

Abstract
Billions of US dollars are invested every year by the pharmaceutical industry in drug development, with the aim of introducing new drugs that are effective and have minimal side effects. Thirty percent of in-pipeline drugs are excluded in an early phase of preclinical and clinical screening owing to cardiovascular safety concerns, and several lead molecules that pass the early safety screening make it to market but are later withdrawn owing to severe cardiac side effects. Although the current drug safety screening methodologies can identify some cardiotoxic drug candidates, they cannot accurately represent the human heart in many aspects, including genomics, transcriptomics, and patient- or population-specific cardiotoxicity. Despite some limitations, human induced pluripotent stem cell–derived cardiomyocytes (hiPSC-CMs) are a powerful and evolving technology that has been shown to recapitulate many attributes of human cardiomyocytes and their drug responses. In this review, we discuss the potential impact of the inclusion of the hiPSC-CM platform in premarket candidate drug screening
Keywords: human induced pluripotent stem cells, differentiation, cardiac, cardiomyocyte, cardiotoxicity, pharmacogenomics, chemotherapy
INTRODUCTION
During the process of drug development, detection of toxicity to the heart is the leading cause for halting further progression of lead compounds. Many drugs have been associated with various cardiovascular side effects, including disturbances in ventricular de- or repolarization leading to lethal arrhythmias and cardiomyocyte injury leading to heart failure. About 30% of potential drug candidates are discontinued during the clinical development phase owing to safety concerns; the majority of these are excluded because of adverse cardiovascular reactions (1,55). Genetic, epigenetic, and nongenetic environmental factors can also affect drug toxicity. Genetic variants that have a deleterious effect in one ethnic group may have no effect in another ethnic group. As a result, cardiotoxicities may only be discovered late in drug development, and in some cases only after the drug has been brought to market, with enormous clinical and economic burdens on the health care system. Two major factors have been responsible for this situation: First, existing methodologies for assessing cardiotoxicity, which employ models such as ion channel–overexpressing cells or animal models, are often inaccurate for detecting human cardiac drug responses; and second, some rare, patient-specific drug responses are difficult or impossible to discover in the small cohorts of patients studied during Phase II and Phase III clinical trials. To overcome these limitations, a system must be developed that can recapitulate patient- or population-specific drug response, being both specific and sensitive enough to minimize false positives and false negatives.
DRUG-INDUCED CARDIOTOXICITY
Adverse drug reactions (ADRs) are a significant cause of morbidity and mortality worldwide. The number of ADRs reported by the US Food and Drug Administration (FDA) Adverse Event Reporting System has increased 4-fold, from 335,751 in 2006 to 1,204,685 in 2014 (2). Cardiac ADRs include disturbances in ventricular repolarization and QT interval, arrhythmias, bradycardia, tachycardia, decreases in left ventricular ejection fraction, and congestive heart failure. Making the situation even more complex, drug induced cardiovascular adverse events may present either as early-onset acute events detected shortly after the initiation of treatment or as delayed-onset chronic events, which take longer to progress to the point of detection and therefore might only be discovered during postmarket monitoring.
Notably, nearly 2,000 marketed drugs have been associated with cardiovascular side effects, including drugs with both cardiovascular and noncardiovascular indications (3) (Figure 1a). Anticancer agents in particular are associated with undesired cardiac side effects with incidence rates of 8–26% for doxorubicin, 7–28% for trastuzumab, 5–30% for paclitaxel, and 2–35% for tyrosine kinase inhibitors (TKIs) (4). High-dose corticosteroids are associated with atrial fibrillation in 11% of patients, most likely due to direct effects on cell membranes causing an increase in potassium efflux (5, 6). Using the Tennessee Medicaid insurance database, Ray et al. (7, 8) showed that the antibiotic erythromycin and tricyclic antidepressants are associated with sudden cardiac death (SCD). The risk of SCD increases significantly upon coadministration of erythromycin with a CYP3A4 inhibitor such as verapamil or diltiazem, CYP3A4 being the primary enzyme responsible for erythromycin metabolism.
Figure 1.

(a) On-market drugs and cardiovascular adverse events. Data adapted from the SIDER 4.1: Side Effect Resource (http://sideeffects.embl.de) database of drugs and adverse drug reactions (3). (b) Drug withdrawal due to serious cardiovascular adverse reactions. Sixty-three drugs in different therapeutic classes were withdrawn from the market between 1953 and 2013 owing to serious cardiotoxic effects.
DRUG DEVELOPMENT AND POSTMARKETING DRUG WITHDRAWAL
The failure rates in drug development due to safety issues during Phase II and Phase III trials are 22% and 35%, respectively. This increase in the failure rate from Phase II (shorter follow-up) to Phase III (longer follow-up) likely occurs because many drug-induced toxicities develop and progress with time. The vast majority of drug-induced toxicities occur at low frequencies, making it difficult to get a robust signal when examining only a few hundred to a few thousand patients, as recommended in the FDA guidelines for Phase III clinical trials (1). Accordingly, current methodologies for premarket screening can detect only toxicities that are common and develop within relatively short intervals, increasing the probability of overlooking adverse effects. The high incidence of postmarketing drug withdrawal emphasizes the urgent need to develop preclinical and clinical drug screening schemes that reliably identify adverse effects during lead compound development to avoid the unnecessary clinical and economic burdens associated with postmarketing ADRs. Achieving this goal will also help enhance postmarketing drug safety monitoring through pharmacovigilance.
A total of 462 drugs were withdrawn from the market between 1953 and 2013, 63 (14%) because of serious cardiovascular adverse events (9) (Figure 1b). This long list includes psychostimulants such as fenfluramine, sibutramine, benfluorex, and dexfenfluramine; nonsteroidal anti-inflammatory drugs such as valdecoxib, rofecoxib, alphacetylmethadol, and parecoxib; antiarrhythmics such as adenosine phosphate, bepridil, and encainide; and antipsychotics such as sertindole. Several of these have been on the market for an extended time, including benfluorex, available for 33 years (1976–2009), with its first drug-induced cardiotoxicity (DIC) reported in 2003; bepridil, available for 23 years (1981–2004), with its first DIC reported in 1982; and rofecoxib, available for 5 years (1999–2004), with its first DIC reported in 2002 (9). A study examining ADRs in 8,208,960 hospitalized Medicare patients showed the cost of managing complex ADRs (hospitalization, medical monitoring, and prescribing additional drugs to treat toxicity) was over $300,000,000 (10). The existing approaches for premarketing safety screening for in-pipeline candidate drug molecules are clearly insufficient.
SAFETY SCREENING ASSAYS IN DRUG DEVELOPMENT
Several efforts have been made to develop clinical and preclinical cardiotoxicity screening approaches over the past decade, starting with the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) guidelines ICH S7B and ICH E14.
ICH S7B describes the evaluation of a drug’s potential to delay ventricular repolarization and comprises two main assays. First, an in vitro delayed rectifier potassium current (IKr) functional assay tests the effect of a drug on the voltage-gated potassium channel Kv11.1 (formerly known as human ether-à-go-go, or hERG) using cell lines that heterologously express hERG. Second, an in vivo QT assay tests the effect of a drug on the electrocardiographic QT interval using animal models. Its companion guideline, ICH E14, includes the clinical electrocardiographic evaluation of QT/QTc interval prolongation and proarrhythmic potential in humans in clinical studies.
The sensitivity and specificity of the in vitro hERG assay are 64–82% and 75–88%, respectively, suggesting that even in the best-case scenario, many tested drugs that are actually noncardiotoxic would be discarded at this stage (11, 12). Similarly, a large proportion of tested drugs that may cause deleterious adverse cardiac events can pass this screening stage and potentially reach the market. Assaying hERG currents in noncardiac cells that heterologously express hERG may not accurately recapitulate the electrophysiology of adult human cardiomyocytes. Furthermore, the substantial physiological differences between humans and animals constitute a serious limitation for the use of animals to model arrhythmia risk (13).
The ICH S7B/E14 approach focuses primarily on assaying the effect of drugs on reducing the hERG current and their effect on QT prolongation as surrogates of arrhythmogenic risk. However, these assays are suboptimal markers because (a) hERG is not the only ion channel that regulates ventricular repolarization (14, 15), and (b) QT interval prolongation alone is not an ideal marker for clinical arrhythmia risk (16).
Drug safety screening has evolved during the past decade in an effort to fill the gaps left by the ICH S7B/E14 approach. This led the FDA to propose the Comprehensive in vitro Proarrhythmia Assay (CiPA) project in 2013, with three basic aims: (a) examine the effect of a drug on seven ionic currents that contribute to regulating ventricular repolarization, (b) integrate data from the seven ion channel assays in the first aim to build an in silico human ventricular tissue model, and (c) use commercially available human induced pluripotent stem cell–derived cardiomyocytes (hiPSC-CMs) to confirm arrhythmia risk by examining the effect of drugs on electrical activity in hiPSC-CMs. According to the latest CiPA progress report, 28 drugs classified as being at high, intermediate, and low risk of inducing torsades de pointes (TdP) and with known cardiac electrophysiological effects have been selected for ongoing CiPA calibration and validation (17).
PATIENT-SPECIFIC EFFECTS AND PHARMACOGENOMICS
The ultimate goal of precision medicine is to tailor treatments to fit specific populations, sub-populations, or even individual patients such that each patient receives treatment that provides the highest therapeutic efficacy with the lowest risk of side effects. However, differential patient-specific reactions to a particular drug are driven by interactions between genetic, epigenetic, and environmental factors. Inherited polymorphisms in drug metabolizing enzymes and transporters can alter their expression, activity, or both, influencing drug pharmacokinetics. Similarly, inherited polymorphisms in target enzymes, transporters, ion channels, and receptors can influence drug pharmacodynamics. Epigenetic alterations, including DNA methylation, histone modification, microRNAs, mRNA instability, and nucleosome positioning, have been shown to be associated with cardiovascular diseases including atherosclerosis and hypertension (18, 19). Environmental and nongenetic factors including body mass index and behavioral patterns, especially those in early life, also modulate the risk of cardiovascular disease (18, 19).
Analyzing 12,500 electrocardiograms, Gallagher et al. (20) showed that the corrected QT interval differs considerably in normal healthy individuals, ranging from 335 ms to more than 460 ms. These findings highlight that individual variability in cardiac repolarization irrespective of disease, administered drugs, or both. Several ion channels, ion transporters, and channel-interacting protein-encoding genes are implicated in altering cardiac electrical activity and repolarization, and several genetic polymorphisms directly alter electrical activity (21, 22).
Common genetic variants in ion channels can increase or decrease the QT interval by 1–4 ms per allele, whereas rare polymorphisms can alter QT interval by up to 100 ms. A genome-wide association study (GWAS) performed by the QT Interval–International GWAS Consortium (QT-IGC) examining >100,000 patients of European ancestry for 2.5 million single-nucleotide polymorphisms (SNPs) identified 35 loci associated with altered QT interval (23). QT-IGC identified 68 independent SNPs, distributed over 35 loci across the genome. Examination of the predictive value of 67 of the 68 identified SNPs in a cohort of 13,105 African American patients revealed that only 9 of 67 SNPs were associated with QT interval (23). Although the effect of genetic variants in European and African populations was reasonably correlated, approximately 8% of identified SNPs had effects on QT interval in different directions in patients of African ancestry compared to patients of European ancestry (23). Thus, a SNP that is a good biomarker for QT prolongation in one population may have no effect or be associated with QT shortening in another. Nongenetic covariates can influence QT intervals, including gender, age, obesity, heart rate, and concomitant drugs; accordingly, the level of significance of each genetic marker must be adjusted to the effect of these covariates (24, 25).
An illustrative example of a population-specific drug response is the combination of isosorbide dinitrate and hydralazine (BiDil), used to treat heart failure in African American patients. The African American Heart Failure Trial (A-HeFT I) demonstrated decreased mortality in African American patients treated with BiDil compared to placebo, whereas there was no difference in survival in Caucasians. Interestingly, genotype TT of SNP rs5443 in the G-protein beta-3 subunit (GNB3) has been associated with greater BiDil therapeutic effect. rs5443-TT is the predominant genotype in African Americans (~50%), whereas it is rare (~10%) in Caucasians (26). Thus, there is a direct relationship between population-specific common SNPs and population-specific drug responses that should be considered when screening potential drug candidates.
Taken together, these issues emphasize the importance of developing a patient-focused cardiotoxicity screening model that can (a) simulate patient-specific exposure to genomic, epigenomic, and environmental modulators and subsequently recapitulate a patient- or population-dependent drug response; (b) be scaled up to thousands of patients to identify cardiotoxic phenotypes in a statistically meaningful manner; (c) be used to assay different types of DIC; and (d) provide monitoring of cardiotoxicity phenotypes without the need for frequent patient clinical encounters.
HUMAN INDUCED PLURIPOTENT STEM CELL–DERIVED CARDIOMYOCYTES
Cardiomyocyte Differentiation
Current methods of differentiating hiPSCs into cardiomyocytes stem from established knowledge of cell signaling events during embryonic cardiac development. All protocols begin with mesodermal induction of the pluripotent cells, accomplished via activation of the WNT, activin/NODAL, and/or bone morphogenetic protein (BMP) pathways in the first 1–2 days of differentiation (27–31). WNT is commonly activated using CHIR99021, along with various combinations of the growth factors activin A and BMP4 (27, 29–33). Cardiac specification of the mesodermally committed cells is achieved by inhibiting WNT with small-molecule inhibitors such as Wnt-C59 (27, 31, 34) or the protein Dickkopf WNT signaling pathway inhibitor 1 (DKK1) (33). Following these steps, the committed cardiac progenitor cells largely continue their differentiation to cardiomyocytes with no further modifications to the basal media, although some groups have found the addition of vascular endothelial growth factor A (VEGFA) or fibroblast growth factor 2 (FGF2) to the media to be beneficial (33, 34).
The first reports of beating cardiomyocytes from human embryonic stem cells (hESCs) were from spherical aggregates termed embryoid bodies (EBs), of which approximately 8% of cells would spontaneously contract (35). Subsequent efforts identified growth factors capable of increasing the percentage of beating EBs to over 50% (36–38). However, production of cardiomyocytes from EBs was very labor intensive, so attempts to differentiate monolayers of hESCs followed. This was first demonstrated by Laflamme et al. (39), who applied the sequential addition of the activin A and BMP4, although efficiency was relatively low at 30%. The addition of WNT inhibition following mesodermal induction improved efficiency to over 50% (40), which was further improved to 75–95% when growth factors were replaced with small-molecule inhibitors (27, 41, 42).
Recognizing the importance of optimal culture conditions for efficient and consistent cardiomyocyte production, and that human- or animal-derived products in the protocol will impede progress toward FDA approval of these cells for therapeutic purposes, the field has made great progress toward chemically defining all media used and eliminating animal-derived products (e.g., serum and bovine serum albumin) from the media. Xu et al. (43) performed the first successful differentiation in EBs in chemically defined differentiation media. Burridge et al. (44) improved the efficiency of beating EB generation by optimizing the protocol and using recombinant human albumin to replace a critical component of serum. However, both approaches still relied on the use of animal-derived knockout serum replacement or mouse embryonic fibroblast–conditioned media for maintenance of pluripotent cells prior to differentiation. The introduction of E8 pluripotency media and chemically defined media for monolayer differentiation (27) made it possible to generate hiPSC-CMs in fully chemically defined and xeno-free conditions. However, the use of small amounts of serum may be beneficial for cardiomyocyte differentiation, maintaining cells in long-term culture, and possibly maturing cardiomyocytes to a more adult-like phenotype. Although xeno-free, chemically defined conditions are necessary for human therapeutic applications, drug screening assays do not preclude the use of serum in cardiomyocyte cultures. Indeed, a more robust and mature in vitro model would be advantageous for such applications.
High-Throughput Human Induced Pluripotent Stem Cell–Derived Cardiomyocyte Differentiation
hiPSC-CM differentiation protocols have been designed with consideration for future scale-up, with the knowledge that EBs are compatible with existing large-scale bioreactors commonly used in commercial mammalian cell production (37). Despite this, monolayer protocols that do not have the complexity of EB formation have proved popular and provide simple assessment of differentiation efficacy (39). The primary barrier to high-throughput differentiation is that monolayer differentiation, commonly performed in 12- and 6-well plates, has significantly less reproducibility when larger surface areas are used. Efforts to differentiate hiPSCs to cardiomyocytes in flasks have required selection of one or two hiPSC lines and line-specific optimization to overcome this issue (45). This lack of scalability has been attributed to the reliance on the paracrine effect for mesoderm induction, the difficulty in seeding cells evenly in large adherent cultures, and the high cost of media and factors, many of which have been overcome (27, 46). Finally, researchers have recently made progress with cardiac differentiation in stirred suspension vessels (47), as hiPSCs easily adapt to suspension-based culture using either cell aggregates (EBs) or attachment to matrix-coated microcarriers (48, 49). Currently, these are considerably less efficient for hiPSC growth. To improve cell purity, commercial hiPSC-CM production uses a cardiac-specific promotor such as MYH6 linked to an antibiotic resistance gene. With these commercial cells becoming more generally available, this genetic modification has now become acceptable within the field.
Cardiomyocyte Subtype Specification
The aforementioned differentiation protocols all result in mixed populations of electrophysiologically variable cardiomyocytes, with most cells representing immature ventricular cardiomyocyte phenotypes, whereas ~15–20% resemble atrial and 5% resemble nodal cells by patch clamp analysis (27). Ideally, the use of hiPSC-CMs for drug efficacy and toxicity screening should involve relatively pure populations of the cardiomyocyte subtype of interest, as electrical heterogeneity in in vitro preparations could skew results, particularly where atrial or ventricular arrhythmias are concerned. Efforts to modify differentiation conditions to direct hiPSCs to become specific cardiac subtypes have been met with moderate success. One approach has been to use reporter gene constructs to select specific cell types, such as myosin light chain 2 (MYL2)-GFP for ventricular myocytes (50) or sarcolipin (SLN)-tdTomato to isolate atrial cells (51). Another approach involves modifying differentiation strategies to drive hiPSCs preferentially toward one subtype. Inhibition of neuregulin with AG1478 increases the nodal population (52); retinoic acid inhibition with BMS-189453 increases the ventricular population (53); and retinoic acid activation increases the atrial population (53). Retinoic acid increases atrial-specific ion channels and allows for the in vitro testing of drugs that specifically target atrial channels as potential treatments for atrial arrhythmias (54).
Cardiomyocyte Maturation
hiPSC-CMs derived by existing protocols represent a developmentally immature, embryonic, or fetal-like state (56). That these cells lack maturity has been reviewed extensively elsewhere (57–62) and is based on the requirement for glycolytic metabolism, maximum diastolic potential of approximately −50 to −60 mV, cellular size and circularity, lack of sarcomeric alignment (63), reduced fractional shortening, lack of transverse (t) tubules, automaticity, and the expression of fetal genes and isoforms of many calcium-handling and contractile proteins, such as TNNI1, rather than the mature TNNI3 (64). Despite this, hiPSC-CMs have been shown to express all the ion channels of mature left ventricular cardiomyocytes (65), albeit some, such as KCNJ2 (IK1), at a low level (66). hiPSC-CMs have also been shown to have relative calcium transients similar to those of mouse and rabbit CMs (56), although with a lower rise and decline rate (67) and with a negative force-frequency response (68), suggesting reduced SERCA function.
hiPSC-CMs undergo some degree of maturation when researchers simply extend cell culture to >80 days (69, 70). Additionally, when engrafted in the rat heart, hiPSC-CMs can mature functionally (71) and structurally (72), proving that under suitable conditions, these cells are capable of maturation. Numerous techniques that do not involve prolonged growth have been employed to improve maturation, including electrical stimulation (73), stretch or mechanical loading (74, 75), micropatterning (76, 77), and growth as part of an engineered heart tissue (78). Particular success has been shown when hiPSC-CMs are patterned to a length:width ratio similar to that of adult cardiomyocytes (7:1) and on substrates of stiffness similar to that of the human heart (10 kPa) (79).
Another maturation technique is to alter soluble factors [e.g., triiodothyronine (80, 81), dexamethasone (82), insulin, or insulin-like growth factor-1 (79, 83, 84)], all of which have been shown to enhance the electrophysiological properties, bioenergetics, and contractile force of hiPSC-CMs. Other media supplements, such as L-carnitine, taurine, and creatine phosphate (85), may also augment maturation. Adrenergic stimulation, despite playing a key role in maturation of the heart, merely induces hypertrophy, not maturation in hiPSC-CMs (86). Finally, transfection of microRNAs such as miR-499 (87) and let-7 (88) may also play a future role.
One of the most difficult challenges in maturing hiPSC-CMs has been the transition of metabolism from the use of glucose via anaerobic glycolysis to the use of fatty acids to produce acetyl-CoA via beta oxidation, typical of mature cardiomyocytes (89–91). Preventing hiPSC-CMs from using glucose (known as the Crabtree effect) is particularly important for cardiotoxicity analysis, as drugs that impair mitochondrial function will have attenuated deleterious effects in glycolysis-dependent cells (92).
The degree to which hiPSC-CM maturation is necessary for testing drug responses and disease phenotypes is largely dependent on the specific effect or toxicity being tested. For common electrophysiological phenotypes such as congenital long QT syndrome (93) and perhaps for some cardiomyopathies, existing levels of hiPSC-CM maturation appear to be suitable (94, 95). For more complex disease phenotypes, including many cardiomyopathies, further maturation will be necessary to recapitulate the clinical phenotype (79, 84). However, for testing drugs for toxicity involving contractile function or structural remodeling, the degree to which cardiotoxicity signaling pathways in hiPSC-CMs recapitulate those in adult cardiomyocytes is critical.
High-Throughput Analysis of Human Induced Pluripotent Stem Cell–Derived Cardiomyocytes
Now that successful cardiac differentiation in moderate quantity is feasible, existing techniques for analysis of cardiomyocyte phenotypes need to be updated with more high-throughput methodologies (Figure 2). One of the first steps has been the transfer to 384-well plates (96), requiring suitable cardiomyocyte dissociation and replating protocols to be developed. Once in 384-well format, industry standard techniques for measuring cardiomyocyte drug response, such as luminescent measurement of viability, caspase activity and apoptosis, and reactive oxygen species production, can be used (97). For cell dye–based assays in which drug toxicity might skew analysis of surviving cells, such as mitochondrial membrane potential and superoxide production, high-throughput flow cytometry can be performed in at least a 96-well format (31). Many of the major demonstrations of hiPSC-CM phenotype modulation have come from simple cell fluorescent imaging, such as changes in cell size or proportions (84, 94, 95), lipogenesis (79), and sarcomere alignment. For these assays, automated high-content imaging would be most suitable for high-throughput analysis. This technique could also be combined with fluorescent gene tagging systems commonly employed to assess drug effects on specific gene expression (98).
Figure 2.
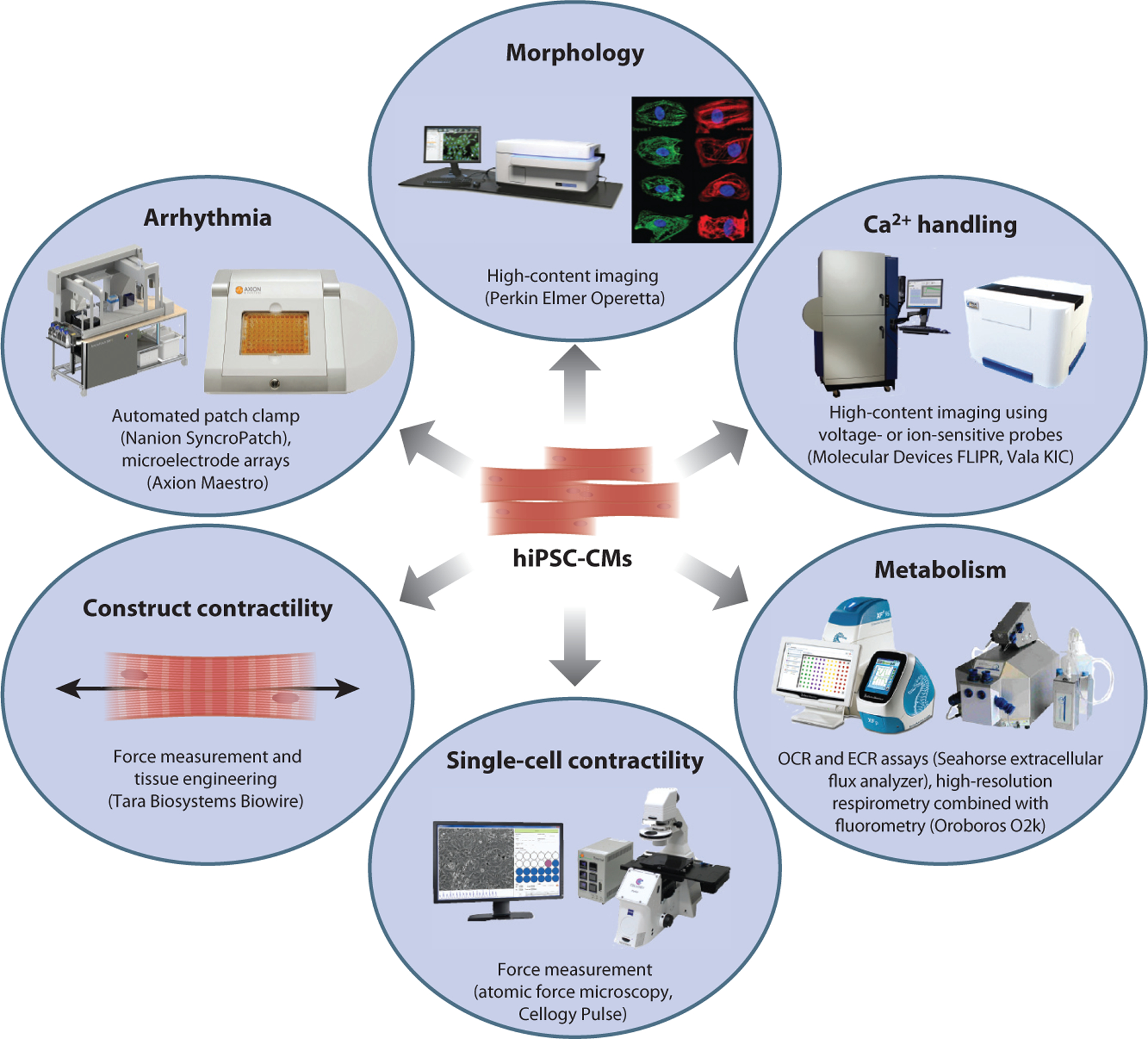
Six major phenotypic areas that can be assessed in human induced pluripotent stem cell–derived cardiomyocytes (hiPSC-CMs) and high-throughput methods that can be applied. Other abbreviations: ECR, extracellular acidification rate; OCR, oxygen consumption rate.
For measurement of electrophysiological phenotypes, patch clamp is the gold standard, yet it is very low throughput (<10 cells per day). Equipment developed for high-throughput automated patch clamp, such as that involving microfluidics, is not suitable for hiPSC-CMs owing to the variable cell sizes and debris associated with dissociation. Newer systems capable of recording from 48 cells simultaneously have proved successful for hiPSC-CMs (99). An alternative to patch clamp is the use of planar microelectrode arrays, which, although they record only extracellular field potentials, have nonetheless been widely used in the hiPSC-CM field (100) and specifically in the CiPA Phase II study. A third method for assessing electrophysiological parameters is the application of calcium imaging, using calcium-sensitive dyes such as Fluo-4 and Fura-2 or genetically encoded calcium indicators such as R-GECO, GCaMP3f, and GCaMP6f (101, 102).
Recently, high-throughput platforms suitable for hiPSC-CM calcium and voltage imaging have been described (103). Finally, the high-throughput measurement of metabolic parameters such as mitochondrial function and substrate use is possible in up to 96-well format using the Seahorse extracellular flux analyzer.
Thus, next-generation high-throughput analysis techniques make it possible to assay a wide variety of cellular phenotype and drug responses in hiPSC-CMs. By tailoring assays to expected disease phenotypes, it will be possible to rapidly assess drug efficacy and toxicity driven by a specific mechanism.
DRUG-INDUCED CARDIOTOXICITY: FROM TORSADES DE POINTES TO CONTRACTILE AND STRUCTURAL CARDIOTOXICITY
Electrophysiology: Torsades de Pointes
The most prominent drug cardiotoxicity—and the one that has halted the development of several drugs in clinical trials—is the induction of ventricular arrhythmias caused by prolongation of the ventricular action potential. Clinically, this manifests as prolongation of the QT interval on a surface electrocardiogram and increased risk for the unstable ventricular arrhythmia TdP, a form of ventricular tachycardia. On a cellular level, drug-induced QT prolongation and arrhythmia risk is most commonly due to prolongation of phase 3 of the ventricular action potential owing to decreased activity of IKr (104, 105). Accordingly, this channel, encoded by the KCNH2 gene (hERG), has been a major focus of preclinical drug toxicity screening assays, as described above. However, effects on other ion channels can similarly prolong the action potential, including enhanced inward sodium or calcium currents or inhibition of the other outward potassium delayed rectifier currents, IKs or IKur (104, 105). Associated with these perturbations in repolarization is an increase in early afterdepolarizations (EADs), which are thought to be due to increases in inward sodium or calcium currents during phase 2 or phase 3 of the action potential (106). When late EADs bring the membrane potential back above threshold, another action potential can be triggered, potentially resulting in reentry and TdP, which can further degenerate into ventricular fibrillation and SCD.
Mechanisms of Contractile and Structural Cardiotoxicity
Although the risk of arrhythmias such as TdP has been a major focus of preclinical and clinical phase testing in recent years, several other forms of cardiotoxicity can result in significant morbidity and mortality. These can be categorized as contractile or structural toxicity (defined broadly as reduced cell viability or morphological damage) and include target-mediated pharmacologic effects on the heart as well as target-independent effects. Such cellular disruption may lead to structural damage to cardiomyocytes (107) and other cardiac cell types via mitochondrial damage (108), oxidative stress (107), or activation of DNA damage response pathways (109). Furthermore, endothelial, fibroblast, or smooth muscle cell dysfunction (107); cardiac fibrosis; and initiation of apoptosis or necrosis may also occur. Structural cardiotoxicity is particularly problematic with chemotherapeutic agents such as anthracyclines and TKIs. hiPSC-CMs have been used to elucidate the mechanisms involved, as they recapitulate the cardiotoxicities of drugs in vitro (97), providing a unique human model for mechanistic studies.
Dysregulation of calcium handling.
Tight regulation of intracellular calcium concentrations is central to the proper electrical and contractile function of the heart. Disturbances in calcium regulation are thought to be one of the mechanisms of contractile toxicity induced by the anthracycline doxorubicin (110). Comparing hiPSC-CMs from patients with a history of doxorubicin cardiotoxicity (DOXTOX) to those from patients who did not develop cardiotoxicity after doxorubicin treatment (DOX) and to healthy (untreated) controls, Burridge et al. (97) have shown significantly larger calcium transient decay times with reduced calcium transient amplitude and time to peak signal in DOXTOX cells. This is consistent with rodent models of DOXTOX (111–113) in which calcium dysregulation is mediated via oxidative stress (111, 112). Similarly, the cardiotoxic TKIs nilotinib and vandetanib also prolonged calcium transient duration and decreased the beat rate in hiPSC-CMs (96). Decreased calcium transients likely play a critical role in drug-induced contractile dysfunction, as was demonstrated in adult mouse ventricular cardiomyocytes exposed to sunitinib (114). We have observed decreased calcium transients with a corresponding decrease in sarcomere shortening on acute exposure to sunitinib.
The above data from hiPSC-CMs are consistent with existing animal models of calcium dysregulation in DIC, but hiPSC-CMs, at present, remain immature and for the most part lack developed t-tubules. Therefore, their ability to accurately model calcium handling perturbations in DIC may be limited until better methods to stimulate hiPSC-CM maturation are developed. Nevertheless, the studies presented here suggest that basic insights into calcium disturbances can still be gained from hiPSC-CMs, even with present limitations.
Cell death, reactive oxygen species, and DNA damage.
Several drugs have been shown to result in decreased cardiomyocyte viability—particularly chemotherapeutics, which are designed to be cytotoxic. The loss of cardiomyocytes may involve apoptosis, necrosis, or both, depending on the drug in question (109). In the case of anthracycline cardiotoxicity, cardiomyocyte loss is primarily due to an increase in programmed cell death (97). We have demonstrated that hiPSC-CMs from patients who developed DOXTOX show more prominent activation of apoptosis at lower concentrations of doxorubicin as compared to hiPSC-CMs from untreated controls or patients who did not develop toxicity (97), suggesting that an individual’s predilection for cardiac cytotoxicity transfers directly to their hiPSC-CMs.
Doxorubicin-induced apoptosis in cardiomyocytes is thought to be driven by multiple mechanisms, the most important of which may be oxidative stress (115). Doherty et al. (116) demonstrated an increase in superoxide generation on exposure to crizotinib and nilotinib, but not sunitinib or erlotinib, suggesting different mechanisms of cardiotoxicity even within the class of TKIs. In our studies, 24 h of doxorubicin exposure resulted in a >2-fold increase in ROS in DOXTOX hiPSC-CMs compared to DOX hiPSC-CMs, as well as a decrease in levels of the antioxidant glutathione (97). Mitochondrial superoxide levels were significantly higher in DOXTOX cells, whereas mitochondrial membrane potential was decreased, consistent with increased mitochondrial ROS.
Independent of oxidative stress, doxorubicin may directly activate caspase-3/7 as another means of apoptosis induction (117). Doherty et al. (116) similarly observed an increase in caspase-3/7 activation in cells exposed to the TKIs crizotinib and nilotinib, but not sunitinib or erlotinib. In agreement with these data, screening of 21 TKIs using hiPSC-CMs demonstrated that crizotinib and nilotinib were among the drugs most associated with cytotoxicity, whereas sunitinib and erlotinib caused less cytotoxicity (96).
Binding of doxorubicin to topoisomerase II-α (TOP2A), an enzyme involved in DNA replication and management of DNA supercoils, is thought to be responsible for its antitumor effects (109, 118). In contrast, doxorubicin binds to another TOP2 isoform found in the heart, topoisomerase II-β (TOP2B), forming TOP2B-doxorubicin-DNA complexes that can result in DNA double-strand breaks (118). Consistent with this mechanism, hiPSC-CMs from DOXTOX patients showed a significant increase in double-stranded DNA damage (97). Thus, a similar mechanism that underlies doxorubicin cytotoxicity in tumors likely also plays some role in cardiomyocyte death.
Noncardiomyocyte cell dysfunction within the heart.
Notwithstanding the significant role of toxicities on cardiomyocytes in producing drug-induced cardiac disease, noncardiomyocyte cells in the heart may also be subject to toxicities from pharmaceuticals and can have a profound impact on cardiac function. In fact, postmarket realization of such toxicities has resulted in some of the most infamous drug failures in recent years. As an example, normal endothelial cell function is critical to maintain proper coronary blood flow in the face of varying hemodynamic conditions and myocardial demands. The effects of endothelial cell dysfunction can be life threatening, resulting in atherosclerosis, cardiac ischemia, or acute atherothrombosis. The COX2 inhibitor rofecoxib was withdrawn from the market after it was found to increase the risk of thromboembolic events and myocardial infarction, an effect that may have been mediated by a shift in the balance between prothrombotic thromboxane from platelets and antithrombotic prostacyclin production by arterial endothelial cells upon inhibition of the COX2 enzyme (119). These toxicities were not identified during preclinical drug development owing in part to the lack of a model to screen for such effects in human cells. Endothelial differentiation of hiPSCs (120, 121) may provide the necessary screening platform, allowing for assays to assess endothelial cell viability as well as production of nitric oxide and antithrombotic factors following drug exposure.
The development of valvular heart disease has been associated with multiple medications, including the migraine prophylactics methysergide and ergotamine; the appetite suppressants fenfluramine, dexfenfluramine, and benfluorex; and the ergot-based dopamine agonists pergolide and cabergoline used to treat Parkinson’s disease (122, 123). Patients treated with these drugs have an increased risk of regurgitant valvular disease, which is histopathologically similar to carcinoid heart disease (123), a paraneoplastic syndrome caused by increased circulating serotonin levels as a result of overproduction by a neuroendocrine tumor. The changes in the valve tissue may be related to an agonist interaction of the offending drugs with the 5-hydroxytryptamine type 2B serotonin receptor, which produces mitogenic effects on fibroblasts and myofibroblasts (124–126).
CONCLUSIONS
With billions of dollars invested in R&D and thousands of drugs in the pipeline, pharmaceutical companies constantly seek novel therapeutics that demonstrate superior efficacy or are associated with fewer and less significant side effects. Yet drug discovery and development have faced serious issues with imperfect preclinical and clinical drug screening, leading to either early exclusion of potential drug candidates or postmarketing withdrawal. Importantly, the most commonly used preclinical and clinical screening approaches, including ICH S7B/E14 and CiPA, fail to fully recapitulate patient- or population-dependent drug responses.
Cardiotoxicity is a well-established side effect associated with thousands of drugs originally approved to treat both cardiovascular and noncardiovascular diseases. Genetic polymorphisms, epigenetic regulation, and environmental factors all contribute to interindividual variability across populations in susceptibility to drug-induced cardiotoxic events and represent a major challenge in drug safety screening. Patient-specific hiPSC-CMs provide a reliable model to study human heart tissue, as they share common genomic and transcriptomic profiles. In addition, hiPSC-CMs recapitulate native cardiomyocyte electrophysiological, biochemical, contractile, and beating activity (127–129), albeit with several important differences (for the current generation of these cells) compared to primary adult human cardiomyocytes. Despite these limitations, patient-derived hiPSC-CMs have the potential to be used to model many types of cardiotoxicity and have been successfully employed to study basal mechanisms and to provide fundamental and mechanistic understanding of a wide variety of cardiovascular diseases, including long QT syndrome (93, 130), LEOPARD syndrome (131), Timothy syndrome (132), arrhythmogenic right ventricular cardiomyopathy (79), dilated cardiomyopathy (95), Barth syndrome (90), and diabetic cardiomyopathy (133). Methods continue to evolve, including human somatic cell reprogramming to hiPSCs, pluripotent culture, hiPSC differentiation to cardiomyocytes, bioreactor-enhanced scalability of hiPSC-CM production, and high-throughput differentiation and phenotypic analysis. This, coupled with major advances in the field of quantitative genomics, whole-genome and targeted locus sequencing, and high-content analysis tools, has led to the use of hiPSCs-CMs to recapitulate patient-specific pharmacological drug responses. Generating a large patient-derived hiPSCs-CM biobank for various populations, such as Caucasians, Asians, African Americans, or even patients with specific genomic characteristics, followed by assessing a drug candidate’s efficacy and safety in each of these populations, will allow us to accurately identify drugs that are effective or toxic in certain populations or in specific individuals at earlier stages in drug development. We propose that inclusion of a patient-derived hiPSC-CM model in drug safety screening platforms (Figure 3) will fill many of the gaps in currently used systems, providing data that will aid in identifying population-specific cardiotoxicities and allowing development of the safest and most effective drug candidates.
Figure 3.

Application of human induced pluripotent stem cell–derived cardiomyocytes (hiPSC-CMs) in the drug discovery pipeline and possible modalities by which hiPSC-CMs can be used during drug discovery.
ACKNOWLEDGMENTS
This work was supported by the US National Institutes of Health (NIH) grant R00 HL121177 and an Innovation Development Progress Foundation Research Innovation Challenge Grant (P.W.B.); NIH F32 Postdoctoral Fellowship (A.J.T.S.); NIH R01 HL128170, NIH R01 HL126527, and NIH R24 HL117756 (J.C.W.); and NIH HL123655 (D.B.).
Glossary
- ADR
adverse drug reaction
- TKI
tyrosine kinase inhibitor
- DIC
drug-induced cardiotoxicity
- ICH
International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use
- IKr
rectifier potassium current
- hERG
human ether-à-go-go
- CiPA
Comprehensive in vitro Proarrhythmia Assay
- hiPSC
human induced pluripotent stem cell
- CM
cardiomyocyte
- TdP
torsades de pointes
- SNP
single-nucleotide polymorphism
- BMP
bone morphogenetic protein
- hESC
human embryonic stem cell
- EB
embryoid body
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Arrowsmith J, Miller P. 2013. Trial watch: phase II and phase III attrition rates 2011–2012. Nat. Rev. Drug Discov 12:569. [DOI] [PubMed] [Google Scholar]
- 2.US Food Drug Admin. 2015. Reports received and reports entered into FAERS by year In FDA Adverse Event Reporting System (FAERS). Silver Spring, MD: US Food Drug Admin; https://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Surveillance/AdverseDrugEffects/ucm070434.htm. [Google Scholar]
- 3.Kuhn M, Letunic I, Jensen LJ, Bork P. 2016. The SIDER database of drugs and side effects. Nucleic Acids Res. 44:D1075–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Magdy T, Burmeister BT, Burridge PW. 2016. Validating the pharmacogenomics of chemotherapy-induced cardiotoxicity: What is missing? Pharmacol. Ther 168:113–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van der Hooft CS, Heeringa J, Brusselle GG, Hofman A, Witteman JC, et al. 2006. Corticosteroids and the risk of atrial fibrillation. Arch. Intern. Med 166:1016–20 [DOI] [PubMed] [Google Scholar]
- 6.Wilkinson GS, Baillargeon J, Kuo YF, Freeman JL, Goodwin JS. 2010. Atrial fibrillation and stroke associated with intravenous bisphosphonate therapy in older patients with cancer. J. Clin. Oncol 28:4898–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ray WA, Meredith S, Thapa PB, Hall K, Murray KT. 2004. Cyclic antidepressants and the risk of sudden cardiac death. Clin. Pharmacol. Ther 75:234–41 [DOI] [PubMed] [Google Scholar]
- 8.Ray WA, Murray KT, Meredith S, Narasimhulu SS, Hall K, Stein CM. 2004. Oral erythromycin and the risk of sudden death from cardiac causes. N. Engl. J. Med 351:1089–96 [DOI] [PubMed] [Google Scholar]
- 9.Onakpoya IJ, Heneghan CJ, Aronson JK. 2016. Post-marketing withdrawal of 462 medicinal products because of adverse drug reactions: a systematic review of the world literature. BMC Med. 14:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bond CA, Raehl CL. 2006. Adverse drug reactions in United States hospitals. Pharmacotherapy 26:601–8 [DOI] [PubMed] [Google Scholar]
- 11.Gintant G 2011. An evaluation of hERG current assay performance: translating preclinical safety studies to clinical QT prolongation. Pharmacol. Ther 129:109–19 [DOI] [PubMed] [Google Scholar]
- 12.Wallis RM. 2010. Integrated risk assessment and predictive value to humans of non-clinical repolarization assays. Br. J. Pharmacol 159:115–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gintant G, Sager PT, Stockbridge N. 2016. Evolution of strategies to improve preclinical cardiac safety testing. Nat. Rev. Drug Discov 15:457–71 [DOI] [PubMed] [Google Scholar]
- 14.Kramer J, Obejero-Paz CA, Myatt G, Kuryshev YA, Bruening-Wright A, et al. 2013. MICE models: superior to the HERG model in predicting Torsade de Pointes. Sci. Rep 3:2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mirams GR, Davies MR, Brough SJ, Bridgland-Taylor MH, Cui Y, et al. 2014. Prediction of Thorough QT study results using action potential simulations based on ion channel screens. J. Pharmacol. Toxicol. Methods 70:246–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Opstal JM, Schoenmakers M, Verduyn SC, de Groot SHM, Leunissen JDM, et al. 2001. Chronic amiodarone evokes no torsade de pointes arrhythmias despite QT lengthening in an animal model of acquired long-QT syndrome. Circulation 104:2722–27 [DOI] [PubMed] [Google Scholar]
- 17.Colatsky T, Fermini B, Gintant G, Pierson JB, Sager P, et al. 2016. The Comprehensive in Vitro Proarrhythmia Assay (CiPA) initiative—update on progress. J. Pharmacol. Toxicol. Methods 81:15–20 [DOI] [PubMed] [Google Scholar]
- 18.Handy DE, Castro R, Loscalzo J. 2011. Epigenetic modifications: basic mechanisms and role in cardiovascular disease. Circulation 123:2145–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Loscalzo J, Handy DE. 2014. Epigenetic modifications: basic mechanisms and role in cardiovascular disease (2013 Grover Conference series). Pulmonary Circ. 4:169–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gallagher MM, Magliano G, Yap YG, Padula M, Morgia V, et al. 2006. Distribution and prognostic significance of QT intervals in the lowest half centile in 12,012 apparently healthy persons. Am. J. Cardiol 98:933–35 [DOI] [PubMed] [Google Scholar]
- 21.Ackerman MJ, Mohler PJ. 2010. Defining a new paradigm for human arrhythmia syndromes: phenotypic manifestations of gene mutations in ion channel–and transporter-associated proteins. Circ. Res 107:457–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen IY, Matsa E, Wu JC. 2016. Induced pluripotent stem cells: at the heart of cardiovascular precision medicine. Nat. Rev. Cardiol 13:333–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arking DE, Pulit SL, Crotti L, van der Harst P, Munroe PB, et al. 2014. Genetic association study of QT interval highlights role for calcium signaling pathways in myocardial repolarization. Nat. Genet 46:826–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Omran J, Firwana B, Koerber S, Bostick B, Alpert MA. 2016. Effect of obesity and weight loss on ventricular repolarization: a systematic review and meta-analysis. Obes. Rev 17:520–30 [DOI] [PubMed] [Google Scholar]
- 25.Rabkin SW. 2015. Impact of age and sex on QT prolongation in patients receiving psychotropics. Can. J. Psychiatry 60:206–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McNamara DM, Taylor AL, Tam SW, Worcel M, Yancy CW, et al. 2014. G-protein beta-3 subunit genotype predicts enhanced benefit of fixed-dose isosorbide dinitrate and hydralazine: results of A-HeFT. JACC Heart Failure 2:551–57 [DOI] [PubMed] [Google Scholar]
- 27.Burridge PW, Matsa E, Shukla P, Lin ZC, Churko JM, et al. 2014. Chemically defined generation of human cardiomyocytes. Nat. Methods 11:855–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mordwinkin NM, Burridge PW, Wu JC. 2013. A review of human pluripotent stem cell-derived cardiomyocytes for high-throughput drug discovery, cardiotoxicity screening, and publication standards. J. Cardiovasc. Transl. Res 6:22–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van den Berg CW, Elliott DA, Braam SR, Mummery CL, Davis RP. 2016. Differentiation of human pluripotent stem cells to cardiomyocytes under defined conditions. Methods Mol. Biol 1353:163–80 [DOI] [PubMed] [Google Scholar]
- 30.Aguilar JS, Begum AN, Alvarez J, Zhang XB, Hong Y, Hao J. 2015. Directed cardiomyogenesis of human pluripotent stem cells by modulating Wnt/β-catenin and BMP signalling with small molecules. Biochem. J 469:235–41 [DOI] [PubMed] [Google Scholar]
- 31.Burridge PW, Holmström A, Wu JC. 2015. Chemically defined culture and cardiomyocyte differentiation of human pluripotent stem cells. Curr. Protocols Hum. Genet 87:21.3.1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Minami I, Yamada K, Otsuji TG, Yamamoto T, Shen Y, et al. 2012. A small molecule that promotes cardiac differentiation of human pluripotent stem cells under defined, cytokine- and xeno-free conditions. Cell Rep. 2:1448–60 [DOI] [PubMed] [Google Scholar]
- 33.Willems E, Cabral-Teixeira J, Schade D, Cai W, Reeves P, et al. 2012. Small molecule-mediated TGF-β type II receptor degradation promotes cardiomyogenesis in embryonic stem cells. Cell Stem Cell 11:242–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang M, Schulte JS, Heinick A, Piccini I, Rao J, et al. 2015. Universal cardiac induction of human pluripotent stem cells in two and three-dimensional formats: implications for in vitro maturation. Stem Cells 33:1456–69 [DOI] [PubMed] [Google Scholar]
- 35.Kehat I, Kenyagin-Karsenti D, Snir M, Segev H, Amit M, et al. 2001. Human embryonic stem cells can differentiate into myocytes with structural and functional properties of cardiomyocytes. J. Clin. Investig 108:407–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yao S, Chen S, Clark J, Hao E, Beattie GM, et al. 2006. Long-term self-renewal and directed differentiation of human embryonic stem cells in chemically defined conditions. PNAS 103:6907–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burridge PW, Anderson D, Priddle H, Barbadillo Muñoz MD, Chamberlain S, et al. 2007. Improved human embryonic stem cell embryoid body homogeneity and cardiomyocyte differentiation from a novel V-96 plate aggregation system highlights interline variability. Stem Cells 25:929–38 [DOI] [PubMed] [Google Scholar]
- 38.Yang L, Soonpaa MH, Adler ED, Roepke TK, Kattman SJ, et al. 2008. Human cardiovascular progenitor cells develop from a KDR+ embryonic-stem-cell-derived population. Nature 453:524–28 [DOI] [PubMed] [Google Scholar]
- 39.Laflamme MA, Chen KY, Naumova AV, Muskheli V, Fugate JA, et al. 2007. Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat. Biotechnol 25:1015–24 [DOI] [PubMed] [Google Scholar]
- 40.Uosaki H, Fukushima H, Takeuchi A, Matsuoka S, Nakatsuji N, et al. 2011. Efficient and scalable purification of cardiomyocytes from human embryonic and induced pluripotent stem cells by VCAM1 surface expression. PLOS ONE 6:e23657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gonzalez R, Lee JW, Schultz PG. 2011. Stepwise chemically induced cardiomyocyte specification of human embryonic stem cells. Angew. Chem. Int. Ed. Engl 50:11181–85 [DOI] [PubMed] [Google Scholar]
- 42.Lian X, Hsiao C, Wilson G, Zhu K, Hazeltine LB, et al. 2012. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. PNAS 109:E1848–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu XQ, Graichen R, Soo SY, Balakrishnan T, Rahmat SN, et al. 2008. Chemically defined medium supporting cardiomyocyte differentiation of human embryonic stem cells. Differentiation 76:958–70 [DOI] [PubMed] [Google Scholar]
- 44.Burridge PW, Thompson S, Millrod MA, Weinberg S, Yuan X, et al. 2011. A universal system for highly efficient cardiac differentiation of human induced pluripotent stem cells that eliminates interline variability. PLOS ONE 6:e18293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chong JJ, Yang X, Don CW, Minami E, Liu YW, et al. 2014. Human embryonic-stem-cell-derived cardiomyocytes regenerate non-human primate hearts. Nature 510:273–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen G, Gulbranson DR, Hou Z, Bolin JM, Ruotti V, et al. 2011. Chemically defined conditions for human iPSC derivation and culture. Nat. Methods 8:424–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kempf H, Andree B, Zweigerdt R. 2016. Large-scale production of human pluripotent stem cell derived cardiomyocytes. Adv. Drug Deliv. Rev 96:18–30 [DOI] [PubMed] [Google Scholar]
- 48.Lam AT, Chen AK, Ting SQ, Reuveny S, Oh SK. 2016. Integrated processes for expansion and differentiation of human pluripotent stem cells in suspended microcarriers cultures. Biochem. Biophys. Res. Commun 473:764–68 [DOI] [PubMed] [Google Scholar]
- 49.Lecina M, Ting S, Choo A, Reuveny S, Oh S. 2010. Scalable platform for human embryonic stem cell differentiation to cardiomyocytes in suspended microcarrier cultures. Tissue Eng. Part C Methods 16:1609–19 [DOI] [PubMed] [Google Scholar]
- 50.Bizy A, Guerrero-Serna G, Hu B, Ponce-Balbuena D, Willis BC, et al. 2013. Myosin light chain 2-based selection of human iPSC-derived early ventricular cardiac myocytes. Stem Cell Res. 11:1335–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Josowitz R, Lu J, Falce C, D’Souza SL, Wu M, et al. 2014. Identification and purification of human induced pluripotent stem cell-derived atrial-like cardiomyocytes based on sarcolipin expression. PLOS ONE 9:e101316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhu WZ, Xie Y, Moyes KW, Gold JD, Askari B, Laflamme MA. 2010. Neuregulin/ErbB signaling regulates cardiac subtype specification in differentiating human embryonic stem cells. Circ. Res 107:776–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang Q, Jiang J, Han P, Yuan Q, Zhang J, et al. 2011. Direct differentiation of atrial and ventricular myocytes from human embryonic stem cells by alternating retinoid signals. Cell Res. 21:579–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Devalla HD, Schwach V, Ford JW, Milnes JT, El-Haou S, et al. 2015. Atrial-like cardiomyocytes from human pluripotent stem cells are a robust preclinical model for assessing atrial-selective pharmacology. EMBO Mol. Med 7:394–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smith AS, Macadangdang J, Leung W, Laflamme MA, Kim DH. 2017. Human iPSC-derived cardiomyocytes and tissue engineering strategies for disease modeling and drug screening. Biotechnol. Adv 35:77–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hwang HS, Kryshtal DO, Feaster TK, Sanchez-Freire V, Zhang J, et al. 2015. Human induced pluripotent stem cell (hiPSC) derived cardiomyocytes to understand and test cardiac calcium handling: a glass half full. J. Mol. Cell. Cardiol 89:379–80 [DOI] [PubMed] [Google Scholar]
- 57.Yang X, Pabon L, Murry CE. 2014. Engineering adolescence: maturation of human pluripotent stem cell–derived cardiomyocytes. Circ. Res 114:511–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Denning C, Borgdorff V, Crutchley J, Firth KS, George V, et al. 2016. Cardiomyocytes from human pluripotent stem cells: from laboratory curiosity to industrial biomedical platform. Biochim. Biophys. Acta 1863:1728–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhu R, Blazeski A, Poon E, Costa KD, Tung L, Boheler KR. 2014. Physical developmental cues for the maturation of human pluripotent stem cell-derived cardiomyocytes. Stem Cell Res. Ther 5:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Veerman CC, Kosmidis G, Mummery CL, Casini S, Verkerk AO, Bellin M. 2015. Immaturity of human stem-cell-derived cardiomyocytes in culture: fatal flaw or soluble problem? Stem Cells Dev. 24:1035–52 [DOI] [PubMed] [Google Scholar]
- 61.Robertson C, Tran DD, George SC. 2013. Concise review: maturation phases of human pluripotent stem cell-derived cardiomyocytes. Stem Cells 31:829–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Keung W, Boheler KR, Li RA. 2014. Developmental cues for the maturation of metabolic, electrophysiological and calcium handling properties of human pluripotent stem cell-derived cardiomyocytes. Stem Cell Res. Ther 5:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bedada FB, Wheelwright M, Metzger JM. 2016. Maturation status of sarcomere structure and function in human iPSC-derived cardiac myocytes. Biochim. Biophys. Acta 1863:1829–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bedada FB, Chan SSK, Metzger SK, Zhang L, Zhang J, et al. 2014. Acquisition of a quantitative, stoichiometrically conserved ratiometric marker of maturation status in stem cell-derived cardiac myocytes. Stem Cell Rep. 3:594–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liang P, Lan F, Lee AS, Gong T, Sanchez-Freire V, et al. 2013. Drug screening using a library of human induced pluripotent stem cell-derived cardiomyocytes reveals disease-specific patterns of cardiotoxicity. Circulation 127:1677–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lieu DK, Fu JD, Chiamvimonvat N, Tung KC, McNerney GP, et al. 2013. Mechanism-based facilitated maturation of human pluripotent stem cell-derived cardiomyocytes. Circ. Arrhythmia Electrophysiol 6:191–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rao C, Prodromakis T, Kolker L, Chaudhry UA, Trantidou T, et al. 2013. The effect of microgrooved culture substrates on calcium cycling of cardiac myocytes derived from human induced pluripotent stem cells. Biomaterials 34:2399–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Germanguz I, Sedan O, Zeevi-Levin N, Shtrichman R, Barak E, et al. 2011. Molecular characterization and functional properties of cardiomyocytes derived from human inducible pluripotent stem cells. J. Cell. Mol. Med 15:38–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lundy SD, Zhu WZ, Regnier M, Laflamme MA. 2013. Structural and functional maturation of cardiomyocytes derived from human pluripotent stem cells. Stem Cells Dev. 22:1991–2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jung G, Fajardo G, Ribeiro AJ, Kooiker KB, Coronado M, et al. 2016. Time-dependent evolution of functional versus remodeling signaling in induced pluripotent stem cell-derived cardiomyocytes and induced maturation with biomechanical stimulation. FASEB J. 30:1464–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cho GS, Lee DI, Tampakakis E, Murphy S, Andersen P, et al. 2017. Neonatal transplantation confers maturation of PSC-derived cardiomyocytes conducive to modeling cardiomyopathy. Cell Rep. 18:571–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kadota S, Pabon L, Reinecke H, Murry CE. 2017. In vivo maturation of human induced pluripotent stem cell-derived cardiomyocytes in neonatal and adult rat hearts. Stem Cell Rep. 8:278–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nunes SS, Miklas JW, Liu J, Aschar-Sobbi R, Xiao Y, et al. 2013. Biowire: a platform for maturation of human pluripotent stem cell–derived cardiomyocytes. Nat. Methods 10:781–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tulloch NL, Muskheli V, Razumova MV, Korte FS, Regnier M, et al. 2011. Growth of engineered human myocardium with mechanical loading and vascular coculture. Circ. Res 109:47–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ruan JL, Tulloch NL, Saiget M, Paige SL, Razumova MV, et al. 2015. Mechanical stress promotes maturation of human myocardium from pluripotent stem cell-derived progenitors. Stem Cells 33:2148–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Carson D, Hnilova M, Yang X, Nemeth CL, Tsui JH, et al. 2016. Nanotopography-induced structural anisotropy and sarcomere development in human cardiomyocytes derived from induced pluripotent stem cells. ACS Appl. Mater. Interfaces 8:21923–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ribeiro AJ, Ang YS, Fu JD, Rivas RN, Mohamed TM, et al. 2015. Contractility of single cardiomyocytes differentiated from pluripotent stem cells depends on physiological shape and substrate stiffness. PNAS 112:12705–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tiburcy M, Hudson JE, Balfanz P, Schlick SF, Meyer T, et al. 2017. Defined engineered human myocardium with advanced maturation for applications in heart failure modelling and repair. Circulation 135:1832–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kim C, Wong J, Wen J, Wang S, Wang C, et al. 2013. Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature 494:105–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yang X, Rodriguez M, Pabon L, Fischer KA, Reinecke H, et al. 2014. Tri-iodo-L-thyronine promotes the maturation of human cardiomyocytes-derived from induced pluripotent stem cells. J. Mol. Cell. Cardiol 72:296–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ivashchenko CY, Pipes GC, Lozinskaya IM, Lin Z, Xiaoping X, et al. 2013. Human-induced pluripotent stem cell-derived cardiomyocytes exhibit temporal changes in phenotype. Am. J. Physiol. Heart Circ. Physiol 305:H913–22 [DOI] [PubMed] [Google Scholar]
- 82.Kosmidis G, Bellin M, Ribeiro MC, van Meer B, Ward-van Oostwaard D, et al. 2015. Altered calcium handling and increased contraction force in human embryonic stem cell derived cardiomyocytes following short term dexamethasone exposure. Biochem. Biophys. Res. Commun 467:998–1005 [DOI] [PubMed] [Google Scholar]
- 83.Wen JY, Wei CY, Shah K, Wong J, Wang C, Chen HS. 2015. Maturation-based model of arrhythmogenic right ventricular dysplasia using patient-specific induced pluripotent stem cells. Circ. J 79:1402–8 [DOI] [PubMed] [Google Scholar]
- 84.Birket MJ, Ribeiro MC, Kosmidis G, Ward D, Leitoguinho AR, et al. 2015. Contractile defect caused by mutation in MYBPC3 revealed under conditions optimized for human PSC-cardiomyocyte function. Cell Rep 13:733–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Xu C, He JQ, Kamp TJ, Police S, Hao X, et al. 2006. Human embryonic stem cell-derived cardiomyocytes can be maintained in defined medium without serum. Stem Cells Dev. 15:931–41 [DOI] [PubMed] [Google Scholar]
- 86.Földes G, Matsa E, Kriston-Vizi J, Leja T, Amisten S, et al. 2014. Aberrant α-adrenergic hypertrophic response in cardiomyocytes from human induced pluripotent cells. Stem Cell Rep. 3:905–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fu JD, Rushing SN, Lieu DK, Chan CW, Kong CW, et al. 2011. Distinct roles of microRNA-1 and −499 in ventricular specification and functional maturation of human embryonic stem cell-derived cardiomyocytes. PLOS ONE 6:e27417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kuppusamy KT, Jones DC, Sperber H, Madan A, Fischer KA, et al. 2015. Let-7 family of microRNA is required for maturation and adult-like metabolism in stem cell-derived cardiomyocytes. PNAS 112:E2785–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rana P, Anson B, Engle S, Will Y. 2012. Characterization of human-induced pluripotent stem cell–derived cardiomyocytes: bioenergetics and utilization in safety screening. Toxicol. Sci 130:117–31 [DOI] [PubMed] [Google Scholar]
- 90.Wang G, McCain ML, Yang L, He A, Pasqualini FS, et al. 2014. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat. Med 20:616–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Poon E, Keung W, Liang Y, Ramalingam R, Yan B, et al. 2015. Proteomic analysis of human pluripotent stem cell-derived, fetal, and adult ventricular cardiomyocytes reveals pathways crucial for cardiac metabolism and maturation. Circ. Cardiovasc. Genet 8:427–36 [DOI] [PubMed] [Google Scholar]
- 92.Marroquin LD, Hynes J, Dykens JA, Jamieson JD, Will Y. 2007. Circumventing the Crabtree effect: replacing media glucose with galactose increases susceptibility of HepG2 cells to mitochondrial toxicants. Toxicol. Sci 97:539–47 [DOI] [PubMed] [Google Scholar]
- 93.Itzhaki I, Maizels L, Huber I, Zwi-Dantsis L, Caspi O, et al. 2011. Modelling the long QT syndrome with induced pluripotent stem cells. Nature 471:225–29 [DOI] [PubMed] [Google Scholar]
- 94.Lan F, Lee AS, Liang P, Sanchez-Freire V, Nguyen PK, et al. 2013. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell Stem Cell 12:101–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sun N, Yazawa M, Liu J, Han L, Sanchez-Freire V, et al. 2012. Patient-specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Sci. Transl. Med 4:130ra47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sharma A, Burridge PW, McKeithan WL, Serrano R, Shukla P, et al. 2017. High-throughput screening of tyrosine kinase inhibitor cardiotoxicity with human induced pluripotent stem cells. Sci. Transl. Med 9:eaaf2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Burridge PW, Li YF, Matsa E, Wu H, Ong SG, et al. 2016. Human induced pluripotent stem cell–derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin-induced cardiotoxicity. Nat. Med 22:547–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Crivat G, Taraska JW. 2012. Imaging proteins inside cells with fluorescent tags. Trends Biotechnol. 30:8–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rajamohan D, Kalra S, Duc Hoang M, George V, Staniforth A, et al. 2016. Automated electrophysiological and pharmacological evaluation of human pluripotent stem cell-derived cardiomyocytes. Stem Cells Dev. 25:439–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Navarrete EG, Liang P, Lan F, Sanchez-Freire V, Simmons C, et al. 2013. Screening drug-induced arrhythmia using human induced pluripotent stem cell–derived cardiomyocytes and low-impedance microelectrode arrays. Circulation 128:S3–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Helassa N, Zhang XH, Conte I, Scaringi J, Esposito E, et al. 2015. Fast-response calmodulin-based fluorescent indicators reveal rapid intracellular calcium dynamics. Sci. Rep 5:15978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, et al. 2013. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499:295–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cerignoli F, Charlot D, Whittaker R, Ingermanson R, Gehalot P, et al. 2012. High throughput measurement of Ca2+ dynamics for drug risk assessment in human stem cell-derived cardiomyocytes by kinetic image cytometry. J. Pharmacol. Toxicol. Methods 66:246–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Roden DM. 2008. Cellular basis of drug-induced torsades de pointes. Br. J. Pharmacol 154:1502–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lester RM, Olbertz J. 2016. Early drug development: assessment of proarrhythmic risk and cardiovascular safety. Expert Rev. Clin. Pharmacol 9:1611–18 [DOI] [PubMed] [Google Scholar]
- 106.Kramer DB, Zimetbaum PJ. 2011. Long-QT syndrome. Cardiol. Rev 19:217–25 107. [DOI] [PubMed] [Google Scholar]
- 107.Cross MJ, Berridge BR, Clements PJ, Cove-Smith L, Force TL, et al. 2015. Physiological, pharmacological and toxicological considerations of drug-induced structural cardiac injury. Br. J. Pharmacol 172:957–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Varga ZV, Ferdinandy P, Liaudet L, Pacher P. 2015. Drug-induced mitochondrial dysfunction and cardiotoxicity. Am. J. Physiol. Heart Circ. Physiol 309:H1453–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mercurio V, Pirozzi F, Lazzarini E, Marone G, Rizzo P, et al. 2016. Models of heart failure based on the cardiotoxicity of anticancer drugs. J. Card. Fail 22:449–58 [DOI] [PubMed] [Google Scholar]
- 110.Hanna AD, Lam A, Tham S, Dulhunty AF, Beard NA. 2014. Adverse effects of doxorubicin and its metabolic product on cardiac RyR2 and SERCA2A. Mol. Pharmacol 86:438–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sag CM, Kohler AC, Anderson ME, Backs J, Maier LS. 2011. CaMKII-dependent SR Ca leak contributes to doxorubicin-induced impaired Ca handling in isolated cardiac myocytes. J. Mol. Cell. Cardiol 51:749–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kim SY, Kim SJ, Kim BJ, Rah SY, Chung SM, et al. 2006. Doxorubicin-induced reactive oxygen species generation and intracellular Ca2+ increase are reciprocally modulated in rat cardiomyocytes. Exp. Mol. Med 38:535–45 [DOI] [PubMed] [Google Scholar]
- 113.Wang GX, Wang YX, Zhou XB, Korth M. 2001. Effects of doxorubicinol on excitation–contraction coupling in guinea pig ventricular myocytes. Eur. J. Pharmacol 423:99–107 [DOI] [PubMed] [Google Scholar]
- 114.Rainer PP, Doleschal B, Kirk JA, Sivakumaran V, Saad Z, et al. 2012. Sunitinib causes dose-dependent negative functional effects on myocardium and cardiomyocytes. BJU Int. 110:1455–62 [DOI] [PubMed] [Google Scholar]
- 115.Octavia Y, Tocchetti CG, Gabrielson KL, Janssens S, Crijns HJ, Moens AL. 2012. Doxorubicin-induced cardiomyopathy: from molecular mechanisms to therapeutic strategies. J. Mol. Cell Cardiol 52:1213–25 [DOI] [PubMed] [Google Scholar]
- 116.Doherty KR, Wappel RL, Talbert DR, Trusk PB, Moran DM, et al. 2013. Multi-parameter in vitro toxicity testing of crizotinib, sunitinib, erlotinib, and nilotinib in human cardiomyocytes. Toxicol. Appl. Pharmacol 272:245–55 [DOI] [PubMed] [Google Scholar]
- 117.Ueno M, Kakinuma Y, Yuhki K, Murakoshi N, Iemitsu M, et al. 2006. Doxorubicin induces apoptosis by activation of caspase-3 in cultured cardiomyocytes in vitro and rat cardiac ventricles in vivo. J. Pharmacol. Sci 101:151–58 [DOI] [PubMed] [Google Scholar]
- 118.Lyu YL, Kerrigan JE, Lin CP, Azarova AM, Tsai YC, et al. 2007. Topoisomerase IIβ–mediated DNA double-strand breaks: implications in doxorubicin cardiotoxicity and prevention by dexrazoxane. Cancer Res. 67:8839–46 [DOI] [PubMed] [Google Scholar]
- 119.Anwar A, Anwar IJ, Delafontaine P. 2015. Elevation of cardiovascular risk by non-steroidal anti-inflammatory drugs. Trends Cardiovasc. Med 25:726–35 [DOI] [PubMed] [Google Scholar]
- 120.Li Z, Hu S, Ghosh Z, Han Z, Wu JC. 2011. Functional characterization and expression profiling of human induced pluripotent stem cell- and embryonic stem cell-derived endothelial cells. Stem Cells Dev. 20:1701–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.White MP, Rufaihah AJ, Liu L, Ghebremariam YT, Ivey KN, et al. 2013. Limited gene expression variation in human embryonic stem cell and induced pluripotent stem cell-derived endothelial cells. Stem Cells 31:92–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bhattacharyya S, Schapira AH, Mikhailidis DP, Davar J. 2009. Drug-induced fibrotic valvular heart disease. Lancet 374:577–85 [DOI] [PubMed] [Google Scholar]
- 123.Cosyns B, Droogmans S, Rosenhek R, Lancellotti P. 2013. Drug-induced valvular heart disease. Heart 99:7–12 [DOI] [PubMed] [Google Scholar]
- 124.Fitzgerald LW, Burn TC, Brown BS, Patterson JP, Corjay MH, et al. 2000. Possible role of valvular serotonin 5-HT2B receptors in the cardiopathy associated with fenfluramine. Mol. Pharmacol 57:75–81 [PubMed] [Google Scholar]
- 125.Nebigil CG, Launay JM, Hickel P, Tournois C, Maroteaux L. 2000. 5-Hydroxytryptamine 2B receptor regulates cell-cycle progression: cross-talk with tyrosine kinase pathways. PNAS 97:2591–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rothman RB, Baumann MH, Savage JE, Rauser L, McBride A, et al. 2000. Evidence for possible involvement of 5-HT2B receptors in the cardiac valvulopathy associated with fenfluramine and other serotonergic medications. Circulation 102:2836–41 [DOI] [PubMed] [Google Scholar]
- 127.Babiarz JE, Ravon M, Sridhar S, Ravindran P, Swanson B, et al. 2012. Determination of the human cardiomyocyte mRNA and miRNA differentiation network by fine-scale profiling. Stem Cells Dev. 21:1956–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ma J, Guo L, Fiene SJ, Anson BD, Thomson JA, et al. 2011. High purity human-induced pluripotent stem cell-derived cardiomyocytes: electrophysiological properties of action potentials and ionic currents. Am. J. Physiol. Heart Circ. Physiol 301:H2006–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Puppala D, Collis LP, Sun SZ, Bonato V, Chen X, et al. 2013. Comparative gene expression profiling in human-induced pluripotent stem cell–derived cardiocytes and human and cynomolgus heart tissue. Toxicol. Sci 131:292–301 [DOI] [PubMed] [Google Scholar]
- 130.Malan D, Zhang M, Stallmeyer B, Müller J, Fleischmann BK, et al. 2016. Human iPS cell model of type 3 long QT syndrome recapitulates drug-based phenotype correction. Basic Res. Cardiol 111:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Carvajal-Vergara X, Sevilla A, D’Souza SL, Ang Y-S, Schaniel C, et al. 2010. Patient-specific induced pluripotent stem cell derived models of LEOPARD syndrome. Nature 465:808–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Yazawa M, Hsueh B, Jia X, Pasca AM, Bernstein JA, et al. 2011. Using iPS cells to investigate cardiac phenotypes in patients with Timothy Syndrome. Nature 471:230–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Drawnel FM, Boccardo S, Prummer M, Delobel F, Graff A, et al. 2014. Disease modeling and phenotypic drug screening for diabetic cardiomyopathy using human induced pluripotent stem cells. Cell Rep. 9:810–21 [DOI] [PubMed] [Google Scholar]