

Abstract
The recent emergence of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and the ensuing global pandemic has presented a health emergency of unprecedented magnitude. Recent clinical data has highlighted that coronavirus disease 2019 (COVID-19) is associated with a significant risk of thrombotic complications ranging from microvascular thrombosis, venous thromboembolic disease, and stroke. Importantly, thrombotic complications are markers of severe COVID-19 and are associated with multiorgan failure and increased mortality. The evidence to date supports the concept that the thrombotic manifestations of severe COVID-19 are due to the ability of SARS-CoV-2 to invade endothelial cells via ACE-2 (angiotensin-converting enzyme 2), which is expressed on the endothelial cell surface. However, in patients with COVID-19 the subsequent endothelial inflammation, complement activation, thrombin generation, platelet, and leukocyte recruitment, and the initiation of innate and adaptive immune responses culminate in immunothrombosis, ultimately causing (micro)thrombotic complications, such as deep vein thrombosis, pulmonary embolism, and stroke. Accordingly, the activation of coagulation (eg, as measured with plasma D-dimer) and thrombocytopenia have emerged as prognostic markers in COVID-19. Given thrombotic complications are central determinants of the high mortality rate in COVID-19, strategies to prevent thrombosis are of critical importance. Several antithrombotic drugs have been proposed as potential therapies to prevent COVID-19-associated thrombosis, including heparin, FXII inhibitors, fibrinolytic drugs, nafamostat, and dipyridamole, many of which also possess pleiotropic anti-inflammatory or antiviral effects. The growing awareness and mechanistic understanding of the prothrombotic state of COVID-19 patients are driving efforts to more stringent diagnostic screening for thrombotic complications and to the early institution of antithrombotic drugs, for both the prevention and therapy of thrombotic complications. The shifting paradigm of diagnostic and treatment strategies holds significant promise to reduce the burden of thrombotic complications and ultimately improve the prognosis for patients with COVID-19.
Keywords: coronavirus, mortality, stroke, thrombosis, viruses
The rapidly evolving coronavirus disease 2019 (COVID-19) global pandemic is one of the greatest public health challenges since the Spanish flu pandemic over 100 years ago. Since the discovery of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which causes COVID-19, millions of cases have been diagnosed worldwide resulting in hundreds of thousands of deaths. With no current effective therapy or vaccine for COVID-19, the enormous number of cases and deaths due to COVID-19 is likely to continue to increase. In this regard, although the adverse effects of COVID-19 were initially considered to primarily affect the respiratory tract by causing pneumonia and acute respiratory distress syndrome (ARDS), it has now become apparent that COVID-19 is associated with a prothrombotic state, which can manifest as microvascular thrombosis, venous, or arterial thrombosis, the presence of which usually portends an adverse prognosis. Thus, there is an urgent need to understand how SARS-CoV-2 induces a prothrombotic state to develop effective therapeutic approaches for COVID-19-associated thrombosis. This review will detail the significant emerging clinical findings of the prothrombotic status of patients suffering from COVID-19. We then provide an overview of the inherent links between the immune response and thrombosis, and how these mechanisms may help explain the prothrombotic effects of SARS-CoV-2. Finally, we will use this data to explore available therapeutic approaches for the prevention and treatment of COVID-19-associated thrombosis. Given the relative lack of data to date demonstrating the precise mechanisms of the link between SARS-CoV-2 infection, thrombosis, and clinical outcomes, we also highlight experimental data from other viral infections and thromboinflammatory disorders that are likely relevant to SARS-CoV-2 infection. Further research is urgently required to clearly define how SARS-CoV-2 infection results in thrombotic complications and how this impacts the disease course and severity. Therefore, this review will hopefully spark further interest and research, and importantly raise awareness of the importance of anticoagulation in patients with COVID-19 for both the prevention and treatment of thrombosis.
Coronaviruses Causing Human Disease
Since the beginning of the 21st century, 3 beta coronaviruses have been found to cause severe respiratory illness in humans, including SARS-CoV, MERS-CoV (Middle Eastern respiratory syndrome coronavirus), and now SARS-CoV-2. Coronaviruses reside in many animal hosts and can adapt to different species, including humans. The proximal origin of SARS-CoV and SARS-CoV-2 is thought to be bats, while the reservoir host for MERS-CoV is the dromedary camel.1–3 Previous data suggest that in order for coronaviruses to exhibit efficient zoonotic transmission, the virus must undergo natural selection in an animal host (ie, intermediary species) such that it acquires features affording tropism to human tissue. To date, the intermediate host for SARS-CoV-2 is not known, however, a closely related coronavirus to SARS-CoV-2 has been identified in Malayan pangolins.4 Moreover, it remains to be established whether SARS-CoV-2 has acquired genomic changes within an intermediate animal host before the occurrence of human transmission, or whether a SARS-CoV-2 progenitor may have undergone genomic changes during undetected human-to-human transmission, ultimately resulting in the current pandemic.3
In humans, these coronaviruses gain entry into host cells by way of their transmembrane spike (S) GP (glycoprotein), which comprises an S1 and S2 subunit (Figure 1). The S1 subunit is responsible for binding to the host cell receptor, and the S2 subunit assists with viral and host cell fusion.1 While the MERS-CoV S GP binds to DPP-4 (dipeptidyl peptidase 4) expressed on epithelial tissue, both SARS-CoV-2 and SARS-CoV bind to human cells via the ACE-2 (angiotensin-converting enzyme 2) receptor.5 In addition to having different amino acid residues within the RBD (receptor-binding domain) of the S protein compared to SARS-CoV, another novel structural feature of SARS-CoV-2, is the presence of a polybasic furin cleavage site, at the junction of S1 and S2. Cleavage of the S protein by cellular proteases, including furin and TMPRSS-2 (transmembrane protease serine 2), is speculated to play an important role in the infectivity and host range of SARS-CoV-2, although its functional significance regarding how this feature may mediate SAR-CoV-2 transmissibility and pathogenicity is yet be fully elucidated.1 In the context of COVID-19-associated thrombosis, understanding how these structural differences of SARS-CoV-2 may relate to the prothrombotic phenotype of COVID-19 is likely to be of fundamental importance given the significantly higher rates of thrombosis observed in SARS-CoV-2, compared with SARS-CoV and MERS-CoV infections.
Figure 1.
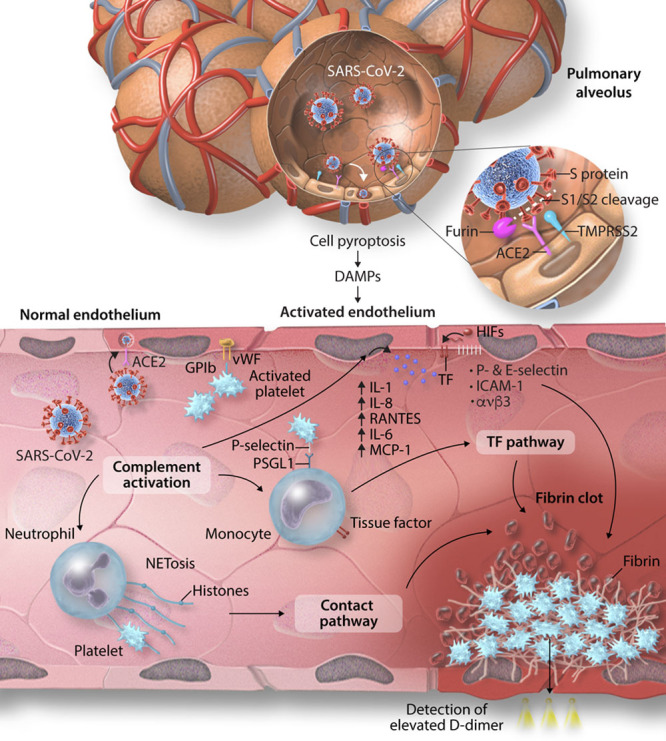
Proposed mechanisms of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) transmission, and coronavirus disease 2019 (COVID-19)-associated thrombosis. SARS-CoV-2 gains entry to host lung epithelial cells by the binding of the transmembrane spike (S) glycoprotein to ACE-2 (angiotensin-converting enzyme 2). The S1 subunit of the S protein binds to ACE-2 and mediates viral attachment. Proteolytic cleavage of the S protein at the S1/2 junction by the proteases, furin, and TMPRSS-2 (transmembrane protease serine 2), facilitates viral entry. SARS-CoV-2 can also directly invade endothelial expressed ACE-2. Infected cells undergo pyroptosis leading to the release of danger-associated molecular patterns (DAMPs) and triggering the release of proinflammatory cytokines and chemokines. The activated endothelium upregulates the expression of VWF (von Willebrand factor) and adhesion molecules including ICAM (intercellular adhesion molecule)-1, αvβ3, P-selectin and E-selectin leading to recruitment of platelets and leukocytes and complement activation. Neutrophils release neutrophil extracellular traps (NETS), causing direct activation of the contact pathway. Complement activation potentiates these mechanisms by increasing endothelial and monocyte tissue factor (TF), further platelet activation and amplifies endothelial inflammation, which increases production of proinflammatory cytokines from the endothelium including IL (interleukin)-1, IL-8, RANTES (regulated on activation, normal T-cell expressed and secreted), IL-6, and MCP (monocyte chemoattractant protein)-1. The hypoxic environment can induce HIFs (hypoxia-inducible factors) which upregulates endothelial TF expression. These mechanisms ultimately lead to the unchecked generation of thrombin, resulting in thrombus formation. The fibrin degradation product, D-dimer, which is a marker of coagulation activation, appears to be a strong prognostic marker associated with high mortality in patients with COVID-19.
SARS-CoV-2 and Its Impact on Coagulation
An emerging threat of the COVID-19 pandemic is the propensity of SARS-CoV-2 to cause microvascular, venous, and arterial thrombosis, and thereby exacerbating organ injury. Patients with severe COVID-19 appear to have a hyperinflammatory response, which is linked to the development of ARDS and multiorgan failure.6 However, it is now well appreciated that the innate immune response and thrombotic response are closely linked (Figure 2) and accordingly in patients with severe COVID-19 there is a correlation with elevated acute phase reactants, such as fibrinogen and CRP (C-reactive protein), which may contribute to COVID-associated hypercoagulability.7–9 Moreover, these biomarkers and acute phase reactants are associated with adverse clinical outcomes and increased severity of infection.7,9,10 In addition, abnormal coagulation parameters, including prolonged prothrombin time and activated partial thromboplastin time, are found to be associated with a higher mortality from COVID-19, demonstrating the significance of coagulation abnormalities in this population.3
Figure 2.
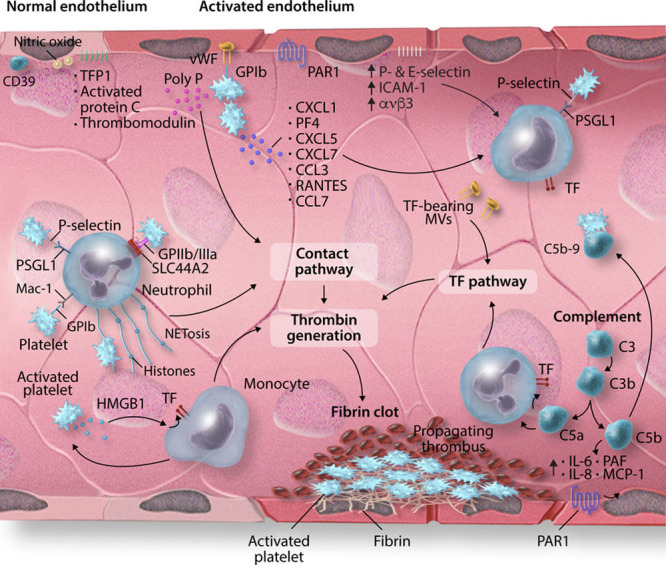
Mechanisms regulating immunothrombosis. In vascular homeostasis, the endothelium possesses anti-inflammatory and antithrombotic properties due to the expression of CD39, nitric oxide (NO), and prostacyclin in addition to the natural anticoagulants, TFPI (tissue factor pathway inhibitor), activated protein C, and thrombomodulin. In the setting of infection or inflammation, endothelial cells upregulate the expression of VWF (von Willebrand factor) and adhesion molecules such as ICAM (intercellular adhesion molecule)-1, αvβ3, P-selectin and E-selectin, promoting the adhesion of leukocytes and platelets. Activated platelets release chemokines CXCL1, PF (platelet factor)-4, CXCL5, CXCL7, CCL3, RANTES (regulated on activation, normal T-cell expressed and secreted), and CCL7 to enhance leukocyte recruitment. Leukocytes interact with platelets via several receptor/ligand pairs. These include platelet P-selectin binding to its cognate receptor PSGL (P-selectin glycoprotein)-1 on leukocytes, GP-Ib on platelets interacting with Mac-1 on monocytes and neutrophils, and GPIIb/IIIa binding to SLC44A2/CTL-2 on neutrophils. Activated platelets release polyphosphate (polyP), which activates the contact pathway, and HMGB (high mobility group box)-1, which enhances monocyte recruitment and monocyte tissue factor (TF) expression, thereby amplifying thrombin generation by way of the TF pathway of coagulation. Neutrophils release neutrophil extracellular traps (NETs) which promote thrombosis via activation of the contact pathway and the binding and activation of platelets. Finally, complement activation leads to the recruitment of leukocytes and upregulates TF expression, amplifies platelet activation and upregulates endothelial expression of proinflammatory cytokines, including IL (interleukin)-1, IL-6, IL-8, and MCP (monocyte chemoattractant protein)-1. These mechanisms result in excess thrombin generation, which potentiates the activation of platelets, leukocytes, and endothelium via PARs (protease-activated receptors) and culminates in a fibrin clot.
Plasma D-dimer measurement is emerging as a direct prognostic marker in COVID-19. In this regard, D-dimer, a fibrin degradation product released when plasmin cleaves cross-linked fibrin, appears to be higher in patients with severe COVID-19 when compared with nonsevere disease.11–15 Subsequently, further studies have highlighted that patients who do not survive COVID-19 have an elevated D-dimer level, and D-dimer continues to increase during admission before death.10,12,16,17
Interplay Between Ethnicity, Coagulation, and Thrombosis
Epidemiological data have demonstrated that race and ethnicity influence the risk of developing venous thromboembolism (VTE), with Blacks found to have the highest rate of incident VTE when compared with other ethnic groups, and Asians and Pacific Islanders having a lower rate of VTE than Whites.18,19 The underlying mechanisms for these racial differences are likely complex and to date remain to be fully understood. Previous data have suggested that higher Factor VIII levels, VWF (von Willebrand Factor) levels and lower Protein C levels observed in African populations may account for some of the racial differences in thrombotic risk.20 In addition, recent data have highlighted important racial differences in platelet reactivity to thrombin, with platelets from Black patients commonly exhibiting polymorphisms in the PAR (protease-activated receptor)-4 that are associated with a markedly enhanced aggregation response to thrombin.21
Interestingly, however, studies evaluating Chinese and White populations have demonstrated that abnormal coagulation parameters are associated with severe infection and poor prognosis from COVID-19 regardless of ethnicity.10,12,16,22 Additionally, as discussed below, rates of thrombosis in COVID-19 appear increased regardless of the ethnic group studied, but the recognized and striking differences between racial groups are important to consider in the context of the SARS-CoV-2 pandemic.
COVID-19 and Venous Thrombosis
Current research has highlighted that rates of VTE in patients with COVID-19 appear markedly increased. Indeed, the cumulative incidence of VTE is reported to be between 25% and 49% of patients with severe COVID-19 with pulmonary embolism being the most common thrombotic complication.23–25 Importantly, even with the use of VTE thromboprophylaxis, the risk of VTE appears to remain elevated.24,26 Interestingly, when compared with non-COVID ARDS, patients with COVID-19 ARDS have a substantially increased rate of pulmonary embolism diagnosis (2.1% versus 11.7%, respectively, P=0.008) and patients diagnosed with a thrombotic complication have more than a 5-fold increase in all-cause mortality.25,27 Recently, 2 autopsy series have revealed that in patients with COVID-19 who were not clinically suspected to have VTE ante mortem, deep vein thrombosis, or pulmonary embolism was a common finding at autopsy (reported in 7/12 [58%] and 11/11 [100%], respectively).28,29 Furthermore, a small study found massive pulmonary embolism to be the direct cause of death in over 30% of patients, highlighting the important interplay between COVID-19 and venous thrombosis.29
COVID-19 and Arterial Thrombosis
In addition to VTE, there are emerging reports that the rate of arterial thromboembolism is increased in patients with COVID-19. Indeed, several groups have now reported rates of acute ischemic stroke of between 3% and 5% in patients admitted to hospital with COVID-19 and, in addition, preliminary reports suggest that these ischemic strokes are affecting younger people without traditional cardiovascular risk factors.24,30,31
Furthermore, acute myocardial injury commonly complicates the clinical course of severe COVID-19 and is reported in up to 20% of hospital presentations with an adverse impact on mortality.32 The underlying cause of myocardial injury is not yet evident, but it is speculated that in addition to potential damage from hypoxia or severe sepsis, SARS-CoV-2 may directly damage myocardial tissue by way of ACE-2 signaling, which is discussed further below.33 As such, controlling for underlying cardiovascular risk factors in patients with COVID-19 remains a priority due to the association with increased severity of disease, in addition to a 5-fold increase in mortality in patients with underlying cardiovascular disease.34,35
COVID-19 and Microvascular Thrombosis
In addition to macrovascular complications, there is a strong association of COVID-19 with microvascular thrombosis. Pulmonary microvascular thrombosis has been previously described in autopsies as a complication of severe ARDS and has subsequently been reported to complicate ARDS during outbreaks of other coronaviruses, including SARS-CoV and MERS-CoV.36–41 However, this feature appears more pronounced in severe SARS-CoV-2 infection, with histology from patients with COVID-19-associated respiratory failure demonstrating a 9-fold increase in the prevalence of alveolar-capillary microthrombi when compared with patients with influenza.42 In this regard, autopsy findings have shown that, in addition to the expected features of diffuse alveolar damage found in ARDS, platelet-fibrin thrombi are a common microscopic finding in the small pulmonary vasculature, occurring in 80% to 100% of lungs examined at autopsy.43–45 In combination with other reported thrombotic events, the emerging microvascular thrombotic complications indicate a strong interaction between the SARS-CoV-2 and coagulation. Below, we review the underlying known mechanisms for thrombosis and the growing body of work that implicates SARS-CoV-2 as a key player driving many molecular processes underpinning pathological thrombosis.
COVID-19 and the Endothelium—Hijacking the Guardian of Vascular Integrity
The primary site of human infection with SARS-CoV-2 is the lung, where S1 attaches with high affinity to ACE-2 expressed on lung alveolar type II epithelial cells (Figure 1).1,2,5 The replication and release of virus from infected cells results in pyroptosis, a highly inflammatory form of cell death that is a common feature among many cytopathic viruses. PRR (Pattern recognition receptors), such as TLR (Toll-like receptors), subsequently detect the release of pathogen-associated molecular patterns, and intracellular danger-associated molecular patterns, such as ATP and DNA, which evokes an intense inflammatory reaction and triggers the release of proinflammatory chemokines and cytokines from neighboring cells (Figure 1).46 Strikingly, SARS-CoV-2 has been found to contain multiple single-stranded RNA fragments that can be recognized by TLR-7 and TLR-8, similar in numbers to MERS-CoV but more than SARS-CoV.47 The recognition of SARS-CoV-2 by these receptors may contribute to innate immune hyperactivation and the cytokine storm often seen in severe COVID-19.
In the setting of SARS-CoV-2 infection, the vascular endothelium appears to be targeted directly by the virus as well as the milieu of proinflammatory cytokines elicited by the immune response.48 ACE-2 is also expressed widely in extrapulmonary sites including blood vessels, heart, kidney, and intestine.49,50 Accordingly, viral RNA has been detected in urine and stool samples from patients with COVID-19.51,52 In this regard, it is currently hypothesized that viral entry via endothelial-expressed ACE-2 represents a mechanism by which the virus can enter and infect other tissues. Recent experimental data have highlighted the ability of COVID-19 to infect human endothelial cells. Indeed, human blood vessel organoids, which closely resemble human capillaries, contain viral RNA after exposure to COVID-19 in vitro.53 Strikingly, the application of human recombinant soluble ACE-2 markedly inhibited viral infection in human capillary organoids, pointing to a direct role for endothelial ACE-2 in viral uptake in blood vessels. Intriguingly, the uptake of SARS-CoV-2 into cells is associated with the downregulation of ACE-2 expression.54,55 This appears to play an important role in promoting a proinflammatory and prothrombotic milieu since ACE-2 plays a pivotal role in regulating the RAS (renin-angiotensin system), by degrading angiotensin II to angiotensin 1–7. While angiotensin II induces vasoconstriction and promotes a proinflammatory/prothrombotic phenotype, angiotensin 1–7 opposes these effects via binding to the MAS receptor, which is widely expressed including on endothelial cells and platelets.55
Interestingly, patients with SARS-CoV-2 have been demonstrated to have increased levels of IL (interleukin)-6, IL-1β, IFN (interferon)-γ, MCP-1 (monocyte chemoattractant protein 1), MIP (macrophage inflammatory protein), and IP10 (CXCL10).11 These proinflammatory cytokines can disrupt endothelial function and integrity leading to the release of VWF, upregulation of adhesion molecules, such as ICAM (intercellular adhesion molecule)-1, integrin αvβ3, P- and E-selectin, and the production of endothelial cytokines and chemokines.56,57 Thus, the endothelium adopts a proinflammatory and procoagulant phenotype that supports the recruitment of platelets and leukocytes.58,59 Furthermore, these changes are associated with the downregulation of the natural anticoagulants and corresponding upregulation of endothelial tissue factor (TF, CD142).57 Moreover, a common finding in patients with COVID-19 is profound hypoxia. Although not formally tested in the context of SARS-CoV-2 infection, hypoxia is also likely to induce a prothrombotic endothelial phenotype since vascular hypoxia induces the activation of HIFs (hypoxia-inducible transcription factors) that have been demonstrated to upregulate endothelial TF expression while downregulating the natural anticoagulants, Protein S and TFPI (tissue factor pathway inhibitor), thus imparting a procoagulant endothelial phenotype.60
A recent case series of autopsy sections from patients with COVID-19 aligns with these experimental findings described above and highlight the ability of COVID-19 to infect endothelial cells and cause endothelial injury. Sections of lung, kidney, and intestine from patients with COVID-19 demonstrate evidence of viral inclusion within endothelial cells, in addition to histological evidence of endothelial inflammation and cell death.48 These data point to a direct role of COVID-19 in promoting systemic microvascular dysfunction which potentially promotes a prothrombotic state.53
Platelets—First Responders in Innate Immunity
Platelets are the second most abundant cell in the circulation with >1 trillion patrolling the vasculature at any one time. Therefore, in addition to their well-appreciated role in mediating hemostasis, they serve important immune functions.61,62 Platelets are equipped with a number of receptors and granular proteins affording them important roles in the immune response. Indeed, activated platelets release a number of chemokines from their alpha and dense granules that facilitate leukocyte recruitment to sites of vascular injury/inflammation. These include CXCL1, PF-4 (platelet factor 4/CXCL4), CXCL5, NAP-2 (CXCL7), and CCL3, RANTES (regulated on activation, normal T-cell expressed and secreted) (CCL5), and CCL7.63 The release of these chemokines by platelets enhances leukocyte recruitment and adhesion to platelet thrombi and also serves to modulate leukocyte functional responses. Moreover, while lacking a nucleus, platelets express IL-1β mRNA that can be translated and secreted as mature IL-1β by activated platelets.64 Further supporting the recruitment of leukocytes, platelets express a range of receptors that facilitate leukocyte adhesion. Activated platelets express P-selectin which allows leukocyte tethering via leukocyte expressed PSGL-1 (P-selectin glycoprotein ligand 1), while GP-Ib facilitates stable adhesion via binding to the leukocyte integrin Mac-1.65,66 More recently, platelets have been observed to have the unique ability to migrate, scavenge, and bundle bacteria up, thereby presenting them to neutrophils for phagocytosis and triggering the release of neutrophil extracellular traps (NETs), which can be induced by the P-selectin/PSGL-1 interaction as well as the recently described platelet GPIIb/IIIa (αIIbβ3, CD41/CD61) interaction with SLC44A2 (CTL-2) on neutrophils.67–69 Activated platelets further support coagulation by the release of inorganic polyphosphate (PolyP) from platelet dense granules which drives intravascular thrombin formation due to its ability to activate FXII.70 Last, activated platelets release microvesicles, small particles ranging in size from 100 to 1000 nm, that are enriched in lipid and can carry a range of cargo including cytokines and chemokines, such as IL-1β, CXCL4, CXCL7, and CCL5, growth factors, microRNA, and mitochondria.71 Moreover, CRP has recently been demonstrated to bind platelet microvesicles and activate complement via binding C1q.72 Thus, the release of platelet microvesicles can regulate immune responses by mediating a range of target cells, particularly leukocytes and endothelial cells.
COVID-19 and Platelets—Sentinels of Viral Infection and Modulators of Adaptive Immunity
The most well-studied effect of SARS-CoV-2 on platelets pertains to its effect on platelet number. Thrombocytopenia has been reported as a common finding in severe COVID-19 with a recent meta-analysis demonstrating that thrombocytopenia was associated with a 5-fold increased risk of severe disease.14,16,73,74 The degree of thrombocytopenia in severe COVID-19 is usually mild and does not appear to be a common feature in nonsevere COVID-19.73 Whether thrombocytopenia is a direct effect of SARS-CoV-2 or multifactorial in the setting of severe illness remains to be elucidated.
To date, there is a relative paucity of data regarding how SARS-CoV-2 may influence platelet function and potentially contribute to the prothrombotic phenotype observed in patients with COVID-19. However, there is now a large body of evidence demonstrating that viruses can directly interact with platelets thus potentially modulating their thrombotic and inflammatory function.75,76 Indeed, platelets are endowed with a large repertoire of adhesion receptors and immune receptors such as TLRs, FcγRIIa, and CXC/CCL receptors in addition to possessing the ability to engulf virions. Influenza, like SARS-CoV-2, is a single-stranded RNA virus which has been demonstrated to activate platelets via TLR-7 and the FcγRIIa receptor. The activation of platelet TLR-7 by viral RNA can evoke a range of functional responses which support thrombus formation, including platelet degranulation, the formation of platelet-leukocyte aggregates and platelet C3 release which in turn stimulates NETosis.75–78 In addition, previous work focusing on platelet activation by viruses has demonstrated that HSV-1 (herpes simplex virus 1) can induce thrombi via FcγRIIa on platelets and that the influenza strain H1N1 can activate platelets, both in a FcγRIIa- and thrombin-dependent fashion, thus leading to platelet GPIIb/IIIa activation, arachidonic acid metabolism, and platelet microvesicle release.79,80 Corresponding mouse studies have emphasized an important role of platelets in H1N1 infection since mice infected with sublethal doses of H1N1 demonstrated large numbers of activated platelets in their lung tissue in addition to platelet-leukocyte aggregates and microvascular thrombosis.81 Interestingly, inhibition of the major platelet adhesion receptor, GPIIb/IIIa, resulted in a protection from fatal influenza and a reduction in lung inflammation and microvascular thrombosis.81 Moreover, recent studies have demonstrated that platelets from patients with influenza contain virus particles, while human platelets appear to have the ability to internalize the influenza virus.77 Thus, a key outstanding area of research regarding the effects of SARS-CoV-2 and thrombosis pertains to how this virus interacts with platelets to mediate platelet number and function. In this regard, understanding if SARS-CoV-2 can be internalized and activate platelet TLR-7 and whether the ACE-2 receptor is expressed on platelets remain core unanswered questions. This is likely to be critical to understanding the link between COVID-19 and the prothrombotic phenotype given the key role of platelets in co-ordinating the thromboinflammatory response.
A prominent clinical feature of severe COVID-19 is the occurrence of respiratory decompensation typically 1 week after initial symptom onset.82 This clinical trajectory appears to correlate with the time course of the adaptive immune response to SARS-CoV-2, with T- and B-cell responses to SARS-CoV-2 being detected ≈1 week after symptom onset.46 Given it is postulated that this respiratory decompensation is driven by pulmonary microvascular thrombosis, understanding the intersection between thrombosis and the adaptive immune response to SARS-CoV-2 is of significant interest. These interactions are likely important since platelets have previously been demonstrated to be key modulators of adaptive immunity in the context of a range of inflammatory disorders. Platelets play an important role in regulating T-cell trafficking by facilitating the recruitment of T lymphocytes to sites of vascular injury or infection.83 Moreover, platelets enhance T-cell functional responses via multiple mechanisms. For example, platelets express major histocompatibility complex class 1 molecules and can, therefore, directly activate T cells in an major histocompatibility complex class 1 dependent manner,84 while platelet-derived PF-4 and serotonin can regulate Th17 expansion85 and naïve T-cell activation and proliferation,86 respectively. The importance of platelets in mediating T-cell recruitment and trafficking is underscored by data demonstrating that platelets play a fundamental role in the accumulation of cytotoxic T cells, and virus-induced organ damage, in a murine model of acute viral hepatitis.87 Akin to the bidirectional relationship between platelets and other leukocyte subsets, the activation of T cells amplifies platelet aggregation with more recent data highlighting an important role for T cells recruited to sites of venous thrombosis in orchestrating the inflammatory response and subsequent thrombus resolution.88,89
In addition to mediating T-cell functions, platelets can increase B-cell immunoglobulin (Ig)G1, IgG2, and IgG3 production.90 This is potentially significant in the setting of COVID-19 since the development of antiphospholipid antibodies, such as anti-β2 glycoprotein IgG antibodies, have been described in association with thrombosis in patients with COVID-19.91 In addition, a cohort study from France describes a large percentage of patients with severe COVID-19 demonstrate a detectable lupus anticoagulant.27 However, whether the presence of antiphospholipid antibodies in COVID-19 plays a role in COVID-19-associated thrombosis or represents merely an association remains to be established. Nevertheless, delineating how platelets modulate the adaptive immune response to SARS-CoV-2 and the role adaptive immunity plays in mediating thrombosis and organ injury will be crucial in uncovering novel therapeutic strategies.
Leukocytes—Innate Immune Cells as Actors in Thrombosis
The recruitment of leukocytes to a thrombus not only aids the immune response but also serves to facilitate platelet activation and coagulation.92 Activated platelets facilitate neutrophils to release NETs, which are a matrix of DNA decorated with neutrophils granule proteins, such as MPO (myeloperoxidase), cathepsin G, and neutrophil elastase.93,94 NETs appear to be a primary defence mechanism to invading pathogens and as such have been demonstrated to trap and kill bacteria and fungi in addition to sequestering viruses.94,95 However, NETs have also been implicated in the pathogenesis of arterial and venous thrombosis in addition to a range of pathologies, including disseminated intravascular coagulation, sepsis, preeclampsia, vasculitis, and systemic lupus erythematosus.96,97 In the context of thrombosis, the release of the highly negatively charged NETs can trap erythrocytes and activate platelets.98 Moreover, NETs serve as a scaffold and potent activator of coagulation via multiple mechanisms. Indeed, neutrophil elastase associated with NETs degrades the anticoagulant protein, TFPI, thereby enhancing TF activity and thrombin generation.96 Histones activate platelets via TLR-2 and TLR-4 and enhance fibrin formation, while the contact activation system, via FXII activation, is initiated by negatively charged DNA.96,99 The activation of FXII not only initiates coagulation but also has important proinflammatory properties by way of its ability to liberate the generation of bradykinin, upregulate neutrophil function, and the generation of monocyte-derived proinflammatory cytokines.100
Activated leukocytes, principally monocytes, and neutrophils, upregulate TF in response to proinflammatory cytokines, predominantly IL-6, and subsequently act to enhance coagulation.101,102 Additionally, monocytes provide a major source of intravascular TF by way of monocyte-derived, TF-bearing microvesicles.103,104 The major danger-associated molecular pattern, HMGB (high mobility group box protein)-1 released by platelets during inflammation has also been shown to be modified by leukocytes, thus facilitating its ability to co-ordinate thrombosis.105 Indeed, platelet-derived HMGB-1 is oxidized by activated monocyte-derived reactive oxygen species, thereby imparting HMGB-1s prothrombotic potential acting in a paracrine fashion to enhance platelet activation, monocyte TF expression and NET formation, and ultimately thrombin generation.105 Interestingly, it has been highlighted that type 1 interferons, a key family of widely expressed cytokines required for the co-ordinated innate immune response to bacterial and viral pathogens, enhance the release of HMBG1 thus enhancing the procoagulant activity of TF.106
COVID-19 and Leukocytes—Adding Fuel to the Thromboinflammatory Fire
The formation of NETs from activated neutrophils represents an innate immune response that aims to corral and kill invading pathogens. However, while having an important immune function, the Janus face of NETosis is the exacerbation of tissue injury, inflammation, and intravascular thrombosis. In the context of respiratory viruses, H1N1 infection has been linked to the excessive formation NETs in a mouse model of viral pneumonitis.95 Moreover, the infiltration of neutrophils at sites of infection and the risk of ARDS development has been demonstrated in a range of other pandemic respiratory viruses, such as H1N1, SARS-CoV, and MERS-CoV.107,108 There is now data demonstrating that patients with COVID-19 exhibit elevated levels of NETs in serum as measured by cell-free DNA, MPO-DNA, and citrullinated histone H3. The levels of MPO-DNA and citrullinated histone H3 were higher in patients receiving mechanical ventilation compared with those not requiring any supplemental oxygen suggesting that levels of NETs correlate with disease severity.109 Interestingly, the finding that the level of NETs correlated with elevations in D-dimer and sera from patients with COVID-19 could induce NETosis in purified neutrophils suggests that NETs, by way of their ability to generate thrombin, via activation of FXII, and activate platelets, may play an important role in mediating the prothrombotic phenotype observed in COVID-19.
One of the striking observations in severe COVID-19 is the correlation between inflammatory markers, such as CRP, ferritin and IL-6 and a raised D-dimer, the latter reporting on an activated coagulation pathway. Although a full discussion regarding the pathological hyperinflammatory response observed in severe COVID-19 is beyond the scope of this review, there is mounting evidence that monocytes/macrophages play a central role in this process.104 This is perhaps best highlighted by the observation that patients with severe COVID-19 in ICU exhibit a significant expansion of CD14+CD16+ monocytes producing IL-6 compared with nonhospitalized patients with COVID-19. As discussed above, this is particularly relevant in the context of thrombosis given the bidirectional relationship between monocytes and coagulation. First, activated platelets form heterotypic aggregates with monocytes in the setting of infection and inflammation. Platelet-monocyte aggregates may play a role in mechanical occlusion of the microvasculature in addition to altering platelet and monocyte functional responses.110,111 In this regard, platelets induce monocyte NF-κB (nuclear factor κ light-chain-enhancer of activated B cells) signaling and thereby modulate monocyte effector functions and induce the synthesis of the proinflammatory cytokines IL-8 and MCP-1.112 Moreover, platelet P-selectin induces monocyte TF expression and upregulates the expression of monocyte Mac-1 (αMβ2, CD11b/CD18).113,114 The platelet-derived chemokines, PF-4 and CXCL-12, enhance monocyte survival and facilitate differentiation into macrophages,115,116 while activated platelets also enhance the phagocytic activity of macrophages.117 A consistent feature of COVID-19 is the finding that patients with existing cardiovascular risk factors are at an increased risk of severe COVID-19 and have higher fatality rates from the disease.34,35 It is noteworthy that oxidized phospholipids, which are strongly associated with atherosclerosis, have been detected in the lungs of patients with SARS-CoV.118,119 OxPLs (oxidised phospholipids) are generated by reactive oxygen species and play important roles in regulating inflammation given their demonstrated roles in promoting monocyte TF production, monocyte activation, and endothelial cell activation.104 The oxidized phospholipid, oxPAPC, has been demonstrated to play a central role in the pathogenesis of a murine model of SARS-induced acute lung injury in a process linked to monocyte/macrophage activation via the TLR-4-TRIF-TRAF-6-NF-κB pathway.119
COVID-19 and Complement—Connecting Innate Immunity and Thrombosis
The complement cascade is another important arm of the innate immune system that has been implicated in COVID-19-associated thrombosis. Activation of the complement system, and vascular deposition of complement components, is linked to a range of thrombotic microangiopathies, including antiphospholipid syndrome and atypical hemolytic uremic syndrome.120 Recent histological findings from biopsies obtained from patients with severe COVID-19 have demonstrated the extensive deposition of the terminal complement components, C5b-9 (membrane attack complex) and C4d, in addition to the MASP-2 (mannose-binding lectin-associated serine protease) within the microvasculature.121 Strikingly, these vessels largely exhibited microvascular thrombi and, furthermore, the SARS-CoV-2 spike glycoprotein colocalized with C5b-9 and C4d in a subset of cases.121 These findings appear consistent with a recent Chinese study where lung sections from patients with severe COVID-19 all revealed strong complement staining, while patients with severe COVID-19 have also been described to have elevated serum C5a levels.122 Mechanistically, it has been suggested that the nucleocapsid (N) protein of SARS-CoV-2 binds to MASP-2 thereby triggering complement activation and amplifying tissue injury. The activation of the complement pathway leads to enhanced production of endothelial cytokines, such as IL-1, IL-8, RANTES, IL-6, and MCP-1, and also upregulates the expression of important endothelial adhesion molecules, such as P-selectin and VWF.120,123 Moreover, the anaphylatoxins C5a and C3a, act to facilitate the recruitment and activation of monocytes and neutrophils. Complement activation also directly induces a prothrombotic phenotype via C5a induction of neutrophil TF expression, C5b-7 monocyte TF expression, and C5b-8/C5b-9 mediated platelet activation.124,125 Furthermore, CRP has been described as a complement pathway activator and the correlation of its plasma level with COVID-19 severity highlights another potential link between immune and complement system.72 These observations have attracted major interest in exploring the role of complement directed therapies in COVID-19 patients, in particular, in COVID-19 mediated thrombosis.126
Thrombin—at the Intersection of Thrombosis and Inflammation
The canonical role of thrombin is the enzymatic cleavage of fibrinogen to fibrin to ensure efficient hemostasis. However, thrombin also exerts pleiotropic proinflammatory effects on endothelial cells, platelets, and leukocytes (Figure 3). In the context of viral infection, the coagulation cascade is activated as a host defence mechanism to try and limit pathogen spread. Indeed, a number of viruses have been demonstrated to directly activate coagulation, including HIV, Ebola, Coxsackie virus, and Dengue.127 Previous data have highlighted that a major mechanism by which viral infection can induce a procoagulant phenotype pertains to the upregulation of TF expression by virus-infected endothelial cells and endothelial TLR-3 signaling in response to virus-derived pathogen-associated molecular patterns.128,129 However, excessive activation of coagulation and thrombin generation can result in deleterious consequences. Thrombin also signals via the PARs, which are widely distributed on a broad range of cells, including platelets, epithelial cells, immune cells, astrocytes, and neurons.130 There are 4 members of the PAR receptors, PAR 1–4, all of which are G protein-coupled receptors. Thrombin efficiently cleaves and activates PAR-1, -3, and -4.131,132 Thus, in addition to being a potent platelet agonist, thrombin also activates immune effector and endothelial cells.133,134 Indeed, thrombin-induced activation of leukocytes results in the production of several cytokines and growth factors while also upregulating leukocyte adhesive function. Thrombin-induced endothelial activation, via PAR-1, induces the production of a range of proinflammatory cytokines and chemokines, including IL-6, IL-8, PAF (platelet-activating factor), and MCP-1, while also triggering the expression of the proadhesive molecules ICAM-1, E- and P-selectin.133–135 Accordingly, PAR signaling has been demonstrated to modulate the immune response to a number of viruses.127 In this regard, the inhibition of coagulation with an FVIIa/TF nematode anticoagulant protein in a model of Ebola virus reduced D-dimer levels, intravascular thrombosis, IL-6, and MCP-1 concentrations and improved survival in treated animals.136 Therefore, the disruption of the normal regulatory mechanisms governing thrombin production and activity occurs in the face of viral sepsis and the ensuing thromboinflammatory response. Thus, given the raised D-dimer levels observed in patients with severe COVID-19, it is likely that dysregulation of the thrombin production is a feature of the prothrombotic phenotype associated with SARS-CoV-2 infection.
Figure 3.

Proposed role of thrombin in coronavirus disease 2019 (COVID-19)-associated immunothrombosis. Infection of endothelial cells by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and liberation of viral danger-associated molecular pattern (DAMPs) results in endothelial activation with the upregulation of tissue factor (TF) and adhesion molecule expression in addition to endothelial cytokine production. This leads to the recruitment and activation of leukocytes and platelets. Activated leukocytes release neutrophil extracellular traps (NETs) and monocyte-derived, TF-bearing microvesicles (MV). Activated platelets release polyP from dense granules. This initiates intravascular thrombin generation via the TF and contact pathways of coagulation. Thrombin exerts its thrombotic effect by activating platelets through the platelet PAR (protease-activated receptor)-1/4 in addition to mediating the cleavage of fibrinogen to fibrin. Furthermore, thrombin possesses proinflammatory functions due to its ability to activate endothelial cells and leukocytes. Thrombin activates endothelial cells via the endothelial PAR-1 receptor, leading to upregulation of IL-6, IL-8, PAF (platelet-activating factor), and MCP (monocyte chemoattractant protein)-1 in addition to the adhesion molecules P-selectin, E-selectin and ICAM (intercellular adhesion molecule)-1, all of which serve to increase leukocyte recruitment and activation. Similarly, monocyte and T-cell functions are enhanced by thrombin activation of monocyte and T-cell expressed PAR-1 and PAR-3. The resultant endothelial cell, platelet, and leukocyte interactions establish a positive feedback loop, which further promulgates ongoing thrombin generation leading to immunothrombosis. This thrombotic phenotype likely results in the clinical manifestations seen in COVID-19, including pulmonary embolism (PE), microvascular thrombosis, ischemic stroke, and deep vein thrombosis (DVT).
Targeting Thrombosis for the Treatment of COVID-19
The high rates of thrombotic complications observed in patients with severe COVID-19, combined with the current lack of an effective therapy for SARS-CoV-2, has led to significant interest regarding the use of antithrombotics for patients with COVID-19. In this regard, antithrombotic drugs which possess anti-inflammatory and/or antiviral properties have already entered clinical trials and will be discussed below (Figure 4).
Figure 4.
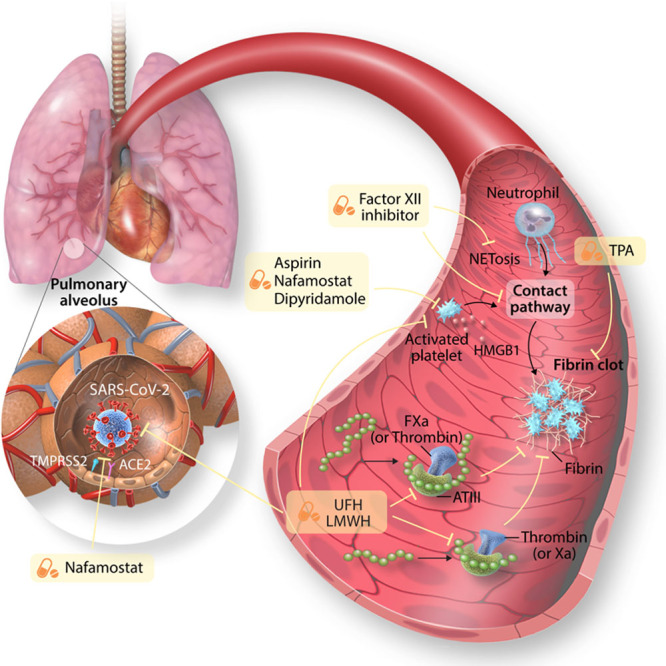
Treatments for targeting coronavirus disease 2019 (COVID-19)-associated thrombosis. Heparins, including unfractionated heparin (UFH) and low molecular weight heparin (LMWH), bind antithrombin (AT), and potentiate the inhibitory effect of AT on coagulation factors Xa and thrombin. Furthermore, UFH may have antiviral effects by having the ability to bind the receptor-binding domain of the S protein of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in addition to potentially acting as a decoy for naturally expressed heparan sulfate thus reducing the ability of the virus to bind to and invade cells. The putative anti-inflammatory effects of UFH is related to its ability to bind danger-associated molecular pattern (DAMPs). Inhibitors of FXII block the contact factor pathway of coagulation, initiated by NETs, and also appear to have pleiotropic anti-inflammatory effects. Antiplatelet agents, such as dipyridamole, nafamostat, and aspirin inhibit platelet activation, which can inhibit NETosis and the release of platelet-derived DAMPs such as HMGB (high mobility group box)-1. Nafamostat may inhibit the TMPRSS-2 (transmembrane protease serine 2) and therefore impede viral entry. Fibrinolytics, such as tPA (tissue-type plasminogen activator), degrade cross-linked fibrin.
Anticoagulation
The strikingly increased prevalence of thrombotic complications has led to many hospitals instituting the routine administration of strict VTE prophylaxis with either low molecular weight heparin or unfractionated heparin and also resulting in several groups using intermediate or full-dose anticoagulation to prevent these thromboembolic phenomena.137,138 Preliminary reports have suggested that low molecular weight heparin treatment reduces mortality in COVID-19 patients with an elevated D-dimer or elevated sepsis-induced coagulopathy score.139 The heparins are widely used anticoagulants which inhibit coagulation by the antithrombin mediated inhibition of FXa or thrombin. However, aside from their effects on anticoagulation, the heparins appear to have pleiotropic effects that may provide unique advantages in the context of viral infection, including anti-inflammatory effects by way of their ability to bind to danger-associated molecular patterns, such as HMGB-1, and proinflammatory cytokines.140,141 Unfractionated heparin has a similar structure as heparan sulfate, the sulfated polysaccharide present on cell surfaces and extracellular matrix, with repeating disaccharide units. In contrast, low molecular weight heparin is a modified heparin with shorter polysaccharide chains. Intriguingly, heparan sulfate has previously implicated as a docking site for a number of viruses, thus facilitating cellular attachment of the virus and subsequent viral infection.142,143 Indeed, heparin, acting as a decoy receptor for naturally expressed heparan sulfate, has been shown to inhibit the binding of the SARS coronavirus to cells in vitro.144 While ACE-2 is the major receptor that mediates viral entry into cells, heparan sulfate has previously been demonstrated to play an important role in facilitating coronavirus entry into cells by functioning as an adhesion receptor for the virus on the cell surface thereby allowing the increased density of viral particles on the cell surface which then facilitates ACE-2-mediated cell entry.145 Recent studies have directly investigated the link between heparin and SARS-CoV-2 and suggest that heparin can directly bind with the spike protein of the SARS-CoV-2 virus and, therefore, may have antiviral properties.146
Another interesting aspect of how the SARS-CoV-2 virus may be influenced by the use of heparin as anticoagulant is the previous observation that FXa can cleave the spike protein of SARS-CoV to render it more efficient in invading cells.147 However, to date, this aspect remains to be investigated with SARS-CoV-2. These data have led some groups to propose specifically engineered heparins for the treatment of COVID-19, while clinical trials are underway to investigate the effects of low dose, and full-dose, anticoagulation with heparin on clinical outcomes for patients with COVID-19 (URL: https://www.clinicaltrials.gov. Unique identifiers: NCT04344756 and NCT04345848).142 It is important to note that given the potential for heparin therapeutics to cause heparin-induced thrombocytopenia, but reassuringly, despite both thrombocytopenia and thrombosis being common complications of COVID-19, the development of heparin-induced thrombocytopenia appears limited to individual case reports.148
Inhibition of the Contact Factor Activation Pathway
The inhibition of FXII as means to inhibit the thromboinflammatory response incited by severe COVID-19 appears an attractive and rational therapeutic target. Activation of the contact pathway is initiated by FXII activation by negatively charged molecules such as NETs, which are increased in severe COVID-19 patients, and platelet-derived PolyP.100 This leads to the generation of thrombin and the activation of bradykinin, of which the latter in particular results in downstream complement activation and the production of inflammatory cytokines.149 Importantly, therapeutic inhibition of FXIIa demonstrates protection from occlusive thrombus formation in animal models of thrombosis without impeding hemostasis.150 Treatment with an inhibitory FXIIa antibody in animal models of systemic inflammatory response syndrome, using a lethal E coli challenge, have shown that FXIIa inhibition improves survival and decreases a number of inflammatory markers including complement activation, neutrophil degranulation, and IL-6.151–153 Importantly, the potential safety of FXII inhibitors is highlighted by the fact that FXII-deficient individuals display no bleeding phenotype, and there is no known effect on immune function.154 A FXIIa inhibitory antibody has recently entered clinical trials for the treatment of hereditary angioedema and therefore represents a promising novel combination of antithrombotic and anti-inflammatory drugs (URL: https://www.clinicaltrials.gov. Unique identifier: NCT03712228).
Fibrinolysis as a Novel Therapy in COVID-19
Other therapies targeting coagulation are being examined regarding their potential therapeutic utility in COVID-19, including fibrinolytic agents such as tPA (tissue-type plasminogen activator). tPA converts the inactive enzyme plasminogen to plasmin leading to the subsequent breakdown of cross-linked fibrin. The interest in tPA has emerged from the concept that ARDS is characterized by significant local inflammatory reaction in addition to a hypofibrinolytic state. As discussed previously, severe COVID-19 can result in ARDS, which promotes mass upregulation of inflammatory cytokines and leukocytes, and can result in extensive thrombin generation, fibrin formation, and microvascular thrombosis.155
The use of systemic tPA in preclinical and human models of ARDS demonstrates improved oxygenation, and a case report of tPA use in severe COVID-19 showed an initial improvement in oxygenation during tPA infusion, but this effect was lost after tPA therapy ceased.156–158 However, the benefits of tPA may be offset by the considerable risk of major bleeding seen in nonstroke clinical trials,159 which has led to the consideration of nebulized therapy to increase local concentrations and reduce systemic coagulation effects. Recent clinical trial data suggest that, in severe ARDS, nebulized fibrinolytic therapy is associated with improved oxygenation and ventilatory parameters, which may be another plausible treatment option in COVID-19.155
Antiplatelet Agents as Potential Therapeutics for COVID-19
A recent meta-analysis has demonstrated that antiplatelet therapy is associated with improved mortality from sepsis.160 Interestingly, the majority of data linking antiplatelet therapy with these improved clinical outcomes relates to aspirin.161 In this regard, recent experimental data have highlighted that targeting platelets with aspirin prevents intravascular coagulation and neutrophil-mediated microvascular thrombosis in a mouse model of bacterial sepsis.162 However, to date, the effect of aspirin in COVID-19 remains to be established. Several other therapies with antiplatelet effects, including dipyridamole and nafamostat, are currently being evaluated for their potential role in reducing the severity of COVID-19. The antiplatelet agent dipyridamole has recently been reported to suppress SARS-CoV-2 replication in vitro, with some early data suggesting that use of adjunct dipyridamole may improve the clinical course in severe COVID-19.163 Finally, the role of nafamostat, a serine protease inhibitor which has antiplatelet effects and is currently marketed in Asia for the treatment of disseminated intravascular coagulation and pancreatitis, is being evaluated.164 Nafamostat is known to inhibit TMPRSS-2, and in preclinical studies, serine protease inhibitors are able to block SARS-CoV-2 infection of lung cells. Prior studies of nafamostat demonstrated that the drug has potential in blocking MERS-CoV infection in vitro, suggesting a potential role for this drug in reducing the burden of severe COVID-19.5,165 The RACONA Study (URL: https://www.clinicaltrials.gov. Unique identifier: NCT04352400) is a double-blind trial set to evaluate the efficacy of this agent in severe COVID-19. Nonetheless, it remains to be seen if any of these investigational therapies by virtue of their antiplatelet or antiviral effects will play a substantial role in the treatment of COVID-19.
Summary and Conclusions
The emergence of the SARS-CoV-2 virus and the ensuing global pandemic has presented unprecedented healthcare and economic challenges. A key clinical feature of severe COVID-19 seems to be a highly prothrombotic state, which is linked to excess rates of arterial, venous and microvascular thrombosis, and adverse clinical outcomes. Emerging evidence suggests that SARS-CoV-2 can infect endothelial cells with the associated immune response and ensuing activation of inflammatory pathways resulting in dysregulation of the endothelium, leukocyte activation, NET generation, complement deposition, and platelet consumption. These pathways can conspire to unleash a prothrombotic state (immunothrombosis), which may result in significant thrombotic complications. Accordingly, there remains much interest, and an urgent clinical need, to precisely delineate the mechanisms by which SARS-CoV-2 infection causes thrombotic complications in the hope that new insights into this process will yield novel therapeutic approaches. This is especially pressing in the absence of an effective therapy or vaccine for SARS-CoV-2. Thus, in the context of COVID-19 the intersection of innate and adaptive immunity, inflammation, and thrombosis has been thrust into the global spotlight, and the challenge is now to urgently find therapeutic concepts to disrupt this unholy trinity in the face of a global pandemic. The growing awareness and understanding of thrombotic complications in patients with SARS-CoV-2 infection will contribute to a more rigorous diagnostic approach resulting in the earlier detection of thrombotic events. Importantly, the shift towards the early institution of antithrombotic therapy for the prevention and treatment of COVID-19-associated thrombosis will hopefully translate into improved outcomes for patients with COVID-19.
Sources of Funding
J.D. McFadyen is supported by a National Health and Medical Research Council (NHMRC) and Heart Foundation Early Career Fellowship. H. Stevens is supported by the Monash University RTP Scholarship, Wheaton Family Scholarship, and HSANZ New Investigator Scholarship. K. Peter is supported by an NHMRC Senior Principal Research Fellowship.
Disclosures
None.
Footnotes
Nonstandard Abbreviations and Acronyms
- ACE-2
- angiotensin-converting enzyme 2
- ARDS
- acute respiratory distress syndrome
- COVID-19
- coronavirus disease 2019
- CRP
- C-reactive protein
- DPP-4
- dipeptidyl peptidase 4
- GP
- glycoprotein
- HIFs
- hypoxia-inducible transcription factors
- HMGB
- high mobility group box protein
- HSV-1
- herpes simplex virus 1
- ICAM-1
- intercellular adhesion molecule 1
- IFN
- interferon
- IL
- interleukin
- MCP-1
- monocyte chemoattractant protein 1
- MERS-CoV
- Middle Eastern respiratory syndrome coronavirus
- MIP
- macrophage inflammatory protein
- MPO
- myeloperoxidase
- NETs
- neutrophil extracellular traps
- NF-κB
- nuclear factor κ light-chain-enhancer of activated B cells
- PAF
- platelet-activating factor
- PAR-4
- protease-activated receptor 4
- PRR
- pattern recognition receptors
- PSGL-1
- P-selectin glycoprotein ligand 1
- RANTES
- regulated on activation, normal T-cell expressed and secreted
- RBD
- receptor-binding domain
- SARS-CoV-2
- severe acute respiratory syndrome coronavirus 2
- TFPI
- tissue factor pathway inhibitor
- TLR
- Toll-like receptors
- TMPRSS-2
- transmembrane protease serine 2
- tPA
- tissue-type plasminogen activator
- VTE
- venous thromboembolism
- VWF
- von Willebrand Factor
J.D.M. and H.S. contributed equally to this article.
For Sources of Funding and Disclosures, see page 584.
References
- 1.Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020181281–292.e6doi: 10.1016/j.cell.2020.02.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, Si HR, Zhu Y, Li B, Huang CL, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020579270–273doi: 10.1038/s41586-020-2012-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF. The proximal origin of SARS-CoV-2. Nat Med 202026450–452doi: 10.1038/s41591-020-0820-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lam TT, Jia N, Zhang YW, Shum MH, Jiang JF, Zhu HC, Tong YG, Shi YX, Ni XB, Liao YS, et al. Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature. 2020 doi: 10.1038/s41586-020-2169-0. doi: 10.1038/s41586-020-2169-0. [DOI] [PubMed] [Google Scholar]
- 5.Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu NH, Nitsche A, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020181271–280.e8doi: 10.1016/j.cell.2020.02.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jose RJ, Manuel A. COVID-19 cytokine storm: the interplay between inflammation and coagulation. Lancet Respir Med 20208e46–e47doi: 10.1016/S2213-2600(20)30216-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu F, Li L, Xu M, Wu J, Luo D, Zhu Y, Li B, Song X, Zhou X. Prognostic value of interleukin-6, C-reactive protein, and procalcitonin in patients with COVID-19. J Clin Virol. 2020;127:104370. doi: 10.1016/j.jcv.2020.104370. doi: 10.1016/j.jcv.2020.104370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maier CL, Truong AD, Auld SC, Polly DM, Tanksley CL, Duncan A. COVID-19-associated hyperviscosity: a link between inflammation and thrombophilia? Lancet 20203951758–1759doi: 10.1016/S0140-6736(20)31209-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giamarellos-Bourboulis EJ, Netea MG, Rovina N, Akinosoglou K, Antoniadou A, Antonakos N, Damoraki G, Gkavogianni T, Adami ME, Katsaounou P, et al. Complex immune dysregulation in COVID-19 patients with severe respiratory failure. Cell Host Microbe 202027992–1000.e3doi: 10.1016/j.chom.2020.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tang N, Li D, Wang X, Sun Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost 202018844–847doi: 10.1111/jth.14768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020395497–506doi: 10.1016/S0140-6736(20)30183-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, Wang B, Xiang H, Cheng Z, Xiong Y, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA 20203231061–1069doi: 10.1001/jama.2020.1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang JJ, Dong X, Cao YY, Yuan YD, Yang YB, Yan YQ, Akdis CA, Gao YD. Clinical characteristics of 140 patients infected with SARS-CoV-2 in Wuhan, China. Allergy. 2020 doi: 10.1111/all.14238. doi: 10.1111/all.14238. [DOI] [PubMed] [Google Scholar]
- 14.Guan WJ, Ni ZY, Hu Y, Liang WH, Ou CQ, He JX, Liu L, Shan H, Lei CL, Hui DSC, et al. ; China Medical Treatment Expert Group for Covid-19 Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med 20203821708–1720doi: 10.1056/NEJMoa2002032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lippi G, Favaloro EJ. D-dimer is associated with severity of coronavirus disease 2019: a pooled analysis. Thromb Haemost 2020120876–878doi: 10.1055/s-0040-1709650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, Xiang J, Wang Y, Song B, Gu X, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 20203951054–1062doi: 10.1016/S0140-6736(20)30566-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang L, Yan X, Fan Q, Liu H, Liu X, Liu Z, Zhang Z. D-dimer levels on admission to predict in-hospital mortality in patients with Covid-19. J Thromb Haemost 2020181324–1329doi: 10.1111/jth.14859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liao S, Woulfe T, Hyder S, Merriman E, Simpson D, Chunilal S. Incidence of venous thromboembolism in different ethnic groups: a regional direct comparison study. J Thromb Haemost 201412214–219doi: 10.1111/jth.12464 [DOI] [PubMed] [Google Scholar]
- 19.White RH, Keenan CR. Effects of race and ethnicity on the incidence of venous thromboembolism. Thromb Res 2009123 Suppl 4S11–S17doi: 10.1016/S0049-3848(09)70136-7 [DOI] [PubMed] [Google Scholar]
- 20.Zakai NA, McClure LA. Racial differences in venous thromboembolism. J Thromb Haemost 201191877–1882doi: 10.1111/j.1538-7836.2011.04443.x [DOI] [PubMed] [Google Scholar]
- 21.Edelstein LC, Simon LM, Montoya RT, Holinstat M, Chen ES, Bergeron A, Kong X, Nagalla S, Mohandas N, Cohen DE, et al. Racial differences in human platelet PAR4 reactivity reflect expression of PCTP and miR-376c. Nat Med 2013191609–1616doi: 10.1038/nm.3385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fogarty H, Townsend L, Ni Cheallaigh C, Bergin C, Martin-Loeches I, Browne P, Bacon CL, Gaule R, Gillett A, Byrne M, et al. COVID19 coagulopathy in Caucasian patients. Br J Haematol 20201891044–1049doi: 10.1111/bjh.16749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cui S, Chen S, Li X, Liu S, Wang F. Prevalence of venous thromboembolism in patients with severe novel coronavirus pneumonia. J Thromb Haemost 2020181421–1424doi: 10.1111/jth.14830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klok FA, Kruip MJHA, van der Meer NJM, Arbous MS, Gommers DAMPJ, Kant KM, Kaptein FHJ, van Paassen J, Stals MAM, Huisman MV, et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res 2020191145–147doi: 10.1016/j.thromres.2020.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klok FA, Kruip MJHA, van der Meer NJM, Arbous MS, Gommers D, Kant KM, Kaptein FHJ, van Paassen J, Stals MAM, Huisman MV, et al. Confirmation of the high cumulative incidence of thrombotic complications in critically ill ICU patients with COVID-19: an updated analysis. Thromb Res 2020191148–150doi: 10.1016/j.thromres.2020.04.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Middeldorp S, Coppens M, van Haaps TF, Foppen M, Vlaar AP, Muller MCA, Bouman CCS, Beenen LFM, Kootte RS, Heijmans J, et al. Incidence of venous thromboembolism in hospitalized patients with covid-19. J Thromb Haemost. 2020 doi: 10.1111/jth.14888. doi: 10.1111/jth.14888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Helms J, Tacquard C, Severac F, Leonard-Lorant I, Ohana M, Delabranche X, Merdji H, Clere-Jehl R, Schenck M, Fagot Gandet F, et al. ; CRICS TRIGGERSEP Group (Clinical Research in Intensive Care and Sepsis Trial Group for Global Evaluation and Research in Sepsis) High risk of thrombosis in patients with severe SARS-CoV-2 infection: a multicenter prospective cohort study. Intensive Care Med 2020461089–1098doi: 10.1007/s00134-020-06062-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lax SF, Skok K, Zechner P, Kessler H, Kaufmann N, Koelblinger C, Vander K, Bargfrieder U, Trauner M. Pulmonary arterial thrombosis in COVID-19 with fatal outcome: results from a prospective, single-center, clinicopathologic case series. Ann Intern Med. 2020:M20-2566. doi: 10.7326/M20-2566. doi: 10.7326/M20-2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wichmann D, Sperhake JP, Lutgehetmann M, Steurer S, Edler C, Heinemann A, Heinrich F, Mushumba H, Kniep I, Schroder AS, et al. Autopsy findings and venous thromboembolism in patients with COVID-19: a prospective cohort study. Ann Intern Med. 2020:M20-2003. doi: 10.7326/M20-2003. doi: 10.7326/M20-2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mao L, Jin H, Wang M, Hu Y, Chen S, He Q, Chang J, Hong C, Zhou Y, Wang D, Miao X, et al. Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China. JAMA Neurology 2020771–9doi: 10.1001/jamaneurol.2020.1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oxley TJ, Mocco J, Majidi S, Kellner CP, Shoirah H, Singh IP, De Leacy RA, Shigematsu T, Ladner TR, Yaeger KA, et al. Large-vessel stroke as a presenting feature of COVID-19 in the young. N Engl J Med. 2020;382:e60. doi: 10.1056/NEJMc2009787. doi: 10.1056/NEJMc2009787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shi S, Qin M, Shen B, Cai Y, Liu T, Yang F, Gong W, Liu X, Liang J, Zhao Q, et al. Association of cardiac injury with mortality in hospitalized patients with COVID-19 in Wuhan, China. JAMA Cardiol. 2020:e200950. doi: 10.1001/jamacardio.2020.0950. doi: 10.1001/jamacardio.2020.0950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zheng YY, Ma YT, Zhang JY, Xie X. COVID-19 and the cardiovascular system. Nat Rev Cardiol 202017259–260doi: 10.1038/s41569-020-0360-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang J, Zheng Y, Gou X, Pu K, Chen Z, Guo Q, Ji R, Wang H, Wang Y, Zhou Y. Prevalence of comorbidities and its effects in patients infected with SARS-CoV-2: a systematic review and meta-analysis. Int J Infect Dis 20209491–95doi: 10.1016/j.ijid.2020.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu Z, McGoogan JM. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: Summary of a report of 72314 cases from the Chinese center for disease control and prevention. JAMA. 2020 doi: 10.1001/jama.2020.2648. doi: 10.1001/jama.2020.2648. [DOI] [PubMed] [Google Scholar]
- 36.Lang ZW, Zhang LJ, Zhang SJ, Meng X, Li JQ, Song CZ, Sun L, Zhou YS, Dwyer DE. A clinicopathological study of three cases of severe acute respiratory syndrome (SARS). Pathology 200335526–531doi: 10.1080/00313020310001619118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ding Y, Wang H, Shen H, Li Z, Geng J, Han H, Cai J, Li X, Kang W, Weng D, et al. The clinical pathology of severe acute respiratory syndrome (SARS): a report from China. J Pathol 2003200282–289doi: 10.1002/path.1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hwang DM, Chamberlain DW, Poutanen SM, Low DE, Asa SL, Butany J. Pulmonary pathology of severe acute respiratory syndrome in Toronto. Mod Pathol 2005181–10doi: 10.1038/modpathol.3800247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Franks TJ, Chong PY, Chui P, Galvin JR, Lourens RM, Reid AH, Selbs E, McEvoy CP, Hayden CD, Fukuoka J, et al. Lung pathology of severe acute respiratory syndrome (SARS): a study of 8 autopsy cases from Singapore. Hum Pathol 200334743–748doi: 10.1016/s0046-8177(03)00367-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li K, Wohlford-Lenane C, Perlman S, Zhao J, Jewell AK, Reznikov LR, Gibson-Corley KN, Meyerholz DK, McCray PB., Jr. Middle east respiratory syndrome coronavirus causes multiple organ damage and lethal disease in mice transgenic for human dipeptidyl peptidase 4. J Infect Dis 2016213712–722doi: 10.1093/infdis/jiv499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tomashefski JF, Jr, Davies P, Boggis C, Greene R, Zapol WM, Reid LM. The pulmonary vascular lesions of the adult respiratory distress syndrome. Am J Pathol 1983112112–126 [PMC free article] [PubMed] [Google Scholar]
- 42.Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, Vanstapel A, Werlein C, Stark H, Tzankov A, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in covid-19. N Engl J Med. 2020 doi: 10.1056/NEJMoa2015432. doi: 10.1056/NEJMoa2015432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fox SE, Akmatbekov A, Harbert JL, Li G, Quincy Brown J, Vander Heide RS. Pulmonary and cardiac pathology in African American patients with COVID-19: an autopsy series from New Orleans. Lancet Respir Med. 2020:S2213-2600(20)30243-5. doi: 10.1016/S2213-2600(20)30243-5. doi: 10.1016/S2213-2600(20)30243-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carsana L, Sonzogni A, Nasr A, Rossi RS, Pellegrinelli A, Zerbi P, Rech R, Colombo R, Antinori S, Corbellino M, et al. Pulmonary post-mortem findings in a series of COVID-19 cases from northern Italy: a two-centre descriptive study. Lancet Infect Dis. 2020:S1473-3099(20)30434-5. doi: 10.1016/S1473-3099(20)30434-5. doi: 10.1016/S1473-3099(20)30434-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dolhnikoff M, Duarte-Neto AN, de Almeida Monteiro RA, da Silva LFF, de Oliveira EP, Saldiva PHN, Mauad T, Negri EM. Pathological evidence of pulmonary thrombotic phenomena in severe COVID-19. J Thromb Haemost 2020181517–1519doi: 10.1111/jth.14844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tay MZ, Poh CM, Rénia L, MacAry PA, Ng LFP. The trinity of COVID-19: immunity, inflammation and intervention. Nat Rev Immunol 202020363–374doi: 10.1038/s41577-020-0311-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moreno-Eutimio MA, López-Macías C, Pastelin-Palacios R. Bioinformatic analysis and identification of single-stranded RNA sequences recognized by TLR7/8 in the SARS-CoV-2, SARS-CoV, and MERS-CoV genomes. Microbes Infect 202022226–229doi: 10.1016/j.micinf.2020.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, Mehra MR, Schuepbach RA, Ruschitzka F, Moch H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 20203951417–1418doi: 10.1016/S0140-6736(20)30937-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Crackower MA, Sarao R, Oudit GY, Yagil C, Kozieradzki I, Scanga SE, Oliveira-dos-Santos AJ, da Costa J, Zhang L, Pei Y, et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 2002417822–828doi: 10.1038/nature00786 [DOI] [PubMed] [Google Scholar]
- 50.Danilczyk U, Penninger JM. Angiotensin-converting enzyme II in the heart and the kidney. Circ Res 200698463–471doi: 10.1161/01.RES.0000205761.22353.5f [DOI] [PubMed] [Google Scholar]
- 51.Chen Y, Chen L, Deng Q, Zhang G, Wu K, Ni L, Yang Y, Liu B, Wang W, Wei C, et al. The presence of SARS-CoV-2 RNA in the feces of COVID-19 patients. J Med Virol 202092833–840doi: 10.1002/jmv.25825 [DOI] [PubMed] [Google Scholar]
- 52.Peng L, Liu J, Xu W, Luo Q, Chen D, Lei Z, Huang Z, Li X, Deng K, Lin B, et al. Sars-cov-2 can be detected in urine, blood, anal swabs, and oropharyngeal swabs specimens. J Med Virol. 2020 doi: 10.1002/jmv.25936. doi: 10.1002/jmv.25936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Monteil V, Kwon H, Prado P, Hagelkrüys A, Wimmer RA, Stahl M, Leopoldi A, Garreta E, Hurtado Del Pozo C, Prosper F, et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell 2020181905–913.e7doi: 10.1016/j.cell.2020.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang H, Penninger JM, Li Y, Zhong N, Slutsky AS. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med 202046586–590doi: 10.1007/s00134-020-05985-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Verdecchia P, Cavallini C, Spanevello A, Angeli F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur J Intern Med 20207614–20doi: 10.1016/j.ejim.2020.04.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kayal S, Jaïs JP, Aguini N, Chaudière J, Labrousse J. Elevated circulating E-selectin, intercellular adhesion molecule 1, and von Willebrand factor in patients with severe infection. Am J Respir Crit Care Med 1998157776–784doi: 10.1164/ajrccm.157.3.9705034 [DOI] [PubMed] [Google Scholar]
- 57.Aird WC. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood 20031013765–3777doi: 10.1182/blood-2002-06-1887 [DOI] [PubMed] [Google Scholar]
- 58.Massberg S, Enders G, Leiderer R, Eisenmenger S, Vestweber D, Krombach F, Messmer K. Platelet-endothelial cell interactions during ischemia/reperfusion: the role of P-selectin. Blood 199892507–515 [PubMed] [Google Scholar]
- 59.Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell 199476301–314doi: 10.1016/0092-8674(94)90337-9 [DOI] [PubMed] [Google Scholar]
- 60.Gupta N, Zhao YY, Evans CE. The stimulation of thrombosis by hypoxia. Thromb Res 201918177–83doi: 10.1016/j.thromres.2019.07.013 [DOI] [PubMed] [Google Scholar]
- 61.Semple JW, Italiano JE, Jr, Freedman J. Platelets and the immune continuum. Nat Rev Immunol 201111264–274doi: 10.1038/nri2956 [DOI] [PubMed] [Google Scholar]
- 62.McFadyen JD, Kaplan ZS. Platelets are not just for clots. Transfus Med Rev 201529110–119doi: 10.1016/j.tmrv.2014.11.006 [DOI] [PubMed] [Google Scholar]
- 63.Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol 20131334–45doi: 10.1038/nri3345 [DOI] [PubMed] [Google Scholar]
- 64.Lindemann S, Tolley ND, Dixon DA, McIntyre TM, Prescott SM, Zimmerman GA, Weyrich AS. Activated platelets mediate inflammatory signaling by regulated interleukin 1beta synthesis. J Cell Biol 2001154485–490doi: 10.1083/jcb.200105058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Palabrica T, Lobb R, Furie BC, Aronovitz M, Benjamin C, Hsu YM, Sajer SA, Furie B. Leukocyte accumulation promoting fibrin deposition is mediated in vivo by P-selectin on adherent platelets. Nature 1992359848–851doi: 10.1038/359848a0 [DOI] [PubMed] [Google Scholar]
- 66.Simon DI, Chen Z, Xu H, Li CQ, Dong Jf, McIntire LV, Ballantyne CM, Zhang L, Furman MI, Berndt MC, et al. Platelet glycoprotein ibalpha is a counterreceptor for the leukocyte integrin Mac-1 (CD11b/CD18). J Exp Med 2000192193–204doi: 10.1084/jem.192.2.193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gaertner F, Ahmad Z, Rosenberger G, Fan S, Nicolai L, Busch B, Yavuz G, Luckner M, Ishikawa-Ankerhold H, Hennel R, et al. Migrating platelets are mechano-scavengers that collect and bundle bacteria. Cell 20171711368–1382.e23doi: 10.1016/j.cell.2017.11.001 [DOI] [PubMed] [Google Scholar]
- 68.Etulain J, Martinod K, Wong SL, Cifuni SM, Schattner M, Wagner DD. P-selectin promotes neutrophil extracellular trap formation in mice. Blood 2015126242–246doi: 10.1182/blood-2015-01-624023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Constantinescu-Bercu A, Grassi L, Frontini M, Salles C, II, Woollard K, Crawley JT. Activated alphaiibbeta3 on platelets mediates flow-dependent netosis via slc44a2. Elife. 2020;9:e53353. doi: 10.7554/eLife.53353. doi: 10.7554/eLife.53353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Müller F, Mutch NJ, Schenk WA, Smith SA, Esterl L, Spronk HM, Schmidbauer S, Gahl WA, Morrissey JH, Renné T. Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo. Cell 20091391143–1156doi: 10.1016/j.cell.2009.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zaldivia MTK, McFadyen JD, Lim B, Wang X, Peter K. Platelet-derived microvesicles in cardiovascular diseases. Front Cardiovasc Med. 2017;4:74. doi: 10.3389/fcvm.2017.00074. doi: 10.3389/fcvm.2017.00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Braig D, Nero TL, Koch HG, Kaiser B, Wang X, Thiele JR, Morton CJ, Zeller J, Kiefer J, Potempa LA, et al. Transitional changes in the CRP structure lead to the exposure of proinflammatory binding sites. Nat Commun. 2017;8:14188. doi: 10.1038/ncomms14188. doi: 10.1038/ncomms14188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lippi G, Plebani M, Henry BM. Thrombocytopenia is associated with severe coronavirus disease 2019 (COVID-19) infections: a meta-analysis. Clin Chim Acta 2020506145–148doi: 10.1016/j.cca.2020.03.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang X, Yang Q, Wang Y, Wu Y, Xu J, Yu Y, Shang Y. Thrombocytopenia and its association with mortality in patients with COVID-19. J Thromb Haemost 2020181469–1472doi: 10.1111/jth.14848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Koupenova M, Clancy L, Corkrey HA, Freedman JE. Circulating platelets as mediators of immunity, inflammation, and thrombosis. Circ Res 2018122337–351doi: 10.1161/CIRCRESAHA.117.310795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Assinger A. Platelets and infection - an emerging role of platelets in viral infection. Front Immunol. 2014;5:649. doi: 10.3389/fimmu.2014.00649. doi: 10.3389/fimmu.2014.00649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Koupenova M, Corkrey HA, Vitseva O, Manni G, Pang CJ, Clancy L, Yao C, Rade J, Levy D, Wang JP, et al. The role of platelets in mediating a response to human influenza infection. Nat Commun. 2019;10:1780. doi: 10.1038/s41467-019-09607-x. doi: 10.1038/s41467-019-09607-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Koupenova M, Vitseva O, MacKay CR, Beaulieu LM, Benjamin EJ, Mick E, Kurt-Jones EA, Ravid K, Freedman JE. Platelet-TLR7 mediates host survival and platelet count during viral infection in the absence of platelet-dependent thrombosis. Blood 2014124791–802doi: 10.1182/blood-2013-11-536003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Boilard E, Paré G, Rousseau M, Cloutier N, Dubuc I, Lévesque T, Borgeat P, Flamand L. Influenza virus H1N1 activates platelets through FcγRIIA signaling and thrombin generation. Blood 20141232854–2863doi: 10.1182/blood-2013-07-515536 [DOI] [PubMed] [Google Scholar]
- 80.Cloutier N, Allaeys I, Marcoux G, Machlus KR, Mailhot B, Zufferey A, Levesque T, Becker Y, Tessandier N, Melki I, et al. Platelets release pathogenic serotonin and return to circulation after immune complex-mediated sequestration. Proc Natl Acad Sci U S A 2018115E1550–E1559doi: 10.1073/pnas.1720553115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lê VB, Schneider JG, Boergeling Y, Berri F, Ducatez M, Guerin JL, Adrian I, Errazuriz-Cerda E, Frasquilho S, Antunes L, et al. Platelet activation and aggregation promote lung inflammation and influenza virus pathogenesis. Am J Respir Crit Care Med 2015191804–819doi: 10.1164/rccm.201406-1031OC [DOI] [PubMed] [Google Scholar]
- 82.Berlin DA, Gulick RM, Martinez FJ. Severe covid-19. N Engl J Med. 2020 doi: 10.1056/NEJMcp2009575. [DOI] [PubMed] [Google Scholar]
- 83.Hu H, Zhu L, Huang Z, Ji Q, Chatterjee M, Zhang W, Li N. Platelets enhance lymphocyte adhesion and infiltration into arterial thrombus. Thromb Haemost 20101041184–1192doi: 10.1160/TH10-05-0308 [DOI] [PubMed] [Google Scholar]
- 84.Chapman LM, Aggrey AA, Field DJ, Srivastava K, Ture S, Yui K, Topham DJ, Baldwin WM, 3rd, Morrell CN. Platelets present antigen in the context of MHC class I. J Immunol 2012189916–923doi: 10.4049/jimmunol.1200580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shi G, Field DJ, Ko KA, Ture S, Srivastava K, Levy S, Kowalska MA, Poncz M, Fowell DJ, Morrell CN. Platelet factor 4 limits Th17 differentiation and cardiac allograft rejection. J Clin Invest 2014124543–552doi: 10.1172/JCI71858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.León-Ponte M, Ahern GP, O’Connell PJ. Serotonin provides an accessory signal to enhance T-cell activation by signaling through the 5-HT7 receptor. Blood 20071093139–3146doi: 10.1182/blood-2006-10-052787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Iannacone M, Sitia G, Isogawa M, Marchese P, Castro MG, Lowenstein PR, Chisari FV, Ruggeri ZM, Guidotti LG. Platelets mediate cytotoxic T lymphocyte-induced liver damage. Nat Med 2005111167–1169doi: 10.1038/nm1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li N, Ji Q, Hjemdahl P. Platelet-lymphocyte conjugation differs between lymphocyte subpopulations. J Thromb Haemost 20064874–881doi: 10.1111/j.1538-7836.2006.01817.x [DOI] [PubMed] [Google Scholar]
- 89.Luther N, Shahneh F, Brähler M, Krebs F, Jäckel S, Subramaniam S, Stanger C, Schönfelder T, Kleis-Fischer B, Reinhardt C, et al. Innate effector-memory T-cell activation regulates post-thrombotic vein wall inflammation and thrombus resolution. Circ Res 20161191286–1295doi: 10.1161/CIRCRESAHA.116.309301 [DOI] [PubMed] [Google Scholar]
- 90.Cognasse F, Hamzeh-Cognasse H, Lafarge S, Chavarin P, Cogné M, Richard Y, Garraud O. Human platelets can activate peripheral blood B cells and increase production of immunoglobulins. Exp Hematol 2007351376–1387doi: 10.1016/j.exphem.2007.05.021 [DOI] [PubMed] [Google Scholar]
- 91.Zhang Y, Xiao M, Zhang S, Xia P, Cao W, Jiang W, Chen H, Ding X, Zhao H, Zhang H, et al. Coagulopathy and antiphospholipid antibodies in patients with covid-19. N Engl J Med. 2020;382:e38. doi: 10.1056/NEJMc2007575. doi: 10.1056/NEJMc2007575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Swystun LL, Liaw PC. The role of leukocytes in thrombosis. Blood 2016128753–762doi: 10.1182/blood-2016-05-718114 [DOI] [PubMed] [Google Scholar]
- 93.Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, Patel KD, Chakrabarti S, McAvoy E, Sinclair GD, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med 200713463–469doi: 10.1038/nm1565 [DOI] [PubMed] [Google Scholar]
- 94.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science 20043031532–1535doi: 10.1126/science.1092385 [DOI] [PubMed] [Google Scholar]
- 95.Narasaraju T, Yang E, Samy RP, Ng HH, Poh WP, Liew AA, Phoon MC, van Rooijen N, Chow VT. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am J Pathol 2011179199–210doi: 10.1016/j.ajpath.2011.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Massberg S, Grahl L, von Bruehl ML, Manukyan D, Pfeiler S, Goosmann C, Brinkmann V, Lorenz M, Bidzhekov K, Khandagale AB, et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med 201016887–896doi: 10.1038/nm.2184 [DOI] [PubMed] [Google Scholar]
- 97.Martinod K, Wagner DD. Thrombosis: tangled up in NETs. Blood 20141232768–2776doi: 10.1182/blood-2013-10-463646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD, Jr, Wrobleski SK, Wakefield TW, Hartwig JH, Wagner DD. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A 201010715880–15885doi: 10.1073/pnas.1005743107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Semeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NL, Esmon CT. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood 20111181952–1961doi: 10.1182/blood-2011-03-343061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Maas C, Renné T. Coagulation factor XII in thrombosis and inflammation. Blood 20181311903–1909doi: 10.1182/blood-2017-04-569111 [DOI] [PubMed] [Google Scholar]
- 101.Müller I, Klocke A, Alex M, Kotzsch M, Luther T, Morgenstern E, Zieseniss S, Zahler S, Preissner K, Engelmann B. Intravascular tissue factor initiates coagulation via circulating microvesicles and platelets. FASEB J 200317476–478doi: 10.1096/fj.02-0574fje [DOI] [PubMed] [Google Scholar]
- 102.Giesen PL, Rauch U, Bohrmann B, Kling D, Roqué M, Fallon JT, Badimon JJ, Himber J, Riederer MA, Nemerson Y. Blood-borne tissue factor: another view of thrombosis. Proc Natl Acad Sci U S A 1999962311–2315doi: 10.1073/pnas.96.5.2311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Owens AP, 3rd, Mackman N. Microparticles in hemostasis and thrombosis. Circ Res 20111081284–1297doi: 10.1161/CIRCRESAHA.110.233056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Merad M, Martin JC. Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat Rev Immunol 202020355–362doi: 10.1038/s41577-020-0331-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Stark K, Philippi V, Stockhausen S, Busse J, Antonelli A, Miller M, Schubert I, Hoseinpour P, Chandraratne S, von Brühl ML, et al. Disulfide HMGB1 derived from platelets coordinates venous thrombosis in mice. Blood 20161282435–2449doi: 10.1182/blood-2016-04-710632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yang X, Cheng X, Tang Y, Qiu X, Wang Z, Fu G, Wu J, Kang H, Wang J, Wang H, et al. The role of type 1 interferons in coagulation induced by gram-negative bacteria. Blood 20201351087–1100doi: 10.1182/blood.2019002282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Perlman S, Dandekar AA. Immunopathogenesis of coronavirus infections: implications for SARS. Nat Rev Immunol 20055917–927doi: 10.1038/nri1732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Blondonnet R, Constantin JM, Sapin V, Jabaudon M. A pathophysiologic approach to biomarkers in acute respiratory distress syndrome. Dis Markers. 2016;2016:3501373. doi: 10.1155/2016/3501373. doi: 10.1155/2016/3501373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zuo Y, Yalavarthi S, Shi H, Gockman K, Zuo M, Madison JA, Blair C, Weber A, Barnes BJ, Egeblad M, et al. Neutrophil extracellular traps in covid-19. JCI Insight. 2020;5:e138999. doi: 10.1172/jci.insight.138999. doi: 10.1172/jci.insight.138999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Freedman JE, Loscalzo J. Platelet-monocyte aggregates: bridging thrombosis and inflammation. Circulation 20021052130–2132doi: 10.1161/01.cir.0000017140.26466.f5 [DOI] [PubMed] [Google Scholar]
- 111.Bozza FA, Shah AM, Weyrich AS, Zimmerman GA. Amicus or adversary: platelets in lung biology, acute injury, and inflammation. Am J Respir Cell Mol Biol 200940123–134doi: 10.1165/rcmb.2008-0241TR [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Dale DC, Boxer L, Liles WC. The phagocytes: neutrophils and monocytes. Blood 2008112935–945doi: 10.1182/blood-2007-12-077917 [DOI] [PubMed] [Google Scholar]
- 113.Celi A, Pellegrini G, Lorenzet R, De Blasi A, Ready N, Furie BC, Furie B. P-selectin induces the expression of tissue factor on monocytes. Proc Natl Acad Sci U S A 1994918767–8771doi: 10.1073/pnas.91.19.8767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Neumann FJ, Zohlnhöfer D, Fakhoury L, Ott I, Gawaz M, Schömig A. Effect of glycoprotein IIb/IIIa receptor blockade on platelet-leukocyte interaction and surface expression of the leukocyte integrin Mac-1 in acute myocardial infarction. J Am Coll Cardiol 1999341420–1426doi: 10.1016/s0735-1097(99)00350-2 [DOI] [PubMed] [Google Scholar]
- 115.Chatterjee M, von Ungern-Sternberg SN, Seizer P, Schlegel F, Büttcher M, Sindhu NA, Müller S, Mack A, Gawaz M. Platelet-derived CXCL12 regulates monocyte function, survival, differentiation into macrophages and foam cells through differential involvement of CXCR4-CXCR7. Cell Death Dis. 2015;6:e1989. doi: 10.1038/cddis.2015.233. doi: 10.1038/cddis.2015.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Scheuerer B, Ernst M, Dürrbaum-Landmann I, Fleischer J, Grage-Griebenow E, Brandt E, Flad HD, Petersen F. The CXC-chemokine platelet factor 4 promotes monocyte survival and induces monocyte differentiation into macrophages. Blood 2000951158–1166 [PubMed] [Google Scholar]
- 117.Ali RA, Wuescher LM, Dona KR, Worth RG. Platelets mediate host defense against staphylococcus aureus through direct bactericidal activity and by enhancing macrophage activities. J Immunol 2017198344–351doi: 10.4049/jimmunol.1601178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tsimikas S, Brilakis ES, Miller ER, McConnell JP, Lennon RJ, Kornman KS, Witztum JL, Berger PB. Oxidized phospholipids, Lp(a) lipoprotein, and coronary artery disease. N Engl J Med 200535346–57doi: 10.1056/NEJMoa043175 [DOI] [PubMed] [Google Scholar]
- 119.Imai Y, Kuba K, Neely GG, Yaghubian-Malhami R, Perkmann T, van Loo G, Ermolaeva M, Veldhuizen R, Leung YH, Wang H, et al. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 2008133235–249doi: 10.1016/j.cell.2008.02.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Foley JH, Conway EM. Cross talk pathways between coagulation and inflammation. Circ Res 20161181392–1408doi: 10.1161/CIRCRESAHA.116.306853 [DOI] [PubMed] [Google Scholar]
- 121.Magro C, Mulvey JJ, Berlin D, Nuovo G, Salvatore S, Harp J, Baxter-Stoltzfus A, Laurence J. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Transl Res 20202201–13doi: 10.1016/j.trsl.2020.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gao T, Hu M, Zhang X, Li H, Zhu L, Liu H, Dong Q, Zhang Z, Wang Z, Hu Y, et al. Highly pathogenic coronavirus n protein aggravates lung injury by masp-2-mediated complement over-activation. medRxiv. 2020 2020.2003.2029.20041962. [Google Scholar]
- 123.Foreman KE, Vaporciyan AA, Bonish BK, Jones ML, Johnson KJ, Glovsky MM, Eddy SM, Ward PA. C5a-induced expression of P-selectin in endothelial cells. J Clin Invest 1994941147–1155doi: 10.1172/JCI117430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ritis K, Doumas M, Mastellos D, Micheli A, Giaglis S, Magotti P, Rafail S, Kartalis G, Sideras P, Lambris JD. A novel C5a receptor-tissue factor cross-talk in neutrophils links innate immunity to coagulation pathways. J Immunol 20061774794–4802doi: 10.4049/jimmunol.177.7.4794 [DOI] [PubMed] [Google Scholar]
- 125.Wiedmer T, Esmon CT, Sims PJ. Complement proteins C5b-9 stimulate procoagulant activity through platelet prothrombinase. Blood 198668875–880 [PubMed] [Google Scholar]
- 126.Campbell CM, Kahwash R. Will complement inhibition be the new target in treating COVID-19-related systemic thrombosis? Circulation 20201411739–1741doi: 10.1161/CIRCULATIONAHA.120.047419 [DOI] [PubMed] [Google Scholar]
- 127.Antoniak S, Mackman N. Multiple roles of the coagulation protease cascade during virus infection. Blood 20141232605–2613doi: 10.1182/blood-2013-09-526277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Key NS, Vercellotti GM, Winkelmann JC, Moldow CF, Goodman JL, Esmon NL, Esmon CT, Jacob HS. Infection of vascular endothelial cells with herpes simplex virus enhances tissue factor activity and reduces thrombomodulin expression. Proc Natl Acad Sci U S A 1990877095–7099doi: 10.1073/pnas.87.18.7095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Shibamiya A, Hersemeyer K, Schmidt Wöll T, Sedding D, Daniel JM, Bauer S, Koyama T, Preissner KT, Kanse SM. A key role for Toll-like receptor-3 in disrupting the hemostasis balance on endothelial cells. Blood 2009113714–722doi: 10.1182/blood-2008-02-137901 [DOI] [PubMed] [Google Scholar]
- 130.Coughlin SR. Thrombin signalling and protease-activated receptors. Nature 2000407258–264doi: 10.1038/35025229 [DOI] [PubMed] [Google Scholar]
- 131.Camerer E, Huang W, Coughlin SR. Tissue factor- and factor X-dependent activation of protease-activated receptor 2 by factor VIIa. Proc Natl Acad Sci U S A 2000975255–5260doi: 10.1073/pnas.97.10.5255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Coughlin SR, Camerer E. PARticipation in inflammation. J Clin Invest 200311125–27doi: 10.1172/JCI17564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Chen D, Carpenter A, Abrahams J, Chambers RC, Lechler RI, McVey JH, Dorling A. Protease-activated receptor 1 activation is necessary for monocyte chemoattractant protein 1-dependent leukocyte recruitment in vivo. J Exp Med 20082051739–1746doi: 10.1084/jem.20071427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sugama Y, Tiruppathi C, offakidevi K, Andersen TT, Fenton JW, 2nd, Malik AB. Thrombin-induced expression of endothelial P-selectin and intercellular adhesion molecule-1: a mechanism for stabilizing neutrophil adhesion. J Cell Biol 1992119935–944doi: 10.1083/jcb.119.4.935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kaplanski G, Marin V, Fabrigoule M, Boulay V, Benoliel AM, Bongrand P, Kaplanski S, Farnarier C. Thrombin-activated human endothelial cells support monocyte adhesion in vitro following expression of intercellular adhesion molecule-1 (ICAM-1; CD54) and vascular cell adhesion molecule-1 (VCAM-1; CD106). Blood 1998921259–1267 [PubMed] [Google Scholar]
- 136.Geisbert TW, Hensley LE, Jahrling PB, Larsen T, Geisbert JB, Paragas J, Young HA, Fredeking TM, Rote WE, Vlasuk GP. Treatment of Ebola virus infection with a recombinant inhibitor of factor VIIa/tissue factor: a study in rhesus monkeys. Lancet 20033621953–1958doi: 10.1016/S0140-6736(03)15012-X [DOI] [PubMed] [Google Scholar]
- 137.Bikdeli B, Madhavan MV, Jimenez D, Chuich T, Dreyfus I, Driggin E, Nigoghossian C, Ageno W, Madjid M, Guo Y, et al. ; Global COVID-19 Thrombosis Collaborative Group, Endorsed by the ISTH, NATF, ESVM, and the IUA, Supported by the ESC Working Group on Pulmonary Circulation and Right Ventricular Function COVID-19 and thrombotic or thromboembolic disease: implications for prevention, antithrombotic therapy, and follow-up: JACC State-of-the-art review. J Am Coll Cardiol 2020752950–2973doi: 10.1016/j.jacc.2020.04.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Connors JM, Levy JH. COVID-19 and its implications for thrombosis and anticoagulation. Blood 20201352033–2040doi: 10.1182/blood.2020006000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Tang N, Bai H, Chen X, Gong J, Li D, Sun Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J Thromb Haemost 2020181094–1099doi: 10.1111/jth.14817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Arnold K, Xu Y, Sparkenbaugh EM, Li M, Han X, Zhang X, Xia K, Piegore M, Zhang F, Zhang X, et al. Design of anti-inflammatory heparan sulfate to protect against acetaminophen-induced acute liver failure. Sci Transl Med. 2020;12:eaav8075. doi: 10.1126/scitranslmed.aav8075. doi: 10.1126/scitranslmed.aav8075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Poterucha TJ, Libby P, Goldhaber SZ. More than an anticoagulant: Do heparins have direct anti-inflammatory effects? Thromb Haemost 2017117437–444doi: 10.1160/TH16-08-0620 [DOI] [PubMed] [Google Scholar]
- 142.Liu J, Thorp SC. Cell surface heparan sulfate and its roles in assisting viral infections. Med Res Rev 2002221–25doi: 10.1002/med.1026 [DOI] [PubMed] [Google Scholar]
- 143.Shukla D, Spear PG. Herpesviruses and heparan sulfate: an intimate relationship in aid of viral entry. J Clin Invest 2001108503–510doi: 10.1172/JCI13799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lang J, Yang N, Deng J, Liu K, Yang P, Zhang G, Jiang C. Inhibition of SARS pseudovirus cell entry by lactoferrin binding to heparan sulfate proteoglycans. PLoS One. 2011;6:e23710. doi: 10.1371/journal.pone.0023710. doi: 10.1371/journal.pone.0023710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Milewska A, Zarebski M, Nowak P, Stozek K, Potempa J, Pyrc K. Human coronavirus NL63 utilizes heparan sulfate proteoglycans for attachment to target cells. J Virol 20148813221–13230doi: 10.1128/JVI.02078-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Mycroft-West C, Su D, Elli S, Guimond S, Miller G, Turnbull J, Yates E, Guerrini M, Fernig D, Lima M, Skidmore M. The 2019 coronavirus (SARS-cov-2) surface protein (spike) s1 receptor binding domain undergoes conformational change upon heparin binding. bioRxiv. 2020 [Google Scholar]
- 147.Belouzard S, Chu VC, Whittaker GR. Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proc Natl Acad Sci U S A 20091065871–5876doi: 10.1073/pnas.0809524106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Riker RR, May TL, Fraser GL, Gagnon DJ, Bandara M, Zemrak W, Seder DB. Heparin-induced thrombocytopenia with thrombosis in covid-19 adult respiratory distress syndrome. Research and Practice in Thrombosis and Haemostasis. doi: 10.1002/rth2.12390. n/a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Renné T, Stavrou EX. Roles of factor XII in innate immunity. Front Immunol. 2019;10:2011. doi: 10.3389/fimmu.2019.02011. doi: 10.3389/fimmu.2019.02011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Larsson M, Rayzman V, Nolte MW, Nickel KF, Björkqvist J, Jämsä A, Hardy MP, Fries M, Schmidbauer S, Hedenqvist P, et al. A factor XIIa inhibitory antibody provides thromboprotection in extracorporeal circulation without increasing bleeding risk. Sci Transl Med. 2014;6:222ra17. doi: 10.1126/scitranslmed.3006804. doi: 10.1126/scitranslmed.3006804. [DOI] [PubMed] [Google Scholar]
- 151.Pixley RA, De La Cadena R, Page JD, Kaufman N, Wyshock EG, Chang A, Taylor FB, Jr, Colman RW. The contact system contributes to hypotension but not disseminated intravascular coagulation in lethal bacteremia. In vivo use of a monoclonal anti-factor XII antibody to block contact activation in baboons. J Clin Invest 19939161–68doi: 10.1172/JCI116201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Shatzel JJ, DeLoughery EP, Lorentz CU, Tucker EI, Aslan JE, Hinds MT, Gailani D, Weitz JI, McCarty OJT, Gruber A. The contact activation system as a potential therapeutic target in patients with COVID-19. Res Pract Thromb Haemost 20204500–505doi: 10.1002/rth2.12349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Jansen PM, Pixley RA, Brouwer M, de Jong IW, Chang AC, Hack CE, Taylor FB, Jr, Colman RW. Inhibition of factor XII in septic baboons attenuates the activation of complement and fibrinolytic systems and reduces the release of interleukin-6 and neutrophil elastase. Blood 1996872337–2344 [PubMed] [Google Scholar]
- 154.Renné T, Schmaier AH, Nickel KF, Blombäck M, Maas C. In vivo roles of factor XII. Blood 20121204296–4303doi: 10.1182/blood-2012-07-292094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Whyte CS, Morrow GB, Mitchell JL, Chowdary P, Mutch NJ. Fibrinolytic abnormalities in acute respiratory distress syndrome (ARDS) and versatility of thrombolytic drugs to treat covid-19. J Thromb Haemost 2020181548–1555doi: 10.1111/jth.14872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wang J, Hajizadeh N, Moore EE, McIntyre RC, Moore PK, Veress LA, Yaffe MB, Moore HB, Barrett CD. Tissue plasminogen activator (tpa) treatment for covid-19 associated acute respiratory distress syndrome (ARDS): a case series. J Thromb Haemost 2020181752–1755doi: 10.1111/jth.14828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Liu C, Ma Y, Su Z, Zhao R, Zhao X, Nie HG, Xu P, Zhu L, Zhang M, Li X, et al. Meta-analysis of preclinical studies of fibrinolytic therapy for acute lung injury. Front Immunol. 2018;9:1898. doi: 10.3389/fimmu.2018.01898. doi: 10.3389/fimmu.2018.01898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Hardaway RM, Harke H, Tyroch AH, Williams CH, Vazquez Y, Krause GF. Treatment of severe acute respiratory distress syndrome: a final report on a phase I study. Am Surg 200167377–382 [PubMed] [Google Scholar]
- 159.Meyer G, Vicaut E, Danays T, Agnelli G, Becattini C, Beyer-Westendorf J, Bluhmki E, Bouvaist H, Brenner B, Couturaud F, et al. ; PEITHO Investigators Fibrinolysis for patients with intermediate-risk pulmonary embolism. N Engl J Med 20143701402–1411doi: 10.1056/NEJMoa1302097 [DOI] [PubMed] [Google Scholar]
- 160.Wang L, Li H, Gu X, Wang Z, Liu S, Chen L. Effect of antiplatelet therapy on acute respiratory distress syndrome and mortality in critically ill patients: a meta-analysis. PLoS One. 2016;11:e0154754. doi: 10.1371/journal.pone.0154754. doi: 10.1371/journal.pone.0154754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ouyang Y, Wang Y, Liu B, Ma X, Ding R. Effects of antiplatelet therapy on the mortality rate of patients with sepsis: A meta-analysis. J Crit Care 201950162–168doi: 10.1016/j.jcrc.2018.12.004 [DOI] [PubMed] [Google Scholar]
- 162.Carestia A, Davis RP, Grosjean H, Lau MW, Jenne CN. Acetylsalicylic acid inhibits intravascular coagulation during Staphylococcus aureus-induced sepsis in mice. Blood 20201351281–1286doi: 10.1182/blood.2019002783 [DOI] [PubMed] [Google Scholar]
- 163.Liu X, Li Z, Liu S, Sun J, Chen Z, Jiang M, Zhang Q, Wei Y, Wang X, Huang YY, et al. Potential therapeutic effects of dipyridamole in the severely ill patients with covid-19. Acta Pharm Sin B. 2020 doi: 10.1016/j.apsb.2020.04.008. doi: 10.1016/j.apsb.2020.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Asakura H, Ogawa H. Potential of heparin and nafamostat combination therapy for COVID-19. J Thromb Haemost 2020181521–1522doi: 10.1111/jth.14858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Yamamoto M, Matsuyama S, Li X, Takeda M, Kawaguchi Y, Inoue JI, Matsuda Z. Identification of nafamostat as a potent inhibitor of middle east respiratory syndrome coronavirus S protein-mediated membrane fusion using the split-protein-based cell-cell fusion assay. Antimicrob Agents Chemother 2016606532–6539doi: 10.1128/AAC.01043-16 [DOI] [PMC free article] [PubMed] [Google Scholar]