

Abstract
INTRODUCTION:
Colorectal cancer arises in a multistep process of carcinogenesis from normal mucosa. The earliest precursor might be a morphologically inconspicuous precancerous field, harboring cancer-associated mutations.
METHODS:
We systematically analyzed genetic alterations in 77 tissue samples from 30 patients with sporadic colorectal neoplasms (18 large adenomas and 12 adenocarcinomas) and matched adjacent normal mucosa (N = 30), as well as normal rectal tissue (N = 17). We profiled mutations associated with colorectal cancer by targeted sequencing of 46 genetic loci using 157 custom amplicons and a median depth of 42,655 reads per loci.
RESULTS:
Multiple mutations were found in colorectal neoplasms, most frequently in APC, KRAS, and TP53. In a subgroup of 11 of 30 patients, alterations were also detected in non-neoplastic mucosa. These mutations were divergent from those in matched neoplasms. The total alteration count and the allele frequency of mutations were higher in neoplasms compared with those in adjacent tissues. We found that younger patients (≤70 years) are less likely affected by mutations in non-neoplastic mucosa than older patients (>70 years, P = 0.013), although no association was found for other variables, including type, location and differentiation of neoplasia, and previous history of polyps.
DISCUSSION:
Our data show that cancer-associated mutations can be found in non-neoplastic tissues in a subgroup of patients with colorectal neoplasms. Further studies are needed to specify the risk of occurrence and recurrence of neoplasia in this patient population.
INTRODUCTION
Colorectal cancer (CRC) has precursor lesions that can be detected and removed by endoscopy (1–3), thereby efficiently reducing disease-associated mortality (4,5). However, there is a broad spectrum of individual risk profiles ranging from no or few diminutive polyps up to advanced lesions including adenocarcinomas (6,7). This demands stratification strategies for surveillance that complement established predictors of adenoma recurrence, which focus on the number of lesions or grade of dysplasia (8,9). A potential strategy for risk stratification is based on the concept of field cancerization, which postulates that environmental factors can induce cancer-associated alterations in larger areas of the intestine (10,11). In such a scenario, individuals with field cancerization are at higher risk of developing recurrent or multiple adenomas and should therefore undergo intensified screening. Several markers of field cancerization have been identified in the normal colon mucosa, including epigenetic alterations (12–14), and transcriptional changes (15,16). Furthermore, an increased vascularization or aberrant crypt foci were identified as biomarkers (17,18). However, despite the predictive value of these markers, genetic alterations in tumor suppressors and oncogenes remain the critical events that drive carcinogenesis in CRC. Large cancer genomics studies showed that these alterations recurrently occur in a well-defined set of genes in CRC (19). Given the high phenotypical penetrance of hereditary CRC syndromes, the presence of genetic mutations in cancer-associated genes can be considered a bona fide marker of field cancerization.
In this study, we investigated whether mutations in a set of frequently altered genes in CRC can be found in non-neoplastic mucosa adjacent to colorectal lesions. To this end, we endoscopically collected paired tissues from colorectal adenomas or adenocarcinomas and surrounding macroscopically normal mucosa and performed targeted deep sequencing of DNA from the samples using a custom amplicon panel. We demonstrated that cancer-associated mutations can be found in non-neoplastic tissue in a subgroup of patients. The identified alterations were distinct from mutations found in matched neoplastic lesions, occurred at lower allele frequencies, and preferentially in elderly patients.
METHODS
Ethics approval
Collection of tissue biopsies and DNA analysis were approved by the Medical Ethics Committee II of the Medical Faculty Mannheim, Heidelberg University (reference no. 2013-632N-MA, 2014-633N-MA, and 2016-607N-MA), and were performed in accordance with the Treaty of Helsinki. All patients gave written informed consent before tissue sampling.
Patient cohort and tissue sampling
Patients with large colorectal adenomas (>1 cm) or adenocarcinoma were included in the study. Colorectal lesions were endoscopically diagnosed in external hospitals or practices and referred to the Central Interdisciplinary Endoscopy Unit of Mannheim University Hospital, Heidelberg University, for tissue sampling or adenoma removal. Tissue samples were endoscopically obtained from neoplastic lesions and adjacent macroscopically normal tissue (distance to the border of the lesion >2 cm). Additional samples from the rectum were obtained when adenomas were located in the colon (compare Supplementary Methods, Supplementary Digital Content 1, http://links.lww.com/CTG/A329 and Supplementary Tables 1 and 2, Supplementary Digital Content 1, http://links.lww.com/CTG/A329).
Design of amplicon panel and library preparation
Sequencing libraries were prepared with a custom panel (Tru-Seq custom library kit; Illumina, San Diego, CA) according to the manufacturer's protocol and sequenced with a MiSeq (Illumina). Targeted regions included the most commonly mutated hot spots in CRC as defined by the COSMIC (20) database.
Statistical analysis
Allele frequency and mutational counts were compared using a 2-sided Student T-test. Associations of mutations in non-neoplastic mucosa were calculated using the χ2 tests or Fisher exact tests. All tests were 2-sided with a significance level of P = 0.05.
Additional methods
A description of additional methods can be found in the Supplementary Methods section.
RESULTS
To investigate whether cancer-associated mutations occur in non-dysplastic colorectal mucosa, we analyzed paired tissue samples from large adenomas or adenocarcinomas, their surrounding normal mucosa and samples from the rectum, focusing on the most frequently mutated genetic loci in CRC. We used an amplicon sequencing approach with a high sequencing depth to detect mutational events also at low allelic frequencies in normal tissues (Figure 1a). We designed 157 amplicons covering hot spots in 46 cancer-associated genes according to the COSMIC database (20) (see Supplementary Table 3, Supplementary Digital Content 1, http://links.lww.com/CTG/A329). These genes are associated with major cellular processes and signaling pathways altered in CRC (see Supplementary Figure 1, Supplementary Digital Content 1, http://links.lww.com/CTG/A329). Of all genomic regions, 102 could be sequenced with sufficient depth (>100 average mapped aligned reads per amplicon per sample), with a median sequencing depth per amplicon region among all samples of 42,655 reads (range: 359–621,749 reads, see Supplementary Figure 2, Supplementary Digital Content 1, http://links.lww.com/CTG/A329). After processing of sequencing data, we identified 88 mutational events (see Supplementary Table 4, Supplementary Digital Content 1, http://links.lww.com/CTG/A329).
Figure 1.
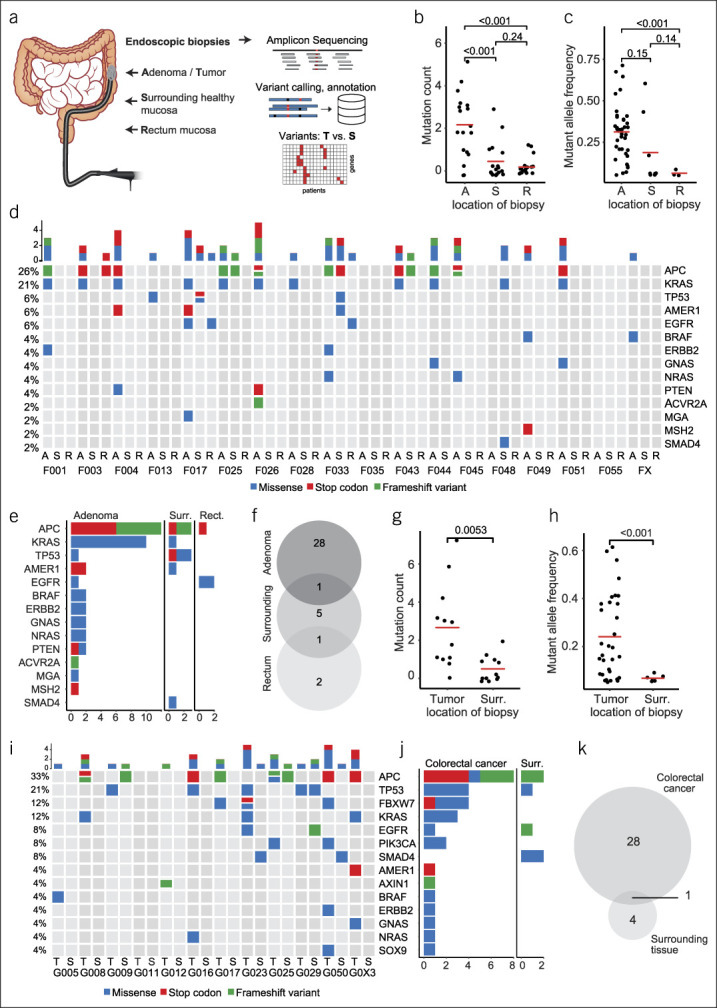
Systematic mutational profiling of colorectal neoplasms and adjacent normal colorectal mucosa. (a) Schematic representation of biopsy acquisition and experimental workflow. Patients with either large colorectal adenomas or colorectal cancer (CRC) were recruited. Biopsies were obtained using a forceps via colonoscopy from the neoplasia, adjacent normal mucosa, and in case of adenomas also from healthy rectum mucosa. DNA isolated from all samples underwent amplicon sequencing of frequently mutated loci in CRC-associated genes and subsequent bioinformatics analysis. (b) Mutation count in adenoma, surrounding mucosa and rectum mucosa samples. (c) Fraction of mutated alleles detected in adenoma, surrounding mucosa and rectum mucosa samples. (d) Oncoprint of all mutations identified in adenomas (A), surrounding mucosa (S) and rectal mucosa (R) from 20 patients. Additional rectal mucosa samples were not analyzed in 3 patients with rectal adenomas. (e) Frequency of mutations by cancer-gene stratified by location of biopsy (adenoma, surr., surrounding, rect., rectum) (f) Venn diagram showing the concordance of mutations in adenomas, surrounding healthy mucosa and rectal mucosa. (g) Mutation count in CRC and surrounding mucosa. (h) Fraction of mutated alleles detected in CRCs and surrounding mucosa. (i) Oncoprint of all mutations identified in CRCs (T = tumor) and surrounding mucosa (S = surrounding). (j) Frequency of mutations by cancer-gene, stratified by location of biopsy (CRC vs surr., surrounding) (k) Venn diagram showing the concordance of mutations in CRC and surrounding healthy mucosa.
In 18 large colorectal adenomas and their surrounding normal tissue, as well as 17 biopsies of rectal mucosa, a total of 50 cancer-associated mutations (37 unique mutations) were identified. The total alteration count was significantly higher in adenomas compared with adjacent tissues (Figure 1b). Mutant allele frequency varied greatly between genetic alterations (range: 5.1%–71.3%), and a trend toward a higher frequency in adenomas compared with non-neoplastic mucosa was observed (Figure 1c). Mutation count and allele frequencies did not differ between adenomas with high-grade and low-grade dysplasia (see Supplementary Figure 3A, B, Supplementary Digital Content 1, http://links.lww.com/CTG/A329). The most frequently altered genes in patients with adenomas were APC, KRAS, TP53, AMER1, and EGFR (Figure 1d,e). Both sessile serrated adenomas, which were located in the ascending colon, harbored a BRAF V600E mutation. The concordance of mutations from adenomas and normal mucosa was low, showing only one match between adenoma and surrounding tissue and one between adenoma and rectum (Figure 1f).
A total of 38 mutations (33 unique) were identified in 11 adenocarcinoma and 5 adjacent tissues (of 12 sequenced pairs). The mutation count and mutant allele frequencies were significantly higher in tumor samples compared biopsies from surrounding tissues. The allele frequency in tumors varied between 5.7% and 61.4% compared with 5.7% and 9.1% in adjacent tissues (Figure 1g,h). The most frequently altered genes were APC, TP53, FBXW7, and KRAS (Figure 1i,j). Comparison of mutations in adenocarcinomas and adjacent tissues showed only one overlapping mutation in a matched pair (Figure 1k).
We noticed that mutations in non-neoplastic mucosa occurred more likely in elderly patients (>70 years) (P = 0.013, χ2 tests, Table 1). Other variables, including sex, type of neoplasia (cancer vs adenoma), neoplasia location, or history of polyps were not associated with mutations in non-neoplastic mucosa.
Table 1.
Associations of mutations in non-neoplastic mucosa with clinical variables

DISCUSSION
In this study, we systematically analyzed cancer-associated mutations in sporadic colorectal neoplasms, surrounding mucosa and corresponding rectum mucosa. Adenomas and adenocarcinomas in our cohort most frequently harbored mutations in APC, KRAS, and TP53, which is similar to results from cancer genomics studies (19). Interestingly, only a subgroup of the analyzed patients had mutations in non-neoplastic mucosa, and we noticed an association of the occurrence of non-neoplastic driver mutations with increased age. This may be explained by higher chances of exposure to carcinogens and subsequent accumulation of mutations with time. It is in line with previous observations of higher transition rates from adenoma to cancer in higher age groups (21).
Surprisingly, the mutations found in mucosa adjacent to neoplasms were divergent from those identified in the corresponding tumors. This finding suggests that, on a genetic level, there is not a homogeneous precancerous field. Rather, our data suggest a stochastic process in which single, dispersed stem cell clones acquire distinct mutations by chance and thus become more susceptible to additional genetic alterations ultimately leading to carcinogenesis. These findings are in concordance with previous studies (22–26). Recently, Lee-Six et al. (22) isolated and analyzed normal colon crypts from 42 individuals by whole genome sequencing, thereby generating a high-resolution map of genomic alterations in normal colon mucosa. In accordance with our data, they identified cancer-associated mutations in normal colorectal mucosa cells. Potential driver mutations were detected in about 1% of crypts in middle-aged persons. Genetic analyses of matched colorectal neoplasms were not performed in this work. In a different approach, Di et al. (24) analyzed synchronous sporadic CRCs from 15 patients by whole genome sequencing. They found significant variation in somatic mutations and copy number alterations of synchronous cancers, supporting independent cancer-initiating and driving events. Similarly, Hawthorn et al. (23) observed chromosomal aberrations in tumor-adjacent tissues that occurred at lower frequencies compared with carcinomas and were heterogeneously distributed within the normal mucosa.
There are several limitations to our study. First, we obtained only one biopsy from the adjacent tissue and rectum. It is therefore possible that we missed non-neoplastic mutations because of sampling error. In addition, we analyzed our samples by panel sequencing, only covering small parts of the genome harboring the most frequent cancer-associated mutations, thereby potentially missing alterations in other regions, copy number variations, or epigenetic changes. Finally, the question whether patients with driver mutations in non-neoplastic tissue comprise a subgroup with higher risk for developing recurrent adenomas cannot be answered with the current study because of its exploratory design and small sample size.
In conclusion, we found that a subgroup of patients with sporadic colorectal neoplasia harbors potential cancer driver mutations in non-neoplastic mucosa, demanding further studies to analyze the risk of developing recurrent neoplasia in this specific group.
CONFLICTS OF INTEREST
Guarantor of the article: Johannes Betge, MD.
Specific author contributions: Tianzuo Zhan, MD, Sebastian Belle, MD, and Johannes Betge, MD, contributed equally to this work. Tianzuo Zhan, MD and Sebastian Belle, MD are shared first authors. Concept and design: T.Z., S.B., J.B. Acquisition of data: T.Z.; S.B., S.H., T.M., G.K., T.G., and J.B. Analysis and interpretation of data: T.Z., S.B., E.V., S.H., T.M., G.K., T.G., M.B., M.P.E., and J.B. Drafting of manuscript: T.Z. and J.B. Critical revision of the manuscript for important intellectual content: S.B., E.V., S.H., T.M., T.G., M.B., and M.P.E. Statistical analysis: T.Z., E.V., and J.B. Obtained funding: M.B. and M.P.E. All authors approved the final manuscript.
Financial support: T.Z. was supported by the Clinician Scientist program “Interfaces and Interventions in Complex Chronic Conditions” by the German Research Foundation (DFG) (EB 187/8-1). J.B. was supported by the “Translational Physician Scientist (TRAPS)” program of the Medical Faculty Mannheim, Heidelberg University and the State of Baden-Württemberg. T.Z., M.B., and M.P.E. were supported by the grant “Biology of Frailty—Sonderlinie Medizin” from the State of Baden-Württemberg.
Potential competing interests: We have read and understood the ICMJE policy on declaration of conflicts of interests and declare we have no conflicts of interest.
Supplementary Material
ACKNOWLEDGEMENTS
We thank all patients who participated in this study. We thank all clinicians and nurses from the Department of Medicine II and the Central Interdisciplinary Endoscopy of University Hospital Mannheim, who helped with recruiting patients, informed consent, and sample acquisition.
Study Highlights.
WHAT IS KNOWN
✓ CRC arises from normal mucosa in a multistep process.
✓ CRC displays typical, recurrent mutations.
WHAT IS NEW HERE
✓ About 1/3 of patients had cancer-typic mutations in non-neoplastic mucosa.
✓ Older patients were more likely affected than younger patients.
TRANSLATIONAL IMPACT
✓ Non-neoplastic mutations might be associated with cancer in a subgroup of patients.
✓ Further studies may quantify the risk of neoplasia when mutations in non-neoplastic mucosa are present.
Footnotes
SUPPLEMENTARY MATERIAL accompanies this paper at http://links.lww.com/CTG/A329
REFERENCES
- 1.Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68(6):394–424. [DOI] [PubMed] [Google Scholar]
- 2.Shaukat A, Mongin SJ, Geisser MS, et al. Long-term mortality after screening for colorectal cancer. N Engl J Med 2013;369(12):1106–14. [DOI] [PubMed] [Google Scholar]
- 3.Lieberman DA, Weiss DG, Bond JH, et al. Use of colonoscopy to screen asymptomatic adults for colorectal cancer. N Engl J Med 2000;343(3):162–8. [DOI] [PubMed] [Google Scholar]
- 4.Zauber AG, Winawer SJ, O'Brien MJ, et al. Colonoscopic polypectomy and long-term prevention of colorectal-cancer deaths. N Engl J Med 2012;366(8):687–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pox CP, Altenhofen L, Brenner H, et al. Efficacy of a nationwide screening colonoscopy program for colorectal cancer. Gastroenterology 2012;142(7):1460–7.e2. [DOI] [PubMed] [Google Scholar]
- 6.Neugut AI, Jacobson JS, Ahsan H, et al. Incidence and recurrence rates of colorectal adenomas: A prospective study. Gastroenterology 1995;108(2):402–8. [DOI] [PubMed] [Google Scholar]
- 7.Yamaji Y, Mitsushima T, Ikuma H, et al. Incidence and recurrence rates of colorectal adenomas estimated by annually repeated colonoscopies on asymptomatic Japanese. Gut 2004;53(4):568–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van Stolk RU, Beck GJ, Baron JA, et al. Adenoma characteristics at first colonoscopy as predictors of adenoma recurrence and characteristics at follow-up. Gastroenterology 1998;115(1):13–8. [DOI] [PubMed] [Google Scholar]
- 9.Lieberman DA, Weiss DG, Harford WV, et al. Five-year colon surveillance after screening colonoscopy. Gastroenterology 2007;133(4):1077–85. [DOI] [PubMed] [Google Scholar]
- 10.Lochhead P, Chan AT, Nishihara R, et al. Etiologic field effect: Reappraisal of the field effect concept in cancer predisposition and progression. Mod Pathol 2015;28(1):14–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Curtius K, Wright NA, Graham TA. An evolutionary perspective on field cancerization. Nat Rev Cancer 2017;18(1):19–32. [DOI] [PubMed] [Google Scholar]
- 12.Kamiyama H, Suzuki K, Maeda T, et al. DNA demethylation in normal colon tissue predicts predisposition to multiple cancers. Oncogene 2012;31(48):5029–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Milicic A, Harrison LA, Goodlad RA, et al. Ectopic expression of P-cadherin correlates with promoter hypomethylation early in colorectal carcinogenesis and enhanced intestinal crypt fission in vivo. Cancer Res 2008;68(19):7760–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shen L, Kondo Y, Rosner GL, et al. MGMT promoter methylation and field defect in sporadic colorectal cancer. J Natl Cancer Inst 2005;97(18):1330–8. [DOI] [PubMed] [Google Scholar]
- 15.Hao CY, Moore DH, Chiu YSY, et al. Altered gene expression in normal colonic mucosa of individuals with polyps of the colon. Dis Colon Rectum 2005;48(12):2329–35. [DOI] [PubMed] [Google Scholar]
- 16.Dela Cruz M, Ledbetter S, Chowdhury S, et al. Metabolic reprogramming of the premalignant colonic mucosa is an early event in carcinogenesis. Oncotarget 2017;8(13):20543–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Backman V, Roy HK. Light-scattering technologies for field carcinogenesis detection: A modality for endoscopic prescreening. Gastroenterology 2011;140(1):35–41.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roy HK, Gomes A, Turzhitsky V, et al. Spectroscopic microvascular blood detection from the endoscopically normal colonic mucosa: Biomarker for neoplasia risk. Gastroenterology 2008;135(4):1069–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muzny DM, Bainbridge MN, Chang K, et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012;487(7407):330–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Forbes SA, Beare D, Boutselakis H, et al. COSMIC: Somatic cancer genetics at high-resolution. Nucleic Acids Res 2017;45(D1):D777–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brenner H, Hoffmeister M, Stegmaier C, et al. Risk of progression of advanced adenomas to colorectal cancer by age and sex: Estimates based on 840 149 screening colonoscopies. Gut 2007;56(11):1585–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee-Six H, Olafsson S, Ellis P, et al. The landscape of somatic mutation in normal colorectal epithelial cells. Nature 2019;574(7779):532–7. [DOI] [PubMed] [Google Scholar]
- 23.Hawthorn L, Lan L, Mojica W. Evidence for field effect cancerization in colorectal cancer. Genomics 2014;103(2-3):211–21. [DOI] [PubMed] [Google Scholar]
- 24.Di J, Yang H, Jiang B, et al. Whole exome sequencing reveals intertumor heterogeneity and distinct genetic origins of sporadic synchronous colorectal cancer. Int J Cancer 2018;142(5):927–39. [DOI] [PubMed] [Google Scholar]
- 25.Zauber P, Marotta S, Sabbath-Solitare M. Molecular genetic changes in benign colorectal tumors synchronous with microsatellite unstable carcinomas do not support a field defect. Int J Mol Epidemiol Genet 2017;8(3):27–39. [PMC free article] [PubMed] [Google Scholar]
- 26.Snippert HJ, Schepers AG, Van Es JH, et al. Biased competition between Lgr5 intestinal stem cells driven by oncogenic mutation induces clonal expansion. EMBO Rep 2014;15(1):62–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.