice fed HP diet. A direct role of AMPK in increased expression of at least two urea cycle genes, CPS1 and OTC, was confirmed in human primary hepatocytes treated with AMPK activator AICAR.
The decreased abundance of the Nags protein in response to HP diet was unexpected and may indicate a different regulatory mechanism for expression of the Nags gene. Enzymatic activity of mammalian recombinant NAGS doubles upon binding of L-arginine (Caldovic et al 2006; Haskins et al 2008) and intake of this amino acid would be higher in mice fed HP diet. Therefore, it is possible that higher intake of L-arginine in mice fed HP diet activated Nags and resulted in higher production of the NAG for activation of Cps1 despite lower abundance of the Nags enzyme. While this mechanism may be responsible for short-term regulation of urea production it may not explain adaptive changes in expression of urea cycle genes in response to dietary protein intake.
Integration of transcription and protein profiling data was needed because neither approach alone pointed to a signaling pathway that could be responsible for changes in expression of urea cycle genes and abundance of urea cycle enzymes in response to increased dietary protein intake. This is not surprising since changes in dietary protein intake are more likely to trigger changes in activity and phosphorylation status of various signaling proteins than their abundance or expression of their mRNA. Combined mRNA and protein expression analysis identified a coordinated decrease in the abundance of the key enzymes in the fatty acid and steroid biosynthesis in mice fed HP diet, a process that is regulated by AMPK signaling (Beg and Brewer 1982; Ha et al 1994; An et al 2007; Hindupur et al 2015). This suggested that AMPK might also regulate expression of the urea cycle genes and enzymes in response to dietary protein intake while increased expression of the CPS1 and OTC mRNA in primary human hepatocytes treated with AICAR, an activator of AMPK, supports its role in regulation of expression of at least two urea cycle genes. Because the fractions of carbohydrates and proteins were varied in the diets of experimental animals, it is possible that regulation of urea cycle genes and enzymes was in response to low carbohydrate instead of high protein intake. However, that is unlikely because expression of urea cycle genes did not change in mice that were fed diets with varied content of carbohydrates and fat (Do et al 2011; Ichimura et al 2012).
AMP is a product of the reaction catalyzed by the urea cycle enzyme ASS that directly activates AMPK (Madiraju et al 2016); therefore, increased AMP production after a HP meal could lead to activation of AMPK and increased expression of urea cycle enzymes. Additionally, the patterns of the AMPK Thr172 phosphorylation differed in mice fed either HP or LP diet and suggested activation of AMPK signaling after an HP meal. These results and regulation of urea cycle gene expression by cAMP and protein kinase A (McLean and Novello 1965; Snodgrass et al 1978; Gebhardt and Mecke 1979; Lin et al 1982; Morris et al 1987; Nebes and Morris 1988; Ulbright and Snodgrass 1993; Hazra et al 2008) suggest that cAMP and AMPK signaling pathways act in synergy to regulate expression of at least two urea cycle genes. Synergistic action of cAMP and AMPK signaling has been shown to regulate glycogen, fatty acid and cholesterol metabolism in the liver (Winz et al 1994; Kimball et al 2004; Hurley et al 2006; Homolya et al 2014)
A role for AMPK in the regulation of urea cycle enzymes opens up new prospects for treatment of patients with partial defects of UCD, who have diminished capacity to produce urea and represent about 70% of all UCD patients (Batshaw et al 2014; Kim et al 2016). Protein restricted diet is a mainstay treatment for patients with UCD because it lowers production of ammonia in their bodies. Because our bodies can sense the amount of dietary protein intake and adjust expression of urea cycle genes and enzymes accordingly, the low protein diet can put these patients at increased risk of hyperammonemia due to further decrease of their already low capacity to produce urea. A drug treatment targeted at increasing expression of urea cycle genes and enzymes, including the partially defective one, despite a low protein diet, could benefit patients with partial UCD by increasing their capacity to produce urea and decreasing the risk of hyperammonemia. Because of its central role in cellular energy metabolism, AMPK has been implicated in diseases such as diabetes, cancer and obesity, and has been a target for drug development (Kim et al 2016) that also might benefit patients with partial deficiencies of urea cycle enzymes.
Supplementary Material
Synopsis:
Integrated transcriptional and proteomic profiling of the liver mRNA and proteins from mice fed different protein content diets revealed that AMPK signaling pathway regulates expression of urea cycle enzymes.
Acknowledgements
This work was supported by the Public Health Service grants K01DK076846 and R01DK064913 from the National Institute of Diabetes Digestive and Kidney Diseases, National Institutes of Health, Department of Health and Human Services.
Abbreviations:
- HP
high protein
- LP
low protein
- AMPK
AMP-activated protein kinase
- AICAR
5-aminoimidazole-4-carboxamide ribonucleotide
- NAGS
N-acetylglutamate synthase
- CPS1
carbamoylphosphate synthetase
- OTC
ornithine transcarbamoylase
- ASS
argininosuccinate synthetase
- ASL
argininosuccinate lyase
- ARG1
arginase 1
- ORNT
ornithine/citrulline transporter
- UCD
urea cycle disorders
- CREB
cAMP response element binding transcription factor
- SILAM
stable isotope labeling by amino acids in mammals
- Accα
acetyl CoA carboxylase α
- Fasn
fatty acid synthase
- Hmgcr
3-hydroxy-3-methyl-glutaryl-coenzyme A reductase
- mTor
mammalian target of rapamycin
- 4E-BP1
eukaryotic translation initiation factor 4E binding protein 1
- PCCA
propionyl-CoA carboxylase subunit α
- PCCB
propionyl-CoA carboxylase subunit β
- BCKDHA
branched chain keto acid dehydrogenase E1 α polypeptide
- BCKDHB
branched chain keto acid dehydrogenase E1 subunit β
- MUT
mutase
Footnotes
Conflict of Interest:
Sandra Kirsch Heibel declares that she has no conflict of interest.
Peter J McGuire declares that he has no conflict of interest.
Nantaporn Haskins declares that she has no conflict of interest.
Himani Datta Majumdar declares that she has no conflict of interest.
Sree Rayavarapu declares that he has no conflict of interest.
Kanneboyina Nagaraju declares that he has no conflict of interest.
Yetrib Hathout declares that he has no conflict of interest.
Kristy Brown declares that she has no conflict of interest.
Mendel Tuchman declares that he has no conflict of interest.
Ljubica Caldovic declares that she has no conflict of interest.
Animal Rights:
All institutional and national guidelines for the care and use of laboratory animals were followed.
References
- Adam S, Almeida MF, Assoun M, et al. (2013) Dietary management of urea cycle disorders: European practice. Mol Genet Metab 110: 439–445. [DOI] [PubMed] [Google Scholar]
- Adam S, Champion H, Daly A, et al. (2012) Dietary management of urea cycle disorders: UK practice. J Hum Nutr Diet 25: 398–404. [DOI] [PubMed] [Google Scholar]
- Ah Mew N, Simpson KL, Gropman AL, Lanpher BC, Chapman KA, Summar ML (1993) Urea Cycle Disorders Overview In Adam MP, Ardinger HH, Pagon RA et al. eds. GeneReviews® Seattle (WA). [PubMed] [Google Scholar]
- An Z, Wang H, Song P, Zhang M, Geng X, Zou MH (2007) Nicotine-induced activation of AMP-activated protein kinase inhibits fatty acid synthase in 3T3L1 adipocytes: a role for oxidant stress. J Biol Chem 282: 26793–26801. [DOI] [PubMed] [Google Scholar]
- Barber MC, Price NT, Travers MT (2005) Structure and regulation of acetyl-CoA carboxylase genes of metazoa. Biochim Biophys Acta 1733: 1–28. [DOI] [PubMed] [Google Scholar]
- Batshaw ML, Tuchman M, Summar M, Seminara J (2014) A longitudinal study of urea cycle disorders. Mol Genet Metab 113: 127–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beg ZH, Brewer HB Jr. (1982) Modulation of rat liver 3-hydroxy-3-methylglutaryl-CoA reductase activity by reversible phosphorylation. Fed Proc 41: 2634–2638. [PubMed] [Google Scholar]
- Ben-Ari Z, Dalal A, Morry A, et al. (2010) Adult-onset ornithine transcarbamylase (OTC) deficiency unmasked by the Atkins’ diet. J Hepatol 52: 292–295. [DOI] [PubMed] [Google Scholar]
- Brusilow SW, Horwich AL (2001) Urea Cycle Enzymes In Scriver CR, Beaudet AL, Sly WS, Valle D eds. The Metabolic & Molecular Bases of Inherited Disease: McGraw-Hill, 1909–1963. [Google Scholar]
- Caldovic L, Lopez GY, Haskins N, et al. (2006) Biochemical properties of recombinant human and mouse N-acetylglutamate synthase. Mol Genet Metab 87: 226–232. [DOI] [PubMed] [Google Scholar]
- Caldovic L, Morizono H, Panglao MG, et al. (2005) Late onset N-acetylglutamate synthase deficiency caused by hypomorphic alleles. Hum Mutat 25: 293–298. [DOI] [PubMed] [Google Scholar]
- Christoffels VM, Grange T, Kaestner KH, et al. (1998) Glucocorticoid receptor, C/EBP, HNF3, and protein kinase A coordinately activate the glucocorticoid response unit of the carbamoylphosphate synthetase I gene. Mol Cell Biol 18: 6305–6315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoffels VM, Habets PE, Das AT, et al. (2000) A single regulatory module of the carbamoylphosphate synthetase I gene executes its hepatic program of expression. J Biol Chem 275: 40020–40027. [DOI] [PubMed] [Google Scholar]
- Christoffels VM, van den Hoff MJ, Lamers MC, et al. (1996) The upstream regulatory region of the carbamoyl-phosphate synthetase I gene controls its tissue-specific, developmental, and hormonal regulation in vivo. J Biol Chem 271: 31243–31250. [DOI] [PubMed] [Google Scholar]
- Das TK, Waterlow JC (1974) The rate of adaptation of urea cycle enzymes, aminotransferases and glutamic dehydrogenase to changes in dietary protein intake. Br J Nutr 32: 353–373. [DOI] [PubMed] [Google Scholar]
- DeBose-Boyd RA (2008) Feedback regulation of cholesterol synthesis: sterol-accelerated ubiquitination and degradation of HMG CoA reductase. Cell Res 18: 609–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do GM, Oh HY, Kwon EY, et al. (2011) Long-term adaptation of global transcription and metabolism in the liver of high-fat diet-fed C57BL/6J mice.Mol Nutr Food Res 55 Suppl 2: S173–185. [DOI] [PubMed] [Google Scholar]
- Emerling BM, Viollet B, Tormos KV, Chandel NS (2007) Compound C inhibits hypoxic activation of HIF-1 independent of AMPK. FEBS Lett 581: 5727–5731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer LG, Parbu-Patel A, Carling D (2002) Protein kinase inhibitors block the stimulation of the AMP-activated protein kinase by 5-amino-4-imidazolecarboxamide riboside. FEBS Lett 531: 189–192. [DOI] [PubMed] [Google Scholar]
- Gebhardt R, Mecke D (1979) Permissive effect of dexamethasone on glucagon induction of urea-cycle enzymes in perifused primary monolayer cultures of rat hepatocytes. Eur J Biochem 97: 29–35. [DOI] [PubMed] [Google Scholar]
- Gramignoli R, Green ML, Tahan V, et al. (2012) Development and application of purified tissue dissociation enzyme mixtures for human hepatocyte isolation. Cell Transplant 21: 1245–1260. [DOI] [PubMed] [Google Scholar]
- Ha J, Daniel S, Broyles SS, Kim KH (1994) Critical phosphorylation sites for acetyl-CoA carboxylase activity. J Biol Chem 269: 22162–22168. [PubMed] [Google Scholar]
- Haeberle J, Boddaert N, Burlina A, et al. (2012) Suggested Guidelines for the Diagnosis and Management of Urea Cycle Disorders. Orphanet J Rare Dis 7: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haskins N, Panglao M, Qu Q, et al. (2008) Inversion of allosteric effect of arginine on N-acetylglutamate synthase, a molecular marker for evolution of tetrapods. BMC Biochem 9: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazra A, DuBois DC, Almon RR, Snyder GH, Jusko WJ (2008) Pharmacodynamic modeling of acute and chronic effects of methylprednisolone on hepatic urea cycle genes in rats. Gene Regul Syst Bio 2: 1–19. [PMC free article] [PubMed] [Google Scholar]
- Heibel SK, Lopez GY, Panglao M, et al. (2012) Transcriptional regulation of N-acetylglutamate synthase. PLoS One 7: e29527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindupur SK, Gonzalez A, Hall MN (2015) The opposing actions of target of rapamycin and AMP-activated protein kinase in cell growth control. Cold Spring Harb Perspect Biol 7: a019141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homolya L, Fu D, Sengupta P, et al. (2014) LKB1/AMPK and PKA control ABCB11 trafficking and polarization in hepatocytes. PLoS One 9: e91921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley RL, Barre LK, Wood SD, et al. (2006) Regulation of AMP-activated protein kinase by multisite phosphorylation in response to agents that elevate cellular cAMP. J Biol Chem 281: 36662–36672. [DOI] [PubMed] [Google Scholar]
- Husson A, Renouf S, Fairand A, Buquet C, Benamar M, Vaillant R (1990) Expression of argininosuccinate lyase mRNA in foetal hepatocytes. Regulation by glucocorticoids and insulin. Eur J Biochem 192: 677–681. [DOI] [PubMed] [Google Scholar]
- Ichimura A, Hirasawa A, Poulain-Godefroy O, et al. (2012) Dysfunction of lipid sensor GPR120 leads to obesity in both mouse and human. Nature 483: 350–354. [DOI] [PubMed] [Google Scholar]
- Kim J, Yang G, Kim Y, Kim J, Ha J (2016) AMPK activators: mechanisms of action and physiological activities. Exp Mol Med 48: e224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimball SR, Siegfried BA, Jefferson LS (2004) Glucagon represses signaling through the mammalian target of rapamycin in rat liver by activating AMP-activated protein kinase. J Biol Chem 279: 54103–54109. [DOI] [PubMed] [Google Scholar]
- Lin RC, Snodgrass PJ, Rabier D (1982) Induction of urea cycle enzymes by glucagon and dexamethasone in monolayer cultures of adult rat hepatocytes. J Biol Chem 257: 5061–5067. [PubMed] [Google Scholar]
- Madiraju AK, Alves T, Zhao X, et al. (2016) Argininosuccinate synthetase regulates hepatic AMPK linking protein catabolism and ureagenesis to hepatic lipid metabolism. Proc Natl Acad Sci U S A 113: E3423–3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manoli I, Sloan JL, Venditti CP (1993) Isolated Methylmalonic Acidemia In Adam MP, Ardinger HH, Pagon RA et al. eds. GeneReviews® Seattle (WA). [PubMed] [Google Scholar]
- Marcus N, Scheuerman O, Hoffer V, Zilbershot-Fink E, Reiter J, Garty BZ (2008) Stupor in an adolescent following Yom Kippur fast, due to late-onset ornithine transcarbamylase deficiency. Isr Med Assoc J 10: 395–396. [PubMed] [Google Scholar]
- Martinez de Lapiscina EH, Aguirre ME, Giadas TC, Alvarez MT (2012) 5-Fluorouracil induced hyperammonemic encephalophathy: etiopathologic correlation. Can J Neurol Sci 39: 553–554. [PubMed] [Google Scholar]
- McLean P, Novello F (1965) Influence of Pancreatic Hormones on Enzymes Concerned with Urea Synthesis in Rat Liver. Biochem J 94: 410–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer AJ, Lamers WH, Chamuleau RAFM (1990) Nitrogen Metabolism and Ornithine Cycle Function. Physiological Reviews 70: 701–748. [DOI] [PubMed] [Google Scholar]
- Morris SM Jr. (1987) Thyroxine elicits divergent changes in mRNA levels for two urea cycle enzymes and one gluconeogenic enzyme in tadpole liver. Arch Biochem Biophys 259: 144–148. [DOI] [PubMed] [Google Scholar]
- Morris SM Jr., Moncman CL, Rand KD, Dizikes, Cederbaum SD, O’Brien WE (1987) Regulation of mRNA levels for five urea cycle enzymes in rat liver by diet, cyclic AMP, and glucocorticoids. Arch Biochem Biophys 256: 343–353. [DOI] [PubMed] [Google Scholar]
- Nebes VL, Morris SM Jr. (1988) Regulation of messenger ribonucleic acid levels for five urea cycle enzymes in cultured rat hepatocytes. Requirements for cyclic adenosine monophosphate, glucocorticoids, and ongoing protein synthesis. Mol Endocrinol 2: 444–451. [DOI] [PubMed] [Google Scholar]
- Nurmohamed S, Weenink A, Moeniralam H, Visser C, Bemelman F (2007) Hyperammonemia in generalized Mycobacterium genavense infection after renal transplantation. Am J Transplant 7: 722–723. [DOI] [PubMed] [Google Scholar]
- Nuzum CT, Snodgrass PJ (1971) Urea cycle enzyme adaptation to dietary protein in primates. Science 172: 1042–1043. [DOI] [PubMed] [Google Scholar]
- Paddon-Jones D, Westman E, Mattes RD, Wolfe RR, Astrup A, Westerterp-Plantenga M (2008) Protein, weight management, and satiety. Am J Clin Nutr 87: 1558S–1561S. [DOI] [PubMed] [Google Scholar]
- Panlaqui OM, Tran K, Johns A, McGill J, White H (2008) Acute hyperammonemic encephalopathy in adult onset ornithine transcarbamylase deficiency. Intensive Care Med 34: 1922–1924. [DOI] [PubMed] [Google Scholar]
- Saheki T, Hosoya M, Fujinami S, Katsunuma T (1982) Regulation of urea synthesis: changes in the concentration of ornithine in the liver corresponding to changes in urea synthesis. Adv Exp Med Biol 153: 255–263. [DOI] [PubMed] [Google Scholar]
- Schimke RT (1962) Adaptive characteristics of urea cycle enzymes in the rat. J Biol Chem 237: 459–468. [PubMed] [Google Scholar]
- Schimke RT (1962) Differential effects of fasting and protein-free diets on levels of urea cycle enzymes in rat liver. J Biol Chem 237: 1921–1924. [PubMed] [Google Scholar]
- Schimke RT (1963) Studies on factors affecting the levels of urea cycle enzymes in rat liver. J Biol Chem 238: 1012–1018. [PubMed] [Google Scholar]
- Shchelochkov OA, Carrillo N, Venditti C (1993) Propionic Acidemia In Adam MP, Ardinger HH, Pagon RA et al. eds. GeneReviews® Seattle (WA). [PubMed] [Google Scholar]
- Snodgrass PJ, Lin RC, Muller WA, Aoki TT (1978) Induction of urea cycle enzymes of rat liver by glucagon. J Biol Chem 253: 2748–2753. [PubMed] [Google Scholar]
- Strauss KA, Puffenberger EG, Morton DH (1993) Maple Syrup Urine Disease In Adam MP, Ardinger HH, Pagon RA et al. eds. GeneReviews® Seattle (WA). [PubMed] [Google Scholar]
- Ulbright C, Snodgrass PJ (1993) Coordinate induction of the urea cycle enzymes by glucagon and dexamethasone is accomplished by three different mechanisms. Arch Biochem Biophys 301: 237–243. [DOI] [PubMed] [Google Scholar]
- Veldhorst M, Smeets A, Soenen S, et al. (2008) Protein-induced satiety: effects and mechanisms of different proteins. Physiol Behav 94: 300–307. [DOI] [PubMed] [Google Scholar]
- Winz R, Hess D, Aebersold R, Brownsey RW (1994) Unique structural features and differential phosphorylation of the 280-kDa component (isozyme) of rat liver acetyl-CoA carboxylase. J Biol Chem 269: 14438–14445. [PubMed] [Google Scholar]
- Yeh KH, Cheng AL (1997) High-dose 5-fluorouracil infusional therapy is associated with hyperammonaemia, lactic acidosis and encephalopathy. Br J Cancer 75: 464–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.