

Abstract
Since the first high-resolution structure of the nucleosome was reported in 1997, the available information on chromatin structure has increased exponentially. Here, we review insights derived from cutting-edge biophysical and structural approaches applied to the study of nucleosome dynamics and nucleosome-binding factors, with a focus on the experimental advances driving the research. In addition, we highlight emerging challenges in nucleosome structural biology.
Graphical Abstract
Introduction
In all eukaryotes, DNA is compacted into chromatin. Viewed under the electron microscope, chromatin structure appears as “beads on a string”1 in which each “bead” represents the basic repeating unit of chromatin, the nucleosome2. Initial structural information on the nucleosome came as crystal structures of the nucleosome at low resolution3 and of the histone octamer in the absence of DNA at 3.2 Å4. The first high-resolution (2.8 Å) crystal structure of the nucleosome was determined a few years later5, providing detailed information on how nucleosomal DNA is deformed by an intricate arrangement of histones.
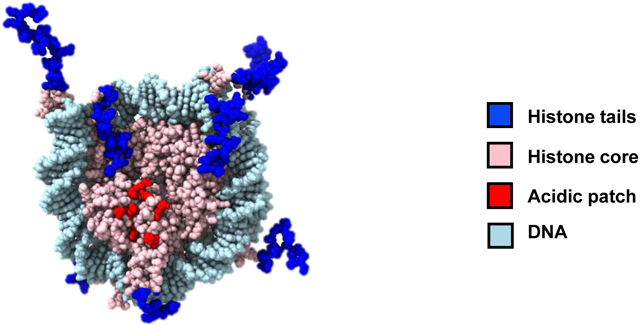
In a canonical nucleosome, an octamer of two copies each of the four core histones (H3, H4, H2A and H2B) is wrapped by 145-147 base pairs (bp) of DNA in a left-handed supercoil (Figure 1). The main contacts between histones and DNA are made through structurally conserved histone-fold domains which organize ~120 bp of DNA, while the remaining ~13 bp of DNA at each end are bound by the N-terminal alpha-helix (α-N) that is unique to H35. These latter interactions are important for maintaining the stability of the nucleosome6. In established terminology, each location where the major groove faces the histone octamer is designated as a “super helix location” (or SHL), numbered from SHL 0 at the nucleosomal dyad to SHL +/−7 for the very last region of histone-bound DNA5 (Figure 1c).
Figure 1: Nucleosome structure.

A: Nucleosome disc view, model derived from PDB entries 1KX576 and 1ZBB109 (DNA from 1ZBB, histone octamer core from 1KX5).
B: Electrostatic potential of the nucleosome surface (electrostatic potential calculated from PDB 1KX5, using APBS within PyMOL version 2.2.0).
C: Nucleosomal DNA and linker DNA (from PDB entry 1ZBB). Along the 2-fold axis, nucleosomal DNA (145-147 bp) can be divided into two “gyres” (about 72 bp each). The super-helical location (SHL) designation represents the position of each major groove facing inward. The dyad (center of the nucleosomal DNA) is defined as position 0. The numbers “1-7” highlight the SHL on DNA. Linker DNA is the extra-nucleosomal DNA which locates next to the entry/exit site of nucleosomal DNA.
Inside the histone core, histones H3 and H4 form a symmetric hetero-tetramer through a four-helix bundle structure between two H3 molecules. Two H2A-H2B dimers interact with the (H3-H4)2 tetramer through multiple interactions including a similar four-helix bundle structure between H2B and H4, and additional interactions between the H2A docking domain and (H3-H4). The two H2A chains form a very small interface formed by their L1 loops in the nucleosome core.
Taken together, these interactions render the nucleosome very stable, but it is not the static “disc” suggested by the crystal structure. Rather, nucleosomes are highly dynamic both in terms of composition and conformation. For example, nucleosomes can transition between different states of post-translational modification (PTMs) and histone variant composition, which subtly alters their structure and thus their interaction properties. Nucleosomes also display intrinsic structural dynamics that manifest in rapid DNA unwrapping and rewrapping, also called “DNA breathing”. Moreover, as the basic units of chromatin and the main carriers of epigenetic marks, nucleosomes interact with hundreds of proteins, which affect nucleosome structure and dynamics. During the past decade, a combination of x-ray crystallography, single-particle cryo-electron microscopy (cryoEM) and nuclear magnetic resonance (NMR) has provided information on how chromatin-binding proteins interface with nucleosomes. Together, these studies advance our understanding of how nucleosomes and the plethora of nuclear factors they interact with regulate access to the DNA they organize. Here, we review recent progress in biophysical studies of DNA unwrapping and nucleosome dynamics as well as structures of chromatin-binding proteins in complex with nucleosomes, illuminating how these molecular machines gain access to DNA and carry out their various biological functions.
Biophysical studies of nucleosome dynamics
Thermodynamics and kinetics of nucleosome unwrapping is shown by FRET
An important aspect of nucleosome dynamics is the spontaneous unwrapping of DNA from the histone octamer. For the initial thermodynamic characterization of this phenomenon, Li & Widom7 used a FRET system that allowed measurement of an equilibrium constant of nucleosome unwrapping (Keq) of ~0.02–0.1, meaning that nucleosomes are partially unwrapped 2 to 10% of the time (Figure 2a). Transient unwrapping exposes protein binding sites in nucleosomal DNA that are otherwise buried, and upon protein binding the unwrapping equilibrium shifts, facilitating more unwrapping. This explains how sequence-specific DNA binding can occur in the presence of nucleosomes. Importantly, nucleosomes within a nucleosomal array undergo unwrapping similarly to single, isolated nucleosomes, and their DNA is equally accessible to DNA-binding proteins8,9, suggesting that spontaneous site exposure can facilitate protein binding in vivo.
Figure 2: Dynamics of nucleosome unwrapping.

A: CryoEM structures of a canonical nucleosome (PDB entry 6ESF24) and a partially unwrapped nucleosome (PDB entry 6ESH24). Rate constants of spontaneous unwrapping and rewrapping, determined by stopped-flow spectroscopy, are indicated.
B: Location of post-translational modifications that have been studied for their effect on nucleosome unwrapping: at the DNA entry/exit site, at SHL +/−3 (about 35 bp into the nucleosome) and at the dyad. The effects of these post-translational modifications on nucleosome dynamics are indicated. From PDB entry 1KX576.
A complementary kinetic study allowed determination of the rate constants of spontaneous DNA unwrapping (kunwrap = ~4 s−1) and rewrapping (krewrap = ~20-90 s−1)10. These rate constants imply that the lifetime of the wrapped state is ~250 ms, while the lifetime of the unwrapped state is in the range of ~10-50 ms. Although the conditions under which these experiments were performed are necessarily different from the conditions in the nucleus, spontaneous unwrapping likely allows to access sites occupied by nucleosomes on a time scale compatible with biological responses, and independently of chromatin remodeling factors.
It has long been hypothesized that histone PTMs effect at least some of their biological functions by changing the dynamic behavior of nucleosomes, and thus similar studies have investigated the effects of histone PTMs on unwrapping. For example, incorporation of a fully synthetic histone H3 with acetylated lysine 56 (H3K56Ac), a PTM involved in regulation of transcription and DNA repair and located near the DNA entry/exit site, shifts the unwrapping equilibrium towards unwrapped nucleosomes with a 2-fold effect on Keq11. Analysis of the effects of H3K56Ac on the rate constants of unwrapping and rewrapping demonstrated that its presence increases kunwrap by 2 to 3-fold compared to unmodified nucleosome12, indicating that changes in unwrapping equilibrium arise from changes in the rate of unwrapping rather than rewrapping. Thus, H3K56Ac facilitates protein binding to internal sites in nucleosomal DNA by increased site exposure.
Nucleosomes often bear multiple PTMs in vivo, which motivated unwrapping equilibrium studies of nucleosomes with defined combinations of PTMs13,14. Overall, PTMs located near the dyad influence histone release from DNA during mechanical disassembly using magnetic tweezers, but do not modulate unwrapping, while PTMs near the DNA entry/exit site and SHL +/−3 favor unwrapping by 2 to 3-fold compared to unmodified nucleosomes13 (Figure 2b). Certain combinations of PTMs also display strong synergistic effects14. Two recent reviews provide further discussion on the effects of histone PTMs on nucleosome dynamics15 and on strategies for chemical synthesis of histones with defined PTMs16.
How does DNA sequence affect nucleosome unwrapping? Nucleosomes can form on almost any DNA sequence, but some sequences exert a stronger positioning effect than others. For example, sequences with AA, TT or TA dinucleotides spaced by 10 bp bend more easily and display higher affinity for the histone octamer. Two such “nucleosome positioning” sequences are commonly used for in vitro experiments that require homogeneously positioned nucleosomes: the naturally occurring 5S rRNA gene sequence17 and the Widom 601 sequence18, which was selected in vitro by directed evolution for its affinity for the (H3-H4)2 tetramer. Comparison indicated that sequence variations at the DNA entry/exit site are sufficient to significantly modulate unwrapping12. The effects of PTMs and DNA sequence at the entry/exit site on unwrapping are additive, allowing for finely tuned control of unwrapping of any given nucleosome governed by its genomic location and its PTM status. Given that ~30% of transcription factor (TF) binding sites in S. cerevisiae are located in the entry/exit region of a nucleosome, spontaneous nucleosome unwrapping could be an intrinsic regulatory mechanism of TF binding12.
A systematic equilibrium study of the salt-dependence of nucleosome stability19 comparing different DNA sequences and histones from different organisms showed that DNA sequence has a stronger influence on nucleosome stability than the histones, and that disassembly intermediates are the same regardless of histone and DNA composition. An equivalent systematic kinetic study has not been performed yet but would be informative to better understand how unwrapping propensity correlates with DNA sequence.
SAXS and single-molecule reveal the mechanism of unwrapping
The dynamics of unwrapping have also been characterized by SAXS, using salt-induced destabilization of nucleosomes. This was done both at equilibrium and in kinetic measurements with a coupled stopped-flow system20, using contrast matching to distinguish DNA from protein (Box 2). These technically challenging experiments demonstrated that the 5S nucleosome unwraps rapidly (within milliseconds after reaching 2 M NaCl), whereas the 601 nucleosome displays a partially unwrapped intermediate that is stable for about 200 ms. This is in apparent contradiction with the observation that the 5S sequence has a slower rate constant of spontaneous unwrapping12, suggesting that salt affects different nucleosomes differently.
Box 2: Structural dynamics methods.
Four experimental methods have been commonly used to probe nucleosome structural dynamics and interactions: Small-Angle X-ray Scattering (SAXS), Fluorescence Resonance Energy Transfer (FRET), Nuclear Magnetic Resonance (NMR) and Hydrogen-Deuterium Exchange coupled with Mass Spectrometry (HDX-MS).
SAXS is mostly used for “structure estimation” because the parameters derived from scattering curves (radius of gyration and maximal intramolecular distance) are sensitive to global shape and conformation80. In comparative studies, conformational differences between various complexes can be determined81-84,14. Ab initio shape reconstruction provides low-resolution information, although for protein-DNA complexes this is complicated by different electron density, and therefore scattering, of the two components. Contrast matching exploits this property to obtain additional information: the buffer electron density is adjusted to match the average protein electron density, thereby masking scattering from protein components and isolating scattering contributions of DNA85. SAXS shines as a structural dynamics method, when combined with a stopped-flow system for kinetic measurements of global conformational changes on a millisecond time scale20,22. SAXS requires high sample concentration and is strongly affected by aggregation, posing a double constraint on sample preparation.
FRET is a highly sensitive and specific distance probe. The first use of FRET applied to nucleosomes involved fluorescently labeling the two DNA ends to study linker DNA dynamics86. Labeling of histones followed quickly87, and the combined labeling of histones and DNA has enabled a large number of FRET-based studies of nucleosome dynamics, including DNA unwrapping and histone release, reviewed elsewhere88.
NMR allows measurements of protein dynamics and interactions at the single residue level. It requires large amounts of isotopically-labeled protein, which still is expensive and challenging. Isotopic labeling strategies have been reviewed recently, both generic89 and specifically developed for nucleosomes90. NMR is uniquely capable of mapping protein interactions at the level of single residues even in disordered regions, or transient complexes91,27,66, which makes it complementary to other methods delivering static structures. Current progress of NMR for the study of larger complexes and transient interactions92, and of solid-state NMR93,94, are all relevant to the study of nucleosome dynamics and interactions with nucleosome-binding factors.
HDX-MS allows detection of changes in solvent accessibility of backbone amide groups, which occur upon conformational changes and intermolecular interactions. There is no size limitation, and lower sample requirements make HDX-MS complementary to NMR for the study of structural dynamics and interactions at a residue-level resolution (recently reviewed95). Mass spectrometry can also determine binding affinities of protein/DNA and protein/nucleosome interactions on a proteome-wide basis and in cell extracts96, complementary to targeted in vitro studies.
The SAXS data were best explained by ensemble modeling with a pool of DNA structures unwrapped to different degrees, suggesting that both the 601 and 5S sequences unwrap asymmetrically. This was later confirmed by single-molecule force spectroscopy21. Specifically, it was shown that asymmetric unwrapping under tension is governed by DNA flexibility in the inner turn: flexible sequences can tolerate being bent in a nucleosomal conformation and unwrap at higher forces, while less flexible sequences unwrap more easily at lower forces. Moreover, unwrapping of the stiff side stabilizes the flexible side, possibly amplifying small differences in sequence flexibility into a more pronounced asymmetry of the two sides of a nucleosome.
Contrast matching time-resolved (TR) SAXS was also combined with TR-FRET to monitor both DNA unwrapping and H2A-H2B dimer release on the same time scale during salt-induced partial disassembly22. Release of one H2A-H2B dimer occurs within 30 s after initiation of salt-induced disassembly, while the second dimer is only released within 5 min. Thus, under low-salt conditions and in absence of nucleosome-binding factors, spontaneous H2A-H2B release is an extremely rare event. In vivo, H2A-H2B release likely only happens in the presence of histone chaperones, to allow targeted regulation of DNA accessibility.
Unwrapping was also directly visualized, originally by AFM23 and more recently by cryoEM24. The latter study confirmed that unwrapping of the 601 sequence is preferentially asymmetric and that H2A-H2B release is rare under low-salt conditions (observation of hexasomes required a higher salt concentration). It was also estimated that H2A-H2B release only occurs when ~40 bp of DNA have unwrapped, indicating that DNA must dissociate from the entire H2A-H2B dimer to allow its release. Intriguingly, there is evidence for the existence of an overlapping dinucleosome25 in which DNA wraps around one histone octamer plus one histone hexamer (i.e. an octamer lacking one H2A-H2B dimer), and for the existence of partially unwrapped subnucleosomal particles in vivo26. Both were hypothesized as possible products of ATP-dependent chromatin remodelers.
NMR and HDX-MS reveal histone dynamics and interacting surfaces
DNA unwrapping and histone release are phenomena that are well suited to be monitored by FRET, given the dramatic conformational or compositional changes of the nucleosome during these processes. However, more subtle structural dynamics of histones are also an important part of nucleosome dynamics, and their study requires different experimental approaches, such as NMR and HDX-MS. Here we discuss two examples that exemplify the strengths of both techniques.
Methyl-TROSY NMR, pioneered for the nucleosome in a tour de force effort by the Kay lab27, was used to reveal subtle conformational dynamics of H4 residues buried at the H3-H4 interface when the nucleosome was bound to the ATP-dependent chromatin remodeling factor Snf228. The structural plasticity of the histone octamer is functionally important, since its destabilization facilitated remodeling, while constraining it with inter-histone crosslinks hindered remodeling. The second approach, HDX-MS, was used to investigate how nucleosomes containing the H3 variant CENP-A interact with two centromere-specific binding-factors to maintain centromeres29. NMR and HDX-MS have also been used to characterize the effects of histone point mutations on octamer structural dynamics30 and to study the dynamics of histone tails31-33.
Structural studies of nucleosome-binding factor complexes
Structure and dynamics are intrinsically regulated and are also affected by nucleosome-binding proteins recognizing nucleosomes in different contexts. For example, histone tail PTMs can be “read”, “written” or “erased” by protein factors, and histone variants can be incorporated or removed by specific chaperones. Nucleosome-binding proteins can stabilize or reposition the nucleosome and promote or inhibit higher-order assemblies of chromatin. Many structural studies have shed light on how these proteins are recruited to the nucleosome, and how they affect nucleosome structure and dynamics. Nucleosomes provides many unique structural features for protein recruitment, and indeed, these factors target either nucleosomal or extra-nucleosomal DNA, the histone core surface, histone tails, or combinations of the above.
Recognition of nucleosomal DNA
Nucleosomal DNA has a much higher curvature than free DNA and is partially occluded by histones and the second gyre of DNA. The unique features of nucleosomal DNA were exploited in the design of synthetic DNA binding reagents that specifically recognize nucleosomes, for example dimeric pyrrole-imidazole polyamide molecules designed in the Dervan lab34. This polyamide clamp recognizes the “super-groove” formed by two DNA gyres of the nucleosome in a site-specific manner34, and suggests a mechanism by which remodeling might move DNA by facilitating naturally occurring “twist diffusion”35.
Nucleosomal DNA is also specifically recognized by viral integrases (for example HIV-integrase), a class of proteins that insert viral DNA into nucleosomal DNA36. The cryoEM map of the prototype foamy virus (PFV) intasome (including integrase and viral DNA) at 7.8 Å resolution37 allowed unambiguous docking of the intasome and nucleosome crystal structures (Figure 3b), showing that the intasome invades DNA gyres at SHL +/−3.5, where DNA exhibits the highest curvature, and contacts the H2A C-terminal helix as well as H2B (Figure 3b)37. The gyre close to the intasome/H2A interface is captured, lifted and deformed by the intasome.
Figure 3: Structures of chromatin-binding factor complexes.

PDB entries of the structures shown in the figure. “1” indicates structures obtained by crystallography. “2” indicates structural models built from single-particle cryoEM maps. “2*” represents docking models generated to interpret cryoEM maps.
A: Proteins targeting the surface of nucleosome: a1, small protein fragments or polypeptides recognizing the acidic patch on the nucleosome surface; a2, proteins recognizing both the acidic patch and epigenetic marks on the nucleosome surface; a3, proteins binding to both histones and nucleosomal DNA on the nucleosome surface (the acidic patch also plays an important role in complex a3.1 but not in a3.2).
B: Proteins invading nucleosomal DNA gyres.
C: Proteins interacting with linker DNA.
Both polyamide clamp and intasome structures provide insights into how architecture of nucleosomal DNA can be read out by interacting factors. The most biologically important class of proteins that recognize DNA sequence are transcription factors. Most of these require their recognition sequence to be nucleosome-free. However, a class of transcription factors termed pioneer factors, exemplified by FoxA, specifically target binding sites within nucleosomal DNA and establish subsequent cooperative interactions with non-pioneer factors38. No structure of any pioneer factors in complex with a nucleosome is as yet available, despite one documented attempt for FoxA39, and it will be exciting to see at molecular detail how sequence information is decoded by these proteins in its “natural” nucleosomal context.
Linker DNA, the histone-free DNA that connects nucleosomes, is recognized by linker histone, a small group of small basic proteins (structurally unrelated to the four core histones) that re-organize the linker DNA to promote chromatin compaction40. Recent effort from several labs show that H1 simultaneously interacts with DNA at the dyad as well as with the two linker DNA arms, thereby making the nucleosome more compact (Figure 3c)41, and contributing to chromatin compaction42.
Docking onto the histone surface through the acidic patch
The histone core surface constitutes ~40% of total solvent accessible surface area (not accounting for the histone tails) in a nucleosome. The “acidic patch” on H2A-H2B has emerged as the most prominent feature recognized by most chromatin-binding proteins (Figure 1b). The acidic patch comprises six amino acid residues of H2A (in human H2A: Glu56, Glu61, Glu64, Asp90, Glu91, Glu92) and two residues of H2B (in human H2B: Glu105, Glu113) that together create a highly negatively charged groove.
A peptide derived from the latency-associated nuclear antigen (LANA) of Kaposi’s sarcoma–associated herpesvirus (KSHV) was the first protein shown to specifically recognize the acidic patch43. The side chain of Arginine, later termed Arginine anchor, is critical for the acidic patch docking. It was shown subsequently that other viral proteins also recognize this highly conserved surface through an Arginine anchor (Figure 3 a1). The nucleosome-binding domain of Human cytomegalovirus (hCMV) immediate early 1 (IE1) protein and the C-terminal sequence motif CBS (chromatin-binding sequence) of prototype foamy virus (PFV) structural protein GAG employ nearly the same binding mode as the LANA peptide (Figure 3 a1)44,45. Incidentally, the acidic patch plays an important role in chromatin compaction through its interaction with the H4 tail of a neighboring nucleosome46, and it was hypothesized that these viral proteins might use this property to regulate the higher order structure of chromatin by targeting the acidic patch44,47.
In contrast to these viral proteins that only target the acidic patch, many other nucleosome interacting proteins require additional contacts (Figure 3). For example, RCC1 (Regulator of Chromosome Condensation) additionally interacts with nucleosomal DNA facing the histone core through a DNA-binding loop and its N-terminal region (Figure 3 a3.1)48. The Sir3 (Silent information regulator) BAH (Bromo-Associated Homology) domain simultaneously recognizes the H2A/H2B acidic patch, H4 tail and a surface area comprised of amino acids from H3 and H4 (Figure 3 a3.1)49.
The reading and writing of histone tail PTMs
Histone tails are biologically important features for nucleosome recognition for the purpose of establishing and decoding PTMs that reside on them. In many cases, nucleosomal DNA and/or the acidic patch further fortifies these interactions. For example, the Spt-Ada-Gcn5 acetyltransferase (SAGA) deubiquitinating (DUB) module “reads” mono-ubiquitinated H2BK120 (ubH2B). While the catalytic lobe of the DUB module exclusively interacts with the H2B C-terminal helix containing the conjugated ubiquitin50, a basic zinc finger domain in the DUB module docks onto the H2A/H2B acidic patch (Figure 3 a2). A second example for this dual recognition mode is the tumor suppressor protein 53BP1, a reader of both H2AK15Ub and H4K20Me251. Single-particle cryoEM revealed that the “ubiquitination-dependent recruitment motif” (UDR) of 53BP1 is sandwiched between ubiquitin and the nucleosome surface and includes the H2B C-terminal helix where ubiquitin is located, as well as the acidic patch52. Both SAGA DUB module and 53BP1 adopt a “recognize and dock” mode to ensure specificity of PTM recognition (Figure 3 a2).
“Writers” deposit PTMs on specific amino acids in histone tails, which requires accurate recognition of tail residues. PRC1 (Polycomb repressive complex 1) ubiquitinates histone H2A on K119, a residue located in the C-terminal tail53. The crystal structure of the nucleosome in complex with the ubiquitination module of PRC1 shows that the active site cleft of PRC1 is positioned over the C-terminal tail of H2A near the target residue K119 in the context of the nucleosome, and several other surface features including the acidic patch, C-terminal end of the α1 helix of H3, as well as DNA support the interaction (Figure 3 a3.1). Thus, the spatial organization of the histone tail combined with other nucleosomal features ensure the specificity of histone modification, which could be a common principle for many “writers”.
Polycomb repressive complex 2 (PRC2) serves both as a “reader” and “writer” of histone PTM H3K27Me3. The cryoEM structure of PRC2 bound to a dinucleosome54 beautifully illustrates how PRC2 recognizes H3K27Me3 on one nucleosome and binds to the unmodified H3 tail of the other nucleosome through two different domains (Figure 3c). The structural organization of PRC2 subunits and their unique recognition of histone tails with and without histone PTMs match its functions in reading and writing. Interacting with DNA at the entry/exit site might be a common feature when a protein complex acts on more than one nucleosomes.
Recognition of nucleosome containing histone variants
Just like histone PTMs, histone variants serve as epigenetic marks as “special histones for special occasions”. The centromeric histone H3 variant CENP-A defines the centromere by recruiting kinetochore proteins55. CENP-C and CENP-N, two inner kinetochore proteins, are “readers” of CENP-A nucleosomes. Employing similar strategies as PTM readers, CENP-C specifically recognizes the unique motif on C-terminal tail of CENP-A as well as the acidic patch (Figure 3 a2)56. In contrast, CENP-N (which acts in conjunction with CENP-C) recognizes a CENP-A-specific loop on the nucleosome surface as well as forms an extensive interface with the adjacent nucleosomal DNA (Figure 3 a3.2)57-59. These multivalent contacts between inner kinetochore proteins and the centromeric nucleosome deliver high fidelity of kinetochore assembly at the centromere.
Invading and remodeling nucleosome by multiple recognitions
SWI/SNF, ISWI, CHD, and INO80 are ATP-dependent chromatin remodeling factors that reposition or restructure the nucleosome through ATP hydrolysis60. Recently, a deluge of structures has provided unprecedented insight into the mechanism by which these complexes function. In order to move the DNA relative to the histone core, these machines need to bind both histone and DNA at distinct sites on the nucleosome. Snf2 (the ATPase motor common to the SWI/SNF family of remodelers) from Saccharomyces cerevisiae interacts with one DNA gyre near the nucleosomal dyad (at SHL +2; Figure 1c) through its primary DNA-binding domain, while the other DNA gyre is contacted by the secondary DNA binding domain at the DNA exit site at SHL −6 (Figure 3b). The H4 tail, which is essential for Snf2 function, also directly interacts with Snf2, further stabilizing the interaction61. Embracing or invading DNA gyres is a key signature for nucleosome remodeling.
The structure of the Chd1-nucleosome complex in the presence of an ATP analogue, solved by single particle cryoEM, displays a similar principle. The Chd1 ATPase domain adopts a similar binding mode as Snf2 by docking onto SHL +2 and the H4 tail62,63, but additionally contacts the DNA at SHL +1 and the detached DNA at SHL −7 (Figure 3b). Unlike the partially closed ATPase domain observed in the Snf2-nucleosome complex (in the absence of ATP), the Chd1-nucleosome complex in the presence of an ATP analogue shows an entirely closed conformation as well as a one base pair offset on DNA in the direction of translocation. Since hydrolysis of ATP results in the dissociation of ADP from the ATPase domain to reset it to the pre-translocation state, these two structures provide insight into how ATP hydrolysis might drive remodeling.
The ATPase of INO80 does not embrace DNA at SHL +2, but rather at SHL −6, and disrupts the H2A/DNA interaction by unwrapping about 15 bp of DNA (Figure 3b)64,65. This unique binding mode enables INO80 to pump DNA into the nucleosome, forming a DNA loop. Other subunits of INO80 grip the DNA at SHL −2 and SHL −3 as well as the acidic patches on both sides of the nucleosome, and this serves as a counter grip for the ATPase. These interactions provide an anchor on the histone octamer during translocation and likely prevent complete unwrapping of nucleosomal DNA. The different binding modes observed for Snf2 and INO80 result in different remodeling mechanisms.
Recent structures from X-ray crystallography and single particle cryoEM provide deep insight into the structural basis of nucleosome recognition and consequences for nucleosome structure and dynamics. However, structural characterization of interactions between flexible or dynamic regions has remained challenging. NMR provides a powerful complementary method. For example, RNF169, the reader of H2A[K13Ub, K15Ub], has a disordered region at the C-terminus of a helix that binds to the canonical site in ubiquitin, which could not be observed in the cryoEM map66. The methyl-TROSY NMR spectra exhibit clear chemical shift changes for residues in the flexible region of RNF169 which interact with the acidic patch. The structure of RNF169 ubiquitin-dependent recruitment module 2 (UDM2)-ubNucleosome was modeled from molecular dynamics simulations constrained by chemical shift perturbations (CSPs), mutagenesis data and the cryoEM map, in an example of the power of hybrid approaches to tackle difficult questions in structural biology.
Concluding remarks
The nucleosome is no longer considered a simple barrier that blocks access to DNA during transcription and replication. Rather, it serves as a dynamic platform linking and integrating many biological processes. Therefore, investigating the structural dynamics of nucleosomes is key to understand how they regulate genome accessibility. Current methods including single-molecule FRET, SAXS and AFM provide detailed mechanistic insight into the dynamic behavior of this complicated assembly. These approaches also shed light on how PTMs and histone variants intrinsically affect nucleosome structure and dynamics. With the same approaches, the effects of chromatin-binding proteins on nucleosomes are also evaluated.
Recent technical progress has made single-particle cryoEM a powerful and feasible tool for structural studies of nucleosome-binding factors in complex with nucleosomes. However, accurate de novo building of atomic models for nucleosome-binding proteins only based on cryoEM maps is still challenging. The fitting of available crystal structures into cryoEM maps is the most popular method of obtaining structural information on macromolecular assemblies. Considering the limitations of crystallography and cryoEM for factors with large disordered regions, NMR fills a niche to characterize the dynamic properties of these complexes. Integrative structural biology approaches combining crystallography, cryoEM, NMR, and molecular dynamics simulations will provide a comprehensive understanding of chromatin-binding proteins and their dynamic interactions with nucleosomes. Structure determination (Box 1) and methods probing structural dynamics (Boxes 2 and 3) are now frequently used in combination to provide integrated models of the structure and dynamics of nucleosome complexes, as exemplified by recent complementary studies of the chromatin remodelers Chd167,68,62, INO8064,65,69 and Snf261,28,70.
Box 1: Structure determination methods.
Electron microscopy (EM) has been used since the earliest days of chromatin structural biology, famously uncovering the “beads on a string” structure of the 10 nm chromatin fiber from negatively stained chromatin spreads74,75. Due to the limited resolution of EM at that time, nucleosome structural biology has been dominated by X-ray crystallography. A major advantage of crystallography is its potential to obtain near-atomic resolution (for nucleosomes 1.9 Å, PDB entry 1KX576), but this requires well-diffracting crystals and the phase problem must be solved. While the latter is no longer a bottleneck for nucleosomes, obtaining well-diffracting crystals is still challenging due to high sample consumption, and because many nucleosome complexes are dynamic and populate different conformational states. An intrinsic limitation of crystallography is that it provides very little information about structural dynamics.
The recent “resolution revolution” in cryoEM77 eliminated these limitations, while creating new ones. CryoEM consumes less sample and gives intermediate results more rapidly, providing rational ways to optimize sample and grid preparation. A single dataset can reveal several conformations. However, not all complexes remain stable upon vitrification, and picking and classifying particles from noisy images is a computational challenge. Access to high-end instrumentation can also be limiting. Nevertheless, the nucleosome has proved to be a tractable target for single-particle cryoEM. In 2016, a cryoEM map of a nucleosome reached a resolution of better than 4 Å78, a significant improvement over previous cryoEM maps that were limited to ~7 Å resolution. The application of cryoEM to nucleosomes and chromatin has recently been reviewed79.
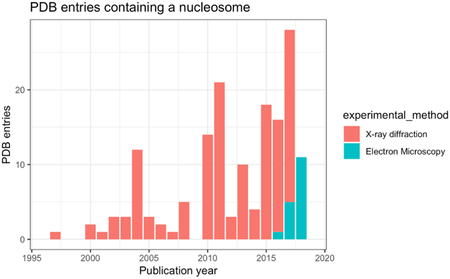
At the time of writing this review, in 2018 alone, eleven cryoEM structures of nucleosome complexes have been deposited in the PDB (along with a few unmodeled cryoEM maps in the EMDB). CryoEM is becoming the default method in nucleosome structural biology, for its typical resolution range of 4 to 10 Å is often sufficient for the large complexes for which higher resolution structures of subunits or domains have been solved by crystallography. Crystallography will remain the method of choice for high-resolution (< 3 Å) studies of smaller components, or of structures with small-molecule compounds. Together, these two complementary methods are a winning team to tackle the most difficult problems in structural biology.
Box 3: Single-molecule methods.
Single-molecule methods can uncover transient intermediates that would be undetectable in an ensemble. These methods fall into three categories: detection, manipulation and imaging.
Single-molecule detection always involves fluorescent labeling to achieve the necessary sensitivity. Either confocal fluorescence microscopes or total internal reflection fluorescence (TIRF) microscopes are used. Detection of a single fluorophore provides information about translational and rotational diffusion and the method can monitor binding events if they affect the properties of the fluorophore. More precise mechanistic information can be obtained by using an intramolecular FRET pair and detecting single-molecule FRET bursts generated by a conformational change, happening either spontaneously or triggered by a binding event. With an appropriate labeling strategy, single-molecule FRET can detect and distinguish subtle conformational changes in nucleosomes, for example DNA sliding or breathing across gyres97. Measurements of at least three distances between known locations and a part of a macromolecular complex whose location is unknown allows to pinpoint by triangulation the possible locations of the unknown part (nano-positioning system)98.
Single-molecule manipulation is achieved with optical or magnetic tweezers. In both cases, one DNA end is tethered to a surface and the other end is tethered to a bead. In an optical tweezer setup, the bead is held by a laser beam, and the surface is moved to exert tension99. In a magnetic tweezer setup, this bead is a magnet, and tension100 or torsion101 is applied by a tunable electromagnet. Both setups enable force spectroscopy measurements, monitoring DNA length as a function of increasing tension, or disruption of histone/DNA interactions during DNA unzipping at constant force102. Single-molecule manipulation can be combined with FRET detection in a powerful approach for monitoring changes of specific intramolecular distances as a function of increasing force21.
Atomic Force Microscopy (AFM) allows single-molecule imaging and manipulation. It has been used to study nucleosomes and nucleosome-binding factors, with recent examples including a dinucleosome103, linker histone H1104 and CENP-A nucleosome105. Recently, scanning speed has increased enough to allow time-resolved imaging106,107. A more detailed review of single-molecule methods applied to nucleosomes was published elsewhere108.
Research focused on single nucleosomes provides only a limited perspective. It is still unclear whether PTMs, histone variants and chromatin-binding proteins affect chromatin arrays in the same way as they do single nucleosomes. This represents the next frontier in chromatin structural biology. Current work on chromatin usually involves reconstitution of nucleosome arrays in which every nucleosome has the same composition. Innovative methods have to be developed to assemble nucleosomal arrays in which each nucleosome has a determined set of PTMs, histone variants or binding factors at defined positions. Similar to protein structure and function, which is determined by amino acid sequence, chromatin structure and function might also be determined by the “sequence” of nucleosomes carrying various modifications or histone variants. Thus, evaluating and visualizing nucleosome structure and dynamics in a more natural context will be critical to understand the molecular basis of how nucleosomes behave and are recognized within nuclear chromatin. Ambitious research in this direction has already started71-73.
Acknowledgments
We apologize to the many researchers whose work we could not cite due to space constraints. We thank our colleagues Jonathan W. Markert and Dr. Uma M. Muthurajan for critical reading of this review. Research in the Luger Lab is supported by the Howard Hughes Medical Institute and by the NIH (CA218255).
References
- 1.Woodcock CLF, Safer JP & Stanchfield JE Structural repeating units in chromatin. I. Evidence for their general occurrence. Exp. Cell Res 97, 101–110 (1976). [DOI] [PubMed] [Google Scholar]
- 2.Kornberg RD Chromatin Structure: A Repeating Unit of Histones and DNA. Science (80-. ). 184, 868–871 (1974). [DOI] [PubMed] [Google Scholar]
- 3.Richmond TJ, Finch JT, Rushton B, Rhodes D & Klug A Structure of the nucleosome core particle at 7 resolution. Nature 311, 532–537 (1984). [DOI] [PubMed] [Google Scholar]
- 4.Arents G, Burlingame RW, Wang BC, Love WE & Moudrianakis EN The nucleosomal core histone octamer at 3.1 A resolution: a tripartite protein assembly and a left-handed superhelix. Proc. Natl. Acad. Sci 88, 10148–10152 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luger K, Mäder AW, Richmond RK, Sargent DF & Richmond TJ Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389, 251–260 (1997).First high-resolution structure of the nucleosome.
- 6.Iwasaki W et al. Contribution of histone N-terminal tails to the structure and stability of nucleosomes. FEBS Open Bio 3, 363–9 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li G & Widom J Nucleosomes facilitate their own invasion. Nat. Struct. Mol. Biol 11, 763–769 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Poirier MG, Bussiek M, Langowski J & Widom J Spontaneous Access to DNA Target Sites in Folded Chromatin Fibers. J. Mol. Biol 379, 772–786 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Poirier MG, Oh E, Tims HS & Widom J Dynamics and function of compact nucleosome arrays. Nat. Struct. Mol. Biol 16, 938–944 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li G, Levitus M, Bustamante C & Widom J Rapid spontaneous accessibility of nucleosomal DNA. Nat. Struct. Mol. Biol 12, 46–53 (2005).First determination of the rate constants of spontaneous nucleosome unwrapping and rewrapping.
- 11.Shimko JC, North JA, Bruns AN, Poirier MG & Ottesen JJ Preparation of Fully Synthetic Histone H3 Reveals That Acetyl-Lysine 56 Facilitates Protein Binding Within Nucleosomes. J. Mol. Biol 408, 187–204 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.North JA et al. Regulation of the nucleosome unwrapping rate controls DNA accessibility. Nucleic Acids Res. 40, 10215–10227 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simon M et al. Histone fold modifications control nucleosome unwrapping and disassembly. Proc. Natl. Acad. Sci 108, 12711–12716 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brehove M et al. Histone Core Phosphorylation Regulates DNA Accessibility. J. Biol. Chem 290, 22612–22621 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bowman GD & Poirier MG Post-Translational Modifications of Histones That Influence Nucleosome Dynamics. Chem. Rev 115, 2274–2295 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nadal S, Raj R, Mohammed S & Davis BG Synthetic post-translational modification of histones. Curr. Opin. Chem. Biol 45, 35–47 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Dong F, Hansen JC & Holde KE van. DNA and protein determinants of nucleosome positioning on sea urchin 5S rRNA gene sequences in vitro. Proc. Natl. Acad. Sci 87, 5724–5728 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lowary P . & Widom J New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol 276, 19–42 (1998). [DOI] [PubMed] [Google Scholar]
- 19.Tóth K et al. Histone- and DNA sequence-dependent stability of nucleosomes studied by single-pair FRET. Cytom. Part A 83, 839–846 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Chen Y et al. Revealing transient structures of nucleosomes as DNA unwinds. Nucleic Acids Res. 42, 8767–8776 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ngo TTM, Zhang Q, Zhou R, Yodh JG & Ha T Asymmetric Unwrapping of Nucleosomes under Tension Directed by DNA Local Flexibility. Cell 160, 1135–1144 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Y et al. Asymmetric unwrapping of nucleosomal DNA propagates asymmetric opening and dissociation of the histone core. Proc. Natl. Acad. Sci 114, 334–339 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shlyakhtenko LS, Lushnikov AY & Lyubchenko YL Dynamics of Nucleosomes Revealed by Time-Lapse Atomic Force Microscopy. Biochemistry 48, 7842–7848 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bilokapic S, Strauss M & Halic M Histone octamer rearranges to adapt to DNA unwrapping. Nat. Struct. Mol. Biol 25, 101 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kato D et al. Crystal structure of the overlapping dinucleosome composed of hexasome and octasome. Science (80-. ). 356, 205–208 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Henikoff JG, Belsky JA, Krassovsky K, MacAlpine DM & Henikoff S Epigenome characterization at single base-pair resolution. Proc. Natl. Acad. Sci 108, 18318–18323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kato H et al. Architecture of the high mobility group nucleosomal protein 2-nucleosome complex as revealed by methyl-based NMR. Proc. Natl. Acad. Sci 108, 12283–12288 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sinha KK, Gross JD & Narlikar GJ Distortion of histone octamer core promotes nucleosome mobilization by a chromatin remodeler. Science (80-. ). 355, eaaa3761 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo LY et al. Centromeres are maintained by fastening CENP-A to DNA and directing an arginine anchor-dependent nucleosome transition. Nat. Commun 8, 15775 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kitevski-LeBlanc JL et al. Investigating the Dynamics of Destabilized Nucleosomes Using Methyl-TROSY NMR. J. Am. Chem. Soc (2018). doi: 10.1021/jacs.8b00931 [DOI] [PubMed] [Google Scholar]
- 31.Zhou B-R et al. Histone H4 K16Q Mutation, an Acetylation Mimic, Causes Structural Disorder of Its N-Terminal Basic Patch in the Nucleosome. J. Mol. Biol 421, 30–37 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gao M et al. Histone H3 and H4 N-Terminal Tails in Nucleosome Arrays at Cellular Concentrations Probed by Magic Angle Spinning NMR Spectroscopy. J. Am. Chem. Soc 135, 15278–15281 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karch KR et al. Hydrogen-deuterium exchange coupled to top- and middle-down mass spectrometry enables high-resolution measurements of histone tail dynamics before and after nucleosome assembly. bioRxiv 310177 (2018). doi: 10.1101/310177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Edayathumangalam RS, Weyermann P, Gottesfeld JM, Dervan PB & Luger K Molecular recognition of the nucleosomal ‘supergroove’. Proc. Natl. Acad. Sci. U. S. A 101, 6864–6869 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Edayathumangalam RS, Weyermann P, Dervan PB, Gottesfeld JM & Luger K Nucleosomes in solution exist as a mixture of twist-defect states. J. Mol. Biol 345, 103–114 (2005). [DOI] [PubMed] [Google Scholar]
- 36.Pryciak PM & Varmus HE Nucleosomes, DNA-binding proteins, and DNA sequence modulate retroviral integration target site selection. Cell 69, 769–780 (1992). [DOI] [PubMed] [Google Scholar]
- 37.Maskell DP et al. Structural basis for retroviral integration into nucleosomes. Nature 523, 366–369 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zaret KS, Lerner J & Iwafuchi-Doi M Chromatin Scanning by Dynamic Binding of Pioneer Factors. Mol. Cell 62, 665–667 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takizawa Y et al. Cryo-EM structure of the nucleosome containing the ALB1 enhancer DNA sequence. Open Biol. 8, 170255 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tremethick DJ Higher-Order Structures of Chromatin: The Elusive 30 nm Fiber. Cell 128, 651–654 (2007). [DOI] [PubMed] [Google Scholar]
- 41.Bednar J et al. Structure and Dynamics of a 197 bp Nucleosome in Complex with Linker Histone H1. Mol. Cell 66, 384–397.e8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Song F et al. Cryo-EM Study of the Chromatin Fiber Reveals a Double Helix Twisted by Tetranucleosomal Units. Science (80-. ). 344, 376–380 (2014). [DOI] [PubMed] [Google Scholar]
- 43.Barbera AJ et al. The Nucleosomal Surface as a Docking Station for Kaposi’s Sarcoma Herpesvirus LANA. Science (80-. ). 311, 856–861 (2006).First crystal structure of a chromatin-binding factor in complex with the nucleosome.
- 44.Fang Q et al. Human cytomegalovirus IE1 protein alters the higher-order chromatin structure by targeting the acidic patch of the nucleosome. Elife 5, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lesbats P et al. Structural basis for spumavirus GAG tethering to chromatin. Proc. Natl. Acad. Sci 114, 5509–5514 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kalashnikova AA, Porter-Goff ME, Muthurajan UM, Luger K & Hansen JC The role of the nucleosome acidic patch in modulating higher order chromatin structure. J. R. Soc. Interface 10, 20121022–20121022 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chodaparambil JV et al. A charged and contoured surface on the nucleosome regulates chromatin compaction. Nat. Struct. Mol. Biol 14, 1105–1107 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Makde RD, England JR, Yennawar HP & Tan S Structure of RCC1 chromatin factor bound to the nucleosome core particle. Nature 467, 562–566 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Armache K-J, Garlick JD, Canzio D, Narlikar GJ & Kingston RE Structural Basis of Silencing: Sir3 BAH Domain in Complex with a Nucleosome at 3.0 Å Resolution. Science (80-. ). 334, 977–982 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morgan MT et al. Structural basis for histone H2B deubiquitination by the SAGA DUB module. Science (80-. ). 351, 725–728 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fradet-Turcotte A et al. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature 499, 50–54 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wilson MD et al. The structural basis of modified nucleosome recognition by 53BP1. Nature 536, 100–103 (2016).First near-atomic resolution structure of a chromatin-binding factor in complex with nucleosome by single-particle cryoEM.
- 53.McGinty RK, Henrici RC & Tan S Crystal structure of the PRC1 ubiquitylation module bound to the nucleosome. Nature 514, 591–596 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Poepsel S, Kasinath V & Nogales E Cryo-EM structures of PRC2 simultaneously engaged with two functionally distinct nucleosomes. Nat. Struct. Mol. Biol 25, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stellfox ME, Bailey AO & Foltz DR Putting CENP-A in its place. Cell. Mol. Life Sci 70, 387–406 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kato H et al. A Conserved Mechanism for Centromeric Nucleosome Recognition by Centromere Protein CENP-C. Science (80-. ). 340, 1110–1113 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pentakota S et al. Decoding the centromeric nucleosome through CENP-N. Elife 6, e33442 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chittori Sagar, Hong Jingjun, Saunders Hayden, Feng Hanqiao, Ghirlando Rodolfo, Kelly Alexander E., Bai Yawen, S. S. Structural mechanisms of centromeric nucleosome recognition by the kinetochore protein CENP-N. Science (80-. ). (2017). doi: 10.1126/science.aar2781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tian T et al. Molecular basis for CENP-N recognition of CENP-A nucleosome on the human kinetochore. Cell Res. 1–5 (2018). doi: 10.1038/cr.2018.13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Clapier CR & Cairns BR The Biology of Chromatin Remodeling Complexes. Annu. Rev. Biochem 78, 273–304 (2009). [DOI] [PubMed] [Google Scholar]
- 61.Liu X, Li M, Xia X, Li X & Chen Z Mechanism of chromatin remodelling revealed by the Snf2-nucleosome structure. Nature 544, 440–445 (2017). [DOI] [PubMed] [Google Scholar]
- 62.Farnung L, Vos SM, Wigge C & Cramer P Nucleosome–Chd1 structure and implications for chromatin remodelling. Nature 550, 539–542 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sundaramoorthy R, Hughes A, El-Mkami H, Norman D & Owen-Hughes T Structure of the chromatin remodelling enzyme Chd1 bound to a ubiquitinylated nucleosome. bioRxiv 290874 (2018). doi: 10.1101/290874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Eustermann S et al. Structural basis for ATP-dependent chromatin remodelling by the INO80 complex. Nature 556, 386–390 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ayala R et al. Structure and regulation of the human INO80–nucleosome complex. Nature 556, 391–395 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kitevski-LeBlanc J et al. The RNF168 paralog RNF169 defines a new class of ubiquitylated histone reader involved in the response to DNA damage. Elife 6, e23872 (2017).Complementary use of NMR and cryoEM.
- 67.Sundaramoorthy R et al. Structural reorganization of the chromatin remodeling enzyme Chd1 upon engagement with nucleosomes. Elife 6, e22510 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tokuda JM et al. The ATPase motor of the Chd1 chromatin remodeler stimulates DNA unwrapping from the nucleosome. Nucleic Acids Res. 46, 4978–4990 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schwarz M et al. Single-molecule nucleosome remodeling by INO80 and effects of histone tails. FEBS Lett. 592, 318–331 [DOI] [PubMed] [Google Scholar]
- 70.Gamarra N, Johnson SL, Trnka MJ, Burlingame AL & Narlikar GJ The nucleosomal acidic patch relieves auto-inhibition by the ISWI remodeler SNF2h. Elife 7, e35322 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ou HD et al. ChromEMT: Visualizing 3D chromatin structure and compaction in interphase and mitotic cells. Science (80-. ). 357, eaag0025 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dekker J et al. The 4D nucleome project. Nature 549, 219–226 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Eltsov M et al. Nucleosome conformational variability in solution and in interphase nuclei evidenced by cryo-electron microscopy of vitreous sections. Nucleic Acids Res. (2018). doi: 10.1093/nar/gky670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Oudet P, Gross-Bellard M & Chambon P Electron microscopic and biochemical evidence that chromatin structure is a repeating unit. Cell 4, 281–300 (1975). [DOI] [PubMed] [Google Scholar]
- 75.Frado L-LY, Annunziato AT & Woodcock CLF Structural repeating units in chromatin III. A comparison of chromatin subunits from vertebrate, ciliate and angiosperm species. Biochim. Biophys. Acta - Nucleic Acids Protein Synth 475, 514–520 (1977). [DOI] [PubMed] [Google Scholar]
- 76.Davey CA, Sargent DF, Luger K, Maeder AW & Richmond TJ Solvent Mediated Interactions in the Structure of the Nucleosome Core Particle at 1.9Å Resolution. J. Mol. Biol 319, 1097–1113 (2002). [DOI] [PubMed] [Google Scholar]
- 77.Bai X, McMullan G & Scheres SHW How cryo-EM is revolutionizing structural biology. Trends Biochem. Sci 40, 49–57 (2015). [DOI] [PubMed] [Google Scholar]
- 78.Chua EYD et al. 3.9 Å structure of the nucleosome core particle determined by phase-plate cryo-EM. Nucleic Acids Res. 44, 8013–8019 (2016).First high-resolution (better than 4.0 Å) single-particle cryoEM map of a nucleosome.
- 79.Wilson MD & Costa A Cryo-electron microscopy of chromatin biology. Acta Crystallogr. Sect. D Struct. Biol 73, 541–548 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Receveur-Brechot V & Durand D How Random are Intrinsically Disordered Proteins? A Small Angle Scattering Perspective. Curr. Protein Pept. Sci 13, 55–75 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bertin A, Durand D, Renouard M, Livolant F & Mangenot S H2A and H2B tails are essential to properly reconstitute nucleosome core particles. Eur. Biophys. J 36, 1083–1094 (2007). [DOI] [PubMed] [Google Scholar]
- 82.Yang C et al. Biophysical analysis and small-angle X-ray scattering-derived structures of MeCP2–nucleosome complexes. Nucleic Acids Res. 39, 4122–4135 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Arimura Y, Tachiwana H, Oda T, Sato M & Kurumizaka H Structural Analysis of the Hexasome, Lacking One Histone H2A/H2B Dimer from the Conventional Nucleosome. Biochemistry 51, 3302–3309 (2012). [DOI] [PubMed] [Google Scholar]
- 84.Sugiyama M et al. Solution structure of variant H2A.Z.1 nucleosome investigated by small-angle X-ray and neutron scatterings. Biochem. Biophys. Reports 4, 28–32 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tokuda JM, Pabit SA & Pollack L Protein–DNA and ion–DNA interactions revealed through contrast variation SAXS. Biophys. Rev 8, 139–149 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tóth K, Brun N & Langowski J Trajectory of Nucleosomal Linker DNA Studied by Fluorescence Resonance Energy Transfer. Biochemistry 40, 6921–6928 (2001). [DOI] [PubMed] [Google Scholar]
- 87.Park Y-J, Dyer PN, Tremethick DJ & Luger K A new fluorescence resonance energy transfer approach demonstrates that the histone variant H2AZ stabilizes the histone octamer within the nucleosome. J. Biol. Chem 279, 24274–82 (2004). [DOI] [PubMed] [Google Scholar]
- 88.Buning R & van Noort J Single-pair FRET experiments on nucleosome conformational dynamics. Biochimie 92, 1729–1740 (2010). [DOI] [PubMed] [Google Scholar]
- 89.Zhang H & van Ingen H Isotope-labeling strategies for solution NMR studies of macromolecular assemblies. Curr. Opin. Struct. Biol 38, 75–82 (2016). [DOI] [PubMed] [Google Scholar]
- 90.Liokatis S Reconstitution of Nucleosomes with Differentially Isotope-labeled Sister Histones. JoVE (Journal Vis. Exp e55349–e55349 (2017). doi: 10.3791/55349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hansen DF, Feng H, Zhou Z, Bai Y & Kay LE Selective Characterization of Microsecond Motions in Proteins by NMR Relaxation. J. Am. Chem. Soc 131, 16257–16265 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kay LE New Views of Functionally Dynamic Proteins by Solution NMR Spectroscopy. J. Mol. Biol 428, 323–331 (2016). [DOI] [PubMed] [Google Scholar]
- 93.Xiang S et al. Site-Specific Studies of Nucleosome Interactions by Solid-State NMR Spectroscopy. Angew. Chemie Int. Ed 57, 4571–4575 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shi X et al. Structure and Dynamics in the Nucleosome Revealed by Solid-State NMR. Angew. Chemie Int. Ed 57, 9734–9738 (2018). [DOI] [PubMed] [Google Scholar]
- 95.Oganesyan I, Lento C & Wilson DJ Contemporary hydrogen deuterium exchange mass spectrometry. Methods 144, 27–42 (2018). [DOI] [PubMed] [Google Scholar]
- 96.Makowski MM et al. Global profiling of protein–DNA and protein–nucleosome binding affinities using quantitative mass spectrometry. Nat. Commun 9, 1653 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Falk SJ et al. CENP-C directs a structural transition of CENP-A nucleosomes mainly through sliding of DNA gyres. Nat. Struct. Mol. Biol 23, 204–8 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Muschielok A et al. A nano-positioning system for macromolecular structural analysis. Nat. Methods 5, 965–971 (2008). [DOI] [PubMed] [Google Scholar]
- 99.Mihardja S, Spakowitz AJ, Zhang Y & Bustamante C Effect of force on mononucleosomal dynamics. Proc. Natl. Acad. Sci 103, 15871–15876 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chien F-T & van der Heijden T Characterization of Nucleosome Unwrapping within Chromatin Fibers Using Magnetic Tweezers. Biophys. J 107, 373–383 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Vlijm R, Kim SH, De Zwart PL , Dalal Y & Dekker C The supercoiling state of DNA determines the handedness of both H3 and CENP-A nucleosomes. Nanoscale 9, 1862–1870 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hall MA et al. High-resolution dynamic mapping of histone-DNA interactions in a nucleosome. Nat. Struct. Mol. Biol 16, 124–129 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Filenko NA, Palets DB & Lyubchenko YL Structure and Dynamics of Dinucleosomes Assessed by Atomic Force Microscopy. J. Amino Acids (2012). doi: 10.1155/2012/650840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.White AE, Hieb AR & Luger K A quantitative investigation of linker histone interactions with nucleosomes and chromatin. Sci. Rep 6, 19122 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Stumme-Diers MP, Banerjee S, Hashemi M, Sun Z & Lyubchenko YL Nanoscale dynamics of centromere nucleosomes and the critical roles of CENP-A. Nucleic Acids Res. (2017). doi: 10.1093/nar/gkx933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Miyagi A, Ando T & Lyubchenko YL Dynamics of Nucleosomes Assessed with Time-Lapse High-Speed Atomic Force Microscopy. Biochemistry 50, 7901–7908 (2011). [DOI] [PubMed] [Google Scholar]
- 107.Katan AJ, Vlijm R, Lusser A & Dekker C Dynamics of Nucleosomal Structures Measured by High-Speed Atomic Force Microscopy. Small 11, 976–984 (2015). [DOI] [PubMed] [Google Scholar]
- 108.Ordu O, Lusser A & Dekker NH Recent insights from in vitro single-molecule studies into nucleosome structure and dynamics. Biophys. Rev 8, 33–49 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Schalch T, Duda S, Sargent DF & Richmond TJ X-ray structure of a tetranucleosome and its implications for the chromatin fibre. Nature 436, 138–141 (2005). [DOI] [PubMed] [Google Scholar]