

Summary:
Approximately one-third of the world’s population suffers from allergies1. Allergen exposure crosslinks mast cell- and basophil-bound immunoglobulin E (IgE), triggering the release of inflammatory mediators, including histamine2. Although IgE is absolutely required for allergies, it is not understood why total and allergen-specific IgE concentrations do not reproducibly correlate with allergic disease3–5. It is well-established that glycosylation of IgG dictates its effector function and has disease-specific patterns. However, whether IgE glycans differ in disease states or impact biological activity is completely unknown6. We therefore unbiasedly examined glycosylation patterns of total IgE from peanut-allergic and non-atopic individuals. This revealed an increase in sialic acid content on total IgE from peanut-allergic individuals compared to non-atopic subjects. Sialic acid removal from IgE attenuated effector cell degranulation and anaphylaxis in multiple functional models of allergic disease. Therapeutic interventions, including sialic acid removal from cell-bound IgE with a FcεRI targeted-neuraminidase, or administration of asialylated IgE, markedly reduced anaphylaxis. Together, these results establish IgE glycosylation, and specifically sialylation, as an important regulator of allergic disease.
IgE antibodies bind to the surface of mast cells or basophils that express the IgE high affinity receptor, FcεRI2. Subsequent exposure to allergen crosslinks cell-bound IgE, leading to cellular activation and release of allergic mediators including histamine, prostaglandins, and leukotrienes2. This cascade culminates in the canonical symptoms of allergic diseases, the most severe of which is anaphylaxis. While IgE that recognizes otherwise innocuous allergens is well established as the causative agent of most allergic diseases1,2, clinical allergy diagnostics remain relatively inaccurate3–5, and curative therapies, including oral immunotherapy, are cumbersome, and only partially effective7,8. Further, allergen-specific IgE is detected in many people who do not experience allergic symptoms3,5. Thus, while IgE is absolutely necessary for triggering the allergic cascade, it is not clear how IgE causes allergic disease in some circumstances and not others.
The composition of the single N-linked glycan on IgG antibodies profoundly influences its biological activity, and impacts the outcome of many diseases, including Dengue hemorrhagic fever9, Mycobacterium tuberculosis latency10, Influenza vaccination11, rheumatoid arthritis6,12, and granulomatosis with polyangiitis13,14. There are seven asparagine (N)-linked glycosylation sites distributed across the heavy chains of human IgE (hIgE)6,15. However, whether particular IgE glycans are associated with allergic diseases, or impact IgE function, is unknown. IgE is the least abundant antibody class in circulation, and, as such, analysis of hIgE glycosylation has been restricted to samples from subjects with myelomas, hyper IgE syndromes, hyperimmune syndromes pooled from multiple donors, or recombinant IgE15–18. These studies revealed a single N-linked oligomannose glycan at N394 on IgE, N383 is unoccupied, and the remaining five sites are occupied by complex antennary glycans (Fig. 1a). Previously, the importance of glycans to IgE biology has been examined through glycosidase-treatments17,19 and mutation of glycosylation sites17,20. This revealed the N394 oligomannose was required for appropriate IgE folding and FcεRI binding17,20 to initiate effector functions.
Fig. 1 |. Glycan composition of non-atopic and allergic IgE.
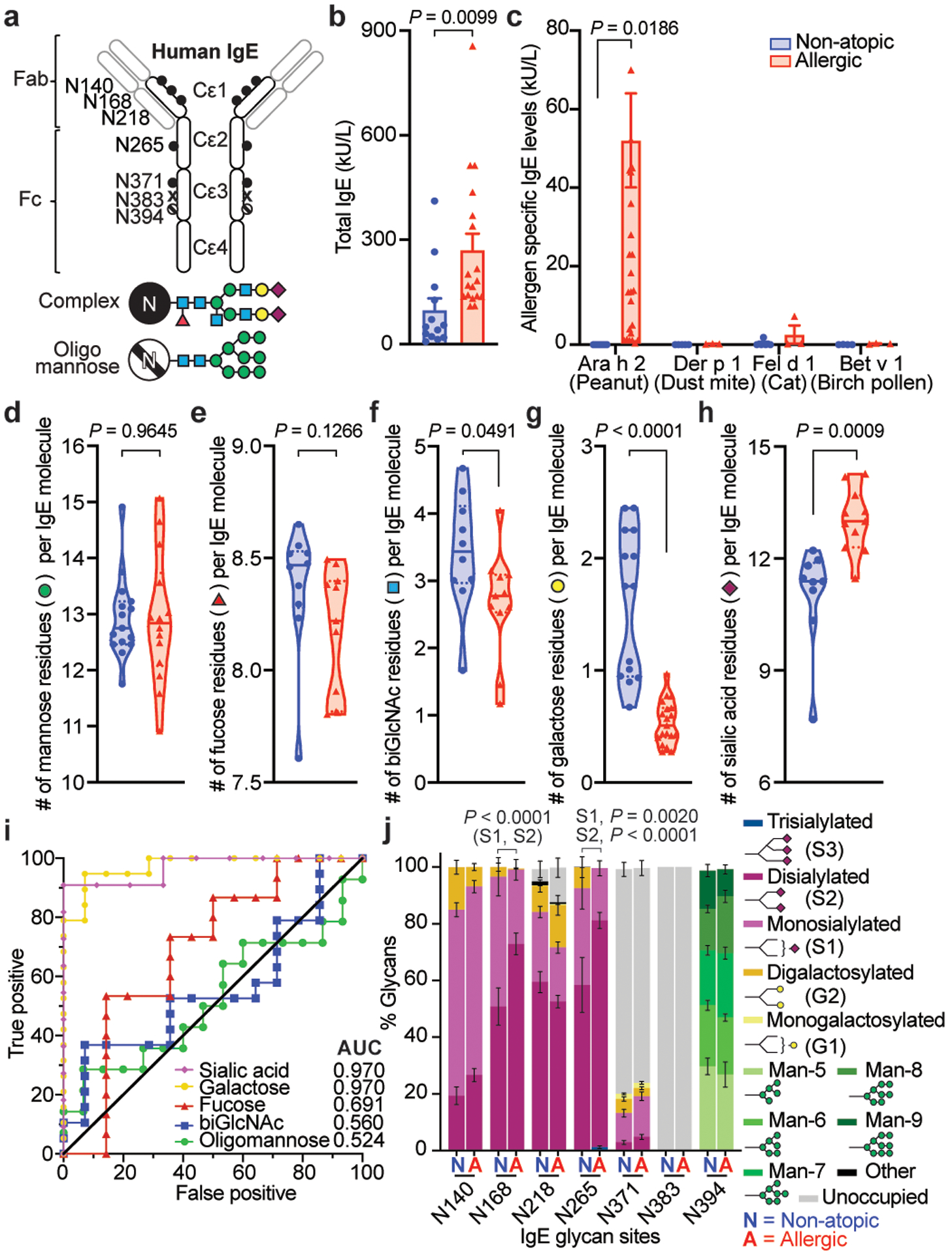
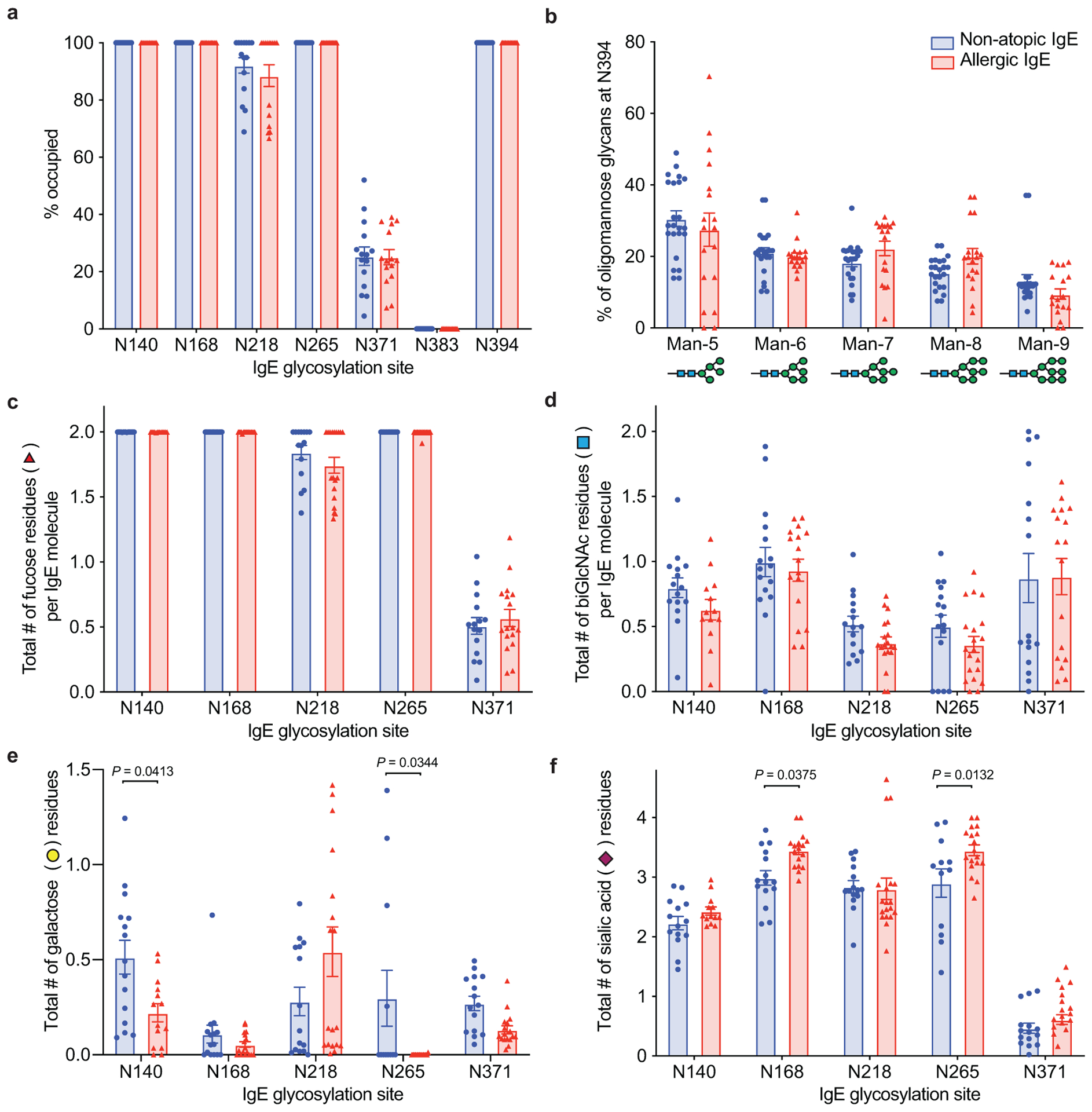
a, Human IgE N-linked glycosylation sites: complex biantennary glycans closed circles, oligomannose hatched circles, unoccupied X; blue squares, GlcNAc; green circles, mannose; red triangle, fucose; yellow circles, galactose; maroon diamonds, sialic acid. b, c, Total IgE titers (b) and allergen-specific IgE levels (c) in non-atopic (blue, n = 17) and allergic (red, n = 13) subjects. d-h, gMS quantified glycan moieties per IgE molecule in non-atopic (blue) and peanut allergic (red) individuals; mannose (d, non-atopic n = 15, allergic n = 14), fucose (e, non-atopic n = 10, allergic n = 11), biGlcNAc (f, non-atopic n = 10, allergic n = 11), galactose (g, non-atopic n = 14, allergic n = 19), and sialic acid (h, non-atopic n = 9, allergic n = 11). i, ROC for total IgE glycan moieties isolated from allergic versus non-atopic subjects. Sialic acid (non-atopic n = 9, allergic n = 11); galactose (non-atopic n = 14, allergic n = 19); fucose (non-atopic n = 14, allergic n = 15); biGlcNAc (non-atopic n = 14, allergic n = 19); oligomannose (non-atopic n = 15, allergic n = 14). j, gMS analysis of site-specific N-glycan structures on total IgE from non-atopic (N; N140 n = 11, N168 n = 13, N218 n = 11, N265, N371, N394 n = 12) and allergic (A; N140 n = 11, N168 n = 15, N218 n = 17, N265 n = 18, N371 n = 14, N394 n = 12) individuals. Representative glycan structures per group are detailed in Extended Data Fig. 1h. Data are mean ± s.e.m. (b, c, j), median (solid line) and interquartile range (dotted line) (d-h); two-tailed unpaired t-test (b, d-h), two-way ANOVA with Sidak’s (c) or Tukey’s multiple comparison test (j).
Here, we asked whether allergic disease-specific glycosylation patterns existed for IgE, and if so, whether those patterns influenced IgE biological activity. Subjects reporting no history of atopy, with low total IgE titers, and little IgE reactivity to peanut allergen (Ara h 2), birch tree pollen allergen (Bet v 1), house dust mite allergen (Der p 1), or cat allergen (Fel d 1) were categorized as non-atopic (Fig. 1b, c; Extended Data Fig. 1a; Extended Data Table 1). Peanut allergic subjects reported multiple atopies, had approximately two-fold higher total IgE titers, with reactivity to peanut allergen (Ara h 2) but not to other tested allergens, and were confirmed by clinician-supervised oral challenge (Fig. 1b, c; Extended Data Fig. 1a; Extended Data Table 1)8. We sensitized human LAD2 mast cells with similar amounts of total IgE enriched from the sera of these cohorts and activated the cells by anti-IgE crosslinking. Intriguingly, less degranulation, as measured by β-hexosaminidase release, was observed in mast cells sensitized with IgE isolated from sera of non-atopic individuals compared peanut allergic patients (Extended Data Fig. 1c), despite similar surface IgE loading (Extended Data Fig. 1d, e). This suggested intrinsic functional differences between non-atopic and allergic IgE, independent of allergen specificity.
Next, the N-glycans on total IgE enriched from these two cohorts were analyzed by mass spectrometry16,18,21. Mannose content on oligomannose moieties was similar between total non-atopic and allergic IgE (Fig. 1d). Fucose, N-acetyl glucosamine (GlcNAc), galactose, and sialic acid can be attached to complex glycans (Fig. 1a). While total fucose content was similar between non-atopic and allergic IgE (Fig. 1e), significantly increased levels of bisecting GlcNAc (biGlcNAc) and terminal galactose were found on non-atopic IgE (Fig. 1f, g) whereas increased terminal sialylation was detected on allergic IgE (Fig. 1h).
To determine whether the glycan differences on total IgE were predictive of allergic disease, we assessed the variable glycan content on non-atopic and allergic IgE using Receiver Operating Characteristics (ROC) curves (Fig. 1i). Galactose and sialic acid content of IgE were uniquely strong predictors of allergic disease. Of note, differences in IgE sialylation were not sex- or age-dependent (Extended Data Fig. 1f, g). Glycosylation site analysis showed N140, N168, N265 and N394 of IgE were fully occupied by N-linked glycans, N218 and N371 partially occupied (75% and 30% respectively), and N383 completely unoccupied (Fig. 1j; Extended Data Fig. 4a), consistent with previous results15,16,18. N394 oligomannose structures (Fig. 1j; Extended Data Fig. 4b), and N140, N168, N265, and N371 fucose and biGlcNAc content was similar between samples (Fig. 1j; Extended Data Fig. 4c, d). However, N140 and N265 complex glycans terminating in galactose were enriched on non-atopic IgE, while terminal sialic acid, particularly disialylated glycans, were significantly enriched at N168 and N265 on allergic IgE (Fig. 1j; Extended Data Fig. 1h, 4e, 4f). Together, these results reveal specific glycosylation patterns distinguish allergic from non-atopic total IgE.
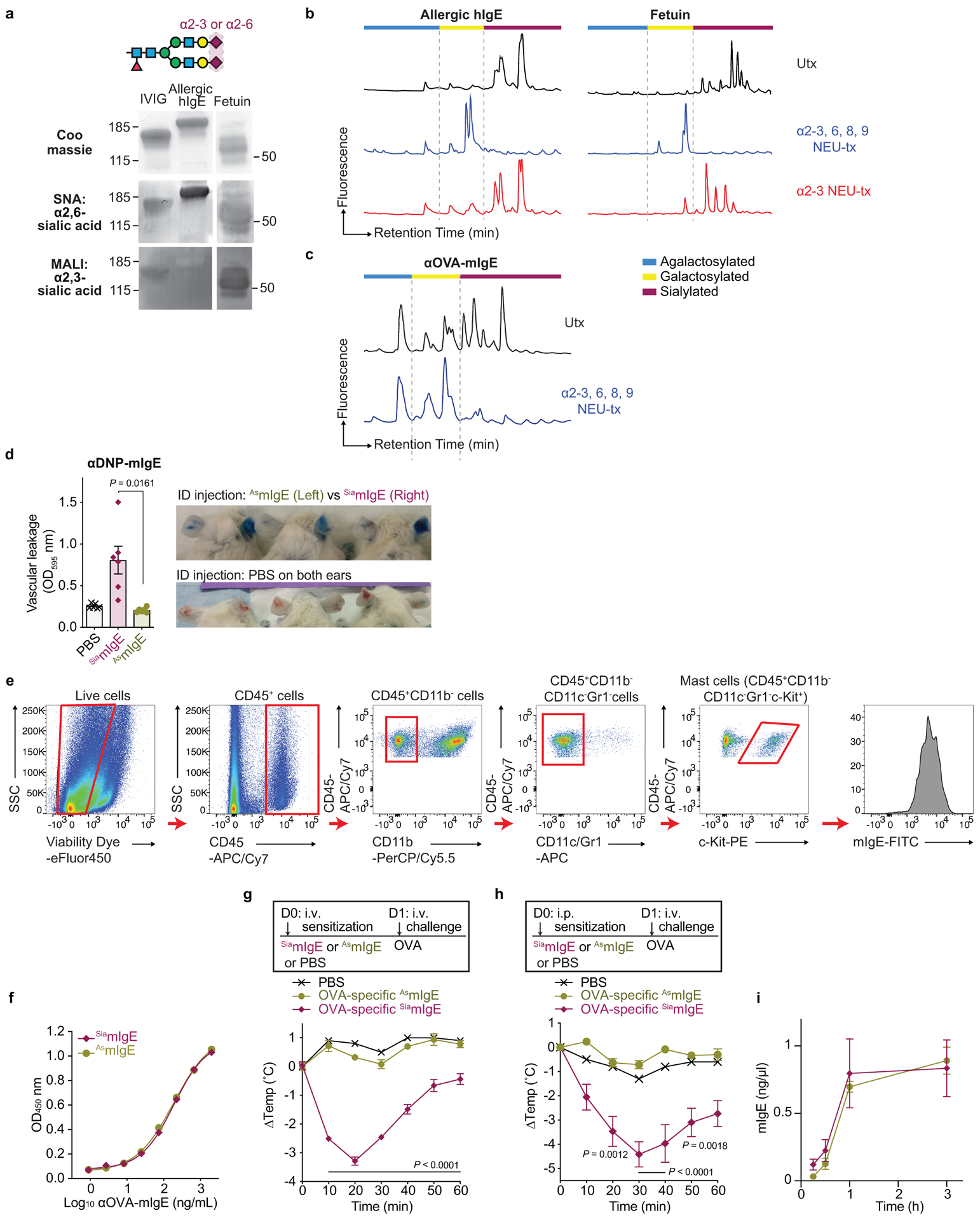
Sialylation has been implicated in regulating multiple antibody classes, including IgG1 anti-inflammatory activity22, IgA nephropathy and influenza neutralization23,24, IgM-induced inhibitory signaling on B and T cells25,26. Sialic acid was attached in α2,6 linkages on hIgE and mouse IgE (mIgE) as determined by neuraminidase (NEU) digestion assays and lectin blotting (Extended Data Fig. 5a–c, Fig. 2a), consistent with previous studies6,15,17. Thus, we treated mIgE with NEU, or buffer, to generate mIgE of identical allergen-specificity differing only in sialic acid content (Fig. 2a). In a model of passive cutaneous anaphylaxis (PCA), mice were sensitized with PBS, OVA-specific sialylated-mIgE (SiamIgE), or OVA-specific asialylated-IgE (AsmIgE) intradermally in the ears. The next day, the mice were challenged with allergen, OVA, in Evan’s blue dye intravenously. Forty minutes after challenge, the amount of blue dye in the ear was quantified as a surrogate of histamine-mediated vascular leakage. PBS-injection elicited little blue dye accumulation in the ear injection site, while significant blue coloration was observed in SiamIgE-sensitized ears (Fig. 2b; Extended Data Fig. 5d). Strikingly, AsmIgE-sensitized ears exhibited markedly reduced blue coloration, indicative of attenuated anaphylaxis (Fig. 2b; Extended Data Fig. 5d). To confirm sialic acid removal was responsible for reduced PCA reactivity, sialic acid was reattached to AsmIgE in vitro (Re-siamIgE). Re-siamIgE triggered a robust PCA reaction (Fig. 2b), demonstrating that IgE sialylation impacts the magnitude of anaphylaxis. Flow cytometry analysis of mast cells recovered from the mouse ears revealed no differences in IgE loading following sensitization with SiamIgE or AsmIgE (Fig. 2c; Extended Data Fig. 5e) and SiamIgE or AsmIgE bound allergen similarly as determined by ELISA (Extended Data Fig. 5f). Thus, attenuated cutaneous anaphylaxis by AsmIgE was independent of IgE loading on mast cells in vivo or allergen recognition.
Fig. 2 |. Sialic acid removal attenuates IgE.
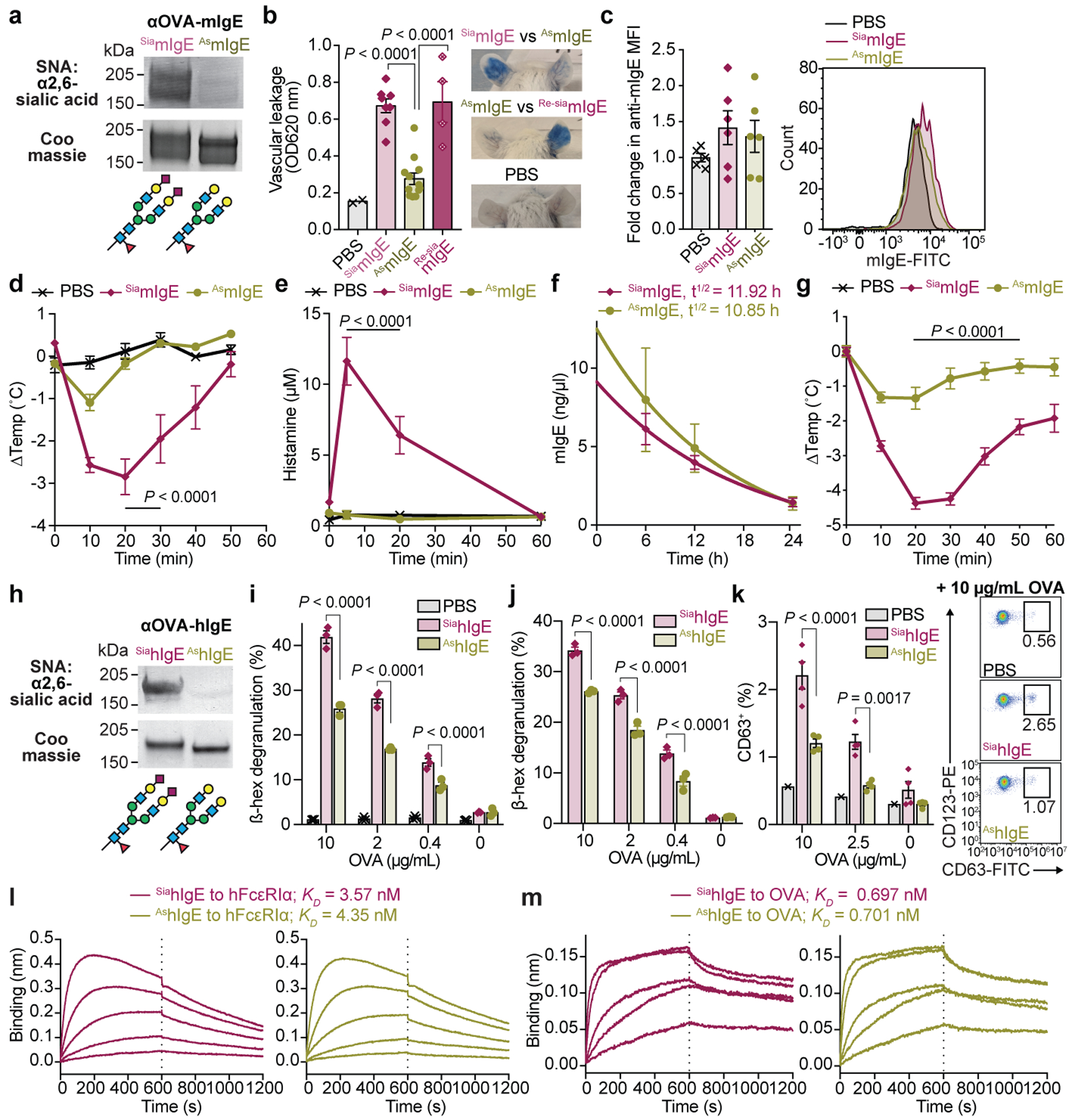
a, SNA lectin blot and Coomassie-stained gel of OVA-specific SiamIgE and AsmIgE. b, Left, quantification of ear blue coloration and right, representative ear images following OVA-induced PCA by PBS, OVA-specific SiamIgE, AsmIgE, or Re-SiamIgE (n = 2, 8, 12, 4 mouse ears respectively). c, Left, mean fluorescence intensity (MFI) and right, representative histograms of anti-mIgE determined by FACS on dermal mouse ears mast cells following sensitization by PBS, OVA-specific SiamIgE, or AsmIgE (n = 5, 6, 6 mouse ears, respectively from two independent experiments). d, e, Temperature change (d) and serum histamine (e) following DNP-induced PSA in mice sensitized with PBS, DNP-specific SiamIgE, or AsmIgE (n = 3, 5, 5 mice respectively). f, Serum levels of DNP-specific SiamIgE (n = 4 mice) or AsmIgE (n = 5 mice) after intraperitoneal administration. g, Temperature change following PFA elicited by oral TNP-OVA administration in mice sensitized with PBS, TNP-specific SiamIgE, or AsmIgE (n = 2, 4, 4 respectively). h, SNA lectin blot and Coomassie-stained gel of OVA-specific SiahIgE and AshIgE. i-k, OVA-induced degranulation in LAD2 mast cells (i), peripheral blood mononuclear cell-derived human mast cells (j), or basophils (k) sensitized with PBS (for i (n = 3), k (n = 1)), OVA-specific SiahIgE or AshIgE (for i, n = 3; j, n = 3; k, n = 4). n = technical replicates and are representative of three biologically independent experiments. l, m, Binding kinetics of OVA-specific SiahIgE or AshIgE to hFcεRIα (l) or OVA (m). Data are mean ± s.e.m. (b-g, i-k) and are representative of two (f) or three independent experiments (a, b, d, e, g-m). One-way ANOVA with Tukey’s (b), or two-way ANOVA with Tukey’s (d, e, i, k) or Sidak’s (g, j) multiple comparison test. For gel source data, see Supplementary Figure 1.
Next, we systemically sensitized mice with SiamIgE, AsmIgE, or PBS and challenged with allergen the following day in a model of passive systemic anaphylaxis (PSA). SiamIgE-sensitized mice elicited a robust anaphylactic response underscored by significant temperature loss 20 minutes after allergen challenge (Fig. 2d; Extended Data Fig. 5g, h). However, minimal temperature drop was observed in AsmIgE- or PBS-sensitized mice (Fig. 2d; Extended Data Fig. 5g, h). Consistently, a systemic increase in histamine was detected in SiamIgE-sensitized animals following challenge, but not in AsmIgE- or PBS-treated mice (Fig. 2e). Asialylated glycoproteins have decreased serum half-life27, and we therefore compared the levels of SiamIgE and AsmIgE in circulation following systemic administration. However, sialic acid removal had little effect on IgE half-life (Fig. 2f, Extended Data Fig. 5i). To extend these findings to a model of passive food allergy, we sensitized mice systemically with PBS, SiamIgE or AsmIgE, and challenged with allergen orally the following day. SiamIgE-, but not AsmIgE- or PBS-sensitization resulted in a significant temperature loss following oral allergen challenge (Fig. 2g).
We next asked whether sialylation similarly regulated hIgE, and sensitized human LAD2 mast cells with PBS, sialylated or asialylated hIgE (SiahIgE and AshIgE, respectively, Fig. 2h). The cells were stimulated with allergen, and degranulation quantified by β-hexosaminidase release assays. AshIgE-sensitized cells had markedly reduced degranulation following allergen challenge, compared to SiahIgE-sensitized cells (Fig. 2i). LAD2 mast cells after sensitization was examined by flow cytometry and revealed comparable hIgE loading following SiahIgE or AshIgE sensitization (Extended Data Fig. 6a). Similar findings were observed in human mast cells derived from primary peripheral blood CD34+ cell culture, where AshIgE-sensitized cells had markedly reduced allergen-specific degranulation compared to SiahIgE-sensitized cells (Fig. 2j; Extended Data Fig. 6b). In parallel, primary basophils were sensitized with PBS, SiahIgE and AshIgE and stimulated with allergen (Extended Data Fig. 6c). AshIgE-sensitized basophils elicited reduced degranulation after allergen stimulation as measured by surface staining of the granule marker, CD63, compared to basophils sensitized with SiahIgE (Fig. 2k). Although mast cell loading was similar between mouse and human SiaIgE and AsIgE (Fig. 2c, Extended Data Fig. 6a), we asked whether sialylation altered binding kinetics of hIgE to its receptor, FcεRI. Bio-layer interferometry (BLI) assays revealed no difference in SiahIgE and AshIgE interactions with FcεRI (Fig. 2l). Sialylation also did not alter IgE binding to the allergen (Fig. 2m). Thus, removing sialic acid from IgE attenuates its effector functions in vivo and in vitro, while binding to allergen, mast cells and FcεRI remained intact.
Because sialylation does not alter IgE interactions to allergen and receptor, we examined whether signaling downstream of FcεRI was affected. LAD2 mast cells sensitized with SiahIgE or AshIgE were stimulated with allergen and cellular lysates collected at defined intervals. Western blotting of mast cell lysates for Syk revealed reduced phosphorylation in cells sensitized with AshIgE at 5 and 30 minutes after stimulation (Fig. 3a). Similarly, calcium flux was reduced in AshIgE-sensitized LAD2 mast cells following allergen stimulation compared to SiahIgE-sensitized cells (Fig. 3b). We then asked whether a surrogate asialylated glycoprotein could attenuate anaphylaxis similarly to asialylated IgE. LAD2 mast cells were sensitized with SiahIgE, and supplemented with either sialylated fetuin (SiaFetuin) or asialylated fetuin (AsFetuin; Extended Data Fig. 5b). Quantifying allergen-specific degranulation revealed that addition of sialylated fetuin had no effect, while asialylated fetuin inhibited allergen-induced mast cell degranulation (Fig. 3c). Together, these results suggest that sialic acid removal exposes an inhibitory glycan that dampens FcεRI signaling.
Fig. 3 |. Modulating IgE sialylation and anaphylaxis.
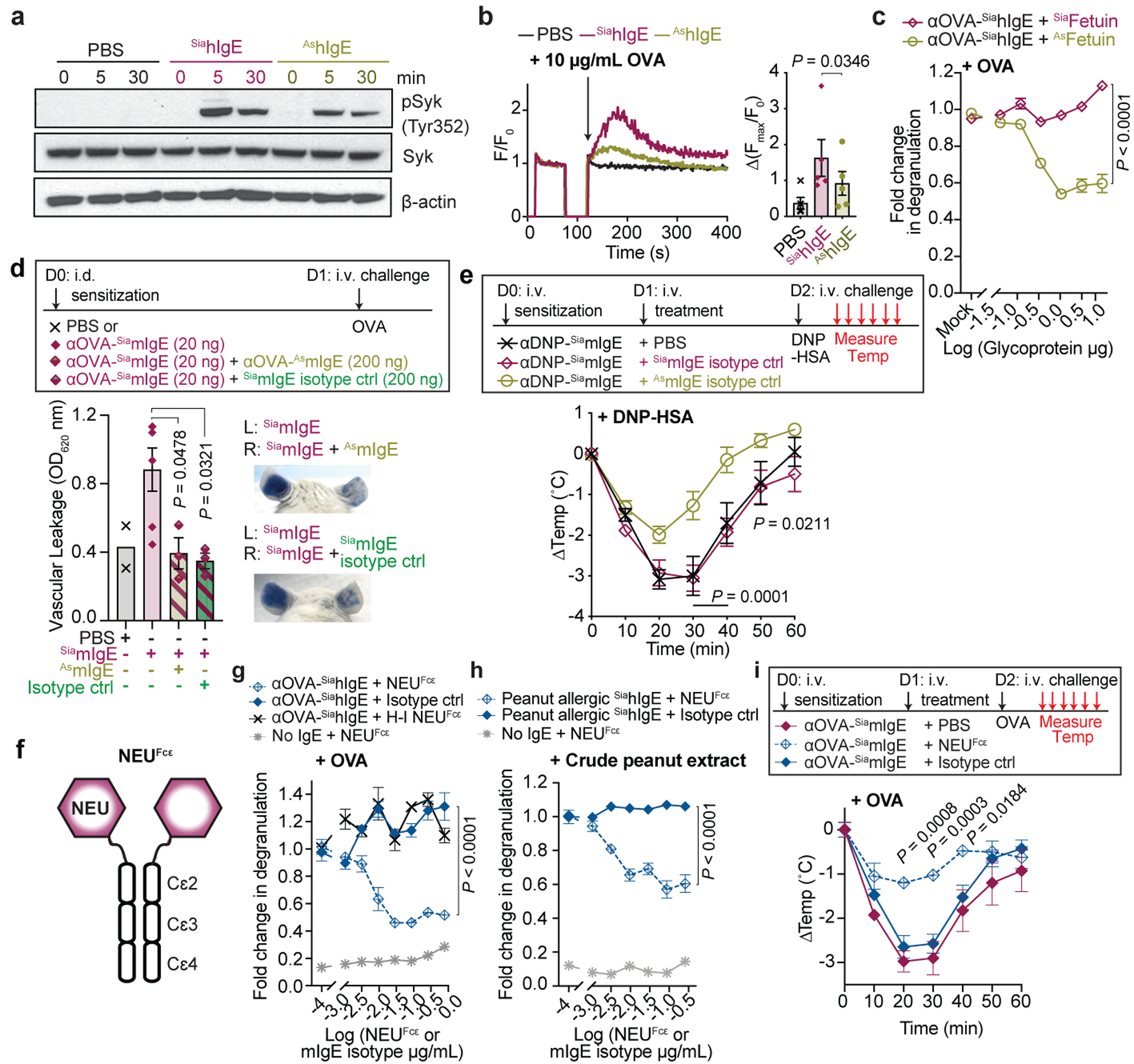
a, Immunoblots of pSyk, total Syk, and β-actin in LAD2 mast cells sensitized with PBS, OVA-specific SiahIgE or AshIgE after OVA stimulation. b, OVA-induced Ca2+ flux traces (left) and maximum values (right) in LAD2 cells sensitized with PBS (black), OVA-specific SiahIgE (maroon) or AshIgE (gold). n = 5 biologically independent samples from three independent experiments. c, OVA-elicited degranulation in LAD2 cells sensitized with OVA-specific SiahIgE and treated with SiaFetuin (maroon) or AsFetuin (gold). n = 3 technical replicates. d, Quantification of ear blue coloration (left) and representative images (right) following OVA-induced PCA in mice sensitized with PBS, OVA-specific SiamIgE, both OVA-specific SiamIgE + AsmIgE, or both OVA-specific SiamIgE + mIgE isotype control (n = 2, 6, 3, 3 mice ears, respectively). e, Temperature change following DNP-induced PSA in mice receiving DNP-specific SiamIgE on day 0 and PBS, OVA-specific SiamIgE or AsmIgE on day 1 (n = 6, 7, 7, respectively from two independent experiments). f, Schematic of NEUFcε. g, OVA-induced degranulation in LAD2 cells sensitized with OVA-specific SiahIgE and treated with PBS, NEUFcε, heat-inactivated NEUFcε (H-I NEUFcε) or IgE isotype control. h, Peanut-induced degranulation in LAD2 cells sensitized with peanut-allergic SiahIgE treated with PBS, NEUFcε, or IgE isotype control. g, h, n = 3 technical replicates. i, Temperature changes following OVA-induced PSA in mice receiving OVA-specific SiamIgE on day 0 and PBS, NEUFcε, or IgE isotype control on day 1. n = 4 mice per group. Data are mean ± s.e.m. (b-e, g-i) and are representative of two (a, d) and three (c, g-i) independent experiments. Two-tailed paired t-test (b), one-way ANOVA with Tukey’s (d), or two-way ANOVA with Sidak’s (c) or Tukey’s multiple comparison test (e, g-i). For gel source data, see Supplementary Figure 1.
These observations indicated that AsIgE could actively inhibit anaphylaxis in vivo. We therefore sensitized mice intradermally in the ears with PBS, OVA-specific SiamIgE, a combination of OVA-specific SiamIgE and ten-fold more OVA-specific AsmIgE, or a combination of OVA-specific SiamIgE and ten-fold more TNP-specific SiamIgE isotype control. The next day mice were challenged with OVA and blue coloration of the ears quantified. Extensive vascular leakage occurred in ears sensitized with OVA-specific SiamIgE alone (Fig. 3d). However, co-sensitization of OVA-specific SiamIgE with either OVA-specific AsmIgE or TNP-specific SiamIgE both resulted in significantly reduced vascular leakage (Fig. 3d). Next, mice were systemically sensitized by DNP-specific SiamIgE on day 0, and PBS, OVA-specific SiamIgE, or OVA-specific AsmIgE on day 1, and challenged with DNP-HSA on day 2. Intriguingly, mice that were sensitized with DNP-specific SiamIgE on day 0 and PBS or OVA-specific SiamIgE on day 1 exhibited robust temperature loss after allergen challenge. However, DNP-specific SiamIgE-sensitized mice that received OVA-specific AsmIgE on day 1 had significantly attenuated temperature loss upon allergen challenge (Fig. 3e). Systemic challenge of these treatment groups with OVA revealed that only sensitization with OVA-specific SiamIgE resulted in temperature drop, while all other groups were unaffected (Extended Data Fig. 7a). These results suggest that AsmIgE attenuates anaphylaxis by occupying FcεRI, but can actively dampen systemic anaphylaxis.
As sialic acid removal attenuated IgE effector functions, we explored whether targeting sialic acid on IgE-bearing cells represents a viable strategy for attenuating allergic inflammation. Thus, we genetically fused a neuraminidase to the N-terminus of IgE Fc Cε2–4 domains (NEUFcε, Fig. 3f; Extended Data Fig. 7b) to direct sialic acid removal specifically to IgE-bearing cells. This fusion protein retained binding to FcεRI (Extended Data Fig. 7c), could be loaded on mast cells (Extended Data Fig. 7d), and had neuraminidase activity (Extended Data Fig. 7e–h). LAD2 mast cells were sensitized with OVA-specific SiahIgE, and then incubated briefly with increasing concentrations of NEUFcε, heat-inactivated NEUFcε, or an IgE isotype to control for FcεRI occupancy, and stimulated with OVA. Remarkably, treatment with NEUFcε, but not heat-inactivated NEUFcε nor the isotype control attenuated OVA-induced degranulation in a dose-dependent manner (Fig. 3g). To extend our findings to allergic hIgE from peanut allergic patients, we sensitized LAD2 mast cells with peanut allergic SiahIgE and treated with NEUFcε, or an IgE isotype control. Consistently, allergen-induced degranulation was significantly attenuated by NEUFcε treatment of peanut allergic SiahIgE-sensitized cells compared to IgE isotype control treatment (Fig. 3h). Unsensitized LAD2 mast cells treated with NEUFcε did not degranulate (no IgE + NEUFcε, Fig. 3g, h), indicating NEUFcε treatment does not stimulate mast cells. We next explored the therapeutic potential of modulating sialic acid content in vivo. Mice were sensitized systemically with SiamIgE on day 0, received PBS, NEUFcε, or IgE isotype control treatment on day 1. The following day, the mice were challenged systemically with allergen, and core body temperature measured. SiamIgE-sensitized mice that received PBS or isotype control exhibited robust drops in temperature (Fig. 3i). Remarkably, NEUFcε treatment significantly attenuated allergen-induced temperature drop (Fig. 3i), providing evidence of the therapeutic potential of targeting sialic acid on IgE-bearing cells.
IgE-mediated allergic diseases are multifactorial, with a broad range of clinical presentations, and paradoxically, many individuals produce allergen-specific IgE without manifestion of disease. Further, there is a high rate of false positive test results for food allergens3,5,8,28. Many non-mutually exclusive mechanisms for this discrepancy exist, including differences in IgE affinity or epitope diversity for allergens, mast cell numbers, FcεRI expression levels, Syk signaling, allergen-specific IgG antibodies, anti-IgE antibodies, and regulatory T cells numbers29. Here we demonstrate that sialic acid content on total IgE distinguishes peanut-allergic and non-atopic IgE. Further, allergic reactions are attenuated through removal of sialic acid from IgE or administration of asialylated glycoproteins. Although IgE sialic acid content and its role in other contexts is unknown, we propose sialylation is an additional factor that regulates its biology. Thus, exploitation of the IgE-sialylation axis presents a compelling diagnostic and therapeutic strategy.
Methods
IgE Antibodies
All human samples were collected under IRB approved protocols by MGH and Research Blood Components (Boston, MA), including informed consent obtained in accordance with relevant ethical regulations. Serum samples were obtained from peanut-allergic individuals prior to treatment. Peanut allergy was confirmed by clinical history, allergen-specific IgE screening, and double-blind placebo-controlled oral challenge (PNOIT2, NCT01750879, Extended Data Table 1)8. Non-atopic adults were recruited on the basis of self-identification as non-allergic donors. Non-atopy was confirmed by clinical history, and allergen-specific IgE screening (Extended Data Table 1). Total IgE, Ara h 2-specific IgE, Fel d 1-specific IgE, Der p 1-specific IgE, and Bet v 1-specific IgE were determined by ImmunoCap Assay (Phalleon, Thermo Scientific) according to manufacturer’s protocols. Primary IgE was enriched from serum samples by serially depleting IgG by protein G agarose (GE Healthcare) followed by anti-IgE conjugated NHS-beads (GE Healthcare). IgE purity was confirmed by protein electrophoresis and coomassie gel staining. Recombinant OVA-specific IgE was generated as described17. Briefly, cDNA sequences for generating OVA-specific heavy ε and light κ chain of mouse and human IgE17 were cloned into pcDNA3.4 using restriction enzyme sites XbaI and AgeI. To generate recombinant OVA-specific mouse or human IgE, plasmids containing OVA-specific heavy and light chain were transiently co-transfected at 1:1 ratio using Expi293 Expression System Kit (Life Technologies) according to the manufacturer’s protocol. The cells expressing IgE were selected by addition of 400 μg/mL G418 in the culture media for two weeks and maintained before expanding to a larger scale production. OVA-specific IgE was purified from cell culture supernatant by OVA-coupled agarose beads17.
ELISAs
Sandwich ELISA for quantifying mIgE and OVA-specific binding were conducted as previously described17. Briefly, 96-well Nunc plates were coated with goat polyclonal anti–mouse IgE (Bethyl Laboratories) or OVA and blocked with BSA in PBS (1% BSA for mIgE and 2% for OVA) prior to sample incubation. Samples were probed with goat polyclonal anti–mouse IgE-HRP (2 ng/ml; Bethyl Laboratories). The reactions were detected by 3,3,5,5- tetramethylbenzidine (TMB; Thermo Fisher Scientific), stopped by 2 M sulfuric acid, and the absorbance measured at 450 nm.
Glycopeptide Mass Spectrometry and Glycan Analysis
Site specific glycosylation was quantified for IgE isolated from non-allergic donors and from peanut allergic donors using nano LC-MS/MS following enzymatic digestion of the proteins as described previously, with minor modifications16–18 (Extended Data Table 2). The isolated polyclonal primary hIgE and myeloma hIgE (Sigma Aldrich AG30P) was prepared for proteolysis by denaturing the protein in 6M guanidine HCl followed by reduction with dithiothreitol and alkylation with iodoacetamide followed by dialysis into 25mM ammonium bicarbonate pH 7.8. Proteolysis was done with either trypsin to quantify N218, N371 and N394 or chymotrypsin to quantify N140, N168 and N265. For the tryptic digest IgE was incubated with trypsin (Trypsin Gold Promega) at a 1:50 enzyme to substrate ratio overnight at 37C. For the chymotryptic digest IgE was incubated with chymotrypsin (Sequencing Grade Promega) at a 1:100 enzyme to substrate ratio for 4 hours at 25C. Both enzymes were quenched with formic acid added to 2% w/w. The separation was performed on a Thermo EasySpray C18 nLC column 0.75umx50cm using water and acetonitrile with 0.1% formic acid for mobile phase A and mobile phase B respectively. A linear gradient from 1% to 35% mobile phase B was run over 75 minutes. Mass spectra were recorded on a Thermo Q Exactive mass spectrometer operated in positive mode using data independent acquisition (DIA) targeting the masses shown in table SX and SY. Glycopeptides were quantified based on the extracted ion area of the Y1 ion (Extended Data Fig. 2). The relative abundance was calculated for all identified glycan species for each site. Myeloma IgE (Sigma Aldrich AG30P) was run prior to paired sample sets to monitor retention time shifts and ensure consistency in the analytical results across the sample set. The percentage of glycan moieties at each site was calculated using the relative abundance of each glycan. For example, if a particular site was determined to have 60% monosialylated, fucosylated glycans (A1F), and 40% of disialylated, fucosylated glycans (A2F), the number of sialic acids at one site would be 1.4 (0.6×1+0.4×2), and total 2.8 sialic acids per molecule accounting for two sites.
Generation of NEUFcε
The neuraminidase fusion protein was designed by fusing a kappa light chain secretion signal sequence and the sialidase gene from Arthrobacter urefaciens (EC 3.2.1.18, gene AU10430. Stop codon of the AU104 was omitted, instead, a short flexible linker peptide (GGGGGG), mouse IgE Cε2, Cε3, Cε4, and His6-tag was inserted to the C-terminus of the sialidase. The gene was codon optimized for human and synthesized by GenScript. The protein of 288 kDa was then produced by WuXi biologics. Sialidase activity of NEUFcε was determined by the level of p-nitrophenol released from 250 μM 2-O-(p-Nitrophenyl)-α-D-N-acetylneuraminic acid (Sigma) in 100 mM sodium phosphate (pH 5.5) for 10 min at 37°C. The reaction was terminated by adding 0.5 M sodium carbonate and the absorbance quantified at 405 nm.
Mice
Five- to six-week-old female BALB/c mice were purchased from the Jackson Laboratory and used in these studies. All mice were housed in specific pathogen-free conditions according to the National Institutes of Health (NIH), and all animal experiments were conducted under protocols approved by the MGH IACUC, and in compliance with appropriate ethical regulations. For all experiments, age- and sex-matched mice were randomized allocating to experimental group, with 4–5 mice per group, and repeated three independent times. No statistical method was used to determine sample size.
Passive Cutaneous Anaphylaxis (PCA) was conducted as previously described17. In brief, monoclonal SiamIgE or AsmIgE specific for OVA or dinitrophenyl (DNP, clone SPE-7; Sigma-Aldrich) was injected intradermally in the mice ears. For experiments where OVA-specific AsmIgE was added to OVA-specific SiamIgE, a mIgE isotype control (clone MEA-36, Biolegend) was included. The next day mice were intravenously challenged with PBS containing 125 μg OVA (Sigma-Aldrich) or DNP-Human Serum Albumin (DNP-HSA; Sigma-Aldrich) and 2% Evans blue dye in PBS. 45 min after challenge, the ears were excised and minced before incubation in N,N-dimethyl-formamide (EMD Millipore) at 55°C for 3 h. The degree of blue dye in the ears was quantitated by the absorbance at 595 nm.
Passive Systemic Anaphylaxis (PSA) was elicited as previously described with minor modifications31,32. Briefly, mice were injected intravenously with monoclonal mIgE specific for OVA or DNP (clone SPE-7; Sigma-Aldrich) in PBS and challenged the next day intravenously with PBS containing 1 mg OVA (Sigma-Aldrich) or DNP-HSA (Sigma-Aldrich). For examining the therapeutic potential of AsmIgE, mice that had been injected intravenously with 10 μg DNP-specific mIgE (clone SPE-7; Sigma-Aldrich) the first day were injected intravenously with PBS, 20 μg OVA-specific SiamIgE or 20 μg OVA-specific AsmIgE the next day and challenged with 1 mg DNP-HSA or OVA (Sigma-Aldrich) the third day. For testing the therapeutic potential of NEUFcε, mice injected intravenously with 10 μg OVA-specific mIgE the first day were further injected intravenously with PBS, 100 μg NEUFcε or 100 μg mIgE isotype control (clone MEA-36, Biolegend) the next day and challenged with 1 mg OVA (Sigma-Aldrich) the third day. Core temperature was recorded at the baseline and every 10 min after the allergen challenge in a blinded manner by a rectal microprobe thermometer (Physitemp). Histamine in the blood was quantified by histamine enzyme immunoassay kit (SPI-Bio) according to the manufacturer’s protocol. Briefly, histamine in the blood was derivatized and incubated with plate precoated with monoclonal anti-histamine antibodies and histamine-AchE tracer at 4°C for 24 h. The plate was then washed and developed with Ellman’s reagent and the absorbance measured at 405 nm.
Passive Food Anaphylaxis (PFA) was elicited by adapting PSA described above. Briefly, mice injected intravenously with 20 μg monoclonal mIgE specific for TNP (clone MEA-36; Biolegend) in PBS the first day were administered with 20 mg TNP-OVA in PBS (Biosearch Technologies) by oral gavage the next day. Core temperature was recorded at the baseline and every 10 min after the challenge by a rectal microprobe thermometer (Physitemp) in a blinded manner.
To determine in vivo half-lives of SiamIgE or AsmIgE, mice were injected intraperitoneally with 30 μg DNP-specific SiamIgE or AsmIgE and the blood collected at the indicated times after injection into a Microtainer blood collection tube with clot actiator/SST gel (BD Diagonistics). The level of mIgE was quantified by mIgE ELISA described above.
Basophil activation tests
Basophil activation was performed as previously described33. Buffy coats of human blood from healthy, de-identified, consenting donors were obtained from the MGH Blood Transfusion Service. Peripheral blood mononuclear cells (PBMCs) were separated from buffy coats by a density gradient centrifugation using Ficoll Paque Plus (GE Healthcare) and resuspended in 0.5% BSA in RPMI 1640 Media (GE Healthcare). PBMCs were incubated for 2 min with ice-cold lactic acid buffer (13.4 mM lactate, 140 mM NaCl, 5 mM KCl, pH 3.9) to remove endogenous human IgE on the cell surface prior to neutralization by 12% Tris (pH 8). Cells were then washed and incubated 1 hour at 37°C with 1 μg OVA-specific SiahIgE or AshIgE per 1 × 106 cells in basophil activation buffer (0.5% BSA, 2 mM CaCl2 and 2 mM MgCl2 in RPMI 1640 Media). Sensitized cells were washed and resuspended in basophil activation buffer supplemented with 10 ng/mL human IL3 (PeproTech) prior to 30 min OVA activation. Activation was stopped by addition of ice-cold 0.2 M EDTA in FACS buffer. Cells were washed and resuspended in FACS buffer prior to antibody staining (Extended Data Table 3) for activation marker (LAMP-3; CD63+) on basophils (CD123+HLADR−).
Human mast cell culture and degranulation
Human LAD2 mast cell line was a generous gift of Dr. Metcalfe (NIAID, NIH) and was maintained as previously described17,34. Briefly, LAD2 cells were cultured in StemPro-34 SFM medium (Life Technologies) supplemented with 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 100 ng/ml recombinant human stem cell factor (PeproTech). The cells were hemi-depleted each week with fresh medium and maintained at 2–5 × 105 cells/ml at 37°C and 5% CO2.
Primary human mast cells were generated as previously described35. Briefly, PBMC were separated from buffy coats as described above before isolation of CD34+ pluripotent hematopoietic cells by EasySep™ Human Whole Blood CD34 Positive Selection Kit II (STEMCELL Technologies). CD34+ cells were cultured in StemPro-34 SFM medium (Life Technologies) supplemented with 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 50 ng/mL recombinant human stem cell factor (PeproTech), 50 ng/mL human IL-6 (PeproTech), and with 10 ng/mL human IL-3 (PeproTech) in the first week. The cells were cultured in similar culture media but without human IL-3 after the first week to mature for 10 weeks. Cultured mast cells were confirmed by FACS staining of CD45+, c-KIT+, FcεRI+.
Degranulation assays were performed as previously described (Shade, 2015), LAD2 or peripheral blood-derived human mast cells were sensitized overnight with 1 μg/mL OVA-specific hIgE or 50 ng/mL peanut-allergic hIgE. The following day, the cells were pelleted by centrifugation, resuspended in HEPES buffer, plated in 96-well plates, and stimulated with allergen OVA or crude peanut extract at defined concentrations. Upon allergen challenge, mast cell degranulation was determined by the amount of substrate p-nitrophenyl N-acetyl-β-D-glucosamide digested by β-hexosaminidase release from the mast cell granules at the absorbance of 405 nm. To assess the effect of sialic acid removal on IgE-bound mast cells, IgE-sensitized LAD2 cells were treated with NEUFcε, heat-inactivated NEUFcε, mIgE isotype control (clone MEA-36, Biolegend) for 20 min before allergen challenge. To inactivate NEUFcε, the enzyme was heated at 95°C for 10 min. To determine whether addition of a surrogate asialylated glycoprotein could recapitulate the phenotype of sialic acid removal from IgE, LAD2 cells sensitized with OVA-specific SiahIgE were incubate with sialylated fetuin (SiaFetuin) or asialylated fetuin (AsFetuin) at defined amount for 20 min before allergen challenge.
Crude peanut extract preparation
Unsalted dry-roasted peanuts (Blanched Jumbo Runner cultivar; Planters) were ground to a smooth paste, followed by washing with 20 volumes of cold acetone, filtered using Whatman paper, and dried as previously described17. Protein was extracted by agitating the peanut flour overnight with PBS containing protease inhibitor cocktail without EDTA (Roche). The peanut protein extracts were collected as the supernatant after centrifugation at 24,000 × g for 30 min.
IgE Glycosylation Engineering
To remove sialic acids on IgE, IgE was digested with Glyko Sialidase A (recombinant from Arthrobacter urefaciens expressed in E. Coli; Prozyme) at 37°C for 72 h according to the manufacturer’s instructions. To re-sialylate AsmIgE by in vitro sialylation reaction, AsmIgE was incubated with human alpha-2,6 sialyltransferase 1 (ST6GAL1; generously provided by Harry Meade, LFB-USA) at a ratio of 20 μg AsmIgE per μg of ST6GAL1 and 5 mM Cytidine-5’-monophospho-N-acetylneuraminic acid (CMP-Neu5Ac2; Nacalai USA) in the sialylation buffer (150 mM NaCl, 20 mM HEPES, pH7.4) overnight at room temperature as previously described36. Following reactions, OVA-specific SiaIgE or AsIgE were purified by OVA-coupled beads to remove glycosylation modifying enzymes as described17. All digestion or sialylation reactions were verified by lectin blotting or HPLC.
Protein gel stain and lectin blotting
Equal amounts of SiaIgE or AsIgE were resolved on 3–8% Tris-Acetate protein gels (Life Technologies) in SDS-PAGE under nonreducing conditions. For protein stain, gels were incubated in AcquaStain Protein Gel Stain (Bulldog Bio) for 1 h at room temperature and destained in distilled water. For lectin blotting, the protocol was conducted as described17. Briefly, after resolved proteins on the gel were transferred to Immobilon-PSQ polyvinylidene difluoride membranes (Millipore Sigma), the membranes were blocked with 0.2% BSA in TBS for 1 hour at room temp, washed in TBS, followed by incubation with biotinylated Sambucus nigra lectin (SNA; 0.4 μg/ml; Vector Laboratories) in TBS with 0.1 M Ca2+ and 0.1 M Mg2+ for 1 hour at room temp to determine the level of terminal α2,6 sialic acids on N-linked glycans of proteins. The membrane was then washed in TBS and incubated with alkaline phosphatase conjugated goat anti-biotin (1:5000 dilution; Vector Laboratories) in TBS for 1 hour at room temp. Sialylated proteins on membranes were visualized by incubation with 1-Step NBT/BCIP plus Suppressor Substrate Solution.
Flow cytometry
Details of antibodies used for surface allergen staining are listed in Extended Data Table 3. For staining for mouse cells, suspension cells were incubated with anti–mouse CD16/CD32 (clone 2.4G2, BD Biosciences) prior to antibody staining. Cells were incubated in FACS buffer with desired staining antibodies for 20 minutes at 4°C. Cells were then washed in FACS buffer before being acquired by an LSRII flow cytometer (BD Biosciences) or CytoFLEX (Beckman Coulter). Data were analyzed using FlowJo software version 10.4 software (Tree Star). To quantify OVA-specific hIgE loading following sensitization, PBS or 1 μg/mL OVA-specific SiahIgE or AshIgE were incubated with 2.5 × 105 cells/mL LAD2 mast cells overnight before wash with FACS buffer and stained with anti-c-Kit antibody and OVA-A647. To quantify native hIgE loading on LAD2 mast cells, cells were sensitized with 32 ng total non-atopic or allergic hIgE overnight before washed in FACS buffer and stained with anti-c-Kit and anti-hIgE antibodies. To quantify dermal mast cell IgE loading, single cell suspensions were generated from mouse ears as described17. Ears were intradermally injected with 40 ng OVA-specific SiamIgE or AsmIgE. The following day, ears were removed, separated into dorsal and ventral halves, and minced before incubation in DMEM containing 2% FCS, 1% HEPES, 500 units/mL Collagenase type 4 (Worthington), 0.5 mg/mL hyaluronidase (Sigma) and DNase I (Roche) at 37°C for 1 h at 180 RPM. The digested sample was then subjected to disruption by Gentle MACS and filtered through a 70 μm cell strainer followed by a 40 μm cell strainer in FACS buffer (2 mM EDTA and 0.5% Bovine Serum Albumins (BSA) in PBS). mIgE loading was detected by FACS using anti-mIgE antibodies on dermal mast cells (CD45+CD11b−CD11c−Gr1−c-Kit+).
Biolayer interferometric assays for binding
Binding kinetics and affinity of protein interaction studies were performed by the Octet K2 system (Molecular Devices) using Octet buffer (PBS with 0.025% Tween and 1% BSA). For measuring hFcεRIα interaction, ligand 0.25 μg/mL His-tagged hFcεRIα (Acro Biosystems) was loaded onto Anti-Penta-HIS (HIS1K) Biosensors (Molecular Devices). For OVA interaction, ligand 100 μg/mL OVA was immobilized onto Amine Reactive Second-Generation (AR2G) Biosensors in 10 mM sodium acetate, pH 5 using EDC/Sulfo-NHS based chemistry. Association of analyte OVA-specific SiahIgE or AshIgE was performed in 3-fold serial dilution from 90 to 1 nM or NEUFcε in 3-fold serial dilution from 24 to 0.3 nM in Octet buffer. Analyte dissociation was measured in Octet buffer. Analysis of binding kinetic parameters were performed by Octet data analysis software 10.0 using interaction of ligand-loaded biosensor with no analyte during association phase as the reference sensor.
Immunoblotting for Syk Signaling
1.5 × 106 LAD2 cells were sensitized with PBS or 1 μg/mL OVA-specific SiahIgE or AshIgE. Sensitized cells were washed and resuspended in HEPES buffer the next day followed by 10 μg/mL OVA stimulation at 37°C for indicated times. Cells were immediately centrifuged after OVA stimulation and the cell pellets lysed in ice-cold lysis buffer for 30 min on ice (RIPA buffer (Boston BioProducts), 1x Halt Protease Inhibitor Cocktail (Thermo Scientific), 1x Halt™ Phosphatase Inhibitor Cocktail (Thermo Scientific) and 2.5 mM EDTA). After incubation on ice, lysed pellets were passed rapidly through a 27G needle on ice and centrifuged at 15,000 RPM at 4°C for 15 min to clear the membrane and nuclei. The protein concentration was quantified using Pierce BCA Protein Assay kit (Thermo Scientific) and 20 μg of protein lysate was loaded per well on 4–12% Bis-Tris protein gels (Life Technologies) in SDS-PAGE under denaturing and reducing conditions. Briefly, after protein transferred to PVDF membranes described as above, the membranes were blocked with 5% milk in TBS with 0.1% Tween (TBST) for 1 hour at room temp, washed in TBST, followed by incubation with 1:2000 Rabbit anti-Phospho-Syk (Tyr352) Antibody (Cell Signaling Techology) in 5% BSA in TBST overnight at 4°C. The membrane was then washed in TBST before incubating with anti-rabbit-HRP for 1 hour at room temp and washed in TBST again followed by chemiluminescent detection using Immobilon Western Chemiluminescent HRP Substrate (Millipore Sigma). To detect total Syk on the membrane, after chemiluminescent detection using autoradiography film, the membrane was stripped by incubating in stripping buffer (2% SDS and 0.1 M β-mercaptoethanol in Tris buffer) at 50°C for 30 min. The stripped membranes were then blocked, washed as above and then incubated with 1:2000 Rabbit anti-Syk Antibody (Cell Signaling Techology) for 2 h in 5% BSA in TBST at room temp before incubating with 1:30,000 anti-rabbit-HRP for 1 hour at room temp. To probe for β-Actin, the stripped membranes were incubated with 1:150,000 anti-β-Actin HRP (Santa Cruz Biotechnology) for 1 hour at room temp, washed and signal determined by chemiluminescent detection.
Calcium flux
5 × 105 LAD2 cells were sensitized overnight with PBS or 500 ng/mL OVA-specific SiahIgE or AshIgE. Next day, sensitized cells were washed before loading with 2 μM Fluo-4-AM (Invitrogen) at 37°C in HEPES buffer for 20 minutes. After loading, the cells were washed and resuspended in HEPES buffer. Fluorescence was filtered through the 530/30 band pass filter and collected in FL-1/FITC. Baseline Ca2+ fluorescence levels were recorded for 1 minute on the Accuri C6 (BD Biosciences) before the addition of indicated allergen or buffer to each sample. At the end of allergen stimulation, cells were added 2 μM Ca2+ ionophore A23187 (Sigma) as a positive control.
Statistical analyses
Results are shown as mean ± s.e.m. for all except for the gMS quantified glycan residues per IgE molecule (Fig. 1d–h) where results are presented as median and interquartile range. The number of mice used in each experiment is indicated in the figure legends. Visual examination of the data distribution as well as normality testing demonstrated that all variables appeared to be normally distributed. Statistical analyses were performed using Prism 8 (GraphPad Software) with un-paired and paired Student’s t test for assessing two unmatched and matched groups, respectively, two-way ANOVA with Sidak’s multiple comparison test for comparing two groups of multiple conditions, and one-way or two-way ANOVA with Tukey’s multiple comparison test for three or more groups. The P values denoted throughout the manuscript highlight biologically relevant comparisons. Accuracy of individual IgE glycan moieties capacity to distinguish allergic IgE was analyzed by receiver operating characteristic (ROC) curves by Prism 8 (GraphPad Software). Area under each ROC curve (AUC) was calculated for each glycan moiety. AUC was interpreted as follows, where a maximum AUC of 1 indicates the specific glycan moiety is able to distinguish allergic IgE from non-allergic IgE. An AUC of 0.5 indicates the differentiation capacity of a specific glycan moiety is poor.
Extended Data
Extended Data Fig. 1 |. Characterization of non-atopic and allergic human IgE.
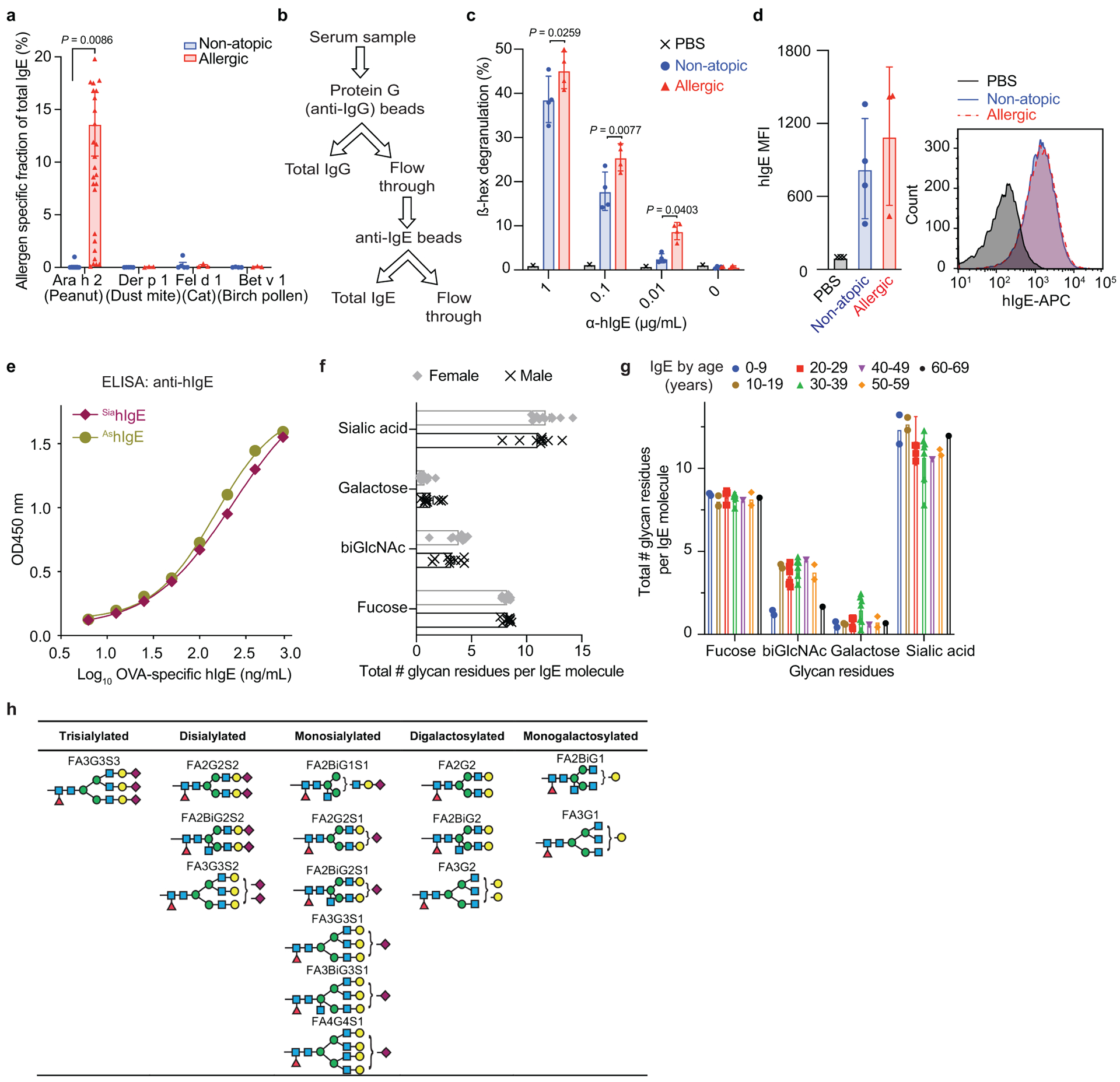
a, Allergen-specific IgE levels for Ara h 2 (peanut: non-atopic n = 11, allergic n = 30), Der p 1 (dust mite: n = 5, 3), Fel d 1 (cat: n = 5, 3), and Bet v 1 (birch pollen: n = 4, 3). b, Strategy for enriching IgE from human sera. c, Quantified degranulation of human LAD2 mast cells sensitized with PBS, non-atopic, or allergic IgE and stimulated by anti-human IgE (PBS n = 1, non-atopic n = 4, allergic n = 4). d, Quantified MFI (left) and representative histograms (right) of anti-hIgE FACS staining on human LAD2 mast cells sensitized with PBS, non-atopic, or allergic hIgE (PBS n = 3, non-atopic n = 4, allergic n = 3). e, Anti-hIgE from Extended Fig. 1c, d, binds similarly to SiahIgE and AshIgE as determined by hIgE ELISA assays. n = 2 technical replicates per group and are representative of three experiments. f, g, IgE glycan distribution by sex (f; n = 9 males, n = 12 females) and age (g; 0–9 (n = 2), 10–19 (n = 2), 20–29 (n = 6), 30–39 (n = 7), 40–49 (n = 1), 50–59 (n = 2), 60–69 (n = 1)). h, Representative structures of complex N-glycans in Fig. 1j. Data are mean ± s.e.m. (a, c, d, f, g). P values were determined by two-tailed unpaired t-test (d, f) or two-way ANOVA with Sidak’s multiple comparison test (a, c, g). n represents biologically independent serum samples (a, c, d, f, g).
Extended Data Fig. 2 |. N-linked glycans observed on native human IgE.
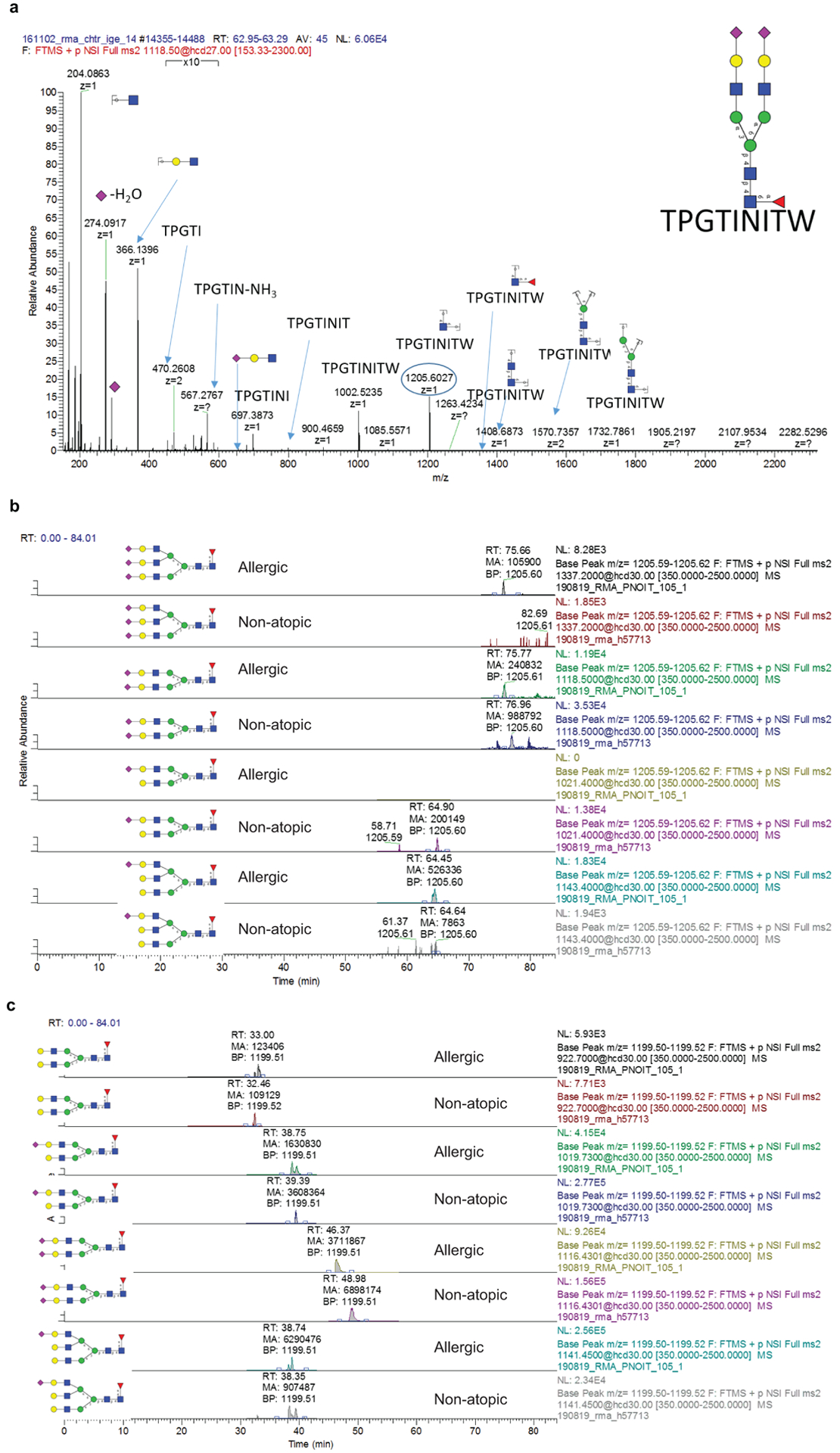
a, Representative MS/MS spectrum for N265 A2F glycopeptide showing B and Y ions from glycosidic bond cleavage as well as B ions from peptide bond cleavage. The Y1 ion used for quantification of glycopeptides is circled (n = 18 biologically independent samples). b, Extracted ion chromatograms for IgE N265 sialylation variants from an allergic patient and non-allergic donor. c, Extracted ion chromatograms for IgE N168 sialylation variants from an allergic patient and non-allergic donor.
Extended Data Fig. 3 |. N-linked glycans observed on IgE myeloma standard.
a,b, Extracted ion chromatograms for site-specific N-glycosylation from chymotryptic (a) or tryptic (b) digest of the IgE myeloma sample used as a standard.
Extended Data Fig. 4 |. Site-specific characterization of resolved IgE glycans from non-atopic and allergic individuals.
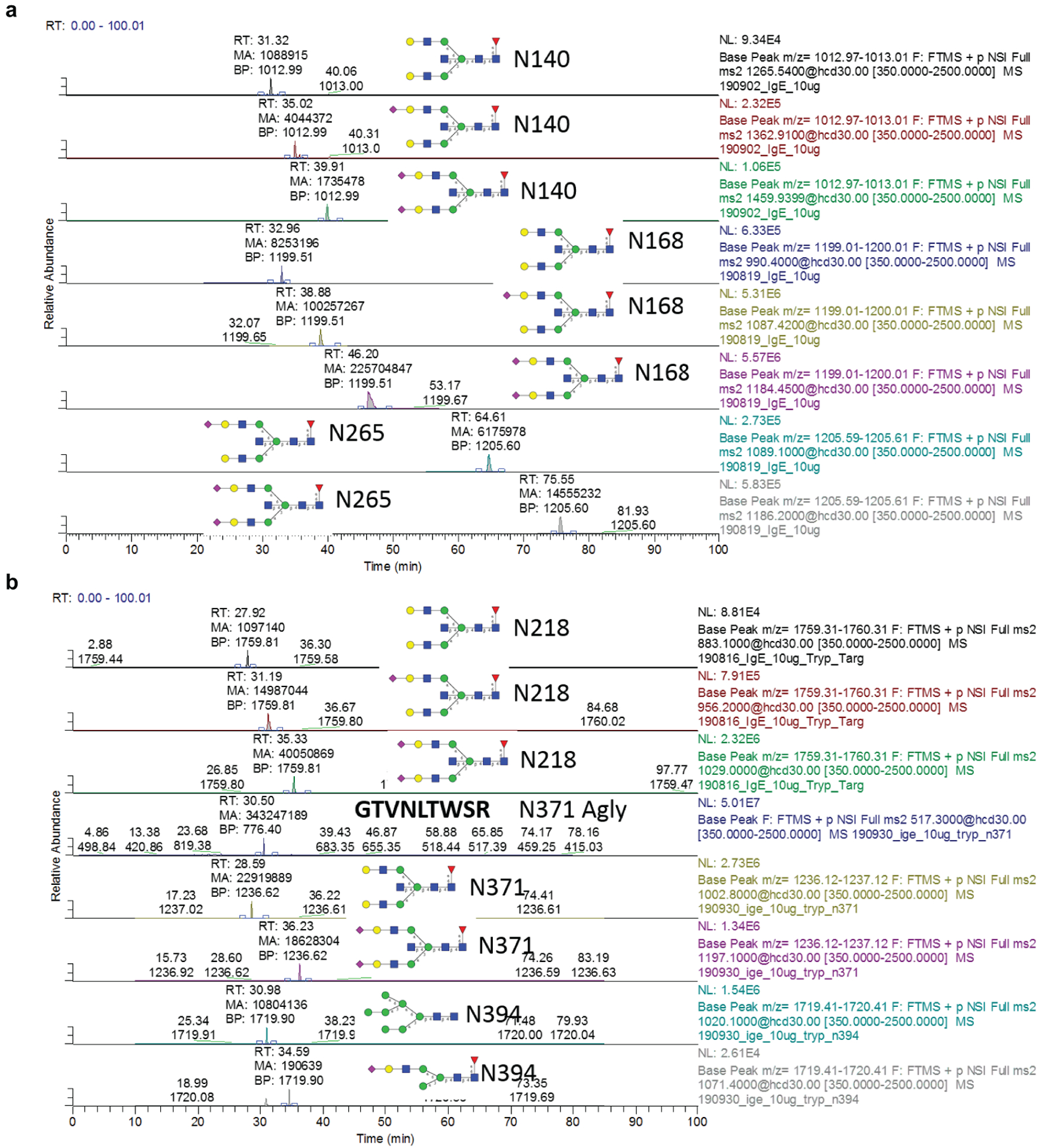
a, Occupancy of N-linked glycosylation sites; N140 (non-atopic n = 15, allergic n = 13), N168 (n = 16, 14), N218 (n = 15, 15), N265 (n = 12, 15), N371 (n = 15, 15), N383 (n = 16, 15), N394 (n = 13, 16). b, Percentage of oligomannose moieties at N394 (n = 23, 18). c, Number of fucose residues; N140 (n = 15, 13), N168 (n = 15, 17), N218 (n = 15, 19), N265 (n = 12, 18), N371 (n = 15, 17). d, Number of biGlcNAc residues; N140 (n = 15, 13), N168 (n = 16, 17), N218 (n = 15, 19), N265 (n = 16, 20), N371 (n = 16, 17). e, Number of galactose residues; N140 (n = 15, 14), N168 (n = 15, 17), N218 (n = 15, 19), N265 (n = 12, 19), N371 (n = 15, 17). f, Number of sialic acid residues; N140 (n = 14, 13), N168 (n = 15, 13), N218 (n = 15, 17), N265 (n = 12, 19), N371 (n = 15, 17). Data plotted are mean ± s.e.m. P values determined by two-way ANOVA with Sidak’s multiple comparison test. n represents biologically independent serum samples (a-f).
Extended Data Fig. 5 |. IgE sialic acid removal.
a, Protein gel stain and lectin blots of IVIG, native human IgE purified from allergic patients, and fetuin. b, HPLC glycan traces of undigested, or allergic human IgE or fetuin digested with sialidase from Arthrobacter ureafaciens for releasing α2,3-, α2,6-, α2,8- and α2,9-linked sialic acids or sialidase from Streptococcus pneumoniae for releasing α2,3-linked sialic acids. c, HPLC glycan traces of undigested or recombinant OVA-specific mIgE digested with sialidase from Arthrobacter ureafaciens. d, Quantitation of vascular leakage by Evan’s blue dye (left, n = 6 mouse ears per group) and representative ear images (right) after PCA in mice sensitized with PBS, or SiamIgE and AsmIgE specific for DNP. e, Gating strategy for IgE loading on mouse skin ear mast cells. Representative FACS plots used to identify mast cells in mouse ears and determine IgE levels on mouse ear mast cells. SSC, side scatter. f, Binding of OVA-specific SiamIgE and AsmIgE to OVA as determined by ELISA. n = 2 technical replicates per group and are representative of three biologically independent experiments. g, h, OVA-elicited systemic anaphylaxis as measured by temperature drop in mice sensitized with PBS, OVA-specific SiamIgE (n = 4 for g and 6 for h) or AsmIgE (n = 5 for g and 6 for h) by intravenous (g) or intraperitoneal (h) injection. i, Serum levels of DNP-specific SiamIgE (n = 4) and AsmIgE (n = 3) in mice at defined times after systemically administration as determined by ELISA. Data are mean ± s.e.m. and are representative of three experiments. P values determined by two-tailed paired t-test (d), or two-way ANOVA with Tukey’s multiple comparison test (g, h).
Extended Data Fig. 6 |. FACS analysis of human LAD2 mast cell loading of SiahIgE and AshIgE, phenotypic staining of PBMC-derived mast cells, and activation in primary basophils.
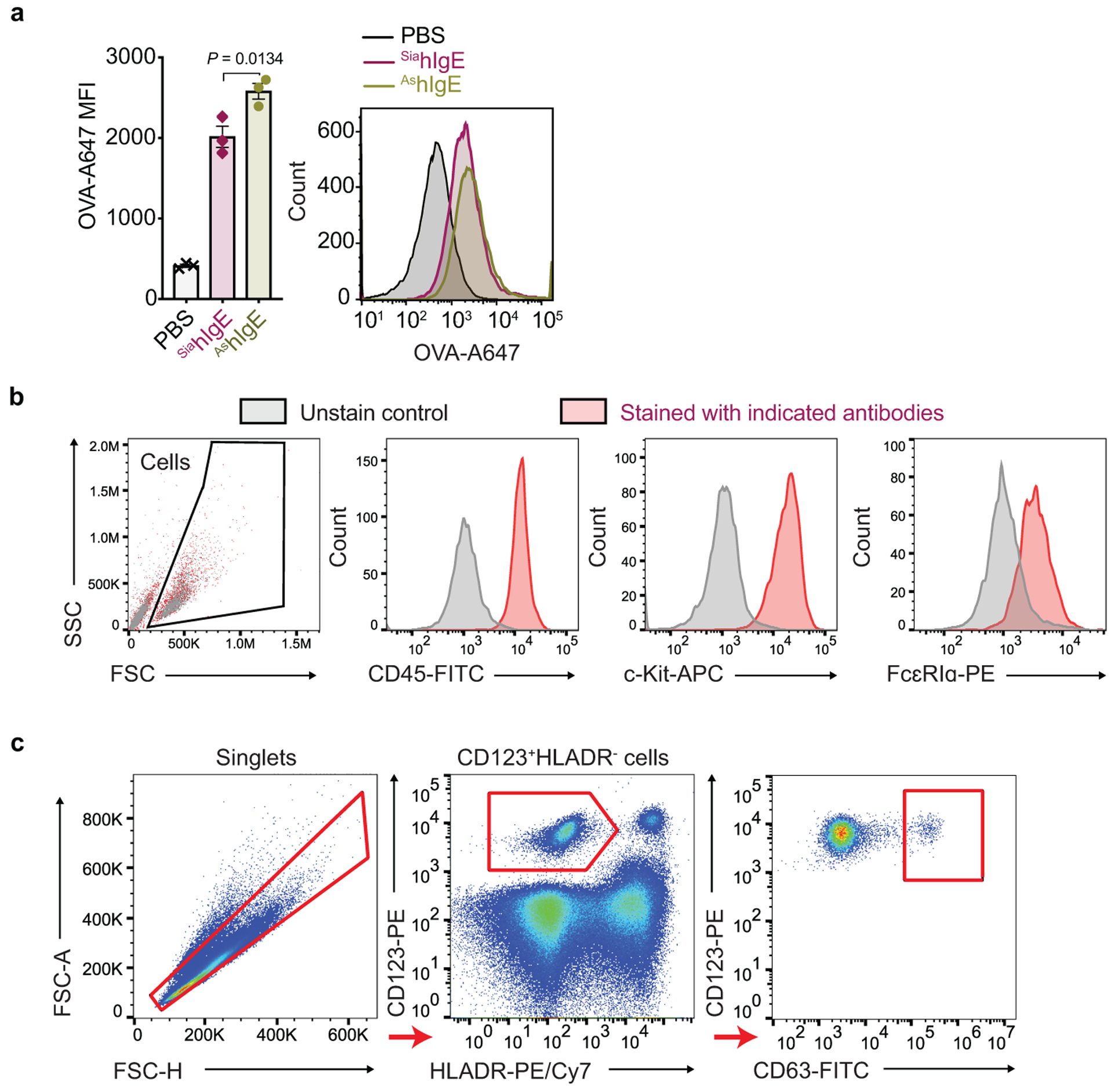
a, MFI (left) and representative histogram (right) of surface-bound hIgE on LAD2 mast cells following sensitization with PBS, OVA-specific SiahIgE or AshIgE (n = 3 technical replicates per group). Data are mean ± s.e.m. and are representative of three independent experiments. One-way ANOVA with Tukey’s multiple comparison test. b, Representative phenotypic staining by FACS of primary human mast cells from peripheral blood-derived CD34+ pluripotent hematopoietic cells (n = 2 technical replicates per group). c, Gating strategy for basophil activation assay. Representative FACS plots used to determine basophil activation from PBMC.
Extended Data Fig. 7 |. PSA for IgE isotype controls and characterization of NEUFcε.
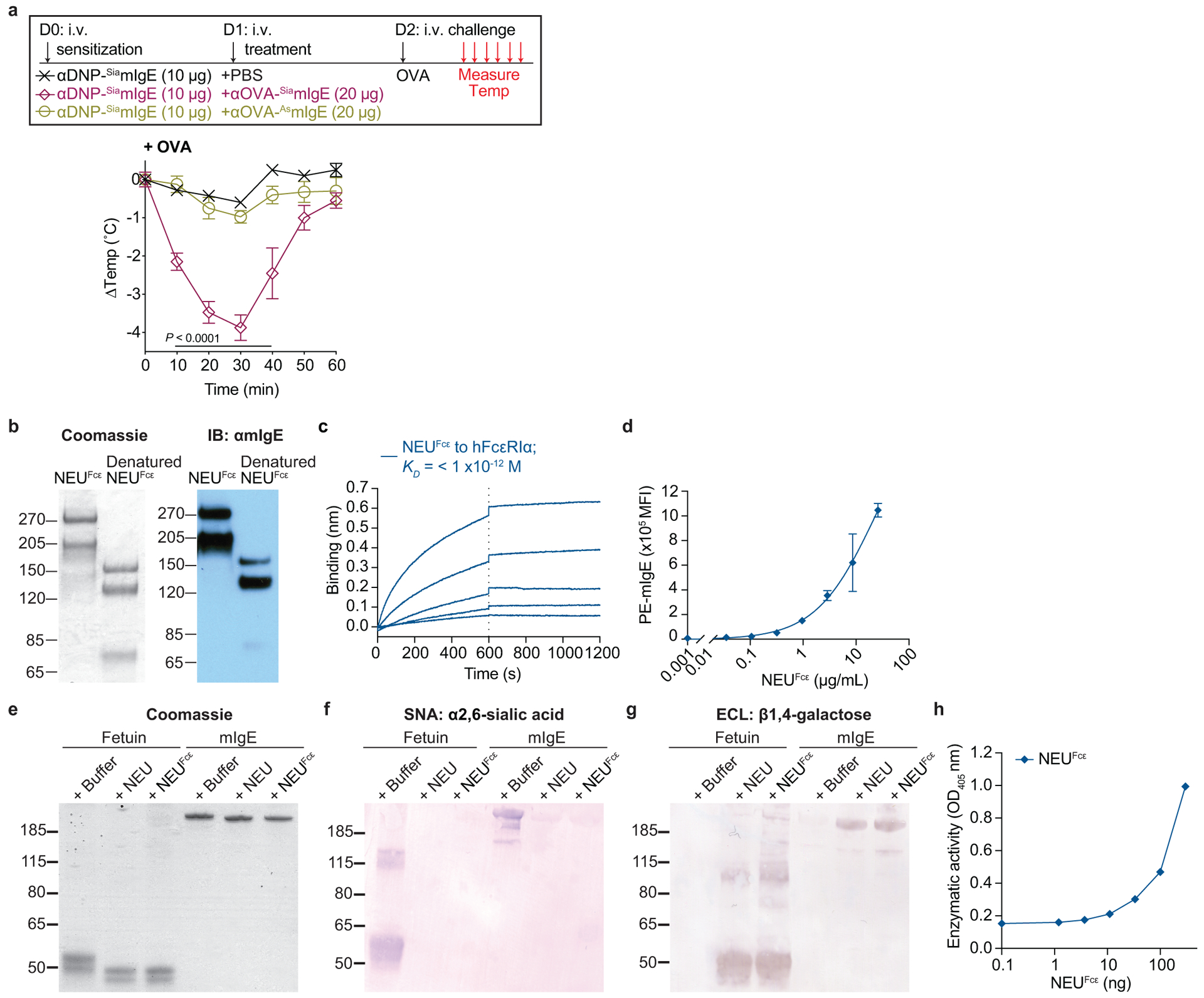
a, Temperature change following OVA-induced PSA in mice receiving DNP-specific SiamIgE on day 0 and PBS, OVA-specific SiamIgE, or OVA-specific AsmIgE isotype controls from Fig. 3e on day 1. n = 4 mice for all groups. Two-way ANOVA with Tukey’s multiple comparison test. b, Protein gel stain (left) and immunoblot for mIgE (right) of native and denatured NEUFcε. c, Binding kinetics of analyte NEUFcε to ligand hFcεRIα on biosensor. Analytes kinetics were performed with 3-fold serial dilution of analyte from 26.2 to 0.32 nM. d, MFI of surface-bound NEUFcε on LAD2 mast cells following 30 min sensitization at 37°C by FACS analysis (n = 3 technical replicates per group and are representative from two independent experiments). e-h, Neuraminidase activity of NEUFcε determined by digestion of mIgE or fetuin overnight (e-g) and detection of protein loading by coomassie (e), terminal α2,6-sialic acid by SNA (f), and terminal galactose by ECL (g) or by the amount of substrate 2-O-(p-Nitrophenyl)-α-D-N-acetylneuraminic acid digested by NEUFcε in a colorimetric assay (h, n = 3 technical replicates per group and are representative from two independent experiments). Data are mean ± s.e.m.
Extended Data Table 1 |.
Patient Demographic Data
| Sample # | Diagnosis | Gender | Age | Other Atopies* |
|---|---|---|---|---|
| 73 | Allergic | M | 22 | Yes |
| 10 | Allergic | F | 25 | Yes |
| 22 | Allergic | F | 19 | Yes |
| 34 | Allergic | F | 30 | Yes |
| 51 | Allergic | F | 15 | Yes |
| 60 | Allergic | F | 40 | Yes |
| 61 | Allergic | F | 27 | Yes |
| 67 | Allergic | F | 52 | Yes |
| 84 | Allergic | M | 22 | Yes |
| 97 | Allergic | F | 36 | Yes |
| 24 | Allergic | M | 36 | Yes |
| 33 | Allergic | F | 30 | Yes |
| 34 | Allergic | F | 16 | Yes |
| 69 | Allergic | M | 15 | Yes |
| 80 | Allergic | F | 8 | Yes |
| 95 | Allergic | M | 8 | Yes |
| 97 | Allergic | F | 36 | Yes |
| 100 | Allergic | F | 22 | Yes |
| 105 | Allergic | F | 22 | Yes |
| 111 | Allergic | F | 22 | Yes |
| 106 | Allergic | F | 32 | Yes |
| 149 | Non-atopic | M | 27 | No |
| 349 | Non-atopic | F | 35 | No |
| 241 | Non-atopic | M | 29 | No |
| 528 | Non-atopic | M | 38 | No |
| 53208 | Non-atopic | M | 36 | No |
| 53209 | Non-atopic | M | 37 | No |
| 53210 | Non-atopic | M | 39 | No |
| 53211 | Non-atopic | F | 31 | No |
| 53195 | Non-atopic | M | 60 | No |
| 57543 | Non-atopic | M | 69 | No |
| 57544 | Non-atopic | F | 27 | No |
| 57546 | Non-atopic | M | 22 | No |
| 57699 | Non-atopic | M | 58 | No |
| 57713 | Non-atopic | M | 32 | No |
| 57714 | Non-atopic | M | 28 | |
| 56986 | Non-atopic | M | 29 | |
| 56988 | Non-atopic | F | 50 | |
| 57527 | Non-atopic | F | 29 |
Extended Data Table 2 |.
Targeted mass list for IgE glycosylation sites N140, N168 and N265 glycopeptides from the chymotryptic digest and N218, N371 and N394 glycopeptides from the tryptic digest
| FACS assay | Target | Clone | Fluorochromes | Vendor | Dilution |
|---|---|---|---|---|---|
| IgE loading on mouse skin mast cells | Mouse CD45 | 30-F11 | APC/Cyanine7 | Biolegend | 1:400 |
| Mouse/human CD11b | M1/70 | PerCP/Cyanine5.5 | Biolegend | 1:100 | |
| Mouse CD11c | N418 | FITC | Biolegend | 1:100 | |
| Mouse Ly-6G/Ly-6C (Gr-1) | RB6–8C5 | FITC | Biolegend | 1:100 | |
| Mouse CD117 (c-Kit) | 2B8 | APC | Biolegend | 1:100 | |
| Mouse IgE | RME-1 | PE | Biolegend | 1:100 | |
| IgE loading on human LAD2 cells | Human CD117 (c-kit) | 104D2 | PE | Biolegend | 1:400 |
| Ovalbumin | A647 | Invitrogen | 1:400 of 2 mg/mL stock | ||
| Human IgE | MHE-18 | APC | Biolegend | 1:400 | |
| Basophil activation tests | Human HLA-DR | L243 | PE/Cy7 | Biolegend | 1:35 |
| Human CD123 | 6H6 | PE | Biolegend | 1:30 | |
| Human CD63 | H5C6 | FITC | Biolegend | 1:30 | |
| Phenotype staining for human mast cells | Human CD117 (c-kit) | 104D2 | APC | Biolegend | 1:400 |
| Human CD45 | HI30 | FITC | Biolegend | 1:100 | |
| Human FcεRIα | AER-37 (CRA-1) | PE | Biolegend | 1:400 | |
| General | Viability | eFluor450 | eBioscience | 1:500 | |
| ELISA assay | Target | Catalog # | Conjugation | Vendor | Dilution |
| Mouse IgE ELISA | mlgE | A90–115A | No | Bethyl | 1:200 |
| mlgE | A90–115P | HRP | Bethyl | 1:30,000 | |
| Human IgE ELISA | hlgE | A80–108A | No | Bethyl | 1:200 |
| hlgE | A80–108P | HRP | Bethyl | 1:30,000 | |
| Immunoblotting | Target | Catalog # | Species/conjugation | Vendor | Dilution |
| Immunoblotting for Syk | Phospho-Syk | 2701 | Rabbit | Cell Signaling Technology | 1:2000 |
| Total | 2712 | Rabbit | Cell Signaling Technology | 1:2000 | |
| Secondary antibody | Rabbit IgG | W4011 | HRP | Promega | 1:30,000 |
| Immunoblotting for Actin | Actin | SC-47778 HRP | HRP | Santa Cruz Biotechnology | 1:50,000 |
| Passive Anaphylaxis | Species | Clone | Antigen specificity | Vendor | |
| IgE | Mouse | SPE-7 | dinitrophenyl (DNP) | Sigma-Aldrich | |
| IgE | Mouse | MEA-36 | Trinitrophenyl (TNP) | Biolegend |
Extended Data Table 3 |.
Pertinent information for commercial antibody reagents
| Mass [m/z] | CS [z] | Polarity | Start [min] | End [min] | (N)CE | Comment |
|---|---|---|---|---|---|---|
| 1197.84000 | 3 | Positive | 23.00 | 30.00 | 27 | IgE N140 G2F |
| 1295.22000 | 3 | Positive | 28.00 | 34.00 | 27 | IgE N140 A1F |
| 1265.54000 | 3 | Positive | 25.00 | 33.00 | 27 | IgE N140 G2F+BglcNAc |
| 1362.91000 | 3 | Positive | 29.00 | 34.00 | 27 | IgE N140 A1F+BGlcNAc |
| 1391.91000 | 3 | Positive | 32.00 | 37.00 | 27 | IgE N140 A2F |
| 1459.94000 | 3 | Positive | 32.00 | 37.00 | 27 | IgE N140 A2F+BglcNAc |
| 1416.94000 | 3 | Positive | 28.00 | 34.00 | 27 | IgE N140 A1F+LAcNAc |
| 1513.97000 | 3 | Positive | 32.00 | 37.00 | 27 | IgE N140 A2F+LacNAc |
| 1319.57000 | 3 | Positive | 23.00 | 30.00 | 27 | IgE N140 G2F+LacNAc |
| 922.70000 | 3 | Positive | 30.00 | 34.00 | 27 | IgE N168 G2F |
| 990.40000 | 3 | Positive | 30.00 | 34.00 | 27 | IgE N168 G2F+BGlcNAc |
| 1019.73000 | 3 | Positive | 36.00 | 40.00 | 27 | IgE N168 A1F |
| 1087.42000 | 3 | Positive | 36.00 | 40.00 | 27 | IgE N168 A1F+BGlcNAc |
| 1184.45000 | 3 | Positive | 42.00 | 50.00 | 27 | IgE N168 A2F+BGlcNAc |
| 1116.43000 | 3 | Positive | 42.00 | 50.00 | 27 | IgE N168 A2F |
| 1141.45000 | 3 | Positive | 36.00 | 40.00 | 27 | IgE N168 A1F+LAcNAc |
| 1238.48000 | 3 | Positive | 42.00 | 50.00 | 27 | IgE N168 A2F+LAcNAc |
| 1044.43000 | 3 | Positive | 30.00 | 34.00 | 27 | IgE N168 G2F+LAcNAc |
| 501.80000 | 2 | Positive | 45.00 | 55.00 | 27 | IgE N265 Agly |
| 1337.20000 | 3 | Positive | 65.00 | 75.00 | 27 | IgE N265 A3F |
| 992.10000 | 3 | Positive | 35.00 | 55.00 | 27 | IgE N265 G2F+GlcNAc |
| 1118.50000 | 3 | Positive | 60.00 | 65.00 | 27 | IgE N265 A2F |
| 1186.20000 | 3 | Positive | 60.00 | 65.00 | 27 | IgE N265 A2F+GlcNAc |
| 1089.10000 | 3 | Positive | 52.00 | 58.00 | 27 | IgE N265 A1F+GlcNAc |
| 924.40000 | 3 | Positive | 35.00 | 55.00 | 27 | IgE N265 G2F |
| 1021.40000 | 3 | Positive | 52.00 | 58.00 | 27 | IgE N265 A1F |
| 1143.40000 | 3 | Positive | 52.00 | 58.00 | 27 | IgE N265 A1F+LacNAc |
| 1240.50000 | 3 | Positive | 60.00 | 65.00 | 27 | IgE N265 A2F+LacNAc |
| 905.40000 | 4 | Positive | 27.00 | 32.00 | 27 | N218 A1F |
| 883.10000 | 4 | Positive | 24.00 | 30.00 | 27 | N218 G2F+GlcNAc |
| 1231.50000 | 3 | Positive | 23.00 | 30.00 | 27 | N218 G3F |
| 519.60000 | 3 | Positive | 39.00 | 55.00 | 27 | Agly N218 |
| 1029.00000 | 4 | Positive | 48.00 | 63.00 | 27 | N218 A2F+GlcNAc |
| 1142.20000 | 4 | Positive | 35.00 | 45.00 | 27 | N218 A3F |
| 1123.15000 | 3 | Positive | 24.00 | 30.00 | 27 | N218 G1F+GlcNAc |
| 1069.10000 | 4 | Positive | 30.00 | 38.00 | 27 | N218 A2F+LacNAc |
| 956.20000 | 4 | Positive | 27.00 | 33.00 | 27 | N218 A1F+GlcNAc |
| 996.70000 | 4 | Positive | 27.00 | 33.00 | 27 | N218 A1F+LacNAc |
| 1109.13000 | 3 | Positive | 24.00 | 30.00 | 27 | N218 G2F |
| 978.20000 | 4 | Positive | 30.00 | 38.00 | 27 | N218 A2F |
| 1002.80000 | 3 | Positive | 30.00 | 37.00 | 27 | N371 G2F+GlcNAc |
| 1099.80000 | 3 | Positive | 35.00 | 40.00 | 27 | N371 A1F+GlcNAc |
| 1197.10000 | 3 | Positive | 40.00 | 50.00 | 27 | N371 A2F+GlcnAc |
| 948.80000 | 3 | Positive | 30.00 | 38.00 | 27 | N371 G1F+GlcNAc |
| 1153.80000 | 3 | Positive | 35.00 | 40.00 | 27 | N371 A1F+LacNAc |
| 1032.80000 | 3 | Positive | 34.00 | 39.00 | 27 | N371 A1F |
| 1045.80000 | 3 | Positive | 35.00 | 41.00 | 27 | N371 G1F+GlcNAc+NeuAc |
| 1129.80000 | 3 | Positive | 40.00 | 50.00 | 27 | N371 A2F |
| 935.10000 | 3 | Positive | 30.00 | 35.00 | 27 | N371 G2F+GlcNAc |
| 1056.80000 | 3 | Positive | 30.00 | 35.00 | 27 | N371 G3F |
| 517.30000 | 2 | Positive | 30.00 | 38.00 | 27 | N371 Agly |
| 912.10000 | 3 | Positive | 34.00 | 40.00 | 27 | N394 HM5 |
| 966.10000 | 3 | Positive | 34.00 | 40.00 | 27 | N394 HM6 |
| 1020.10000 | 3 | Positive | 34.00 | 40.00 | 27 | N394 HM7 |
| 1074.20000 | 3 | Positive | 34.00 | 40.00 | 27 | N394 HM8 |
| 1128.20000 | 3 | Positive | 34.00 | 40.00 | 27 | N394 HM9 |
| 1071.40000 | 3 | Positive | 38.00 | 45.00 | 27 | N394 A1F-LAcNAc |
| 1179.50000 | 3 | Positive | 38.00 | 45.00 | 27 | N394 3,6,1,1,0 |
| 1130.50000 | 3 | Positive | 38.00 | 45.00 | 27 | N394 3,6,0,1,0 |
Supplementary Material
Acknowledgements
The authors are grateful to Kate Jeffrey, Fredrik Wermeling, and Andrew Luster for constructive comments, Takara Stanley and Douglas Hayden (Harvard Catalyst) for biostatistical analysis guidance, Neal Smith for technical assistance, and to the individuals participating in our studies. This work was supported by grants from NIH (DP2AR068272, R01AI139669) and FARE to RMA, NIH T32 and K12 fellowships (T32AR007258, K12HL141953) to KTS, NIH NIAID K23 grant (K23AI121491) to SP, NIH NIAID U19 (5U19 AI095261-03) to WS, and a Sanofi iAward to MEC.
Footnotes
Competing interests
The authors declare no competing interests.
Data Availability
Source data tables are provided for Figs. 1–3 and Extended Data Figs. 1, 4–7. Full scans of all uncropped protein gel stains, lectin or western blots with size marker indication presented in this manuscript are shown in the Supplementary Information. All other data supporting the findings in this study are available from the corresponding author upon request.
References
- 1.Gupta RS et al. Prevalence and Severity of Food Allergies Among US Adults. JAMA Netw Open 2, e185630, doi: 10.1001/jamanetworkopen.2018.5630 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gould HJ & Sutton BJ IgE in allergy and asthma today. Nature Reviews Immunology 8, 205–217, doi: 10.1038/nri2273 (2008). [DOI] [PubMed] [Google Scholar]
- 3.Bird JA, Crain M & Varshney P Food allergen panel testing often results in misdiagnosis of food allergy. J Pediatr 166, 97–100, doi: 10.1016/j.jpeds.2014.07.062 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Du Toit G et al. Randomized trial of peanut consumption in infants at risk for peanut allergy. The New England Journal of Medicine 372, 803–813, doi: 10.1056/NEJMoa1414850 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peters RL, Gurrin LC & Allen KJ The predictive value of skin prick testing for challenge-proven food allergy: a systematic review. Pediatr Allergy Immunol 23, 347–352, doi: 10.1111/j.1399-3038.2011.01237.x (2012). [DOI] [PubMed] [Google Scholar]
- 6.Arnold JN, Wormald MR, Sim RB, Rudd PM & Dwek RA The impact of glycosylation on the biological function and structure of human immunoglobulins. Annual Review of Immunology 25, 21–50 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Begin P et al. Phase 1 results of safety and tolerability in a rush oral immunotherapy protocol to multiple foods using Omalizumab. Allergy, Asthma, and Clinical Immunology : official journal of the Canadian Society of Allergy and Clinical Immunology 10, 7, doi: 10.1186/1710-1492-10-7 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Patil SU et al. Peanut oral immunotherapy transiently expands circulating Ara h 2-specific B cells with a homologous repertoire in unrelated subjects. The Journal of Allergy and Clinical Immunology 136, 125–134 e112, doi: 10.1016/j.jaci.2015.03.026 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang TT et al. IgG antibodies to dengue enhanced for FcgammaRIIIA binding determine disease severity. Science 355, 395–398, doi: 10.1126/science.aai8128 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu LL et al. A Functional Role for Antibodies in Tuberculosis. Cell 167, 433–443 e414, doi: 10.1016/j.cell.2016.08.072 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang TT et al. Anti-HA Glycoforms Drive B Cell Affinity Selection and Determine Influenza Vaccine Efficacy. Cell 162, 160–169, doi: 10.1016/j.cell.2015.06.026 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parekh RB et al. Association of rheumatoid arthritis and primary osteoarthritis with changes in the glycosylation pattern of total serum IgG. Nature 316, 452–457, doi: 10.1038/316452a0 (1985). [DOI] [PubMed] [Google Scholar]
- 13.Espy C et al. Sialylation levels of anti-proteinase 3 antibodies are associated with the activity of granulomatosis with polyangiitis (Wegener’s). Arthritis and Rheumatism 63, 2105–2115, doi: 10.1002/art.30362 (2011). [DOI] [PubMed] [Google Scholar]
- 14.Wang TT & Ravetch JV Functional diversification of IgGs through Fc glycosylation. The Journal of Clinical Investigation 129, 3492–3498, doi: 10.1172/JCI130029 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arnold JN et al. The glycosylation of human serum IgD and IgE and the accessibility of identified oligomannose structures for interaction with mannan-binding lectin. Journal of Immunology 173, 6831–6840 (2004). [DOI] [PubMed] [Google Scholar]
- 16.Plomp R et al. Site-Specific N-Glycosylation Analysis of Human Immunoglobulin E. Journal of Proteome Research, doi: 10.1021/pr400714w (2013). [DOI] [PubMed] [Google Scholar]
- 17.Shade KT et al. A single glycan on IgE is indispensable for initiation of anaphylaxis. The Journal of Experimental Medicine 212, 457–467, doi: 10.1084/jem.20142182 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu G et al. Glycoproteomic studies of IgE from a novel hyper IgE syndrome linked to PGM3 mutation. Glycoconj J 33, 447–456, doi: 10.1007/s10719-015-9638-y (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamazaki T et al. Receptor-destroying enzyme (RDE) from Vibrio cholerae modulates IgE activity and reduces the initiation of anaphylaxis. The Journal of Biological Chemistry 294, 6659–6669, doi: 10.1074/jbc.RA118.006375 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jabs F et al. Trapping IgE in a closed conformation by mimicking CD23 binding prevents and disrupts FcepsilonRI interaction. Nature Communications 9, 7, doi: 10.1038/s41467-017-02312-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wuhrer M et al. Glycosylation profiling of immunoglobulin G (IgG) subclasses from human serum. Proteomics 7, 4070–4081, doi: 10.1002/pmic.200700289 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Nimmerjahn F & Ravetch JV Anti-inflammatory actions of intravenous immunoglobulin. Annual Review of Immunology 26, 513–533 (2008). [DOI] [PubMed] [Google Scholar]
- 23.Ding JX et al. Aberrant sialylation of serum IgA1 was associated with prognosis of patients with IgA nephropathy. Clinical Immunol 125, 268–274, doi: 10.1016/j.clim.2007.08.009 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Maurer MA et al. Glycosylation of Human IgA Directly Inhibits Influenza A and Other Sialic-Acid-Binding Viruses. Cell Reports 23, 90–99, doi: 10.1016/j.celrep.2018.03.027 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Colucci M et al. Sialylation of N-linked glycans influences the immunomodulatory effects of IgM on T cells. Journal of Immunology 194, 151–157, doi: 10.4049/jimmunol.1402025 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Collins BE, Smith BA, Bengtson P & Paulson JC Ablation of CD22 in ligand-deficient mice restores B cell receptor signaling. Nature Immunology 7, 199–206, doi: 10.1038/ni1283 (2006). [DOI] [PubMed] [Google Scholar]
- 27.Abe K, Yamamoto K & Sinohara H Clearance of desialylated mouse alpha-macroglobulin and murinoglobulin in the mouse. J Biochem 108, 726–729, doi: 10.1093/oxfordjournals.jbchem.a123272 (1990). [DOI] [PubMed] [Google Scholar]
- 28.Fleischer DM et al. Oral food challenges in children with a diagnosis of food allergy. J Pediatr 158, 578–583 e571, doi: 10.1016/j.jpeds.2010.09.027 (2011). [DOI] [PubMed] [Google Scholar]
- 29.Sampath V et al. Deciphering the black box of food allergy mechanisms. Ann Allergy Asthma Immunol 118, 21–27, doi: 10.1016/j.anai.2016.10.017 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Christensen S & Egebjerg J Cloning, expression and characterization of a sialidase gene from Arthrobacter ureafaciens. Biotechnol Appl Biochem 41, 225–231, doi: 10.1042/BA20040144 (2005). [DOI] [PubMed] [Google Scholar]
- 31.Dombrowicz D et al. Anaphylaxis mediated through a humanized high affinity IgE receptor. Journal of immunology 157, 1645–1651 (1996). [PubMed] [Google Scholar]
- 32.Dombrowicz D, Flamand V, Brigman KK, Koller BH & Kinet JP Abolition of anaphylaxis by targeted disruption of the high affinity immunoglobulin E receptor alpha chain gene. Cell 75, 969–976 (1993). [DOI] [PubMed] [Google Scholar]
- 33.MacGlashan D Jr., Lavens-Phillips S & Katsushi M IgE-mediated desensitization in human basophils and mast cells. Front Biosci 3, d746–756 (1998). [DOI] [PubMed] [Google Scholar]
- 34.Kirshenbaum AS et al. Characterization of novel stem cell factor responsive human mast cell lines LAD 1 and 2 established from a patient with mast cell sarcoma/leukemia; activation following aggregation of FcepsilonRI or FcgammaRI. Leukemia research 27, 677–682 (2003). [DOI] [PubMed] [Google Scholar]
- 35.Bandara G, Metcalfe DD & Kirshenbaum AS Growth of human mast cells from bone marrow and peripheral blood-derived CD34(+) pluripotent hematopoietic cells. Methods in molecular biology 1220, 155–162, doi: 10.1007/978-1-4939-1568-2_10 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anthony RM et al. Recapitulation of IVIG anti-inflammatory activity with a recombinant IgG Fc. Science 320, 373–376 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.