

SUMMARY
Regional specification is critical for skin development, regeneration, and evolution. The contribution of epigenetics in this process remains unknown. Here, using avian epidermis, we find two major strategies regulate β-keratin gene clusters. (1) Over the body, macro-regional specificities (scales, feathers, claws, etc.) established by typical enhancers control five subclusters located within the epidermal differentiation complex on chromosome 25; (2) within a feather, micro-regional specificities are orchestrated by temporospatial chromatin looping of the feather β-keratin gene cluster on chromosome 27. Analyses suggest a three-factor model for regional specification: competence factors (e.g., AP1) make chromatin accessible, regional specifiers (e.g., Zic1) target specific genome regions, and chromatin regulators (e.g., CTCF and SATBs) establish looping configurations. Gene perturbations disrupt morphogenesis and histo-differentiation. This chicken skin paradigm advances our understanding of how regulation of big gene clusters can set up a two-dimensional body surface map.
Graphical Abstract
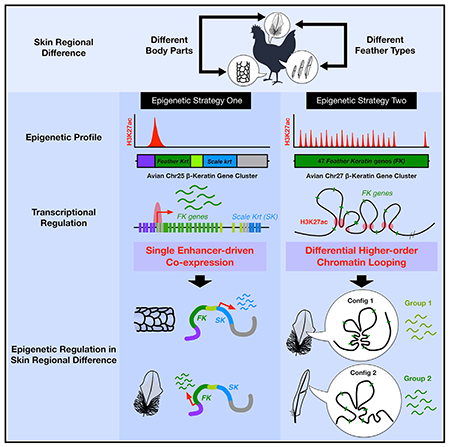
In Brief
Skin has regionally unique structure. Working in chicken skin, Liang et al. show that cross-body differences are controlled by enhancer-driven uniform expression of subclustered genes, while within-feather differences are controlled by intra-cluster chromatin looping. This illustrates how gene clusters are regulated to generate temporospatial complexity in skin structure.
INTRODUCTION
Skin on the body surface forms regional specificity (e.g., hairs, glands, feathers, and scales) to provide diverse functions (Chuong et al., 2012; Lu et al., 2016; Yu et al., 2018). Changes in skin appendages also occur in different life stages to adapt to the environment and fulfill physiological needs. During development, skin progenitors undergo global epigenetic programing and differentiate into different cell types, producing region-specific appendages in different body parts. Over evolutionary time, new skin appendages are formed whose characteristics define newly emergent animal classes. The most dramatic example is the evolution of feathers during the dinosaur-to-bird evolution (Chang et al., 2019; Dhouailly et al., 2019; Xu et al., 2014). While comparative genomics (Greenwold et al., 2014; Lowe et al., 2015) and developmental biology approaches (Chen et al., 2015; Wu et al., 2018a) have been applied to study avian skin regional specification, the role of epigenetic mechanisms in establishing regional differences remains unknown.
Skin appendage formation requires input from both epidermal and dermal components (Dhouailly, 1975; Rinn et al., 2008). Because chicken skin shows obvious regional differences and experimental accessibility, we focus on the chicken epidermis to study how region-specific epidermal genes are controlled. Among them, the Keratin (Krt) gene family constituting the outer layer of the skin is the largest and most representative region-specific gene family, α-keratin (α-krt) genes form intermediate 2filaments, the major keratinocyte cytoskeleton (Fuchs and Cleveland, 1998). β-keratins (α-krt), also named corneous α-proteins, are small structural proteins that evolved differently from members of the epidermal differentiation complex to strengthen krt stiffness (Greenwold and Sawyer, 2010; Holthaus et al., 2018). α-krt genes are grouped into type I (acidic) and type II (basic to neutral) Krts arranged into distinct gene clusters located on different chromosomes (Wang et al., 2016). Type I and type II α-krt gene clusters are located on chromosomes (Chr) 17 and 12 in humans and Chr 11 and 15 in mice (Hesse et al., 2004). Chickens have about 33 putative α-krt and 149 β-krt genes (Ng et al., 2014). There are two major β-krt gene clusters located on Chr 25 and 27. Interestingly, the chicken β-krt gene cluster located on Chr 25 (Chr25 β-krt cluster) is embedded within the chicken epidermal differentiation complex (EDC), but human, mouse, and chicken α-krt gene clusters are separated from EDC loci. Moreover, the Chr25 β-krt cluster is organized in five subclusters (Presland et al., 1989a; Presland et al., 1989b; Wu et al., 2015). Each subcluster (about 3–16 genes) is differentially enriched in keratinocytes of different skin regions (feathers, scales, claws, etc.); whereas, the chicken β-krt gene cluster located on Chr 27 (Chr27 β-krt cluster) contains 48 clustered genes, which are differentially expressed exclusively in feathers but with different expression patterns within feathers. (Ng et al., 2014; Wu et al., 2015). Thus, correspondence between the body regional topographic map and genomic organization of β-krt clusters offers a wonderful opportunity to study the epigenetic mechanisms regulating skin regional specification.
Here, using the avian Krt system as a paradigm, we show that skin regional specification is established through two different epigenetic strategies. “Macro” regional skin specificity is regulated by expressing β-krts on Chr25 in different body regions (e.g., feathers versus scales) through the differential regulation of typical enhancers. “Micro” regional specificity is set up within the feather follicle by mechanisms that lead to differential higher-order looping configurations conferring differential Chr27 β-krt expression. In this cluster, we found previously unidentified consensus elements from 38 H3K27ac-marked regions that act as looping anchor candidates to bring together chromatin regulators (e.g., CCCTC-binding factor [CTCF] and Krüppel Like Factor 4 [KLF4]), competence factors (e.g., Activator Protein-1 [AP1]), and region-specific transcription factors (e.g., Zic1). Together they provide numerous combinatorial looping configuration possibilities to regulate β-krt expression patterns. This study provides the epigenetic basis of how regional specificity is set up via a three-dimensional (3D) chromatin looping of feather β-krt clusters and suggests fundamental principles of how evolutionary complexities arose by the co-regulation of multiplex gene clusters in duplicated genomes. The findings here also provide a possible genomic explanation on how the large krt repertoire can be generated for the evolution of complex feather bio-architectures (Chang et al., 2019).
RESULTS
Two Distinct Epigenetic Modes of β-Keratin Gene Cluster Regulation Are Revealed by Transcriptional and Histone Modification Profiling of Avian Epidermis
To understand the transcriptional and epigenetic control of Krt clusters during embryonic skin specification, we examined region-specific expression patterns and quantified both gene expression and histone modifications of major Krt clusters at three stages of skin development. We used embryonic day 7 (E7) dorsal back epidermis, which shows no region-specific development, E9 dorsal back, and leg epidermis, which show early feather specification on the back but no specification on the legs, as well as E14 dorsal back and leg epidermis, which show completely specified feather and scale epidermis, respectively (Figure 1A). We performed RNA-seq and ChIP-seq with antibodies against histone H3 lysine 27 acetylation (H3K27ac), H3 lysine 4 mono-methylation (H3K4me1), and H3 lysine 4 trimethylation (H3K4me3).
Figure 1. Distinct Transcription and Epigenetic Landscapes of Avian β-keratin Gene Clusters in Skin Regional Specification.

(A) Schematic of different avian skin developmental stages used in the study. Embryonic tissue stages were taken from feather (green) and scale producing (blue) regions (E7, E9, and E14). Short vertical lines represent individual clustered genes.
(B) Schematic of two major avian β-krt clusters on Chr25 and Chr27. Chr25 β-krt cluster contains five subclusters located within the chicken epidermal differentiation complex (EDC). FK, feather Krt; FL, feather-like; SK, scale Krt; Ktn, keratinocyte Krt. Arrows under subclusters represent orientations of the subclusters based on the general orientation of clustered genes. →, sense; ←, antisense.
(C) y axis of RNA-seq tracks represents normalized read coverages of 1 × sequencing depth (RPGC). y axis of H3K27ac tracks represents a linear scale fold enrichment (FE) of ChIP-enriched/input genomic DNA signal intensity. The yellow boxes represent super-enhancers (SE) analyzed by HOMER using ChIP-seq against H3K27ac and H3K4me1 marks.
(C′) Enlargement of the FK gene subcluster and atypical enhancer (TE; red triangles) at its 5′ end.
(D) Profiles of RNA-seq and H3K27ac marks on Chr27 β-krt cluster during embryonic skin patterning. Red triangles indicate H3K27ac peaks.
(D′) A closer look at the 5′-end of the Chr27 β-krt cluster.
(E) Summary of epigenetic landscapes and basic information of β-krt clusters.
The first surprise came when we compared the RNA-seq and ChIP-seq profiles from different stages. Although clusters on Chr25 and Chr27 contain the same sub-type of β-krt genes, Feather Keratin (FK) (Figure 1B), they are differentially regulated showing two different histone modification landscapes. In E14 feather-bearing skin, the Chr25 Feather Krt cluster located within the chicken EDC contains a single typical enhancer (TE) characterized by HOMER enhancer analysis (findPeak command–typical and–style super) using H3K27ac and H3K4me1 marks (Figures 1C, 1C′, S1A, and S1A′). These marks were gradually established 5′ to the FK subcluster start site from E7 to E14 as were two super-enhancers (SE) located at both ends of the whole EDC. In contrast, individual H3K27ac and H3K4me1 signals were found among FK genes on the Chr27 cluster in feather but not in scale-bearing regions (Figures 1D, 1D′, S1B, and S1B′). We further analyzed the genomic location of the 6-Kb typical enhancer on Chr25 (Figure S1C) and found it contains both promoter (H3K27ac and lower H3K4me1/H3K4me3 ratio) and putative enhancer (H3K27ac and higher H3K4me1/H3K4me3 ratio) characteristics (Andersson and Sandelin, 2020). No similarly enriched histone marks exist around other FK genes on the Chr25 cluster (dash-box from FK2 to FK5, Figure 1C′). The individual active chromatin marks on the Chr27 β-krt cluster were not expected to serve as promoters since their genomic locations are far (5–13Kb) from the FK gene transcription start sites (TSSs) (Figure S1D) (Andersson and Sandelin, 2020). Interestingly, through RNA-seq results, we found a highly expressed EDC gene called Scaffoldin (SCFN) in E14 scale compared to feather-bearing skin (Figures 1C and S1F, black box). However, its function in scale specification is as yet unknown.
Previous studies in the mouse β-globin cluster (Dean, 2006) and Hox gene clusters (Andrey et al., 2013) demonstrated that 3D gene cluster organization is established based on long-range chromatin interactions at hubs with sequences enriched for acetylated histone marks. Our results (Figure 1E) imply a possibility that two fundamental epigenetic modes act to regulate multiplex β-krt clusters (two-mode): (1) a single enhancer-mediates the simultaneous transcription of specific β-krt subclusters (Figure S1G), and (2) intra-cluster higher-order chromatin looping controls differential β-krt gene expression within a cluster (Figure S1H).
Unexpected Finding that the 38 Putative Enhancers within β-Keratin Cluster on Chromosome 27 Contain CTCF and KLF4 Binding Motifs Embedded within Consensus Sequences
During development, undifferentiated epidermal progenitors undergo chromatin reorganization orchestrated by a group of chromatin-associated architectural proteins that bring distant genes close together so they can be co-regulated at correct places and times (Botchkarev et al., 2012; Fessing et al., 2011). Facilitating the formation of cell/tissue-specific chromatin conformation requires chromatin regulators such as CTCF (Hanssen et al., 2017), KLF4 (Di Giammartino et al., 2019; Wei et al., 2013), and AT-rich sequence binding proteins (SATBs) (Cai et al., 2003).
The second surprise in our study was revealed when we performed multiple sequencing alignment of the 38 selected H3K27ac regions on Chr27 β-krt cluster using Clustal Omega (Madeira et al., 2019). The results show almost every candidate anchor contained three consensus sequences (CS-1/2/3) that were not present in the typical enhancer of the Chr25 FK subcluster (Figures 2A, S2A, and S2A’ ; Table S1). We then used MATCH, TRANSFAC, and HOMER to predict consistent transcription-factor-binding motifs within the 38 candidate anchors. Binding motifs of CTCF and KLF4 were significantly enriched within CS-1 (Figures 2A, S2A, and S2A′). Besides CTCF and KLF4, we also found SATBs as candidates for chromatin organization. We previously showed that SATB2 was differentially expressed in feathers compared to scales (Wu et al., 2018b). SATBs serve as nuclear scaffolds that help form tissue-specific chromatin architectures (Cai et al., 2003). Moreover, SATB1 protein is essential for the specific spatial organization of the central domain of the EDC locus containing genes activated during terminal keratinocyte differentiation in the epidermis (Fessing et al., 2011).
Figure 2. Identification of Consensus Sequences in Selected β-keratin Gene Clusters H3K27ac Regions that Are Targeted by CTCF and KLF4.
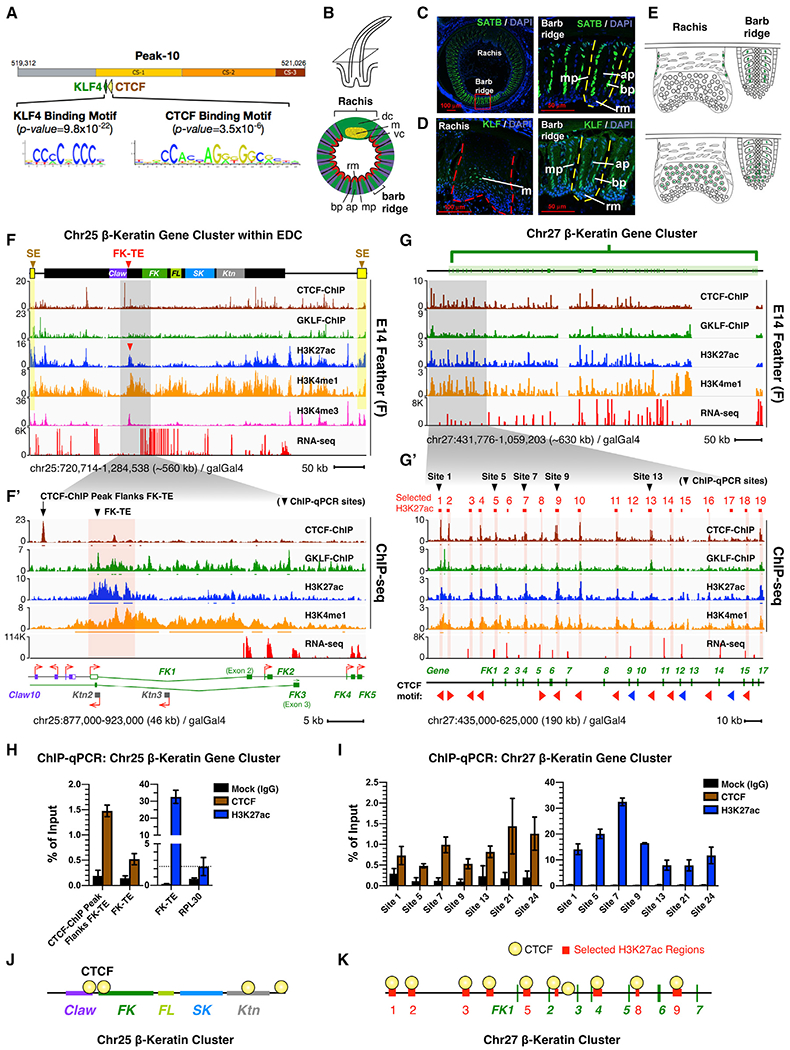
(A) Discovery of Chr27 β-krt cluster consensus sequences (CS) 1/2/3 enriched for CTCF and KLF4 binding motifs at selected H3K27ac regions. Motif prediction by HOMER and MATCH.
(B) Schematic drawing of a growth phase feather follicle and a cross-section. Axial plate, ap; barbule plate, bp; dorsal cortex, dc; marginal plate, mp; medulla, m; ramus zone, rm; ventral cortex, vc.
(C and D) IM of an E21 flight feather follicle with antibodies to (C) SATBs and (D) KLFs. Left panel, rachis; right panel, barb ridge.
(E) Schematic drawing of data from (C and D). Detailed IM results are shown in Figures S2D–S2H.
(F) Profiles of CTCF-/KLFs-/H3K27ac-ChIP and RNA-seq on the Chr25 β-krt cluster embedded within the chicken EDC.
(F′) Enlargement of Feather Krt gene subcluster and a TE at its 5′-end.
(G) Profiles of CTCF-/KLFs-/H3K27ac-ChIP and RNA-seq on the chicken Chr27 β-krt cluster.
(G′) Enlargement of the 5′-end of the Chr27 β-krt cluster. Triangles pointing left or right indicate orientations of CTCF-binding motifs. CTCF-ChIP peaks with (red) and without (blue) predicted CTCF-binding motifs.
(H and I) ChIP-qPCR analyses of CTCF and H3K27ac at the FK-TE and selected H3K27ac regions in chick E14 feathers. qPCR primers, black arrows in panels (F′) and (G′). Data (mean ± SEM) are representative of two independent ChIP experiments. RPL30 primers served as positive control.
(J) Schematic of CTCF-targeting at the FK-TE on the Chr25 β-krt cluster.
(K) Schematic of CTCF-targeting at H3K27ac-marked regions of the Chr27 β-krt cluster.
To validate if CTCF, KLF4, and SATB2 functioned in chicken skin, we first examined their expression using in situ hybridization (ISH). CTCF and KLF4 were expressed within feather buds at E7, E8, E9, and E12; whereas, SATB2 was expressed in both E12 feather- and scale-bearing skins (Figures S2B and S2C). We further use immunostaining to examine their expression patterns in developing flight feather follicles (Figures 2B–2E and S2D–S2H) whose filaments include a rachis and multiple barb ridges (Figure 2B). In the developing rachis, CTCF is highly expressed in the medulla and ventral cortex (Figure S2F, second row). KLFs are expressed in a similar way but absent in the ventral cortex (Figure S2F, third row). In contrast, SATBs is absent in the medulla but expressed in some cortex region (Figure S2F, first row). In the developing barb, both CTCF and KLFs are mainly expressed in the barbule plate cells and ramus region (Figure S2G, second and third rows). SATBs expression is restricted to barbule plate cells only (Figure S2G, first row). In feather filaments, CTCF/KLF4 and SATB2 are expressed in different epithelial compartments (Figure S2H), implying their unique contributions to skin appendage morphogenesis.
To test if CTCF and KLF4 target the Chr25 and Chr27 β-krt clusters, we performed ChIP-seq with antibodies against CTCF and KLF4 in E14 feather filaments and leg scale skin (Figures S2I–S2K). The results show:
CTCF targeted the H3K27ac regions of the Chr27 β-krt cluster in feather filaments (Figures 2G and 2G′, top track), suggesting it plays an active role in feather-bearing regions. Since CTCF facilitates chromatin looping through the loop-loop extrusion mechanism (Nichols and Corces, 2015), we examined the orientations of CTCF motifs found on the selected H3K27ac regions of the Chr27 β-krt cluster. Reverse-oriented CTCF motifs were located at H3K27ac (Ac) peak numbers 1/2 and 8/9 (Ac Pk1/2 and Pk8/9; red numbers) (Figure 2G′). For the Chr25 cluster, we observed that, unlike its role in intra-cluster looping, CTCF did not bind within the FK subcluster but rather targeted junctions between subclusters (Figures 2F and 2F′, top track).
KLF4 also targeted some selected H3K27ac regions of the Chr27 β-krt cluster but at lower abundance than CTCF (Figures 2G and 2G′; second track). However, the KLF4-binding profile on the Chr25 β-krt cluster differed from the CTCF-binding profile. In feather-bearing skins, KLF4 targeted within the FK cluster body rather than at subcluster junctions, indicating these chromatin regulators have functional differences in the same Krt cluster (Figures 2F and 2F′). ChIP-qPCR results further validated the enrichment of H3K27ac and CTCF on the FK-TE and selected H3K27ac regions (Figures 2H and 2I). Taken together, these results demonstrate the putative FK-TE on the Chr25 Krt cluster and the selected H3K27ac regions on the Chr27 Krt cluster are targeted by CTCF (Figures 2J and 2K) and KLF4 in chicken E14 feather filaments.
Capture-C Analyses Show Higher-Order Chromatin Looping in β-Keratin Clusters Occurs between Subclusters in Chr25 but within the Subcluster in Chr27
To test the hypothesis that intra-cluster chromatin looping controls differential FK gene expression of the Chr27 β-krt cluster (Figure S1H), we used Next-Generation Capture-C (Hughes et al., 2014), a chromosome conformation capture-based technique that identifies all genomic regions interacting with specific sites of interest (baits) (Figure 3A). Capture-C has the most suitable resolution, around 100 to 500 bp, and sensitivity to examine chromatin interactions within the size range of the β-krt clusters (Figures 1B and 1E). We used dorsal skin epidermis (experimental group) and whole brain (control group) collected at different developmental stages (E7 and E14). To prevent missing possible looping anchors, we manually selected 38 genomic regions marked by H3K27ac signals and named them sequentially as candidate anchors, H3K27ac Peak 1–38 (Pk1-Pk38) (horizontal red lines in Figure 3B; Table S2).
Figure 3. Temporospatial Chromatin Looping of the Chr27 β-Keratin Gene Cluster during Skin Development.
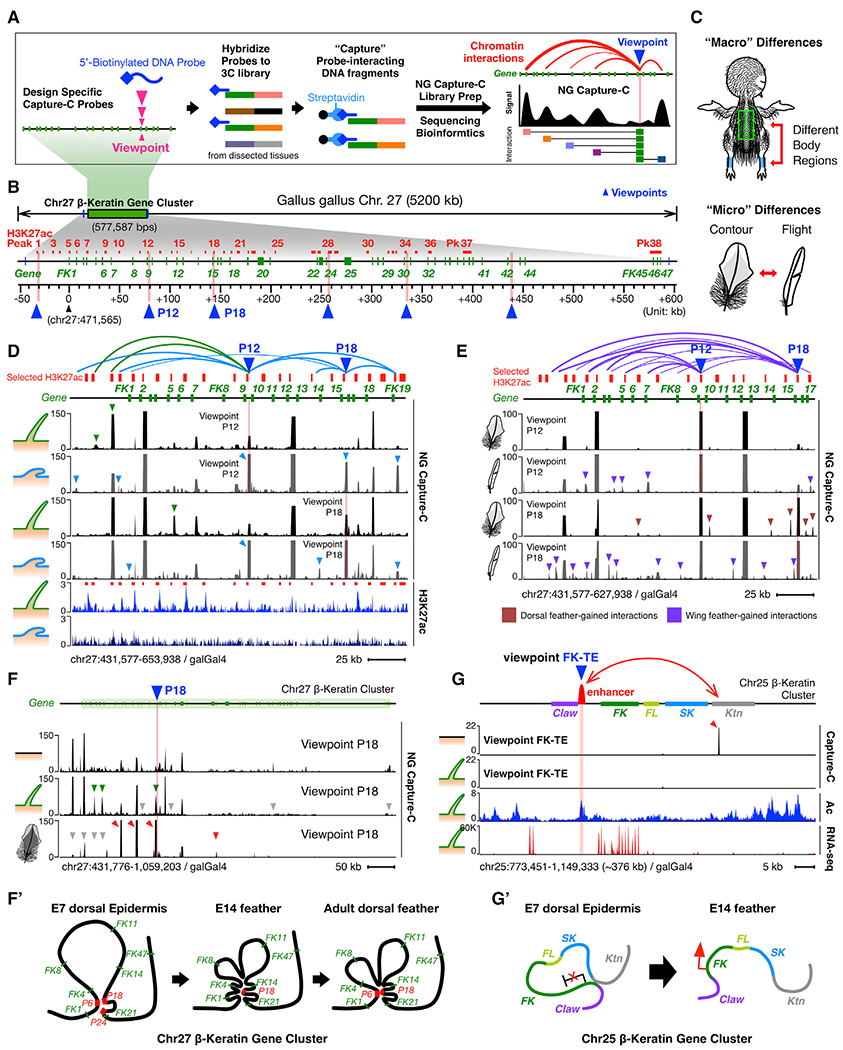
(A) Schematic of NG Capture-C experiment and its data presentation.
(B) Schematic of selected H3K27ac regions as candidate looping anchors at the Chr27 β-krt cluster. Red lines, selected H3K27ac regions; pink bars and blue triangles, bait sites (viewpoints) used in NG Capture-C experiments.
(C) Schematic of experimental design for comparison of skin “macro” and “micro” differences.
(D) NG Capture-C interactions of different skin regions (feather filaments and scale epidermis) from the same-aged chicks (E14).
(E) NG Capture-C interactions of different feather types (dorsal contour feather barbs and flight feather barbs) from the same-aged adult chickens.
(F) NG Capture-C interactions of different aged skins (E7, E14, and adult) from the same dorsal back region. Green triangles, feather-specific chromatin interactions; red triangles, adult contour feather-specific chromatin interactions.
(F′)Schematic of dynamic intra-cluster chromatin looping of Chr27 β-krt cluster during feather-skin development.
(G) NG Capture-C interactions of FK-TE at the Chr25 β-krt cluster.
(G′) Schematic of dynamic and inter-subcluster chromatin looping at the Chr25 β-krt cluster.
To design Capture-C baits, we first generated duplicate 3C libraries using a restriction enzyme Dpn-II for each tissue and examined digestion efficiencies of 38 DpnII sites at selected H3K27ac regions. We used six baits located at/around Pk1, Pk12, Pk18, Pk28, and FK42 cluster (highlighted as vertical pink bars and blue triangles in Figure 3B; Table S3) because they are: (1) with >60% digestion efficiencies in both feather and scale tissues and (2) distributed throughout the whole Chr27 β-krt (see Method Details). Only consistent interactions appearing in both duplicates with over 20 captured reads are considered true interactions. We performed principal component analysis (PCA) and checked the Spearman correlation of the duplicates to verify the reproduction of Capture-C results (Figures S3A and S3B; Table S4). We then summarized all true Capture-C interactions in Table S5 and categorized them as interactions (>20 captured reads/site), strong/stable interactions (>100 captured reads/site), and very strong/stable interactions (>1,000 captured reads/site). Using bait at candidate anchor Pk18 as a demonstration Capture-C viewpoint (P18, blue triangle), we observed increasing captured signals in the E14 feather filaments compared to E14 brain, indicating intra-cluster interactions take place between Pk18 and the other genomic regions within Chr27 β-krt cluster (Figure S3C, red arrows). This design provided chromatin interactions with validated Dpn-II sites at the selected H3K27ac regions, enabling clear comparison among different skin regions, but it did not provide exhaustive interactions within the whole cluster, and other interactions may also contribute to the regulation of Krt clusters.
Chromatin-Looping Configurations in β-Keratin Clusters Are Temporospatial-Specific
If chromatin looping is functionally specific, the configuration should correlate with temporospatial β-krt expression in skin appendages in vivo. We first evaluated whether there is region-specific looping of β-krt genes within clusters. Since skin is afforded additional functions by the presence of diverse skin appendages in different regions, we classified “regional differences” into macro and micro differences, indicating differences across different body parts and within an appendage, respectively (Figure 3C). To compare macro-skin regional differences, we generated skin epidermis 3C libraries for Capture-C from the same-stage embryos but different body parts (E14 dorsal back feather filaments and E14 leg scale epidermis). Using Pk1, Pk12, Pk18, and Pk28 as Capture-C viewpoints, we observed significantly increasing chromatin interactions of Pk15-Pk12 and Pk15-Pk18 (captured reads >1,000) as well as Pk12-Pk18 and Pk12-Pk22 (captured reads >100), suggesting compaction of the Chr27 β-krt cluster in E14 scale epidermis compared to feather-bearing regions (Figures 3D, S3D, S3F, and S3F′; Table S5).
To examine micro-skin regional differences, we used barb branches from adult chicken dorsal and wing feathers. The Capture-C results show that compared to adult wing feather barb branches, dorsal feather barb branches lack not only the very strong interaction of Pk12-Pk6 but also many intra-cluster interactions within the Chr27 β-krt cluster (highlighted in purple arrows in Figures 3E and S3E). To verify Capture-C-determined chromatin interactions, we generated EcoRI-digested 3C libraries using E14 feather and scale skins, performed 3C-PCR, and sequenced the interacting DNA fragments. The 3C-PCR results confirmed the interaction of Peak-12 and Peak-9 (Pk12-Pk9) and Peak-12 and Peak-6 (Pk12-Pk6; Figures S3H–S3J).
To examine if chromatin looping is dynamic during skin development, we collected different staged epidermis from the same dorsal back region, including E7, E14, and adult dorsal back feather barbs. We observed a gradually established chromatin looping of the Chr27 β-krt cluster from E7 to E14 (Figure 3F, top two tracks). Furthermore, adult dorsal back feather barb branches had distinct chromatin interactions compared to embryonic stages (Figure 3F bottom two tracks, quantified in Figure S3K), suggesting dynamic intra-cluster chromatin interactions occur throughout the whole life of skin epidermis (Figures 3F and S3L).
To further understand possible interacting chromatin regions with the Chr25 Feather Keratin typical enhancer (FK-TE), we performed Capture-C using baits designed at the feather-specific typical enhancer (Figure 3G). The results show there was no intra-cluster interaction within the FK subcluster but rather an “inter-cluster” interaction between FK and Keratinocyte Krt (Ktn) subclusters (Figure 3G, top track, viewpoint FK-TE). This result suggested that, in the enhancer-mediated mode, a subcluster is controlled as a unit and looped together through specific enhancers (Figure 3G′).
In Situ Hybridization Shows Co-expression of β-Keratin Genes Located within the Same Chromatin Loop
If the chromatin-looping configuration is specific, β-krt genes present within a chromatin loop might be co-expressed in the same regions of developing feather filaments. Figure S4A shows a drawing of a longitudinal feather follicle section depicting its structure with the stem cells (red circles) located above the dermal papilla (green) near the base of the feather. The feather backbone (rachis) and barbs are also shown. In Figure S4B, a cross-section shows the rachis with its cortex and medulla as well as the barb ridge, which gives rise to branches. ISH was performed using specific probes for both Chr25 and Chr27 β-krt genes (Figures 4A, 4B, and S4B–S4D). We first demonstrated that FK genes from both clusters possess region-specific expression in E14 feather-bearing skins (Figure 4A, Chr25 FK3/12 were detected in feathers but not scales, and Figure S4B). We found two distinct FK gene expression distributions from the two clusters, which matched the two epigenetic modes–(1) a uniform distribution of all FK genes on Chr25 (Figures 4A, 4A′, S4B, and S4E) and (2) differentially grouped distributions of FK genes on Chr27 where Chr27-FK1 and -FK12 genes within a loop show the same expression pattern, whereas -FK39 and -FK45 in another loop show a different expression pattern (Figures 4B, 4B′, S4C, and S4D). These results indicate that temporospatial chromatin looping of the β-krt cluster correlates with β-krt expression. In the enhancer-mediated mode (Figure 4C), a subcluster is controlled as a unit and looped together through specific enhancers controlled by CTCF (Figure 4C), generating macro-differences of different body regions. In the intra-cluster looping mode, differential chromatin looping controlled by CTCF and/or KLF4 at different sub-locations elaborate micro-differences within a feather (Figures 4D and S4E).
Figure 4. β-Keratin Expression Patterns Are the Same for Those within Feather Subclusters on Chr25 or Co-expressed for Those within the Same Chromatin Loops on Chr27.
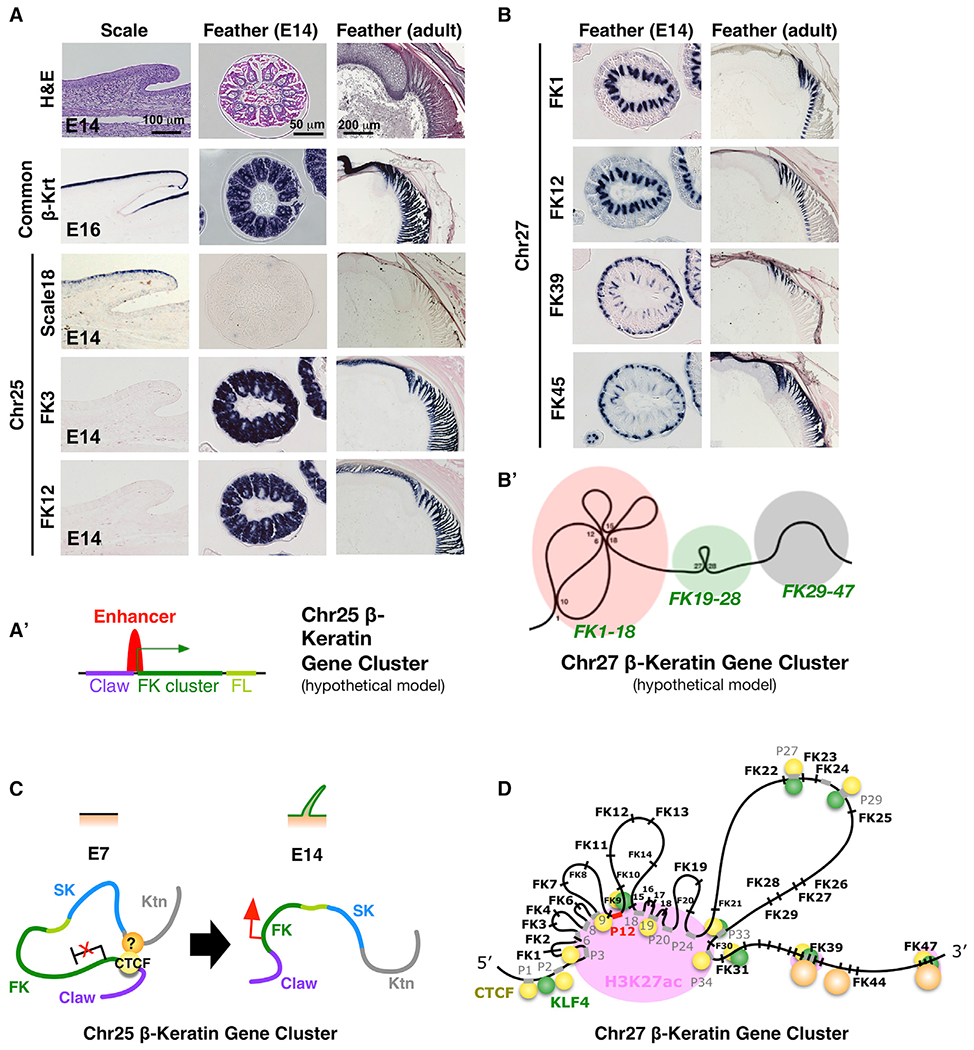
(A) Expression of Chr25 β-krt genes in embryonic scale (left column), embryonic feather (middle column), and adult feather (right column). First row, H&E staining. Second row, common β-krt. Third to fifth rows, Scale Krt 18, FK3, and FK12 from Chr25, respectively. Note FK3 and FK12 show the same expression pattern. (A′) Schematic drawing of single enhancer-driven co-expression of subclustered genes on the Chr25 β-krt cluster.
(B) Expression of Chr27 FK genes in embryonic (left column) and adult feathers (right column). Note FK1 and FK12 show the same expression pattern in both embryonic and adult feathers, whereas FK39 and FK45 show the same expression pattern in embryonic feathers and different patterns in adult feathers.
(B′) Schematic drawing of intra-cluster chromatin looping of Chr27 β-krt cluster.
(C and D) Schematic of the two-mode epigenetic regulation – enhancer-driven co-expression of the whole subclustered FK genes on the Chr25 β-krt cluster (C) or CTCF/KLF4-mediated temporospatial chromatin looping of the Chr27 β-krt cluster (D).
A Three-Factor Hypothesis for the Specification of Region-Specific β-Keratin Expression
If co-expressed β-krts are present in the same chromatin loop, we want to know how specific chromatin loops are configured. Learning from other organ systems and the regulation of Hox gene clusters, we assumed that groups of DNA-binding factors act cooperatively as either pioneer factors to prime specific genomic regions for future chromatin re-modeling (Biddie et al., 2011; Phanstiel et al., 2017) or as co-factors to work with chromatin regulators to configure chromatin (Jerković et al., 2017). Here we defined “chromatin regulators” as chromatin-associated proteins that function to organize 3D chromatin looping. To this end, we hypothesized regulatory elements important to this process will be co-localized in looping anchors. Therefore, we designed an analytical strategy to obtain genomic elements co-occupied by both CTCF and H3K27ac marks in different skin regions, meaning all possible region-specific CTCF-looped anchors. Then we searched for enriched motifs in these elements (Figure 5A). We identified feather-specific as well as scale-specific DNA-binding transcription factors (feather-TXs and scale-TXs) that have the potential to facilitate region-specific chromatin looping with CTCF (Figure 5B).
Figure 5. Identification of DNA-Binding Factors that Co-occupy Region-Specific Regulatory Elements.
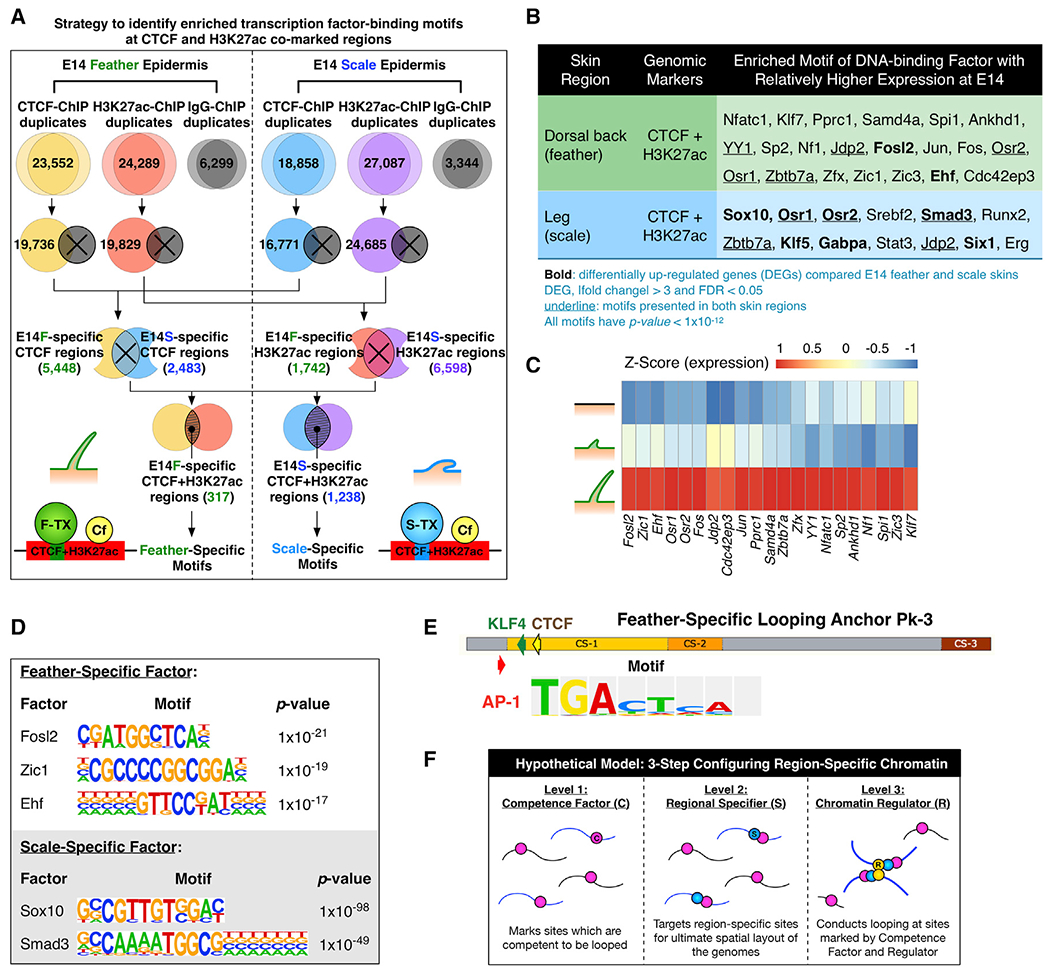
(A) Schematic depicting the analysis strategy used to identify motifs of region-specific transcription factors in CTCF/H3K27ac co-marked chromatin.
(B) HOMER de novo motif discovery showing enriched motifs of DNA-binding factors at CTCF and H3K27ac co-marked chromatin in E14 feathers and scales. Expression of the listed factors was validated using RNA-seq results.
(C) Normalized expression of skin region-specific DNA-binding factors during skin specification. Z score, [RPKM-(mean RPKM)/standard deviation]. The order of factors was sorted by differential expression.
(D) Lists of skin-region-specific binding motifs.
(E) Positions of an AP1 factor binding motif in feather-specific looping anchor Pk3 of the Chr27 β-krt cluster.
(F) The three-factor hypothesis elucidating how region-specific chromatin looping is established – competence factor, regional specifier, and chromatin regulators.
Among the enriched motifs, we found several DNA-binding factors with region-specific expression. The major ones are Zic1 and Ehf in feathers as well as Sox10 and Smad3 in scales (Figure 5D). Among feather-specific TXs, we noted Zic1 because of its relatively high expression in E14 compared to E7 and E9 skins (Figure 5C). Furthermore, it has been reported to serve as a master scale-to-feather converter (Wu et al., 2018b), implying its role in determining feather-skin specification (Figure S5). On the other hand, among scale-specific TXs, Smad3 and Osr1/2 drew our attention (Figure 5B). Since Smad3 was reported to mediate chromatin organization with CTCF (Bergström et al., 2010) and have a profound impact on skin morphogenesis (Buschke et al., 2011), we proposed one of its functions might be involved in organizing region-specific chromatin.
We also observed an AP1 transcription factor, JDP2, was present in both feather and scale regions, whereas other AP1 family members, such as Fosl2, Jun, and Fos, were specifically present in feather samples and had relatively higher expression at E14 (Figure 5C). To further evaluate this, we examined the feather-specific looping anchor Pk3 (Figure 2B), which contained an AP1 binding motif (Figure 5E). Since AP1 transcription factors, Jun and Fos, are reported to have pioneer functions in skin specification (Biddie et al., 2011; Jerković et al., 2017), we postulated that AP1 factors might function to prime potential genomic sites for future looping.
Understanding how region- or organ-specific chromatin organizations are established has long been an important unsolved issue. Roles of general chromatin regulators, such as CTCF and STAB2, have been addressed but how these general regulators determine their region-/organ-specific targets temporospatially is still puzzling. Through our motif discovery and clues from the literature, we propose a three-factor working model consisting of competence factors, regional specifiers, and chromatin regulators (Figure 5F). Competence factors, such as AP1, mark all potential sites that are accessible to serve as anchors for chromatin looping; regional specifiers, such as Zic1, are expressed temporospatially in feathers and target only region-specific sites for future chromatin looping; chromatin regulators, such as SATB2 and CTCF, build chromatin loops through protein-protein and/or protein-DNA interactions. They work together to set up regional specific β-krt expression.
Functional Analysis of β-Keratin Expression in a Feather Filament and Perturbation of the Putative Feather-Specific Transcription Factor Zic1 and Chromatin Regulator SATB2
To evaluate this three-factor model in skin development in vivo, we evaluated examples from each category. Level 1–competence factor. We first examined the expression of two competence factors, FOSL2 and JUN, in developing E9 and E14 chick skin via ISH (Figure 6A). The results show positive signals of both FOSL2 and JUN in feather-bearing skins at E9 and E14. Whereas a low FOSL2 signal is present in E14 scale-bearing skin, confirming that FOSL2 is differentially expressed in feathers compared to scales (Figure 5B). To explore the role of AP1 factors in the regulation of β-krt expression, we used an alternative approach to examine chromatin occupancy of AP1 factors after exposing scale-forming skin to several factors that can cause scale-to-feather conversion: Sox2, Grem1, Spry2, and β-catenin (Wu et al., 2018b). We surmise that during the scale-to-feather conversion, in order to reconfigure feather-specific loops, chromatin must become accessible to both competence factors and feather specifiers. To test this hypothesis, we used the Assay for Transposase Accessible Chromatin with high-throughput sequencing (ATAC-seq). Indeed, motifs of AP1 factors such as Atf1, c-Jun, and JDP2 are enriched on differential transposase accessible regions (DARs) in Sox2-/Grem1-/Spry2-/RA-/β-catenin-converted skins compared to untreated skins. Moreover, we also identified motifs of the feather specifier, Zic1, and chromatin regulators, CTCF and KLF4, present in the DARs (Figure 6B; Tables S8 and S9), suggesting all three factors–competence factors, regional specifiers, and chromatin regulators–are involved in the feather re-specification.
Figure 6. Functional Evaluation of the Three-Factor Hypothesis for Region-Specific β-Keratin Expression.
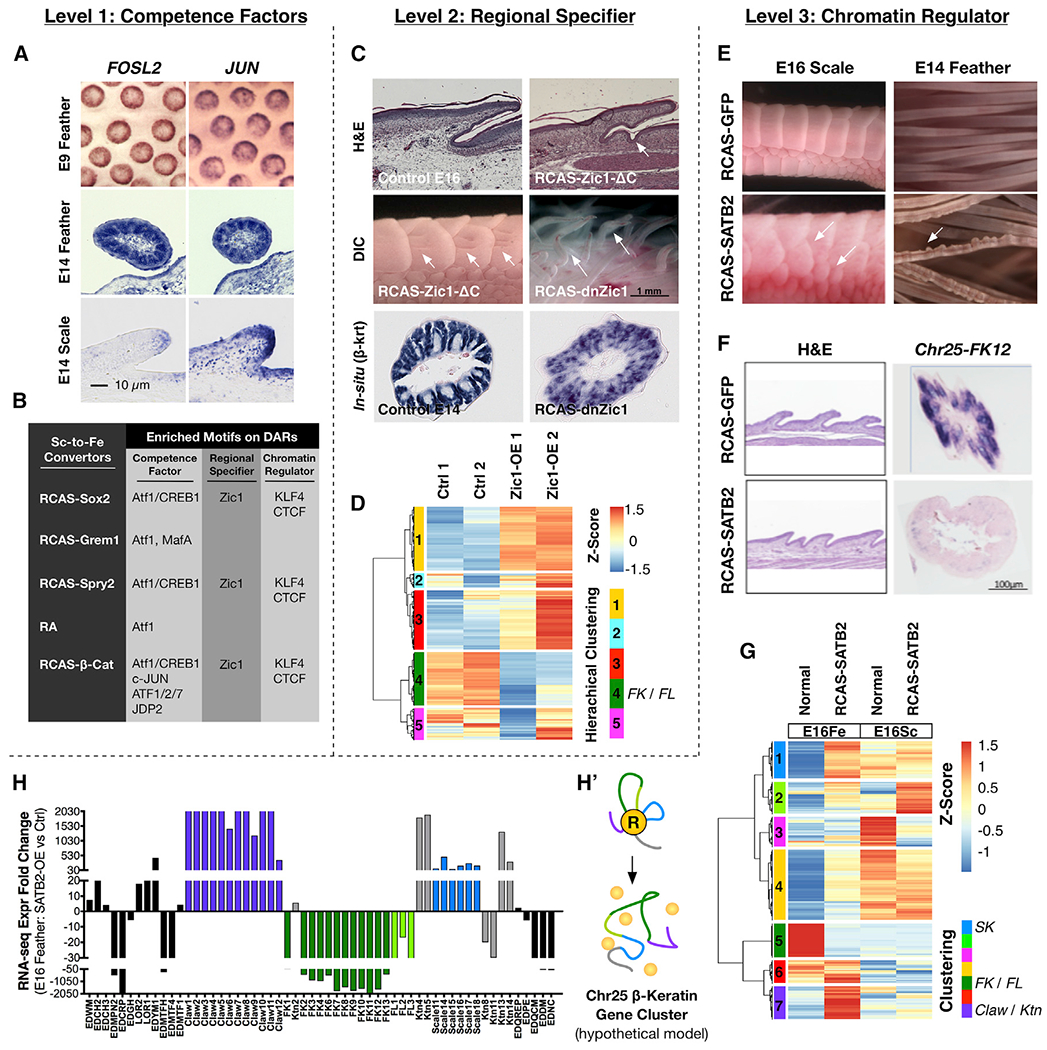
(A) ISH of FOSL2 and JUN genes at E9 and E14 feathers and scales.
(B) A summary table showing enriched motifs of the three factors on DARs under scale-to-feather conversion.
(C) Expression of a constitutively active form (RCAS-Zic1-ΔC) induced invagination in the scale hinge region, and barb ridge-like invaginations on the scale surface; both are seen in scale-to-feather transition. A dominant-negative form of Zic1 (RCAS-dnZic1) led to deformed feather filaments accompanied by disrupted β-krt expression.
(D) Normalized expression of DEGs from E14 RCAS-Zic1-infected feathers compared to its control groups. Z score, [RPKM-(mean RPKM)/standard deviation]. The DEGs were further clustered based on similarities of their expression patterns. Hierarchical Cluster 4 (green) includes FK genes.
(E) Over-expression of SATB2 in chick E14 and E16 embryos led to irregular scale formation and abnormal beaded structures in feather filaments.
(F) Aberrant Krt gene expression in irregularly sized scales after ectopic SATB2 expression. ISH shows Chr25-FK12 disappears from feather filaments.
(G) Normalized expression of DEGs from E16 RCAS-SATB2-infected feather (E16Fe) and scale epidermis (E16Sc) compared to their control groups. DEGs were further clustered by expression pattern similarities. Hierarchical Cluster 1 (blue) includes Scale Krt (SK) genes; Hierarchical Cluster 5 (green) includes FK and Feather-Like Krt (FL) genes; Hierarchical Cluster 6 (purple) includes Claw and keratinocyte (Ktn) Krt genes.
(H) EDC gene and β-krt gene fold-changes after misexpression of SATB2.
(H′) A hypothetical schematic showing disruption of the Chr25 β-krt cluster chromatin structure after SATB2 misexpression.
Level 2–regional specifier. We chose Zic1 as an example. Earlier we showed Zic1 is expressed in the feather and not the scale region. We expected that misexpression of Zic1 would lead toward homeotic skin appendages and aberrant Krt expression patterns. We used multiple ways to perturb Zic1 function by over-expressing a constitutively active (RCAS-Zic1-ΔC) (Wu et al., 2018b) and a dominant-negative form of Zic1 (RCAS-dnZic1). For RCAS-Zic1-ΔC, we found it caused (1) invagination at the edge of the scale, an event that occurs in feather follicle formation, and (2) invagination of the scale surface to form barb ridge-like structures; both events are characteristic of feathers (Wu et al., 2018b).
RCAS-dnZic1 caused an irregular feather filament shape with flattened areas (Figure 6C). By comparing ISH and RNA-seq results with those of normal E16 feather-bearing skins, over-expression of dnZic1 significantly altered β-krt gene expression profiles (Figures 6C, 6D, and S6A), producing a significant down-regulation of the whole FK subclustered genes (Figure 6D, hierarchical clustering). Moreover, EDC gene expression was also altered (Table S6). These results demonstrate that the regional specifier, Zic1, is responsible for establishing proper skin morphology and the region-specific expression of the Krt clusters and EDC genes.
Level 3–chromatin regulator. We expected that misexpression of one of the chromatin regulators (Figure 2) would lead to failure in morphogenesis and defective histo-differentiation. We performed functional experiments by over-expressing SATB2 in feather- and scale-forming regions. We found irregular scales were formed in the scale-forming region; in feather filaments, we found beaded deformities (Figure 6E). In addition, over-expression of SATB2 significantly altered Krt gene-expression profiles as determined by ISH and RNA-seq (Figures 6F, 6G, and S6B–S6C’). The results indicate significant up-regulation of the whole Claw and Scale Krt subclustered genes but down-regulation of the whole FK subclustered genes in the RCAS-SATB2-infected feather-bearing skins (Figure 6G, hierarchical clustering). Moreover, EDC gene expression was also altered (Table S6.2). These results suggest that proper balance of distinct chromatin regulators is needed for proper temporospatial Krt cluster expression, while perturbation in their milieu might result in abnormal gene transcription and tissue morphogenesis. Taken together, the results suggest that proper expression of components in the three-factor hypothesis is required for normal β-krt gene expression and skin specification (Figure 7).
Figure 7. The Three-Factor Hypothesis of How Region-Specific β-Keratin Expression May Be Established via the Two-Mode Epigenetic Strategy.
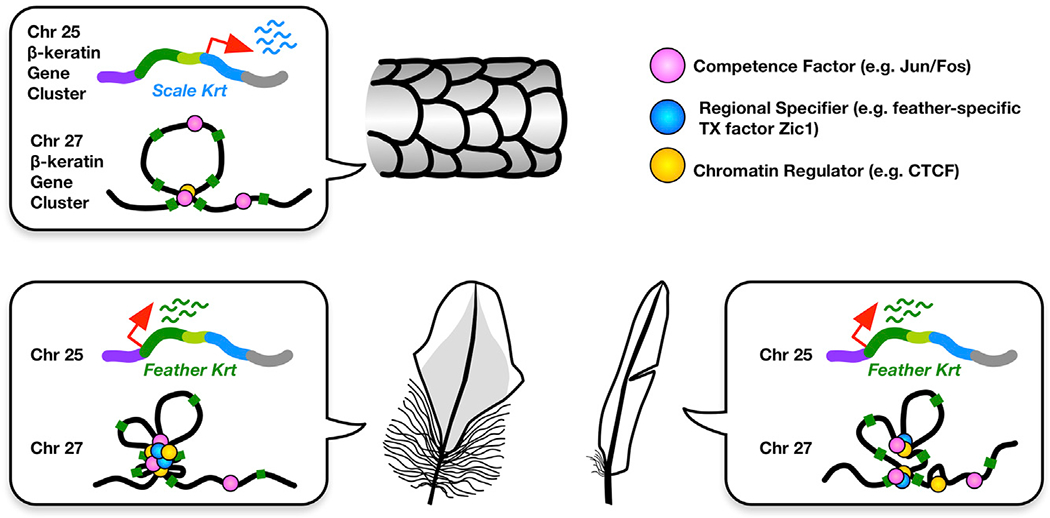
Throughout the body, the differences are controlled by the enhancer-driven uniform expression of subclustered genes. Whereas within feather differences are controlled by intra-cluster chromatin looping.
DISCUSSION
Our current results and those from the literature have demonstrated region-specific β-krt gene expression (Greenwold and Sawyer, 2010; Knapp et al., 1993) (Figures 1C, 1D, and S6B). A feather β-krt is also expressed specifically in pennaceous barbules of the contour feather (Kowata et al., 2014). How different β-krt clusters are regulated epigenetically is still unknown (Chuong et al., 2013). Using recent omics approaches, we profiled histone modification landscapes together with the temporospatial transcription of major avian β-krt clusters. Our results led us to propose that two fundamental epigenetic modes apply to Krt gene clusters. These include (1) a single enhancer driving the uniform expression of the whole Krt subcluster and (2) an intra-cluster chromatin looping that elaborates differential expression of grouped Krt genes. The two-mode regulation explained ISH results showing different expression patterns (Figures 4A and 4B) while Capture-C results further demonstrated not only intra-cluster (Figures 3C–3E) but also inter-cluster chromatin looping (Figure 3F) configure the Krt gene family. These results provide molecular evidence adding 3D genome complexities to Krt gene regulation and reveal true genomic contacts of Krt clusters in the context of skin regional specification.
Region-specific and tissue-specific chromatin conformations may not be at the scale of differences observed in topologically associating domains (TADs). Previous studies showed that both mouse type-I and type-II α-krt clusters are organized within individual TADs (Dixon et al., 2012), and 5C analysis further revealed that the mouse EDC is organized into separate TADs, which possess cell-type-specific genomic contacts (Poterlowicz et al., 2017). Since the Chr25 β-krt cluster is arranged within the EDC, the potential interactions between the Chr25 Krt locus and the EDC locus will need to be analyzed further in the context of region-specific appendage differentiation. Results here inspire the concept of intra- and inter-cluster chromatin looping during skin development: dynamic regulation of single-clustered Krt genes might be controlled at the intra-TAD level (Berlivet et al., 2013) while co-regulation of different Krt clusters might be mediated by inter-TAD looping.
It has been a technical challenge to collect sufficient and relatively pure cell populations from tissues to apply more 3D genome technologies in order to obtain a genome-wide level understanding of the multiplex Krt cluster. Once epigenetic technologies using fewer cells become available, techniques like Hi-ChIP and HiC will reveal new insights of global looping with Krt clusters. Advancements in technologies such as CRISPR/Cas9 systems may help to answer how the selective use of anchoring sites can generate diversity in β-krt expression required in feather bio-architecture. Our study applied these epigenetic technologies to study Krt cluster regulation in macro-level tissue patterning at the genomic level and shows a path forward for future research exploring tissue specification in the context of global genome mapping.
Clustered genes are widespread in genomes, 17.6% of human annotated genes and 27% of mouse genes (Yi et al., 2007), whether there are general rules to control gene clusters is unclear. We have documented two mechanisms of epigenetic strategies in chicken Krt clusters. Chromatin looping seems to be an effective epigenetic mechanism, which produces an almost limitless combinatorial complexity from a limited number of genes. HoxA-D gene clusters show intra-cluster chromatin looping but not necessarily in enhancer or promoter regions (Ferraiuolo et al., 2010) as was seen in the Chr27 β-krt clusters described here. Whereas, Protocadherin (Pdch) gene cluster regulation (Guo et al., 2012) uses promoter choice of the starting exon and cis-alternative pre-mRNA splicing (Tasic et al., 2002; Wang et al., 2002). While each of these clusters appears to utilize chromatin regulators, such as CTCF, to construct their loops, how these regulators determine loop specificity in different scenarios is unknown. Also, CTCF may help to loop, but other interactions may still be present. To explain loop specificity, we propose a three-factor working hypothesis: competence factors first mark all possible looping anchors, regional specifiers are up-regulated in specific skin regions, and then guide chromatin regulators to configure region-specific looping anchors.
We first identified candidate chromatin regulators, CTCF and KLF4, which target the putative FK-TE and selected H3K27ac regions (Figure 2). We next predicted the concurrent binding motifs of pioneer factors and region-specific DNA-binding factors on CTCF-H3K27ac (Figure 5A). We identified transcription factor AP1 family members JUN and FOSL2 as putative feather competence factors (Figure 5B). We focused on AP1 transcription factors because they are significantly enriched and up-regulated in feather-bearing skins and they regulate chromatin structures and expression of mouse EDC genes in the skin (Oh et al., 2014). Moreover, AP1 was reported as a pioneer factor that primed and repositioned genomic regions for glucocorticoid receptor binding in response to hormone (Biddie et al., 2011). In macrophage development, the AP1 bound to active H3K27ac-marked loop hubs during macrophage specification (Phanstiel et al., 2017). AP1 factors also are required for the specification of human epidermal keratinocytes, supporting our hypothesis that regional specifiers are involved in determining AP1 targets (Rossi et al., 1998). JUN also activates β-krt gene expression in avian fibroblasts, providing a direct link between AP1 factors and transcriptional regulation of Krt clusters (Hartl and Bister, 1995). These reports strongly suggest that AP1 functions as a competence factor targeting chromatin anchors for future loop formation. API and CTCF-binding motifs were also significantly enriched in the Hox gene clusters (Jerković et al., 2017). These independent findings, in different organ systems, support the three-factor hypothesis; future studies will be required to further develop and revise the model.
Chromatin regulators, CTCF/KLF4 and SATB2 may play different roles in Krt gene expression. Region-specific chromatin interactions of Chr27 β-krt in feather and scale epidermis are revealed by the difference in Capture-C results. The scale-specific chromatin conformation may lead to the silencing of FK genes. Over-expression of SATB2 suppresses Chr25 FK in the feather filament–direct binding remains to be investigated. These results suggest different chromatin configurations can result in the activation, repression, constitutive silencing, or pausing of transcription. The future challenges are to determine what mechanisms establish the skin-region-specific chromatin configurations, and what factors are used to read the configurations and affect expression patterns.
The ability of the integument, at the interface of an organism and its environment, to generate regional specificity is key to successful adaptation (Lai and Chuong, 2016). This can be appreciated from Evo-Devo and comparative genomic studies (Greenwold et al., 2014; Lowe et al., 2015; Wu et al., 2004). Here, we show how temporal and regional control of β-krt clusters are epigenetically regulated with unique regulatory strategies. Macro-regional differences are controlled by typical and super-enhancers on Chr25. To venture into new eco-spaces, a large spectrum of intra-feather rigidities and flexibilities, representing micro-within-feather regional specificity, were required for successful feather evolution (Greenwold and Sawyer, 2011). The FK cluster on Chr27 evolved chromatin-looping mechanisms to generate enormous combinatorial possibilities with a large spectrum of biophysical properties to cope with the rapid evolution of feathers. The complexity of Krt cluster regulation is probably involved in diversifying tissue types within individuals as well as between species during evolution.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact and Materials Availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Cheng-Ming Chuong (cmchuong@usc.edu).
Data and Code Availability
The raw and processed data of RNA-seq, ChIP-seq, and NG Capture-C reported in this paper is available at NCBI GEO database, accession GSE136224.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Chicken Model
Fertilized pathogen-free White Leghorn chicken eggs were purchased from Charles River Laboratories. Eggs were incubated at 37.5°C in GQF HOVA-Bator Thermal Air Incubators with automatic turner until embryos reached the desired developmental stages. Chick embryonic age 7 days (E7, also known as HH31), 9 days (E9/HH35), 14 days (E14/HH40), and 16 days (E16/HH42) were used. Hatched chickens were housed in the USC Department of Animal. The USC Institutional Animal Care and Use Committee (IACUC) approved our work with chickens, under protocol number 11903. All experiments conform to the relevant regulatory standards.
METHOD DETAILS
Biological Specimen Collection and Cell Preparation
All procedures were done on ice exclude those with specific notes. Dorsal back feather and leg scale skins were dissected from chick E14 embryos. To collect non-keratinized feather filament epidermis, we first trimmed Kized parts off a feather filament (around 2/3 from a filament tip) with a micro-scissor and then pulled each remaining feather filament base off the skin under a dissecting microscope. Only ~2 mm of feather filament bases were kept and washed in 1X cold PBS. To collect leg (reticular) scale epidermis, a skin was submerged in 2XCMF solution containing 0.25% EDTA for 15-20 min on ice and then the epidermis was carefully peeled off from the dermis. To dissociate cells, feather filament bases and leg scale epidermis were incubated in 0.35% Collagenase Type I (Worthington, Cat. LS004196) prepared in HBSSat 37°Cforan hour with rotating and pipetting occasionally to disaggregate cells/tissues. The dissociated cells were then filtered by a 70 μm and then a 40 μm cell strainers. 2X volume of chicken cell culture medium (DMEM with 10% FBS, 2% chicken serum, and 1% P/S) was added to eliminate the collagenase reaction and cells were pelleted down by centrifugation at 300 x g for 10 min at 4°C. Cells were washed in 1X cold PBS containing 2% BSA twice and cell numbers were count. In general, we could collect ~8x107 dissociated feather base epithelium cells and ~5x107 dissociated scale epithelium cells from 19 E14 embryos. It required 6 hours for two researchers to process 10 embryos.
Paraffin Section, In Situ Hybridization and Immunostaining
Chicken embryos were collected, fixed in 4% paraformaldehyde at 4°C overnight for in situ hybridization (ISH). For IM, frozen sections of the E21 wing flight feather were prepared. Adjacent sections were used to perform H&E staining and IM. ISH and IM were followed the procedures described by Jiang et al (Jiang et al., 1998). To generate specific β-keratin antisense RNA probes, we used the three prime untranslated regions (3’-UTRs) as PCR template targets. PCR primers for each Keratin form are list below: Chr25-FK3, 5’-aggaagcccaaagtctgaca (forward), 5’-tctggtacaggttgcaatgg (backward); Chr25-FK12, 5’-tgaggtggacatcctgtgaa (forward), 5’-acaatgggatgcctgacttc (backward); Chr27-FK1, 5’-cccggagaaattcatccata (forward), 5’-ccaagtctgagctcatgcaa (backward); Chr27-FK12, 5’-ggtgctggatgaagaaggaa (forward), 5’-aggctccacagtgcaagatt (backward); Chr27-FK39, 5’-ctggacggcagaacagactc (forward), 5’-agagagcagcaggagcagag (backward); Chr25-FK45, 5’-attgacagacctgcagtgga (forward), 5’-cagctgtccatctgcctttt (backward); the PCR product was inserted into the pDrive plasmid (Qiagen). Diluted eosin was used for faint counter-staining. Leica TCS SP8 confocal system with an inverted Leica DMI8 microscope equipped with a motorized scanning stage was used.
Antibodies
From Abcam, Inc: rabbit polyclonal anti-H3K27ac (ab4729), anti-H3K4me1 (ab8895), and anti-H3K4me3 (ab8580) antibodies. From Aviva Systems Biology: rabbit polyclonal anti-hCTCF-N-terminal antibody (ARP38820_P050) which shared 86% immunogen sequence identity to chicken CTCF. From Santa Cruz Biotechnology: ChIP-seq control anti-IgG antibody (sc2712) and mouse monoclonal anti-hGKLF/EKLF/LKLF antibody clone F-8 (sc-166238 X) which shared 97% immunogen sequence identity to chicken KLF4. From Cell Signaling Technology, Inc: ChIP-qPCR control normal rabbit IgG (#2729). From Proteintech Group, Inc: rabbit polyclonal anti-SATB1/2 antibody (15400-1-AP). Usage: The antibody amounts used in the ChIP-seq experiments were 2 μg of anti-H3K27ac, 2.5 μg of anti-CTCF, 5 μg of anti-KLFs, 5μg of anti-H3K4me1, and 5 μg of anti-IgG control antibodies per IP.
RNA Sequencing (RNA-seq)
Each group had two replicates.
RNA Extraction
Total RNAs of indicated tissues were extracted followed by the standard Trizol extraction protocol (Invitrogen, Cat.15596026). RNA purity was accessed via A260/A280 ratio and RNA integrity was accessed by electrophoresis, comparing 28S and 18S intensities.
RNA-Seq Library Preparation
RNA-seq libraries were generated using TruSeq RNA Prep Kit v2 (Illumina, RS-122-2001/-2002). In brief, 1 μg of total RNA was used to prepare one cDNA library. This kit uses oligo-dT affinity beads in the first step to purify mRNA and other RNAs containing poly-A sequences. After library amplification, 0.8X AMPure XP bead (Beckman, A63880) clean-up was performed for size selection and removal of adapter-dimers.
Sequencing
After AMPure XP bead clean-up, cDNA libraries were submitted to USC Molecular Genomic Core (MGC) and libraries with a size of 300-400 bp determined by using Bioanalyzer DNA 1000 Kit (Agilent) were sequenced on the NextSeq500 platform using the single-end 50-bp protocol (Illumina).
Chromatin Immunoprecipitation Sequencing (ChIP-seq)
The protocol was adapted from Dr. Ruchi Bajpai’s laboratory (Bajpai et al., 2010). Each group had two replicates.
ChIP
All procedures were done on ice exclude those with specific notes. To prepare one ChIP-seq library, 5-9x107 dissociated cells were freshly prepared according to the Cell preparation section and resuspended in 5 ml of cold PBS with 2% BSA. Cells were cross-linked by adding 500 μl of Fixative solution (11% formaldehyde, 50 mM HEPES-KOH pH=7.5, 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA) and incubating at RT for 14 min. Formaldehyde was quenched by adding 250 μl of 2M glycine and rotating at RT for 5 min. Cross-linked cells were pelleted at 300 x g for 5 min and washed in 10 ml of cold PBS with 1 mM PMSF for 4 times. The washed cells were further lysed in 5 ml of Lysis Buffer 1 (50 mM HEPES-KOH, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP-40, 0.25% Triton X-100) containing 100 μl of 50X protease inhibitor cocktail (Roche, Cat. 11873580001), rotated for 10 min, and pelleted down at 300 x g for 5 min. The chromatin pellet was resuspended in 5 ml of Lysis Buffer 2 (10 mM Tris-HCl pH=8, 200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA) with 100 μl of 50X Protease Inhibitor Cocktail, incubated at RT for 10 min with gently rocking, and pelleted at 300 x g for 5 min. The chromatin pellet was then resuspended in 2 ml of Lysis Buffer 3 (10 mM Tris-HCl pH=8, 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% Na-deoxycholate, 0.5% N-lauroylsarcosine) with 60 μl of 50X Protease Inhibitor Cocktail and aliquoted into sonication tubes (Diagenode, Cat. M-50050). Chromatin was fragmented using sonication under Diagenode Bioruptor: high amplitude, pulse for 30 sec on and 30 sec off for a total 25 min elapsed time. After fragmentation, the chromatin solution was centrifuged at 2x104 x g for 10 min. Sheared chromatin in the supernatant was transferred into a new tube containing 1/10 volume of 10% Triton X-100 and centrifuged at 2x104 x g for another 10 min. 300 μl of sheared chromatin was used for validation of chromatin shearing efficiency and 50 μl of sheared chromatin was saved at −80°C as an input gDNA control. Sheared chromatin with an average size of 0.5-2 Kb was proceeded to the next IP step. For IP, 2-5 μg of antibody was added into 300 μl of sheared chromatin solution and rotated at 4°C for 16 hours. For antibody amounts used in the study, please see the Antibodies section. After IP, 50 μl of 1:1 Dynabeads Protein A (In nitrogen Cat. 100-02D) and Protein G (Invitrogen Cat. 100-04D) mixture was washed with Block Solution (0.5% BSA in 1X PBS) for three times and added into the chromatin-antibody mixture. The chromatin-antibody-beads mixture was then rotated at 4°C for another 16 hours. After IP, the mixture was collected using a magnetic stand, washed in cold RIPA Wash Buffer (50 mM HEPES-KOH pH=7.6, 500 mM LiCl, 1 mM EDTA, 1% NP-40, 0.7% Na-deoxycholate) for 4 times and in TE buffer (10 mM Tris pH=8, 50 mM NaCl) once. Buffers were discarded and the beads with chromatin were collected by centrifugation at 960 x g for 3 min followed by using a magnetic stand. IP-pull-downed (IPed) chromatin was then eluted in 210 μL of Elution Buffer (50 mM Tris-HCl, pH=8. 10 mM EDTA, 1% SDS, stored at RT) on a 65°C hot plate for 15 min. The beads were pelleted by centrifugation and a magnetic stand. IPed chromatin in the supernatant was collected into a new tube and reverse-crosslinked at 65°C for 16 hours together with the tube of input gDNA control. Proteins and RNA were digested by addition of RNase A (0.2 μg/ml) at 37°C for 2 hours and Protease K (0.2 μg/ml) at 55°C for another 2 hours. ChIPed-DNA was further purified using the traditional phenol-chloroform extraction method and finally resuspended in 10 mM Tris-HCl (pH=8) at the concentration of 100 ng/μl.
ChIP-seq Library Preparation
280-1000 ng of ChIPed-DNA and input gDNA control were used for one library preparation. For end fill-in, NEBNext Ultra End Repair/dA-Tailing Module (New England Biolabs, Cat. E7442S) was used. For adapter ligation, NEBNext Ultra Ligation Module (New England Biolabs, Cat. E7445S) was used. 1X AMPure XP bead clean-up was performed to purify libraries after adapter ligation. For library amplification, KAPA HiFi HotStart ReadyMix (Kapa Biosystems, Cat. KK2601) was used. We used 6 cycles for input gDNA control and 12 cycles for ChIPed-DNA. After 0.8X AMPure XP bead clean-up, libraries were submitted to USC MGC.
Sequencing
Libraries were sequenced on the NextSeq500 platform using the single-end 50-bp protocol (Illumina) for H3K27ac-ChIP and 75-bp protocol for the others.
Chromatin Immunoprecipitation Coupled with Quantification PCR (ChIP-qPCR)
Each ChIP group had two independent biological replicates and each qPCR had two technique replicates. To prepare one ChIP reaction, minimum 1x107 dissociated cells were prepared according to the Cell preparation section. SimpleChIP Enzymatic Chromatin IP Kit Agarose Beads (Cell Signaling Cat. #9002) was used to prepare ChIPed-DNA and SimpleChIP Universal qPCR Master Mix (Cell Signaling Cat. #88989) was used to perform qPCR. Antibodies used in ChIP-qPCR were listed in the Antibodies section. qPCR was performed on the Mx3000P qPCR system from Agilent Technology. ChIP-qPCR data were normalized by the percent input method [% of Input = 100x2(ΔCt)]. Delta Ct (ΔCt) = Ct (Adjusted input) − Ct (Test Sample). Adjusted Input (100%) = Ct of 2% Input-5.64. Primer sequences were listed in Table S3.
Next Generation Capture-C (NG Capture-C)
The protocol was adapted from Dr. Wange Lu’s laboratory (Davies et al., 2017, 2016). Each group had two replicates.
3C Library Generation
To construct one 3C library, 9x105-1.4x108 dissociated cells from embryonic tissues or 2x105-2.4x106 dissociated cells from adult tissues were used. Dissociated cells were freshly prepared, resuspended in 20 ml of PBS, and cross-linked by addition of 1.14 ml of 37% formaldehyde at RT for 10 min. The reaction was quenched by addition of 1.2 ml of 2.5M glycine at RT for 5 min, pelleted at 300 x g for 5 min, and washed in 1 ml of cold PBS. Cross-linked cells were then lysed in 2 ml of cold Lysis Buffer (10 mM Tris-HCl pH=7.5, 10 mM NaCl, 5 mM MgCl2, 0.1 mM EGTA, 1X protease inhibitor cocktail, 0.2% NP-40) on ice for 10 min. The cells were then homogenized using a tissue grind pestle (Kimble, Cat. 885302-0002) for 20 slow strokes on ice. The nuclei were pelleted by 2,200 x g for 5 min at 4°C and washed in 800 μl of 1.2X cold NEBuffer DpnII (NEB, Cat. B0543) once. The nuclei pellet was resuspended 500 μl of 1.2X cold NEBuffer DpnII with 0.3% SDS and incubated with shaking at 950 r.p.m. at 37°C for an hour. 50 μl of 20% Triton X-100 was added into the tube and the mixture was incubated with shaking at 950 r.p.m. at 37°C for an extra hour. After addition of 40 μl of nuclease-free water, 10 μl of the mixture was saved as undigested control (UND), 300 U of DpnII destruction enzyme (NEB, Cat. R0543) was added into the remaining mixture, and incubated with end-to-end rotation at 37°C for 16 hours. Another 300 U of DpnII destruction enzyme was added and the reaction was incubated at 37°C for another 4 hours. The digestion reaction was stopped by incubating at 65°C for 20 min. 10 μl of digested chromatin was saved as digested control (DIG). The DpnII-digested chromatin was then transferred into a 15-ml tube containing 1X T4 ligase buffer (50 mM Tris-HCl pH=7.5, 10 mM MgCl2, 10 mM DTT) and 4000 U of T4 DNA ligase (NEB, Cat. M0202) to a final volume was 5 ml, and incubated at 16°C for 16 hours. The ligation reaction was then stopped by adding 110 μl of 0.5 M EDTA. To obtain the final DpnII-diested 3C (3C-DpnII) library, DNA of ligated chromatin was extracted using RNase A, Protease K, the phenol-chloroform method, and finally stored in 10 mM Tris buffer (pH=8) at −80°C. To examine the digestion efficiency of designed sites for capture baits, DNA of previous saved UND and DIG was extracted and qPCR was perform using paired primers of the target positions and control positions which were without DpnII sites. Formula for calculation of Digestion % is 100x[1-(DIGtarget/UNDtarget)/(DIGctrl/UNDctrl)]. Only the sites with over 60% digestion efficiency in both feather and scale tissues were used as bait-targeting sites for future Capture-C and 3C-PCR. In order to best cover the whole Chr27 β-krt gene cluster, we used bait-targeting sites near-evenly distributed across the cluster (total=~578 Kb): the bait at Pk1 (Pk1-bait) is located at 5’-end of the cluster; Pk12-bait is ~107 Kb downstream of Pk1; Pk18-bait is ~64 Kb downstream of Pk12; Pk28-bait is ~114 Kb downstream of Pk18; Pk34-bait is ~73 Kb downstream of Pk28; FK42-bait is ~100 Kb downstream of Pk34.
Capture Probe Design
The 120-bp biotinated capture probes were designed according to the publication (Davies et al., 2016), following the CapSequm website (http://apps.molbiol.ox.ac.uk/CaptureC/cgi-bin/CapSequm.cgi?rm=mode_25). Capture probes were finally filtered based on repeat content using a Multi-Image Genome browser (McGowan et al., 2013).
Target Enrichment (Double Capture)
The 3C-DpnII DNA was sheared by sonication to 200-300 bp as described in the ChIP-seq section. The sheared DNA was purified using 1.8X AMPure XP beads. 5 μg of sheared 3C-DpnII DNA was used in adaptor ligation for pre-capture library using NEBNext DNA Library Prep Master Mix Set for Illumina (NEB, Cat. E6040). Adaptors were from NEBNext Multiplex Oligos for Illumina (NEB, Cat. E7500). The indexed 3C DNA was amplified using a primer pair of P5 (5’-aatgatacggcgaccaccga) and P7 (5’-caagcagaagacggcatacgagat) and purified using 1.8X AMPure XP beads. To perform the first capture, SeqCap EZ Hybridization and Wash Kit (Roche, Cat. 05634261001) was used. For one library, the indexed sequencing library, 5 μg of a Cot-1 DNA equivalent chicken Hybloc DNA (Applied Genetics Lab, Cat. CHB), 1 μl of 1xGen Universal Blocking Oligo–TS-p5 (1 nmole/μl), and 1 μl of 1xGen Universal Blocking Oligo–TS-p7(6nt) (1 nmole/μl) were mixed well and dried completely in a low retention PCR tube (Axygen, Cat. PCR-02-L-C). The dried pellet was then resuspended in Nimblegen Hybridization Buffer and Component A, mixed with 2 μl of a capture probe pool (3 pmole in total), and incubated for hybridization at 47°C for 72 hours. After the first hybridization, biotinylated-probe-hybridized DNA (captured DNA) was mixed with 40 μl of prepared streptavidin magnetic beads (Invitrogen, Cat. 65001) and incubated at 47°C for 45 min. The mixture was further washed using the SeqCap Wash Kit. The captured DNA was then purified using 1.8X AMPure XP beads and eluted in final 44 μl of nuclease-free water and amplified using the P5/P7 primer pair for 12 cycles. This 1st-captured DNA was finally cleaned up with 1.8X AMPure XP beads and eluted in 25 μl of water. To perform the second capture, the 1st-captured DNA was mixed with components described in the first capture part and incubated for hybridization at 47°C for 24 hours. The double-captured DNA was then washed, pulled down by using the streptavidin beads, purified, amplified, and cleaned up as described before. The final 25 μl of double-captured DNA was submitted to USC MGC.
Sequencing
Libraries were sequenced on the NextSeq500 platform using the paired-end 75-bp protocol (Illumina).
Chromatin Conformation Capture PCR (3C-PCR)
The protocol was adapted from the publication (Naumova et al., 2012).
Single Cell Preparation and Crosslinking
Desired chicken skin regions were freshly dissected, washed, and dissociated as described in the Biological Specimen Collection and Cell Preparation section. For one 3C library, 1x107-1x108 dissociated cells were suspended in 45 ml of the warm chicken cell culture medium (DMEM with 10% FBS, 2% chicken serum, and 1% P/S) and cross-linked by the addition of 1.25 ml of 37% formaldehyde and tumbled at RT for 10 min. The reaction was quenched with the addition of 2.5 ml of 2.5M ice-cold glycine at RT for 15 min and cells were pelleted at 800 x g at 4°C for 10 min. The supernatant was removed completely. Cell Lysis. Cross-linked cells were lysed in 2 ml of cold Lysis Buffer (10 mM Tris-HCl pH=7.5, 10 mM NaCl, 5 mM MgCl2, 0.1 mM EGTA, 1X protease inhibitor cocktail, 0.2% NP-40) on an end-to-end rocker at 4°C for 30 min. The lysate was transferred into two 1.5-ml tubes and two nuclear pellets were collected by centrifugation at 2,200 x g at 4°C for 5 min.
Digestion (Works for HindIII-HF, NEB # R3104S)
The two nuclear pellets were suspended in 1 ml of 1.2X restriction enzyme buffer (for HindIII-HF, use SmartCut Buffer from NEB Cat. B7204S) and distributed into twenty 1.5-ml tubes (50 μl per tube). An additional 312 μl of 1X restriction enzyme buffer and 38 μl of 1% SDS were added into each tube. (Please note: % of SDS depends on the samples and researchers have to test the best condition of their samples.) The solution was mixed well by pipetting, incubated at 65°C for exactly 10 min, and then snapped cool on ice for 3 min. 44 μl of 10% (v/v) Triton X-100 was added into each tube and mixed well by pipetting. For 1x107 cells from a specific chicken skin region (one sample but 20 tubes now), 80 μl of the chromatin solution was saved as undigested genomic DNA control (UNDctrl) at −20°C. 400 units of restriction enzyme HindIII-HF (NEB, Cat. R3104S) was added into each tube, mixed well by pipetting, and incubated on an end-to-end rocker at 37°C for 16 hours. After the digestion, 80 μl of the chromatin solution was saved as digested genomic DNA control (DIGctrl) at −20°C.
Check for Digestion Efficiency
To extract the control DNA, 10 μl of nuclease-free H2O, 6 μl of 5M NaCl, and 4 μl of Protease K (10 mg/ml) were added into each 80 μl of UNDctrl and DIGctrl tube, mixed and incubated at 65°C for two hours. 100 μl of phenol-chloroform was added and the solution were mixed vigorously for 2 min. An aqueous layer (around 100 μl) was then revealed by centrifugation at 12,000 r.p.m. for 5 min and carefully transferred into a new 1.5-ml tube containing 0.1X 3M NaOAc (pH=5.2) and 3X volume of 100% ethanol. The solution was mixed and kept at −80°C for 20 min. DNA was then participated by centrifugation at 12,000 r.p.m. at 4°C for 30 min, washed with 1 ml of 70% ethanol, and finally suspended in total 30 μl H2O. To examine the HindIII digestion efficiency, we ran qPCR using Un-/Digested genomic DNA samples (UNDctrl and DIG ctrl) with specific primer pairs designed spanning HindIII sites around selected H3K27ac regions on the Chr27 β-krt gene cluster. Also, we designed primer pairs, which are not spanning any HindIII cutting site, as internal control pairs (Int Ctrl). Please find sequences of the primer pairs in Table S3. For each qPCR reaction, we added: 4.5 μl of 1/60-diluted UNDctrl or DIGctrl DNA as template, 10 μM forward primer, 10 μM reverse primer, and 5 μl of 2X SYBR Green/ROX qPCR Master Mix (Thermo Scientific, Cat. K0223); parameters of qPCR were: 95°C 10 min start-up, followed by 40 cycles of 95°C 3 min / 60°C 30 sec, and finally followed by 95°C 15 min / 60°C 1 min / 95°C 15 min dissociation curve. Ct values were used to calculate digestion efficiencies (% efficiency=100-100/2^[(CtR-CtC) Digested-(CtR-CtC) Undigested)]. R=samples and C=internal controls. Digestion efficiencies should be above 80% but 60-70% is acceptable. Only the sites with over 60% digestion efficiency in both feather and scale tissues were used for 3C-PCR.
Ligation
After digesting for 16 hours, the reaction was stopped by adding 86 μl of 10% SDS, mixed, and incubated at 65°C for 30 min. The 20 tubes were then snapped cooled on ice for 3 min. The digested chromatin solution was transferred into a 15-ml tube containing 7.61 ml of Ligation Cocktail [745 μl of 10% Triton X-100, 745 μl of 10X T4 DNA Ligation Buffer (500 mM Tris-HCl pH=7.5, 100 mM MgCl2, 100 mM DTT), 80 μl of 10 mg/ml BSA, 80 μl of 100mM ATP, and 5,960 μl of H2O]. 20-30 units of T4 DNA ligase diluted in 1X T4 Ligation Buffer were added into each 15-ml tubes and the tube was incubated at 16°C for 4 hours and then at RT for one extra hour. After ligation, 30 μl of 10 mg/ml Protease K was added and the solution was incubated at 65°C for 16 hours.
DNA Purification
30 μl of 10 mg/ml RNase A was added and the solution was incubated at 37°C for 45 min. 7 ml of phenol-chloroform was added and the solution was mixed vigorously. An aqueous layer (around 8 ml) was revealed by centrifugation at 2,200 x g at RT for 15 min and carefully transferred into a new 50-ml tube containing 7 ml of nuclease-free H2O, 1.5 ml of 3M NaOAc (pH=5.2), and 35 ml of 100% ethanol. The solution was mixed well, distributed into two ultracentrifuge tubes (50 ml per tube), and kept at −80°C for one hour. DNA was pelleted by centrifugation at 12,000 r.p.m. at 4°C for 30 min. DNA pellets from different tubes were combined, washed with 20 ml of 70% ethanol, and finally resolved in total 600 μl of 10 mM Tris-HCl (pH=7.4). The DNA concentration was measured.
PCR
We prepared each PCR reaction as the following: 10 mg of template (purified DNA after HindIII digestion and ligation), 2.5 μl of DMSO (final conc. 5%), 0.5 μl of 10 mM bait forward primer, 0.5 μl of 10 mM target forward primer, 25 μl of 2X PCR Master Green Mix, and nuclear-free H2O until final volume to be 50 μl. The parameters of PCR were: 95°C 15 min start-up, followed by 35 cycles of 95°C 30 sec / 60°C 30 sec / 72°C 5 min, followed by 72°C 5 min extension, and finally stored at 4°C. 25 μl of the PCR product was used for electrophoresis. PCR fragments with accurate sizes of chromatin interactions were collected from the gel, cloned (Qiagen, #231124), and sequenced.
Experimental Controls
There were key control sets for 3C-PCR experiments (Davies et al., 2016): (1) Primer efficiency– to normalize different primer efficiencies, a control library was made from 4 chicken BAC clones, clone #CH261-64J24, # CH261-154N1, # CH261-61E3, and # CH261-160M5 (the BACPAC Resources Center at BACPAC Genomics, Inc.). (2) Internal primers were used to normalize for amount of template added in each qPCR reaction. Primers are designed in undigested regions. (3) Digestion efficiency check–restriction sites are cut thoroughly by comparing aliquots of undigested and digested 3C template. (4) Sample purity–to ensure amplicons are not artifacts amplified by a dirty 3C template, serial dilutions of the template are set up and 3C performed using bait/target primer sets to see if Ct values decrease in accordance to dilution factor.
Assay for Transposase-Accessible Chromatin using sequencing (ATAC-seq)
The protocol was adapted from Dr. Howard Chang’s laboratory (Buenrostro et al., 2013). Each group had two biological replicates. All procedures were done on ice exclude those with specific notes.
Tn5 Transposition
RCAS-infected skins were dissected and cut into small (1mm x 1mm) pieces. Tissues were washed in iced-cold 1X PBS once and lysed in iced-cold 400 μl of ATAC lysis buffer (10 mM Tris-HCl pH=7.4, 10 mM NaCl, 3 mM MgCl2, and freshly added 0.1% IGEPAL CA-630) for 30 min on an end-to-end rocker at 4°C. The clear lysate was carefully transferred to a new pre-chilled tube and nuclei were pelleted by 500 x g at 4°C for 10 min. For each replicate, ~1x104 cells were used. We estimated cell numbers from calculating genomic DNA contents of the lysate. Chromatin was transposed by adding 2.5 μl of Tn5 transposase in total 50 μl of 1X TD buffer (Illumina, Cat. FC-121-1030) at 37°C for 30 min. The transposed DNA was purified using a MinElute PCR Purification Kit (QIAGEN, Cat. 28004).
Library Preparation
The transposed DNA fragments were amplified for 5 cycles using NEBNext High-Fidelity 2X PCR Mater Mix (NEB, Cat. M0541S). To obtain numbers of additional amplification cycles priorto saturation, 5 μl out of the 50 μl PCR product was used in real-time qPCR and a number of cycle that was corresponded to ¼ of maximum fluorescent intensity was used for the rest 45 μl of the PCR sample. After library amplification, 1X AMPure XP bead (Beckman, A63880) clean-up was performed for size selection and removal of adapter-dimers.
Sequencing
After AMPure XP bead clean-up, cDNA libraries were submitted to USC Molecular Genomic Core (MGC) and libraries with the standard ATAC-seq ladders determined by using Bioanalyzer DNA 1000 Kit (Agilent) were sequenced on the NextSeq500 platform using the paired-end 40-bp protocol (Illumina).
Bioinformatic Analysis
The RNA-seq and ChIP-seq analysis was conducted using Chuong lab local-installed Galaxy platform (Afgan et al., 2018; Blankenberg et al., 2014, 2010b), version 18.5. Default parameters were used exclude those with specific notes. In general, we used FastQC (Dr. Simon Andrews at Babraham Institute) for pre-alignment QC and Qualimap 2 (Okonechnikov et al., 2016) for post-alignment QC. To calculate correlation between replicates, we used DeepTools (Ramírez et al., 2014) multiBigwigSummary and plotCorrelation. We summarized QC results in Table S4 and bioinformatic analysis pipelines including softwares for manuscript preparation in Table S7.
RNA-seq Analysis
The RNA-seq analysis pipeline was adapted according to the publication (Trapnell et al., 2012).
Manipulation
Tools—fastq_groomer (Blankenberg et al., 2010a) and fastq_trimmer_by_quality (window size 3; min score >= 20)—were used.
Mapping
Tool—TopHat2 (Kim et al., 2013)—was used
Quantification and Differentially Expressed Gene Analysis
Tools—Cufflinks package (Trapnell et al., 2010)–including Cufflinks, Cuffmerge, Cuffquant, and Cuffdiff—were used. Deeptools (Ramírez et al., 2014) bamCoverage was used to normalize the reads to 1X sequencing depth and generate bigWig files of read coverages.
Gene Ontology Analysis
The pathway enrichment analysis was generated through the use of Ingenuity Pathway Analysis (IPA, QIAGEN Inc.) and Partek Genomics Suite software, version 6.6, build 6.14.0514 (Partek Inc.).
ChIP-seq Analysis
Manipulation
Tools—fastq_groomer, fastq_quality_filter, fastq_trimmer_by_quality (window size 3; min score >= 20)—were used.
Mapping
Tool—bowtie2 (Langmead and Salzberg, 2012) (Genome: UCSC galGal4, –very-sensitive)—was used.
Filtering
Tools—samtool_filter2 (Li et al., 2009) (-q 1), samtools_sort, samtools_rmdup (BAM is single-end)—were used.
Peak-Calling
Tools—MACS2 (Feng et al., 2012; Zhang et al., 2008) (for H3K27ac-ChIP, –gsize 1065365425 –nomodel –extsize 147 –bdg –broad –broad-cutoff 0.1; for CTCF/KLF-ChIP, –gsize 1065365425 –nomodel –extsize value obtained from macs2_predictd tool) and macs2_bdgcmp (-m FE and -m logLR -p 0.00001)—were used. To normalize MACS2 results from different libraries for comparison, we followed a “Build Signal Track” instruction from https://github.com/taoliu/MACS/wiki/Build-Signal-Track#Run_MACS2_bdgcmp_to_generate_foldenrichment_and_logLR_track.
Convert Format
Tool—wig_to_bigWig—was used.
Typical-/Super-Enhancer Analysis
To find typical and super-enhancers, HOMER (Heinz et al., 2010) findPeak command was used with options of -style super and -typical TE. To validate sample duplication quality, please see a PCA plot of H3K27ac of E14 feather- and scale-bearing skins in Figure S1E.
NG Capture-C Analysis
Double Capture-C results were analyzed according to the publication (Davies et al., 2016). The analysis pipeline was well documented and can be downloaded from “https://github.com/Hughes-Genome-Group/captureC/releases/download/VS2.0/UserManualforCaptureCanalysis.pdf”. In brief, 1) download the CaptureC analyser package including CCanalyser3.pl, dpngenome3_1.pl, dpnII2E.pl from “https://github.com/Hughes-Genome-Group/captureC/releases“, 2) generate in silico DpnII digested chicken genome ver. Ensemble galGal4 Release 72 with alpha and beta keratins, 3) perform adapter trimmer using the tool trim_galore (Dr. Felix Krueger at the Babraham Institute), 4) Merge overlapping reads by using the tool FLASH (Magoč and Salzberg, 2011), 5) in silico DpnII digest the FLASH-merged reads; 6) install Bowtie 1.2 (Langmead et al., 2009), 7) build up bowtie index using the original genome used in the Step 2, 8) align digested FLASH-merged reads (output of Step 5) using Bowtie1.2, and 9) run CCanalyser3.pl. To validate NG Capture-C quality, we performed Principle Component Analysis (PCA) and checked the Spearman correlation of the duplicates (data not show) and confirmed that the captured reads were over the 10,000 fragments threshold, indicating the results were reliable (Table S4).
ATAC-seq Analysis
Manipulation
Tools—fastq_groomer, fastq_quality_filter, fastq_trimmer_by_quality (window size 3; min score >= 20)—were used.
Mapping
Tool—bowtie2 (–very-sensitive -maxins 2000)—was used.
Peak-calling
Tools—Partek Genomic Suite (maximum average fragment size 200; window size 200; peak cut-off FDR <0.001)—were used.
Differential Transposase Accessible Region (DAR) Detection
Tool — Partek Genomic Suite—was used. We first obtained consistent peaks between two replicates of the same group. And then found differential peaks by comparing consistent peaks between groups. For example: consistent peaks of RCAS-Sox2 replicates versus consistent peaks of RCAS-GFP repilicates. Thus, we obtained DARs of RCAS-Sox2, DARs of RCAS-GFP, and their common peaks.
Motif Prediction
Tools—HOMER (Heinz et al., 2010) findMotifsGenome command, TRANSFAC (Matys et al., 2006) and MATCH (Kel et al., 2003)—were used.
Graph/Display Data
Bigwig files were uploaded onto the UCSC Genome Browser (Kent et al., 2002), served as input files for pyGenomeTracks (Ramírez et al., 2018) and Integrative Genomics Viewer (IGV) (Robinson et al., 2011) to generate tracks for visualizing. Barcharts and line plots were generated by using GraphPad Prism version 7.0a for Mac OS X (GraphPad Software Inc., San Diego, United States of America).
Construction of RCAS-SATB2 Wild-Type, RCAS-Zic1 Wild-Type and Dominant-Negative Forms
For cloning of Zic1, PCR was performed by using primer set (forward, 5’-ggggacaagtttgtacaaaaaagcaggcttcaccatgcttctggatgctggaccgca; backward, 5’-ggggaccactttgtacaagaaagctgggtcttacacgtaccattcgttaaaat) to generate RCAS-Zic1 wild-type form. Another set of primers (forward, 5’-ggggacaagtttgtacaaaaaagcaggcttcaccatggcgggggccttcttccg; backward, 5’- ggggaccactttgtacaagaaagctgggtcttacacgtaccattcgttaaaat) was used to generate Zic1 dominant-negative form, which is a truncated Zic1 missing N-termimal (Nakata et al., 2000). For cloning of SATB2, PCR was performed by using primer set (forward, 5’- ggggacaagtttgtacaaaaaagcaggcttcaccatggagcggaggagcgaga; backward, 5’- ggggaccactttgtacaagaaagctgggtcctatctctggtcgatgtctgc) to generate RCAS-SATB2 wild-type form. cDNA from E9 embryonic chicken skin was used as template. These forms were cloned into RCAS using Gateway system (Loftus et al., 2001). Virus was made according to Jiang et al. (Jiang et al., 1998) and concentrated by ultracentrifugation. The constitutive active form of Zic1, RCAS-Zic1-deltaC (ΔC), was from (Wu et al., 2018b).
Gene Misexpression
For viral mediated functional studies, 4-10 μl of concentrated RCAS viruses was injected to the amniotic cavity at E3 (stage 18) and samples were collected at E14 (stage 40)-E15 (stage 41) or E16 (stage 42).
QUANTIFICATION AND STATISTICAL ANALYSIS
For RNA-seq, ChIP-seq, NG Capture-C, and ATAC-seq, two biological replicates were used except H3K4me3-ChIP. For ChIP-qPCR, two biological replicates for each ChIP group and two technical replicates for qPCR were used. Each biological replicate for omic studies combined specific skin regions from at least 6 chicken embryos. Spearman’s correlation coefficient (Figures S2J and S2K) was used to calculate the correlation between replicates. ρ (rho or as rs) = 0.6-0.79 represents a strong correlation. For immunohistochemistry, immunofluorescence, and in-situ hybridization, at least three biological replicates were used. ChIP-qPCR data (Figures 2H and 2I) were normalized by the percent input method [% of Input = 100x2(ΔCt)]. Delta Ct (ΔCt) = Ct (Adjusted input) – Ct (Test Sample). Adjusted Input (100%) = Ct of 2% Input-5.64. For quantification of NG Capture-C (Figures S3F and S3K), normalized read counts were calculated from: captured reads with their genomic locations reported in gff files were normalized by their capture-containing reads entering the analysis listed in statistics reports from CCanalyser3.pl (CC3) outputs. Mean values of normalized read counts from two replicates were used to plot the final charts. Statistical analysis and graphical representations were generated by using GraphPad Prism version 7.0a for Mac OS X (GraphPad Software Inc.). To obtain consistent and differential ChIP-seq peaks from replicates (Figure 5), Partek Genomics Suite software (Partek Inc.) with peak cut-off FDR 0.001 was used. To generate hierarchical clustering of differentially expressed genes (DEG) for Figures 6D and 6G, z-scores were calculated from [RPKM-(mean RPKM)/Standard Deviation]. The DEGs were further clustered based on similarities of their expression patterns (Chen et al., 2016). In MACS2 callpeak and Cufflinks analysis, minimum False Discovery Rate (FDR/q-value) cutoff for peak detection was 0.05. All tests of significance were defined as p-value < 0.05.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Histone H3 (acetyl K27) antibody - ChIP Grade | Abcam plc. | Cat#ab4729 |
| Anti-Histone H3K4me1 antibody - ChIP Grade | Abcam plc. | Cat#ab8895 |
| Anti-Histone H3K4me3 antibody - ChIP Grade | Abcam plc. | Cat#ab8580 |
| CTCF Antibody - N-terminal region | Aviva Systems Biology Corp. | Cat#ARP38820_P050 |
| Normal Rabbit IgG | Cell Signaling Technology, Inc. | Cat#2729 |
| Anti-IgG antibody | Santa Cruz Biotechnology | Cat#sc2712 |
| GKLF/EKLF/LKLF Antibody (F-8): | Santa Cruz Biotechnology | Cat#sc-166238X |
| Rabbit anti-SATB1/2 antibody | Proteintech Group, Inc. | Cat#15400-1-AP |
| Bacterial and Virus Strains | ||
| RCAS-Zic1 Wild-type | This paper | N/A |
| RCAS-Zic1 Dominant-negative | This paper | N/A |
| RCAS-SATB2 Wild-type | This paper | N/A |
| RCAS-Zic1 -deltaC | Wu et al., 2018b | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| TRIzol RNA Isolation Reagents | Invitrogen (Thermo Fisher Scientific Inc.) | Cat#15596026 |
| AMPure XP beads | Beckman Coulter, Inc. | Cat#A63880 |
| Collagenase Type 1 | Worthington Biochemical Corp. | Cat#LS004196 |
| Dynabeads Protein A | Invitrogen (Thermo Fisher Scientific Inc.) | Cat#100-02D |
| Protein G | Invitrogen (Thermo Fisher Scientific Inc.) | Cat#100-04D |
| DpnII destruction enzyme | New England Biolabs | Cat#R0543 |
| Streptavidin magnetic beads | Invitrogen (Thermo Fisher Scientific Inc.) | Cat#65001 |
| TDE1 Tagment DNA Enzyme | Illumina, Inc. | Cat#15027865 |
| TD Tagment DNA Buffer | Illumina, Inc. | Cat#15027866 |
| NEBNext High-Fidelity 2X PCR Mater Mix | New England Biolabs | Cat# M0541S |
| Critical Commercial Assays | ||
| TruSeq RNA Prep Kit v2 | Illumina, Inc. | Cat#RS-122-2001 Cat#RS-122-2002 |
| NEBNext Ultra End Repair/dA-Tailing Module | New England Biolabs | Cat#E7442S |
| NEBNext Ultra Ligation Module | New England Biolabs | Cat#E7445S |
| KAPA HiFi HotStart ReadyMix | Kapa Biosystems, Inc. | Cat#KK2601 |
| SimpleChIP Enzymatic Chromatin IP Kit Agarose Beads | Cell Signaling Technology, Inc. | Cat#9002 |
| SimpleChIP Universal qPCR Master Mix | Cell Signaling Technology, Inc. | Cat#88989 |
| NEBNext DNA Library Prep Master Mix Set for Illumina | New England Biolabs | Cat#E6040 |
| NEBNext Multiplex Oligos for Illumina | New England Biolabs | Cat#E7500 |
| SeqCap EZ Hybridization and Wash Kit | F. Hoffmann-La Roche Ltd | Cat#05634261001 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO: GSE136224 |
| Chicken UCSC Genome Browser assembly ID: galGal4 (GG4) | Genomic Institute, University of California Santa Cruz | http://hgdownload.soe.ucsc.edu/goldenPath/galGal4/bigZips/galGal4.fa.gz |
| Experimental Models: Organisms/Strains | ||
| Gallus gallus (chicken): white leghorn strain | Charles River Laboratories | SPAFAS |
| Oligonucleotides | ||
| Cot-1 DNA equivalent chicken Hybloc DNA | Applied Genetics Lab | Cat. #CHB |
| 1xGen Universal Blocking Oligo—TS-p5 | IDT | N/A (contact IDT) |
| 1xGen Universal Blocking Oligo—TS-p7(6nt) | IDT | N/A (contact IDT) |
| ISH Probe: Chr25-FK3 FW 5’-aggaagcccaaagtctgaca-3’ |
This paper | N/A |
| ISH Probe: Chr25-FK3 RV 5’-tctggtacaggttgcaatgg-3’ |
This paper | N/A |
| ISH Probe: Chr25-FK12 FW 5’-tgaggtggacatcctgtgaa-3’ |
This paper | N/A |
| ISH Probe: Chr25-FK12 RV 5’-acaatgggatgcctgacttc-3’ |
This paper | N/A |
| ISH Probe: Chr27-FK1 FW 5’-cccggagaaattcatccata-3’ |
This paper | N/A |
| ISH Probe: Chr27-FK1 RV 5’-ccaagtctgagctcatgcaa-3’ |
This paper | N/A |
| ISH Probe: Chr27-FK12 FW 5’-ggtgctggatgaagaaggaa-3’ |
This paper | N/A |
| ISH Probe: Chr27-FK12 RV 5’-aggctccacagtgcaagatt-3’ |
This paper | N/A |
| ISH Probe: Chr27-FK39 FW 5’-ctggacggcagaacagactc-3’ |
This paper | N/A |
| ISH Probe: Chr27-FK39 RV 5’-agagagcagcaggagcagag-3’ |
This paper | N/A |
| ISH Probe: Chr25-FK45 FW 5’-attgacagacctgcagtgga-3’ |
This paper | N/A |
| ISH Probe: Chr25-FK45 RV 5’-cagctgtccatctgcctttt-3’ |
This paper | N/A |
| Primer: P5 5’-aatgatacggcgaccaccga-3’ |
This paper | N/A |
| Primer: P7 5’-caagcagaagacggcatacgagat-3’ |
This paper | N/A |
| Primer: RCAS-Zic1 wild-type form FW 5’-ggggacaagtttgtacaaaaaagcaggcttcaccatgcttctggatgctggaccgca-3’ |
This paper | N/A |
| Primer: RCAS-Zic1 wild-type form RV 5’-ggggaccactttgtacaagaaagctgggtcttacacgtaccattcgttaaaat-3’ |
This paper | N/A |
| Primer: RCAS-Zic1 dominant-negative form FW5’-ggggacaagtttgtacaaaaaagcaggcttcaccatggcgggggccttcttccg-3’ |
This paper | N/A |
| Primer: RCAS-Zic1 dominant-negative form RV 5’-ggggaccactttgtacaagaaagctgggtcttacacgtaccattcgttaaaat-3’ |
This paper | N/A |
| Primer: RCAS-SATB2 FW 5’-ggggacaagtttgtacaaaaaagcaggcttcaccatggagcggaggagcgaga-3’ |
This paper | N/A |
| Primer: RCAS-SATB2 RV 5’-ggggaccactttgtacaagaaagctgggtcctatctctggtcgatgtctgc-3’ |
This paper | N/A |
| Probes for NG Capture-C | This paper | Table S3 |
| Primers for 3C-PCR | This paper | Table S3 |
| Primers for ChIP-qPCR | This paper | Table S3 |
| Recombinant DNA | ||
| Chicken BAC clone: galgal4 chr27:421,977-608,328 | BACPAC Resources Center, BACPAC Genomics, Inc. | CH261-64J24 |
| Chicken BAC clone: galgal4 chr27:567,425-727,740 | BACPAC Resources Center, BACPAC Genomics, Inc. | CH261-154N1 |
| Chicken BAC clone: galgal4 chr27:695,282-892,074 | BACPAC Resources Center, BACPAC Genomics, Inc. | CH261-61E3 |
| Chicken BAC clone: galgal4 chr27:864,805-1,067,678 | BACPAC Resources Center, BACPAC Genomics, Inc. | CH261-160M5 |
| Chicken BAC clone: galgal4 chr25:942,403-1,159,746 | BACPAC Resources Center, BACPAC Genomics, Inc. | CH261-170A15 |
| Chicken BAC clone: galgal4 chr25:835,251-960,765 | BACPAC Resources Center, BACPAC Genomics, Inc. | CH261-27I14 |
| Chicken BAC clone: galgal4 chr25:780,190-942,274 | BACPAC Resources Center, BACPAC Genomics, Inc. | CH261-178I11 |
| Software and Algorithms | ||
| TopHat2 | Kim et al., 2013 | http://ccb.jhu.edu/software/tophat/downloads/ |
| Cufflinks package | Trapnell et al., 2010 | http://cole-trapnell-lab.github.io/cufflinks/install/ |
| Deeptools | Ramírez et al., 2014 | https://deeptools.readthedocs.io/en/develop/content/installation.html |
| Ingenuity Pathway Analysis (IPA) | QIAGEN Inc. | https://analysis.ingenuity.com/pa/installer/select |
| Partek Genomics Suite (PGS) | Partek Inc. | https://documentation.partek.com/display/PGS/Node+Locked+Installation |
| Bowtie 1.2 | Langmead et al., 2009 | https://sourceforge.net/projects/bowtie-bio/files/bowtie/1.2.0/ |
| Bowtie 2 | Langmead and Salzberg, 2012 | https://sourceforge.net/projects/bowtie-bio/files/bowtie2/2.3.5.1/ |
| Samtools | Li et al., 2009 | http://www.htslib.org/download/ |
| MACS2 | Feng et al., 2012; Zhang et al., 2008 | https://github.com/taoliu/MACS/wiki/Install-macs2 |
| HOMER | Heinz et al., 2010 | http://homer.ucsd.edu/homer/download.html |
| CaptureC analyser package: CCanalyser3.pl dpngenome3_1.pl dpnII2E.pl |
Davies et al., 2016 |
https://github.com/Hughes-Genome-Group/captureC/releases |
| Trim Galore! | Dr. Felix Krueger at the Babraham Institute | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ |
| FLASH (Fast Length Adjustment of SHort reads) | Magoc and Salzberg, 2011 | http://ccb.jhu.edu/software/FLASH/ |
| MATCH: a weight matrix-based program for predicting transcription factor binding sites | Kel et al., 2003 | http://gene-regulation.com/pub/programs.html |
| pyGenomeTracks | Ramírez et al., 2018 | https://github.com/deeptools/pyGenomeTracks |
| Integrative Genomics Viewer (IGV) | Robinson et al., 2011 | https://software.broadinstitute.org/software/igv/download |
| GraphPad Prism | GraphPad Software Inc. | https://www.graphpad.com/scientific-software/prism/ |
| OmniGraffle | The Omni Group | https://www.omnigroup.com/omnigraffle |
| RNA-seq analysis pipeline | This paper | Table S7 |
| ChIP-seq analysis pipeline | This paper | Table S7 |
| Software for manuscript preparation | This paper | Table S7 |
| Other | ||
| Tissue grind pestle | Kimble Chase Life Science and Research Products, LLC. | Cat#885302-0002 |
Highlights.
Two epigenetic modes control two β-keratin gene clusters during skin specification
One enhancer co-activates all subclustered Keratin genes on the Chr25 cluster
3D chromatin looping mediates differential Keratin expression of the Chr27 cluster
3-factor model explains how to establish region-specific chromatin configuration
ACKNOWLEDGMENTS
This work is supported by NIH NIAMS Arthritis and Musculoskeletal and Skin Diseases (AR47364 and AR60306) and Institute of General Medical Sciences (GM125322). The work is also supported by a contract from China Medical University and Hospital in Taiwan (CMU) (USC grant number 5351285884). Y.-C. Liang and C.-K.C. are supported by Postdoctoral Research Abroad Program, Ministry of Science and Technology (MOST), Taiwan (102-2917-I-564-002-A1 and 107-2917-I-564-024). S.T. is supported by NIH T90 (5T90DE021982), USC Craniofacial Biology Ph.D. Program, Study Abroad Scholarship (1040120022) MOST. We thank USC Molecular Genomic Core for conducting next-generation sequencing and the USC Norris Medical Library Bioinformatics Service for consultation. Confocal microscopy was performed by the Cell and Tissue Imaging Core of the USC Research Center for Liver Diseases, NIH grant no. P30 DK048522. We thank all colleagues from the Chuong laboratory for discussion. We thank CMU Integrative Stem Cell Center Director Dr. Shih-Chieh Hung for support and colleagues in CMU for discussion. We appreciate Drs. Vlad Botchkarev, Howard Chang, and Ruchi Bajpai for inspiring discussions. We are grateful to Dr. Elena Ezhkova and Dr. Terumi Kohwi-Shigematsu for critical reading of the manuscript.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.devcel.2020.05.007.
DECLARATION OF INTERESTS
Cheng-Ming Chuong is a paid science advisor of the China Medical University in Taiwan.
REFERENCES
- Afgan E, Baker D, Batut B, van den Beek M, Bouvier D, Cech M, Chilton J, Clements D, Coraor N, Gmning BA, et al. (2018). The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 46, W537–W544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson R, and Sandelin A (2020). Determinants of enhancer and promoter activities of regulatory elements. Nat. Rev. Genet 21, 71–87. [DOI] [PubMed] [Google Scholar]
- Andrey G, Montavon T, Mascrez B, Gonzalez F, Noordermeer D, Leleu M, Trono D, Spitz F, and Duboule D (2013). A switch between topological domains underlies HoxD genes collinearity in mouse limbs. Science 340, 1234167. [DOI] [PubMed] [Google Scholar]
- Bajpai R, Chen DA, Rada-Iglesias A, Zhang J, Xiong Y, Helms J, Chang CP, Zhao Y, Swigut T, and Wysocka J (2010). CHD7 cooperates with PBAFto control multipotent neural crest formation. Nature 463, 958–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergström R, Savary K, Morén A, Guibert S, Heldin CH, Ohlsson R, and Moustakas A (2010). Transforming growth factor beta promotes complexes between Smad proteins and the CCCTC-binding factor on the H19 imprinting control region chromatin. J. Biol. Chem 285, 19727–19737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlivet S, Paquette D, Dumouchel A, Langlais D, Dostie J, and Kmita M (2013). Clustering of tissue-specific sub-TADs accompanies the regulation of HoxA genes in developing limbs. PLoS Genet. 9, e1004018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biddie SC, John S, Sabo PJ, Thurman RE, Johnson TA, Schiltz RL, Miranda TB, Sung MH, Trump S, Lightman SL, et al. (2011). Transcription factor AP1 potentiates chromatin accessibility and glucocorticoid receptor binding. Mol. Cell 43, 145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenberg D, Gordon A, Von Kuster G, Coraor N, Taylor J, and Nekrutenko A; Galaxy Team (2010a). Manipulation of FASTQ data with Galaxy. Bioinformatics 26, 1783–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenberg D, Von Kuster G, Bouvier E, Baker D, Afgan E, Stoler N, Galaxy Team, Taylor J, and Nekrutenko A (2014). Dissemination of scientific software with Galaxy ToolShed. Genome Biol. 15, 403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenberg D, Von Kuster G, Coraor N, Ananda G, Lazarus R, Mangan M, Nekrutenko A, and Taylor J (2010b). Galaxy: a web-based genome analysis tool for experimentalists. Curr Protoc Mol Biol. 10, 11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botchkarev VA, Gdula MR, Mardaryev AN, Sharov AA, and Fessing MY (2012). Epigenetic regulation of gene expression in keratinocytes. J. Invest. Dermatol 132, 2505–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buschke S, Stark HJ, Cerezo A, Prätzel-Wunder S, Boehnke K, Kollar J, Langbein L, Heldin CH, and Boukamp P (2011). A decisive function of transforming growth factor-β/Smad signaling in tissue morphogenesis and differentiation of human HaCaT keratinocytes. Mol. Biol. Cell 22, 782–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai S, Han HJ, and Kohwi-Shigematsu T (2003). Tissue-specific nuclear architecture and gene expression regulated by SATB1. Nat. Genet 34, 42–51. [DOI] [PubMed] [Google Scholar]
- Chang WL, Wu H, Chiu YK, Wang S, Jiang TX, Luo ZL, Lin YC, Li A, Hsu JT, Huang HL, et al. (2019). The making of a flight feather: bio-architectural principles and adaptation. Cell 179, 1409–1423. e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CF, Foley J, Tang PC, Li A, Jiang TX, Wu P, Widelitz RB, and Chuong CM (2015). Development, regeneration, and evolution of feathers. Annu. Rev. Anim. Biosci 3, 169–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CK, Ng CS, Wu SM, Chen JJ, Cheng PL, Wu P, Lu MY, Chen DR, Chuong CM, Cheng HC, et al. (2016). Regulatory differences in natal down development between altricial zebra finch and precocial chicken. Mol. Biol. Evol 33, 2030–2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuong CM, Randall VA, Widelitz RB, Wu P, and Jiang TX (2012). Physiological regeneration of skin appendages and implications for regenerative medicine. Physiol. (Bethesda) 27, 61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuong CM, Yeh CY, Jiang TX, and Widelitz R (2013). Module-based complexity formation: periodic patterning in feathers and hairs. Wiley Interdiscip. Rev. Dev. Biol 2, 97–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies JO, Oudelaar AM, Higgs DR, and Hughes JR (2017). How best to identify chromosomal interactions: a comparison of approaches. Nat. Methods 14, 125–134. [DOI] [PubMed] [Google Scholar]
- Davies JO, Telenius JM, McGowan SJ, Roberts NA, Taylor S, Higgs DR, and Hughes JR (2016). Multiplexed analysis of chromo some conformation at vastly improved sensitivity. Nat. Methods 13, 74–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean A (2006). On a chromosome far, far away: LCRs and gene expression. Trends Genet. 22, 38–45. [DOI] [PubMed] [Google Scholar]
- Dhouailly D (1975). Formation of cutaneous appendages in dermo-epidermal recombinations between reptiles, birds and mammals. Wilehm Roux Arch. Dev. Biol 177, 323–340. [DOI] [PubMed] [Google Scholar]
- Dhouailly D, Godefroit P, Martin T, Nonchev S, Caraguel F, and Oftedal O (2019). Getting to the root of scales, feather and hair: as deep as Odontodes? Exp. Dermatol 28, 503–508. [DOI] [PubMed] [Google Scholar]
- Di Giammartino DC, Kloetgen A, Polyzos A, Liu Y, Kim D, Murphy D, Abuhashem A, Cavaliere P, Aronson B, Shah V, et al. (2019). KLF4 is involved in the organization and regulation of pluripotency-associated three-dimensional enhancer networks. Nat. Cell Biol 21, 1179–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, and Ren B (2012). Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Liu T, Qin B, Zhang Y, and Liu XS (2012). Identifying ChIP-seq enrichment using MACS. Nat. Protoc 7, 1728–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraiuolo MA, Rousseau M, Miyamoto C, Shenker S, Wang XQ, Nadler M, Blanchette M, and Dostie J (2010). The three-dimensional architecture of Hox cluster silencing. Nucleic Acids Res. 38, 7472–7484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fessing MY, Mardaryev AN, Gdula MR, Sharov AA, Sharova TY, Rapisarda V, Gordon KB, Smorodchenko AD, Poterlowicz K, Ferone G, et al. (2011). p63 regulates Satb1 to control tissue-specific chromatin re-modeling during development of the epidermis. J. Cell Biol 194, 825–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs E, and Cleveland DW (1998). A structural scaffolding of intermediate filaments in health and disease. Science 279, 514–519. [DOI] [PubMed] [Google Scholar]
- Greenwold MJ, Bao W, Jarvis ED, Hu H, Li C, Gilbert MT, Zhang G, and Sawyer RH (2014). Dynamic evolution of the alpha (α) and beta (β) keratins has accompanied integument diversification and the adaptation of birds into novel lifestyles. BMC Evol. Biol 14, 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwold MJ, and Sawyer RH (2010). Genomic organization and molecular phylogenies of the beta (beta) keratin multigene family in the chicken (Gallus gallus) and zebra finch (Taeniopygia guttata): implications for feather evolution. BMC Evol. Biol 10, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwold MJ, and Sawyer RH (2011). Linking the molecular evolution of avian beta (β) keratins to the evolution of feathers. J. Exp. Zool. B Mol. Dev. Evol 316, 609–616. [DOI] [PubMed] [Google Scholar]
- Guo Y, Monahan K, Wu H, Gertz J, Varley KE, Li W, Myers RM, Maniatis T, and Wu Q (2012). CTCF/cohesin-mediated DNA looping is required for protocadherin alpha promoter choice. Proc. Natl. Acad. Sci. USA 109, 21081–21086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanssen LLP, Kassouf MT, Oudelaar AM, Biggs D, Preece C, Downes DJ, Gosden M, Sharpe JA, Sloane-Stanley JA, Hughes JR, et al. (2017). Tissue-specific CTCF-cohesin-mediated chromatin architecture delimits enhancer interactions and function in vivo. Nat. Cell Biol 19, 952–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl M, and Bister K (1995). Specific activation in jun-transformed avian fibroblasts of a gene (bkj) related to the avian beta-keratin gene family. Proc. Natl. Acad. Sci. USA 92, 11731–11735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesse M, Zimek A, Weber K, and Magin TM (2004). Comprehensive analysis of keratin geneclusters in humans and rodents. Eur. J. Cell Biol 83, 19–26. [DOI] [PubMed] [Google Scholar]
- Holthaus KB, Eckhart L, Dalla Valle L, and Alibardi L (2018). Review: Evolution and diversification of corneous beta-proteins, the characteristic epidermal proteins of reptiles and birds. J. Exp. Zool. B Mol. Dev. Evol 330, 438–453. [DOI] [PubMed] [Google Scholar]
- Hughes JR, Roberts N, McGowan S, Hay D, Giannoulatou E, Lynch M, De Gobbi M, Taylor S, Gibbons R, and Higgs DR (2014). Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. Nat. Genet 46, 205–212. [DOI] [PubMed] [Google Scholar]
- Jerković I, Ibrahim DM, Andrey G, Haas S, Hansen P, Janetzki C, González Navarrete I, Robinson PN, Hecht J, and Mundlos S (2017). Genome-wide binding of posterior HOXA/D transcription factors reveals sub-grouping and association with CTCF. PLoS Genet. 13, e1006567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang TX, Stott NS, Widelitz RB, and Chuong CM (1998). Current methods in the study of avian skin appendages In Molecular Basis of Epithelial Appendage Morphogenesis, Chuong CM, ed. (Landes Bioscience; )), pp. 395–408. [Google Scholar]
- Kel AE, Gössling E, Reuter I, Cheremushkin E, Kel-Margoulis OV, and Wingender E (2003). MATCH: a tool for searching transcription factor binding sites in DNA sequences. Nucleic Acids Res. 31, 3576–3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, and Haussler D (2002). The human genome browser at UCSC. Genome Res. 12, 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp LW, Shames RB, Barnes GL, and Sawyer RH (1993). Region-specific patterns of beta keratin expression during avian skin development. Dev. Dyn 196, 283–290. [DOI] [PubMed] [Google Scholar]
- Kowata K, Nakaoka M, Nishio K, Fukao A, Satoh A, Ogoshi M, Takahashi S, Tsudzuki M, and Takeuchi S (2014). Identification of a feather beta-keratin gene exclusively expressed in pennaceous barbule cells of contour feathers in chicken. Gene 542, 23–28. [DOI] [PubMed] [Google Scholar]
- Lai YC, and Chuong CM (2016). The “tao” of integuments. Science 354, 1533–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The sequence alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loftus SK, Larson DM, Watkins-Chow D, Church DM, and Pavan WJ (2001). Generation of RCAS vectors useful for functional genomic analyses. DNA Res. 8, 221–226. [DOI] [PubMed] [Google Scholar]
- Lowe CB, Clarke JA, Baker AJ, Haussler D, and Edwards SV (2015). Feather development genes and associated regulatory innovation predate the origin of Dinosauria. Mol. Biol. Evol 32, 23–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu CP, Polak L, Keyes BE, and Fuchs E (2016). Spatiotemporal antagonism in mesenchymal-epithelial signaling in sweat versus hair fate decision. Science 354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeira F, Park YM, Lee J, Buso N, Gur T, Madhusoodanan N, Basutkar P, Tivey ARN, Potter SC, Finn RD, and Lopez R (2019). The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 47, W636–W641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magoč T, and Salzberg SL (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M, Hornischer K, et al. (2006). TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 34, D108–D110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan SJ, Hughes JR, Han ZP, and Taylor S (2013). MIG: multi-image genome viewer. Bioinformatics 29, 2477–2478. [DOI] [PubMed] [Google Scholar]
- Nakata K, Koyabu Y, Aruga J, and Mikoshiba K (2000). A novel member of the Xenopus Zic family, Zic5, mediates neural crest development. Mech. Dev 99, 83–91. [DOI] [PubMed] [Google Scholar]
- Naumova N, Smith EM, Zhan Y, and Dekker J (2012). Analysis of long-range chromatin interactions using chromosome conformation capture. Methods 58, 192–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng CS, Wu P, Fan WL, Yan J, Chen CK, Lai YT, Wu SM, Mao CT, Chen JJ, Lu MY, et al. (2014). Genomic organization, transcriptomic analysis, and functional characterization of avian alpha- and beta-keratins in diverse feather forms. Genome Biol. Evol 6, 2258–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols MH, and Corces VG (2015). A CTCF code for 3D genome architecture. Cell 162, 703–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh IY, Albea DM, Goodwin ZA, Quiggle AM, Baker BP, Guggisberg AM, Geahlen JH, Kroner GM, and de Guzman Strong C (2014). Regulation of the dynamic chromatin architecture of the epidermal differentiation complex is mediated by a c-Jun/AP-1-modulated enhancer. J. Invest. Dermatol 134, 2371–2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okonechnikov K, Conesa A, and García-Alcalde F (2016). Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 32, 292–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phanstiel DH, Van Bortle K, Spacek D, Hess GT, Shamim MS, Machol i., Love MI, Aiden EL, Bassik MC, and Snyder MP (2017). Static and dynamic DNA loops form AP-1-bound activation hubs during macrophage development. Mol. Cell 67, 1037–1048. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poterlowicz K, Yarker JL, Malashchuk I, Lajoie BR, Mardaryev AN, Gdula MR, Sharov AA, Kohwi-Shigematsu T, Botchkarev VA, and Fessing MY (2017). 5C analysis of the epidermal differentiation complex locus reveals distinct chromatin interaction networks between gene-rich and gene-poor TADs in skin epithelial cells. PLoS Genet. 13, e1006966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presland RB, Gregg K, Molloy PL, Morris CP, Crocker LA, and Rogers GE (1989a). Avian keratin genes I. A molecular analysis of the structure and expression of a group of feather keratin genes. J. Mol. Biol 209, 549–559. [DOI] [PubMed] [Google Scholar]
- Presland RB, Whitbread LA, and Rogers GE (1989b). Avian keratin genes. II. Chromosomal arrangement and close linkage of three gene families. J. Mol. Biol 209, 561–576. [DOI] [PubMed] [Google Scholar]
- Ramírez F, Bhardwaj V, Arrigoni L, Lam KC, Grüning BA, Villaveces J, Habermann B, Akhtar A, and Manke T (2018). High-resolution TADs reveal DNA sequences underlying genome organization in flies. Nat. Commun 9, 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Dündar F, Diehl S, Grüning BA, and Manke T (2014). deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42, W187–W191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinn JL, Wang JK, Allen N, Brugmann SA, Mikels AJ, Liu H, Ridky TW, Stadler HS, Nusse R, Helms JA, and Chang HY (2008). A dermal HOX transcriptional program regulates site-specific epidermal fate. Genes Dev. 22, 303–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat. Biotechnol 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi A, Jang SI, Ceci R, Steinert PM, and Markova NG (1998). Effect of AP1 transcription factors on the regulation of transcription in normal human epidermal keratinocytes. J. Invest. Dermatol 110, 34–40. [DOI] [PubMed] [Google Scholar]
- Tasic B, Nabholz CE, Baldwin KK, Kim Y, Rueckert EH, Ribich SA, Cramer P, Wu Q, Axel R, and Maniatis T (2002). Promoter choice determines splice site selection in protocadherin alpha and gamma pre-mRNA splicing. Mol. Cell 10, 21–33. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, and Pachter L (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, and Pachter L (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Yang W, McKittrick J, and Meyers MA (2016). Keratin: structure, mechanical properties, occurrence in biological organisms, and efforts at bio-inspiration. Prog. Mater. Sci 76, 229–318. [Google Scholar]
- Wang X, Su H, and Bradley A (2002). Molecular mechanisms governing Pcdh-gammagene expression: evidence for a multiple promoter and cis-alternative splicing model. Genes Dev. 16, 1890–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Z, Gao F, Kim S, Yang H, Lyu J, An W, Wang K, and Lu W (2013). Klf4 organizes long-range chromosomal interactions with the oct4 locus in re-programming and pluripotency. Cell Stem Cell 13, 36–47. [DOI] [PubMed] [Google Scholar]
- Wu P, Hou L, Plikus M, Hughes M, Scehnet J, Suksaweang S, Widelitz R, Jiang TX, and Chuong CM (2004). Evo-Devo of amniote integuments and appendages. Int. J. Dev. Biol 48, 249–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P, Lai YC, Widelitz R, and Chuong CM (2018a). Comprehensive molecular and cellular studies suggest avian scutate scales are secondarily derived from feathers, and more distant from reptilian scales. Sci. Rep 8, 16766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P, Ng CS, Yan J, Lai YC, Chen CK, Lai YT, Wu SM, Chen JJ, Luo W, Widelitz RB, et al. (2015). Topographical mapping of alpha- and beta-keratins on developing chicken skin integuments: functional interaction and evolutionary perspectives. Proc. Natl. Acad. Sci. USA 112, E6770–E6779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P, Yan J, Lai YC, Ng CS, Li A, Jiang X, Elsey RM, Widelitz R, Bajpai R, Li WH, and Chuong CM (2018b). Multiple regulatory modules are required for scale-to-feather conversion. Mol. Biol. Evol 35, 417–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Zhou Z, Dudley R, Mackem S, Chuong CM, Erickson GM, and Varricchio DJ (2014). An integrative approach to understanding bird origins. Science 346, 1253293. [DOI] [PubMed] [Google Scholar]
- Yi G, Sze SH, and Thon MR (2007). Identifying clusters of functionally related genes in genomes. Bioinformatics 23, 1053–1060. [DOI] [PubMed] [Google Scholar]
- Yu Z, Jiang K, Xu Z, Huang H, Qian N, Lu Z, Chen D, Di R, Yuan T, Du Z, et al. (2018). Hoxc-dependent mesenchymal niche heterogeneity drives regional hair follicle regeneration. Cell Stem Cell 23, 487–500.e6. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw and processed data of RNA-seq, ChIP-seq, and NG Capture-C reported in this paper is available at NCBI GEO database, accession GSE136224.