

Supplemental Digital Content is available in the text.
Keywords: blood-brain barrier, endothelium, extracellular matrix, glucose transport, glycolysis, homeostasis
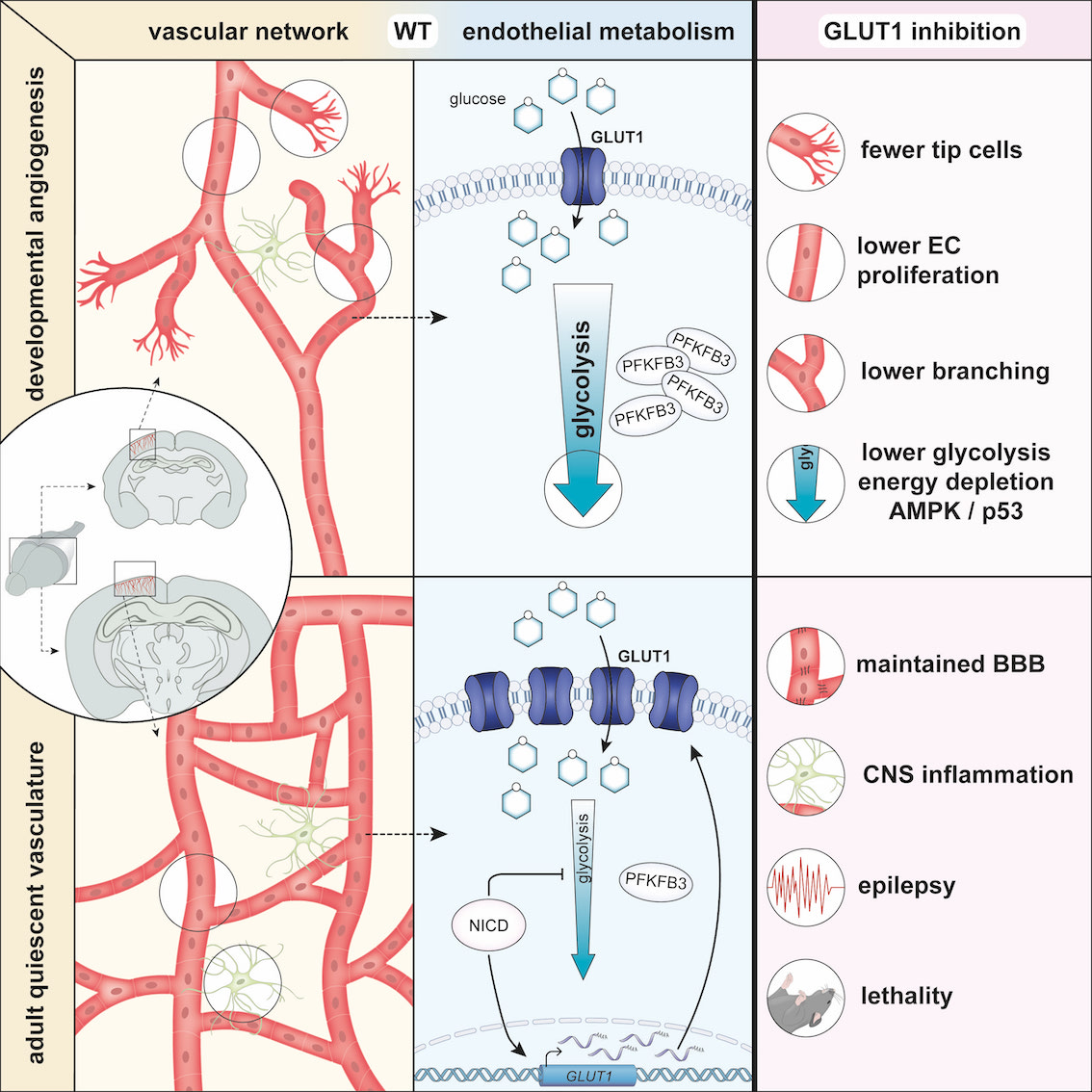
Abstract
Rationale:
Endothelial cells (ECs) are highly glycolytic and generate the majority of their energy via the breakdown of glucose to lactate. At the same time, a main role of ECs is to allow the transport of glucose to the surrounding tissues. GLUT1 (glucose transporter isoform 1/Slc2a1) is highly expressed in ECs of the central nervous system (CNS) and is often implicated in blood-brain barrier (BBB) dysfunction, but whether and how GLUT1 controls EC metabolism and function is poorly understood.
Objective:
We evaluated the role of GLUT1 in endothelial metabolism and function during postnatal CNS development as well as at the adult BBB.
Methods and Results:
Inhibition of GLUT1 decreases EC glucose uptake and glycolysis, leading to energy depletion and the activation of the cellular energy sensor AMPK (AMP-activated protein kinase), and decreases EC proliferation without affecting migration. Deletion of GLUT1 from the developing postnatal retinal endothelium reduces retinal EC proliferation and lowers vascular outgrowth, without affecting the number of tip cells. In contrast, in the brain, we observed a lower number of tip cells in addition to reduced brain EC proliferation, indicating that within the CNS, organotypic differences in EC metabolism exist. Interestingly, when ECs become quiescent, endothelial glycolysis is repressed, and GLUT1 expression increases in a Notch-dependent fashion. GLUT1 deletion from quiescent adult ECs leads to severe seizures, accompanied by neuronal loss and CNS inflammation. Strikingly, this does not coincide with BBB leakiness, altered expression of genes crucial for BBB barrier functioning nor reduced vascular function. Instead, we found a selective activation of inflammatory and extracellular matrix related gene sets.
Conclusions:
GLUT1 is the main glucose transporter in ECs and becomes uncoupled from glycolysis during quiescence in a Notch-dependent manner. It is crucial for developmental CNS angiogenesis and adult CNS homeostasis but does not affect BBB barrier function.
In This Issue, see p 449
To grow as well as to sustain a healthy life, tissues require optimal delivery of oxygen and nutrients. These are transported throughout the body by an ingenious network of blood vessels, formed by aligned endothelial cells (ECs). Oxygen and nutrient scarcity triggers tissue vascularization and to meet metabolic needs of growing tissues, ECs form new blood vessels from existing ones in a process called angiogenesis.1,2 During sprouting angiogenesis, a migrating tip cell leads the new sprout into the oxygen/nutrient scarce area. This tip cell will instruct its neighboring cells to proliferate and form the new vessel tube, thereby adopting a stalk cell phenotype.1,2 Ultimately, restoration of oxygen/nutrient delivery occurs upon fusion of 2 sprouts, ECs subsequently become quiescent again and a mature vessel is formed.1,2 Once this has happened, enhanced nutrient demand by tissues require an appropriate increase in nutrient transport across the endothelium. Remarkable organotypic heterogeneity exists in nutrient preference and how nutrients are exchanged between the blood and tissues: tissues such as the heart and brown adipose tissue require fatty acids, whereas the brain and the exercising muscle favors glucose as main energy substrates.3 Also, while some vascular beds like the liver sinusoidal endothelium passively allow passage of nutrients across discontinuous fenestrated permeable endothelium, other beds like the brain endothelial lining need to actively control nutrient transport across a continuous nonfenestrated tightly sealed endothelium.4
Besides allowing nutrient transport, ECs require nutrients to fuel their own metabolism. ECs possess special metabolic characteristics and generate the majority of their energy from the glycolytic breakdown of glucose to lactate.5,6 Consequently, reducing EC glycolysis through deletion of PFKFB3 (6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3) reduces tip cell migration and stalk cell proliferation.5 Furthermore, ECs are unique because they remain highly glycolytic even under quiescent conditions when other cell types prefer more efficient energy production via oxidative phosphorylation.7 Glucose enters ECs via facilitated diffusion through GLUT1 (glucose transporter isoform 1, encoded by the Glut1/Slc2a1 gene). Mimicking angiogenic conditions in vitro through stimulation with VEGF (vascular endothelial growth factor) increases GLUT1 and glycolysis.5,8 Moreover, highly angiogenic tumor ECs are hyper-glycolytic and have increased Glut1 expression,9 whereas mice carrying a heterozygous deletion of Glut1 have reduced brain vascular density.10 This suggest that GLUT1 expression is coupled to EC glycolysis during angiogenesis and consequently controls angiogenesis.
In contrast with angiogenic ECs, ECs within a mature vessel need to tightly balance glucose transport to supply the surrounding tissues with glucose consumption to fuel their own glycolytic metabolism. How GLUT1 controls glucose transport/metabolism seems to be highly organ-dependent. In particular, endothelial Glut1 (EC-GLUT1) levels at the blood-brain barrier (BBB) are much higher when compared with other organs.11 Also, patients with inactive GLUT1, such as in GLUT1 deficiency syndrome, have reduced glucose transport over the BBB and are clinically diagnosed by lower glucose levels in the cerebrospinal fluid.12 These patients have infantile-onset seizures, delayed neurological development, acquired microcephaly, and movement disorders.12,13 While these data emphasize the crucial role for GLUT1 in glucose transport over the BBB, it is far less understood whether GLUT1 also controls the import of glucose, required to sustain EC metabolism and thus EC function at the BBB. Interestingly, lower GLUT1 levels have been associated to microvascular impairment and BBB dysfunction in patients with Alzheimer14 and lowering GLUT1 levels exacerbated Alzheimer disease in mice.15 However, studies using Glut1 haploinsufficient mouse models have provided conflicting evidence concerning the role of GLUT1 in maintaining the physical integrity of the BBB.10 Moreover, it is not known whether altering GLUT1 levels also affects the expression of other BBB-specific genes such as specialized nutrient and essential molecule transporters. We thus set out to investigate the role of GLUT1 in EC metabolism and function, during developmental central nervous system (CNS) angiogenesis as well as in the adult brain.
Methods
A detailed Methods section can be found in the Data Supplement. Please see the Major Resources Table in the Data Supplement.
The authors declare that the majority of supporting data are presented within this article and in the Data Supplement. Data that are not directly available are available from the corresponding author upon reasonable request.
All sequencing data are deposited in the Gene Expression Omnibus database under accession code GSE141924 and GSE141923.
Results
GLUT1 Controls Glucose Uptake and Glycolysis in ECs
To study GLUT1 in EC glucose metabolism in vitro, we used the highly selective GLUT1 inhibitor BAY-876 (N4-[1-[(4-cyanophenyl)methyl]-5-methyl-3-(trifluoromethyl)-1H-pyrazol-4-yl]-7-fluoro-2,4-quinolinedicarboxamide).16 BAY-876 dose-dependently inhibited 14C-3-O-methylglucose transport into a brain-derived EC line (bEND3; Figure IA in the Data Supplement) reaching near complete inhibition at 20 nmol/L. This inhibition was similar to the reduction obtained using pan-GLUT inhibition by cytochalasin B (Figure 1A). This indicates that GLUT1 is a main glucose transporter and that other GLUTs do not compensate for loss of glucose transport capacity upon GLUT1 inhibition. To confirm that GLUT1-dependent glucose transport is required for endothelial glycolysis, we measured glycolytic flux by assessing 3H-labelled water production originating from D-[5-3H(N)]-glucose. GLUT1 inhibition completely abrogated glycolysis to a similar extent as cytochalasin B (Figure 1B). Consistently, GLUT1 inhibition reduced glycolysis in human brain microvascular and human retinal microvascular ECs (Figure 1C), as well as in ECs isolated from human brain under different pathological conditions, including glioblastoma patients (World Health Organization grade IV astrocytoma ECs), temporal lobectomy, and amygdalohippocampectomy (Figure IB in the Data Supplement). Finally, inhibition of GLUT1 reduced the abundance of glycolytic metabolites (Figure 1D).
Figure 1.
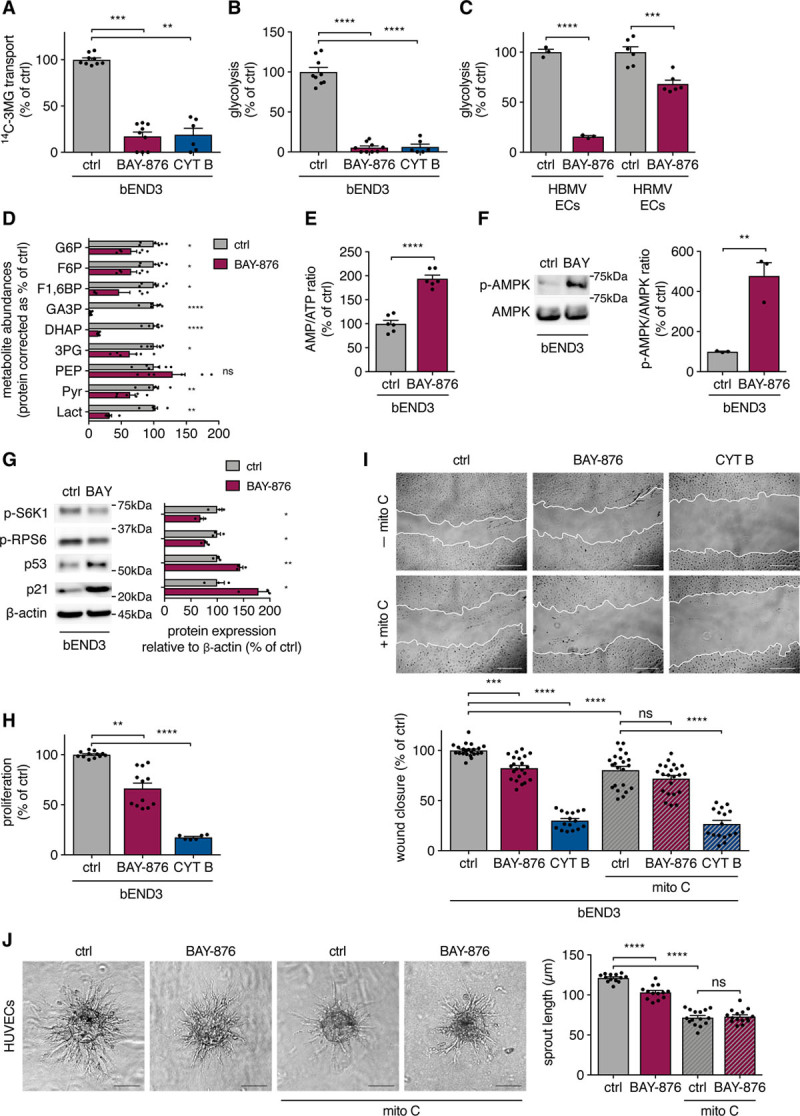
GLUT1 (glucose transporter isoform 1) inhibition impairs endothelial cell (EC) glucose metabolism and proliferation but not migration. A and B, 14C-3-O-methylglucose (3MG) transport (Kruskall-Wallis test and Dunn multiple comparisons test; A) and glycolytic flux (1-way ANOVA and Tukey multiple comparisons test; B) in cells from a brain-derived EC line (bEND3) incubated with 20 nmol/L BAY-876 (N4-[1-[(4-cyanophenyl)methyl]-5-methyl-3-(trifluoromethyl)-1H-pyrazol-4-yl]-7-fluoro-2,4-quinolinedicarboxamide) and 20 µmol/L cytochalasin B (CYT B) vs control. C, Glycolytic flux in human brain microvascular ECs (HBMV ECs) and human retinal microvascular ECs (HRMV ECs) incubated with 20 nmol/L BAY-876 vs control (Student t test). D, Abundances of glycolytic intermediates in bEND3 cells incubated with 20 nmol/L BAY-876 vs control (Student t test or Mann-Whitney U test). E, AMP/ATP ratio in cells from a brain-derived EC line (bEND3) incubated with 20 nmol/L BAY-876 vs control (Student t test). F and G, Western blot of p-AMPK (phospho-AMP-activated protein kinase), AMPK (F) and p-S6K1 (phospho-S6 kinase 1), p-RPS6 (phospho-ribosomal protein S6), p53, and p21 (G) in bEND3 cells incubated with 20 nmol/L BAY-876 vs control (Student t test). H, Proliferation rate of bEND3 cells incubated with 20 nmol/L BAY-876 and 20 µmol/L CYT B vs control (Kruskall-Wallis test and Dunn multiple comparisons test). I, Representative pictures and quantifications of scratch wound closure in bEND3 cells incubated with 20 nmol/L BAY-876 and 20 µmol/L CYT B vs control in conditions with and without mitomycin C (mito C) pretreatment (2-way ANOVA and Tukey multiple comparisons test). J, Representative pictures and quantifications of sprouting human umbilical vein EC (HUVEC) spheroids incubated with 40 nmol/L BAY-876 vs control in conditions with and without mitomycin C pretreatment (2-way ANOVA and Tukey multiple comparisons test). Scale bar=500 µm (I) and 100 µm (J). 3PG indicates 3-phosphoglycerate; AMP, adenosine monophosphate; ATP, adenosine triphosphate; DHAP, dihydroxyacetone phosphate; F1,6BP, fructose 1,6-bisphosphate; F6P, fructose 6-phosphate; G6P, glucose 6-phosphate; GA3P, glyceraldehyde 3-phosphate; Lact, lactate; PEP, phosphoenolpyruvate; and Pyr, pyruvate. *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001.
Since ECs generate most of their ATP via glycolysis,5 we wondered whether GLUT1 inhibition lowered energy availability. GLUT1 inhibition increased the AMP/ATP ratio, leading to activation of AMPK (AMP-activated protein kinase), a main cellular energy sensor which couples cell growth and proliferation to energy availability17 (Figure 1E and 1F). AMPK activation coincided with the inhibition of protein synthesis, illustrated by reduced phosphorylation of mTORC1 (mammalian target of rapamycin complex 1) downstream targets S6K1 (S6 kinase 1) and RPS6 (ribosomal protein S6), as well the induction of cell cycle inhibitors p53/p21,18 which are known AMPK targets (Figure 1G).
We also tested whether GLUT1 controls glucose transport and glycolysis in non-brain ECs, where Glut1 levels are lower.11 GLUT1 inhibition reduced glucose transport and glycolysis in human umbilical vein ECs (Figure IC and ID in the Data Supplement) and in primary mouse ECs isolated from lung and muscle (Figure IE in the Data Supplement). Thus, GLUT1 is the main transporter responsible for glucose uptake in cultured ECs from CNS and non-CNS tissues, and inhibiting GLUT1 profoundly inhibited the glycolytic breakdown of exogenous glucose, leading to AMPK activation.
GLUT1 Inhibition Reduces EC Proliferation But Not Migration
Since GLUT1 inhibition reduced glycolysis and activated AMPK/p53, we wondered whether GLUT1 is required for proliferation and migration. In bEND3 cells, GLUT1 inhibition reduced proliferation by ≈40%, whereas pan-GLUT inhibition using cytochalasin B almost completely blocked proliferation (Figure 1H), suggesting that GLUTs transport solutes, other than glucose, to sustain proliferation. Interestingly, migration (assessed using scratch wound assay) was only mildly delayed upon GLUT1 inhibition, whereas it was severely abrogated upon cytochalasin B treatment. Parallel treatment with the proliferation inhibitor mitomycin C indicated that the reduction in wound closure was due to reduced proliferation, showing that GLUT1 inhibition does not impair EC migration (Figure 1I). Of note, 48 hours of GLUT1 inhibition did not affect cell viability, whereas cytochalasin B treatment compromised cell viability (Figure IIA in the Data Supplement). These data were confirmed in human umbilical vein ECs (Figure IIB through IID in the Data Supplement). Lastly, GLUT1 inhibition reduced sprouting angiogenesis from human umbilical vein EC, human brain microvascular, and human retinal microvascular EC spheroids (Figure 1J and Figure IIE and IIF in the Data Supplement), but spheroids in which proliferation was blocked by mitomycin C did not show impaired sprouting upon GLUT1 inhibition (Figure 1J and Figure IIE and IIF in the Data Supplement). Thus, GLUT1 is required for EC proliferation, but not migration during sprouting angiogenesis.
GLUT1 Expression Increases During Quiescence
Next, we tested whether GLUT1 is actively regulated during induction of quiescence, when ECs change from active proliferation and migration to mature, closely aligned cells which form an active barrier and allow nutrient transport. In vitro, human umbilical vein ECs grown to contact inhibition (CI) function as a model for EC quiescence19 (Figure 2A). Quiescent ECs (ie, CI cells) actively downregulate glycolysis (Figure 2B) through repressing PFKFB3,20,21 which we confirmed (Figure 2C and Figure IIIA in the Data Supplement). Reduced glycolysis, however, remained GLUT1-dependent (Figure 2B). Surprisingly, despite lower glycolysis/PFKFB3, CI cells had higher GLUT1 (Figure 2C and Figure IIIA in the Data Supplement). Notch is a main regulator of cell quiescence and represses glycolysis/PFKFB3 upon CI.5 We found that Notch also controls GLUT1 expression, since stimulating ECs with the Notch ligand Dll4 (delta-like 4; Figure 2D and Figure IIIB in the Data Supplement) as well as overexpressing the Notch intracellular domain increased GLUT1 (Figure 2E and Figure IIIC in the Data Supplement). Moreover, induction of GLUT1 was dependent on γ-secretase activity as treatment with DAPT abolished GLUT1 induction (Figure 2D and Figure IIIB in the Data Supplement). We also overexpressed the flow-sensitive transcription factor KLF2 (Krüppel-like factor 2), which orchestrates a network of genes that promotes EC quiescence in response to flow.22 KLF2 also reduces EC glycolysis by suppressing PFKFB3,21, but we did not find increased GLUT1 protein levels, despite modestly increased Glut1 mRNA levels upon KLF2 overexpression (Figure IIID through IIIE in the Data Supplement). This shows that Notch uncouples glycolysis from GLUT1 levels as soon as quiescence is induced. Based on these findings, we evaluated the role of GLUT1 in postnatal retinal and brain angiogenesis as well as in maintaining EC quiescence.
Figure 2.
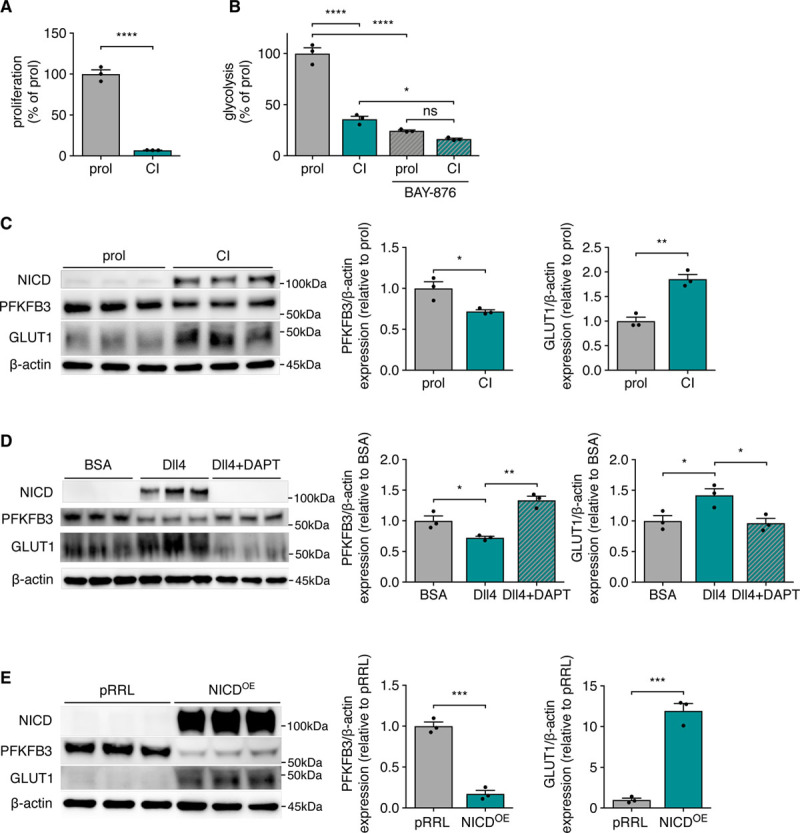
GLUT1 (glucose transporter isoform 1) expression is increased in quiescence and uncoupled from glycolysis. A, Proliferation rate of proliferating (prol) and contact inhibited (CI) human umbilical vein ECs (HUVECs; Student t test). B, Glycolytic flux in proliferating (prol) and contact inhibited (CI) HUVECs incubated with 20 nmol/L BAY-876 (N4-[1-[(4-cyanophenyl)methyl]-5-methyl-3-(trifluoromethyl)-1H-pyrazol-4-yl]-7-fluoro-2,4-quinolinedicarboxamide) vs control (2-way ANOVA and Tukey multiple comparisons test). C, Representative image and quantification of Western blot of Notch intracellular domain (NICD), PFKFB3 (6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3), and GLUT1 protein levels in proliferating vs contact inhibited HUVECs. β-actin is used as loading control (Student t test). D, Representative image and quantification of Western blot of NICD, PFKFB3, and GLUT1 protein levels in HUVECs cultured on BSA or Dll4 (delta-like 4) coated plates with or without γ-secretase inhibitor N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester (DAPT; 20 μmol/L) treatment (1-way ANOVA and Tukey multiple comparisons test). E, Representative image and quantification of Western blot of NICD, PFKFB3, and GLUT1 protein levels in HUVECs with overexpression of NICD vs empty control overexpression vector (pRRL; Student t test). All values are normalized to the control condition (C–E). *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001.
Loss of EC-GLUT1 Controls Postnatal Angiogenesis in Retina and Brain
To study EC-GLUT1 in vivo, we generated endothelial-specific GLUT1 knock-out (GLUT1EC−/−) mice by intercrossing GLUT1lox/lox mice23 with EC-selective Pdgfb.CreERT2 mice24 to generate GLUT1EC−/− mice upon tamoxifen treatment. Tamoxifen treatment led to an almost complete loss of GLUT1 protein in isolated brain ECs (Figure 3A). We confirmed that genetic loss of Glut1 severely reduced glycolysis and proliferation in isolated ECs (Figure 3B and Figure IVA and IVB in the Data Supplement). We subsequently wondered whether GLUT1 controls angiogenesis in vivo and focused on the developing postnatal retina and brain, because angiogenesis in these tissues is controlled by EC glycolysis.5 Neonatal pups were treated with tamoxifen at P1 to P3, and retinal angiogenesis was evaluated at P5 and P6 (Figure 3C). In agreement with our in vitro observations, loss of EC-GLUT1 diminished retinal EC proliferation at P5, measured by the number of EdU+/Erg+ cells (Figure 3D and 3E), leading to delayed vascular plexus outgrowth and reduced branch point density at P6 (Figure 3F through 3H). We did not observe a difference in the number of tip cells, and the number of filopodia per tip cell was even slightly increased (Figure 3I through 3K), which was surprising since tip cells are highly dependent on glycolysis to fuel migration and filopodia formation.5 However, these data are consistent with our in vitro experiments, showing that GLUT1 is not required for EC migration. The absence of any tip cell phenotype in the retina prompted us to evaluate whether GLUT1 is expressed in the vascular forefront of the developing retina. Immunohistochemical staining showed that GLUT1 was abundantly expressed in the hyaloid vessels, but was barely detectable in the primary plexus of neonatal (P6) mice (Figure IVC in the Data Supplement). Thus, even though retinal tip cells are highly glycolytic, they do not require GLUT1.
Figure 3.
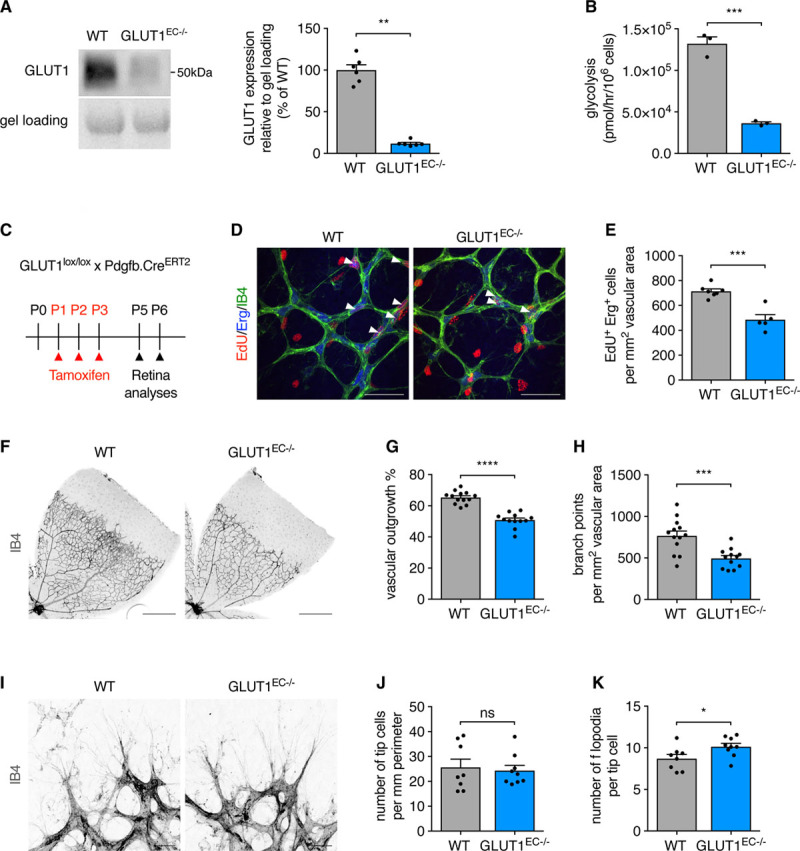
Loss of EC-GLUT1 (endothelial glucose transporter isoform 1) impairs neonatal retinal angiogenesis. A, Representative Western blot and quantification for GLUT1 protein in primary isolated brain ECs from GLUT1EC−/− mice vs wild-type (WT) littermates (Mann-Whitney U test). B, Glycolytic flux in cultured primary isolated mouse ECs from GLUT1EC−/− mice vs WT littermates (Student t test). C, Schematic representation of experimental timing for retina analyses in GLUT1lox/lox×Pdgfb.CreERT2 pups. D and E, Representative pictures (D) and quantification (E) of 5-ethynyl-2’-deoxyuridine (EdU+)/ETS-related gene (Erg+) cells in the primary plexus of P5 GLUT1EC−/− pups vs WT littermates (Student t test). White arrows (D) indicate EdU+/Erg+ cells. F–H, Representative pictures from IB4 (isolectin griffonia simplicifolia B4)-stained flat-mounted retinas from P6 GLUT1EC−/− pups vs WT littermates (F) and quantifications of vascular outgrowth (G) and branch point density (H; Student t test). I–K, Representative pictures from IB4-stained retinal tip cells and filopodia from P6 GLUT1EC−/− pups vs WT littermates (I) and quantifications of tip cell number (J) and filopodia number per tip cell (K) (Student t test or Mann-Whitney U test). Scale bar=50 µm (D), 500 µm (F), and 20 µm (I). *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001.
While GLUT1 levels are low in the neonatal retinal vasculature, GLUT1 is highly expressed in tip cells and filopodia of the developing brain, as shown by GLUT1 staining on brains from P6 pups (Figure IVD in the Data Supplement). Analysis of the brain vasculature in neonatal mice (Figure 4A)25 showed that loss of EC-GLUT1 reduced the number of EdU+/Erg+ proliferating ECs at P5 (Figure 4B and 4C), leading to a diminished vascular length at P6 (Figure 4D and 4E). We also found a striking reduction in the number of tip cells in the brain (Figure 4D and 4F), even though the number of filopodia per tip cell as well as filopodial length were not affected (Figure 4D, 4G, and 4H).
Figure 4.
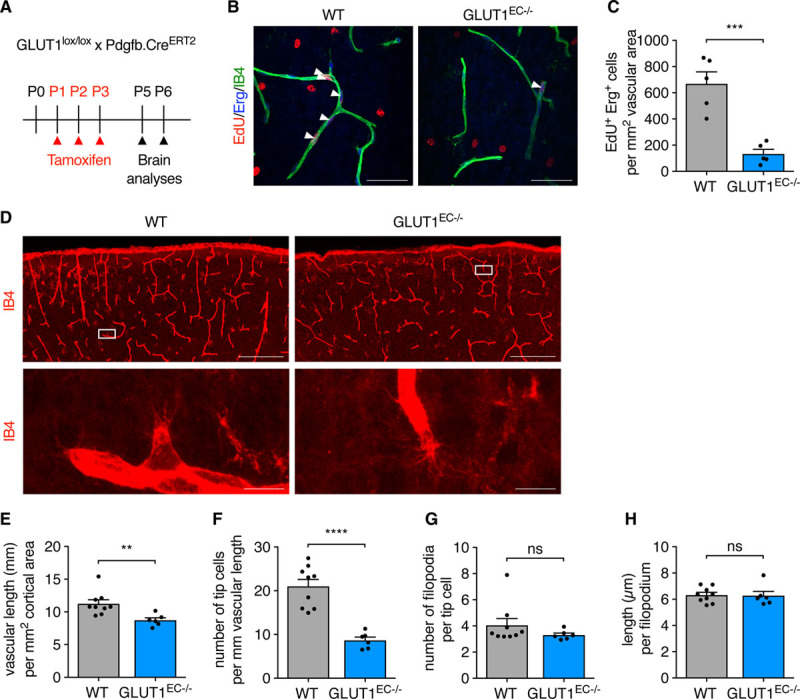
Loss of EC-GLUT1 (endothelial glucose transporter isoform 1) impairs neonatal brain angiogenesis. A, Schematic representation of experimental timing for brain analyses in GLUT1lox/lox×Pdgfb.CreERT2 pups. B and C, Representative pictures (B) and quantification (C) of 5-ethynyl-2’-deoxyuridine (EdU+)/ETS-related gene (Erg+) cells in the primary plexus of P5 GLUT1EC−/− pups vs wild-type (WT) littermates (Student t test). White arrows (B) indicate EdU+/Erg+ cells. D and H, Representative pictures showing the cortical area and a tip cell magnification of IB4 (isolectin griffonia simplicifolia B4)-stained thick brain sections from P6 GLUT1EC−/− pups vs WT littermates (D) and corresponding quantifications of vascular length (E) tip cell number (F) filopodia number per tip cell (G) and filopodial length (H) (Student t test or Mann-Whitney U test). Scale bar=50 µm (B), 200 µm for upper and 10 µm for lower (D). **P<0.01, ***P<0.001, and ****P<0.0001.
Loss of EC-GLUT1 Leads to Progressive Neuronal Loss, CNS Inflammation, and Rapid Lethality
In the adult brain, where ECs are quiescent, GLUT1 regulates glucose transport over the BBB.26 Our observations, showing that GLUT1 controls postnatal brain angiogenesis, prompted us to evaluate whether GLUT1 also controls EC metabolism and function during adulthood. Indeed, endothelial Glut1 expression is higher in brain when compared with other murine organs (Figure 5A).11,27 Also, endothelial Glut1 is higher in adult brain when compared with neonatal brains (Figure 5A),27 a finding reminiscent of increased Glut1 levels upon quiescence-mimicking conditions in vitro (described above). To study the role of EC-GLUT1 in the adult brain independent of developmental effects, we injected adult (>8 week-old) mice with tamoxifen to induce EC-specific loss of GLUT1 and analyzed the mice shortly after the first tamoxifen injection (Figure 5B). Consistent with GLUT1 deficiency syndrome patient data,12 loss of EC-GLUT1 reduced the cerebrospinal fluid/plasma glucose ratio (Figure 5C). GLUT1EC−/− mice also developed a pronounced phenotype, displaying many features of the human GLUT1 deficiency syndrome.12 As soon as 6 days after the first tamoxifen injection, GLUT1EC−/− mice started to show behavioral alterations, characterized by reduced spontaneous movement (Figure 5D) and lack of explorative behavior. From day 4 after the first tamoxifen injection, electrocorticographic abnormalities appeared in GLUT1EC−/− mice which were characterized by multiple sharp spikes or aberrant spike forms (Figure 5E). We observed a gradually increased appearance of spike-wave complexes on the electrocorticography of GLUT1EC−/− mice ultimately leading to clear epileptic seizures or status epilepticus. This confirms previous observations showing that loss of brain EC-GLUT1 suffices to induce epileptic seizures28 and underscores the critical role of EC-GLUT1 in brain metabolism. We counted the number of neurons in the cerebral cortex and in the hippocampus, a region particularly susceptible to seizure-associated damage.29 Loss of EC-GLUT1 did not affect neuronal density up till day 9 and 10 (Figure VA through VF in the Data Supplement), but we detected pycnosis of neuronal nuclei and progressive loss of neurons in some hippocampal (ie, cornu ammonis 1 [CA1] and CA3) but not all (hippocampal dentate gyrus and cortex) brain areas from mice at ≥13 days after the first tamoxifen injection (Figure 5F through 5K). Interestingly, brains from GLUT1EC−/− mice also displayed microgliosis and astrogliosis at day 8 to 10, indicating a state of inflammation and loss of CNS homeostasis that preceded neuronal loss (Figure 5L through 5M). Despite widespread brain inflammation, we did not observe increased recruitment of CD206+ (cluster of differentiation 206, macrophage mannose receptor) macrophages nor increased levels of circulating VEGF (Figure VG and VJ in the Data Supplement). Few days later, the health status of GLUT1EC−/− mice worsened progressively as they lost body weight (−15.8%±1.2% at day 12; P<0.05; n=17). Mice rapidly died within 3 weeks after the first tamoxifen injection (Figure 5N).
Figure 5.
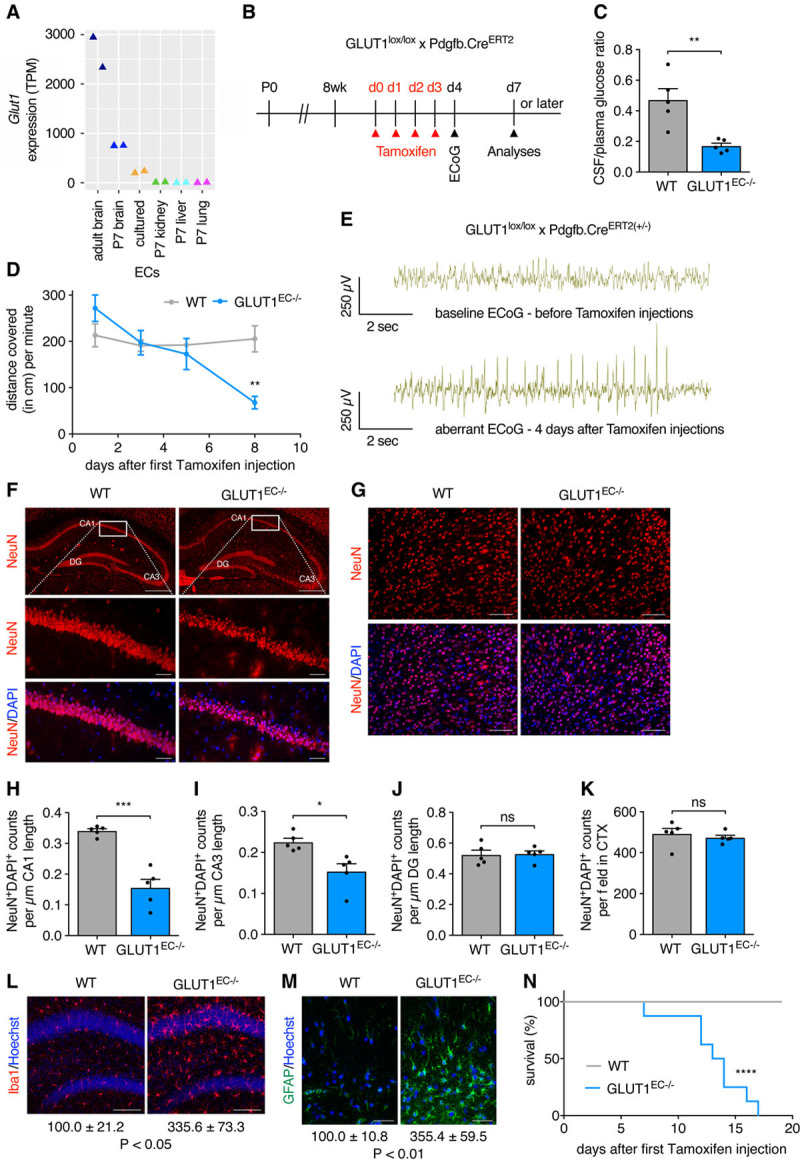
Loss of EC-GLUT1 (endothelial glucose transporter isoform 1) leads to progressive neuronal loss, central nervous system (CNS) inflammation, and rapid lethality. A, Endothelial Glut1 expression levels (transcripts per million [TPM]) in adult mouse brain ECs, cultured brain ECs and ECs from brain, kidney, liver and lung from P7 pups as described in the Vascular Endothelial Cell Trans-omics Resource Database (VECTRDB).27 B, Schematic representation of experimental timing for brain vascular analyses in GLUT1lox/lox×Pdgfb.CreERT2 mice. C, Cerebrospinal fluid (CSF)/plasma glucose ratio in GLUT1EC−/− mice vs wild-type (WT) littermates (Student t test). D, Quantification of spontaneous movement in GLUT1EC−/− mice vs WT littermates at 1, 3, 5, and 8 days after the first tamoxifen injection (n=3 per group; repeated measures ANOVA with Sidak multiple comparisons test). E, Representative electrocorticography (ECoG) excerpts from Cre positive GLUT1lox/lox×Pdgfb.CreERT2 mice, monitored by telemetry-ECoG 24 h/day (n=3 per group) showing the baseline ECoG before the first tamoxifen injection and an aberrant ECoG 4 days after the first tamoxifen injection. F and G, Representative pictures of hippocampal (F) and of cortical (G) NeuN+ (neuronal nuclear protein)/DAPI+ neuronal nuclei in GLUT1EC−/− mice vs WT littermates >13 days after the first tamoxifen injection. H–K, Quantifications of the number of NeuN+/DAPI+ cells in the cornu ammonis 1 (CA1; H), CA3 (I), dentate gyrus (DG; J), and cortical region (K) in GLUT1EC−/− mice vs WT littermates (Student t test). L and M, Representative pictures of Iba1+ microglia (L) and GFAP+ (glial fibrillary acidic protein) astrocytes (M) in the hippocampus of GLUT1EC−/− mice vs WT littermates (n=4 per group). Quantifications show % Iba1+ and % GFAP+ area vs WT (n=4 per group; Student t test). N, Kaplan-Meier survival curve of GLUT1EC−/− mice vs WT littermates (n=8 per group; Log-rank (Mantel-Cox) test). Scale bar=500 µm for upper, 50 µm for lower (F), 50 µm (G), 100 µm (L), and 50 µm (M). *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001.
Loss of EC-GLUT1 Does Not Impair Vascular Function nor BBB Physical Barrier Properties
Against this background, we continued to study whether acute loss of EC-GLUT1 also affects brain EC function and analyzed the structure and function of the capillary network in GLUT1EC−/− mice at day 7 and 8 when electrocorticographic abnormalities are already present (Figure 6A). Vessel density in the cortex (Figure 6B and 6C) and hippocampal areas (Figure VIA through VIF in the Data Supplement) was not affected. Assessment of cerebral blood flow and cerebral blood volume using functional magnetic resonance imaging revealed a 21% increase (though nonsignificant) in cerebral blood flow in GLUT1EC−/− mice, which was accompanied by an increased cerebral blood volume (Figure 6D and 6E). The latter suggests that the vasodilatory response to increased brain activity was preserved in GLUT1EC−/− mice. In addition, vascular reactivity in response to the blood vessel dilator acetazolamide (Diamox),30 expressed as a percentage of baseline cerebral blood volume, was not affected in GLUT1EC−/− mice (Figure 6F).
Figure 6.
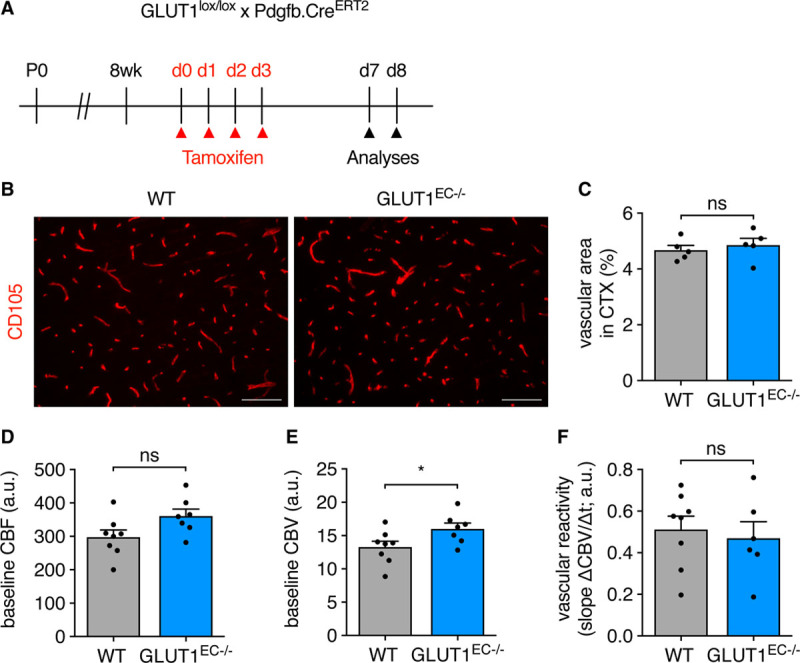
Loss of EC-GLUT1 (endothelial glucose transporter isoform 1) does not impair brain vascular function. A, Schematic representation of experimental timing for brain vascular analyses in adult GLUT1lox/lox×Pdgfb.CreERT2 mice. B and C, Representative pictures of the cluster of differentiation 105-stained cortical vasculature in adult GLUT1EC−/− mice vs wild-type (WT) littermates (B) and quantifications of vascular area (C; Student t test). D–F, Functional magnetic resonance imaging (fMRI) measurements in GLUT1EC−/− mice vs WT littermates for assessment of baseline cerebral blood flow (CBF; D), baseline cerebral blood volume (CBV; E), and for the determination of vascular reactivity, that is, dynamic CBV in response to the injection of the pharmacological vasodilator acetazolamide (F) (Student t test). Scale bar=100 µm (B). *P<0.05.
Subsequently, we evaluated BBB integrity through assessment of vessel permeability. First, we did not detect any increase in brain water content in GLUT1EC−/− mice compared with wild-type (WT) littermates (Figure 7A). Second, there was no accumulation of Evans Blue in the brain (Figure 7B) and spectrophotometric analysis of formamide brain extracts confirmed that this dye—otherwise detectable in WT mice injected with mannitol as a BBB-permeabilizer—was almost undetectable in both WT and GLUT1EC−/− mice (Figure 7C). Third, we also studied whether loss of EC-GLUT1 altered the expression or morphology of BBB tight junctions. Immunostainings for ZO1 (zonula occludens 1) or CLDN5 (claudin-5) did not indicate reduced expression of these proteins in the BBB vessels (Figure 7D through 7G). Moreover, FACS sorted brain ECs from GLUT1EC−/− and WT mice had comparable protein levels of ZO1, OCLN (occludin), and CLDN5, whereas CDH5 (vascular endothelial cadherin) levels showed a minor decrease (Figure 7H). Recognizing that the above described experiments may not detect subtle BBB alterations, we also examined BBB integrity by using the low molecular weight tracer gadolinium (Gd)-DOTA (0.5 kDa). Dynamic contrast-enhanced magnetic resonance imaging on Gd-DOTA injected animals confirmed that BBB integrity was preserved in GLUT1EC−/− mice (Figure 7I). Thus, loss of GLUT1 in ECs during adulthood does not lead to acute alterations in EC junctional proteins and vascular permeability nor alterations in vascular autoregulation.
Figure 7.
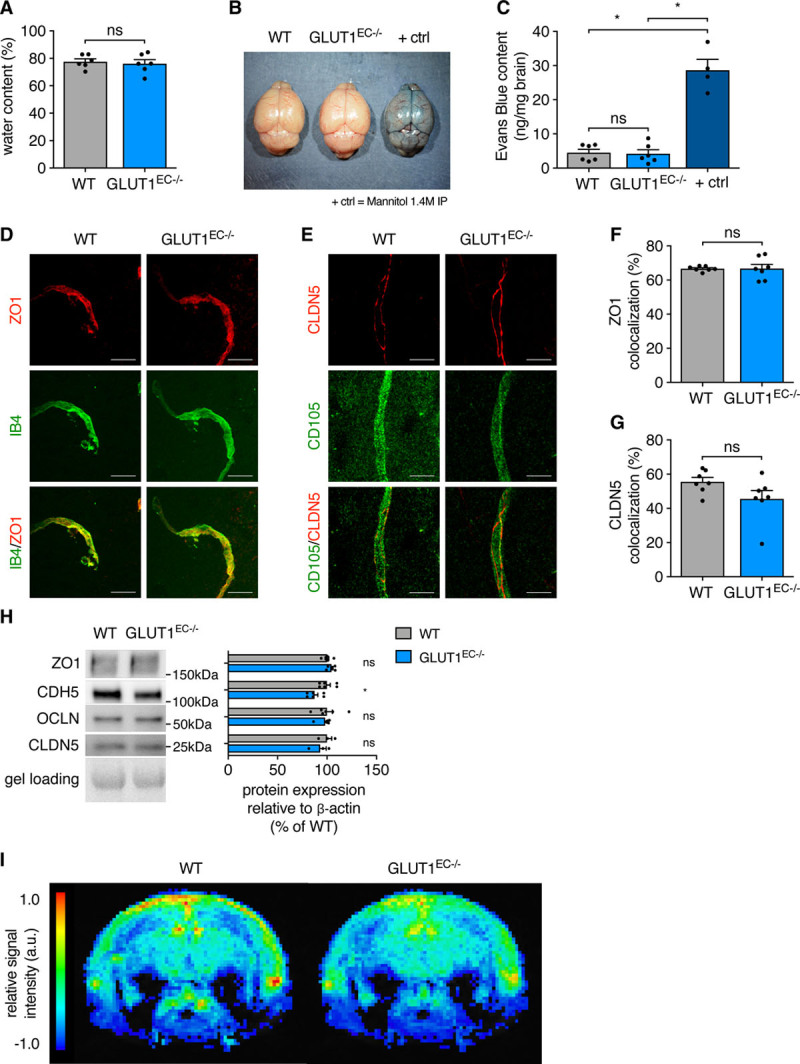
Loss of EC-GLUT1 (endothelial glucose transporter isoform 1) does not impair blood-brain barrier (BBB) physical barrier properties. A, Brain water content in GLUT1EC−/− mice vs wild-type (WT) littermates (Student t test). B and C, Representative pictures from brains of Evans Blue injected GLUT1EC−/− mice vs WT littermates and 1.4 mol/L mannitol-injected WT littermates as BBB-breaching positive controls (B) and quantifications of Evans Blue content from formamide extracted brains (C; Kruskall-Wallis test and Dunn multiple comparisons test). D–G, Representative pictures of IB4 (isolectin griffonia simplicifolia B4)- or cluster of differentiation 105-stained cerebral blood vessels in GLUT1EC−/− mice vs WT littermates, co-stained with the tight junction marker ZO1 (zonula occludens 1; D) or CLDN5 (claudin-5; E) and quantification of vascular (IB4 positive) colocalization analyses with ZO1 (F) and CLDN5 (G) (Student t test). H, Representative Western blots for ZO1, CDH5 (vascular endothelial cadherin), OCLN (occludin), and CLDN5 protein in primary isolated brain ECs from GLUT1EC−/− mice vs WT littermates corrected for gel loading relative to WT (Student t test or Mann-Whitney U test). I, Color coded mean leakage map in GLUT1EC−/− mice (n=7) vs WT littermates (n=8) showing relative signal intensity, calculated from dynamic contrast-enhanced magnetic resonance imaging measurements to evaluate blood-brain barrier permeability. Scale bar=20 µm (D–E). *P<0.05.
Genetic Alterations Induced by Loss of GLUT1 in Brain ECs
To further explore how GLUT1 controls EC function, we compared transcriptomic alterations induced by loss of EC-GLUT1 by performing RNA sequencing analysis on freshly isolated ECs from P6 and adult (9 days after first tamoxifen injection) WT and GLUT1EC−/− brains (Figure 8A, Figure VIIA through VIID in the Data Supplement). At P6, 447 genes were differentially expressed between WT and GLUT1−/− ECs. Subsequent gene set enrichment analysis showed that within the most differentially expressed pathways, several ones were related to p53, confirming our in vitro observations (Figure 8B). Moreover, we did not find consistent alterations in the expression of previously published tip/stalk cell marker genes27 nor genes involved in angiogenesis such as VEGF, Notch, Sonic Hedgehog, SoxF, angiopoietin, and Wnt signaling31 (Figure 8C and 8D), suggesting that GLUT1 controls neonatal angiogenesis via limiting endothelial energy supply rather than affecting molecular pathways involved in developmental angiogenesis.
Figure 8.
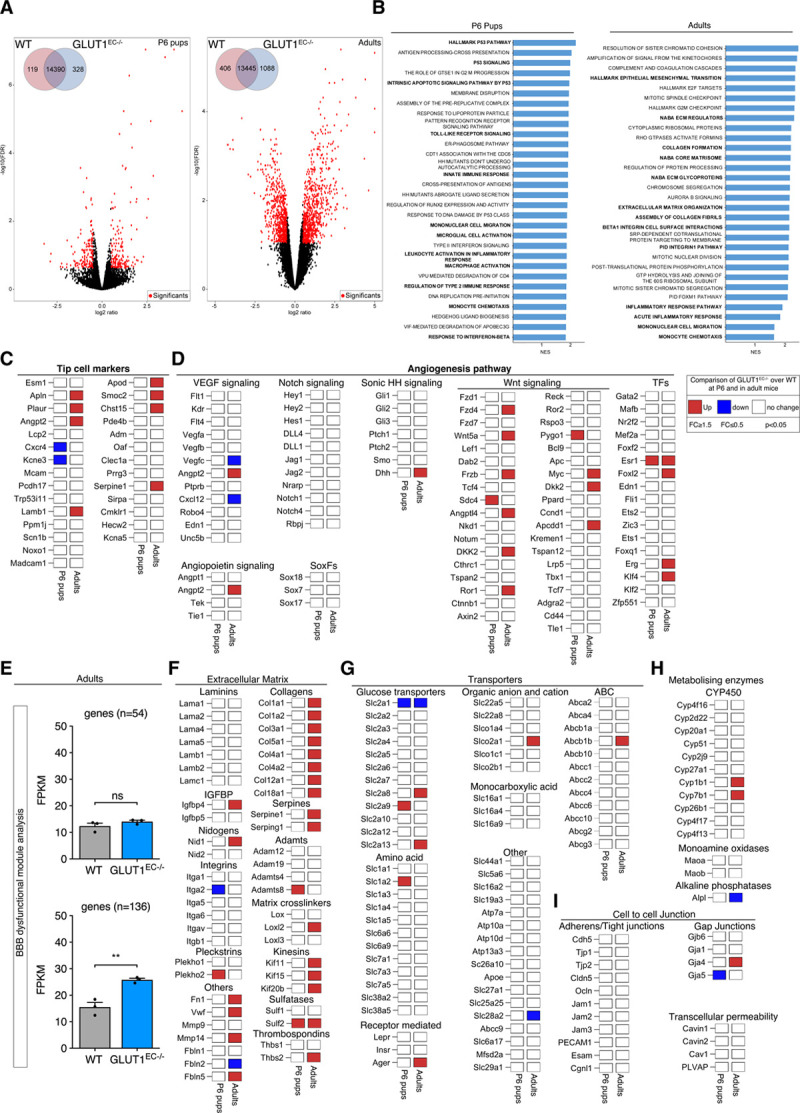
Transcriptional alterations upon loss of GLUT1 (glucose transporter isoform 1) in brain endothelial cells (ECs). A, Volcano plots displaying the magnitude of the differential expression between wild-type (WT) and GLUT1EC−/− brain ECs either at P6 (left; n=4 per genotype) or in adulthood (right; n=3 per genotype). Each dot represents 1 gene that has detectable expression both in WT and GLUT1EC−/− brain ECs. Black dots represent genes that are not altered. Differentially expressed genes are labeled in red. The number of differentially expressed genes between WT and GLUT1EC−/− brain ECs is shown (inset). FDR-values were calculated using Benjamini-Hochberg method; P≤0.01, log ratio ≥0.5). B, Gene set enrichment analysis for both P6 pups (left) or adults (right) showing significantly enriched pathway ranked by normalized enrichment score (NES) and P value. Key pathways highlighted in bold include inflammatory pathways (P6 pups and adults), and p53 and extracellular matrix signaling in P6 pups and adults respectively. C and D, Differential expression for a selected set of tip cell marker genes (C) and angiogenesis related genes (D) at P6 (left column) or in adults (right column) based on criteria defined in the legend box. E, Blood-brain barrier (BBB) dysfunctional modules analysis showing the comparison of the average FPKM values of the core (n=54), as well the compiled core and adjunct (n=136) genes in WT vs GLUT1EC−/− in adulthood (Student t test). F, Differential expression patterns of extracellular matrix related genes including laminins, collagens, serpines, adamts, integrins, matrix cross linkers, and others based on criteria defined in the legend box. G, Differential expression patterns of different glucose, amino acids, monocarboxylic acid, ABC, organic anion, and cation transporters based on criteria defined in the legend box. H, Differential expression of metabolizing enzymes (CYP450, MAOs, and ALPs) implicated in BBB function based on criteria defined in the legend box. I, Differential expression patterns of adherens/tight junctions, gap junctions, and transcellular permeability genes implicated in BBB function based on criteria defined in the legend box (n=3 per genotype for adults; n=4 per genotype for P6 pups). **P<0.01.
In adult brain ECs, 1494 genes were differentially expressed between WT and GLUT1−/− ECs (Figure 8A) but there was no compensatory upregulation of other GLUTs (Figure 8G). Interestingly, gene set enrichment analysis revealed that ECs from GLUT1EC−/− mice were characterized by a robust activation of inflammatory and extracellular matrix pathways (Figure 8B). In fact, also at P6, we picked up enrichment of proinflammatory pathways, showing that the inflammatory state was not restricted to the adult brain. Gene set enrichment analysis did not show altered expression of permeability pathways nor did we find altered expression in EC-cell junction genes (Figure 8B and 8I). We subsequently compared our sequencing data to a recently published BBB dysfunctional module, which compiles a set of genes which are always (core set; n=54 genes) or often (complete set; n=136 genes) altered in mouse models where the BBB has been disrupted.32 Loss of EC-GLUT1 did, however, not activate the expression of the core (n=54) BBB dysfunction gene set and only led to a small increase in the complete set of genes defining the BBB dysfunctional module (Figure 8E). Further investigation of the affected genes within this BBB dysfunctional module showed that they belong to extracellular matrix organization pathways, consistent with their enrichment in the gene set enrichment analysis (Figure 8F).
Finally, besides its role as a physical barrier, the BBB also acts as a location where active transport of metabolites and other compounds takes place as well as an enzymatic barrier due to the presence of a variety of metabolizing enzymes.33–35 We thus compared the expression levels of many genes involved in these processes,31 however, we failed to observe systematic alterations (Figure 8G and 8H). Thus, interfering with a crucial function of the BBB—glucose transport—does not lead to general BBB dysfunction, but rather activates a specific gene set related to, for example, inflammation and extracellular matrix amongst others.
ECs display specific metabolic characteristics, the most striking being their high rate of glycolytic energy production.6,7,36 Interestingly, during angiogenesis, ECs upregulate glycolysis even further.5 While GLUT1 is the main transporter responsible for endothelial glucose uptake, loss of GLUT1 in the retina during postnatal development did not affect the number of tip cells nor tip cell characteristics. This was surprising, because loss of glycolytic regulators5,37 severely impairs retinal tip cell formation. We did not observe GLUT1 protein in retinal tip cells using immunohistochemistry. In contrast, brain tip cells express GLUT1,27,38 and loss of GLUT1 reduced tip cell numbers. This difference between the retina and the brain is striking and suggests that within the CNS, organotypic differences in EC metabolism exist already early on in postnatal development.
It is possible that other transporters than GLUT1 mediate tip cell glucose uptake in the postnatal retina. Indeed, while Glut1 is by far the most abundantly expressed glucose transporter in brain ECs, its expression in ECs isolated from other organs is much lower and similar to other glucose transporters (data not shown). Nonetheless, glycolysis assays in all ECs we tested confirmed the dependence on GLUT1 for driving the glycolytic breakdown of glucose imported from the extracellular space. An alternative source for glycolytic energy production could be glycogen and glycogenolysis could—at least temporarily—feed glycolysis, which would allow tip cells to migrate into areas where glucose availability is scarce. Of note, ECs can store glycogen,39 but whether glycogen breakdown occurs during tip cell migration is not known.
Inhibition of GLUT1 selectively impaired proliferation without changing expression levels of main regulators of developmental angiogenesis nor tip/stalk cell genes. Mechanistically, GLUT1 inhibition reduced energy availability and activated p53/p21-dependent cell cycle arrest, likely due to AMPK activation.18 Interestingly, loss of endothelial pyruvate kinase isoform 2, another rate-limiting glycolytic enzyme, also activated p53/p21 though this occurred independent of its kinase activity.38 This indicates that glycolysis controls EC proliferation via multiple checkpoints and highlights the crucial role for glycolysis during angiogenesis.
When ECs return to quiescence, one of their main functions is to allow optimal nutrient transport to surrounding tissues. While it is well recognized that GLUT1 is required for glucose transport over the BBB, these mechanisms are not well demonstrated in other organs, where paracellular diffusion might be sufficient to account for transport.26 Moreover, it is an outstanding question whether similar mechanisms are involved in the uptake of glucose to fuel endothelial metabolism versus the transport of glucose over the endothelial barrier into the brain. We found that GLUT1 uncouples from glycolysis in quiescent ECs. Indeed, CI increasing Notch activity through Notch intracellular domain overexpression or Dll4 stimulation increased GLUT1 expression. The BBB-specific regulator Wnt/β-catenin also regulates the expression of Glut1.40 Interestingly, EC quiescence coincides with reduced glycolytic flux,5,19,21,41 suggesting that the majority of glucose entering quiescent ECs is transported over the endothelium and that only a minor fraction is used for endothelial metabolism.
GLUT1 has been suggested to have a role in the maintenance of BBB integrity, mainly because lower GLUT1 levels have been associated to microvascular impairment and BBB dysfunction in patients with Alzheimer.14,42 However, it was not clear whether these associations implied causality. Lowering EC-GLUT1 levels in a mouse model of Alzheimer disease worsened disease outcome, and this was associated with reduced vascular density and BBB leakiness.15 On the contrary, previous observations in Glut1 haploinsufficient mice, a mouse model of GLUT1 deficiency syndrome, failed to confirm BBB dysfunction.10 In agreement with the latter, we did not observe loss of vascular barrier function upon acute deletion of GLUT1 in ECs, as indicated by the absence of vessel permeability to exogenous tracers as well as by the similar levels of EC tight junction proteins. We also did not observe altered expression of a core set of genes described as the molecular fingerprint of a leaky BBB.32 Moreover, transcriptional profiling failed to show significant alterations in genes implicated in other BBB functions including metabolite transporters as well as metabolizing enzymes. Furthermore, vascular function was normal in GLUT1EC−/− mice. Thus, our data underscore that reduced accessibility to their preferred metabolic substrate, glucose, does not compromise multiple BBB characteristics of ECs and brain vascular function.
Loss of EC-GLUT1 during adulthood led to severe seizures accompanied by neuronal injury, CNS inflammation, and rapid lethality. A similar phenotype was previously observed upon deletion of β-catenin,43 a well-known upstream regulator of GLUT1 at the BBB.40 Loss of CNS homeostasis in those mice was attributed to loss of tight junctional proteins and BBB leakiness. Even though enhanced BBB leakiness has been observed in several mouse models, it has not consistently resulted into the development of overt epileptic insults. For example, pericyte deficiency as well as loss of Sonic Hedgehog signaling both increase BBB permeability without compromising life span.44,45 Of note, BBB permeability was not compromised at a time when GLUT1EC−/− mice already presented with a clinical phenotype of epileptic seizures and have lost >85% of GLUT1 protein content in brain ECs (±8 days). This shows that vascular leakiness does not induce epileptic seizures in GLUT1EC−/− mice. However, we also observed that at that time, mice already display astrogliosis and microgliosis, features that could induce BBB dysfunction independent of GLUT1. Furthermore, the physical condition of these mice rapidly worsens, and we have observed neuronal death at late stages. We, therefore, cannot exclude that the BBB will become permeable to plasma proteins at later stages, but current data do not support that EC-GLUT1 directly controls BBB function of brain ECs.
GLUT1 deficiency in brain ECs activated an inflammatory gene signature, in postnatal angiogenic as well as in adult quiescent ECs, indicating that it is a consequence of GLUT1 deletion independent of EC fate. It remains to be elucidated whether upregulation of inflammatory pathways in ECs is a primary event induced by loss of EC-GLUT1 or is a secondary event caused by gliosis and seizures. Interestingly, in contrast to previous observations using GLUT1 deletion selectively in brain ECs,28 we did not observe more CD206+ macrophages at the brain vasculature nor changes in circulating VEGF levels. The reasons for this discrepancy remain enigmatic and might be related to differences in the kinetics of disease progression, different genetic background of animals, etc. Since inflammation at the BBB plays a crucial role in the pathogenesis of epilepsy,33,43,46,47 future research should reveal whether the observed severe, lethal phenotype upon loss of EC-GLUT1 is mediated, at least partially, via activating inflammatory pathways within ECs.
Taken together, we show that GLUT1 is the main glucose transporter in ECs and is required for EC glycolysis. Loss of EC-GLUT1 reduces endothelial energy availability and reduces proliferation without affecting migration, thereby delaying developmental angiogenesis. Furthermore, in quiescent ECs, GLUT1 becomes uncoupled from EC glycolysis and loss of EC-GLUT1 during adulthood alters CNS homeostasis leading to epilepsy without altering BBB physical barrier properties and the expression of genes controlling or involved in BBB function.
Acknowledgments
We thank all members of the Laboratory of Angiogenesis and Vascular Metabolism for technical assistance.
Sources of Funding
K. Veys, N.V. Conchinha, and J. Kalucka are supported by the Research Foundation Flanders (Fonds voor Wetenschappelijk Onderzoek [FWO] Vlaanderen). K. Van Laere is senior clinical investigator of the FWO. P. Carmeliet is supported by the VIB TechWatch program, long-term structural Methusalem funding by the Flemish Government, FWO, Foundation against Cancer (2016-078), and European Research Council (ERC) Proof-of-concept (ERC-713758) and Advanced Research Grant (EU-ERC743074). A. Keller is supported by the Swiss Cancer League (KLS-3848-02-2016) and the Swiss Life Jubiläumsstiftung. The research of K.D.B. has received funding from the ERC (ERC-716140). K. De Bock is endowed by the Schulthess foundation.
Disclosures
None.
Supplemental Material
Detailed Methods
Online Figures I–VII
Unedited Gels
Major Resources Table
Statistics Data set
Additional Details RNAseq Data set
Supplementary Material
{kind=link}
Footnotes
Nonstandard Abbreviations and Acronyms
- AMPK
- AMP-activated protein kinase
- BBB
- blood-brain barrier
- CA1
- cornu ammonis 1
- CA3
- cornu ammonis 3
- CD206
- cluster of differentiation 206, macrophage mannose receptor
- CDH5
- vascular endothelial cadherin
- CI
- contact inhibited
- CLDN5
- claudin-5
- CNS
- central nervous system
- DAPT
- γ-secretase inhibitor N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester
- Dll4
- delta-like 4
- EC
- endothelial cell
- EdU
- 5-ethynyl-2’-deoxyuridine
- Erg
- ETS-related gene
- GLUT1
- glucose transporter isoform 1
- KLF2
- Krüppel-like factor 2
- PFKFB3
- 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3
- VEGF
- vascular endothelial growth factor
- WT
- wild-type
- ZO1
- zonula occludens 1
K.V. and Z.F. contributed equally to this article.
For Sources of Funding and Disclosures, see page 481
The Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/CIRCRESAHA.119.316463.
Novelty and Significance
What Is Known?
Endothelial cells (ECs) are highly glycolytic and inhibition of endothelial glycolysis impairs developmental and pathological angiogenesis.
ECs within a mature vessel need to tightly balance glucose consumption to fuel their own metabolism with glucose transport to supply the surrounding tissues.
The GLUT1 (glucose transporter isoform 1) glucose transporter is highly expressed in intact quiescent blood-brain barrier (BBB) ECs and lower GLUT1 levels in ECs impairs glucose transport in GLUT1 deficiency syndrome and is often implicated in BBB dysfunction.
What New Information Does This Article Contribute?
GLUT1 is the main glucose transporter in ECs, controls endothelial glycolysis, and inhibition of GLUT1 leads to energy depletion and lower proliferation in vitro, as well as impaired retinal and brain angiogenesis in vivo.
In quiescent ECs, Notch represses PFKFB3 (6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3) and glycolysis, while at the same time increasing GLUT1 expression, and as such uncouples glucose transport from glycolysis.
GLUT1 deficient mice have a phenotype similar to the human GLUT1 deficiency syndrome including seizures but characterized by an activated expression of inflammatory genes.
ECs are highly glycolytic and inhibition of endothelial glycolysis impairs angiogenesis. Quiescent ECs within a mature vessel are also—although less—glycolytic but need to tightly balance glucose consumption for glycolysis with glucose transport to surrounding tissues. The GLUT1 glucose transporter is highly expressed in quiescent BBB ECs and controls glucose transport over the BBB, however, it was not known whether GLUT1 controls endothelial metabolism and function. We found that GLUT1 is the main glucose transporter in ECs. Inhibition of GLUT1 leads to lower glycolysis, energy depletion, activation of the energy sensor AMPK, and lower proliferation in vitro, as well as impaired retinal and brain angiogenesis in vivo. Also, GLUT1 expression increases in quiescent ECs in a Notch-dependent manner. Notch also represses PFKFB3 and glycolysis in quiescent ECs, and thus uncouples glucose transport from glycolysis. Finally, during adulthood—when ECs are quiescent—GLUT1 deficient mice develop a phenotype reminiscent of the human GLUT1 deficiency syndrome, characterized by seizures. Endothelial GLUT1 deletion did, however, not compromise brain vascular function, BBB barrier function, and other molecular key regulators of BBB function. Instead, we found that GLUT1 deficiency activated an inflammatory gene signature.
References
- 1.Adams RH, Alitalo K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol 20078464–478doi: 10.1038/nrm2183 [DOI] [PubMed] [Google Scholar]
- 2.Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell 2011146873–887doi: 10.1016/j.cell.2011.08.039 [DOI] [PubMed] [Google Scholar]
- 3.Kummitha CM, Kalhan SC, Saidel GM, Lai N. Relating tissue/organ energy expenditure to metabolic fluxes in mouse and human: experimental data integrated with mathematical modeling. Physiol Rep. 2014;2:e12159. doi: 10.14814/phy2.12159. doi: 10.14814/phy2.12159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aird WC. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ Res 2007100158–173doi: 10.1161/01.RES.0000255691.76142.4a [DOI] [PubMed] [Google Scholar]
- 5.De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, Quaegebeur A, Ghesquière B, Cauwenberghs S, Eelen G, et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 2013154651–663doi: 10.1016/j.cell.2013.06.037 [DOI] [PubMed] [Google Scholar]
- 6.Potente M, Carmeliet P. The link between angiogenesis and endothelial metabolism. Annu Rev Physiol 20177943–66doi: 10.1146/annurev-physiol-021115-105134 [DOI] [PubMed] [Google Scholar]
- 7.Fitzgerald G, Soro-Arnaiz I, De Bock K. The warburg effect in endothelialells and its potential as an anti-angiogenic target in cancer. Front Cell Dev Biol. 2018;6:100. doi: 10.3389/fcell.2018.00100. doi: 10.3389/fcell.2018.00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yeh WL, Lin CJ, Fu WM. Enhancement of glucose transporter expression of brain endothelial cells by vascular endothelial growth factor derived from glioma exposed to hypoxia. Mol Pharmacol 200873170–177doi: 10.1124/mol.107.038851 [DOI] [PubMed] [Google Scholar]
- 9.Cantelmo AR, Conradi LC, Brajic A, Goveia J, Kalucka J, Pircher A, Chaturvedi P, Hol J, Thienpont B, Teuwen LA, et al. Inhibition of the glycolytic activator PFKFB3 in endothelium induces tumor vessel normalization, impairs metastasis, and improveshemotherapy. Cancer Cell 201630968–985doi: 10.1016/j.ccell.2016.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tang M, Gao G, Rueda CB, Yu H, Thibodeaux DN, Awano T, Engelstad KM, Sanchez-Quintero MJ, Yang H, Li F, et al. Brain microvasculature defects and glut1 deficiency syndrome averted by early repletion of the glucose transporter-1 protein. Nat Commun. 2017;8:14152. doi: 10.1038/ncomms14152. doi: 10.1038/ncomms14152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nolan DJ, Ginsberg M, Israely E, Palikuqi B, Poulos MG, James D, Ding BS, Schachterle W, Liu Y, Rosenwaks Z, et al. Molecular signatures of tissue-specific microvascular endothelial cell heterogeneity in organ maintenance and regeneration. Dev Cell 201326204–219doi: 10.1016/j.devcel.2013.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Vivo DC, Trifiletti RR, Jacobson RI, Ronen GM, Behmand RA, Harik SI. Defective glucose transport across the blood-brain barrier as a cause of persistent hypoglycorrhachia, seizures, and developmental delay. N Engl J Med 1991325703–709doi: 10.1056/NEJM199109053251006 [DOI] [PubMed] [Google Scholar]
- 13.Seidner G, Alvarez MG, Yeh JI, O’Driscoll KR, Klepper J, Stump TS, Wang D, Spinner NB, Birnbaum MJ, De Vivo DC. GLUT-1 deficiency syndrome caused by haploinsufficiency of the blood-brain barrier hexose carrier. Nat Genet 199818188–191doi: 10.1038/ng0298-188 [DOI] [PubMed] [Google Scholar]
- 14.Kalaria RN, Harik SI. Reduced glucose transporter at the blood-brain barrier and in cerebral cortex in alzheimer disease. J Neurochem 1989531083–1088doi: 10.1111/j.1471-4159.1989.tb07399.x [DOI] [PubMed] [Google Scholar]
- 15.Winkler EA, Nishida Y, Sagare AP, Rege SV, Bell RD, Perlmutter D, Sengillo JD, Hillman S, Kong P, Nelson AR, et al. GLUT1 reductions exacerbate alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat Neurosci 201518521–530doi: 10.1038/nn.3966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Siebeneicher H, Cleve A, Rehwinkel H, Neuhaus R, Heisler I, Müller T, Bauser M, Buchmann B. Identification and optimization of the first highly selective GLUT1 inhibitor BAY-876. ChemMedChem 2016112261–2271doi: 10.1002/cmdc.201600276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hardie DG, Carling D, Carlson M. The AMP-activated/SNF1 protein kinase subfamily: metabolic sensors of the eukaryotic cell? Annu Rev Biochem 199867821–855doi: 10.1146/annurev.biochem.67.1.821 [DOI] [PubMed] [Google Scholar]
- 18.Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell 200518283–293doi: 10.1016/j.molcel.2005.03.027 [DOI] [PubMed] [Google Scholar]
- 19.Kalucka J, Bierhansl L, Conchinha NV, Missiaen R, Elia I, Brüning U, Scheinok S, Treps L, Cantelmo AR, Dubois C, et al. Quiescent endothelialells upregulate fatty acid β-oxidation for vasculoprotection via redox homeostasis. Cell Metab 201828881–894.e13doi: 10.1016/j.cmet.2018.07.016 [DOI] [PubMed] [Google Scholar]
- 20.Schoors S, De Bock K, Cantelmo AR, Georgiadou M, Ghesquière B, Cauwenberghs S, Kuchnio A, Wong BW, Quaegebeur A, Goveia J, et al. Partial and transient reduction of glycolysis by PFKFB3 blockade reduces pathological angiogenesis. Cell Metab 20141937–48doi: 10.1016/j.cmet.2013.11.008 [DOI] [PubMed] [Google Scholar]
- 21.Doddaballapur A, Michalik KM, Manavski Y, Lucas T, Houtkooper RH, You X, Chen W, Zeiher AM, Potente M, Dimmeler S, et al. Laminar shear stress inhibits endothelial cell metabolism via KLF2-mediated repression of PFKFB3. Arterioscler Thromb Vasc Biol 201535137–145doi: 10.1161/ATVBAHA.114.304277 [DOI] [PubMed] [Google Scholar]
- 22.Dekker RJ, Boon RA, Rondaij MG, Kragt A, Volger OL, Elderkamp YW, Meijers JC, Voorberg J, Pannekoek H, Horrevoets AJ. KLF2 provokes a gene expression pattern that establishes functional quiescent differentiation of the endothelium. Blood 20061074354–4363doi: 10.1182/blood-2005-08-3465 [DOI] [PubMed] [Google Scholar]
- 23.Young CD, Lewis AS, Rudolph MC, Ruehle MD, Jackman MR, Yun UJ, Ilkun O, Pereira R, Abel ED, Anderson SM. Modulation of glucose transporter 1 (GLUT1) expression levels alters mouse mammary tumor cell growth in vitro and in vivo. PLoS One. 2011;6:e23205. doi: 10.1371/journal.pone.0023205. doi: 10.1371/journal.pone.0023205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Claxton S, Kostourou V, Jadeja S, Chambon P, Hodivala-Dilke K, Fruttiger M. Efficient, inducible cre-recombinase activation in vascular endothelium. Genesis 20084674–80doi: 10.1002/dvg.20367 [DOI] [PubMed] [Google Scholar]
- 25.Wälchli T, Mateos JM, Weinman O, Babic D, Regli L, Hoerstrup SP, Gerhardt H, Schwab ME, Vogel J. Quantitative assessment of angiogenesis, perfused blood vessels and endothelial tip cells in the postnatal mouse brain. Nat Protoc 20151053–74doi: 10.1038/nprot.2015.002 [DOI] [PubMed] [Google Scholar]
- 26.Yazdani S, Jaldin-Fincati JR, Pereira RVS, Klip A. Endothelial cell barriers: transport of molecules between blood and tissues. Traffic 201920390–403doi: 10.1111/tra.12645 [DOI] [PubMed] [Google Scholar]
- 27.Sabbagh MF, Heng JS, Luo C, Castanon RG, Nery JR, Rattner A, Goff LA, Ecker JR, Nathans J. Transcriptional and epigenomic landscapes of CNS and non-CNS vascular endothelial cells. eLife. 2018;7:e36187. doi: 10.7554/eLife.36187. doi: 10.7554/eLife.36187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jais A, Solas M, Backes H, Chaurasia B, Kleinridders A, Theurich S, Mauer J, Steculorum SM, Hampel B, Goldau J, et al. Myeloid-ell-derived VEGF maintains brain glucose uptake and limitsognitive impairment in obesity. Cell 2016165882–895doi: 10.1016/j.cell.2016.03.033 [DOI] [PubMed] [Google Scholar]
- 29.Briellmann RS, Wellard RM, Jackson GD. Seizure-associated abnormalities in epilepsy: evidence from MR imaging. Epilepsia 200546760–766doi: 10.1111/j.1528-1167.2005.47604.x [DOI] [PubMed] [Google Scholar]
- 30.Schroeter A, Grandjean J, Schlegel F, Saab BJ, Rudin M. Contributions of structural connectivity and cerebrovascular parameters to functional magnetic resonance imaging signals in mice at rest and during sensory paw stimulation. J Cereb Blood Flow Metab 2017372368–2382doi: 10.1177/0271678X16666292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Corada M, Orsenigo F, Bhat GP, Conze LL, Breviario F, Cunha SI, Claesson-Welsh L, Beznoussenko GV, Mironov AA, Bacigaluppi M, et al. Fine-tuning of sox17 and canonical Wnt coordinates the permeability properties of the blood-brain barrier. Circ Res 2019124511–525doi: 10.1161/CIRCRESAHA.118.313316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Munji RN, Soung AL, Weiner GA, Sohet F, Semple BD, Trivedi A, Gimlin K, Kotoda M, Korai M, Aydin S, et al. Profiling the mouse brain endothelial transcriptome in health and disease models reveals a core blood-brain barrier dysfunction module. Nat Neurosci 2019221892–1902doi: 10.1038/s41593-019-0497-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Vliet EA, Aronica E, Gorter JA. Role of blood-brain barrier in temporal lobe epilepsy and pharmacoresistance. Neuroscience 2014277455–473doi: 10.1016/j.neuroscience.2014.07.030 [DOI] [PubMed] [Google Scholar]
- 34.Gorter JA, Aronica E, van Vliet EA. The roof is leaking and a storm is raging: repairing the blood-brain barrier in the fight against epilepsy. Epilepsy Curr 201919177–181doi: 10.1177/1535759719844750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Keller A. Breaking and building the wall: the biology of the blood-brain barrier in health and disease. Swiss Med Wkly. 2013;143:w13892. doi: 10.4414/smw.2013.13892. doi: 10.4414/smw.2013.13892. [DOI] [PubMed] [Google Scholar]
- 36.Rohlenova K, Veys K, Miranda-Santos I, De Bock K, Carmeliet P. Endothelial cell metabolism in health and disease. Trends Cell Biol 201828224–236doi: 10.1016/j.tcb.2017.10.010 [DOI] [PubMed] [Google Scholar]
- 37.Yu P, Wilhelm K, Dubrac A, Tung JK, Alves TC, Fang JS, Xie Y, Zhu J, Chen Z, De Smet F, et al. FGF-dependent metabolic control of vascular development. Nature 2017545224–228doi: 10.1038/nature22322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim J, Kim YH, Kim J, Park DY, Bae H, Lee DH, Kim KH, Hong SP, Jang SP, Kubota Y, et al. YAP/TAZ regulates sprouting angiogenesis and vascular barrier maturation. J Clin Invest 20171273441–3461doi: 10.1172/JCI93825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Amemiya T. Glycogen metabolism in the capillary endothelium. Electron histochemical study of glycogen synthetase and phosphorylase in the pecten capillary of the chick. Acta Histochem 19837393–96doi: 10.1016/S0065-1281(83)80080-4 [DOI] [PubMed] [Google Scholar]
- 40.Daneman R, Agalliu D, Zhou L, Kuhnert F, Kuo CJ, Barres BA. Wnt/Β-catenin signaling is required for CNS, but not non-CNS, angiogenesis. PNAS 2009106641–646doi: 10.1073/pnas.0805165106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wilhelm K, Happel K, Eelen G, Schoors S, Oellerich MF, Lim R, Zimmermann B, Aspalter IM, Franco CA, Boettger T, et al. FOXO1 couples metabolic activity and growth state in the vascular endothelium. Nature 2016529216–220doi: 10.1038/nature16498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Simpson IA, Chundu KR, Davies-Hill T, Honer WG, Davies P. Decreased concentrations of GLUT1 and GLUT3 glucose transporters in the brains of patients with alzheimer’s disease. Ann Neurol 199435546–551doi: 10.1002/ana.410350507 [DOI] [PubMed] [Google Scholar]
- 43.Tran KA, Zhang X, Predescu D, Huang X, Machado RF, Göthert JR, Malik AB, Valyi-Nagy T, Zhao YY. Endothelial β-atenin signaling is required for maintaining adult blood-brain barrier integrity and central nervous system homeostasis. Circulation 2016133177–186doi: 10.1161/CIRCULATIONAHA.115.015982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Armulik A, Genové G, Mäe M, Nisancioglu MH, Wallgard E, Niaudet C, He L, Norlin J, Lindblom P, Strittmatter K, et al. Pericytes regulate the blood-brain barrier. Nature 2010468557–561doi: 10.1038/nature09522 [DOI] [PubMed] [Google Scholar]
- 45.Alvarez JI, Dodelet-Devillers A, Kebir H, Ifergan I, Fabre PJ, Terouz S, Sabbagh M, Wosik K, Bourbonnière L, Bernard M, et al. The hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science 20113341727–1731doi: 10.1126/science.1206936 [DOI] [PubMed] [Google Scholar]
- 46.Fabene PF, Navarro Mora G, Martinello M, Rossi B, Merigo F, Ottoboni L, Bach S, Angiari S, Benati D, Chakir A, et al. A role for leukocyte-endothelial adhesion mechanisms in epilepsy. Nat Med 2008141377–1383doi: 10.1038/nm.1878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bertini G, Bramanti P, Constantin G, Pellitteri M, Radu BM, Radu M, Fabene PF. New players in the neurovascular unit: insights from experimental and clinical epilepsy. Neurochem Int 201363652–659doi: 10.1016/j.neuint.2013.08.001 [DOI] [PubMed] [Google Scholar]
- 48.Reimand J, Isserlin R, Voisin V, Kucera M, Tannus-Lopes C, Rostamianfar A, Wadi L, Meyer M, Wong J, Xu C, et al. Pathway enrichment analysis and visualization of omics data using g: profiler, GSEA, cytoscape and enrichmentMap. Nat Protoc 201914482–517doi: 10.1038/s41596-018-0103-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 200510215545–15550doi: 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cura AJ, Carruthers A. Acute modulation of sugar transport in brain capillary endothelial cell cultures during activation of the metabolic stress pathway. J Biol Chem 201028515430–15439doi: 10.1074/jbc.M110.110593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Veys K, Alvarado-Diaz A, De Bock K. Measuring glycolytic and mitochondrial fluxes in endothelial cells using radioactive tracers. Methods Mol Biol 20191862121–136doi: 10.1007/978-1-4939-8769-6_9 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.