

Abstract
We have previously shown that treatment with a mGluR5 positive allosteric modulator (PAM) is neuroprotective after experimental traumatic brain injury (TBI), limiting post-traumatic neuroinflammation by reducing pro-inflammatory microglial activation and promoting anti-inflammatory and neuroprotective responses. However, the specific molecular mechanisms governing this anti-inflammatory shift in microglia are unknown. Here we show that the mGluR5 PAM, VU0360172 (VuPAM), regulates microglial inflammatory responses through activation of Akt, resulting in the inhibition of GSK-3β. GSK-3β regulates the phosphorylation of CREB, thereby controlling the expression of inflammation-related genes and microglial plasticity. The anti-inflammatory action of VuPAM in microglia is reversed by inhibiting Akt/GSK-3β/CREB signaling. Using a well-characterized TBI model and CX3CR1gfp/+ mice to visualize microglia in vivo, we demonstrate that VuPAM enhances Akt/GSK-3β/CREB signaling in the injured cortex, as well as anti-inflammatory microglial markers. Furthermore, in situ analysis revealed that GFP+ microglia in the cortex of VuPAM treated TBI mice co-express pCREB and the anti-inflammatory microglial phenotype marker YM1. Taken together, our data show that VuPAM decreases pro-inflammatory microglial activation by modulating Akt/GSK-3β/CREB signaling. These findings serve to clarify the potential neuroprotective mechanisms of mGluR5 PAM treatment after TBI, and suggest novel therapeutic targets for post-traumatic neuroinflammation.
Keywords: metabotropic glutamate receptor 5, microglia, positive allosteric modulator, Akt/GSK-3β/CREB signaling, anti-inflammatory, traumatic brain injury
Graphical Abstract

The mGluR5 positive allosteric modulator (PAMs), VU0360172, is neuroprotective when administered after experimental traumatic brain injury (TBI) in mice. Here, using in vitro and in vivo models we determined that VU0360172 stimulates Akt/GSK-3β/CREB signalling to suppress pro-inflammatory microglial responses and concurrently upregulate an anti-inflammatory phenotype. VU0360172, unlike the mGluR5 orthosteric agonist CHPG, had no effect on classical PLC/PKC or NFΚB signalling pathways indicating differences in the downstream signal transduction pathways between orthosteric and allosteric modulators of mGluR5 in microglia. Thus, mGluR5 PAMs stimulate alternative signalling pathways in microglia that contribute to anti-inflammatory and neuroprotective responses in the brain following TBI.
Introduction
Traumatic brain injury (TBI) is a major cause of death and disability in the United States (Taylor et al. 2017). TBI not only causes direct mechanical injury to the brain, but also initiates secondary biochemical and physiological changes that contribute to late brain atrophy and associated neurological deficits (Faden & Loane 2015; Loane & Byrnes 2010; Simon et al. 2017). It has been increasingly recognized, both clinically and experimentally, that neuroinflammation contributes substantially to long-term neurodegeneration and related neuropsychiatric dysfunction (Faden & Loane 2015; Loane & Byrnes 2010; Simon et al. 2017). Like peripheral macrophages, microglia show remarkable plasticity in response to disturbances in homeostasis and local brain injury, and they can acquire either neurotoxic (pro-inflammatory) or neuroprotective (anti-inflammatory) activation states (Kettenmann et al. 2013; Loane & Kumar 2016). Within the context of neuroinflammatory disorders, more recent work suggests that in addition to limiting the pro-inflammatory microglial activation state, therapeutic strategies that promote anti-inflammatory signaling and a neuroprotective microglial phenotype may have greater potential to improve long-term neurological outcomes (Andreou et al. 2017; Kim et al. 2015; Loane & Kumar 2016; Simon et al. 2017).
Metabotropic glutamate receptor 5 (mGluR5) is a G-protein-coupled receptor (GPCR) that has emerged as an important therapeutic target for schizophrenia (Stansley & Conn 2018). We previously demonstrated that selective activation of mGluR5 in microglia by its orthosteric agonist, CHPG, reduces pro-inflammatory microglial activation through a Gαq-coupled signal transduction pathway (Byrnes et al. 2009b). This results in reduced microglial-mediated neurotoxicity in in vitro co-culture models, as well as neuroprotection and enhanced functional recovery in experimental models of traumatic brain and spinal cord injury (Byrnes et al. 2012; Byrnes et al. 2009c; Byrnes et al. 2009a; Loane et al. 2013; Loane et al. 2009b). In contrast to orthosteric agonists, positive allosteric modulators (PAM) bind to sites other than the ligand binding domain of GPCRs and can provide functional selectivity by differentially modulating downstream signaling (Foster & Conn 2017). mGluR5 PAM have been shown to ameliorate fibrinogen-mediated microglial activation and neurodegeneration in vitro (Piers et al. 2011). We showed that the mGluR5 PAM, VU0360172 (VuPAM) limits post-traumatic neuroinflammation and alters microglial phenotypes through mechanisms that differ from orthosteric agonists such as CHPG (Loane et al. 2014). Of note, VuPAM not only decreases pro-inflammatory microglial activation following TBI, similar to CHPG treatment (Byrnes et al. 2012; Loane et al. 2013), but also promotes an anti-inflammatory microglial activation state (Loane et al. 2014). These observations suggest likely differences in downstream signaling between mGluR5 by orthosteric and allosteric modulators.
As mGluR5 is a Gαq-coupled GPCR, its classical downstream signaling is mediated by phosphoinositide hydrolysis and release of Ca2+ from intracellular stores (Stoppel et al. 2017; Lindemann et al. 2011). However, studies indicate that other signal transduction pathways may also play a central role in the modulation of gene expression by mGluR5 (Stoppel et al. 2017; Bhakar et al. 2012; Osterweil et al. 2010; Richter et al. 2015). One alternative pathway activated by mGluR5 is PI3K/Akt (Shah et al. 2012; Batista et al. 2016). Akt, a serine/threonine-specific protein kinase, can regulate diverse physiological functions, ranging from metabolism to cell survival (Manning & Cantley 2007). Akt phosphorylates the constitutively active serine-threonine kinase GSK-3β on Ser9 residue (Beurel et al. 2015), resulting in its inhibition (Atkins et al. 2012). Aberrant GSK-3β activation is associated with neurodegeneration and neuroinflammation, and is known to affect transcription factors involved in inflammatory activation (Maixner & Weng 2013; Eldar-Finkelman & Martinez 2011). Among these transcription factors, CREB - a leucine zipper transcription factor that regulates immune-related genes (Wen et al. 2010) - is activated by Akt signaling through inhibition of GSK-3β (Qiao et al. 2018). However, whether mGluR5 PAM regulate microglial activation through Akt/GSK-3β/CREB signaling has not been examined.
Here, we investigated Akt/GSK-3β/CREB signal transduction pathways in regulating microglial reactivity in response to VuPAM, using both in vitro and in vivo models. We show that VuPAM increases the phosphorylation of Akt, GSK-3β and CREB – thereby reducing pro-inflammatory microglial activation in vitro and following controlled cortical impact in mice. Moreover, pharmacological and dominant negative mutant studies demonstrate that inhibition of Akt/GSK-3β/CREB signaling reverses the anti-inflammatory and neuroprotective actions of VuPAM in microglia.
Materials and Methods
Materials:
N-cyclobutyl-6-[2-(3-fluorophenyl)ethynyl]-3-pyridinecarboxamide hydrochloride (VU0360172; mGluR5 positive allosteric modulator; #4323), (RS)-2-chloro-5-hydroxyphenylglycine (CHPG; mGluR5 orthosteric agonist; #1049), and 3-[(2-Methyl- 4-thiazolyl)ethynyl]pyridine (MTEP; mGluR5 antagonist; #2921) were purchased from Tocris Biosciences (Minneapolis, MN). Wortmannin (PI3K inhibitor, CAS 19545–26-7; # 681675), CCI (CREB/CBP interaction inhibitor, CAS 92–78-4; #217505), AKTi (AKT inhibitor, CAS 612847–09-3, #124005), Rottlerin (PKC inhibitor, CAS 82–08-6, # 557370), and BIO (GSK-3β inhibitor, CAS 667463–62-9, #361550) were from Calbiochem-Merck Millipore (Burlington, MA). DIF-3 (GSK-3β activator, CAS 113411–17-9, #D0567) was from Sigma-Aldrich (St. Louis, MO). Primary antibodies were purchased as indicated: anti-mGluR5 (#ab76316, RRID:AB_1523944), anti-GFAP (#ab7260, RRID:AB_305808) from Abcam (Eugene, OR); anti-p65NFкB (#8242; RRID:AB_10859369), anti-IκB-α (#9246, RRID:AB_2267145), anti-phospho-S133-CREB (#9198; RRID:AB_2561044), anti-CREB (#4820, RRID:AB_1903940), anti-phospho-S9-GSK-3β (#9336, RRID:AB_331405), anti-GSK-3β (#12456, RRID:AB_2636978), anti-phospho-S433-AKT (#9271, RRID:AB_329825), anti-AKT (#9272, RRID:AB_329827), anti-Lamin A/C, (#4777, RRID:AB_10545756) antibodies from Cell Signaling Technology (Danvers, MA); anti-Iba-1 (#019–19741, RRID:AB_839504) from Wako Chemicals (Richmond, VA); anti-Arginase-1 (#610709, RRID:AB_398032), anti-iNOS (#610329, RRID:AB_397719) from BD Transduction Laboratories (San Jose, CA); anti-YM1 (#01404, RRID:AB_528565) from STEMCELL Technologies (Cambridge, MA), anti-β-actin (#A2228, RRID:AB_476697) from Sigma-Aldrich (St. Louis, MO). Alexa Fluor conjugated secondary antibodies (Invitrogen, Carlsbad, CA), and HRP-conjugated secondary antibodies were purchased from Jackson Immuno Research Laboratories (West Grove, PA).
BV2 cell culture
BV2 murine microglia (RRID:CVCL_0182) were cultured in DMEM (Life Technologies, Carlsbad, CA) supplemented with 10% fetal bovine serum (Life Technologies) and 1% penicillin and streptomycin (40 U/mL and 40 µg/mL, respectively; Sigma) at 37°C with 5% CO2, as previously described (Loane et al. 2009b). Cells were split 1:5 until their confluency using 0.05% Trypsin-EDTA (Life Technologies) solution in phosphate buffered saline (PBS). DMEM media was filtered using 0.2-μm filter (Sigma). BV2 microglia were seeded at a density of either 3 × 105 cells/well (6-well plate), 1.5 × 105 cells/well (12-well plate), 0.75 × 105 cells/well (24-well plate) and 1 × 104 cells/well (96-well plate). BV2 cells are not listed in commonly misidentified cell lines provided by the International Cell Line Authentication Committee, and they were not authenticated prior to initiating the study. The maximum number of passages for BV2 cells was set at 15.
Primary microglia and neuronal cultures:
Primary microglia cultures were prepared from cerebral cortices of postnatal day (p) 1 Sprague-Dawley rat pups (Envigo, Indianapolis, IN; RRID:RGD_10395233) as previously described (Loane et al. 2014; Bhat et al. 2018). A total of 60 p1 Sprague-Dawley rat pups were used in experiments. According to the AVMA Guidelines for the Euthanasia of Animals (2013 Edition), narcosis was induced using the gradual fill method for CO2 gas administration using a commercial tank, pressure reducing regulator, and flow meter. The euthanasia chamber was not precharged with CO2. The flow rate was within 20%−30% of the chamber volume per minute. CO2 flow was maintained for a minimum of one minute following respiratory arrest. Decapitation was performed secondarily to assure euthanasia. Then, brains were carefully isolated, and cerebral cortices were collected and freed from meninges in HBSS without Calcium, Magnesium and Phenol Red (Life Technologies). Forebrains were minced and gently dissociated by repeated pipetting in DMEM/F-12 (Life Technologies) and filtered by passing through 70 µm cell strainer (Sigma). Cells were collected by centrifugation (1000 x g, 10 min at 4°C) and resuspended in DMEM/F-12 containing 10% fetal bovine serum and 1% penicillin and streptomycin (40 U/mL and 40 µg/ml, respectively), and cultured on Poly-l-lysine (Sigma) coated 75 cm2 cell culture flask in 5% CO2 at 37 °C. After 12–14 days in culture, microglia were harvested from mixed glia (astrocyte-microglia) cultures by shaking at 200 rpm for 1 h and finally seeded into cell culture plates at the density of either 0.75 × 105 cells/well (24-well plate) or 1.5 × 105 cells/well (12-well plate). On the next day, non-adherent cells were removed by changing the media, and after 1 h cells were used for experiments.
Primary cortical neuronal cultures were prepared from Sprague-Dawley rat embryonic cortices (15–16 d-old embryos; Envigo; RRID:RGD_10395233), as previously described (Sabirzhanov et al. 2014). A total of 20 e15–16 Sprague-Dawley rat pups were used in experiments. Euthanasia was performed according to the AVMA Guidelines for the Euthanasia of Animals (2013 Edition) and as detailed above. Rat pup cortices from 15- to 16-d-old embryos were cleaned from their meninges and blood vessels in Kreb’s-Ringer’s bicarbonate buffer containing 0.3% bovine serum albumin (BSA; Life Technologies, Gaithersburg, MD). Cortices were then minced and dissociated in the same buffer with 1800 U/ml trypsin (Sigma, St. Louis, MO) at 37°C for 20 min. After the addition of 200 U/ml DNase I (Sigma) and 3600 U/ml soybean trypsin inhibitor (Sigma) to the suspension, cells were triturated through a 5 ml pipette. After the tissue was allowed to settle for 5–10 min, the supernatant was collected, and the remaining tissue pellet was retriturated. The combined supernatants were then centrifuged through a 4% BSA layer and the cell pellet was resuspended in neuronal seeding medium, which consisted of neurobasal medium (Life Technologies) supplemented with 1.1% 100X antibiotic-antimycotic solution (Biofluids, Rockville, MD), 25 μM Na-glutamate, 0.5 mM L-glutamine, and 2% B27 supplement (Life Technologies). Cells were seeded at a density of 5 X 105 cells/ml onto poly-D-lysine (70–150 kDa; Sigma)-coated plates (Corning, Corning, NY). All experiments were performed on cultures at 7 days in vitro (DIV).
Treatments:
Primary microglia and BV2 microglia were incubated with or without various concentration of VU0360172 (20 or 50 µM; (Loane et al. 2014)) or MTEP (10 µM, (Byrnes et al. 2009b)) followed by lipopolysaccharide (LPS; from Escherichia coli; 100 ng/mL; Sigma) stimulation for 24 h. In other experiments cells were pre-treated for 30 min with Wortmannin (1 μM; (Mead et al. 2012)), CREB/CBP interaction inhibitor (1 μM; (Alexaki et al. 2018)), AKT inhibitor (1 μM; (Alexaki et al. 2018)), Rottlerin (5 μM; (Bhat et al. 2018)), BIO (5 μM;(Meijer et al. 2003)), or DIF-3 (30 μM; (Takahashi-Yanaga et al. 2006)), followed by VU0360172 (50 μM) treatment and LPS (100 ng/ml) stimulation, respectively.
Cell viability assay:
Cell viability was determined by the tetrazolium salt 3-[4,5-dimethylthiazol-2-yl]-2,5- diphenyltetrazolium bromide (MTT; #11465007001, Sigma-Aldrich) assay as described previously (Bhat et al. 2018). Viability data are expressed as a percentage of surviving cells compared with the control. Cellular toxicity was not found in microglia under any treatment condition (data not shown).
Neurotoxicity assay:
Rat cortical neurons were prepared as described above (Sabirzhanov et al. 2014). Cells were seeded onto poly-D-lysine-coated 12-wells (cell density 1 × 106/cm2) and maintained in serum-free conditions using the B27 supplement. After 6 days in vitro (DIV) primary neurons were treated with conditioned media (1 ml) collected from BV2 microglia. For co-culture experiments BV2 microglia were treated with LPS (100ng/ml) in the presence or absence of VU0360172 (50 µM) or VU0360172 (50 µM) + MTEP (100 µM) for 4 h, media was then replaced with serum free DMEM media and cultured for 20 h. At 24 h, BV2 conditioned media was collected and transferred to before primary neurons and neuronal viability was assessed 24 h later using the MTT assay.
siRNA and cell transfection:
BV2 cells in a 24-well plate were transfected with on-target smart pool siRNA for mGluR5 (50 nM; #108071; Dharmacon, Lafayette, CO) or control, scrambled non-targeting siRNA (50 nM; #D-001810–10-05). mGluR5 siRNAs included: 1) ON-TARGETplus SMARTpool siRNA J-060621–05, Grm5 Target Sequence: AAAGAAGAUGUACGAGGAA, 2) ON-TARGETplus SMARTpool siRNA J-060621–06, Grm5 Target Sequence: AAGAUAUGUCAGCGAAGGA, 3) ON-TARGETplus SMARTpool siRNA J-060621–07, Grm5 Target Sequence: GUGACAAGACUCUAUUCAA, and 4) ON-TARGETplus SMARTpool siRNA J-060621–08, Grm5 Target Sequence: GGGUCUAGCAGGGGAAUUU. All transfections were performed using Lipofectamine RNAimax (Life Technologies) and the reverse transfection method according to manufacturer’s instructions.
For dominant negative mutant CREB studies, BV2 cells seeded in six-well plates were transfected with 5 μg DNA of pCMV-CREB, pCMV-KCREB, or pCMV-CREB-133 plasmids (#631925; Clonetech Inc., Mountain View, CA) using Lipofectamine 3000 Reagent (Life Technologies) according to manufacturer’s instructions and as described previously (Alexaki et al. 2018). pCMV was used as a control (mock), whereas pCMV-KCREB over-expresses CREB containing mutations in its DNA-binding domain, thereby preventing its association with cAMP-regulated enhancer element. pCMV-CREB-133 expresses CREB having a serine to alanine mutation, which blocks the phosphorylation of CREB, thereby preventing transcription. Two days after transfection, 700 μg/ml Geneticin (#G418; Life Technologies) was added to the medium to select transfected clones. The transfected clones were maintained by culturing in G418-suplemented medium (Alexaki et al. 2018).
Western blotting:
To generate whole-cell extracts, cells were washed with ice-cold PBS and lysed in the radioimmunoprecipitation assay buffer (RIPA buffer, Teknova, Hollister, CA), with 3% phosphatase (Phosphatase Inhibitor Cocktail 2 & 3) and protease inhibitor cocktail (Sigma-Aldrich). Protein concentration of the samples was measured using the bicinchoninic acid (BCA) protein assay kit (ThermoFisher Scientific) according to the manufacturer’s instructions. For Western blotting, 25 μg of total protein from each sample was subjected to 5–20% gradient gels for sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were transferred onto nitrocellulose membranes and blocked in 5% BSA in phosphate buffered saline (PBS) containing 0.1% Tween 20 (PBS-T). After blocking, membranes were incubated with primary antibodies, including anti-mGluR5 (1:1000), anti-p65NFкB (1:1000), anti-IκB-α (1:1000), anti-phospho-S133-CREB (1:1000), anti-CREB (total; 1:1000), anti-phospho-S9-GSK-3β(1:1000), anti-GSK-3β (total; 1:1000), anti-phospho-S433-AKT (1:1000), anti-AKT(total; 1:1000), anti-iNOS (1:1000), anti-Arginase1 (1:1000), anti-LaminA/C (1:1000), and anti-β-actin (1:5000). Primary antibodies were diluted 5% BSA or milk in PBS-T. Membranes were incubated with the primary antibody overnight at 4 °C followed by incubation in appropriate HRP-conjugated secondary antibodies (Jackson Immuno Research Laboratories, West Grove, PA) for 1 h at room temperature. After extensive washing (three times for 15 min each in PBS-T), proteins were visualized using Super Signal West Dura Extended Duration Substrate (Thermo Scientific, Rockford, IL). Chemiluminescence was captured using ChemiDoc TM XRS+ System (Bio-Rad; Hercules, CA), and protein bands were quantified by densitometric analysis using Bio-Rad Molecular Imaging software. The data reflect the intensity of target protein band normalized to β-actin endogenous control for each sample (expressed in arbitrary units).
Immunoprecipitation:
For immunoprecipitation studies, aliquots of 200 μg of proteins from each sample were incubated with specific antibodies in the lysis buffer (20 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, containing 1% protease and phosphatase inhibitors) overnight at 4 °C. Thirty μl of protein A/G agarose beads (ThermoFisher Scientific, #20421) were added and the samples were incubated for 2 h at 4 °C. The beads were washed five times with lysis buffer (4 °C), boiled, and separated by SDS-PAGE, transferred onto nitrocellulose membrane for Western blot analysis described above.
Subcellular fractionation:
BV2 microglia were harvested and washed in ice-cold PBS. Cell suspension was centrifuged at 500 g for 15 min at 4 °C. Cell pellet was resuspended for 10 min on ice in the digitonin lysis buffer (20 mM HEPES, pH 7.4, 80 mM KCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 250 mM Sucrose, 200 μg/mL Digitonin and Protease Inhibitor and Phosphatase Inhibitor cocktails (Sigma-Aldrich). The lysate was centrifuged at 1000 g for 5 min at 4 °C to pellet the nuclei. The supernatant was transferred to a new tube and centrifuged at 12 000 g for 10 min at 4 °C. The resulting supernatant, representing the cytosolic fraction, was recovered. The nuclear fraction was prepared in RIPA buffer and subjected to SDS-PAGE.
Nitrite release:
Nitrite levels in cell culture supernatant were measured using a Griess reagent kit (#G7921, Invitrogen, Eugene OR) according to manufacturer’s instruction. Absorbance was measured at 548 nm using a synergy HT multi-mode microplate reader (Biotek, Winooski, VT). The amount of Nitrite (expressed in µM) released by microglia in each sample was calculated from the standard curve.
ELISA:
Cytokine levels in cell culture supernatant were quantified by ELISA using TNF-α and IL-10 specific assays (R&D Systems), according to the manufacturer’s instructions. Cytokine levels were expressed as pg of cytokines/ml.
Animals:
Studies were performed using 30 adult male C57BL/6J (RRID:IMSR_JAX:000664) or B6.129P-CX3CR1tm1Litt/J (CX3CR1gfp/+; Jackson Laboratory; 10–12 weeks old; 22–26 g) mice. All mice were housed in groups of five per cage in specific pathogen‐free conditions (12 h light/dark cycle, 24 ± 1°C and 55 ± 5% humidity) during the experimental period, with ad libitum access to food and water. All surgical procedures were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Maryland School of Medicine (Protocol Number 0318004). Surgical and drug treatment studies in mice were performed between 9 am and 3 pm.
Controlled cortical impact (CCI):
Traumatic brain injury in mice was induced using a custom-designed CCI device as described previously (Loane et al. 2009a). Briefly, C57BL/6J or CX3CR1gfp/+ mice were anesthetized in a chamber containing evaporated mixture of gases 70% N2O and 30% O2 along with isoflurane (induction at 4% and maintenance at 2%). Depth of anesthesia was assessed before initiation of the surgical procedure by monitoring respiration rate, and pedal withdrawal reflexes and body temperature maintained at 37 °C. The head was mounted in a stereotaxic frame and a 10 mm midline incision was made over the skull. After reflecting the skin and fascia, a 5 mm craniotomy was made on the central aspect of the left parietal bone. The impounder tip of the injury device was positioned to the surface of the exposed dura after extending to its full stroke distance (44 mm) and reset to impact the cortical surface. Moderate-severe level injury was induced using an impactor velocity of 6 m/sec and deformation depth of 2 mm. After CCI the incision was closed with interrupted 6–0 silk sutures. Following anesthesia withdrawal, the animal was placed into a heated cage for at least 45 min post-injury to maintain the normal body temperature. The animals were monitored throughout the study. Sham animals underwent the same procedure as injured mice except for craniotomy and cortical impact. A total of 22 mice received CCI and 8 mice underwent sham surgery. No analgesics were used due to their potential to interfere with the anti-inflammatory properties of the drugs being tested (Qian et al. 2007), however local anesthesia using a 0.25% bupivacaine solution (0.1–0.2 mL) was infused into the incision line at least 5 min prior to skin incision and also applied to the surgery site following surgery.
Administration of drug:
For 2-photon imaging studies, CX3CR1gfp/+ were ear tagged and arbitrarily assigned to (A) Vehicle or (B) VuPAM treatment groups prior to surgery/CCI. VuPAM (30 mg/kg in saline + 10 % Tween-80; n=5) or equal volume vehicle (saline + 10 % Tween-80; n=5) were administered by intraperitoneal (i.p.) injection starting at 3 h post-injury, with repeat dosing at 24, 72 and 120 h post-injury. Vehicle-treated sham mice (n=5) were included as baseline controls in the study (Fig. 1). At 7 days post-injury, mice were anesthetized (100 mg/kg sodium pentobarbital, I.P.) and transcardially perfused with ice-cold 0.9% saline (100 ml), followed by perfusion with 300 ml of 4 % paraformaldehyde. Brains were removed and post-fixed in 4% paraformaldehyde overnight, cryoprotected in 30 % sucrose, and were processed for histology. For molecular/biochemical studies, C57BL/6J were ear tagged and arbitrarily assigned to (A) Vehicle or (B) VuPAM treatment groups prior to surgery/CCI. VuPAM (30 mg/kg in saline + 10 % Tween-80; n=6) or equal volume vehicle (saline + 10 % Tween-80; n=6) were administered by I.P. injection starting at 3 h post-injury, with repeat dosing at 24, 72 and 120 h post-injury. Vehicle-treated sham mice (n=3) were included as baseline controls in the study (Fig. 1). At 7 days post-injury, mice were anesthetized (100 mg/kg sodium pentobarbital, I.P.) and transcardially perfused with ice-cold 0.9% saline (100 ml), and ipsilateral hippocampal and cortical tissues were rapidly dissected and snap-frozen on liquid nitrogen for RNA or protein extraction.
Figure 1. Time‐line of study design.

Representative figure showing experimental design and number of animals for each group.
Intravital two-photon microscopy:
For two-photon microscopy experiments, a thinned-skull cranial window next to the 5 mm craniotomy was prepared in sham and TBI CX3CRgfp/+ mice as previously described (Roth et al. 2014). Briefly, the mice were anaesthetized with ketamine (85 mg/kg) and xylazine (13 mg/kg) in PBS and maintained at a core temperature of 37 °C. A metal bracket was secured on skull bone next to a 5 mm craniotomy (positioned at −1.22 mm from bregma), and a cranial window (1 mm x 1 mm; positioned at −0.70 mm from bregma) was quickly thinned to a thickness of ∼30–40 µm. Intravital two-photon imaging of GFP+ microglia was performed using a Leica SP5 II two-photon microscope equipped with a 20 × water-dipping objective (1.0 NA), a PMT-ready Objective Inverter (LSM TECH, Etters, PA), and a Coherent Chameleon Laser tuned to 880 nm (for GFP). Three-dimensional time-lapse movies were captured in z-stacks of 25–30 planes (1.5-μm step size; 3.5 X zoom; 512 × 512) at approximately 90 sec intervals.
Microglial morphological analysis:
All quantitative analyses and processing of 3D imaging data were performed using Imaris 7.0 software (Bitplane), as previously described (Roth et al. 2014). GFP+ microglia were quantified from 512 × 512 × 60 µm (xyz) 3D image stacks obtained at 7 days post-injury from sham, TBI+Veh, and TBI+VuPAM treated CX3CR1gfp/+ mice. GFP+ microglial cells were converted to the surfaces using the Imaris “surface” tool. A minimum of 6 GFP+ microglia/mouse were arbitrarily selected from the perilesional cortex and only cells with all of their processes in the field of view were quantified. Microglial parameters including filament length and filament area were quantified using the Imaris-based automated “filaments” tool. The data obtained was represented on a per cell basis (Roth et al. 2014).
Immunofluorescence imaging:
20 µm coronal brain sections from sham and CCI mice at approximately −1.70 mm from bregma were selected and standard immunostaining techniques were employed. Briefly, 20 µm brain sections were washed three times with 1 x PBS, blocked for 1 h in goat serum containing 0.4 % Triton X-100, and incubated overnight at 4 °C with a combination of primary antibodies, including anti-pCREB (1:1000, Cell Signaling Technology), anti-iNOS (1:1000, BD Transduction Laboratories), and anti-YM1 (1:1000, STEMCELL Technologies). Sections were washed three times with 1 x PBS and incubated with appropriate Alexa Fluor conjugated secondary antibodies (Life Technologies) for 2 h at room temperature. Sections were washed three times with 1 x PBS, counterstained with 4’,6-diamidino-2-phenylindole (DAPI) (1 ug/ml; Sigma-Aldrich), and mounted with glass coverslips using Hydromount solution (National diagnostics, Atlanta, GA). Images were acquired using a fluorescent Nikon Ti-E inverted microscope (Nikon Instrument Inc., Melville, NY), at 10x (Plan Apo 10X NA 0.45) or 20x (Plan APO 20X NA 0.75) magnification. Exposure times were kept constant for all sections in each experiment. All images were quantified using Nikon ND-Elements Software (AR 4.20.01). Co-localization of pCREB, YM1, and CX3CR1 was performed by binary operation intersection followed by thresholding. 12 positive regions of interest near the lesion site per mouse were quantified, and was expressed by Mander’s Overlap Coefficient.
Real-time PCR:
Quantitative gene expression analysis in the cortex of sham and CCI mice was performed using Taqman technology as previously described (Henry et al. 2019). Briefly, total RNA was extracted from cortical tissue using a miRNeasy isolation kit (Qiagen, Valencia, CA) and cDNA was synthesized using High-Capacity cDNA Reverse Transcription Kit (ThermoFisher Scientific; Waltham, MA) as per manufacturer’s protocol. Real-time PCR amplification was carried using Quant Studio™ 5 real-time PCR System (ThermoFisher Scientific) with TaqMan Universal Master Mix II, no UNG (ThermoFisher Scientific). The following Taqman mRNA assays were used: tnf-α, Mm00443258_m1; Il-6, Mm00446190_m1; Il-10, Mm01288386_m1 ; cd68, Mm03047343_m1; cybb (Nox2), Mm01287743_m1; arginase-1, Mm00475988_m1; chil3 (Ym1) Mm00657889_mH; gapdh, Mm99999915_g1. Relative mRNA expression was calculated using the comparative cycle threshold (2−ΔΔ Ct) method and GAPDH served as endogenous control. Relative amount of mRNA was presented as fold change over sham control.
Statistical analysis:
Quantitative data were expressed as mean ± standard errors of the mean (s.e.m.), of at least three independent experiments. Normality testing was performed and data passed the normality test (D’Agostino & Pearson omnibus normality test), and therefore parametric statistical analysis was performed. Data were compared using student’s t-test, one-way analysis of variance (ANOVA) with Tukey post hoc correction (multiple comparisons), or two-way ANOVA followed by Bonferroni post hoc correction. For in vivo studies, mice that were ear tagged and housed five per cage were removed one at a time from the cage and arbitrarily assigned to treatment groups in the following order, a) Sham Vehicle treatment, b) TBI VuPAM treatment, and c) and TBI Vehicle treatment. The procedure was repeated until n=5 was reached for each group. Surgery or CCI was performed followed by drug administration according to this pre-defined code. Blinding of the study was maintained as follows: one researcher performed sham or CCI surgery, and another researcher blinded to surgical condition administered the drugs daily for 3 days. Following euthanasia, all imaging and molecular studies were performed by a third researcher blinded to surgery and treatment groups. Our prior pre-clinical drug treatment studies demonstrated that n=6 animals/group was sufficient to identify neuroprotective effects (Sabirzhanov et al. 2014). Thus, we used an n=6 for VuPAM treatment studies. No exclusion criteria were pre-determined and all animals were included in statistical analyses. The level of significance was set at *p<0.05, **p<0.01, ***p<0.001. Statistical analyses were performed using GraphPad Prism 7 software (GraphPad Software Inc., San Diego, CA, USA, RRID:SCR_002798).
Results
VuPAM reduces pro-inflammatory microglial activation and promotes anti-inflammatory and neuroprotective phenotypes
To test the hypothesis that mGluR5 orthosteric agonists and PAM differentially regulate microglial activation states, we treated BV2 microglia with CHPG (mGluR5 orthosteric agonist; 1 mM) or VU0360172 (mGluR5 PAM, VuPAM; 20 or 50 μM) and stimulated cells with the TLR4 ligand LPS (100 ng/ml) for 24h. LPS increased protein expression of iNOS in cell lysates, and NO and TNF-α in supernatants of BV2 microglia (p<0.01, p<0.001 vs control; Fig. 2a, b, d, e). LPS also modestly increased expression of anti-inflammatory IL-10 protein and decreased expression of Arg-1 (p>0.05 vs control; Fig. 2c, d, f). Pretreatment with VuPAM dose-dependently attenuated LPS-induced pro-inflammatory activation in microglia (iNOS, NO, TNF-α; p<0.05, p<0.01 vs LPS; Fig. 2a, b, d, e), and significantly increased anti-inflammatory IL-10 and Arg-1 protein expression (both p<0.01 vs LPS; Fig. 2c, d, f). Although the orthosteric mGluR5 agonist CHPG prevented pro-inflammatory microglial activation (reduced iNOS, NO, TNF-α; p<0.05, p<0.01 vs LPS; Fig. 2a, b, d, e) it had no effect on anti-inflammatory IL-10 or Arg-1 (Fig. 2c, d, f). In the absence of LPS, VuPAM and CHPG had no effect on NO, TNF-α, or IL-10 protein expression (Fig. 2a–c).
Figure 2. mGluR5 PAM (VuPAM) promotes anti-inflammatory activation of microglia following LPS stimulation.

Stimulation of microglia with LPS (100 ng/ml) increased nitrite production (a), release of TNF-α (b), and induction of iNOS (d,e), with a minor increase in anti-inflammatory IL-10 (c) or Arg-1 (d,f) in BV2 microglia. Pre-treatment of microglia with VuPAM (50 μM), attenuated the LPS-induced nitrite production (a), TNF-α release (b), and iNOS expression (e), and increased IL-10 (c) and Arg-1 (f) protein levels after LPS. CHPG treatment (orthosteric agonist; 1 mM) reduced nitrite production (a), pro-inflammatory TNF-α (b) and iNOS expression (e), without any effect on anti-inflammatory IL-10 (c) or Arg-1. mGluR5 antagonist, MTEP (100 μM), reversed the effects of VuPAM. Treatment with drugs alone had no effect on any of the studied parameters (a-c). Representative Western immunoblots and quantification of iNOS (d,e) and Arg-1 (d,f) protein expression under various treatment conditions. Data represent mean ± SEM, with single data points from independent cell culture preparations (n= 5 for a and b; n= 4 for c; n= 3 for d-f). *p<0.05, **p<0.01 versus control; +p<0.05, ++p<0.01 versus LPS; #p<0.05, ##p<0.01 versus LPS+Vu; one-way ANOVA followed by post hoc Tukey’s test.
To confirm that VuPAM actions are mediated by mGluR5, we co-treated BV2 microglia with VuPAM (50 μM) and MTEP (100 μM), a selective mGluR5 antagonist, prior to LPS stimulation. MTEP co-treatment prevented VuPAM mediated anti-inflammatory activation of microglia (iNOS, NO, TNF-α, IL-10, and Arg-1; p<0.05 vs LPS+Vu (50 μM); Fig. 2a–f). To confirm the specificity of VuPAM for mGluR5, we knocked down mGluR5 protein in BV2 microglia using siRNAs (64% knock-down; p<0.05 vs control siRNA; Fig. 3a). Knocking down mGluR5 reversed the anti-inflammatory effect of VuPAM in LPS-stimulated BV2 microglia as demonstrated by a decrease in IL-10 (p<0.05 vs LPS+Vu) and increase in NO and TNF-α (p<0.01 vs LPS+Vu) protein expression in LPS + VuPAM treated mGluR5 siRNA knock-down microglia (Fig. 3b–d). These data confirmed both the specificity of VuPAM for mGluR5, and its potent anti-inflammatory actions in LPS-stimulated BV2 microglia.
Figure 3. siRNA knockdown of mGluR5 abrogated VuPAM mediated anti-inflammatory activation of microglia following LPS stimulation.
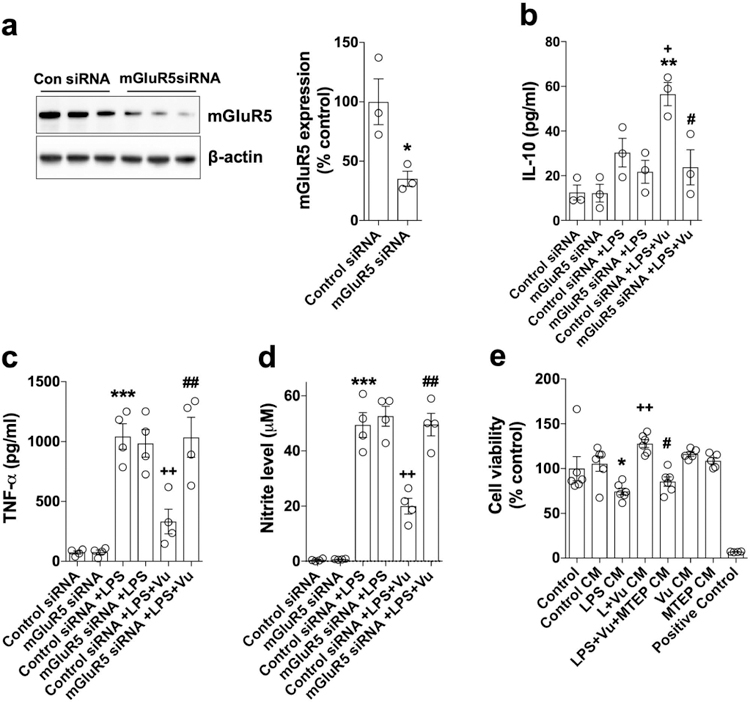
Western immunoblot and quantification of siRNA knockdown of mGluR5 in BV2 microglia (a). VuPAM mediated activation of anti-inflammatory microglia was attenuated in mGluR5 knockdown microglia as shown by decreased IL-10 (b) and increased TNF-α (c) and nitrate (d) levels in VuPAM treated mGluR5 knockdown BV2 microglia. Suppression of LPS induced pro-inflammatory microglia activation by VuPAM (50 μM) prevented neuronal cell death in a co-culture neuronal viability assay (e). Data represent mean ± SEM with single data points from independent cell culture preparations (n= 3 for a and b; n= 4 for c and d; n= 5 for e). *p<0.05, **p<0.01 versus control; +p<0.05, ++p<0.01 versus LPS; #p<0.05, ##p<0.01 versus LPS+Vu; one-way ANOVA followed by post hoc Tukey’s test.
To functionally validate the anti-inflammatory activity of VuPAM, we performed a microglial-mediated neurotoxicity assay using conditioned media from LPS stimulated BV2 microglia that had been pretreated in the presence or absence of VuPAM (50 μM) on the survival of primary cortical neurons. As expected, conditioned media from LPS stimulated microglia induced neuronal cell death, evidenced by a decrease in cell viability of primary cortical neurons (p<0.05 vs control; Fig. 3e). Notably, condition media from the VuPAM treated cells mitigated microglia-mediated neuronal cell death (p<0.05 vs LPS). Furthermore, the neuroprotective effect of VuPAM treatment on neuronal survival was reversed by co-treatment with mGluR5 antagonist, MTEP (p<0.05 vs LPS+Vu; Fig. 3e). Thus, microglia-mediated neurotoxicity studies confirmed the anti-inflammatory and neuroprotective action of VuPAM in microglia.
PI3K/Akt/GSK-3β signaling is critical for VuPAM-mediated anti-inflammatory microglial activation
We previously demonstrated that activation of CHPG stimulates phospholipase C (PLC)/protein kinase C (PKC) signaling that dampens pro-inflammatory microglial activation (Byrnes et al. 2009b). To determine whether VuPAM uses similar signal transduction pathways we co-treated LPS stimulated BV2 microglia with VuPAM (50 μM) and the PKC inhibitor, rottlerin (5 μM). However, the anti-inflammatory effects of VuPAM were not reversed by PKC inhibition using rottlerin (NO, TNF-α, IL-10. p>0.05 vs LPS+Vu; Fig. 4a–c). CHPG was also previously shown to inhibit NFκB signaling in microglia (Loane et al. 2013), therefore we evaluated whether VuPAM modulates this pathway by assessing p-IKB levels in microglia and nuclear expression of NFκB. VuPAM had no inhibitory actions on the NFκB signaling in LPS stimulated BV2 microglia and there was no change in p-IKB expression or nuclear NFкB expression in VuPAM treated microglia (p>0.05 vs LPS; Fig. 4d, e). As expected, the NFкB inhibitor, PDTC (10 μM), reduced p-IKB expression in LPS stimulated microglia demonstrating specific inhibition of NFкB in our system (p<0.05 vs LPS; Fig. 4d). Taken together, these data indicated that mGluR5 orthosteric agonists and mGluR5 PAM use different downstream signal transduction pathways in microglia.
Figure 4. VuPAM does not have an effect on PLC/PKC signaling or NFκB activation in microglia.
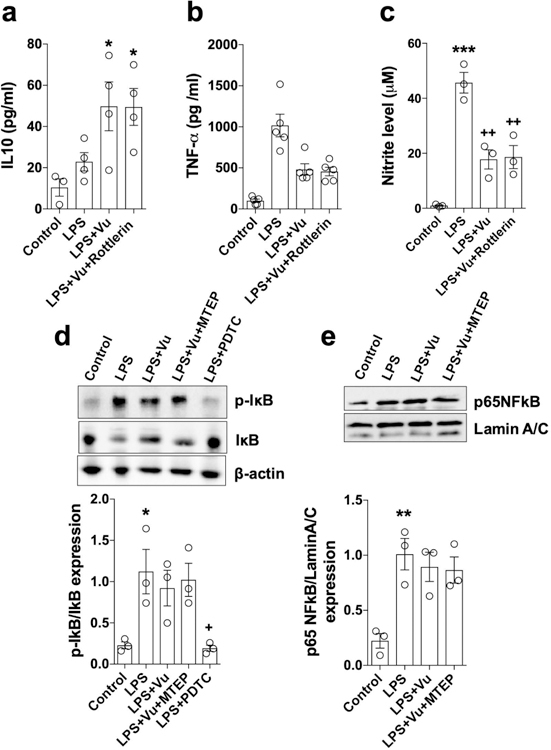
Quantification of anti-inflammatory IL-10 (a), pro-inflammatory TNF-α (b) and nitrate (c) production in culture media of LPS stimulated BV2 microglia at 24 h in the presence of the PKC inhibitor, Rottlerin (5 μM). Representative Western immunoblots and quantification of p-IKB and IKB degradation in BV2 microglia pre-treated with VuPAM (50 μM) and mGluR5 antagonist MTEP (100 μM) 30 min prior to LPS stimulation (d). PDTC (10 μM; NFκB inhibitor) served as a positive control in this experiment. Representative Western immunoblot and quantification of p65-NFκB nuclear translocation in LPS-stimulated BV2 microglia (e). Data represent mean ± SEM with single data points from independent cell culture preparations (n= 4 for a; n= 5 for b; n= 3 for c-e). *p<0.05, **p<0.01 versus control; +p<0.05, ++p<0.01 versus LPS; one-way ANOVA followed by post hoc Tukey’s test.
Since mGluR5 can also activate the PI3K/Akt signal transduction pathway (Shah et al. 2012; Batista et al. 2016), we tested whether PI3K/Akt signaling is involved in VuPAM-mediated anti-inflammatory effects in microglia. Pretreatment of BV2 microglia with VuPAM (50 μM) prior to LPS stimulation increased phosphorylation of Akt on Ser473 in a time dependent manner (Fig. 5a). Peak expression of phospho-Akt was observed at 30 mins post-stimulation (p<0.001; Fig. 5a), and this time point was used for further experiments. VuPAM treatment robustly increased the phosphorylation of Akt when compared to levels in LPS only stimulated microglia (p<0.01; Fig. 5a, b). In contrast, CHPG (1 mM) had no effect on the phosphorylation of Akt in LPS stimulated microglia (p>0.05 vs LPS: Fig. 5b). Co-treatment of BV2 microglia with VuPAM (50 μM) and MTEP (100 μM) prevented Akt phosphorylation induced by the VuPAM in LPS stimulated microglia (p<0.01 vs LPS+Vu; Fig. 5b). These data demonstrate that activation of Akt is an important pathway downstream of mGluR5 stimulated by VuPAM, but not by CHPG. Thus, mGluR5 orthosteric agonists and PAM differ with respect to Akt signaling in microglia.
Figure 5. PI3K/Akt activation promotes VuPAM-mediated suppression of pro-inflammatory microglial activation and up-regulation of anti-inflammatory IL-10 and Arg-1.

Representative Western immunoblots and quantification for Akt activation (0–60 min) following LPS stimulation in the presence or absence of VuPAM (50 μM; a). Representative Western immunoblots and quantification of p-S473 phosphorylation of Akt (b) and expression of iNOS (c) in BV2 microglia pre-treated with VuPAM (50 μM), mGluR5 antagonist MTEP (100 μM), mGluR5 orthosteric agonist CHPG (1 mM), and Wortmannin (1 and 2.5 μM) 30 min prior to LPS stimulation. Quantification of anti-inflammatory IL-10 (d), pro-inflammatory TNF-α (e), and nitrate (f) production in culture media of BV2 microglia at 24 h post-LPS stimulation. Data represent mean ± SEM with single data points from independent cell culture preparations (n= 3 for a-c; n= 4 for d; n= 6 for e and f). *p<0.05, **p<0.01 versus control; +p<0.05, ++p<0.01 versus LPS; #p<0.05, ##p<0.01 versus LPS+Vu; one-way ANOVA followed by post hoc Tukey’s test or two-way ANOVA followed by post hoc Bonferroni test.
To validate the role of PI3K/Akt signaling in anti-inflammatory effects of VuPAM, a PI3K/Akt inhibitor, wortmannin (1 or 2.5 μM), was used in co-treatment studies with VuPAM (50 μM). Wortmannin dose-dependently inhibited VuPAM-induced phosphorylation of Akt in LPS stimulated BV2 microglia (p<0.01 vs LPS+Vu; Fig. 5b). Moreover, inhibition of Akt phosphorylation by wortmannin resulted in the loss of the anti-inflammatory actions of VuPAM in LPS stimulated microglia. Wortmannin co-treatment increased pro-inflammatory iNOS, NO, TNF-α (all p<0.01) expression and decreased anti-inflammatory IL-10 expression (p<0.05) when compared to VuPAM treated LPS stimulated microglia (Fig 5c–f). These studies confirm that Akt phosphorylation is required for the anti-inflammatory actions of VuPAM.
PI3K/Akt inhibits GSK-3β (Atkins et al. 2012), a serine-threonine kinase that is known to regulate the pro-inflammatory phenotype in microglia (Bhat et al. 2018). We next investigated whether inhibition of GSK-3β is downstream of VuPAM induced Akt phosphorylation. Pretreatment of BV2 microglia with VuPAM (50 μM) prior to LPS stimulation increased phosphorylation of GSK-3β on Ser9 in a time dependent manner, thereby inhibiting GSK-3β (Fig. 6a). Peak expression of phospho-GSK-3β was observed at 30 mins post-stimulation (p<0.001 vs LPS; Fig. 6a), and this time point was used for further experiments. Co-treatment of VuPAM (50 μM) with MTEP (100 μM) prevented GSK-3β phosphorylation in LPS stimulated microglia (p<0.05 vs LPS+Vu; Fig. 6b). Moreover, co-treatment of VuPAM (50 μM) with an Akt inhibitor, AKTi (1 μM), prevented GSK-3β phosphorylation in LPS stimulated microglia (p<0.01 vs LPS+Vu; Fig. 6c), demonstrating that GSK-3β is downstream of Akt.
Figure 6. VuPAM treatment abrogated the activation of GSK-3β in LPS stimulated microglia.

Representative Western immunoblots and quantification for GSK-3 activation (0–60 min) following LPS stimulation in the presence or absence of VuPAM (50 μM; a,b). Representative Western immunoblots and quantification of pS9-phosphorylation of GSK-3β in BV2 microglia pre-treated with VuPAM (50 μM; c), mGluR5 antagonist MTEP (100 μM; c), AKT inhibitor (1 μM; d), GSK-3 antagonist BIO (5 μM; e), and GSK-3 activator DIF-3 (30 μM; e) 30 min prior to LPS stimulation. Data represent mean ± SEM with single data points from independent cell culture preparations (n= 3 for a-d). *p<0.05, **p<0.01 versus control; +p<0.05, ++p<0.01 versus LPS; #p<0.05, ##p<0.01 versus LPS+Vu, and LPS+BIO; one-way ANOVA followed by post hoc Tukey’s test or two-way ANOVA followed by post hoc Bonferroni test.
To mimic the effects of inhibiting GSK-3β by VuPAM, we pretreated BV2 microglia with a selective GSK-3β antagonist, BIO (5 μM), prior to LPS stimulation. BIO increased Ser9 phosphorylation of GSK-3β (p<0.01 vs LPS; Fig. 6d), which resulted in decreased expression of pro-inflammatory markers in LPS stimulated microglia (iNOS, NO, TNF-α; p<0.05, p<0.001 vs LPS; Fig. 7a, b, e, f). Furthermore, BIO increased expression of anti-inflammatory IL-10 and Arg-1 (p<0.05 vs LPS; Fig. 7a, c, d) to similar levels as those detected in VuPAM treated LPS microglia.
Figure 7. Inhibition of GSK-3β is required for VuPAM-mediated anti-inflammatory actions in microglia.

Representative Western immunoblots and quantification of iNOS (a, b) and Arg-1 (a, c) protein expression in BV2 microglia under various treatment conditions. Quantification of anti-inflammatory IL-10 (d), pro-inflammatory TNF-α (e), and nitrate (f) production in culture media of BV2 microglia at 24 h post-LPS stimulation under various treatment conditions. Data represent mean ± SEM with single data points from independent cell culture preparations (n= 3 for a-e; n=5 for f). *p<0.05, **p<0.01 versus control; +p<0.05, ++p<0.01 versus LPS; #p<0.05, ##p<0.01 versus LPS+Vu, and LPS+BIO; one-way ANOVA followed by post hoc Tukey’s test.
To determine whether GSK-3β is required for the anti-inflammatory actions of VuPAM, we co-treated BV2 microglia with VuPAM (50 μM) and DIF-3 (30 μM), a GSK-3β activator, prior to stimulation with LPS. Notably, DIF-3 prevented the VuPAM-mediated increase in Ser9 phosphorylation of GSK-3β (p<0.001 vs LPS+Vu; Fig. 6d), and reversed the anti-inflammatory actions of VuPAM in LPS stimulated microglia. When compared to VuPAM treatment, DIF-3 increased expression of pro-inflammatory markers iNOS, NO, and TNF-α (all p<0.05 vs LPS+Vu; Fig. 7a, b, e, f), and decreased expression of anti-inflammatory IL-10 and Arg-1 (p<0.05 vs LPS+Vu; Fig. 7a, c, d) in LPS stimulated microglia. Thus, inhibition of GSK-3β contributes to the anti-inflammatory actions of VuPAM in LPS stimulated microglia. Taken together, our analysis suggests that PI3K/Akt/GSK-3β signaling is critical for VuPAM-mediated anti-inflammatory microglial activation.
CREB activation by Akt/GSK-3β signaling is required for the anti-inflammatory actions of VuPAM
The transcription factor CREB is activated by Akt/GSK-3β signaling (Qiao et al. 2018) to regulate key inflammatory genes, such as TNF-α and IL-10 (Wen et al. 2010; Alexaki et al. 2018). Because VuPAM activated Akt/GSK-3 signaling we investigated whether CREB was involved in development of the anti-inflammatory phenotype in microglia. VuPAM (50 μM) increased the phosphorylation of CREB on Ser133 in LPS stimulated BV2 microglia (p<0.05 vs LPS; Fig. 8a). This effect was reversed not only by the Akt inhibitor, AKTi (1 μM; p<0.01 vs LPS+Vu; Fig. 8a), but also the GSK-3β activator, DIF3 (30 μM; p<0.05 vs LPS+Vu; Fig. 8b), demonstrating that Akt/GSK-3β signaling plays a key role in CREB activation by VuPAM.
Figure 8. VuPAM treatment stimulates the CREB activation and its association with CBP in LPS stimulated microglia.

Representative Western immunoblots and quantification of pS133-phosphorylation of CREB in BV2 microglia pre-treated with VuPAM (50 μM), CBP:CREB interaction inhibitor (CC, 1 μM), AKT inhibitor (1 μM), and GSK-3β activator DIF-3 (30 μM) 30 min prior to LPS stimulation (a, b). Co-immunoprecipitation and reverse co-immunoprecipitation of CBP with pCREB (c, d). BV2 microglial cell lysates were immunoprecipitated with either anti-CBP or anti-CREB antibody under various treatment conditions, and cell pellets were Western immunoblotted with either anti-CREB or anti-CBP antibodies to identify protein interactions. Data represent mean ± SEM with single data points from independent cell culture preparations (n= 3 for a-d). *p<0.05, **p<0.01 versus control; +p<0.05, ++p<0.01 versus LPS; #p<0.05, ##p<0.01 versus LPS+Vu; one way-ANOVA followed by post hoc Tukey’s test.
Activated CREB is known to interact with CREB-binding protein (CBP) to initiate the transcription of inflammatory genes (Wen et al. 2010). Therefore, we co-treated BV2 microglia with VuPAM (50 μM) and the CREB/CBP interaction inhibitor, CC (1 μM), prior to LPS stimulation. CC had no effect on VuPAM mediated phosphorylation of CREB (p>0.05 vs LPS+Vu; Fig. 8a), indicating that CBP is downstream of CREB activation and does not interfere with the phosphorylation of CREB. To confirm the interaction of CREB with CBP, we performed co-immunoprecipitation of CREB using anti-CBP in microglia and found that VuPAM treatment robustly increased the interaction of CREB with CBP (Fig. 8c); notably, this interaction was abrogated by co-treatment of mGluR5 antagonist (MTEP), Akt inhibitor (AKTi), and the CREB:CBP interaction inhibitor (CC; Fig. 8c). Further, reverse co-immunoprecipitation using anti-CREB confirmed that VuPAM increased the interaction of CREB with CBP (Fig. 8d). Once again, the CREB-CBP interaction in microglia following VuPAM treatment was abolished by co-treatment with MTEP, AKTi, and CC (Fig. 8d). The CREB:CBP interaction inhibitor, CC, also abolished the anti-inflammatory effects of VuPAM (50 μM) as demonstrated by increased expression of pro-inflammatory markers in LPS stimulated microglia (iNOS, NO, TNF-α; p<0.01 vs LPS+Vu; Fig. 9a–e), and decreased expression of anti-inflammatory IL-10 and Arg-1 (p<0.01 vs LPS+Vu; Fig. 9a, d, f). These data suggest that activation of CREB and its association with CBP is required for the anti-inflammatory actions of VuPAM in microglia.
Figure 9. Akt/GSK-3 stimulates CREB activation to promote an anti-inflammatory phenotype in microglia following VuPAM treatment.
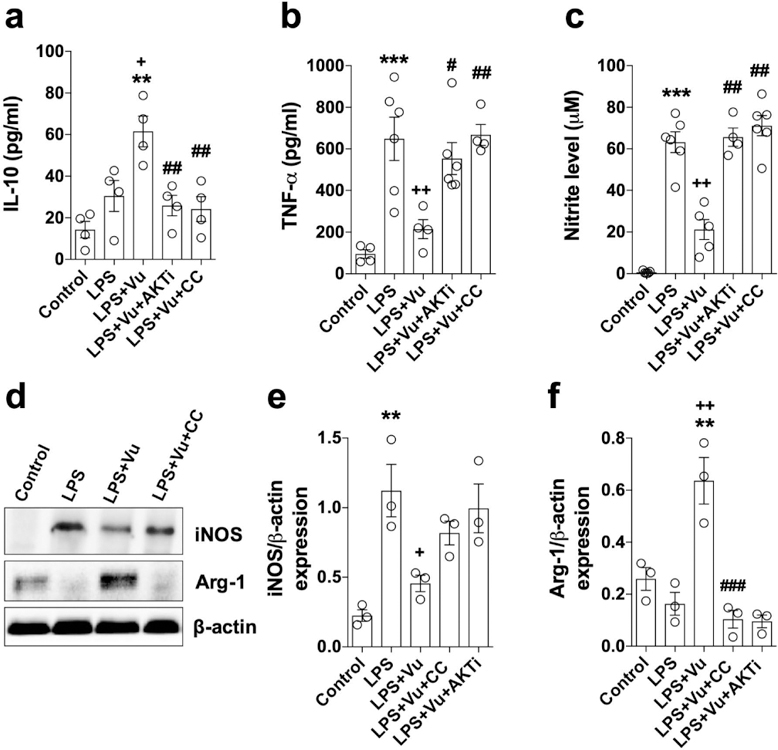
Quantification of anti-inflammatory IL-10 (a), pro-inflammatory TNF-α (b), and nitrate production (c) in culture media of BV2 microglia at 24 h post-LPS stimulation under various treatment conditions. Representative Western immunoblots and quantification of pro-inflammatory iNOS (d,e) and anti-inflammatory Arg-1 (d,f) expression in BV2 microglia under various treatment conditions. Data represent mean ± SEM with single data points from independent cell culture preparations (n= 4 for a; n= 5 for b and c; n= 3 for d-f). *p<0.05, **p<0.01 versus control; +p<0.05, ++p<0.01 versus LPS; #p<0.05, ##p<0.01 versus LPS+Vu; one way-ANOVA followed by post hoc Tukey’s test.
To further validate the role of CREB in promoting the VuPAM-mediated anti-inflammatory phenotype in microglia, we performed dominant negative mutant studies in BV2 microglia transfected with CREB mutant clones, including CREB133 (serine to alanine mutation at Ser133 resulting in blockade of CREB phosphorylation), KCREB (mutation in the DNA-binding domain that prevents CREB’s association with cAMP-regulated enhancer element), or with a mock control vector (pCMV DNA). VuPAM treatment attenuated LPS-induced pro-inflammatory microglial activation in mock control microglia (iNOS, NO, TNF-α; p<0.05 vs LPS; Fig. 10a, b, d, e), and increased expression of IL-10 (p<0.05 vs LPS; Fig. 10c). Significantly, the anti-inflammatory effects of VuPAM treatment were reversed in CREB133 and KCREB microglia (iNOS, NO, TNF-α, IL-10 vs LPS+Vu in mock; Fig. 10a–e), demonstrating an anti-inflammatory transcriptional role of CREB. Therefore, our data indicate that activation of CREB is required for the anti-inflammatory actions of VuPAM in microglia via stimulation of the Akt/GSK-3β/CREB signaling pathway.
Figure 10. CREB is required for the anti-inflammatory actions of VuPAM.

BV2 microglia were transiently transfected with dominant-negative mutant CREB having either a mutation in the DNA-binding domain (KCREB) or in S133 residue (CREB133), or mock control clones (pCMV DNA). After 24 h, cells were pre-treated with VuPAM (50 μM) for 30 min, followed by stimulation with LPS for 24 h. Western immunoblot and quantification of iNOS expression under various treatment conditions (a,b). Quantification of anti-inflammatory IL-10 (c), pro-inflammatory TNF-α (d), and nitrate production (e) in culture media of transiently transfected mock-, KCREB-, and CREB133-mutant BV2 microglia at 24 h post-LPS stimulation. Data represent mean ± SEM with single data points from independent cell culture preparations (n= 3 for a-c; n= 4 for d; n= 6 for e). *p<0.05, **p<0.01, ***p<0.001 versus control; +p<0.05 versus LPS; one-way ANOVA followed by post hoc Tukey’s test.
Akt/GSK-3β/CREB signaling promotes an anti-inflammatory phenotype in VuPAM treated primary cortical microglia
We next sought to confirm the effects of VuPAM on Akt/GSK-3β/CREB signaling in primary cortical microglia derived from neonatal rat pups. Similar to BV2 microglia studies, we determined that VuPAM treatment (50 μM) increased the phosphorylation of Akt (p<0.05 vs LPS; Fig. 11a, b), GSK-3β (p<0.01 vs LPS; Fig. 11a, c), and CREB (p<0.05 vs LPS; Fig. 11a, d) in LPS (100 ng/ml) stimulated primary microglia. Co-treatment of primary microglia with VuPAM (50 μM) and inhibitors of Akt (AKTi; 1 μM), GSK-3β (DIF-3; 30μM), and CREB (CC; 1 μM) attenuated VuPAM-mediated phosphorylation of Akt, GSK-3β, and CREB, in LPS stimulated microglia (p<0.05, p<0.01, p<0.001 vs LPS+Vu; Fig. 11a–d). Furthermore, co-treatment of primary microglia with VuPAM (50 μM) and MTEP (100 μM) prevented activation of Akt, GSK-3β, and CREB in LPS stimulated microglia (p<0.01 vs LPS+Vu; Fig. 11a–d), thereby confirming the specificity of VuPAM for mGluR5 and its impact on downstream signaling pathways.
Figure 11. VuPAM stimulates Akt/GSK-3β/CREB signaling in primary rat cortical microglia.

Representative Western immunoblots and quantification of Akt activation (pS473-phosphorylation, a, b), GSK-3β inhibition (pS9-phosphorylation; a, c), and CREB activation (pS133-phosphorylation; a, d) in primary microglia pre-treated with VuPAM (50 μM) and various inhibitors at 30 min prior to LPS stimulation. Data represent mean ± SEM with single data points from independent cell culture preparations (n= 3 for a-d). *p<0.05, **p<0.01 versus control; +p<0.05, ++p<0.01 versus LPS; #p<0.05, ##p<0.01 versus LPS+Vu; one-way ANOVA followed by post hoc Tukey’s test.
Importantly, activation of Akt/GSK-3β/CREB signaling by VuPAM was associated with an anti-inflammatory phenotype in primary cortical microglia. VuPAM treatment (50 μM) decreased pro-inflammatory iNOS expression (Fig. 12a), and NO (p<0.001 vs LPS; Fig. 12d) and TNF-α (p<0.05 vs LPS; Fig. 12d) release in LPS stimulated primary microglia. Release of anti-inflammatory IL-10 was also increased in primary microglia (p<0.01 vs LPS; Fig. 12b) as was increased expression of phosphorylated CREB (Fig. 12a). As predicted, the anti-inflammatory effects of VuPAM were abrogated by co-treatment with the mGluR5 antagonist, MTEP (100 μM; p<0.05 vs LPS+Vu; Fig. 12a–d). Furthermore, pre-treatment of primary microglia with Akt inhibitor (AKTi), GSK-3β activator (DIF-3), or CREB:CBP interaction inhibitor (CC) abolished the anti-inflammatory effects of VuPAM in LPS stimulated primary microglia (iNOS, NO, TNF-α, IL-10, p<0.05 vs LPS+Vu; Fig. 12a–d). The data obtained in primary cortical microglia confirm that the Akt/GSK-3β/CREB signal transduction pathway is critical for VuPAM-mediated anti-inflammatory actions in microglia.
Figure 12. VuPAM promotes an anti-inflammatory phenotype in primary rat cortical microglia.

Representative fluorescent images of pCREB+ and iNOS+ primary microglia under various treatment conditions (a). Quantification of anti-inflammatory IL-10 (b), pro-inflammatory TNF-α (c), and nitrate production (d) in culture media of primary microglia pre-treated with VuPAM (50 μM) and various inhibitors at 30 min prior to LPS stimulation. Data represent mean ± SEM with single data points from independent cell culture preparations (n= 3 for a; n= 4 for b and c; n= 5 for d). *p<0.05, **p<0.01 versus control; +p<0.05, ++p<0.01 versus LPS; #p<0.05, ##p<0.01 versus LPS+Vu; one-way ANOVA followed by post hoc Tukey’s test.
VuPAM enhances Akt/GSK-3β/CREB signaling in the injured cortex, alters microglial morphology, and reduces pro-inflammatory microglial activation following experimental TBI
We previously demonstrated that VuPAM is neuroprotective in a murine CCI model and results in long-term motor function recovery after brain trauma (Loane et al. 2014). Moreover, VuPAM treatment reduced microglial iNOS expression and up-regulated Arg1 expression at 28 days post-injury (Loane et al. 2014), suggesting that VuPAM can alter microglial pro- and anti-inflammatory phenotypes in vivo. We therefore examined the effect of VuPAM on Akt/GSK-3β/CREB signaling and microglial activation in the cortex of adult male mice subjected to moderate-level CCI. VuPAM (30 mg/kg) or vehicle (saline + 10% Tween-80) was administered systemically (by intraperitoneal injection) to CCI mice starting at 3 hours post-injury, with repeat dosing at 24, 72, and 120 hours post-injury. Vehicle-treated sham mice were included as baseline control animals in all studies. We used CX3CR1gfp/+ mice to assess microglial morphology in vivo using intravital two-photon microscopy (Fig. 13a), and C57BL/6J mice to assess signal transduction pathway in cortex following TBI.
Figure 13. VuPAM partially rescues morphological changes in microglia induced by TBI.

(a) Schematic representation of two-photon microscopy experiment including thin skull preparation adjacent to the craniotomy site in CX3CR1gfp/+ mice. Moderate-level CCI mice were treated with VuPAM (30 mg/kg, IP) or vehicle (saline + 10% Tween-80, IP) starting at 3h post-injury with repeat dosing at 24h, 72h, and 120h post-injury. Intravital two-photon microscopy was performed at 7 days post-injury. (b) Representative images and Imaris-based morphometric analysis of microglia from Sham, TBI and TBI+Vu treated CX3CR1gfp/+ mice at 7 days post-injury. Imaris-based automated “filaments” tool was used to trace and quantify microglial filament length (c) and filament area (d). Data represent mean ± SEM of 6–12 GFP+ microglia/mouse, with n = 4–5 mice/group. *p<0.05, **p<0.01 versus sham; one-way ANOVA followed by post hoc Tukey’s test.
Detailed morphometric analysis of GFP+ microglia in sham, TBI and TBI+VuPAM CX3CR1gfp/+ mice at 7 days post-injury demonstrated that cortical microglia are transformed from a ramified morphology to an activated morphology after TBI. Specifically, microglia in the injured cortex had reduced filament area and length when compared to ramified microglia in sham cortex (p<0.05, p<0.001 vs sham; Fig. 13b–d). Systemic administration of VuPAM to TBI mice altered this morphological transition in regard to filament length, which was not significantly different from sham (Fig. 13c).
Biochemical analysis of cortical tissue at 7 days post-injury revealed that TBI did not increase expression of p-Akt, p-GSK-3β, or p-CREB when compared to levels in sham cortex (p>0.05 vs sham; Fig. 14a–d). However, systemic administration of VuPAM increased Akt/GSK-3β/CREB signaling in the injured cortex as demonstrated by increased expression of p-AKT, p-GSK-3β, and p-CREB in VuPAM-treated TBI mice (p<0.01 vs TBI+Veh; Fig. 14a–d). Next, we assessed pCREB expression in anti-inflammatory YM-1+ microglia in the injured cortex at 7 days post-injury using immunofluorescence staining. Microglia were visualized using the CX3CR1gfp/+ model. Co-localization analysis revealed that in comparison to levels in vehicle-treated TBI mice, VuPAM treatment increased the number of pCREB+/CX3CR1gfp+ microglia (p<0.01 vs TBI+Veh; Fig. 14e–g) and YM-1+/CX3CR1gfp+ microglia (p<0.01 vs TBI+Veh; Fig. 14e, i). Further analysis demonstrated that VuPAM treatment markedly increased the number of pCREB+/YM-1+ microglia in the injured cortex when compared to levels in vehicle-treated TBI mice (p<0.05 vs TBI+Veh; Fig. 14e, k).
Figure 14. VuPAM treatment increases Akt/GSK-3β/CREB signaling in the injured cortex after TBI.

Moderate-level CCI mice were treated with VuPAM (30 mg/kg, IP) or vehicle (saline + 10% Tween-80, IP) starting at 3h post-injury with repeat dosing at 24h, 72h, and 120h post-injury, and cortical tissue was isolated at 7 days post-injury for biochemical analysis of Akt/GSK-3β/CREB signal transduction pathways. Representative Western immunoblots and quantification of Akt activation (pS473-Akt; a,b), GSK-3β inhibition (pS9-GSK-3β; a,c) and CREB activation (pS133-phosphorylation; a,d) in sham, TBI + Vehicle, and TBI + VuPAM mice. Double immunofluorescence staining for pCREB (magenta), YM-1 (red), and CX3CR1 microglia (green) in the injured cortex of sham, TBI + Vehicle, and TBI + VuPAM mice at 7 days post-injury (e). Co-localization analysis of pCREB+/CX3CR1+ (f,g), YM-1+/CX3CR1+ (h,i), and pCREB+/YM-1+ (j,k) microglia in the injured cortex of TBI + Vehicle, and TBI + VuPAM mice at 7 days post-injury. Data represent mean ± SEM. (a-d) Sham (n=3), TBI + Vehicle (n=6) and TBI + VuPAM (n=6). *p<0.05 versus sham; ++p<0.01 versus TBI + Vehicle; one-way ANOVA followed by post hoc Tukey’s test. (e-k) 15–20 sections from n=4/group. *p < 0.05, *p < 0.01 versus TBI + Vehicle; Unpaired t-test.
Finally, we assessed markers of pro- and anti-inflammatory microglial activation in the injured cortex by Western immunoblot and real-time PCR analyses. As expected, TBI induced robust expression of pro-inflammatory microglial activation markers at 7 days post-injury, as demonstrated by increased expression of TNF-α, IL-6, NOX2, CD68 mRNA, and iNOS protein (p<0.05, p<0.01 vs sham; Fig. 15a–f). Notably, systemic administration of VuPAM reduced the expression of all pro-inflammatory markers in injured cortex (p<0.05, p<0.001 vs TBI+Veh; Fig. 15a–f). Moreover, VuPAM administration also promoted an anti-inflammatory activation state in the injured cortex, as demonstrated by increased expression of Arg-1 protein (p<0.01 vs TBI+Veh; Fig. 15e, g), IL-10 mRNA (p<0.01 vs TBI+Veh; Fig. 15h), YM-1 mRNA (Fig. 15i). Taken together, our in vivo data provides substantial evidence that VuPAM treatment activates Akt/GSK-3β/CREB signaling in the injured cortex to promote an anti-inflammatory microglial activation phenotype in the injured brain.
Figure 15. VuPAM promotes an anti-inflammatory neuroinflammatory response in the injured cortex following TBI.

Moderate-level CCI mice were treated with VuPAM (30 mg/kg, IP) or vehicle (saline + 10% Tween-80, IP) starting at 3h post-injury with repeat dosing at 24h, 72h, and 120h post-injury, and cortical tissue was isolated at 7 days post-injury for molecular analysis of post-traumatic neuroinflammation. qPCR measurement of mRNA expression of pro-inflammatory genes TNF-α (a), NOX2 (b), CD68 (c) and IL-6 (d) in the ipsilateral cortex of sham, TBI + Vehicle, and TBI + VuPAM treated mice. Representative Western immunoblots and quantification of pro-inflammatory iNOS (e,f) and anti-inflammatory Arg-1 (e,g). qPCR measurement of mRNA expression of anti-inflammatory genes IL-10 (h) and YM-1 (i). Sham (n=3), TBI + Vehicle (n=6) and TBI + VuPAM (n=6). *p<0.05, **p<0.01 versus sham; +p<0.05, ++p<0.01 versus TBI; one-way ANOVA followed by post hoc Tukey’s test.
Discussion
Microglial-mediated neuroinflammation has been implicated in the pathobiology of age-related neurodegenerative disorders such as Alzheimer’s and Parkinson’s disease, as well as in traumatic brain and spinal cord injury (Colonna & Butovsky 2017; Faden et al. 2016; Simon et al. 2017). Following CNS injury, microglia display functional and phenotypic diversity driven by local environmental cues that can result in cellular neurotoxic or neuroprotective responses (Loane & Kumar 2016). We, and others, have reported that selective activation of mGluR5 in microglia by the orthosteric agonist CHPG reduces microglial-mediated neuroinflammation and is neuroprotective following moderate-severe TBI (Byrnes et al. 2012; Chen et al. 2012a; Chen et al. 2012b; Loane et al. 2013; Wang et al. 2013; Wang et al. 2012). mGluR5 PAM can also reduce pro-inflammatory microglial activation in vitro (Xue et al. 2014), and VU0360172 (VuPAM) not only prevents pro-inflammatory microglial activation after moderate-severe TBI, but can also promote an anti-inflammatory response, which contributes to neuroprotection and long-term functional recovery (Loane et al. 2014). When compared to orthosteric agonists, mGluR5 PAM have superior anti-inflammatory actions due to their increased potency and functional specificity (Foster & Conn 2017). Here, we confirmed that VuPAM is superior to the orthosteric agonist CHPG not only for inhibiting the pro-inflammatory microglial activation state, but also by promoting an anti-inflammatory microglial phenotype. The mGluR5 antagonist MTEP or siRNA knock down of mGluR5 attenuated the anti-inflammatory actions of VuPAM, thereby confirming the specificity of the drug for mGluR5. Thus, differences in stimulation of mGluR5 between the orthosteric agonist and PAM suggest the involvement of different signal transduction pathways which distinctly modulate microglial phenotypic responses.
Allosteric modulation is an important concept in GPCR-based drug discovery. Modulation of endogenous agonists by allosteric ligands offers the advantages of spatial and temporal fine-tuning of receptor activity, increased selectivity and reduced adverse side-effects with the potential to elicit improved clinical outcomes (Nickols & Conn 2014). Allosteric modulators act to alter the conformational state of GPCRs by binding a topographically distinct non-orthosteric site of the receptor, typically found within the heptahelical domain of the GPCR. Pure allosteric modulators do not activate the receptor directly but potentiate or decrease the response to the endogenous orthosteric ligand (i.e. glutamate for mGluRs). Critical for diversity of functional responses, the GPCR conformation is not static but exists in a spectrum of conformational states that can activate a range of signal transduction pathways. Thus, allosteric modulation can bias GPCRs towards conformations that selectively activate a particular signaling pathway over another, and this is referred to as ‘functional selectivity’ or ‘stimulus bias’ (Kenakin & Christopoulos 2013)
As a Gαq GPCR, mGluR5 classically signals through phosphoinositide hydrolysis and release of intracellular Ca2+ (Stoppel et al. 2017; Lindemann et al. 2011). However, recent studies indicate that other signaling pathways may also play a crucial role in mGluR5 functional responses (Stoppel et al. 2017; Bhakar et al. 2012; Osterweil et al. 2010; Richter et al. 2015). For example, when compared to orthosteric agonists, mGluR5 PAM such as CDPPB stimulate signaling through the MAPK/ERK pathway, which results in improved neuronal survival and preserved cognitive function in a mouse model of Huntington’s disease (Doria et al. 2015; Doria et al. 2013). Akt is an alternate signaling pathway downstream of mGluR5 (Shah et al. 2012; Batista et al. 2016), and activation of Akt is neuroprotective in animal models of neurodegeneration, including Parkinson’s disease, Huntington’s disease and amyotrophic lateral sclerosis (Jha et al. 2015; Steidinger et al. 2013). Akt has three different isoforms, of which Akt1 and Akt2 are predominantly expressed by microglia (Chong et al. 2005). Recently, it was demonstrated that DHEA-induced activation of both Akt1 and Akt2 promote an anti-inflammatory microglial phenotype in an LPS model of neuroinflammation (Alexaki et al. 2018). Here, we demonstrated potent activation of Akt in microglia by VuPAM following LPS stimulation. In contrast, CHPG, a classic orthosteric agonist, failed to activate Akt. Crucially, the PI3K inhibitor Wortmannin or the selective Akt inhibitor AKTi reversed VuPAM-mediated anti-inflammatory actions in microglia. Previous studies also reported that inhibition of PI3K/Akt signaling abolished mGluR5 PAM-mediated neuroprotection in models of glutamate excitotoxicity and Huntington’s disease (Batista et al. 2016; Doria et al. 2015; Doria et al. 2013). These observations indicate that activation of Akt is a critical mechanism downstream of mGluR5 that differentially regulates microglial phenotypic responses stimulated by mGluR5 PAM and orthosteric agonists.
Aberrant GSK-3β activation in microglia is associated with chronic neuroinflammation and neurodegeneration (Eldar-Finkelman & Martinez 2011; Maixner & Weng 2013). Akt phosphorylates GSK-3β at Ser9 residue to inhibit its activity (Atkins et al. 2012; Beurel et al. 2015), which can result in a switch between pro- and anti-inflammatory responses in monocytes (Vergadi et al. 2017). We demonstrated that VuPAM activated Akt in microglia, leading to GSK-3β inhibition and a robust increase in anti-inflammatory IL-10 and Arg-1 expression following LPS stimulation. Similarly, the GSK-3 inhibitors BIO or LiCl (data not shown), caused inactivation of GSK-3β and promoted an anti-inflammatory microglial phenotype. In contrast, inhibition of Akt by AKTi or activation of GSK-3β by DIF-3 reversed VuPAM mediated anti-inflammatory activation in microglia. Thus, our data indicate that VuPAM inhibits GSK-3β through activation of PI3K/Akt signaling, causing microglia to switch from a pro-inflammatory to anti-inflammatory phenotype following LPS challenge.
Our study also demonstrated that VuPAM-mediated inhibition of GSK-3β increases phosphorylation of CREB in microglia. These data corroborate prior studies that showed that anti-inflammatory immune activation is regulated by GSK-3β inhibition and activation of CREB (Wen et al. 2010; Jope et al. 2007). NFκB activity is required for transcriptional activation of immune target genes, and is mediated by the interaction of the RelA subunit of NFκB with the CREB coactivator CBP (Ghosh & Hayden 2008; Medzhitov & Horng 2009). NFκB interacts with CBP at the same site as that of phosphorylated CREB. Therefore, NFκB activity is inhibited by phosphorylated CREB through competitive binding to CBP (Ollivier et al. 1996; Parry & Mackman 1997). In our study, VuPAM had no effect on nuclear translocation of p65 NFκB in LPS stimulated microglia but increased phosphorylated CREB in an Akt/GSK-3β-dependent manner. VuPAM also increased binding of CREB to CBP in microglia following LPS stimulation in an Akt/GSK-3β-dependent manner. These data are consistent with a previous study showing that CREB indirectly inhibits NFкB activation by blocking the association of NFкB with CBP to form the NFкB complex, thereby limiting pro-inflammatory activation and simultaneously promoting an anti-inflammatory phenotype during inflammatory conditions (Wen et al. 2010). Thus, VuPAM shifts the balance towards increased interaction of CREB and CBP, while concurrently preventing the NFкB and CBP association in a GSK-3 dependent manner- thereby promoting an anti-inflammatory phenotype in microglia.
mGluR5 PAMs are considered promising neurotherapeutic agents for a number of neurodegenerative and psychiatric disorders (Foster & Conn 2017; Ribeiro et al. 2014; Ribeiro et al. 2017), and novel mGluR5 PAMs are being developed for the treatment of schizophrenia (Niswender & Conn 2010). Earlier studies from our laboratory demonstrated neuroprotective effects of selective mGluR5 activation by either orthosteric agonists or allosteric modulators following traumatic brain or spinal cord injury (Byrnes et al. 2009c; Byrnes et al. 2009a; Loane et al. 2013; Loane et al. 2009b; Loane et al. 2014). The VuPAM used in this study (VU0360172) has an excellent pharmacokinetic profile and readily crosses the blood brain barrier; initial characterization studies demonstrated that VU0360172 was rapidly and significantly absorbed following systemic administration (10 mg/kg; p.o.), as evident from a systemic plasma concentration of 21 μM (Cmax of 7432.98 ng/ml) within 1 hour of dosing (Rodriguez et al. 2010). Moreover, new studies using mGluR5-mediated polyphosphoinositide (PI) hydrolysis functional assays in mice demonstrated that systemic administration (i.p.) of VU0360172 stimulated PI hydrolysis at a dose of 30 mg/kg in hippocampus, striatum, and prefrontal cortex (Zuena et al. 2018). Previously we demonstrated that in addition to suppressing post-traumatic neurotoxic neuroinflammation, VuPAM treatment also upregulates an anti-inflammatory microglial phenotype in the injured cortex, whereas an mGluR5 orthosteric agonist does not (Loane et al. 2014). In the current study, we identified the signal transduction mechanisms that appear to confer VuPAM-mediated neuroprotection after experimental TBI. Thus, systemic administration of VuPAM to mice beginning 3 hours post-injury increased phosphorylation of Akt, GSK-3β and CREB in the injured cortex, as well as reducing markers of pro-inflammatory microglial activation and increased anti-inflammatory IL-10, Arg1, and YM1 expression. Morphological analysis of GFP+ microglia by intravital two-photon imaging demonstrated that VuPAM treatment suppresses the morphological transformation of activated microglia in the injured cortex. VuPAM treatment also increased the expression of pCREB and anti-inflammatory YM1 in GFP+ microglia in the injured cortex. Notably, other pre-clinical TBI studies have implicated the activation of Akt/GSK-3β (Farr et al. 2019; Jiang et al. 2017; Wang et al. 2015) and CREB (Alexaki et al. 2018) signal transduction pathways in the suppression of pro-inflammatory and neurotoxic neuroinflammation, as well as for3 promotion of anti-inflammatory and neuroprotective responses following moderate-severe TBI.
Although we identified key signaling pathways downstream of mGluR5 by which VuPAM regulates anti-inflammatory microglial activation, it is also important to discuss the limitations of our studies. Pharmacological and molecular studies were performed in BV2 microglia, which are limited with regard to microglial phenotypic markers and heterogeneity of responses (Stansley et al. 2012). However, we reproduced key pharmacological experiments in primary rat cortical microglia in order to confirm the essential role of Akt/GSK-3β/CREB signaling in promoting the anti-inflammatory microglial phenotype following VuPAM treatment. Moreover, using a well characterized experimental TBI model and CX3CR1gfp/+ mice to visualize microglia in vivo, we showed that VuPAM enhances Akt/GSK-3β/CREB signaling and markers of anti-inflammatory microglial activation (IL-10, Arg1, YM1) in the injured cortex. In addition, in situ analysis revealed that GFP+ microglia in the cortex of VuPAM treated TBI mice co-express pCREB and the anti-inflammatory microglial phenotype marker, YM1. Another potential limitation was that we performed classical pharmacology (mGluR5 orthosteric versus PAM, mGluR5 antagonist, and inhibitors) in an LPS model of microglial activation, which can have off-target drug effects. To address such concerns, we validated key study components, including mGluR5 selectivity of VuPAM and CREB activation using siRNA knockdown and dominant-negative mutant experiments, respectively.
In conclusion, we provide strong evidence that a mGluR5 PAM (VU0360172) activates specific Akt/GSK-3β/CREB signaling to promote anti-inflammatory microglial activation in vitro and following experimental TBI in mice. These studies provide further support for considering mGluR5 PAMs as neurotherapeutic agents for moderate-to-severe TBI and other neurodegenerative diseases for which microglial-mediated neuroinflammation has been implicated as an important pathobiological mechanism.
Acknowledgments:
We thank Dr. Boris Sabirzhanov for help with primary rat cortical neuron preparations. This work was supported by National Institutes of Health grants R01NS037313 (A.I. Faden), R01NS096002 (B.S. Stoica), R01NS082308 (D.J. Loane), and Science Foundation Ireland grant 17/FRL/4860 (D.J. Loane).
Abbreviations:
- AKTi
AKT inhibitor
- BIO
GSK-3β inhibitor
- BSA
Bovine serum albumin
- CCI
Controlled cortical impact
- CHPG
(RS)-2-chloro-5-hydroxyphenylglycine
- CREB
cAMP response element-binding protein
- DIF-3
GSK-3β activator
- DMEM
Dulbecco’s modified Eagle’s medium
- FBS
Fetal bovine serum
- GPCR
G-protein coupled receptor
- GSK-3β
Glycogen synthase kinase-3β
- HBSS
Hanks’ Balanced Salt Solution
- I.P.
Intraperitoneal
- LPS
Lipopolysaccharide
- mGluR5
Metabotropic glutamate receptor five
- MTEP
3-[(2-Methyl- 4-thiazolyl)ethynyl]pyridine]
- P.O.
Per Os (oral administration)
- PAM
Positive allosteric modulator
- PBS
Phosphate buffered saline
- PKC
Protein kinase C
- PLC
Phospholipase C
- RRID
Research Resource Identifiers
- TBI
Traumatic brain injury
- VU0360172
N-cyclobutyl-6-[2-(3-fluorophenyl)ethynyl]-3-pyridinecarboxamide hydrochloride
- VuPAM
VU0360172
Footnotes
Author Disclosures: No competing financial interests exist.
References
- Alexaki VI, Fodelianaki G, Neuwirth A et al. (2018) DHEA inhibits acute microglia-mediated inflammation through activation of the TrkA-Akt1/2-CREB-Jmjd3 pathway. Mol Psychiatry 23, 1410–1420. [DOI] [PubMed] [Google Scholar]
- Andreou KE, Soto MS, Allen D, Economopoulos V, de Bernardi A, Larkin JR and Sibson NR (2017) Anti-inflammatory Microglia/Macrophages As a Potential Therapeutic Target in Brain Metastasis. Front Oncol 7, 251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins RJ, Dimou J, Paradiso L, Morokoff AP, Kaye AH, Drummond KJ and Hovens CM (2012) Regulation of glycogen synthase kinase-3 beta (GSK-3beta) by the Akt pathway in gliomas. J Clin Neurosci 19, 1558–1563. [DOI] [PubMed] [Google Scholar]
- Batista EM, Doria JG, Ferreira-Vieira TH, Alves-Silva J, Ferguson SS, Moreira FA and Ribeiro FM (2016) Orchestrated activation of mGluR5 and CB1 promotes neuroprotection. Molecular brain 9, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beurel E, Grieco SF and Jope RS (2015) Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol Ther 148, 114–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhakar AL, Dolen G and Bear MF (2012) The pathophysiology of fragile X (and what it teaches us about synapses). Annu Rev Neurosci 35, 417–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat SA, Sood A, Shukla R and Hanif K (2018) AT2R Activation Prevents Microglia Pro-inflammatory Activation in a NOX-Dependent Manner: Inhibition of PKC Activation and p47(phox) Phosphorylation by PP2A. Mol Neurobiol [DOI] [PubMed] [Google Scholar]
- Byrnes KR, Loane DJ and Faden AI (2009a) Metabotropic glutamate receptors as targets for multipotential treatment of neurological disorders. Neurotherapeutics 6, 94–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrnes KR, Loane DJ, Stoica BA, Zhang J and Faden AI (2012) Delayed mGluR5 activation limits neuroinflammation and neurodegeneration after traumatic brain injury. Journal of neuroinflammation 9, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrnes KR, Stoica B, Loane DJ, Riccio A, Davis MI and Faden AI (2009b) Metabotropic glutamate receptor 5 activation inhibits microglial associated inflammation and neurotoxicity. Glia 57, 550–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrnes KR, Stoica B, Riccio A, Pajoohesh-Ganji A, Loane DJ and Faden AI (2009c) Activation of metabotropic glutamate receptor 5 improves recovery after spinal cord injury in rodents. Ann Neurol 66, 63–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Cao L, Dong W, Luo P, Liu W, Qu Y and Fei Z (2012a) Protective effects of mGluR5 positive modulators against traumatic neuronal injury through PKC-dependent activation of MEK/ERK pathway. Neurochemical research 37, 983–990. [DOI] [PubMed] [Google Scholar]
- Chen T, Zhang L, Qu Y, Huo K, Jiang X and Fei Z (2012b) The selective mGluR5 agonist CHPG protects against traumatic brain injury in vitro and in vivo via ERK and Akt pathway. Int J Mol Med 29, 630–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Li F and Maiese K (2005) Activating Akt and the brain’s resources to drive cellular survival and prevent inflammatory injury. Histol Histopathol 20, 299–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colonna M and Butovsky O (2017) Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu Rev Immunol 35, 441–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doria JG, de Souza JM, Andrade JN, Rodrigues HA, Guimaraes IM, Carvalho TG, Guatimosim C, Dobransky T and Ribeiro FM (2015) The mGluR5 positive allosteric modulator, CDPPB, ameliorates pathology and phenotypic signs of a mouse model of Huntington’s disease. Neurobiol Dis 73, 163–173. [DOI] [PubMed] [Google Scholar]
- Doria JG, Silva FR, de Souza JM, Vieira LB, Carvalho TG, Reis HJ, Pereira GS, Dobransky T and Ribeiro FM (2013) Metabotropic glutamate receptor 5 positive allosteric modulators are neuroprotective in a mouse model of Huntington’s disease. British journal of pharmacology 169, 909–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldar-Finkelman H and Martinez A (2011) GSK-3 Inhibitors: Preclinical and Clinical Focus on CNS. Frontiers in molecular neuroscience 4, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faden AI and Loane DJ (2015) Chronic neurodegeneration after traumatic brain injury: Alzheimer disease, chronic traumatic encephalopathy, or persistent neuroinflammation? Neurotherapeutics 12, 143–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faden AI, Wu J, Stoica BA and Loane DJ (2016) Progressive inflammation-mediated neurodegeneration after traumatic brain or spinal cord injury. British journal of pharmacology 173, 681–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farr SA, Niehoff ML, Kumar VB, Roby DA and Morley JE (2019) Inhibition of Glycogen Synthase Kinase 3beta as a Treatment for the Prevention of Cognitive Deficits after a Traumatic Brain Injury. Journal of neurotrauma [DOI] [PubMed] [Google Scholar]
- Foster DJ and Conn PJ (2017) Allosteric Modulation of GPCRs: New Insights and Potential Utility for Treatment of Schizophrenia and Other CNS Disorders. Neuron 94, 431–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S and Hayden MS (2008) New regulators of NF-kappaB in inflammation. Nat Rev Immunol 8, 837–848. [DOI] [PubMed] [Google Scholar]
- Henry RJ, Doran SJ, Barrett JP, Meadows VE, Sabirzhanov B, Stoica BA, Loane DJ and Faden AI (2019) Inhibition of miR-155 Limits Neuroinflammation and Improves Functional Recovery After Experimental Traumatic Brain Injury in Mice. Neurotherapeutics 16, 216–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha SK, Jha NK, Kar R, Ambasta RK and Kumar P (2015) p38 MAPK and PI3K/AKT Signalling Cascades inParkinson’s Disease. Int J Mol Cell Med 4, 67–86. [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Xia QJ, Dong XJ, Hu Y, Chen ZW, Chen K, Wang KH, Liu J and Wang TH (2017) Neuroprotective effect of breviscapine on traumatic brain injury in rats associated with the inhibition of GSK3beta signaling pathway. Brain research 1660, 1–9. [DOI] [PubMed] [Google Scholar]
- Jope RS, Yuskaitis CJ and Beurel E (2007) Glycogen synthase kinase-3 (GSK3): inflammation, diseases, and therapeutics. Neurochemical research 32, 577–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T and Christopoulos A (2013) Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat Rev Drug Discov 12, 205–216. [DOI] [PubMed] [Google Scholar]
- Kettenmann H, Kirchhoff F and Verkhratsky A (2013) Microglia: new roles for the synaptic stripper. Neuron 77, 10–18. [DOI] [PubMed] [Google Scholar]
- Kim JY, Kim N and Yenari MA (2015) Mechanisms and potential therapeutic applications of microglial activation after brain injury. CNS Neurosci Ther 21, 309–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindemann L, Jaeschke G, Michalon A et al. (2011) CTEP: a novel, potent, long-acting, and orally bioavailable metabotropic glutamate receptor 5 inhibitor. The Journal of pharmacology and experimental therapeutics 339, 474–486. [DOI] [PubMed] [Google Scholar]
- Loane DJ and Byrnes KR (2010) Role of microglia in neurotrauma. Neurotherapeutics 7, 366–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loane DJ and Kumar A (2016) Microglia in the TBI brain: The good, the bad, and the dysregulated. Experimental neurology 275 Pt 3, 316–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loane DJ, Pocivavsek A, Moussa CE, Thompson R, Matsuoka Y, Faden AI, Rebeck GW and Burns MP (2009a) Amyloid precursor protein secretases as therapeutic targets for traumatic brain injury. Nature medicine 15, 377–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loane DJ, Stoica BA, Byrnes KR, Jeong W and Faden AI (2013) Activation of mGluR5 and inhibition of NADPH oxidase improves functional recovery after traumatic brain injury. Journal of neurotrauma 30, 403–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loane DJ, Stoica BA, Pajoohesh-Ganji A, Byrnes KR and Faden AI (2009b) Activation of metabotropic glutamate receptor 5 modulates microglial reactivity and neurotoxicity by inhibiting NADPH oxidase. The Journal of biological chemistry 284, 15629–15639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loane DJ, Stoica BA, Tchantchou F, Kumar A, Barrett JP, Akintola T, Xue F, Conn PJ and Faden AI (2014) Novel mGluR5 Positive Allosteric Modulator Improves Functional Recovery, Attenuates Neurodegeneration, and Alters Microglial Polarization after Experimental Traumatic Brain Injury. Neurotherapeutics [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maixner DW and Weng HR (2013) The Role of Glycogen Synthase Kinase 3 Beta in Neuroinflammation and Pain. J Pharm Pharmacol (Los Angel) 1, 001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning BD and Cantley LC (2007) AKT/PKB signaling: navigating downstream. Cell 129, 1261–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mead EL, Mosley A, Eaton S, Dobson L, Heales SJ and Pocock JM (2012) Microglial neurotransmitter receptors trigger superoxide production in microglia; consequences for microglial-neuronal interactions. Journal of neurochemistry 121, 287–301. [DOI] [PubMed] [Google Scholar]
- Medzhitov R and Horng T (2009) Transcriptional control of the inflammatory response. Nat Rev Immunol 9, 692–703. [DOI] [PubMed] [Google Scholar]
- Meijer L, Skaltsounis AL, Magiatis P et al. (2003) GSK-3-selective inhibitors derived from Tyrian purple indirubins. Chem Biol 10, 1255–1266. [DOI] [PubMed] [Google Scholar]
- Nickols HH and Conn PJ (2014) Development of allosteric modulators of GPCRs for treatment of CNS disorders. Neurobiol Dis 61, 55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender CM and Conn PJ (2010) Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol 50, 295–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ollivier V, Parry GC, Cobb RR, de Prost D and Mackman N (1996) Elevated cyclic AMP inhibits NF-kappaB-mediated transcription in human monocytic cells and endothelial cells. The Journal of biological chemistry 271, 20828–20835. [DOI] [PubMed] [Google Scholar]
- Osterweil EK, Krueger DD, Reinhold K and Bear MF (2010) Hypersensitivity to mGluR5 and ERK1/2 leads to excessive protein synthesis in the hippocampus of a mouse model of fragile X syndrome. J Neurosci 30, 15616–15627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry GC and Mackman N (1997) Role of cyclic AMP response element-binding protein in cyclic AMP inhibition of NF-kappaB-mediated transcription. J Immunol 159, 5450–5456. [PubMed] [Google Scholar]
- Piers TM, Heales SJ and Pocock JM (2011) Positive allosteric modulation of metabotropic glutamate receptor 5 down-regulates fibrinogen-activated microglia providing neuronal protection. Neuroscience letters 505, 140–145. [DOI] [PubMed] [Google Scholar]
- Qian L, Tan KS, Wei SJ, Wu HM, Xu Z, Wilson B, Lu RB, Hong JS and Flood PM (2007) Microglia-mediated neurotoxicity is inhibited by morphine through an opioid receptor-independent reduction of NADPH oxidase activity. J Immunol 179, 1198–1209. [DOI] [PubMed] [Google Scholar]
- Qiao X, Gai H, Su R, Deji C, Cui J, Lai J and Zhu Y (2018) PI3K-AKT-GSK3beta-CREB signaling pathway regulates anxiety-like behavior in rats following alcohol withdrawal. J Affect Disord 235, 96–104. [DOI] [PubMed] [Google Scholar]
- Ribeiro FM, Hamilton A, Doria JG, Guimaraes IM, Cregan SP and Ferguson SS (2014) Metabotropic glutamate receptor 5 as a potential therapeutic target in Huntington’s disease. Expert Opin Ther Targets 18, 1293–1304. [DOI] [PubMed] [Google Scholar]
- Ribeiro FM, Vieira LB, Pires RG, Olmo RP and Ferguson SS (2017) Metabotropic glutamate receptors and neurodegenerative diseases. Pharmacol Res 115, 179–191. [DOI] [PubMed] [Google Scholar]
- Richter JD, Bassell GJ and Klann E (2015) Dysregulation and restoration of translational homeostasis in fragile X syndrome. Nature reviews 16, 595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez AL, Grier MD, Jones CK et al. (2010) Discovery of novel allosteric modulators of metabotropic glutamate receptor subtype 5 reveals chemical and functional diversity and in vivo activity in rat behavioral models of anxiolytic and antipsychotic activity. Molecular pharmacology 78, 1105–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth TL, Nayak D, Atanasijevic T, Koretsky AP, Latour LL and McGavern DB (2014) Transcranial amelioration of inflammation and cell death after brain injury. Nature 505, 223–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabirzhanov B, Zhao Z, Stoica BA, Loane DJ, Wu J, Borroto C, Dorsey SG and Faden AI (2014) Downregulation of miR-23a and miR-27a following Experimental Traumatic Brain Injury Induces Neuronal Cell Death through Activation of Proapoptotic Bcl-2 Proteins. J Neurosci 34, 10055–10071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah A, Silverstein PS, Singh DP and Kumar A (2012) Involvement of metabotropic glutamate receptor 5, AKT/PI3K signaling and NF-kappaB pathway in methamphetamine-mediated increase in IL-6 and IL-8 expression in astrocytes. Journal of neuroinflammation 9, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon DW, McGeachy MJ, Bayir H, Clark RS, Loane DJ and Kochanek PM (2017) The far-reaching scope of neuroinflammation after traumatic brain injury. Nat Rev Neurol 13, 171–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stansley B, Post J and Hensley K (2012) A comparative review of cell culture systems for the study of microglial biology in Alzheimer’s disease. Journal of neuroinflammation 9, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stansley BJ and Conn PJ (2018) Neuropharmacological Insight from Allosteric Modulation of mGlu Receptors. Trends Pharmacol Sci 40, 240–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steidinger TU, Slone SR, Ding H, Standaert DG and Yacoubian TA (2013) Angiogenin in Parkinson disease models: role of Akt phosphorylation and evaluation of AAV-mediated angiogenin expression in MPTP treated mice. PLoS One 8, e56092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoppel LJ, Auerbach BD, Senter RK, Preza AR, Lefkowitz RJ and Bear MF (2017) beta-Arrestin2 Couples Metabotropic Glutamate Receptor 5 to Neuronal Protein Synthesis and Is a Potential Target to Treat Fragile X. Cell Rep 18, 2807–2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi-Yanaga F, Mori J, Matsuzaki E, Watanabe Y, Hirata M, Miwa Y, Morimoto S and Sasaguri T (2006) Involvement of GSK-3beta and DYRK1B in differentiation-inducing factor-3-induced phosphorylation of cyclin D1 in HeLa cells. The Journal of biological chemistry 281, 38489–38497. [DOI] [PubMed] [Google Scholar]
- Taylor CA, Bell JM, Breiding MJ and Xu L (2017) Traumatic Brain Injury-Related Emergency Department Visits, Hospitalizations, and Deaths - United States, 2007 and 2013. Morbidity and mortality weekly report. Surveillance summaries 66, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergadi E, Ieronymaki E, Lyroni K, Vaporidi K and Tsatsanis C (2017) Akt Signaling Pathway in Macrophage Activation and M1/M2 Polarization. J Immunol 198, 1006–1014. [DOI] [PubMed] [Google Scholar]
- Wang G, Shi Y, Jiang X et al. (2015) HDAC inhibition prevents white matter injury by modulating microglia/macrophage polarization through the GSK3beta/PTEN/Akt axis. Proc Natl Acad Sci U S A 112, 2853–2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JW, Wang HD, Cong ZX, Zhang XS, Zhou XM and Zhang DD (2013) Activation of metabotropic glutamate receptor 5 reduces the secondary brain injury after traumatic brain injury in rats. Biochemical and biophysical research communications 430, 1016–1021. [DOI] [PubMed] [Google Scholar]
- Wang JW, Wang HD, Zhong WZ, Li N and Cong ZX (2012) Expression and cell distribution of metabotropic glutamate receptor 5 in the rat cortex following traumatic brain injury. Brain research 1464, 73–81. [DOI] [PubMed] [Google Scholar]
- Wen AY, Sakamoto KM and Miller LS (2010) The role of the transcription factor CREB in immune function. J Immunol 185, 6413–6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue F, Stoica BA, Hanscom M, Kabadi SV and Faden AI (2014) Positive allosteric modulators (PAMs) of metabotropic glutamate receptor 5 (mGluR5) attenuate microglial activation. CNS & neurological disorders drug targets 13, 558–566. [DOI] [PubMed] [Google Scholar]
- Zuena AR, Iacovelli L, Orlando R, Di Menna L, Casolini P, Alema GS, Di Cicco G, Battaglia G and Nicoletti F (2018) In Vivo Non-radioactive Assessment of mGlu5 Receptor-Activated Polyphosphoinositide Hydrolysis in Response to Systemic Administration of a Positive Allosteric Modulator. Front Pharmacol 9, 804. [DOI] [PMC free article] [PubMed] [Google Scholar]