

Abstract
Ultraviolet photodissociation (UVPD) has emerged as a promising tool to characterize proteins with regard to not only their primary sequences and post-translational modifications, but also their tertiary structures. In this study, three metal-binding proteins, Staphylococcal nuclease, azurin, and calmodulin, are used to demonstrate the use of UVPD to elucidate metal-binding regions via comparisons between the fragmentation patterns of apo (metal-free) and holo (metal-bound) proteins. The binding of staphylococcal nuclease to calcium was evaluated, in addition to a series of lanthanide(III) ions which are expected to bind in a similar manner as calcium. On the basis of comparative analysis of the UVPD spectra, the binding region for calcium and the lanthanide ions was determined to extend from residues 40–50, aligning with the known crystal structure. Similar analysis was performed for both azurin (interrogating copper and silver binding) and calmodulin (four calcium binding sites). This work demonstrates the utility of UVPD methods for determining and analyzing the metal binding sites of a variety of classes of proteins.
Keywords: protein/metal complexes, ultraviolet photodissociation mass spectrometry
GRAPHICAL ABSTRACT

INTRODUCTION
Metal ions bound to proteins are crucial to many diverse biological processes.1–3 It is estimated that approximately 30–40% of known proteins require metal ions to stabilize their structures in the context of folding and substrate binding or to ensure proper function.4 Metals play a number of roles: (1) they act as Lewis acids, providing reactivity in catalytic processes; (2) they facilitate redox chemistry via multiple oxidation states;1 and (3) they may coordinate multiple sites in a protein and act as cross-linking agents.5 Deciphering all the diverse roles and identities as well as the resulting structures and functions of all metalloproteins in the human proteome remains a significant challenge.6 In addition to the fundamental interest in understanding the biological diversity of metalloproteins, there has also been growing interest in engineered metalloproteins suitable for targeted applications such as cleanup of heavy metals from the environment or for improved photosynthetic or catalytic activity.7,8 In each of these cases, determining the sequences of metal binding motifs in proteins and understanding how they contribute to the selectivity of metal recognition are key problems.
Mass spectrometry has become a powerful and versatile tool in the field of structural biology.9–13 An array of methods, ranging from cross-linking to hydrogen–deuterium exchange reactions and other chemical labeling methods, has advanced the application of mass spectrometry to reveal structures of proteins and protein complexes.14–16 Structural analysis of metal-binding proteins presents numerous challenges to both top-down and bottom-up proteomics strategies because both the denaturing conditions in conventional top-down methods and proteolysis in bottom-up methods result in the loss of metals, as well as any other electrostatically bound species. More recently, the concept of native mass spectrometry has been introduced, in which proteins or complexes are electrosprayed from nondenaturing aqueous solutions containing volatile salts.17–21 Native spray methods can preserve noncovalent interactions and thus have opened the door to using mass spectrometry to investigate a variety of protein complexes, including multimeric proteins and ones bound to nucleic acids, small ligands, cofactors, or metals,18,22–26 Activation of protein complexes in the gas phase allows ligand binding sites to be mapped and provides insight into subunit architecture based on disassembly of the complexes.27,28
In the context of protein–metal and peptide–metal complexes, there have been several studies using both mass spectrometry to probe metal-binding sites and derive structural information.19,29–39 For example, Loo and co-workers showed that the electron ionization dissociation (EID) of superoxide dismutase was sensitive to the bound metal, with variations in fragmentation patterns observed for the apo-, Zn-, and mixed Cu-,Zn-wild type dimers.30 Additionally, they also demonstrated that the binding site of cobalt and manganese could be elucidated for an intrinsically disordered protein, α-synuclein, based on the pattern of metal-containing fragment ions produced by electron capture dissociation (ECD).31 Kelleher and co-workers recently demonstrated that the iron-binding channels of horse spleen ferritin, a ~490 kDa multimeric complex, were accurately mapped based on monitoring the array of c-type fragment ions produced by ECD, thus allowing characterization of two iron binding channels.32 Hartinger and co-workers used electron transfer dissociation (ETD) to determine the binding sites of a denatured metallodrug–protein complex (oxaliplatin bound to ubiquitin).33 Detection of product ions that retained the metal ion led to the confirmation of oxaliplatin binding at sites Met1 and His68.33
Ultraviolet photodissociation (UVPD) has also shown promise for the elucidation of ligand-binding sites in proteins as well as revealing changes in protein conformation as a function of ligand binding.40–44 For example, upon UVPD holo (heme-containing) myoglobin exhibited suppressed backbone fragmentation in the region associated with heme binding.40 In another study, the entire stepwise catalytic cycle of adenylate kinase with respect to conformational changes of the protein between transition states was mapped by UVPD.41 A hallmark of UVPD is the retention of bound ligands by fragment ions released from the protein, an outcome that provides insight in the locations of the ligands. The high energy deposition imparted by absorption of UV photons allows access to direct fragmentation from excited electronic states, a process in which covalent cleavages of the protein backbone may occur faster than disruption of arrays of noncovalent interactions,13,45,46 In contrast to UVPD, collisional activation typically results in disruption of the electrostatic interactions in noncovalent complexes, thus disassembling the complexes but yielding little conformational insight or binding site information.22,40,47,48
In the present study, three metal-binding proteins are analyzed by native MS in order to establish UVPD as a new tool for characterizing metal-binding sites. Staphylococcal nuclease, azurin, and calmodulin were targeted because they are well-studied proteins that bind a range of metal ions, and collectively they demonstrate much of the diversity of metal binding motifs in the proteome. Staphylococcal nuclease (SNase) is a 16 kDa extracellular nuclease of Staphylococcus aureus that contains a calcium binding site crucial for catalyzing the hydrolysis of DNA and RNA at the 5′ position of the phosphodiester bond, producing mono- and dinucleotides.49 The metal center and surrounding secondary structure form the oligonucleotide binding (OB) motif, conserved among many nucleases.50 It has been previously shown that Ca(II) ions can be replaced with lanthanide ions without changing the structure of the protein, though catalytic activity is halted.51 This suggests possible applications in metal capture, recycling, and recovery applications.52,53 Additionally, lanthanides are commonly used as substitutes for calcium ions in spectroscopic studies, as lanthanides often bind to the same sites in proteins as calcium does and with similar coordination chemistry but exhibit luminescent properties.54 Azurin is a 14 kDa bacterial electron transfer protein that shuttles electrons in the cytochrome pathway via a redox cycling between Cu(I) and Cu(II).55–57 Azurin has been shown to be capable of binding to multiple transition metals besides copper, commonly zinc, although mercury, silver, nickel, cadmium, and cobalt have also been reported.57–59 Crystal structures have shown that the metal and oxidation state can affect the structure of azurin.60 One recent study illustrated the ability to modulate the redox properties of azurin via mutation of five residues and using two metals (Cu and Ni), ultimately spanning a 2 V range in reduction potential and offering intriguing opportunities for new biotechnology applications.61 Calmodulin is a highly conserved 16.7 kDa Ca(II)-binding messenger protein, ubiquitous in eukaryotic cells, which responds to changing calcium ion concentrations via structural reorganization and regulates hundreds of different downstream effectors.62–64 The protein is comprised of two pairs of similar Ca(II)-binding helix–loop–helix (EF-hand) motif structures connected by a flexible linker region.65 Despite years of investigation, the relationship between Ca(II) binding and structure of calmodulin is not fully understood. A recent study showed that coordination of calmodulin to other metals such as lanthanides affects the structure of the protein,62 thus motivating our interest in investigating both proteins that have preformed metal binding sites and those whose structures are modulated by metal binding status. In this work, we demonstrate that native electrospray mass spectrometry coupled with UVPD is capable of identifying metal binding sites in proteins, offering a strategy that could be applied to map protein–metal interactions of proteins for which crystal structures are not readily available.
EXPERIMENTAL SECTION
Protein Mutagenesis, Expression, and Purification.
Staphylococcal nuclease (S. aureus), azurin (Pseudomonas aeruginosa), and calmodulin (Arabica punctulata) were expressed and purified as described previously. More details about the expression/purification of each protein are provided in Supporting Information.
CD Spectroscopy.
Circular dichroism (CD) spectroscopy was used to investigate the structures of SNase, azurin, and calmodulin to supplement the MS analysis. CD spectra of apo SNase and holo SNase bound to Ca(II) and several lanthanide ions, apo azurin and holo azurin bound to Cu(II) and Ag(I), and apo calmodulin and holo calmodulin bound to Ca(II) were collected on a JASCO J-815 CD spectrometer (Easton, MD) in a quartz cell with a path length of 0.1 cm. Reported spectra are the average of five scans collected between 190 and 275 nm with a resolution of 0.5 nm.
MS of Protein/Metal Complexes.
Metal acetates (Ca(II), Cu(II), Ag(I), La(III), Pr(III), Nd(III), Gd(III), Dy(III)) were purchased from Sigma-Aldrich and used without further purification. MS/MS analysis of each denatured protein was performed by buffer exchanging each protein into 1:1 methanol/water with 0.5% formic acid using 10 kDa molecular weight cutoff filters (EMD Millipore). Native MS experiments were performed by buffer exchanging each protein into 200 mM ammonium acetate using 10 kDa molecular weight cutoff filters. To generate protein/metal complexes, metal acetates (100-fold excess) were added to each solution after buffer exchange. Adding the metal salts in 100-fold excess enhanced the abundances of the protein/metal complexes, but can also lead to nonspecific metal binding and adduct formation. Activation of loosely bound adducts typically results in separation of the metal from the protein and does not lead to production of diagnostic apo or holo fragment ions. Circular dichroism data helped ensure that the excess metal addition to the solution did not distort the protein structure. All experiments were performed on a Thermo Scientific Orbitrap Elite mass spectrometer (San Jose, CA, USA) equipped with a 193 nm excimer laser (Coherent, Santa Clara, CA, USA) to enable photodissociation in the HCD cell, as described previously.66 ESI was undertaken using Au/Pd-coated static emitters (o.d. = 1.2 mm; pulled to a tip of less than 1 μm o.d.) loaded with a few microliters protein solution. A voltage of 1200 V was applied to the emitter. Mass spectra were collected at an operating resolution of 240 K at m/z 400. Each protein solution was analyzed in the positive ion mode, and the complexes were subjected to both HCD and UVPD for structural characterization. MS/MS spectra were deconvoluted with a signal-to-noise threshold of two using the Xtract algorithm and analyzed with UV-POSIT (uv-posit.cm.utexas.edu), a custom, in-house algorithm to calculate the fragmentation yield of protein backbone ions.67 UV-POSIT allows analysis of the full array of 10 potential backbone cleavage product ions (a, a+1, x, x+1, b, y, y−1, y−2, c, and z ions) as both apo (metal-free) and holo (metal-bound) versions. All N- (an, bn, and cn ions) and C-terminal (xR−n+1, yR−n+1, and zR−n+1 ions) ions, both apo and holo, were summed and plotted against the protein sequence to evaluate subtle differences between the two forms of the protein where n represents the residue number and R represents the total number of amino acids in the protein sequence, as described previously.42 The standard deviation for each measurement is provided on the backbone fragmentation yield histograms for N ≥ 3.
RESULTS AND DISCUSSION
To investigate the ability of UVPD-MS to identify metal binding sites in proteins, we measured the differences in fragmentation of metal-free (apo) and metal-bound (holo) forms of three proteins: SNase, azurin, and calmodulin. Both SNase and azurin have preformed pockets for metal binding; calmodulin reorganizes upon interaction with calcium. Each protein was transferred to the gas phase using native MS conditions in order to preserve the noncovalent interactions between the protein and metal(s). Both higher energy collisional dissociation (HCD) and UVPD were used to characterize the resulting complexes after selecting the most abundant charge state for activation. The resulting MS/MS spectra were analyzed and used to construct both sequence maps and distributions of the backbone cleavages from N-terminus to C-terminus. The latter were normalized relative to the total ion current to allow facile assessment of the changes in fragmentation as a function of the presence and identity of the metal ion. The distributions of the backbone cleavages reveal the propensities for fragmentation throughout the protein as well as changes in fragmentation behavior owing to the presence of the metal ion. On the basis of previous studies of UVPD of protein–ligand complexes, it was found that the enhancement or suppression of backbone cleavages correlated with the degree of intramolecular interactions stabilizing the region and thus inhibiting the separation/detection of fragment ions.40–43 In addition to evaluation of the changes in backbone fragmentation of the holo and apo proteins, the distribution of holo fragment ions, ones that retain the metal ion, were examined. As shown previously, holo fragment ions can be used to pinpoint locations of the metal binding sites.30–33 In essence, charting the absence versus presence of metal-containing product ions originating from cleavages of the backbone extending from the N- or C-terminus provides a means to bracket the binding site. Both of these data analysis strategies (i.e., (i) examination of enhancement or suppression of backbone fragmentation of apo versus holo proteins, and (ii) monitoring metal-containing fragment ions to localize binding sites) are used in a complementary manner in the present study.
Staphylococcal Nuclease (SNase).
Because SNase was purified in the unfolded state from inclusion bodies, protein refolding following purification was confirmed via CD spectroscopy as shown in Figure S1. CD experiments were also conducted to evaluate possible unintended changes in the secondary structure of SNase when bound to different metal ions relative to the apo protein (Figure S2), but there were no differences in the CD spectra for any of the complexes, as expected based on previously reported results.51 Upon ESI of SNase, the 8+ charge state was the most abundant for each of the metal complexes (Ca(II), La(III), Pr(III), Nd(III), Gd(III), Dy(III)) with significantly lower abundance of the 7+ charge state (Figures 1 and S3).
Figure 1.

ESI mass spectra of solutions containing staphylococcal nuclease (SNase): (a) no metal and (b–g) containing metal salts. All solutions contain 120 mM ammonium acetate. The corresponding deconvoluted MS1 spectra are shown in Figure S3.
Each of the 1:1 SNase/metal complexes (8+) was isolated and subjected to 193 nm UVPD. Figure 2 shows the resulting fragmentation patterns for the apoprotein and one representative metal complex (Pr(III)). UVPD of the SNase/metal complexes resulted in both apo fragment ions (no metal retained) and holo fragment ions (ones retaining the metal ion). The type of MS/MS spectra in Figure 2 is commonly observed upon 193 nm UVPD of proteins,40,43 even for the low charge states generated by native MS conditions. High sequence coverages (>65%) are derived from these spectra, as shown by the sequence maps adjacent to the UVPD mass spectra (Figure 2c,d).
Figure 2.
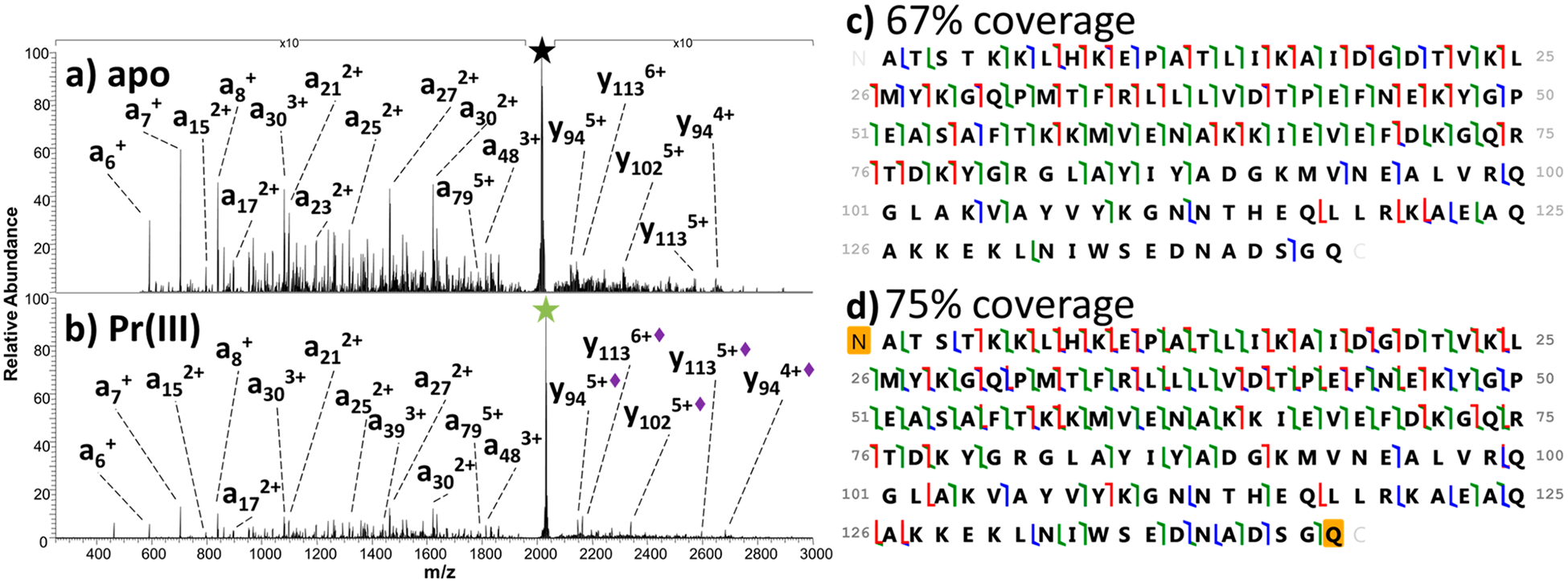
UVPD of (a) apo SNase (8+) and (b) Pr(III)-bound SNase (8+) with some fragment ions labeled. Holo fragment ions (i.e., retaining the metal ion) are denoted with a diamond in the label. (c, d) Sequence coverage maps. Backbone cleavages that result in a/x (green), b/y (blue), and c/z• (red) fragment ions are indicated by the color-coded flags.
The distributions of backbone cleavages from UVPD of apo SNase and complexes containing Ca(II) or Pr(III) are shown in Figure 3. The distribution for apo SNase (Figure 3a) shows extensive fragmentation of the protein backbone with particularly high fragmentation efficiency from residues 26–61. The UVPD distributions obtained for the Ca(II), Pr(III), La(III), Nd(III), Gd(III), and Dy(III)complexes of SNase show similar coverages of the protein backbone, but the efficiency of fragmentation is suppressed in the region from residues Asp40–Asn51.
Figure 3.
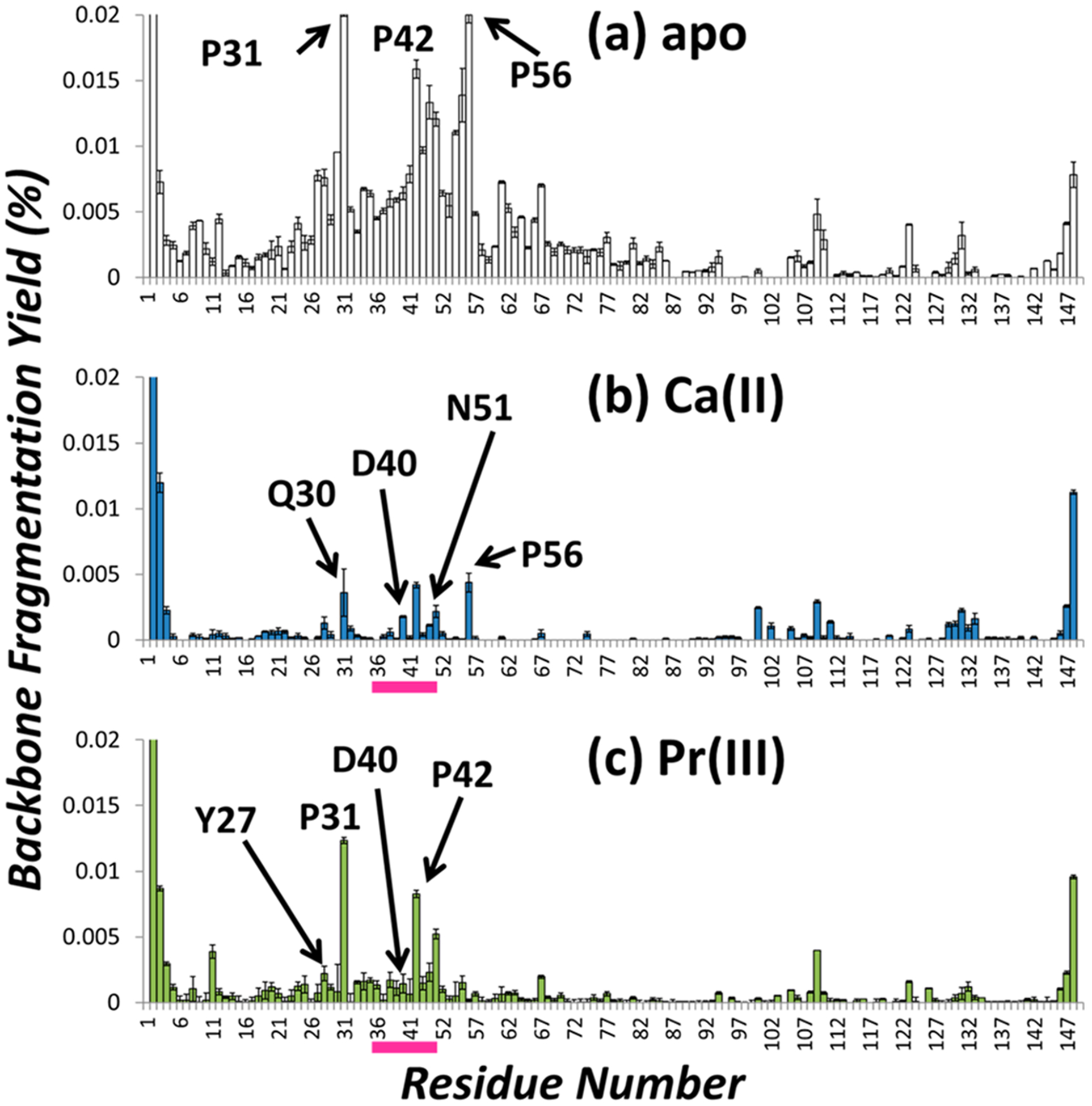
Distribution of backbone cleavages from UVPD of (a) apo SNase (8+), (b) Ca(II)-bound SNase (8+), and (c) Pr(III)-bound SNase (8+), normalized to the TIC. The white bars represent apo fragment ions, and the shaded bars represent holo ions (blue for Ca(II), green for Pr(III)). Selected residues are labeled. The binding sites for Ca(II) and Pr(III) are demarcated by pink bars.
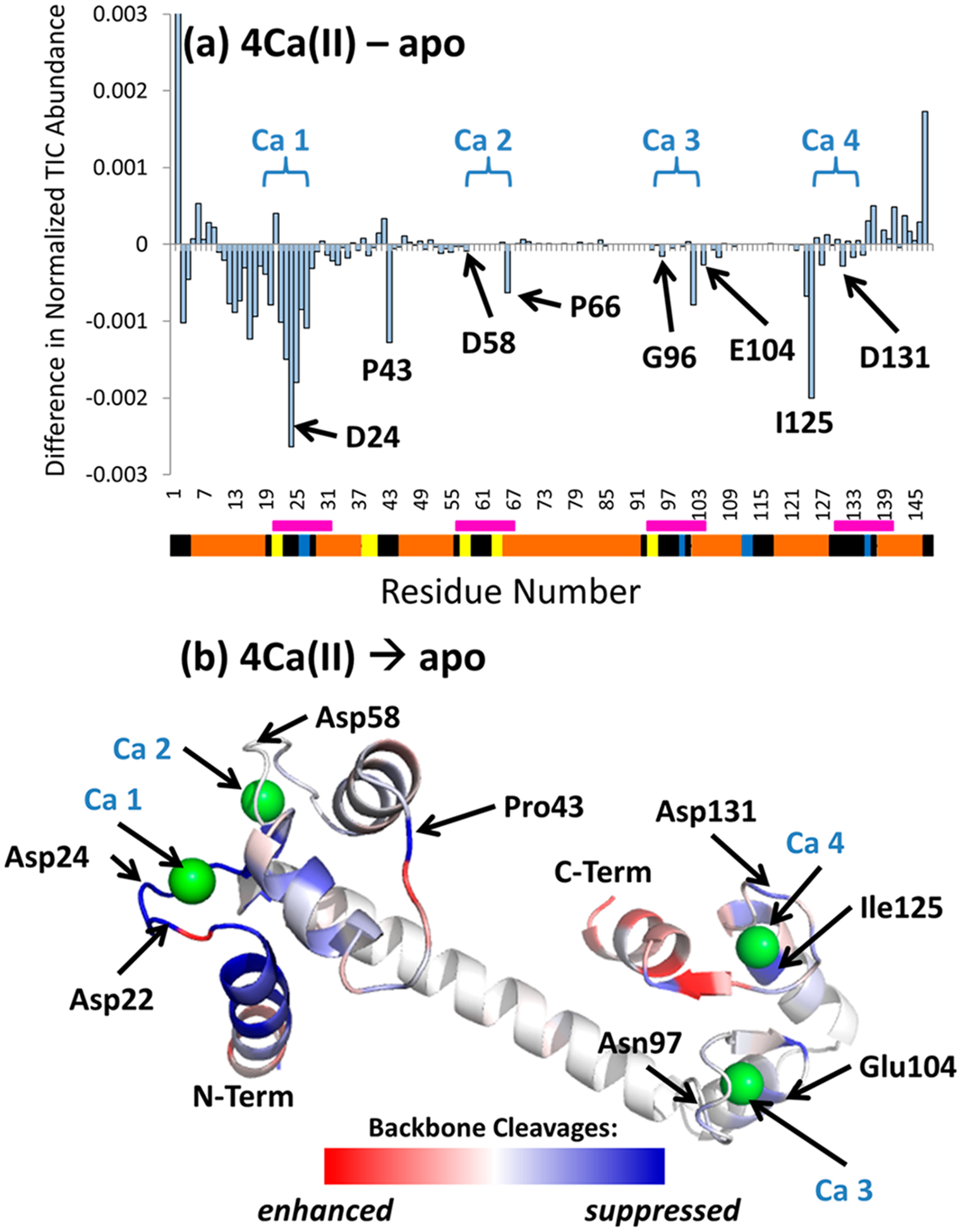
The extent of suppression of fragmentation of the metal complexes along the span of residues from 40 to 51 relative to apo SNase is underscored by the difference plots shown in Figure 4a,b, for the Ca(II) and Pr(III)complexes. The negative portion of the difference plots indicates regions of the protein for which fragmentation is suppressed upon metal complexation. The variation in the efficiency of the backbone cleavages is mapped on the crystal structure of SNase (PDB: 3BDC) in Figure 4c,d, and the binding site for SNase is expanded and labeled in greater detail in Figure S4. In Figure 4c,d, red shading represents enhanced fragmentation, and blue shading represents suppressed fragmentation of the metal complexes relative to apo SNase. For both the Ca(II) and Pr(III) complexes, one notable region of suppression of backbone fragmentation occurs near the known primary metal ion coordination site of the protein (Asp40). There are several other regions of the backbone for which fragmentation is slightly decreased for the metal complexes relative to the apo protein, suggesting an enhancement of structural rigidity upon metal complexation and thus less effective disassembly of that region upon UVPD (allowing less efficient separation and detection of fragments).
Figure 4.

Difference plots for the UVPD backbone cleavage distributions of (a) apo SNase and Ca(II)-bound SNase (summed holo and apo fragment ions) and (b) apo SNase and Pr(III)-bound SNase (summed holo and apo ions) (8+). Negative values represent regions of the protein backbone for which backbone cleavages of the metal complexes are suppressed relative to the apoprotein. The segmented bar beneath the residue numbers displays secondary structural elements: yellow (turn), blue (beta strand), orange (α helix), and black (unassigned). The binding sites for Ca(II) and Pr(III) are demarcated by pink bars. (c, d) The values from the difference plots are superimposed on the crystal structure (PDB: 3BDC) as heat maps to show areas of enhancement (red) and suppression (blue) of fragmentation upon metal binding. Selected residues are labeled. The binding site for SNase is expanded and labeled in greater detail in Figure S4.
Larger variations in the extent of backbone cleavages are also commonly observed very close to the N- or C-termini, as seen for SNase as well as the two other proteins included in this study. Enhanced fragmentation near the termini does not seem to directly correlate with metal binding, and in fact has also been observed for other proteins and using other ion activation methods (data not shown). There seems to be consensus that the termini may have greater conformational flexibility owing to some unravelling, possibly accounting for greater fragmentation. Charge-mediated effects localized near the N-and C-termini may also play a role.
Another region that exhibits suppression of fragmentation for the metal complexes relative to apo SNase occurs around the region from residues Tyr27 to Pro31. There is also consistently decreased fragmentation adjacent to residues Pro31, Pro42, and Pro56 for each SNase/metal complex, suggesting that the proline-effect is suppressed for the metal complexes relative to metal-free SNase. The region of suppressed backbone fragmentation for the metal complexes generally extends from Tyr27 to Pro56 and is attributed to three contributing factors: (1) This region includes a flexible turn region (residues Tyr27 to Gln30) between two antiparallel beta sheets (residues Thr22 to Met26 and Pro31 to Arg35), suggesting that metal binding causes a larger degree of conformational reorganization in this region owing to the greater inherent flexibility of the turn. (2) The complexation of the metal ion in this region of the protein suppresses the potential for fragmentation through newly formed electrostatic interactions between Asp21, Asp40, Thr41, Glu43, and the metal ion. (3) The proline residues at position 31, 42, and 56 are less susceptible to the proline effect upon activation of the metal complexes. Typically, proteins may exhibit enhanced cleavage at the backbone position N-terminal to proline; this preferential cleavage is known as the proline effect.68 Yin and Loo noted Pro-directed fragmentation of the carbonic anhydrase-II:zinc complex which generates a product ion pair at proline sites, with the N-terminal product ion retaining the metal ion after separation of the two fragments.29 For both apo-SNase and its array of metal complexes, there are significant variations in both the fragmentation efficiency around Pro31 and the degree of metal ion retention of the resulting product ions arising from backbone cleavage adjacent to Pro31 upon UVPD, suggesting that there is variability in the interaction of the metal at this site. Fragmentation is consistently suppressed at Pro31 for all the metal complexes, similar to the suppression observed at other proline residues mentioned previously.
The propensity for cleavage adjacent to Pro has been studied extensively, in part because of the frequently observed enhancement of Pro cleavages in MS/MS spectra of peptides69 and in part because of the special cis/trans isomerization of peptide bonds preceding Pro.70 When Pro is preceded by amino acids with branched aliphatic side chains, such as Val, Ile, or Leu, there are significantly greater conformational restrictions that impede rotation of the bond to the cis form and limit adoption of secondary structures in comparison to when Pro is preceded by a more conformationally mobile amino acid such as Gly.69 The net outcome of this variation in conformational flexibility is the enhancement (presence of Val, Ile, Leu) or suppression (the presence of Gly) of backbone cleavages N-terminal to Pro.69 In a similar vein, it was shown that cis/trans isomerization of proline peptide bonds dictated the propensity for backbone cleavages close to Pro, as demonstrated by UVPD of conformer-selected ubiquitin ions.70 In this case, the observation that product ions arising from cleavages near Pro were suppressed for the more compact cis conformers was rationalized based on the greater number of noncovalent contacts around the Pro residues that limited separation of fragment ions formed during UVPD.70 The unifying link between these previous studies is the important role of Pro in mediating conformational flexibility and noncovalent interactions, thus suggesting an analogous role of the metal ion in the present study. Complexation of a metal ion, which promotes formation of multiple contacts with side chains of interacting amino acids, is expected both to significantly alter conformational flexibility and to influence cis/trans isomerization of Pro peptide bonds.71 A combination of these factors may rationalize the suppression of fragmentation in specific regions of the protein/metal complexes upon UVPD, particularly ones encompassing Pro residues.
The distributions of apo and holo fragment ions can also be categorized based on whether they contain the N-terminus (a, b, c ions) or C-terminus (x, y, z ions). These histograms are shown in Figure S5 for each of the SNase:metal complexes. For apo SNase, UVPD produced both N- and C-terminal fragment ions via cleavages throughout the backbone of the protein, as commonly observed for top-down analysis by UVPD. For each of the metal complexes, a majority of the N-terminal ions are apo products, whereas far more of the C-terminal ions retain the metal ion (holo products). C-terminal holo ions are identified consistently until Pro56, along with a small number of low abundance ions from backbone cleavages adjacent to residues Phe61–Asp77 and Glu129–Lys133. Neither of these latter regions is localized near the preformed binding pocket of SNase, implying that either some degree of metal ion scrambling occurs post-photoactivation or that the protein adopts several conformations that are simultaneously sampled and activated. Only a few large N-terminal fragment ions retained the metal ion, such as ones originating from backbone cleavages well past the expected binding site (such as a1006+ and c1096+). The dichotomy between holo fragment ions (mostly long C-terminal ions) and apo fragment ions (mostly short-to-midsized N-terminal ions) may reflect the degree of loss of tertiary structure of the protein after the backbone cleavage occurs. Because the N-terminal ions are shorter, these truncated segments of the protein are less likely to maintain appropriate tertiary structure to retain the metal ion in contrast to the significantly longer C-terminal ions.
For comparison to UVPD, HCD was also performed on apo SNase and the Pr(III) complex to determine whether a similar trend in the suppression of backbone fragmentation was observed. Deconvoluted HCD mass spectra are shown in Figure S6, and the corresponding backbone cleavage distributions are displayed in Figures S7 and S8. While HCD of both the apo and holo SNase yielded satisfactory sequence coverage (averaging over 66%), the difference map did not illuminate structural differences between the two species. Variations in the efficiency of backbone cleavages were observed throughout the protein backbone, including some suppression (notable at Asp19, Asp40, Lys78, and Lys84). HCD of the SNase:Pr(III) complex yielded mainly N-terminal apo product ions with coverage of the protein backbone diminishing past Lys64, and the corresponding holo ion analysis displayed primarily a few C-terminal holo ions. The lower energy, stepwise energization process of HCD restricted the production of sufficient holo product ions needed to characterize the binding sites, making it challenging to rely on the HCD data to provide a clear picture of the metal binding site. These results suggest that UVPD provides a more specific means to examine shifts in the protein conformation attributed to metal coordination, and thus HCD was not evaluated in detail for the other two proteins included in the study.
ETD was also considered as a means to provide additional complementary or Supporting Information about metal binding interactions of proteins. An example is shown in Figure S9 for the 8+ charge state of SNase:Pr(III). ETD primarily led to charge reduction and production of relative few diagnostic sequence ions. The effectiveness of ETD is known to be dependent on the charge density of the precursor ion, and thus the unremarkable performance of ETD for low charge states of the native-like protein/metal complexes in the present study was not unexpected. Far better results have been obtained by using electron ionization dissociation (EID), a method that uses higher energy electrons to cause ionization and electronic excitation of selected precursor ions.30
As a final comparison, the B-factors of apo SNase (PDB: 3SK8) and Ca(II)-bound SNase (PDB: 3BDC) were plotted in Figure S10, along with the difference plot. The B-factors are derived for each α carbon of the protein backbone; a larger value represents a more flexible region of the protein, and a lower value suggests a well-ordered and/or more rigid region.72 According to the B-factors, the Ca(II) binding site of SNase occurs in a region of localized structural rigidity. There does not appear to be significant change in the B-factor between the apo and the Ca(II)-bound SNase structures near the Ca(II)coordination site, confirming that the protein is preorganized prior to metal binding.
Azurin.
Azurin, a bacterial redox protein, has been shown to bind both Cu(II) and Ag(I) in which multiple residues, particularly two imidazoles (His46 and His117), the axial methionine thioether (Met121), and the cysteine thiolate (Cys112) located near the C-terminus, form a single, preorganized binding site.56,73,74 Azurin has been extensively characterized owing to its redox properties and important role in electron transfer.57 The binding site of azurin is ideal for the coordination of Cu(II) (tetra-coordinate) and Cu(I) (tricoordinate), minimizing the reorganization energy between the oxidation states and increasing the rate of electron transfer.75 Ag(I) has been demonstrated to occupy the same binding site (PDB: 3UGE; Gly45 carbonyl at 2.8 Å, His46 imidazole at 2.3 Å, Cys112 thiolate at 2.4 Å, His117 imidazole at 2.3 Å, and Met121 thioether at 3.1 Å)73 nearly isomorphic with Cu(II), suggesting that the preformed binding site is amenable to other metals, even ones that might render the protein inactive.76 It is intriguing to consider the impact of replacing a native metal with a non-native metal with different properties. CD spectra of apo azurin and the Cu(II) and Ag(I) complexes are shown in Figure S11. A slight shift toward more α-helical content (208 nm) was observed for the Ag(I) complex, a feature also evidenced in the crystal structure for Ag(I)-coordinated azurin (PDB: 3UGE).
MS1 spectra of apo-azurin and the copper and silver complexes are shown in Figure S12. Complexes in the 7+ charge state were dominant, with incorporation of one to three Cu(II)ions or two or three Ag(I) ions. Despite significant manipulation of solution conditions and ESI parameters, 1:1 azurin/Ag(I) complexes could not be generated in sufficient abundance for MS/MS characterization. Azurin complexes containing one copper ion or two silver ions were isolated and subjected to UVPD. For analysis of the spectra, holo fragment ions were considered containing the mass of one copper or one silver ion, as only one ion is expected to bind in the pocket according to the crystal structures (PDB: 4AZU for Cu(II)-bound azurin and PDB: 3UGE for Ag(I)-bound azurin). The second and third copper or silver ions are expected to be nonspecifically bound, likely owing to interaction with a peripheral imidazole group as previously observed for azurin and attributed to a similar phenomenon noted for silver complexation of hen egg white lysozyme (PDB: 3RU5).73,77 The ESI mass spectra alone do not allow determination of whether Ag(I) is bound isomorphically as Cu(II). UVPD was used to characterize the complexes (7+), and the resulting sequence maps are shown in Figure S13. Sequence coverages obtained by UVPD ranged from 94% for apo-azurin to 83% for the Cu(II)complex and 81% for the Ag(I) complex.
The distributions of backbone cleavages are shown in Figure S14, showcasing the differences in the behavior of metal-free and metal-bound azurin. Differences plots for the UVPD fragmentation of the Cu(II) and Ag(I) complexes relative to apo azurin are displayed in Figure 5. Fragmentation of much of the protein backbone was slightly or significantly suppressed upon metal complexation, and there were only a few backbone cleavages that were enhanced upon metal binding (such as cleavages adjacent to residues Pro36, Asp98, Phe111, and Pro115 for the Cu(II) complexes and Pro36, Phe97, Cys112, and Pro115 for the Ag(I) complexes). The regions of suppression of backbone cleavages are superimposed on the crystal structures of copper- and silver-coordinated azurin (PDB: 4AZU and 3UGE, respectively) in Figure 5, and expansions of these binding regions are shown in Figure S15.
Figure 5.

Difference plots for the UVPD backbone cleavage yields of azurin and (a) Cu(II)-bound azurin (summed holo and apo fragment ions) and (b) 2Ag(I) complexes (summed holo and apo ions) (7+). Negative values represent regions of the protein backbone for which backbone cleavages of the metal complexes are suppressed relative to the apoprotein. The segmented bar beneath the residue numbers displays secondary structural elements: yellow (turn), blue (beta strand), orange (α helix), and black (unassigned). The binding sites for copper and silver are demarcated by pink bars. (c, d) The values from the difference plots are superimposed on the crystal structures (PDB: 4AZU and 3UGE, respectively) as heat maps to show areas of enhancement (red) and suppression (blue) of fragmentation upon metal binding. Selected residues are labeled. The binding site for azurin is expanded and labeled in greater detail in Figure S15.
The region of greatest suppression covers residues 100–120, ones that are in close proximity or are directly coordinated by the metal ion. Minor suppression of fragmentation is also observed from residues 30–45, which is also in close spatial proximity to the metal ion binding site in the crystal structure. This dual pattern of suppression of backbone fragmentation implies that the observed pattern of product ion suppression is consistent with the reported metal ion binding site of azurin (PDB: 4AZU and 3UGE). The suppression of backbone fragmentation in the region from Ser100 to His117 is more extensive and pronounced for the Ag(I) complexes relative to the Cu(II) complexes. We speculate that this region of suppressed fragmentation may be related to the larger size of Ag(I) (~1.2 Å radius for Ag(I)) compared to Cu(II) (~0.7 Å radius for Cu(II)).73,78 Furthermore, the Ag(I)-bound azurin complex appears to exhibit more generalized suppression of fragmentation throughout the protein backbone, likely owing to the nonspecific interactions with the second Ag(I) ion in the complex.
Calmodulin.
As a final demonstration of the application of UVPD for elucidating the impact of metal binding on protein fragmentation, calmodulin was examined to determine the applicability of UVPD to pinpoint multiple cation binding sites simultaneously, and in this case, the binding sites of four calcium ions to the symmetrical binding motifs of calmodulin. The following residues have been identified in the four binding sites: Site 1: Asp20, Asp22, Asp24, Thr26, Glu31; site 2: Asp56, Asp58, Asn60, Thr62, Glu67; Site 3: Asp93, Asp95, Asn97, Leu99, Glu104 (Site 3); and Site 4: Asp129, Asp131, Asp133, His135, Glu140.62 The MS1 spectra of apo-calmodulin (generated using EDTA to remove all Ca(II) ions from the protein during the cleanup process) and holo-calmodulin (an excess of Ca(II) ions was reintroduced) are shown in Figure S16. The CD spectra of apo- and Ca(II)-loaded calmodulin exhibit no obvious structural differences between the two forms (Figure S17), although it is expected that some degree of reorganization occurs that is not reflected in the secondary structure. The distributions of backbone cleavages across the apo and Ca(II)-loaded protein are shown in Figure S18. Inclusion of one, two, three, or four calcium ions was considered for searching and assignment of all holo fragment ions (Figure S19). Holo product ions containing three or four Ca(II) ions are predominantly large fragment ions that cover three or four binding domains. Product ions containing one and two Ca(II) ions include both N- and C-terminal ions covering the EF-hands at both ends of calmodulin. Interestingly, the α-helical portion of the protein that connects the two EF-hands does not fragment extensively for either the apo- or holo-protein (i.e., residues Phe68 to Phe92).
Sequence maps of the apo- and Ca(II)-bound calmodulin are shown in Figure S20, with sequence coverages of 55% and 43%, respectively. In comparison, a sequence coverage of 71% was obtained via UVPD for denatured calmodulin (1:1 methanol/water with 0.5% formic acid) in the 12+ charge state (data not shown). Sequence coverage of the native protein was lower than observed for other proteins owing to a significant lack of fragmentation about the central helix between the two lobes of the protein and around the helical regions in general. This is a phenomenon that has been observed in prior native mass spectrometry studies which have reported that helical regions are less susceptible to fragmentation.79,80 For example, SNase exhibits three main alpha helical regions: G55–N68, V99–R105, and F122–K134 and azurin exhibits only one main helix, spanning residues M56–S66. Fragmentation is relatively sparse and suppressed for both of these proteins in these alpha helical regions upon UVPD (Figure 3 for SNase; Figure 5 for azurin), similar to the finding for calmodulin. Difference plots of the backbone cleavage efficiencies observed for the holo- versus apo-calmodulin reveal four regions of consistent suppression of fragmentation, corresponding to the four known binding sites of the Ca(II) ions (Figure 6a). These four sites spanned residues Asp19–Thr26 (overlapping with Site 1), Asp53–Glu64 (overlapping with Site 2), Lys94–His104 (overlapping with Site 3), and Ile122–Tyr135 (overlapping with Site 4). Additionally, cleavage adjacent to a proline residue at positions 43 and 66 resulted in a large negative spike in the difference plot, attributed to a diminished proline effect for holocalmodulin.
Figure 6.
(a) Difference plot for the UVPD backbone cleavage yields of apo-calmodulin and the Ca(II) complexes (summed holo and apo fragment ions) (7+). Negative values represent regions of the protein backbone for which backbone cleavages of the metal complexes are suppressed relative to the apoprotein. The segmented bar beneath the residue numbers displays secondary structural elements: yellow (turn), blue (beta strand), orange (α helix), and black (unassigned). The binding sites for the four calcium ions are demarcated by pink bars. (b) The values from the difference plots are superimposed on the crystal structure (PDB: 1CLL) as heat maps to show areas of enhancement (red) and suppression (blue) of fragmentation upon metal binding. Selected residues are labeled for clarity. An expanded region of the Ca 1 binding site illustrating the polar contacts between the metal ion and the protein (side chains) is shown in Figure S21.
The difference plot showing the enhancement/suppression of fragmentation of the Ca(II) complex relative to apo calmodulin was plotted as a heatmap on the crystal structure of calmodulin (Figure 6b, PDB: 1CLL, expanded region of the first calcium binding region showing polar contacts between calcium, and calmodulin is shown in Figure S21). The suppression of fragmentation is generally localized to the regions of the crystal structure known to coordinate the Ca(II) ions, reflecting the structural order induced by metal binding.
A recent study reported variations in the structure of calmodulin as a function of bound metals (Ca(II) versus Tb(III), La(III), and Lu(III)).62 By combining crystallographic data, modeling approaches, and 2D-FTIR spectroscopy, subtle structural changes induced by metal binding were deciphered.62 The metal-binding domains were found to be consistent for the complexes containing the calcium and lanthanide ions, but the specific orientations of amino acids varied upon lanthanide coordination.62 The carboxylate groups in the EF-hands of the calmodulin binding sides were probed in detail, particularly the glutamate residue at position 12, which typically has a bidentate coordination geometry.62 Calmodulin remained functional upon complexation with Ln(III)ions, implying that the net structures were similar to the Ca(II) complexes, but time-dependent 2D IR spectroscopy suggested that the coordination geometry was disrupted upon lanthanide binding.62 For our comparative study, EDTA was used to remove the calcium ions from calmodulin, and then a 100-fold excess of Pr(III) or Gd(III) acetate (by mole) was added. The resulting complexes (7+) contained four metal ions (Figure S22). The backbone fragmentation plots for the Pr(III)- and Gd(III) complexes are displayed in Figure S23 as well as the differences in fragmentation between the lanthanide complexes and apo calmodulin. The fragmentation patterns are similar to that of the Ca(II) complex, showing suppressed fragmentation in many of the same general regions compared to apo calmodulin, particularly around the residues known to be metal binding sites. However, precise coordination geometries, such as monodentate versus bidentate versus bridging versus pseudobridging, at specific carboxylate groups as shown via 2D-FTIR spectroscopy62 cannot be determined by UVPD-MS.
The backbone fragmentation of the complexes containing Pr(III) or 4Gd(III) was compared to the fragmentation of the Ca(II) complexes to examine variations in fragmentation attributed to the nature of the metal ion (see difference plots in Figure S24). Fragmentation along the N-terminal region was more pronounced for the Ca(II) complexes, and fragmentation across the C-terminal region was enhanced for the Pr(III) and Gd(III) complexes. This observation suggests that the lanthanides preferentially coordinate the C-terminus (Site 4), whereas calcium binds more strongly in the N-terminus region (Sites 1 and 2), a finding that might similarly correlate with the switch from bidentate to monodentate coordination geometries noted previously for the different metals.62
For all of the results and comparisons described above, all types of fragment ions arising from each backbone cleavage site were grouped together to show the overall variations in fragmentation throughout the protein. However, it is also interesting to evaluate the specific ion types that contribute to the distributions. Examples of difference plots of each ion type generated by UVPD (a, a + 1, b, c, x, x + 1, y, y – 1, y – 2, and z) for the three proteins are shown in Figures S25–30. The a, x, and y series of ions exhibited the most significant abundances and variations of abundances. The a/x types are most unique to UVPD, thus providing support as to why UVPD might demonstrate fragmentation behavior that is particularly sensitive to structural variations.
CONCLUSION
UVPD was explored as a tool to probe protein topologies in the context of metal binding. Through examining the relative abundances of fragment ions arising from UVPD of apo- versus holo-proteins, differences in relative fragmentation efficiencies were mapped onto the crystal structures. One of the features of UVPD that makes it well-suited for the characterization of protein complexes is the preservation of noncovalent interactions at the same time that covalent backbone bonds are cleaved. The high energy deposition and fast fragmentation process allow electrostatic interactions between a protein and a ligand to be maintained even while covalent bonds are cleaved, a phenomenon essential for the determination of ligand interactions.45,46 For Staphylococcal nuclease, calcium and lanthanide ions bind to the same region of the protein as evidenced by suppressed fragmentation centralized around the known binding site for calcium. Metal complexes of azurin and calmodulin also exhibited suppression of fragmentation near the residues known to coordinate the metals (Ag(I) and Cu(II) for azurin, Ca(II), Pr(III), and Gd(III) for calmodulin). For all three proteins examined, the regions of greatest suppression of fragmentation of the metal complexes relative to the apo protein coincided with the metal binding sites of the protein, demonstrating that UVPD is a promising strategy for pinpointing electrostatic interactions between proteins and metal ions. Evaluation of additional proteins, particularly ones that have preorganized metal binding sites (akin to SNase and azurin) versus ones that undergo larger conformational changes as a result of metal binding (similar to calmodulin), would offer further insight about whether UVPD can be used to discern this level of detail.
Supplementary Material
ACKNOWLEDGMENTS
Funding from the NIH (R01 GM121714 JSB and R01 GM123252 to K.N.D.), NSF (CHE-1807215 and MCB-1714555 L.J.W.), and the Welch Foundation (F-1155 (JSB), F-1722 and H-F-0001 (L.J.W.), and F-1390 to K.N.D.) is acknowledged.
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jasms.0c00066.
Supporting items includes a description of protein preparation, CD spectra and MS1 spectra for each protein and protein complex solution, expanded graphical depictions of the metal binding regions of each protein, distributions of backbone cleavage positions that result in N- and C-terminal product ions for each protein and protein complex, difference plots of backbone cleavage yields for apo versus holo proteins, examples of deconvoluted MS2 spectra of protein and protein complexes, ETD mass spectrum of SNase•Pr(III) complex, B-factor plots for each protein and protein complex, sequence maps of protein, and protein complexes obtained by UVPD (PDF)
Contributor Information
David H. Giles, Division of Chemical Biology and Medicinal Chemistry, University of Texas, Austin, Texas 78712, United States
Whitney A. Fies, Department of Chemistry, University of Texas, Austin, Texas 78712, United States
Kevin N. Dalby, Division of Chemical Biology and Medicinal Chemistry and Graduate Program in Cell and Molecular Biology, University of Texas, Austin, Texas 78712, United States;.
Lauren J. Webb, Department of Chemistry, Institute for Cell and Molecular Biology, and Texas Materials Institute, University of Texas, Austin, Texas 78712, United States;.
Jennifer S. Brodbelt, Department of Chemistry, University of Texas, Austin, Texas 78712, United States;.
REFERENCES
- (1).Liu J; Chakraborty S; Hosseinzadeh P; Yu Y; Tian S; Petrik I; Bhagi A; Lu Y Metalloproteins Containing Cytochrome, Iron-Sulfur, or Copper Redox Centers. Chem. Rev 2014, 114, 4366–4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Bertini PI; Gray HB; Stiefel EI; Valentine JS Biological Inorganic Chemistry: Structure and Reactivity; University Science Books, 2007. [Google Scholar]
- (3).Shi W; Chance MR Metallomics and metalloproteomics. Cell. Mol. Life Sci 2008, 65, 3040–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Shi W; Chance MR Metalloproteomics: forward and reverse approaches in metalloprotein structural and functional characterization. Curr. Opin. Chem. Biol 2011, 15, 144–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Chou C-C; Martin-Martinez FJ; Qin Z; Dennis PB; Gupta MK; Naik RR; Buehler MJ Ion Effect and Metal-Coordinated Cross-Linking for Multiscale Design of Nereis Jaw Inspired Mechanomutable Materials. ACS Nano 2017, 11, 1858–1868. [DOI] [PubMed] [Google Scholar]
- (6).Andreini C; Bertini I; Rosato A Metalloproteomes: A Bioinformatic Approach. Acc. Chem. Res 2009, 42, 1471–1479. [DOI] [PubMed] [Google Scholar]
- (7).Waldron KJ; Robinson NJ How do bacterial cells ensure that metalloproteins get the correct metal? Nat. Rev. Microbiol 2009, 7, 25–35. [DOI] [PubMed] [Google Scholar]
- (8).Lu Y; Yeung N; Sieracki N; Marshall NM Design of functional metalloproteins. Nature 2009, 460, 855–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Heck AJR Native mass spectrometry: a bridge between interactomics and structural biology. Nat. Methods 2008, 5, 927–933. [DOI] [PubMed] [Google Scholar]
- (10).Benesch JL; Ruotolo BT Mass spectrometry: come of age for structural and dynamical biology. Curr. Opin. Struct. Biol 2011, 21, 641–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Jurneczko E; Barran PE How useful is ion mobility mass spectrometry for structural biology? The relationship between protein crystal structures and their collision cross sections in the gas phase. Analyst 2011, 136, 20–28. [DOI] [PubMed] [Google Scholar]
- (12).Aebersold R; Mann M Mass-spectrometric exploration of proteome structure and function. Nature 2016, 537, 347–355. [DOI] [PubMed] [Google Scholar]
- (13).Brodbelt JS Photodissociation mass spectrometry: new tools for characterization of biological molecules. Chem. Soc. Rev 2014, 43, 2757–2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Konermann L; Stocks BB; Pan Y; Tong X Mass spectrometry combined with oxidative labeling for exploring protein structure and folding. Mass Spectrom. Rev 2009, 29, 651–667. [DOI] [PubMed] [Google Scholar]
- (15).Wales TE; Engen JR Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom. Rev 2006, 25, 158–170. [DOI] [PubMed] [Google Scholar]
- (16).Sinz A The advancement of chemical cross-linking and mass spectrometry for structural proteomics: from single proteins to protein interaction networks. Expert Rev. Proteomics 2014, 11, 733–743. [DOI] [PubMed] [Google Scholar]
- (17).Rose RJ; Damoc E; Denisov E; Makarov A; Heck AJR High-sensitivity Orbitrap mass analysis of intact macromolecular assemblies. Nat. Methods 2012, 9, 1084–1086. [DOI] [PubMed] [Google Scholar]
- (18).Li H; Wolff JJ; Van Orden SL; Loo JA Native Top-Down Electrospray Ionization-Mass Spectrometry of 158 kDa Protein Complex by High-Resolution Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Anal. Chem 2014, 86, 317–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Zhang H; Cui W; Wen J; Blankenship RE; Gross ML Native Electrospray and Electron-Capture Dissociation FTICR Mass Spectrometry for Top-Down Studies of Protein Assemblies. Anal. Chem 2011, 83, 5598–5606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).van Duijn E Current limitations in native mass spectrometry based structural biology. J. Am. Soc. Mass Spectrom 2010, 21, 971–978. [DOI] [PubMed] [Google Scholar]
- (21).Lorenzen K; van Duijn E Native Mass Spectrometry as a Tool in Structural Biology. Curr. Protocols Protein Sci 2010, 62, 17.12.1–17.12.17. [DOI] [PubMed] [Google Scholar]
- (22).Belov ME; Damoc E; Denisov E; Compton PD; Horning S; Makarov AA; Kelleher NL From Protein Complexes to Subunit Backbone Fragments: A Multi-stage Approach to Native Mass Spectrometry. Anal. Chem 2013, 85, 11163–11173. [DOI] [PubMed] [Google Scholar]
- (23).Chait BT; Cadene M; Olinares PD; Rout MP; Shi Y Revealing Higher Order Protein Structure Using Mass Spectrometry. J. Am. Soc. Mass Spectrom 2016, 27, 952–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Konermann L; Vahidi S; Sowole MA Mass Spectrometry Methods for Studying Structure and Dynamics of Biological Macromolecules. Anal. Chem 2014, 86, 213–232. [DOI] [PubMed] [Google Scholar]
- (25).Breuker K; Brüschweiler S; Tollinger M Electrostatic Stabilization of a Native Protein Structure in the Gas Phase. Angew. Chem., Int. Ed 2011, 50, 873–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Bellina B; Compagnon I; MacAleese L; Chirot F; Lemoine J; Maître P; Broyer M; Antoine R; Kulesza A; Mitrić R; Bonačić-Koutecký V; Dugourd P Binding motifs of silver in prion octarepeat model peptides: a joint ion mobility, IR and UV spectroscopies, and theoretical approach. Phys. Chem. Chem. Phys 2012, 14, 11433–11440. [DOI] [PubMed] [Google Scholar]
- (27).Gavriilidou AFM; Holding FP; Mayer D; Coyle JE; Veprintsev DB; Zenobi R Native Mass Spectrometry Gives Insight into the Allosteric Binding Mechanism of M2 Pyruvate Kinase to Fructose-1,6-Bisphosphate. Biochemistry 2018, 57, 1685–1689. [DOI] [PubMed] [Google Scholar]
- (28).Song Y; Nelp MT; Bandarian V; Wysocki VH Refining the Structural Model of a Heterohexameric Protein Complex: Surface Induced Dissociation and Ion Mobility Provide Key Connectivity and Topology Information. ACS Cent. Sci 2015, 1, 477–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Yin S; Loo JA Top-down mass spectrometry of supercharged native protein-ligand complexes. Int. J. Mass Spectrom 2011, 300, 118–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Li H; Sheng Y; McGee W; Cammarata M; Holden D; Loo JA Structural Characterization of Native Proteins and Protein Complexes by Electron Ionization Dissociation-Mass Spectrometry. Anal. Chem 2017, 89, 2731–2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Wongkongkathep P; Han JY; Choi TS; Yin S; Kim HI; Loo JA Native Top-Down Mass Spectrometry and Ion Mobility MS for Characterizing the Cobalt and Manganese Metal Binding of α-Synuclein Protein. J. Am. Soc. Mass Spectrom 2018, 29, 1870–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Skinner OS; McAnally MO; Van Duyne RP; Schatz GC; Breuker K; Compton PD; Kelleher NL Native Electron Capture Dissociation Maps to Iron-Binding Channels in Horse Spleen Ferritin. Anal. Chem 2017, 89, 10711–10716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Meier SM; Tsybin YO; Dyson PJ; Keppler BK; Hartinger CG Fragmentation methods on the balance: unambiguous top-down mass spectrometric characterization of oxaliplatin-ubiquitin binding sites. . ChemAnal. Bioanal 2012, 402, 2655–2662. [DOI] [PubMed] [Google Scholar]
- (34).Li H; Snelling JR; Barrow MP; Scrivens JH; Sadler PJ; O’Connor PB Mass Spectrometric Strategies to Improve the Identification of Pt(II)-Modification Sites on Peptides and Proteins. J. Am. Soc. Mass Spectrom 2014, 25, 1217–1227. [DOI] [PubMed] [Google Scholar]
- (35).Li H; Wongkongkathep P; Van Orden SL; Ogorzalek Loo RR; Loo JA Revealing Ligand Binding Sites and Quantifying Subunit Variants of Noncovalent Protein Complexes in a Single Native Top-Down FTICR MS Experiment. J. Am. Soc. Mass Spectrom 2014, 25, 2060–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Erales J; Gontero B; Whitelegge J; Halgand F Mapping of a copper-binding site on the small CP12 chloroplastic protein of Chlamydomonas reinhardtii using top-down mass spectrometry and site-directed mutagenesis. Biochem. J 2009, 419, 75–86. [DOI] [PubMed] [Google Scholar]
- (37).Glover MS; Dilger JM; Zhu F; Clemmer DE The binding of Ca2+, Co2+, Ni2+, Cu2+, and Zn2+ cations to angiotensin I determined by mass spectrometry based techniques. Int. J. Mass Spectrom 2013, 354.–, 318–325. [Google Scholar]
- (38).Banu L; Blagojevic V; Bohme DK Locating Pb2+ and Zn2+ in Zinc Finger-Like Peptides Using Mass Spectrometry. J. Am. Soc. Mass Spectrom 2013, 24, 1534–1542. [DOI] [PubMed] [Google Scholar]
- (39).Martin EM; Kondrat FDL; Stewart AJ; Scrivens JH; Sadler PJ; Blindauer CA Native electrospray mass spectrometry approaches to probe the interaction between zinc and an anti-angiogenic peptide from histidine-rich glycoprotein. Sci. Rep 2018, 8, 8646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Cammarata MB; Brodbelt JS Structural characterization of holo- and apo-myoglobin in the gas phase by ultraviolet photodissociation mass spectrometry. Chem. Sci 2015, 6, 1324–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Mehaffey MR; Cammarata MB; Brodbelt JS Tracking the Catalytic Cycle of Adenylate Kinase by Ultraviolet Photodissociation Mass Spectrometry. Anal. Chem 2018, 90, 839–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Cammarata MB; Thyer R; Rosenberg J; Ellington A; Brodbelt JS Structural Characterization of Dihydrofolate Reductase Complexes by Top-Down Ultraviolet Photodissociation Mass Spectrometry. J. Am. Chem. Soc 2015, 137, 9128–9135. [DOI] [PubMed] [Google Scholar]
- (43).Cammarata M; Thyer R; Lombardo M; Anderson A; Wright D; Ellington AS; Brodbelt J Characterization of trimethoprim resistant E. coli dihydrofolate reductase mutants by mass spectrometry and inhibition by propargyl-linked antifolates. Chemical Science. 2017, 8, 4062–4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Blevins MS; Kim D; Crittenden CM; Hong S; Yeh H-C; Petty JT; Brodbelt JS Footprints of Nanoscale DNA-Silver Cluster Chromophores via Activated-Electron Photodetachment Mass Spectrometry. ACS Nano 2019, 13, 14070–14079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Cui W; Thompson MS; Reilly JP Pathways of Peptide Ion Fragmentation Induced by Vacuum Ultraviolet Light. J. Am. Soc. Mass Spectrom 2005, 16, 1384–1398. [DOI] [PubMed] [Google Scholar]
- (46).Julian RR The Mechanism Behind Top-Down UVPD Experiments: Making Sense of Apparent Contradictions. J. Am. Soc. Mass Spectrom 2017, 28, 1823–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Hall Z; Hernández H; Marsh JA; Teichmann SA; Robinson CV The Role of Salt Bridges, Charge Density, and Subunit Flexibility in Determining Disassembly Routes of Protein Complexes. Structure 2013, 21, 1325–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).O’Brien JP; Li W; Zhang Y; Brodbelt JS Characterization of Native Protein Complexes Using Ultraviolet Photodissociation Mass Spectrometry. J. Am. Chem. Soc 2014, 136, 12920–12928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Heins JN; Suriano JR; Taniuchi H; Anfinsen CB Characterization of a Nuclease Produced by Staphylococcus aureus. J. Biol. Chem 1967, 242, 1016–1020. [PubMed] [Google Scholar]
- (50).Theobald DL; Mitton-Fry RM; Wuttke DS Nucleic Acid Recognition by OB-Fold Proteins. Annu. Rev. Biophys. Biomol. Struct 2003, 32, 115–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Furie B; Eastlake A; Schechter AN; Anfinsen CB The Interaction of the Lanthanide Ions with Staphylococcal Nuclease. J. Biol. Chem 1973, 248, 5821–5825. [PubMed] [Google Scholar]
- (52).Park DM; Brewer A; Reed DW; Lammers LN; Jiao Y Recovery of Rare Earth Elements from Low-Grade Feedstock Leachates Using Engineered Bacteria. Environ. Sci. Technol 2017, 51, 13471–13480. [DOI] [PubMed] [Google Scholar]
- (53).Zhuang W-Q; Fitts JP; Ajo-Franklin CM; Maes S; Alvarez-Cohen L; Hennebel T Recovery of critical metals using biometallurgy. Curr. Opin. Biotechnol 2015, 33, 327–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Brittain HG; Richardson FS; Martin RB Terbium(III) emission as a probe of calcium(II) binding sites in proteins. J. Am. Chem. Soc 1976, 98, 8255–8260. [DOI] [PubMed] [Google Scholar]
- (55).Gray HB; Winkler JR Electron Transfer in Proteins. Annu. Rev. Biochem 1996, 65, 537–561. [DOI] [PubMed] [Google Scholar]
- (56).Nar H; Messerschmidt A; Huber R; van de Kamp M; Canters GW Crystal structure analysis of oxidized Pseudomonas aeruginosa azurin at pH 5·5 and pH 9·0: A pH-induced conformational transition involves a peptide bond flip. J. Mol. Biol 1991, 221, 765–772. [DOI] [PubMed] [Google Scholar]
- (57).Ferapontova EE; Shleev S; Ruzgas T; Stoica L; Christenson A; Tkac J; Yaropolov AI; Gorton L Direct Electrochemistry of Proteins and Enzymes In Paleček E; Scheller F; Wang J, Eds. Perspectives in Bioanalysis; pp 517–598; Elsevier, 2005. [Google Scholar]
- (58).Salgado J; Kroes SJ; Berg A; Moratal JM; Canters GW The Dynamic Properties of the M121H Azurin Metal Site as Studied by NMR of the Paramagnetic Cu(II) and Co(II) Metalloderivatives. J. Biol. Chem 1998, 273, 177–185. [DOI] [PubMed] [Google Scholar]
- (59).Leckner J; Bonander N; Wittung-Stafshede P; Malmström BG; Karlsson BG The effect of the metal ion on the folding energetics of azurin: a comparison of the native, zinc and apoprotein. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol 1997, 1342, 19–27. [DOI] [PubMed] [Google Scholar]
- (60).Cheung K-C; Strange RW; Hasnain SS 3D EXAFS refinement of the Cu site of azurin sheds light on the nature of structural change at the metal centre in an oxidation-reduction process: an integrated approach combining EXAFS and crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr 2000, 56, 697–704. [DOI] [PubMed] [Google Scholar]
- (61).Hosseinzadeh P; Marshall NM; Chaćon KN; Yu Y; Nilges MJ; New SY; Tashkov SA; Blackburn NJ; Lu Y Design of a single protein that spans the entire 2-V range of physiological redox potentials. Proc. Natl. Acad. Sci. U. S. A 2016, 113, 262–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Edington SC; Gonzalez A; Middendorf TR; Halling DB; Aldrich RW; Baiz CR Coordination to lanthanide ions distorts binding site conformation in calmodulin. Proc. Natl. Acad. Sci. U. S. A 2018, 115, E3126–E3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Liddington RC Anthrax: A molecular full nelson. Nature 2002, 415, 373–374. [DOI] [PubMed] [Google Scholar]
- (64).Vetter SW; Leclerc E Novel aspects of calmodulin target recognition and activation. Eur. J. Biochem 2003, 270, 404–414. [DOI] [PubMed] [Google Scholar]
- (65).Chattopadhyaya R; Meador WE; Means AR; Quiocho FA Calmodulin structure refined at 1.7 Å resolution. J. Mol. Biol 1992, 228, 1177–1192. [DOI] [PubMed] [Google Scholar]
- (66).Shaw JB; Li W; Holden DD; Zhang Y; Griep-Raming J; Fellers RT; Early BP; Thomas PM; Kelleher NL; Brodbelt JS Complete Protein Characterization Using Top-Down Mass Spectrometry and Ultraviolet Photodissociation. J. Am. Chem. Soc 2013, 135, 12646–12651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Rosenberg J; Parker WR; Cammarata MB; Brodbelt JS UV-POSIT: Web-Based Tools for Rapid and Facile Structural Interpretation of Ultraviolet Photodissociation (UVPD) Mass Spectra. J. Am. Soc. Mass Spectrom 2018, 29, 1323–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Vaisar T; Urban J Probing Proline Effect in CID of Protonated Peptides. J. Mass Spectrom 1996, 31, 1185–1187. [DOI] [PubMed] [Google Scholar]
- (69).Breci LA; Tabb DL; Yates JR; Wysocki VH Cleavage N-Terminal to Proline: Analysis of a Database of Peptide Tandem Mass Spectra. Anal. Chem 2003, 75, 1963–1971. [DOI] [PubMed] [Google Scholar]
- (70).Warnke S; Baldauf C; Bowers MT; Pagel K; von Helden G Photodissociation of Conformer-Selected Ubiquitin Ions Reveals Site-Specific Cis/Trans Isomerization of Proline Peptide Bonds. J. Am. Chem. Soc 2014, 136, 10308–10314. [DOI] [PubMed] [Google Scholar]
- (71).Baldauf C; Pagel K; Warnke S; von Helden G; Koksch B; Blum V; Scheffler M How Cations Change Peptide Structure. Chem. - Eur. J 2013, 19, 11224–11234. [DOI] [PubMed] [Google Scholar]
- (72).Yuan Z; Bailey TL; Teasdale RD Prediction of protein B-factor profiles. Proteins: Struct., Funct., Genet 2005, 58, 905–912. [DOI] [PubMed] [Google Scholar]
- (73).Panzner MJ; Bilinovich SM; Parker JA; Bladholm EL; Ziegler CJ; Berry SM; Leeper TC Isomorphic deactivation of a Pseudomonas aeruginosa oxidoreductase: The crystal structure of Ag(I) metallated azurin at 1.7Å. J. Inorg. Biochem 2013, 128, 11–16. [DOI] [PubMed] [Google Scholar]
- (74).Adman ET; Stenkamp RE; Sieker LC; Jensen LH A crystallographic model for azurin at 3 Å resolution. J. Mol. Biol 1978, 123, 35–47. [DOI] [PubMed] [Google Scholar]
- (75).Ramirez BE; Malmström BG; Winkler JR; Gray HB The currents of life: the terminal electron-transfer complex of respiration. Proc. Natl. Acad. Sci. U. S. A 1995, 92, 11949–11951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Naro F; Tordi MG; Giacometti GM; Tomei F; Timperio AM; Zolla L Metal binding to Pseudomonas aeruginosa azurin: a kinetic investigation., Zeitschrift fur Naturforschung. C. Z. Naturforsch., C: J. Biosci 2000, 55, 347–354. [DOI] [PubMed] [Google Scholar]
- (77).Panzner MJ; Bilinovich SM; Youngs WJ; Leeper TC Silver metallation of hen egg white lysozyme: X-ray crystal structure and NMR studies. Chem. Commun 2011, 47, 12479–12481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Shannon RD Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr 1976, 32, 751–767. [Google Scholar]
- (79).Kalapothakis JMD; Berezovskaya Y; Zampronio CG; Faull PA; Barran PE; Cooper HJ Unusual ECD fragmentation attributed to gas-phase helix formation in a conformationally dynamic peptide. Chem. Commun 2014, 50, 198–200. [DOI] [PubMed] [Google Scholar]
- (80).Schennach M; Breuker K Probing Protein Structure and Folding in the Gas Phase by Electron Capture Dissociation. J. Am. Soc. Mass Spectrom 2015, 26, 1059–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.