

Abstract
Type 2 diabetes (T2D) and cardiovascular disease (CVD) share many risk factors such as obesity, unhealthy lifestyle, and metabolic syndrome, whose accumulation over years leads to disease onset. However, while lowering plasma low-density lipoprotein cholesterol (LDLC) is cardio-protective, novel evidence have recognised a role for common LDLC-lowering variants (e.g. in HMGCR, PCSK9, and LDLR) and widely used hypocholesterolemic drugs that mimic the effects of some of these variants (statins) in higher risk for T2D. As these conditions decrease plasma LDLC by increasing tissue-uptake of LDL, a role for LDL receptor (LDLR) pathway was proposed. While underlying mechanisms remain to be fully elucidated, work from our lab reported that native LDL directly provoke the dysfunction of human white adipose tissue (WAT) and the activation of WAT NLRP3 (Nucleotide-binding domain and Leucine-rich repeat Receptor, containing a Pyrin domain 3) inflammasome, which play a major role in the etiology of T2D. However, while elevated plasma numbers of apolipoprotein B (apoB)-containing lipoproteins (measured as apoB, mostly as LDL) is associated with WAT dysfunction and related risk factors for T2D in our cohort, this relation was strengthened in regression analysis by lower plasma proprotein convertase subtilisin/kexin type 9 (PCSK9). This supports a central role for upregulated pathway of LDLR and/or other receptors regulated by PCSK9 such as cluster of differentiation 36 (CD36) in LDL-induced anomalies. Targeting receptor-mediated uptake of LDL into WAT may reduce WAT inflammation, WAT dysfunction, and related risk for T2D without increasing the risk for CVD.
Keywords: apoB-lipoproteins, LDLR, CD36, cardiometabolic risk, insulin sensitivity, apoB-to-PCSK9 ratio
Introduction
Every 3 minutes, a new case of diabetes is diagnosed in Canada, mostly type 2 (approximately 90%) increasing cardiovascular death and stroke by 2 to 4 fold[1]. Type 2 diabetes (T2D) is a chronic disease that develops with the accumulation of risk factors, such as unhealthy lifestyle, obesity, age, race, and genetics[1]. This leads to a progressive rise in insulin resistance (IR) in peripheral tissues, which has to be counterbalanced by increased insulin secretion to maintain normal plasma glucose. In time, this is believed to promote β-cell exhaustion and death, hypoinsulinemia, hyperglycemia, and progression to T2D[1–2]. A lower disposition index, which is a measure of lower insulin sensitivity combined with lower insulin secretion, is an independent predictor of the conversion of prediabetes to T2D across many ethnic groups and races[3].
Diabetes increases cardiovascular disease (CVD) risk[1,4], worsens stroke outcomes[4], confers an equal risk to aging 15 years[5], and about 65% to 80% of diabetic patients die from CVD[5]. Diabetes also "erases" the female advantage. While CVD and death are higher in non-diabetic men compared to women, this is reversed with the development of diabetes[4,6]. Importantly, T2D is preventable in high-risk obese subjects by intensive lifestyle intervention (hypocaloric diets and physical activity) and to a lesser extent by metformin (reduction in diabetes incidence compared to placebo of 59% vs. 31%, respectively)[7]. Thus, a better understanding of the multifaceted etiology of T2D may aid in the prevention of T2D and its related co-morbidities.
Elevated plasma number of apolipoprotein B (apoB)-containing lipoproteins and the risk for T2D
Atherogenic apoB-lipoproteins are chylomicrons, very-low-density lipoprotein (VLDL), intermediate-density lipoprotein (IDL), low-density lipoprotein (LDL) and lipoprotein(a). What characterizes these particles is that each particle contains a single molecule of apoB, as apoB48 on intestinal chylomicrons or as apoB100 on hepatic lipoproteins. Thus, plasma apoB represents the number of all apoB-lipoproteins, most of which are in the form of LDL (>90% on average)[8–9]. On the other hand, the lipid content and therefore the size of these particles can vary substantially. When LDL particles are either cholesterol-enriched or cholesterol-depleted, even a direct measure of LDL cholesterol (LDLC) is an inaccurate measure of LDL number, and plasma apoB remains a more accurate measure of LDL number[9–10].
The role of plasma apoB-lipoproteins, mostly LDL, in the etiology and progression of CVD is well established, while their role in the development of T2D has only emerged over the past 15 years. This may be because most clinical studies have focused on the lipid content of apoB-lipoproteins rather than their number. While apoB-lipoproteins are known to induce multiple derangements in inflammatory cascades leading to atherosclerosis[11], large clinical studies that examined the associations between plasma lipids with high sensitivity C-reactive protein (hsCRP), the most used pro-inflammatory marker in terms of clinical utility, concluded that there is little, if any, association between plasma triglycerides (TGs), total cholesterol, LDLC and high-density lipoprotein cholesterol (HDLC) with plasma hsCRP[12–14].
We reported in 2006 that higher plasma apoB predicts higher plasma hsCRP, interleukin-6, orosomucoid, and haptoglobin independent of total fat mass and visceral fat mass in overweight and obese postmenopausal women[15]. Conversely, there was no association between plasma LDLC with these inflammatory markers. However, after adjustment for plasma apoB in regression analysis, the association of plasma LDLC with these inflammatory markers was, when present, in the reverse direction[15]. This suggests that a higher number of small dense LDL (sdLDL) is associated with chronic inflammation in this population[15]. As both CVD and T2D are viewed as chronic inflammatory diseases, we proposed in 2006 that elevated plasma apoB is a promoter and not a mere consequence of IR and T2D[15]. Indeed, emerging epidemiological data since 2007 confirmed that higher plasma apoB predicts the development of T2D 3–10 years before its onset in Turkish[16], Canadian[17], Finnish[18], and Korean[19] populations, independent of traditional risk factors such as central adiposity[16–17], hsCRP[16], fasting glucose and glycated hemoglobin (HbA1C)[17,19].
However, patients with familial hypercho-lesterolemia (FH) with disrupted or absent LDL receptor (LDLR) pathway have lifelong markedly high plasma LDL and risk for CVD but low risk for T2D. A study comparing 25 137 FH patients to 38 183 non-FH family members who participated in the national Dutch screening program reported that the incidence of T2D in FH patients was 1.44% compared to 3.26% in non-FH relatives, after adjusting for age, adiposity, HDLC, TGs, statin use, smoking, CVD, and family relations[20]. Moreover, the more damaging the FH mutation on LDL uptake, the lower was the prevalence of T2D [i.e. LDLR loss-of-function (LOF) mutations were more associated with diabetes risk than APOB mutations][20]. This supports a central role for a functional LDLR pathway in mediating the role of apoB-lipoproteins in the etiology of T2D.
Upregulated LDLR pathway and the risk for T2D
Data analysis emerging since 2010 using randomized control trials and meta-analyses reported that statin therapy, which inhibits 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR), is associated with increased incidence of diabetes by ~12% and that statin-effect was dose-dependent[21–26]. This prompted the FDA in 2012 to include diabetes risk on the label of statins[27]. Statins act by inducing the expression of LDLR, the key step leading to decreased plasma LDLC[28]. Moreover, additional evidence linking upregulated LDLR pathway to T2D risk emerged in 2015 using genetic data of proteins regulating LDLR expression such as proprotein convertase subtilisin/kexin type 9 (PCSK9)[24,29–31]. PCSK9 is a plasma protein primarily of hepatic origin that was identified at Clinical Research Institute of Montreal in 2003 by Drs. Seidah and Chrétien[32]. PCSK9 directly binds to LDLR, after which the complex is internalized and targeted to lysosomal degradation[33–34]. PCSK9 also targets the degradation of other apoB-lipoprotein receptors in hepatic and extrahepatic tissues including white adipose tissue (WAT) and adipocytes, such as VLDLR[35] and scavenger receptor cluster of differentiation 36 (CD36)[36], a receptor for native LDL and VLDL, oxidized LDL (oxLDL) and non-esterified fatty acids (NEFAs)[37–38].
One Mendelian randomization study in approximately 113 000 subjects with average plasma LDLC of 3.36 mmol/L reported that LDL-lowering variants in HMGCR, PCSK9, and LDLR were associated with a 10% to 13% rise in diabetes risk for every 0.26 mmol/L decrease in plasma LDLC associated to these variants with no association to fasting plasma glucose[30]. Moreover, the effects of HMGCR and PCSK9 variants on reducing CVD risk or increasing diabetes risk were additive[30]. Another Mendelian randomization study in 568 448 subjects (average fasting plasma LDLC=3.41 mmol/L, glucose=5.38 mmol/L, and HbA1C=5.5%) reported that for every 1 mmol/L decrease in plasma LDLC attributed to 4 PCSK9 LOF variants, there was an increase of 0.09 mmol/L in fasting plasma glucose and 29% in the prevalence of diabetes[29].
We calculated that the majority of patients with T2D carry at least one allele of these variants. Taking into account the allele frequency of HMGCR allele rs12916 and its odds ratio for T2D, it can be calculated that approximately 84% of T2D patients are carriers of at least one copy of HMGCR allele rs12916[31]. Similar analysis on PCSK9 LOF variants suggests that approximately 77% of patients with T2D carry at least one copy of PCSK9 allele rs2479409[29].
A meta-analysis on the novel hypocholesterolemic agents anti-PCSK9 reported a modest increase in plasma glucose and HbA1C over 78 weeks[39]. On the other hand, the addition of anti-PCSK9 to statin therapy in the FOURIER trial (Further cardiovascular outcomes research with PCSK9 inhibition in subjects with elevated risk) using evolocumab over a median of 2.2 years[40] or in the ODYSSEY Outcomes trial (Evaluation of cardiovascular outcomes after an acute coronary syndrome during treatment with alirocumab) using alirocumab over a median of 2.8 years[41] did not reveal an increase in the risk for T2D compared to statin therapy alone. However, evidence from longer trials targeting PCSK9 is awaited to better parallel the lifelong genetic inhibition of PCSK9[29,42]. A longer trial may be particularly needed if anti-PCSK9 is used in combination with statins, which by themselves are associated with increased risk for T2D in an LDLR-related pathway. Whether anti-PCSK9 has an effect above and beyond that of statins on T2D risk remains to be seen.
Potential mechanisms linking LDL, LDLR, CD36, and PCSK9 to the risk of T2D
WAT dysfunction
WAT dysfunction and IR are believed to play a central role in the pathophysiology of T2D[43–44]. In youth with impaired glucose tolerance, WAT IR is negatively associated with the disposition index[45]. White adipose tissue dysfunction promotes elevated plasma TGs and NEFAs and their influx into other peripheral tissues, such as muscle, liver, and pancreas favoring systemic lipotoxicity, IR, hyperinsulinemia and β-cell exhaustion[43–44]. In the liver, increased NEFAs and TG influx and hepatic IR also promote the lipidation of apoB, which is the key step for the secretion of VLDL particles. This leads to increased plasma TGs and apoB and the exchange of cholesterol in cholesterol-rich lipoproteins (HDL and LDL) for TGs on TG-rich lipoproteins (TRLs, VLDL, and chylomicrons), which decreases both HDL and LDL size and cholesterol content[46]. Accordingly, the atherogenic dyslipoproteinemia that characterizes subjects with IR and T2D is high plasma TGs and apoB, low plasma HDLC, and sdLDL. Plasma LDLC is usually within normal ranges but carried on sdLDL particles[46], which is in line with high frequency of LDLC-lowering (i.e. HMGCR and PCSK9) variants in diabetic patients.
Role of LDL
Our work has established a novel role for native LDL in human WAT dysfunction, fueling a positive feedback loop promoting T2D[47] (Fig. 1). Physiological concentrations of native human LDL decrease the differentiation and function of murine[8] and human[48] preadipocytes in a dose-dependent manner[48]. Moreover, native LDL directly decreases the hydrolysis, uptake, and storage of 3H-labeled-TRLs in human subcutaneous WAT ex vivo over 4 hours, which is a measure of reduced WAT function[8,49]. This is mediated by inhibiting lipoprotein lipase activity and NEFAs uptake and storage, which are key steps needed for efficient plasma clearance of TRLs[8]. Others have also reported that oxLDL increases murine preadipocytes proliferation and decrease cell-apoptotic rate and differentiation[50], a defect that was dependent on CD36[51].
1. Higher plasma LDL promotes subcutaneous WAT dysfunction and related risk factors for T2D.
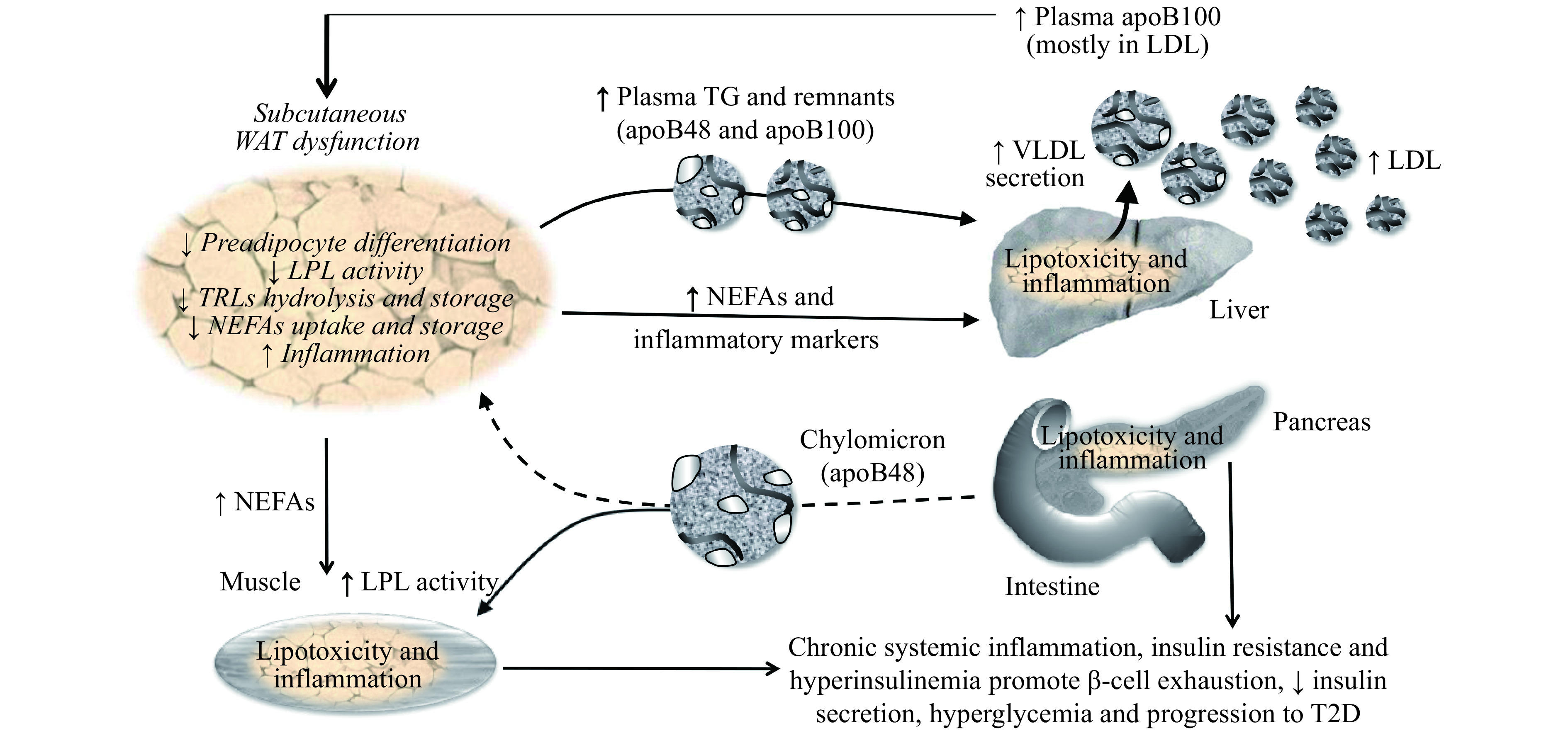
A positive feedback loop exists between WAT dysfunction and elevated plasma apoB that fuels the accumulation of risk factors for T2D, namely WAT dysfunction and inflammation, delayed postprandial plasma clearance of TGs and NEFAs, IR, compensatory hyperinsulinemia and chronic systemic inflammation. Over time, this loop favors β-cell exhaustion, reduced insulin secretion, hyperglycemia and progression to T2D. Solid lines represent activation and dashed lines represent inhibition. Italic text represents reported effects of native LDL on adipocytes and human WAT from our lab. WAT: white adipose tissue; LDL: low-density lipoprotein; VLDL: very-low-density lipoprotein; TRLs: triglyceride-rich lipoproteins; NEFAs: non-esterified fatty acids; LPL: lipoprotein lipase; T2D: type 2 diabetes.
In our research unit, we explore cardiometabolic risk factors in non-diabetic overweight and obese men and postmenopausal women who have low to moderate risk for CVD and are without chronic disease or medication affecting metabolism. In this cohort, plasma apoB, although not plasma total cholesterol or LDLC, is positively associated with WAT dysfunction and related risk factors for T2D, namely delayed postprandial plasma clearance of total and 13C-labeled dietary TGs, glucose-induced hyperinsulinemia, IR and chronic inflammation[8,49,52–54]. Moreover, following a 6-month hypocaloric diet in this cohort, baseline plasma apoB is associated with a post-intervention increase in WAT function and a decrease in glucose-induced insulin secretion independent of plasma total cholesterol, LDLC, and HDLC, sex, and changes in body composition[54]. This suggests that elevated plasma apoB identifies subjects with higher risk factors for T2D, and that targeting these subjects may maximize the benefit of hypocaloric diets to reduce the incidence of T2D among an obese population[54].
Role of plasma PCSK9, LDLR, and CD36
In our cohort, plasma PCSK9 is not associated with any risk factor measured for T2D[53]. However, regression analysis indicated that lower plasma PCSK9 increases the power of a regression model to predict WAT dysfunction and IR once plasma apoB was entered in this model[53]. To underscore the importance of PCSK9 in modulating the effect of apoB-lipoproteins on the risk factors for T2D, we developed the ratio of plasma apoB to plasma PCSK9 (apoB-to-PCSK9)[53]. We showed that a higher plasma apoB-to-PCSK9 ratio was associated with WAT dysfunction, postprandial hypertriglyceridemia, IR, hyperinsulinemia, and elevated plasma interleukin 1 receptor antagonist (IL-1Ra)[53], which is a measure of systemic activation of the IL-1 system[52] that precedes the onset of T2D by 10 years in humans[55]. More recently, we showed that this ratio was also associated with higher WAT surface-expression of LDLR and CD36 and with WAT inflammation[48]. This suggests that reducing plasma PCSK9, which would favor receptor-mediated uptake of apoB-lipoproteins, may accentuate the effects of LDL on WAT inflammation, dysfunction and related risk factors for T2D.
Of note, the effects of receptor-mediated uptake of apoB-lipoproteins on metabolic risk is not limited to WAT. Ceramide-rich LDL and TRL remnants were reported to decrease insulin action in rat L6 muscle cells[56–57]. These effects were paralleled by intramyocyte lipid accumulation and reversed by inhibition of the LDLR family[57]. Similarly, binding and internalization of LDL by β-cell induces cellular dysfunction and apoptosis[58–59]. In vivo, mice lacking Pcsk9 have higher pancreatic cholesterol, reduced insulin secretion and glucose intolerance[60–61] while insulin sensitivity is not affected[61]. Their islets exhibit signs of malformation, apoptosis and inflammation[60]. This phenotype is reversed in Pcsk9/Ldlr double knockout, suggesting the implication of LDLR pathway[61].
The NLRP3 inflammasome and IL-1β system
Macrophages sense microbial or sterile danger-associated molecular patterns, through specialized pattern-recognition receptors, which include membrane-bound Toll-like receptors, CD36 and intracellular Nod-like receptors[62]. While the activation of these receptors is vital in host resistance to infection, chronic activation of the IL-1β pathway is believed to promote CVD and T2D[62–64] and more recently kidney disease[65].
Secretion of IL-1β is regulated by the NLRP3 (Nucleotide-binding domain and Leucine-rich repeat Receptor, containing a Pyrin domain 3) inflammasome, an intracellular complex that is composed of 3 subunits: the scaffolding protein NLRP3, the adaptor protein ASC (apoptosis-associated speck-like protein containing a caspase recruitment domain), and caspase-1[62]. Activation of the inflammasome requires a priming signal via nuclear factor-kappa B (NF-κB) pathway inducing the secretion of an array of inflammatory cytokines and the transcription and translation of NLRP3 and pro-IL-1β (inactive). Common priming signals include cytokines (e.g. IL-1β), microbial lipopolysaccharides[63], oxLDL[66], and palmitate[67]. An activation signal is then needed for the assembly of the NLRP3 inflammasome, activation of pro-caspase-1, cleavage of pro-IL-1β and secretion of IL-1β. Activation signals in primed macrophages and β-cells include adenosine triphosphate[68], glucose[69], palmitate[70], oxLDL[71], ceramide[72], and cholesterol crystals[71]. Activation of the NLRP3 inflammasome is reported via lysosomal disruption and cathepsin B release, K+ efflux and/or generation of reactive oxygen species (ROS[63], which are also implicated in the inflammasome priming[71]).
Targeting IL-1β by IL-1Ra[73] or anti-IL-1β[74] improves plasma glucose, β-cell function, IR, and/or inflammation in T2D patients. More recently in the Canakinumab Antiinflammatory Thrombosis Outcome Study trial, anti-IL-1β therapy over approximately 3.7 years was reported to reduce cardiovascular events in CVD patients (mostly on statins[75]). The risk for T2D was however not affected in this trial and authors concluded that agents with less targeted actions may be needed to inhibit the NF-κB pathway in WAT and reduce the risk for T2D[76].
Role of LDL
Internalized apoB-lipoproteins such as LDL undergo digestion in the lysosomes releasing phospholipids, cholesterol and NEFAs[77]. Accumulation of free cholesterol and phospholipids and loading of macrophages with palmitate induce lysosomal dysfunction and leakage of its content such as Ca2+ and cathepsin B that prime and/or activate the NLRP3 inflammasome[65,77]. While counter-regulatory mechanisms exist in macrophages to block LDL-uptake by LDLR, such as by inhibiting sterol regulatory element-binding protein 2 (SREBP2) transcription factor[78], activation of the NLRP3 inflammasome induces the activation of SREBP2 that increases LDLR expression and NLRP3 inflammasome activation[62,78]. This is expected to disrupt cholesterol homeostasis favoring inflammation and cholesterol accumulation[78]. Recent findings from our lab indicate that incubation of subcutaneous WAT from non-diabetic overweight and obese subjects (without chronic disease) with their own native LDL induces IL-1β secretion over 7 hours[79]. Consistently, plasma apoB, but not lipids, in these subjects is positively associated with IL-1β secretion from their WAT ex vivo[79].
Role of PCSK9, LDLR and CD36
Blood mononuclear cells of subjects with PCSK9 LOF variants have higher postprandial expression of pro-inflammatory cytokines after a high-fat meal, including CD36, IL-1β, and MCP-1 (monocyte chemoattractant protein)[80]. Given that PCSK9 induces the degradation of LDLR, CD36 and other apoB-lipoprotein receptors, reduced PCSK9 plasma or function may favor receptor-mediated uptake of exogenous dietary lipids promoting NLRP3 priming. The scavenger receptor CD36 has been featured as an upstream master regulator of NLRP3 inflammasome in macrophages[71]. Endocytosis of oxLDL by CD36 induces the inflammasome priming and activation while inhibiting macrophage expression of CD36 prevents the rise in serum IL-1β in atherogenic mouse on high-fat/high-cholesterol diet[71]. Moreover in monocytes, inhibiting LDLR and LDL uptake by PCSK9 or anti-LDLR reduces monocyte inflammation and chemotaxis[81].
Recent post-hoc analysis in our cohort of non-diabetic overweight and obese subjects with normal plasma LDLC (<3.5 mmol/L) indicates that subjects with lower than median plasma PCSK9 per sex have higher hip WAT surface-expression of LDLR and CD36, lower WAT function, and higher postprandial WAT NLRP3 inflammasome priming than subjects with higher plasma PCSK9[82]. This suggests that activation of WAT NLRP3 inflammasome may be a mechanism promoting upregulated risk factors for T2D in subjects with lower PCSK9 function or plasma concentration.
Conclusions and future direction
Elevated plasma numbers of apoB-lipoproteins, mostly LDL, promote the risk for T2D. This may be mediated, at least in part by upregulating WAT NLRP3 inflammasome, WAT dysfunction and related risk factors for T2D. However, a functional LDLR pathway may be essential for LDL-induced metabolic abnormalities as highlighted in Fig. 2. As lower plasma PCSK9 or PCSK9 LOF variants may enhance LDL and NEFA uptake via LDLR and/or CD36, they may promote activation of the NLRP3 inflammasome in WAT and other insulin-sensitive tissues. To this end, a higher plasma apoB-to-PCSK9 ratio may be a simple clinical proxy of upregulated LDLR and CD36 pathway and related metabolic risks better than plasma apoB or PCSK9 alone. Finally, as both native and oxLDL are reported to upregulate the NLRP3 inflammasome in tubular endothelial cells[65], internalization of LDL may not only play a role in etiology of T2D but also in its associated complications such as kidney disease.
2. Proposed central role of LDL, PCSK9, LDLR and CD36 in modulating the risk for CVD vs. T2D.
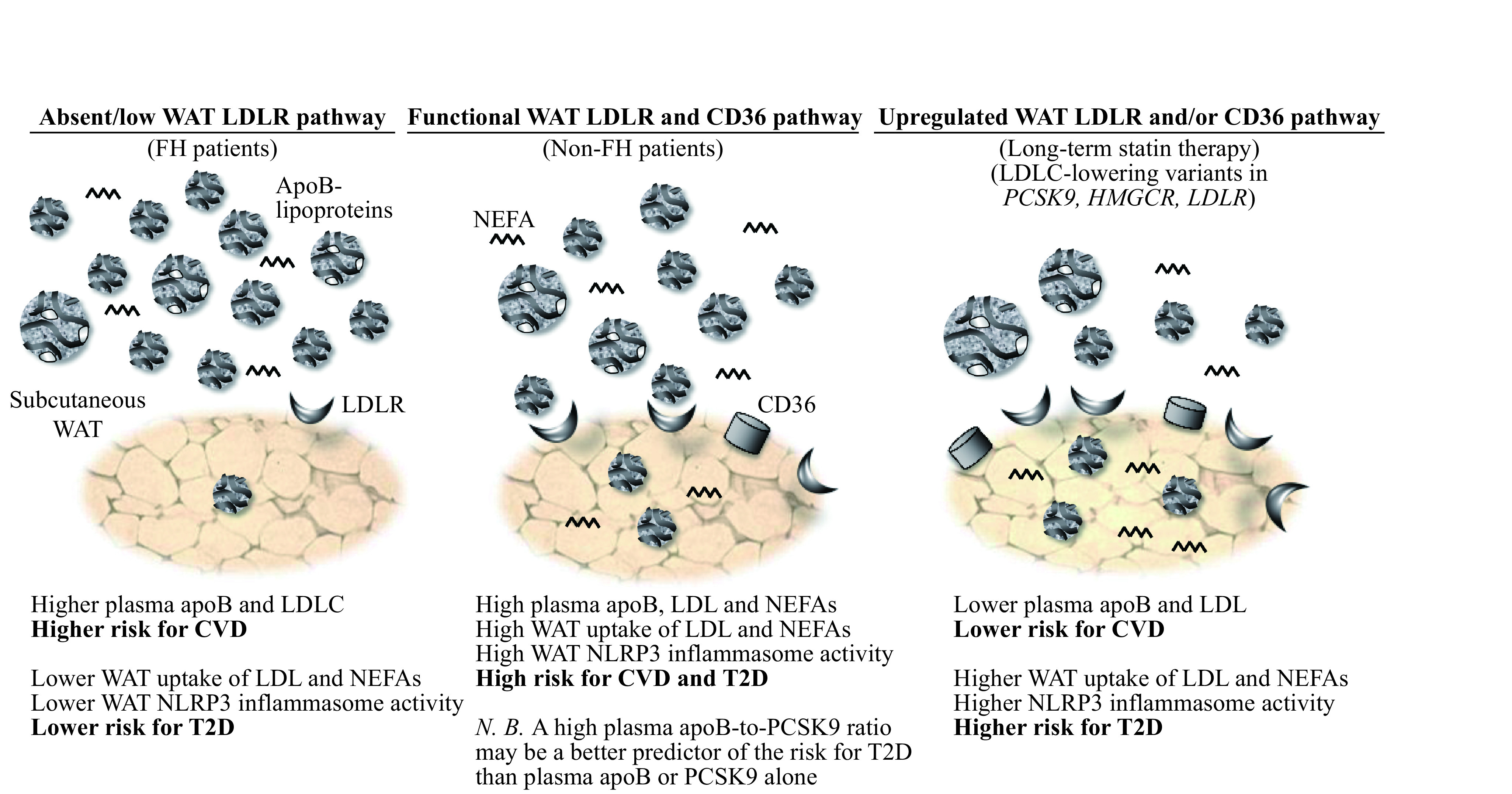
Patients with familial hypercholesterolemia with marked reduction or absence of a functional LDLR pathway have higher risk for CVD but lower risk for T2D despite very high plasma LDL. Conversely, patients with induced (statin therapy) or inherited (LDLC-lowering variants in PCSK9, HMGCR and LDLR) upregulated LDLR pathway have lower risk for CVD but higher risk for T2D despite low plasma LDL and apoB (N.B. CD36 pathway may also be upregulated in subjects with PCSK9 LOF variants). In between these 2 extremities in non-FH patients, the higher plasma apoB, mostly as LDL, the higher is the risk for both CVD and T2D. A higher plasma apoB-to-PCSK9 ratio may indicate a higher risk for T2D than plasma apoB alone as it may predict upregulated WAT surface-expression of LDLR and CD36 and receptor-mediated LDL uptake. WAT: white adipose tissue; FH: familial hypercholesterolemia; LDL: low-density lipoprotein; LDLR: LDL receptor; NEFAs: non-esterified fatty acids; PCSK9: proprotein convertase subtilisin/kexin type 9; CD36: cluster of differentiation 36; HMGCR: 3-hydroxy-3-methylglutaryl-coenzyme A reductase; NLRP3: nucleotide-binding domain and Leucine-rich repeat receptor, containing a Pyrin domain 3; T2D: type 2 diabetes; CVD: cardiovascular disease.
It remains to be investigated if other apoB-lipoprotein receptors regulated by PCSK9 such as VLDLR are also related to WAT NLRP3 inflammasome activity, WAT dysfunction and risk factors for T2D. Moreover, it remains to be investigated whether a less targeted therapy than anti-IL-1β reduces the incidence of T2D. To this end, omega-3 fatty acids, particularly marine source eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) may be a promising therapy as they inhibit the activation of the NF-κB pathway and the NLRP3 inflammasome and the secretion of IL-1β in macrophage[64]. Furthermore, EPA and/or DHA reduce plasma apoB-lipoproteins, primarily by reducing TRL secretion and plasma clearance as reviewed[83]. It was recently reported that while higher plasma NEFAs predicts the risk for T2D over 11 year, this relation was significant in subjects with lower plasma phospholipid content of EPA and DHA only (<75th percentile)[84]. This suggests that EPA∶DHA blunt the inflammation and metabolic dysfunction linked to high plasma NEFAs[84]. Future studies are needed to explore whether EPA and/or DHA may inhibit WAT NLRP3 inflammasome activity and related risk for T2D in humans, and whether they can attenuate statin-related increase in the risk for T2D in CVD patients.
Acknowledgments
This work is funded by Canadian Institutes of Health Research. We thank Dr. Luca Lotta for aid in calculating the frequency of LDLC-lowering variants in a diabetic population.
References
- 1.Diabetes Canada. Diabetes statistics in Canada[EB/OL]. http://www.diabetes.ca/how-you-can-help/advocate/why-federal-leadership-is-essential/diabetes-statistics-in-canada#_ftn1.
- 2.Prentki M, Nolan CJ Islet β cell failure in type 2 diabetes. J Clin Invest. 2006;116(7):1802–1812. doi: 10.1172/JCI29103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lorenzo C, Wagenknecht LE, Rewers MJ, et al Disposition index, glucose effectiveness, and conversion to type 2 diabetes: the Insulin Resistance Atherosclerosis Study (IRAS) Diabetes Care. 2010;33(9):2098–2103. doi: 10.2337/dc10-0165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Idris I, Thomson GA, Sharma JC Diabetes mellitus and stroke. Int J Clin Pract. 2006;60(1):48–56. doi: 10.1111/j.1368-5031.2006.00682.x. [DOI] [PubMed] [Google Scholar]
- 5.Booth GL, Kapral MK, Fung K, et al Relation between age and cardiovascular disease in men and women with diabetes compared with non-diabetic people: a population-based retrospective cohort study. Lancet. 2006;368(9529):29–36. doi: 10.1016/S0140-6736(06)68967-8. [DOI] [PubMed] [Google Scholar]
- 6.Lee WL, Cheung AM, Cape D, et al Impact of diabetes on coronary artery disease in women and men: a meta-analysis of prospective studies. Diabetes Care. 2000;23(7):962–968. doi: 10.2337/diacare.23.7.962. [DOI] [PubMed] [Google Scholar]
- 7.Knowler WC, Barrett-Connor E, Fowler SE, et al Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346(6):393–403. doi: 10.1056/NEJMoa012512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bissonnette S, Salem H, Wassef H, et al Low density lipoprotein delays clearance of triglyceride-rich lipoprotein by human subcutaneous adipose tissue. J Lipid Res. 2013;54(5):1466–1476. doi: 10.1194/jlr.P023176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sniderman AD, St-Pierre AC, Cantin B, et al Concordance/discordance between plasma apolipoprotein B levels and the cholesterol indexes of atherosclerotic risk. Am J Cardiol. 2003;91(10):1173–1177. doi: 10.1016/s0002-9149(03)00262-5. [DOI] [PubMed] [Google Scholar]
- 10.Ramasamy I Update on the laboratory investigation of dyslipidemias. Clin Chim Acta. 2018;479:103–125. doi: 10.1016/j.cca.2018.01.015. [DOI] [PubMed] [Google Scholar]
- 11.Williams KJ, Tabas I Lipoprotein retention—and clues for atheroma regression. Arterioscler Thromb Vasc Biol. 2005;25(8):1536–1540. doi: 10.1161/01.ATV.0000174795.62387.d3. [DOI] [PubMed] [Google Scholar]
- 12.Ridker PM, Rifai N, Rose L, et al Comparison of C-reactive protein and low-density lipoprotein cholesterol levels in the prediction of first cardiovascular events. N Engl J Med. 2002;347(20):1557–1565. doi: 10.1056/NEJMoa021993. [DOI] [PubMed] [Google Scholar]
- 13.Schlitt A, Blankenberg S, Bickel C, et al Prognostic value of lipoproteins and their relation to inflammatory markers among patients with coronary artery disease. Int J Cardiol. 2005;102(3):477–485. doi: 10.1016/j.ijcard.2004.05.056. [DOI] [PubMed] [Google Scholar]
- 14.Albert MA, Glynn RJ, Ridker PM Plasma concentration of C-reactive protein and the calculated framingham coronary heart disease risk score. Circulation. 2003;108(2):161–165. doi: 10.1161/01.CIR.0000080289.72166.CF. [DOI] [PubMed] [Google Scholar]
- 15.Faraj M, Messier L, Bastard JP, et al Apolipoprotein B: a predictor of inflammatory status in postmenopausal overweight and obese women. Diabetologia. 2006;49(7):1637–1646. doi: 10.1007/s00125-006-0259-7. [DOI] [PubMed] [Google Scholar]
- 16.Onat A, Can G, Hergenç G, et al Serum apolipoprotein B predicts dyslipidemia, metabolic syndrome and, in women, hypertension and diabetes, independent of markers of central obesity and inflammation. Int J Obes (Lond) 2007;31(7):1119–1125. doi: 10.1038/sj.ijo.0803552. [DOI] [PubMed] [Google Scholar]
- 17.Ley SH, Harris SB, Connelly PW, et al Association of apolipoprotein B with incident type 2 diabetes in an aboriginal Canadian population. Clin Chem. 2010;56(4):666–670. doi: 10.1373/clinchem.2009.136994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Salomaa V, Havulinna A, Saarela O, et al Thirty-one novel biomarkers as predictors for clinically incident diabetes. PLoS One. 2010;5(4):e10100. doi: 10.1371/journal.pone.0010100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hwang YC, Ahn HY, Park SW, et al Apolipoprotein B and non-HDL cholesterol are more powerful predictors for incident type 2 diabetes than fasting glucose or glycated hemoglobin in subjects with normal glucose tolerance: a 3.3-year retrospective longitudinal study. Acta Diabetol. 2014;51(6):941–946. doi: 10.1007/s00592-014-0587-x. [DOI] [PubMed] [Google Scholar]
- 20.Besseling J, Kastelein JJP, Defesche JC, et al Association between familial hypercholesterolemia and prevalence of type 2 diabetes mellitus. JAMA. 2015;313(10):1029–1036. doi: 10.1001/jama.2015.1206. [DOI] [PubMed] [Google Scholar]
- 21.Sattar N, Preiss D, Murray HM, et al Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. Lancet. 2010;375(9716):735–742. doi: 10.1016/S0140-6736(09)61965-6. [DOI] [PubMed] [Google Scholar]
- 22.Ridker PM, Pradhan A, MacFadyen JG, et al Cardiovascular benefits and diabetes risks of statin therapy in primary prevention: an analysis from the JUPITER trial. Lancet. 2012;380(9841):565–571. doi: 10.1016/S0140-6736(12)61190-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Corrao G, Ibrahim B, Nicotra F, et al Statins and the risk of diabetes: evidence from a large population-based cohort study. Diabetes Care. 2014;37(8):2225–2232. doi: 10.2337/dc13-2215. [DOI] [PubMed] [Google Scholar]
- 24.Swerdlow DI, Preiss D, Kuchenbaecker KB, et al HMG-coenzyme A reductase inhibition, type 2 diabetes, and bodyweight: evidence from genetic analysis and randomised trials. Lancet. 2015;385(9965):351–361. doi: 10.1016/S0140-6736(14)61183-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kato S, Miura M Risk of new‐onset diabetes mellitus during treatment with low‐dose statins in Japan: A retrospective cohort study. J Clin Pharm Ther. 2018;43(4):536–542. doi: 10.1111/jcpt.12675. [DOI] [PubMed] [Google Scholar]
- 26.Thakker D, Nair S, Pagada A, et al Statin use and the risk of developing diabetes: a network meta-analysis. Pharmacoepidemiol Drug Saf. 2016;25(10):1131–1149. doi: 10.1002/pds.4020. [DOI] [PubMed] [Google Scholar]
- 27.FDA Drug Safety Communication: important safety label changes to cholesterol-lowering statin drugs[EB/OL]. (2016-01-19). http://www.fda.gov/Drugs/DrugSafety/ucm293101.htm.
- 28.Preiss D, Sattar N Does the LDL receptor play a role in the risk of developing type 2 diabetes? JAMA. 2015;313(10):1016–1017. doi: 10.1001/jama.2015.1275. [DOI] [PubMed] [Google Scholar]
- 29.Schmidt AF, Swerdlow DI, Holmes MV, et al PCSK9 genetic variants and risk of type 2 diabetes: a Mendelian randomisation study . Lancet Diabetes Endocrinol. 2017;5(2):97–105. doi: 10.1016/S2213-8587(16)30396-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ference BA, Robinson JG, Brook RD, et al Variation in PCSK9 and HMGCR and risk of cardiovascular disease and diabetes . N Engl J Med. 2016;375(22):2144–2153. doi: 10.1056/NEJMoa1604304. [DOI] [PubMed] [Google Scholar]
- 31.Lotta LA, Sharp SJ, Burgess S, et al Association between low-density lipoprotein cholesterol-lowering genetic variants and risk of type 2 diabetes. JAMA. 2016;316(13):1383–1391. doi: 10.1001/jama.2016.14568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seidah NG, Benjannet S, Wickham L, et al The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): Liver regeneration and neuronal differentiation. Proc Natl Acad Sci USA. 2003;100(3):928–933. doi: 10.1073/pnas.0335507100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zaid A, Roubtsova A, Essalmani R, et al Proprotein convertase subtilisin/kexin type 9 (PCSK9): Hepatocyte-specific low-density lipoprotein receptor degradation and critical role in mouse liver regeneration. Hepatology. 2008;48(2):646–654. doi: 10.1002/hep.22354. [DOI] [PubMed] [Google Scholar]
- 34.Zhang DW, Lagace TA, Garuti R, et al Binding of proprotein convertase Subtilisin/Kexin type 9 to epidermal growth factor-like repeat A of low density lipoprotein receptor decreases receptor recycling and increases degradation. J Biol Chem. 2007;282(25):18602–18612. doi: 10.1074/jbc.M702027200. [DOI] [PubMed] [Google Scholar]
- 35.Roubtsova A, Munkonda MN, Awan Z, et al Circulating proprotein convertase Subtilisin/Kexin 9 (PCSK9) regulates VLDLR protein and triglyceride accumulation in visceral adipose tissue. Arterioscler Thromb Vasc Biol. 2011;31(4):785–791. doi: 10.1161/ATVBAHA.110.220988. [DOI] [PubMed] [Google Scholar]
- 36.Demers A, Samami S, Lauzier B, et al PCSK9 induces CD36 degradation and affects long-chain fatty acid uptake and triglyceride metabolism in adipocytes and in mouse liver. Arterioscler Thromb Vasc Biol. 2015;35(12):2517–2525. doi: 10.1161/ATVBAHA.115.306032. [DOI] [PubMed] [Google Scholar]
- 37.Glatz JFC, Luiken JJFP Dynamic role of the transmembrane glycoprotein CD36 (SR-B2) in cellular fatty acid uptake and utilization. J Lipid Res. 2018;59(7):1084–1093. doi: 10.1194/jlr.R082933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Calvo D, Gómez-Coronado D, Suárez Y, et al Human CD36 is a high affinity receptor for the native lipoproteins HDL, LDL, and VLDL. J Lipid Res. 1998;39(4):777–788. [PubMed] [Google Scholar]
- 39.De Carvalho LSF, Campos AM, Sposito AC Proprotein convertase Subtilisin/Kexin type 9 (PCSK9) inhibitors and incident type 2 diabetes: a systematic review and meta-analysis with over 96,000 patient-years. Diabetes Care. 2018;41(2):364–367. doi: 10.2337/dc17-1464. [DOI] [PubMed] [Google Scholar]
- 40.Sabatine MS, Leiter LA, Wiviott SD, et al Cardiovascular safety and efficacy of the PCSK9 inhibitor evolocumab in patients with and without diabetes and the effect of evolocumab on glycaemia and risk of new-onset diabetes: a prespecified analysis of the FOURIER randomised controlled trial. Lancet Diabetes Endocrinol. 2017;5(12):941–950. doi: 10.1016/S2213-8587(17)30313-3. [DOI] [PubMed] [Google Scholar]
- 41.Ray KK, Colhoun HM, Szarek M, et al Effects of alirocumab on cardiovascular and metabolic outcomes after acute coronary syndrome in patients with or without diabetes: a prespecified analysis of the ODYSSEY OUTCOMES randomised controlled trial. Lancet Diabetes Endocrinol. 2019;7(8):618–628. doi: 10.1016/S2213-8587(19)30158-5. [DOI] [PubMed] [Google Scholar]
- 42.Mascitelli L, Goldstein MR Questioning the safety and benefits of evolocumab. Lancet Diabetes Endocrinol. 2018;6(1):11. doi: 10.1016/S2213-8587(17)30397-2. [DOI] [PubMed] [Google Scholar]
- 43.Faraj M, Lu HL, Cianflone K Diabetes, lipids, and adipocyte secretagogues. Biochem Cell Biol. 2004;82(1):170–190. doi: 10.1139/o03-078. [DOI] [PubMed] [Google Scholar]
- 44.Gastaldelli A, Gaggini M, DeFronzo RA Role of adipose tissue insulin resistance in the natural history of type 2 diabetes: results from the San Antonio metabolism study. Diabetes. 2017;66(4):815–822. doi: 10.2337/db16-1167. [DOI] [PubMed] [Google Scholar]
- 45.Kim JY, Nasr A, Tfayli H, et al Increased lipolysis, diminished adipose tissue insulin sensitivity, and impaired β-cell function relative to adipose tissue insulin sensitivity in obese youth with impaired glucose tolerance. Diabetes. 2017;66(12):3085–3090. doi: 10.2337/db17-0551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ginsberg HN, Zhang YL, Hernandez-Ono A Regulation of plasma triglycerides in insulin resistance and diabetes. Arch Med Res. 2005;36(3):232–340. doi: 10.1016/j.arcmed.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 47.Faraj M Au-delà du risque cardiovasculaire: le rôle des lipoprotéines contenant l’apoB athérogènes dans l’étiologie du diabète de type 2. Méd Mal Métabol (in French) 2019;13(2):129–139. [Google Scholar]
- 48.Cyr Y, Bissonnette S, Lamantia V, et al ApoB-lipoproteins and PCSK9 as modulators of human white adipose tissue function and NLRP3 inflammasome activity. Atheroscler Suppl. 2018;32:117. [Google Scholar]
- 49.Lamantia V, Bissonnette S, Wassef H, et al ApoB-lipoproteins and dysfunctional white adipose tissue: relation to risk factors for type 2 diabetes in humans. J Clin Lipidol. 2017;11(1):34–45.e2. doi: 10.1016/j.jacl.2016.09.013. [DOI] [PubMed] [Google Scholar]
- 50.Masella R, Varì R, D'Archivio M, et al Oxidised LDL modulate adipogenesis in 3T3-L1 preadipocytes by affecting the balance between cell proliferation and differentiation. FEBS Lett. 2006;580(10):2421–2429. doi: 10.1016/j.febslet.2006.03.068. [DOI] [PubMed] [Google Scholar]
- 51.Kuniyasu A, Hayashi S, Nakayama H Adipocytes recognize and degrade oxidized low density lipoprotein through CD36. Biochem Biophys Res Commun. 2002;295(2):319–323. doi: 10.1016/S0006-291X(02)00666-6. [DOI] [PubMed] [Google Scholar]
- 52.Bissonnette S, Saint-Pierre N, Lamantia V, et al Plasma IL-1Ra: linking hyperapoB to risk factors for type 2 diabetes independent of obesity in humans. Nutr Diabetes. 2015;5(9):e180. doi: 10.1038/nutd.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wassef H, Bissonnette S, Saint-Pierre N, et al The apoB-to-PCSK9 ratio: a new index for metabolic risk in humans. J Clin Lipidol. 2015;9(5):664–675. doi: 10.1016/j.jacl.2015.06.012. [DOI] [PubMed] [Google Scholar]
- 54.Bissonnette S, Saint-Pierre N, Lamantia V, et al High plasma apolipoprotein B identifies obese subjects who best ameliorate white adipose tissue dysfunction and glucose-induced hyperinsulinemia after a hypocaloric diet. Am J Clin Nutr. 2018;108(1):62–76. doi: 10.1093/ajcn/nqy070. [DOI] [PubMed] [Google Scholar]
- 55.Herder C, Brunner EJ, Rathmann W, et al Elevated levels of the anti-inflammatory interleukin-1 receptor antagonist precede the onset of type 2 diabetes. Diabetes Care. 2009;32(3):421–423. doi: 10.2337/dc08-1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Boon J, Hoy AJ, Stark R, et al Ceramides contained in LDL are elevated in type 2 diabetes and promote inflammation and skeletal muscle insulin resistance. Diabetes. 2013;62(2):401–410. doi: 10.2337/db12-0686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pedrini MT, Kranebitter M, Niederwanger A, et al Human triglyceride-rich lipoproteins impair glucose metabolism and insulin signalling in L6 skeletal muscle cells independently of non-esterified fatty acid levels. Diabetologia. 2005;48(4):756–766. doi: 10.1007/s00125-005-1684-8. [DOI] [PubMed] [Google Scholar]
- 58.Roehrich ME, Mooser V, Lenain V, et al Insulin-secreting β-cell dysfunction induced by human lipoproteins. J Biol Chem. 2003;278(20):18368–18375. doi: 10.1074/jbc.M300102200. [DOI] [PubMed] [Google Scholar]
- 59.Rütti S, Ehses JA, Sibler RA, et al Low- and high-density lipoproteins modulate function, apoptosis, and proliferation of primary human and murine pancreatic β-cells. Endocrinology. 2009;150(10):4521–4530. doi: 10.1210/en.2009-0252. [DOI] [PubMed] [Google Scholar]
- 60.Mbikay M, Sirois F, Mayne J, et al PCSK9-deficient mice exhibit impaired glucose tolerance and pancreatic islet abnormalities. FEBS Lett. 2010;584(4):701–706. doi: 10.1016/j.febslet.2009.12.018. [DOI] [PubMed] [Google Scholar]
- 61.Da Dalt L, Ruscica M, Bonacina F, et al PCSK9 deficiency reduces insulin secretion and promotes glucose intolerance: the role of the low-density lipoprotein receptor. Eur Heart J. 2019;40(4):357–368. doi: 10.1093/eurheartj/ehy357. [DOI] [PubMed] [Google Scholar]
- 62.Skeldon AM, Faraj M, Saleh M Caspases and inflammasomes in metabolic inflammation. Immunol Cell Biol. 2014;92(4):304–313. doi: 10.1038/icb.2014.5. [DOI] [PubMed] [Google Scholar]
- 63.Masters SL, Latz E, O'Neill LAJ The inflammasome in atherosclerosis and type 2 diabetes. Sci Transl Med. 2011;3(81):81ps17. doi: 10.1126/scitranslmed.3001902. [DOI] [PubMed] [Google Scholar]
- 64.Camell C, Goldberg E, Dixit VD Regulation of Nlrp3 inflammasome by dietary metabolites. Semin Immunol. 2015;27(5):334–342. doi: 10.1016/j.smim.2015.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rampanelli E, Orsó E, Ochodnicky P, et al Metabolic injury-induced NLRP3 inflammasome activation dampens phospholipid degradation. Sci Rep. 2017;7(1):2861. doi: 10.1038/s41598-017-01994-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stewart CR, Stuart LM, Wilkinson K, et al CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol. 2010;11(2):155–161. doi: 10.1038/ni.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee JY, Zhao L, Hwang DH Modulation of pattern recognition receptor-mediated inflammation and risk of chronic diseases by dietary fatty acids. Nutr Rev. 2010;68(1):38–61. doi: 10.1111/j.1753-4887.2009.00259.x. [DOI] [PubMed] [Google Scholar]
- 68.Saleh M The machinery of Nod-like receptors: refining the paths to immunity and cell death. Immunol Rev. 2011;243(1):235–246. doi: 10.1111/j.1600-065X.2011.01045.x. [DOI] [PubMed] [Google Scholar]
- 69.Zhou RB, Tardivel A, Thorens B, et al Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11(2):136–140. doi: 10.1038/ni.1831. [DOI] [PubMed] [Google Scholar]
- 70.Wen HT, Gris D, Lei Y, et al Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol. 2011;12(5):408–415. doi: 10.1038/ni.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sheedy FJ, Grebe A, Rayner KJ, et al CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat Immunol. 2013;14(8):812–820. doi: 10.1038/ni.2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vandanmagsar B, Youm YH, Ravussin A, et al The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17(2):179–188. doi: 10.1038/nm.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Larsen CM, Faulenbach M, Vaag A, et al Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med. 2007;356(15):1517–1526. doi: 10.1056/NEJMoa065213. [DOI] [PubMed] [Google Scholar]
- 74.Sloan-Lancaster J, Abu-Raddad E, Polzer J, et al Double-blind, randomized study evaluating the glycemic and anti-inflammatory effects of subcutaneous LY2189102, a neutralizing IL-1β antibody, in patients with type 2 diabetes. Diabetes Care. 2013;36(8):2239–2246. doi: 10.2337/dc12-1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ridker PM, Everett BM, Thuren T, et al Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377(12):1119–1131. doi: 10.1056/NEJMoa1707914. [DOI] [PubMed] [Google Scholar]
- 76.Everett BM, Donath MY, Pradhan AD, et al Anti-inflammatory therapy with canakinumab for the prevention and management of diabetes. J Am Coll Cardiol. 2018;71(21):2392–2401. doi: 10.1016/j.jacc.2018.03.002. [DOI] [PubMed] [Google Scholar]
- 77.Moore KJ, Sheedy FJ, Fisher EA Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. 2013;13(10):709–721. doi: 10.1038/nri3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tall AR, Yvan-Charvet L Cholesterol, inflammation and innate immunity. Nat Rev Immunol. 2015;15(2):104–116. doi: 10.1038/nri3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bissonnette S, Lamantia V, Cyr Y, et al Native LDL are priming signals of white adipose tissue NLRP3 inflammasome in overweight and obese subjects. Atheroscler Suppl. 2018;32:123. [Google Scholar]
- 80.Gagnon A, Ooi TC, Cousins M, et al The anti-adipogenic effect of peripheral blood mononuclear cells is absent with PCSK9 loss-of-function variants. Obesity. 2016;24(11):2384–2391. doi: 10.1002/oby.21656. [DOI] [PubMed] [Google Scholar]
- 81.Grune J, Meyborg H, Bezhaeva T, et al PCSK9 regulates the chemokine receptor CCR2 on monocytes. Biochem Biophys Res Commun. 2017;485(2):312–318. doi: 10.1016/j.bbrc.2017.02.085. [DOI] [PubMed] [Google Scholar]
- 82.Cyr Y, Bissonnette S, Lamantia V, et al Higher adipose tissue surface-expression of LDLR and CD36 and risk factors for T2D in normocholesterolemic subjects with low plasma PCSK9. Atheroscler Suppl. 2018;32:70–71. [Google Scholar]
- 83.Lamantia V, Sniderman A, Faraj M Nutritional management of hyperapoB. Nutr Res Rev. 2016;29(2):202–233. doi: 10.1017/S0954422416000147. [DOI] [PubMed] [Google Scholar]
- 84.Steffen BT, Steffen LM, Zhou X, et al n-3 fatty acids attenuate the risk of diabetes associated with elevated serum nonesterified fatty acids: the multi-ethnic study of atherosclerosis. Diabetes Care. 2015;38(4):575–580. doi: 10.2337/dc14-1919. [DOI] [PMC free article] [PubMed] [Google Scholar]