

Abstract
Objective:
Macrophage activation syndrome (MAS) is a life-threatening complication of systemic juvenile idiopathic arthritis (sJIA) characterized by a vicious cycle of immune amplification that can culminate in overwhelming inflammation and multi-organ failure. The clinical features of MAS overlap with those of active sJIA, complicating early diagnosis and treatment. We evaluated adenosine deaminase 2 (ADA2), a protein of unknown function released principally by monocytes and macrophages, as a novel biomarker of MAS.
Methods:
We established age-based normal ranges of peripheral blood ADA2 activity in 324 healthy children and adults. We compared these ranges with 173 children with inflammatory and immune-mediated diseases, including systemic and non-systemic JIA, Kawasaki disease, pediatric systemic lupus erythematosus, and juvenile dermatomyositis.
Results:
ADA2 elevation beyond the upper limit of normal in children was largely restricted to sJIA with concomitant MAS, a finding confirmed in a validation cohort of sJIA patients with inactive disease, active sJIA without MAS, or sJIA with MAS. ADA2 activity strongly correlated with MAS biomarkers including ferritin, interleukin (IL)-18, and the interferon (IFN)-γ-inducible chemokine CXCL9, but displayed minimal association with the inflammatory markers C-reactive protein and erythrocyte sedimentation rate. Correspondingly, ADA2 paralleled disease activity based on serial measurements in patients with recurrent MAS episodes. IL-18 and IFN-γ elicited ADA2 production by peripheral blood mononuclear cells, and ADA2 was abundant in MAS hemophagocytes.
Conclusions:
These findings collectively identify the utility of plasma ADA2 activity as a biomarker of MAS and lend further support to a pivotal role of macrophage activation in this condition.
Keywords: adenosine deaminase 2, macrophage activation syndrome, biomarker, systemic juvenile idiopathic arthritis
Introduction
Adenosine deaminases (ADA) are a family of enzymes that catalyze the conversion of adenosine to inosine. ADA1 is a ubiquitously-expressed intracellular protein that metabolizes adenosine and 2’-deoxyadenosine as a crucial step in the purine salvage pathway [1]. Deficiency of ADA1 in humans results in defective lymphocyte development and severe combined immunodeficiency [2]. In contrast, ADA2 is a plasma protein secreted primarily by monocytes and macrophages [3, 4]. ADA2 displays much lower substrate affinity compared to ADA1 and its physiologic role remains unclear [5]. Supporting a non-redundant role of the ADA isoenzymes, deficiency of ADA2 (DADA2) is a recently-described autoinflammatory syndrome characterized by childhood-onset stroke, systemic vasculitis, variable immunodeficiency, and hematologic defects [5–7].
In adults, elevated levels of ADA in biological fluids have been described in infections, malignancies, autoimmune diseases and secondary hemophagocytic syndromes [3, 8–11]. However, many early studies focused on ADA1 and little is known about the biology of ADA2. ADA2 levels are higher in children than adults [7]. Whether abnormal ADA2 production is also a hallmark of pediatric inflammatory conditions has not been examined in detail. In this study, we established the normal range of peripheral blood ADA2 activity in healthy children and demonstrated a striking elevation in ADA2 as a sensitive and specific marker of the conversion between active systemic juvenile idiopathic arthritis (sJIA) and sJIA-associated macrophage activation syndrome (MAS), likely reflecting underlying cytokine-driven activation of monocytes and macrophages.
Methods
Human subjects:
These studies were approved by the Institutional Review Boards at Boston Children’s Hospital, Brigham and Women’s Hospital, Cincinnati Children’s Hospital and Children’s Hospital of Pittsburg. Informed consent was provided by participants or legal guardians. Plasma or serum were frozen at − 80 °C and thawed immediately prior to testing. Demographics of patient groups and diagnostic criteria are provided in Supplemental Table S1 and Supplemental Methods, respectively.
Quantification of ADA2 activity:
ADA2 activity was measured in human plasma, serum, or cell culture supernatant by modifying a previously-described automated spectrophotometric assay [12–14], which quantifies the adenosine-dependent generation of ammonia in the presence of a selective inhibitor of ADA1, EHNA (erythro-9-Amino-β-hexyl-α-methyl-9H-purine-9-ethanol hydrochloride). All reagents were purchased from Sigma Aldrich (St. Louis, MO), and the kinetics of each reaction were analyzed using a Synergy Hybrid H1 Microplate Reader (BioTek, Winooski, VT). Additional details are provided in Supplemental Methods.
ELISA, in vitro stimulation, flow cytometry and confocal microscopy:
Please refer to Supplemental Methods for detailed protocol of these studies.
Statistical analysis:
Because ADA2 levels in healthy children were not normally distributed (Supplemental Fig 1C), non-parametric tests were used for statistical analysis. The differences between two groups were analyzed using the Mann-Whitney U test while comparison of multiple groups was performed using the Kruskal-Wallis test. For each disease studied, 1:1 age-matched controls were randomly assigned from the pool of healthy controls. All tests were two-sided, and P < 0.05 was considered significant. For Bonferroni correction of multiple hypothesis testing, α was adjusted to 0.0014 for the correlation matrix. Statistical analyses were performed using Prism 5.0 software (GraphPad Software, La Jolla, CA).
Results
Establishing a reference range of ADA2 activity in healthy children
We first aimed to establish the reference range of plasma ADA2 activity in 324 healthy individuals (174 children and 150 adults). We employed a validated spectrophotometric assay to quantify ADA2 activity in plasma or serum (Figure S1A; see Methods). Confirming the specificity of the assay, DADA2 patients with biallelic ADA2 mutations show a near absence of ADA2 activity, whereas carriers have approximately half-normal plasma ADA2 activity (Figure S1B). The distribution of plasma ADA2 activity in healthy children (under age 18) was skewed towards higher levels (Figure S1C), with a median of 13.0 U/L (interquartile range [IQR]:10.6 – 16.1). The upper limit of normal (ULN), as defined by the 98th percentile, was 27.8 U/L. Comparison of males and females revealed similar ADA2 levels (Figure S1D).
Consistent with previous studies [7, 15], plasma ADA2 activity was higher in children than adults (age 18 and older, 98th percentile 25.7 U/L), with an overall negative correlation with age (Figure 1A). Stratification of healthy children into age categories did not reveal differences in ADA2 levels among groups (Figure 1B; p > 0.05 for all comparisons). While all pediatric categories showed significantly higher median levels compared to healthy adults, considerable variability was displayed within each age group.
Figure 1.
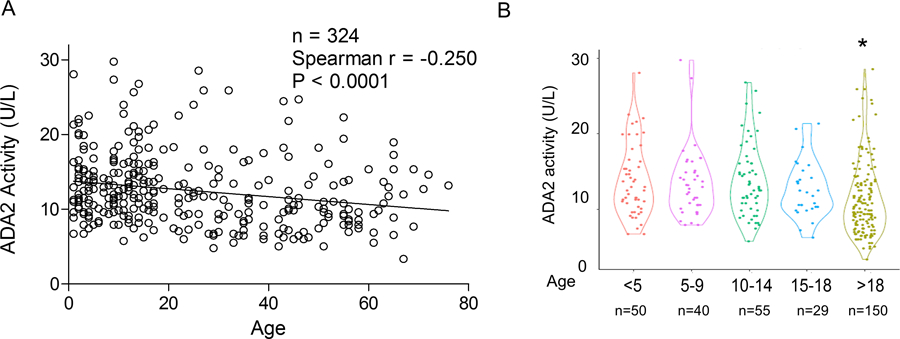
Determination of plasma ADA2 activity in healthy children and adults. A) Correlation between plasma ADA2 activity and age in healthy children (n = 174) and adults (n = 150). B) Violin plot comparing plasma ADA2 activity in healthy individuals stratified by age. * p < 0.05 compared to all other groups.
Evaluation of ADA2 activity in pediatric inflammatory diseases
We compared ADA2 levels in children with various inflammatory conditions with age-matched healthy controls. Demographics of controls and patient groups are provided in Supplemental Table S1. We first studied Kawasaki disease (KD), a highly-inflammatory childhood vasculitis manifested by skin rash, mucositis, extremity swelling, conjunctivitis and lymphadenopathy. All KD samples were collected during the acute phase of disease, prior to treatment. Compared with healthy controls, patients with KD showed similar ADA2 activity (n = 25 per group; Figure 2A). Elevated C-reactive protein (CRP), erythrocyte sedimentation rate (ESR), white blood cell count and platelet count in KD patients did not correlate with ADA2 levels (Figure S2A), establishing that ADA2 is not a general marker of systemic inflammation.
Figure 2.
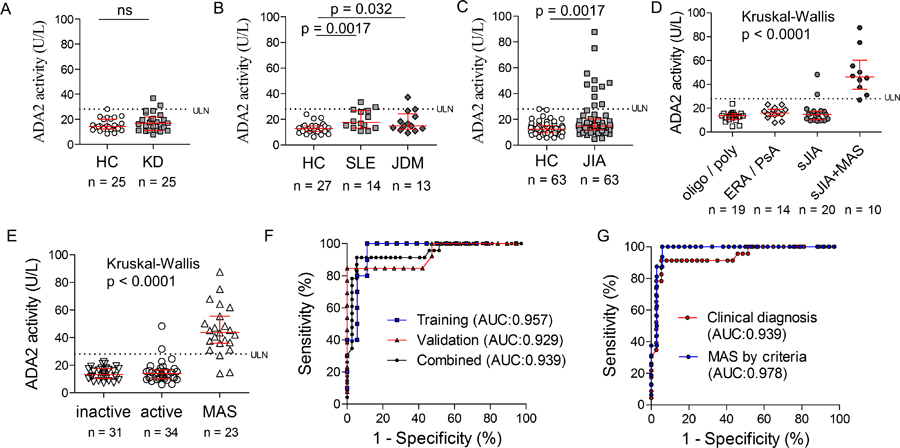
Comparison of ADA2 activity levels in childhood inflammatory diseases. A) Peripheral blood ADA2 levels in patients with Kawasaki disease during the acute phase of illness and age-matched healthy controls (n = 25 per group). B) Peripheral blood ADA2 levels in patients with pSLE (n = 14), patients with JDM (n = 13) and age-matched healthy controls (n = 27). C) Peripheral blood ADA2 levels in all patients with JIA and age-matched healthy controls (n = 63 per group). D) Peripheral blood ADA2 levels in patients with JIA stratified by disease category based on the ILAR classification. E) Stratification of plasma ADA2 levels in sJIA patients by disease activity and MAS (training and validation cohorts combined; n = 88). F) ROC curve of ADA2 in distinguishing active sJIA with or without MAS in training, validation and combined patient cohorts. G) ROC curve of ADA2 in distinguishing active sJIA with or without MAS diagnosed clinically or MAS diagnosed according to the 2016 MAS Classification Criteria. Median and interquartile range are displayed in scatter dot plots.
To examine if ADA2 activity is altered in chronic autoimmune conditions, we studied children with pediatric SLE (pSLE, n=14) and JDM (n=13), most with active disease at the time of sampling (Supplemental Table S1). pSLE patients displayed increased plasma ADA2 compared with age-matched controls (Fig 2B; 17.6 U/L [13.2 – 26.8] vs. 12.8 U/L [10.5 – 15.0], p = 0.0017), although levels remained predominantly below the ULN. ADA2 levels in patients with JDM (14.9 U/L [12.3 – 24.4]) were also mildly increased compared to controls (p=0.032) and statistically indistinguishable from pSLE (p = 0.50). ADA2 levels did not correlate with markers of disease activity in pSLE (complement C3, C4, and ESR; Figure S2B) or JDM (aldolase and LDH; Figure S2C).
Establishing ADA2 as a marker of MAS in systemic JIA
We next investigated ADA2 levels in patients with JIA. While most JIA patients showed plasma ADA2 activity comparable to age-matched controls, a small subset displayed levels well above the upper limit of normal (Fig 2C). JIA can be classified into distinct categories based on the ILAR (International League of Associations for Rheumatology) criteria [16]. Stratification by JIA categories revealed ADA2 activity in children with oligoarticular JIA, polyarticular JIA, enthesitis-related arthritis and psoriatic arthritis comparable to healthy controls (Figure 2D). All 11 patients with ADA2 levels above the ULN shared the diagnosis of systemic JIA, including 9 / 10 cases with MAS at the time of sampling.
Distinct from other forms of childhood arthritis, systemic JIA exhibits quotidian fever, lymphadenopathy, and systemic inflammation accompanied by variable joint involvement. MAS is a life-threatening complication of sJIA characterized by cytokine storm, hemophagocytosis, cytopenias, coagulopathy and multi-organ dysfunction. In contrast to the high levels of ADA2 in patients with MAS, most sJIA patients without MAS showed normal levels of ADA2 (Figure 2D).
To confirm these findings, we measured ADA2 levels in an independent validation cohort of 58 sJIA patients combined from two other institutions. Consistent with our initial observation, patients with MAS displayed significantly higher levels of ADA2 compared to those without MAS, regardless of sJIA disease activity (Figure S3A). Combined analysis of both sJIA cohorts showed that ADA2 levels beyond the ULN distinguished cases of MAS and active sJIA with a sensitivity of 86% and specificity of 94% (Figure 2E). The utility of ADA2 as a biomarker of MAS was supported by receiver operating characteristic (ROC) curve with area under the curve (AUC) of 0.939 (95% confidence interval 0.87–1.00; Figure 2F).
The diagnosis of MAS in these patients was determined clinically by the treating physicians. Review of laboratory data revealed that 15 of the 23 cases fulfilled the 2016 Classification Criteria for MAS [17]. The remaining cases all had hyperferritinemia (> 684 ng/mL) but either did not meet at least 2 of the criteria (n = 4; “No” group) or met one of the minor criteria but did not have complete laboratory parameters to apply the full criteria (n = 4; “Partial” group). Notably, patients who did not meet MAS criteria displayed significantly lower ADA2 levels compared to those who fully or partially met the criteria (Figure S3B). The performance of ADA2 on ROC was enhanced when the MAS group was filtered for patients fulfilling the formal classification criteria (AUC = 0.978; Figure 2G).
ADA2 measurements from healthy controls and all patient groups are displayed in Figure S3C to allow direct comparison. While MAS can also occur with other inflammatory diseases, we did not identify any case of MAS in our KD, SLE or JDM cohorts. Taken together, these data demonstrate the utility of ADA2 as a biomarker for MAS associated with sJIA.
Comparison of ADA2 with other markers of MAS
Next, we compared ADA2 activity in sJIA patients with serologic parameters of inflammation and biomarkers of MAS including ferritin, interleukin (IL)-18 [18, 19], and the interferon γ-inducible chemokine CXCL9 [20], combining training and validation cohorts. A correlation matrix was created based on the Spearman r values of the cross comparisons (Figure 3A). Individual comparisons showed that ADA2 levels correlated well with ferritin (r = 0.532; p < 0.0001), IL-18 (r = 0.723; p < 0.0001), and CXCL9 (r = 0.640; p < 0.0001; Figure 3B). These correlations remained statistically significant after Bonferroni correction. ADA2 activity also correlated well with increased aspartate aminotransferase (AST) levels in MAS (Figure S4A). By contrast, ADA2 activity did not correlate with conventional markers of inflammation including ESR and CRP after adjusting for multiple comparisons (Figure S4B,C).
Figure 3.
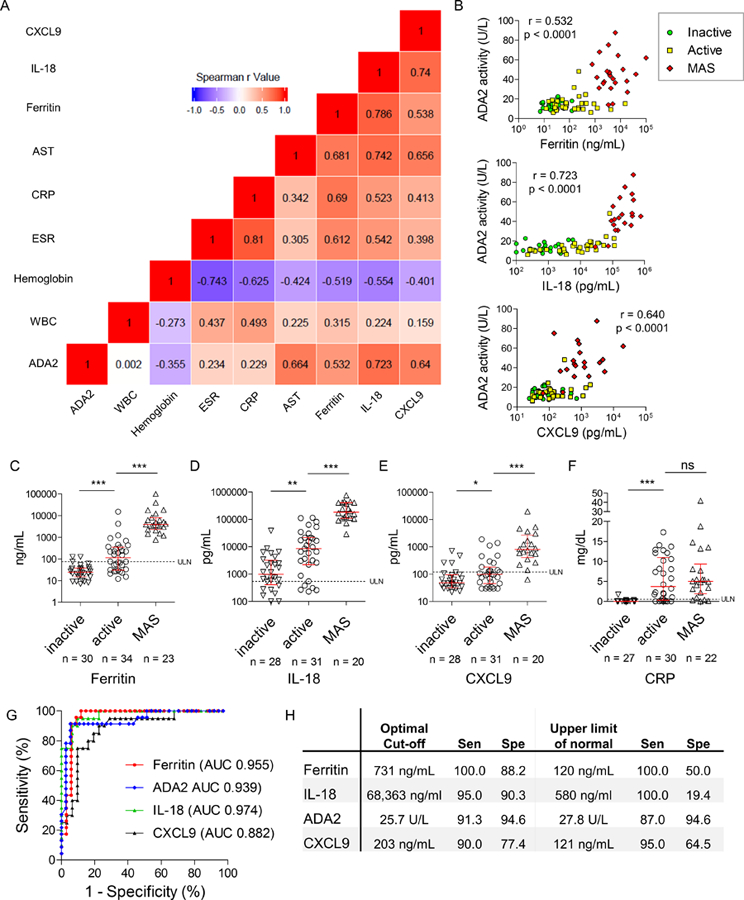
Comparison of ADA2 and other biomarkers of MAS. A) Correlation matrix of ADA2 activity with laboratory parameters in sJIA. Heat-map displays the strength of correlation based on Spearman’s rank correlation coefficient. B) Correlations of ADA2 with serum ferritin, IL-18 and CXCL9. All sJIA patients (inactive, active and MAS groups) were included for calculation of Spearman’s rank correlation coefficient. C-F) Comparison of peripheral blood ferritin, IL-18, CXCL9 and CRP levels in sJIA patient with inactive disease, active disease or MAS. Median and interquartile range are displayed in scatter dot plots. G) ROC curves of ferritin, ADA2, IL-18 and CXCL9. H) Sensitivity (Sen) and specificity (Spe) of MAS biomarkers using the optimized cut-off or upper limit of normal as cut-off. * p < 0.05, ** p< 0.001, *** p<0.0001
Unlike the specific elevation of ADA2 in MAS (Figure 2E), ferritin levels were normal in inactive sJIA and displayed a step-wise increase in active sJIA and MAS groups (Figure 3C). As described by previous studies [18, 19], IL-18 was elevated in the majority of sJIA patients, including many with inactive disease (Figure 3D). Higher levels of IL-18 were observed with active disease and log-fold increases were seen the MAS group. CXCL9 was significantly elevated in the MAS group compared to the active sJIA group (Figure 3E), matching the observations by Bracaglia and colleagues [20]. Only a small difference in CXCL9 levels was observed between the inactive and active sJIA groups.
Despite the different patterns displayed by these markers, all of them were sensitive and specific in discriminating MAS from active sJIA (Figure 3G). Unlike other markers, ADA2 effectively distinguished MAS from active sJIA using the upper limit of normal as cut-off, without the need to optimize the cut-off value (Figure 3H, Supplemental Tables S2 and S3). The recently described ferritin-to-ESR ratio [21] also correlated with ADA2 levels and performed well as an MAS biomarker (Figure S4D–F). In contrast, CRP could differentiate active vs. inactive disease in sJIA but performed poorly as a biomarker of MAS (Figure 3F).
Because features of MAS including ferritin levels may be influenced by biologic therapy [22], we evaluated the relationship between ADA2 activity and treatment choices in MAS. More than half of patients in the combined MAS cohort received IL-1 blockade (anakinra or canakinumab); 1 patient received tocilizumab and 3 patients received combined IL-1 and IL-6 blockade. Overall, we did not find differences in ADA2 activity related to biologic therapy (Figure S4G).
Longitudinal evaluation of ADA2 in patients with recurrent MAS
Serial samples were available from 7 MAS patients, with MAS classification criteria data at each time point. Consistent with our cross-sectional analysis, ADA2 levels were generally higher during confirmed MAS episodes compared to time points without MAS (Figure 4A). As illustrated by a patient with recurrent MAS episodes, longitudinal measures of ADA2 activity trended closely with serum ferritin (Figure 4B).
Figure 4.
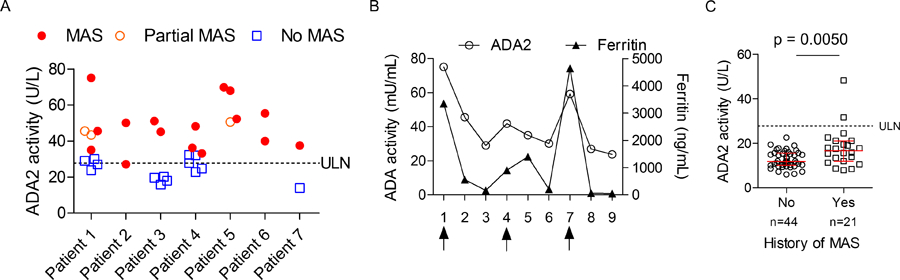
Longitudinal analysis of ADA2 levels in patients with sJIA-associated MAS. A) Display of ADA2 activity measurements from multiple visits in 7 sJIA patients with recurrent MAS. Laboratory parameters from each time point were reviewed to determine the presence of MAS based on the 2016 MAS Classification Criteria. B) Longitudinal display of ADA2 activity and serum ferritin levels in a patient with recurrent MAS. Arrows indicate confirmed MAS episodes based on the Classification Criteria. C) ADA2 activity in patients with inactive and active sJIA (without active MAS) stratified by past history of MAS. Median and interquartile range are displayed in panel C.
Notably, patients with recurrent MAS episodes exhibited ADA2 levels near the ULN even in the absence of MAS (Figure 4A). Expanding on this observation, we asked whether a history of MAS was associated with differences in ADA2 activity in the inactive and active sJIA groups (without MAS at the time of sampling). Among the 65 patients in these two groups (Figure 2E), 21 had a history of at least one MAS episode; these patients collectively displayed significantly higher levels of ADA2 activity compared to the group without a history of MAS (Figure 4C; median 17.5 U/L vs. 11.8 U/L, p=0.005).
Mechanism and cellular source of ADA2 production
The pathophysiology of MAS is reminiscent of primary hemophagocytic lymphohistiocytosis (HLH), wherein the inability to remove activated leukocytes results in a vicious cycle of reciprocal immune activation by NK / T cells and macrophages [23]. Cytokine storm is a hallmark of MAS, and repeated stimulation by Toll-like receptor ligands (TLR) causes excess cytokine production and a MAS-like disease in mice [24]. To understand what drives the expression of ADA2 in MAS, we stimulated peripheral blood mononuclear cells (PBMC) from healthy donors with selected cytokines and TLR ligands. Stimulation with IFNγ, IL-12, IL-18 and TNF-α increased ADA2 activity in the supernatant (Figure 5A). Many of these cytokines, especially IFN-γ (as measured by its proxy CXCL9) and IL-18, are highly elevated in the plasma of patients with MAS [18, 20]. Combining IL-18 and IFN-γ did not further enhance ADA2 production (Figure S5A). In contrast, ADA2 levels were not altered by IL-1β, IL-4, IL-6, IL-10, IFN-α, or transforming growth factor β (TGF-β). While plasma IL-10 and TGF-β levels were elevated in a subset of patients with MAS and the two cytokines correlated with each other, they did not correlate with ADA2 levels (Figure S5B,C). Neither M1/M2 polarization of macrophages nor stimulation with TLR ligands affected ADA2 activity (Figure 5A and S5D), further showing that ADA2 release is closely regulated.
Figure 5.
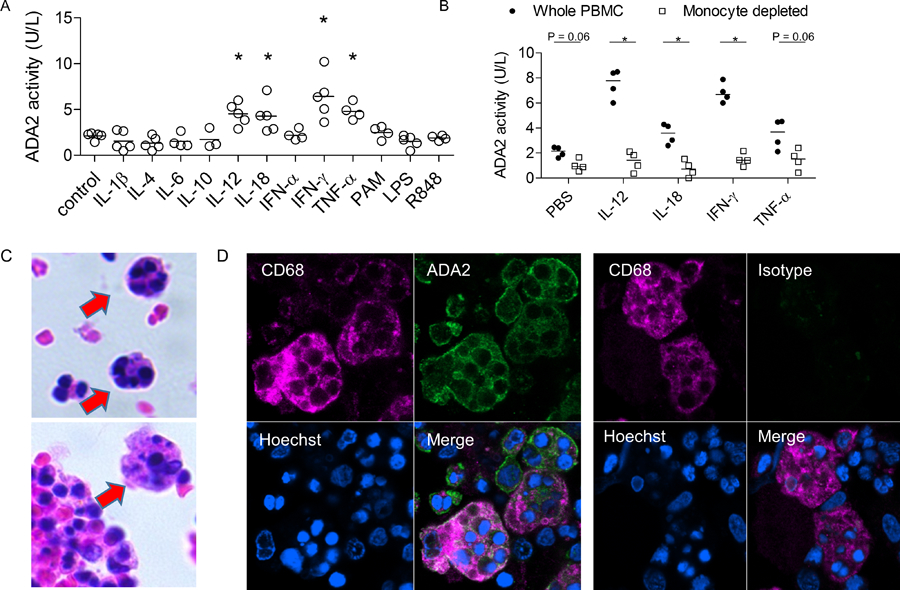
Mechanism and source of ADA2 production in MAS. A) ADA2 activity in the supernatant of healthy donor PBMC stimulated with cytokines or TLR ligands for 5 days. B) ADA2 activity in the supernatant of total donor PBMC or monocyte-depleted PBMC following cytokine stimulation for 5 days. Dots represent results from 3–5 healthy donors per condition. * p < 0.05 compared to unstimulated control (panel A) or compared to whole PBMC (panel B). C) H&E staining of bone marrow aspirate illustrating the presence of hemophagocytes. D) Confocal microscopy of ADA2 staining and isotype staining in CD68+ hemophagocytes.
Early studies found that monocytes and macrophages are primary sources of ADA2 [3, 4]. We performed both monocyte depletion and enrichment studies and confirmed that ADA2 production by PBMC was primary derived from monocytes, with or without cytokine stimulation (Figure 5B and S5E). Thus, ADA2 activity represents a functional readout of monocyte / macrophage activation by cytokines implicated in the pathogenesis of MAS.
Hemophagocytosis by activated macrophages is a hallmark histologic finding of MAS [23]. To investigate whether these tissue macrophages represent a source of ADA2, we performed confocal microscopy of bone marrow from a patient with adult onset Still’s disease and overt MAS. An abundance of hemophagocytes engulfing other leukocytes was evident (H&E staining; Figure 5C). Remarkably, by confocal microscopy, hemophagocytes expressing the macrophage marker CD68 revealed particularly strong expression of ADA2 using polyclonal anti-ADA2 antibodies (Figure 5D). Isotype staining confirmed the specificity of ADA2 staining, and similar results were found using a monoclonal antibody for ADA2 (Figure 5D and S6). These data support monocytes and macrophages, including hemophagocytes, as likely sources of ADA2 in MAS.
Discussion
MAS is a life-threatening complication that occurs in sJIA and other inflammatory conditions. Unopposed activation of immune cells and excess cytokine production results in a vicious cycle of inflammation that can lead to rapid clinical deterioration. Biomarkers of MAS could assist with early detection and therapeutic monitoring of this dangerous complication. Our study now establishes ADA2 as a novel biomarker of MAS in patients with sJIA.
Hallmark findings of MAS include hyperferritinemia, cytopenias, transaminase elevation, and coagulopathy. Activation of T cells, NK cells and myeloid cells in MAS is reflected in markers such as soluble IL-2 receptor (CD25), soluble CD163, IL-18, and the IFN-γ-induced chemokines CXCL9 and CXCL10. In two cohorts of sJIA patients, we found that ADA2 levels were largely normal in both inactive and active sJIA as long as MAS was absent. Using the ULN established in healthy children, we found that ADA2 is a sensitive and specific marker that distinguishes MAS from active sJIA. While elevation of ferritin, IL-18 and CXCL9 are all indicative of MAS, cut-off values well above the ULN are required to distinguish MAS from active sJIA.
ADA2 levels are also increased in the setting of certain infections, including HIV and tuberculosis [8, 25], perhaps in part due to the production of IL-18 and IFN-γ. While ADA2 is a specific marker of MAS in children with sJIA, it remains to be determined whether it can distinguish MAS from infection. Importantly, ADA2 levels were not increased in acute KD, confirming that ADA2 is not simply a marker of systemic inflammation. ADA2 levels were mildly increased in patients with pSLE and JDM compared to healthy controls. The increased ADA2 levels may reflect abnormal monocyte / macrophage activation and cytokine production associated with these diseases, which can also be complicated by MAS [26–28].
The factors that regulate ADA2 production have not been studied [1, 4]. We showed monocytes to be the major producers of ADA2 in PBMC. Using confocal microscopy, we also showed abundant expression of ADA2 in MAS hemophagocytes. Furthermore, we found that stimulation with a specific panel of cytokines increased the production and release of ADA2. Extremely high levels of these cytokines, especially IL-18 and IFN-γ, provides a potential mechanistic explanation for the elevated plasma ADA2 level in sJIA-associated MAS [28].
Whether ADA2 contributes to the pathophysiology of MAS is not clear. The physiologic function of ADA2 remains to be determined. Features of inflammatory vasculitis in patients with DADA2 suggests an immunomodulatory function of ADA2 as well as a role in preventing vasculopathy. Depending on context and receptor utilization, the ADA substrate adenosine can be pro-inflammatory or anti-inflammatory [29]. The reaction product inosine also has immunomodulatory functions [30]. The role of adenosine and inosine in MAS is a topic of interest for future study.
In summary, we establish the normal range of peripheral blood ADA2 activity in children and demonstrate that levels above this range are sensitive and specific for MAS associated with sJIA. The monocyte / macrophage origin of ADA2 and induction by IL-18 and IFN-γ lend further support to the key role of macrophage activation in this life-threatening complication of sJIA.
Supplementary Material
Key Messages.
What is already known about this subject?
Macrophage activation syndrome (MAS) is a life-threatening complication of systemic juvenile idiopathic arthritis (sJIA). Because clinical manifestations of active sJIA and MAS overlap, distinguishing biomarkers are needed for rapid diagnosis and treatment.
What does this study add?
We defined the normal range in peripheral blood of the monocyte / macrophage-secreted protein adenosine deaminase 2 (ADA2) in healthy children and adults.
We found that ADA2 activity above the upper limit of normal distinguishes MAS in sJIA with high sensitivity and specificity, potentially reflect direct stimulation of monocytes and macrophages by cytokines implicated in MAS.
How might this impact on clinical practice or future developments?
Our study demonstrates the utility of peripheral blood ADA2 activity as a direct biomarker of macrophage activation to facilitate rapid diagnosis of MAS in sJIA.
Acknowledgements
We thank the patients and volunteers for participation. We thank N. Ganson and S. Kelly for assistance with developing the spectrophotometric assay for quantitation of ADA2 activity.
Funding Sources
This work was supported by the National Institute of Health / National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) K08-AR074562 (P.Y.L.), K08-AR072075 (G.S.S), R01-AR065538, R01-AR073201, R01-AR075906 and P30-AR070253 (P.A.N.), a Rheumatology Research Foundation Investigator Award (P.Y.L.), a Boston Children’s Hospital Faculty Career Development Award (P.Y.L), and the Fundación Bechara and the Arbuckle Family Fund for Arthritis Research (P.A.N.).
Footnotes
Competing Interests
The authors declare no conflict of interest related to this work.
Ethical approval
These studies were approved by the Institutional Review Boards at Boston Children’s Hospital (P00005723), Cincinnati Children’s Hospital Medical Center (2016–2234), University of Pittsburg (PRO16120025) and Brigham and Women’s Hospital (P000664). Informed consent was provided by participants or legal guardians.
Data Sharing
All data relevant to the study are included in the article or uploaded as supplementary information
Patient and Public Involvement
This research was done without direct patient involvement beyond sample collection. Patients did not participate in designing the study, analyzing the data, or drafting the manuscript.
References
- 1.Ungerer JP, Oosthuizen HM, Bissbort SH, Vermaak WJ. Serum adenosine deaminase: isoenzymes and diagnostic application. Clinical chemistry. 1992. July; 38(7):1322–1326. [PubMed] [Google Scholar]
- 2.Giblett ER, Anderson JE, Cohen F, Pollara B, Meuwissen HJ. Adenosine-deaminase deficiency in two patients with severely impaired cellular immunity. Lancet. 1972. November 18; 2(7786):1067–1069. [DOI] [PubMed] [Google Scholar]
- 3.Ratech H, Martiniuk F, Borer WZ, Rappaport H. Differential expression of adenosine deaminase isozymes in acute leukemia. Blood. 1988. November; 72(5):1627–1632. [PubMed] [Google Scholar]
- 4.Iwaki-Egawa S, Yamamoto T, Watanabe Y. Human plasma adenosine deaminase 2 is secreted by activated monocytes. Biological chemistry. 2006. March; 387(3):319–321. [DOI] [PubMed] [Google Scholar]
- 5.Schrader WP, Pollara B, Meuwissen HJ. Characterization of the residual adenosine deaminating activity in the spleen of a patient with combined immunodeficiency disease and adenosine deaminase deficiency. Proceedings of the National Academy of Sciences of the United States of America. 1978. January; 75(1):446–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Navon Elkan P, Pierce SB, Segel R, Walsh T, Barash J, Padeh S, et al. Mutant adenosine deaminase 2 in a polyarteritis nodosa vasculopathy. The New England journal of medicine. 2014. March 6; 370(10):921–931. [DOI] [PubMed] [Google Scholar]
- 7.Zhou Q, Yang D, Ombrello AK, Zavialov AV, Toro C, Stone DL, et al. Early-onset stroke and vasculopathy associated with mutations in ADA2. The New England journal of medicine. 2014. March 6; 370(10):911–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gakis C, Calia G, Naitana A, Pirino D, Serru G. Serum adenosine deaminase activity in HIV positive subjects. A hypothesis on the significance of ADA2. Panminerva medica. 1989. Jul-Sep; 31(3):107–113. [PubMed] [Google Scholar]
- 9.Stancikova M, Lukac J, Istok R, Cristalli G, Rovensky J. Serum adenosine deaminase activity and its isoenzyme pattern in patients with systemic lupus erythematosus. Clinical and experimental rheumatology. 1998. Sep-Oct; 16(5):583–586. [PubMed] [Google Scholar]
- 10.Sari RA, Taysi S, Yilmaz O, Bakan N. Correlation of serum levels of adenosine deaminase activity and its isoenzymes with disease activity in rheumatoid arthritis. Clinical and experimental rheumatology. 2003. Jan-Feb; 21(1):87–90. [PubMed] [Google Scholar]
- 11.Chen W, Zhang S, Zhang W, Yang X, Xu J, Qiu H, et al. Elevated serum adenosine deaminase levels in secondary hemophagocytic lymphohistiocytosis. International journal of laboratory hematology. 2015. August; 37(4):544–550. [DOI] [PubMed] [Google Scholar]
- 12.Lee PY, Huang Y, Zhou Q, Schnappauf O, Hershfield MS, Li Y, et al. Disrupted N-linked glycosylation as a disease mechanism in deficiency of ADA2. The Journal of allergy and clinical immunology. 2018. October; 142(4):1363–1365 e1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Muraoka T, Katsuramaki T, Shiraishi H, Yokoyama MM. Automated enzymatic measurement of adenosine deaminase isoenzyme activities in serum. Analytical biochemistry. 1990. June; 187(2):268–272. [DOI] [PubMed] [Google Scholar]
- 14.Slaats EH, Asberg EG, van Keimpema AR, Kruijswijk H. A continuous method for the estimation of adenosine deaminase catalytic concentration in pleural effusions with a Hitachi 705 discrete analyser. Journal of clinical chemistry and clinical biochemistry Zeitschrift fur klinische Chemie und klinische Biochemie. 1985. October; 23(10):677–682. [DOI] [PubMed] [Google Scholar]
- 15.Nanthapisal S, Murphy C, Omoyinmi E, Hong Y, Standing A, Berg S, et al. Deficiency of Adenosine Deaminase Type 2: A Description of Phenotype and Genotype in Fifteen Cases. Arthritis Rheumatol. 2016. September; 68(9):2314–2322. [DOI] [PubMed] [Google Scholar]
- 16.Petty RE, Southwood TR, Manners P, Baum J, Glass DN, Goldenberg J, et al. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001. The Journal of rheumatology. 2004. February; 31(2):390–392. [PubMed] [Google Scholar]
- 17.Ravelli A, Minoia F, Davi S, Horne A, Bovis F, Pistorio A, et al. 2016 Classification Criteria for Macrophage Activation Syndrome Complicating Systemic Juvenile Idiopathic Arthritis: A European League Against Rheumatism/American College of Rheumatology/Paediatric Rheumatology International Trials Organisation Collaborative Initiative. Arthritis Rheumatol. 2016. March; 68(3):566–576. [DOI] [PubMed] [Google Scholar]
- 18.Shimizu M, Yokoyama T, Yamada K, Kaneda H, Wada H, Wada T, et al. Distinct cytokine profiles of systemic-onset juvenile idiopathic arthritis-associated macrophage activation syndrome with particular emphasis on the role of interleukin-18 in its pathogenesis. Rheumatology (Oxford). 2010. September; 49(9):1645–1653. [DOI] [PubMed] [Google Scholar]
- 19.Weiss ES, Girard-Guyonvarc’h C, Holzinger D, de Jesus AA, Tariq Z, Picarsic J, et al. Interleukin-18 diagnostically distinguishes and pathogenically promotes human and murine macrophage activation syndrome. Blood. 2018. March 29; 131(13):1442–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bracaglia C, de Graaf K, Pires Marafon D, Guilhot F, Ferlin W, Prencipe G, et al. Elevated circulating levels of interferon-gamma and interferon-gamma-induced chemokines characterise patients with macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. Annals of the rheumatic diseases. 2017. January; 76(1):166–172. [DOI] [PubMed] [Google Scholar]
- 21.Eloseily EMA, Minoia F, Crayne CB, Beukelman T, Ravelli A, Cron RQ. Ferritin to Erythrocyte Sedimentation Rate Ratio: Simple Measure to Identify Macrophage Activation Syndrome in Systemic Juvenile Idiopathic Arthritis. ACR Open Rheumatology. 2019. [DOI] [PMC free article] [PubMed]
- 22.Schulert GS, Minoia F, Bohnsack J, Cron RQ, Hashad S, Kon EPI, et al. Effect of Biologic Therapy on Clinical and Laboratory Features of Macrophage Activation Syndrome Associated With Systemic Juvenile Idiopathic Arthritis. Arthritis care & research. 2018. March; 70(3):409–419. [DOI] [PubMed] [Google Scholar]
- 23.Al-Samkari H, Berliner N. Hemophagocytic Lymphohistiocytosis. Annual review of pathology. 2018. January 24; 13:27–49. [DOI] [PubMed] [Google Scholar]
- 24.Behrens EM, Canna SW, Slade K, Rao S, Kreiger PA, Paessler M, et al. Repeated TLR9 stimulation results in macrophage activation syndrome-like disease in mice. The Journal of clinical investigation. 2011. June; 121(6):2264–2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ungerer JP, Oosthuizen HM, Retief JH, Bissbort SH. Significance of adenosine deaminase activity and its isoenzymes in tuberculous effusions. Chest. 1994. July; 106(1):33–37. [DOI] [PubMed] [Google Scholar]
- 26.Amerio P, Frezzolini A, Abeni D, Teofoli P, Girardelli CR, De Pita O, et al. Increased IL-18 in patients with systemic lupus erythematosus: relations with Th-1, Th-2, pro-inflammatory cytokines and disease activity. IL-18 is a marker of disease activity but does not correlate with pro-inflammatory cytokines. Clinical and experimental rheumatology. 2002. Jul-Aug; 20(4):535–538. [PubMed] [Google Scholar]
- 27.Li Y, Lee PY, Reeves WH. Monocyte and macrophage abnormalities in systemic lupus erythematosus. Archivum immunologiae et therapiae experimentalis. 2010. October; 58(5):355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schulert GS, Grom AA. Pathogenesis of macrophage activation syndrome and potential for cytokine- directed therapies. Annual review of medicine. 2015; 66:145–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hasko G, Linden J, Cronstein B, Pacher P. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nature reviews Drug discovery. 2008. September; 7(9):759–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hasko G, Sitkovsky MV, Szabo C. Immunomodulatory and neuroprotective effects of inosine. Trends in pharmacological sciences. 2004. March; 25(3):152–157. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.