

Abstract
This minireview aims to cover the developments over the past two decades in the chemistry of sulfonium salts. Specifically, insight is provided into the synthetic strategies available for the preparation of these compounds, the different reactivity patterns that are expected depending on their structural features or the reaction conditions applied, and the diversity of organic scaffolds that can thereby be synthesized. Additionally, the pros and cons derived from the use of sulfonium salts are presented and critically compared, when possible, in relation to reagents not based on sulfur but depicting similar reactivity.
Keywords: Chemoselectivity, Coupling reactions, Photocatalysis, Sulfonium salts, Transfer reagents
Quo vadis sulfonium? This minireview compiles the recent developments in the chemistry of sulfonium salts in terms of preparation, reactivity and synthetic utility. Additionally, the pros and cons derived from their use are critically evaluated, when possible, in relation to reagents not based on sulfur but depicting similar reactivity.

1. Introduction
Sulfonium ions are defined as positively charged organosulfur compounds in which the central sulfur atom is bonded to three organic substituents, being their general formula [R3S]+X– (X is a non‐coordinated counter anion).1 In sulfonium salts, the sulfur atom still possesses a stereochemically active free electron pair, which forces sulfur to adopt the pyramidal geometry expected from the application of the VSEPR rules. For this reason, sulfonium salts are not only isoelectronic but also isostructural to typical phosphines (Figure 1a). Similarly to phosphines, their pyramidal inversion barriers are relatively high making this process slow at room temperature (Figure 1b).2 Accordingly, when appropriately substituted, sulfonium salts are chiral at sulfur, and the enantiomeric mixtures can usually be resolved by conventional techniques. Compared with their lighter analogues, oxonium salts, they show higher thermodynamic stability, much more structural diversity, and are easier to handle under atmospheric conditions. In fact, purification by traditional silica gel column chromatography is not infrequent for many sulfonium salts, even though the mixtures of solvents necessary to elute these polar compounds often contain alcohols or acetone.
Figure 1.

Introduction to sulfonium salts.
In general, the reactivity of sulfonium salts is dominated by the positive charge that they bear, which is mainly located at sulfur. Thus, α‐CH deprotonation of alkyl sulfonium salts leads to the formation of sulfur ylides; a class of compounds of demonstrated utility for the synthesis of three‐membered rings such as cyclopropanes, epoxides and aziridines. Moreover, sigmatropic rearrangements among the unsaturated substituents attached to sulfur with concomitant cleavage of a S–C bond are also frequent. These two modes of reactivity have been recently reviewed and will not be covered in this minireview.3
The LUMO or in some cases the LUMO+1 of sulfonium salts often corresponds to a σ*(S–R) orbital of relatively low energy and with a considerable coefficient at the S‐atom (Figure 1c). This makes possible the direct attack of nucleophiles to sulfur followed by ligand coupling, which can also be seen as the reductive elimination of the Nu‐R moiety. Although this process and a typical nucleophilic substitution significantly differ in the mechanism, their product outcomes are identical, and the sulfide moieties behave as excellent leaving groups in these reactions (Figure 1d). In this regard, the reactivity of sulfonium salts shows many similarities with that of hypervalent I(III) reagents.4
An overview on the synthetic potential of sulfonium salts for the functionalization of (elaborated) substrates, together with a critical comparison between their reactivity and scope and that of other reagents is presented in sections 2.3, 2.4, 3.3, 4.3 and 5.3 of this minireview.
The analogy in terms of reactivity between sulfonium salts and (alkyl/aryl)halogenides or other excellent leaving groups also explains the ease with which sulfonium salts undergo oxidative addition of low oxidation‐state metals into one of their R–S bonds. As it will be discussed later along this minireview, this reactivity defines sulfonium salts as suitable electrophilic partners for typical Pd‐ or Ni‐catalyzed coupling reactions.5 Finally, and again as result of their positive charge, one electron reduction of sulfonium salts is a relatively facile process, which ultimately leads to the homolytic fragmentation of one of the C–S bonds and generates a thioether together with an organic radical. This fragmentation paves the way to the use of sulfonium salts as radical precursors.6 However, the synthetic utility of this methodology can only be fully exploited if the chemoselective cleavage of one of the three C–S bonds is achieved.[5a], 7
Rather than an assemblage of detailed information with a complete literature survey, this minireview aims to critically treat selected material to provide the reader not only with a taste about the reactivity of sulfonium salts, but also with a general perspective about their synthetic utility, unsolved problems derived from their use, and possible developments in the area. Having this idea in mind, sulfonium salts are grouped in different sections according to the hybridization of the carbon atoms of the most reactive substituent attached to S (Csp, Csp 2 or Csp 3). Each individual section starts with a description of the synthetic methods already available for the preparation of that specific structure and is followed by a contextualized description of their reactivity and applications.
2. C(sp)‐Substituted Sulfonium Salts
The number of alkynyl‐substituted sulfonium salts 1 already characterized is still quite limited. This is a consequence of the intrinsic shortages of the synthetic methods available for their preparation, which classically relied on the use of I(III)‐reagents or S‐centered radicals.8 Although known since a decade, it has not been until very recently that the reaction of sulfonium bistriflates with TMS‐capped alkynes was recognized as a general method for the synthesis of these compounds.9 In fact, this synthetic strategy has allowed the preparation of a series of S‐(alkynyl)dibezothiophenium salts, as well as other related compounds such as for example S‐(CN) derivatives.10, 11 Previous to the actual renascence of sulfonium salts chemistry, the number of applications of alkynyl‐substituted sulfonium salts was quite reduced, basically limited to their use as cationic polymerization photoinitiators.9 This has now changed, and their capability to act as synthons of alkynyl cations has been even demonstrated in natural product synthesis.12
2.1. Synthesis of Alkynyl‐Substituted Sulfonium Salts
Traditionally, these salts have been obtained by reaction of 1‐alkynyl(aryl)‐λ3‐iodanes of general formula 2 with diaryl sulfides 3. The alkynyl transfer reaction proceeds smoothly and is selective; no aryl transfer product is detected.[8b] Mechanistic studies suggest that the alkyne transfer either progresses via a Michael‐type addition of the sulfide at the β‐acetylenic carbon of 2 with subsequent elimination of the iodobenzene and 1,2‐shift of the R group, or alternatively by attack of the sulfide to the iodo(III) center followed by reductive elimination (Scheme 1a). The operating pathway surely depends on the nature of the R‐substituent at the alkyne. While this route cleanly affords the desired alkynyl‐substituted sulfonium salts 1, its practical utility is severely limited. The preparation of the necessary 1‐alkynyl(aryl)‐λ3‐iodane precursors is synthetically demanding and requires the employment of stoichiometric amounts of peroxides, which discourages their preparation on a large scale.13 In addition, I(III) compounds often show exothermic decompositions upon heating.14
Scheme 1.

Available syntheses for alkynyl‐substituted sulfonium salts 1.
Alternatively, alkynylsulfonium salts can be prepared by reaction of terminal alkynes with 2 equivalents of thianthrenium tetrafluoroborate 4.[8a] The reaction has been described to proceed via double addition of the radical cation to the alkyne to afford dicationic 1,2‐bisadducts 5, which on basic workup are deprotonated and subsequently eliminate a thianthrene unit to deliver the desired salts (Scheme 1b). Alkyl groups of different length are tolerated as substituents at the alkyne moiety; however, again the utility of this synthesis is compromised. Only derivatives of thianthrene radical 4 or structurally similar sulfides, which also are able to stabilize a persistent radical cation, can be obtained through this route.
Arguably, the most practical route to obtain alkynyl sulfonium salts consist in the activation of the respective sulfoxide 6 by an acid anhydride, usually Tf2O, to generate in situ the corresponding S(IV) bistriflate 7. These highly electrophilic intermediates readily react with TMS‐capped alkynes affording the desired salts 1, probably via vinyl triflate derivative 8 (Scheme 1c). While requiring a preliminary functionalization of the alkyne as TMS‐derivative, alkynyl sulfonium salts of diverse structure are satisfactorily prepared through this route even in multigram scale. Interestingly, some of these compounds can be purified by column chromatography and, if kept in a close vial, they show no apparent decomposition after weeks. Our group has made use of this strategy to prepare a complete series of dibenzothiophenium salts 1a–f (Figure 2).10
Figure 2.

S‐(alkynyl)dibenzothiophenium salts 1 prepared from TMS‐alkynes.
2.2. Structure of S‐(Alkynyl) Sulfonium Salts
For this analysis, dibenzothiophenium salt 1a is showcased as illustrative example. As can be observed from its X‐ray structure, in this compound the S1 atom adopts the expected pyramidal geometry (sum of angles around S1 = 302.3°). Also of particular relevance are the O1–S1 contact (3.157 Å), clearly shorter than the sum of the van der Waals radii of the corresponding elements (3.32 Å). This interaction indicates substantial Lewis acidity at the S‐centre and might be considered a model of how nucleophiles approach sulfonium salts under charge control (Figure 3).
Figure 3.

Molecular diagram of the X‐ray structure of 1a (top), ellipsoids represented at 50 % probability; and representation of its HOMO (left) and LUMO+1 (right). Partial charges and Wiberg bond orders are calculated at B3LYP/6‐311G* level.
Natural population analysis at the B3LYP/6‐31G* level additionally indicates that in 1a the sulfur atom bears a nearly complete positive charge (+0.946e), the α‐carbon to sulfur, C1, is negatively charged (–0.343e) while C2 is partially positive charged (+0.182e). Moreover, inspection of the frontier orbitals reveals that the very low‐lying LUMO+1 in 1a corresponds to the π*(C1–C2) bond. This situation also suggests that for an orbital overlap‐controlled process, the incoming nucleophile might add to 1a at C2 in a Michael‐type fashion, affording initial adducts that evolve differently depending on the reaction conditions.[10b]
2.3. Reactivity of S‐(Alkynyl) Sulfonium Salts
In the presence of mild bases such as carbonates, S‐(alkynyl) sulfonium salts react with thiols, sulfonamides and activated methylene groups to afford the desired S‐ (9–11), C‐ (12 and 13) or N‐alkynylated products (14), respectively, in good to excellent yields.[10b] Hence, 1a–f can be considered synthetic equivalents of the [R–C≡C]+ cation. The scope of this transformation is illustrated through a selection of the products that are obtained following this methodology and the synthetic applications developed (Scheme 2a).12 Interestingly, isotope labeling studies reveal that 1a undergoes attack by nucleophiles at the β‐carbon atom of the triple bond, followed by elimination of the dibenzothiophene moiety and subsequent 1,2‐migration of one of the groups at C2. Contrarily, the alkynes having a terminal TIPS moiety were formed via attack of the nucleophile to 1f in the α‐position (Scheme 2b).
Scheme 2.

a) Reactivity of S‐(alkynyl)substituted sulfonium salts: Selected examples. b) Plausible mechanisms operating in the alkyne transfer process.
A priori, the most obvious reagents capable of exhibiting electrophilic alkynylation reactivity are acetylene halides of general formula R–C≡C–X (X = Cl, Br, I); however, these compounds show limited tendency to react in this way and very often undesired additions to the C≡C triple bond take place.15 Only alkynyliodonium salts 16–18, originally prepared by Beringer (in acyclic form)16 and Ochiai (as benziodoxoles),17 and later popularized by Zhdankin,18 Ochiai,19 Stang20 and Waser,21 have demonstrated to be general transfer reagents for electrophilic alkynylation (Figure 4).22 On the other hand, as already mentioned, the use of I(III) derivatives is not always free of inconveniences due to their highly exothermic decompositions on heating.13 Electrophilc alkynylation reagents based on the imidazole‐2‐thione platform 19 have been developed in our group as well; however, the desired alkyne transfer reaction only occurs when an electron‐withdrawing substituent is strategically located at the other terminus of the alkyne moiety.23
Figure 4.
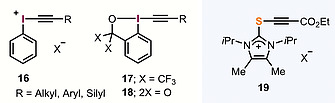
Alternative electrophilc alkynylation reagents available.
2.4. Synthesis and Reactivity of S‐(Cyano) Sulfonium Salts
The synthetic versatility of the cyano group, which is a privileged precursor of both amines and carbonyl groups;24 as well as the prevalence of cyano moieties in natural products,25 pharmaceuticals,26 agrochemicals,27 dyes28 and high performance materials29 has stimulated the development of efficient methodologies for the selective incorporation of CN‐substituents at specific positions of elaborated organic scaffolds.30 However, most of the protocols described still use the inherent nucleophilicity of the cyanide anion to promote the C–CN bond forming event. Disconnection strategies based on the umpolung of the CN moiety are comparatively scarce and their synthetic potential has still not been fully exploited, mainly due to the disadvantages associated with the use of the available electrophilic cyanide sources (Figure 5).31
Figure 5.

Available electrophilic cyanation reagents.
Cyanogen halides 20–22 are very toxic and volatile,32 while N‐cyanosulfonamides 23, N‐cyanobenzotriazole 24 and N‐cyanobenzoimidazole 25 require the use of strong C‐nucleophiles such as organometallic reagents or enolates for the transfer the formal [CN]+ unit.33 It was not until the introduction of cyanobenziodoxone 26 and derivatives by Zhdankin,34 and its intensive use by Waser35 and others36 that metal‐free electrophilic cyanation at non‐functionalized C‐H, N‐H and S‐H positions of organic substrates could be efficiently achieved. Our group recently introduced the first S‐based electrophilic cyanating reagent 28,23, 37 which has a reactivity profile very similar to that of 26.
Interestingly, sulfonium salts are able to mimic hypervalent I(III)‐species in this regard as well, and S‐(cyano) dibenzothiophenium salt 29 also acts as an efficient [CN]+ synthon. This compound has been obtained through a one pot synthesis via activation of sulfoxide 30 with triflic acid anhydride followed by quenching of the in situ generated bistriflate with TMSCN (Scheme 3).11 The X‐ray structure of 29 is also depicted in the scheme.
Scheme 3.

Synthesis and molecular diagram of the X‐ray structure of 29 (ellipsoids represented at 50 % probability).
The profile of 29 regarding substrate scope is quite similar to that of 26–28. Cyano‐transfer reactions to anilines, thiols, silyl enol ethers and electron rich homo‐, hetero‐, and polycyclic aromatics take place at relatively low temperatures (–50 °C → 50 °C), and good yields of compounds 31–34 were obtained without the necessity of any activation or catalyst (Scheme 4).
Scheme 4.

Reaction scope of 29 as electrophilic cyanating reagent.
However, the synthetic relevance of 29 is better observed when intramolecular cyanofunctionalizations are attempted. Specifically, Scheme 5 shows a series of pyrroloindolines 35–37 that are obtained when N‐protected tryptamine derivatives are subjected to the action of 29. Promotion of this kind of cyclization, which delivers cyano‐pyrroloindolines of potential synthetic and/or pharmacological interest, is an example of a reactivity so far exclusively provided by S‐(cyano) sulfonium salts. The development of asymmetric versions of these cyanocyclization reactions and the use of 29 or related sulfonium salts in metal‐catalyzed cyanation reactions are still undiscovered.
Scheme 5.

Cyanocyclizations promoted by 29.
3. C(sp2)‐Substituted Sulfonium Salts: S‐(Alkenyl) Derivatives
In sharp contrasts to S‐(alkynyl)sulfonium salts, S‐alkenyl derivatives 38 have been widely employed as Michael acceptors since decades; being their use as ethylene transfer reagents for the synthesis of cyclopropanes and aziridines one of their most relevant applications in classical synthetic chemistry.38 On the other hand, the range of transformations in which these salts can be involved has been dramatically expanded during the last few years after the recognition that they serve as excellent electrophiles in metal‐catalyzed cross‐coupling reactions.[5b], [5c], 39
3.1. Synthesis of S‐(Alkenyl) Sulfonium Salts
Arguably, the most useful method for the preparation of S‐(alkenyl)sulfonium salts 38 consists of the reaction of mono‐, di‐, or trisubstituted alkenes with in situ generated bistriflates of general formula 7 (Scheme 6a).40 This reaction is tolerant towards the presence of functional groups of different nature such as halogens, ethers, amides, nitriles and sulfonamides. However, alcohols and amines need to be considered with caution. Any ester or amide that might have been formed by direct reaction of the activating anhydride with these groups need to be carefully hydrolysed during the work‐up process. Additionally, Michael‐type reaction between potentially nucleophilic moieties present in the substrate with the newly formed S‐alkenyl sulfonium salt also need to be avoided, and the possibility that undesired Swern‐type oxidations of primary alcohols take place under the reaction conditions applied to obtain S‐(alkenyl)sulfonium salts must not be overlooked. Finally, any substrate containing a particularly electron‐rich aromatic ring needs to be considered with caution for this transformation. Bistriflates of general formula 7 are strong electrophiles and therefore may react via electrophilic aromatic substitution at the ring affording undesired S‐(aryl) sulfonium salts (See Section 4.1.).
Scheme 6.

Synthetic routes for the preparation of S‐alkenyl sulfonium salts 38 and scope of the methods.
Selected examples showcasing the scope of the synthetic method, 38b–g, are depicted in Scheme 6b. Overall, it seems that the synthesis of S‐(alkenyl) sulfonium salts employing the thianthrenium unit is characterized by a broader scope regarding functional group tolerance than the tetrahydrothiophene analogues.39 Finally, it worth mentioning that despite of the already mentioned limitations of this synthetic route, the possibility to obtain sulfonium salts from un‐functionalized alkenes constitutes already a remarkable advantage. The synthesis of hypervalent I(III) reagents 39–40 of similar reactivity require the employment of TMS‐, Bu3Sn‐, (HO)2B‐ or ClCp2Zr‐substituted olefins as starting materials (Scheme 6c).41
The synthetic protocols already described above are not practical for the preparation of the parent S‐(vinyl) salt 38a because the use of ethylene gas would be necessary. This gap is filled by the synthesis developed by Aggarwal and McGarrigle, which is based on the initial alkylation of diphenyl sulfide by reaction with 2‐bromoethyl triflate to form (bromoethyl)(diphenyl)sulfonium triflate 41 followed by elimination of HBr by treatment with potassium bicarbonate (Scheme 6d).42
3.2. Structure of S‐(Alkenyl) Sulfonium Salts
In order to carry out a comparative analysis throughout the entire series of sulfonium salts, compound 38h, based as well as 1a on a dibenzothiophene unit, is showcased as structural model for this family of reagents. In 38h the S1 atom again adopts the expected pyramidal geometry (sum of angles around S1 = 297.8°), but no O1–S1 contacts were detected in this structure. In fact, the triflates anions prefer to interact with the protons of the vinyl rests via C–H···O hydrogen bonds (Figure 6).43
Figure 6.

Molecular diagram of the X‐ray structure of 38h (top), ellipsoids shown at 50 % probability; and representation of its HOMO (left) and LUMO+1 (right). Partial charges and Wiberg bond orders are calculated at B3LYP/6‐311G* level.
Natural population analysis at the B3LYP/6‐31G* level additionally indicates that S1 in 38h is highly charged, bearing a nearly complete positive charge (+0.884e), while the α‐carbon to sulfur, C1, is negatively charged (–0.404e) and C2 can be considered basically neutral (–0.092e). A close look at the LUMO reveals that it corresponds to the π*(C1–C2) bond. This frontier orbital picture perfectly correlates to the one observed in typical Michael‐acceptors; therefore, it is not surprising that all classical reactivity between S‐(alkenyl) sulfonium salts and nucleophiles is initiated by a nucleophilic attack at C2.
3.3. Reactivity of S‐(Alkenyl) Sulfonium Salts
As already pointed, the chemistry of S‐alkenyl sulfonium salts strongly relies on their Michael acceptor character. Note however, that after the initial attack of the nucleophile a sulfonium ylide intermediate is formed, being the subsequent reaction pathway followed by that ylide what determines the kind of products obtained from the reaction. Substituted olefins are gained when the sulfonium ylide reacts with an electrophile and this is followed by elimination of a proton and the sulfide moiety (Pathway A1, Scheme 7a). As representative example of this type of reactivity, Xiao's synthesis of pyrrolo[2,1‐a]isoquinoline 42 is depicted in Scheme 7b. Note that the formation of 42 requires a final oxidation step after elimination of diphenyl sulfide.44 Alternatively, after the attack of the electrophile, the in situ formed S‐(alkyl)sulfonium salt might be prone to react with a second nucleophile via nucleophilic substitution, in which the di‐(aryl/alkyl)sulfide acts as leaving group (Pathway A2, Scheme 7a). This reactivity offers the possibility to link nucleophiles through an ethylene bridge and is especially useful to form cyclic products of several ring sizes. The synthesis of 43 exemplify this reactivity.45 In addition, challenging cyclopropanes 44,46 aziridines 45,47 azetidines and oxetanes48 have been assembled following this route (Scheme 7b).
Scheme 7.

Reactivity scope of S‐alkenyl sulfonium salts.
Finally, another mechanism is operative when the alkenyl group in 38 bears an additional electron withdrawing group on the other alkene terminus. In these cases, nucleophiles attack with inverse regioselectivity (position C2) and the sulfide is substituted by the incoming nucleophile via a Michael addition‐elimination mechanism (Pathway B, Scheme 7a–b).49 N‐Alkenyl indoles such as 46 can be efficiently obtained in this way. Structurally related alkenyl selenonium and alkenyl iodonium salts depict a much stronger preference to follow this reaction pathway; for these compounds this kind of mechanism is operative even in the absence of electron‐withdrawing groups (Scheme 8).50
Scheme 8.

Reactivity of alkenyl iodonium and selenonium salts with conventional nucleophiles.
The seminal report describing the use of S‐(alkenyl) and S‐(aryl) sulfonium salts as electrophilic partners for coupling reactions was published in 1997 by Liebeskind and co‐workers. These authors specifically employed S‐(alkenyl) tetrahydrothiophenium salts as electrophilic partners in Suzuki–Miyaura type transformations and observed that the oxidative addition of the metal occurs regioselectively at the S–C(alkenyl) bond.51 Subsequently, Li, Shen and Lu realized in 2012 that in diaryl(alkenyl) sulfonium salt 38j, the activation of the C–S(alkenyl) bond also takes place selectively over the C–S(aryl) one in the presence of Pd0 catalysts allowing the synthesis of styrene derivatives via Suzuki coupling (Scheme 7b).52 DFT calculations confirmed this observation and indicated the reason for this selectivity. Prior to the oxidative addition step, the Pd0 fragment forms a π complex with the alkenyl rest by donation of some of electron density of the metal into the olefin π*‐orbital. This initial interaction directs the oxidative addition to the C–S(alkenyl) bond. The terminal CF3 group also plays an essential role to achieve chemoselectivity since it lowers the energy of the accepting π*‐orbital, favouring the coordination of the electron rich Pd0 to the olefin (Figure 7).
Figure 7.

Orbital interaction model for the oxidative addition of Pd(0) to S‐(alkenyl)sulfonium salts.
More recently, Procter and Ritter have exploited this reactivity on metal‐catalyzed Negishi, Sonogashira, and Heck reactions (Scheme 7b).[5b], 39 In these cases, where no directing CF3 groups are present, the necessary chemoselectivity is achieved either by replacement of the aryl rests at sulfur by aliphatic ones (tetrahydrothiophene platform), or by tying the aromatic substituents into a cyclic scaffold (thianthrene platform). The reactivity depicted by S‐(alkenyl)sulfonium salts in this context is comparable to that of alkenyl halides. Note however that the formation of salts 38b–g from the corresponding alkenes is highly regioselective, takes place under milder conditions than those required for alkene halogenation, and is still tolerant to a number of functional groups.
4. C(sp2)‐Substituted Sulfonium Salts: S‐(Aryl) Derivatives
Among sulfonium salts, (tri)aryl‐substituted ones are arguably the members of the family that have found more applications from an industrial point of view. These compounds, which are highly photosensitive, are often used as cationic photoinitiators and acid generators in areas as diverse as photolithography, coatings, and ink and adhesive development.53 Sulfonium salts of this structure have also gained increasing attention in organic synthesis. They serve as excellent partners in metal‐catalyzed cross‐coupling reactions,51 and in addition, their ability to generate organic radicals after single electron reduction is well established.6, [7d] It was however only recently when the range of transformations in which these salts are involved dramatically expanded after the recognition that the desired aryl radicals could be generated under the mild reaction conditions offered by photoredox catalysis.[6h], [7d], 54
4.1. Synthesis of S‐(Aryl) Sulfonium Salts
Diaryliodonium salts are able to arylate dialkyl sulfides 3 to afford the corresponding sulfonium salts 51. Unfortunately, harsher conditions are required for diaryl sulfide substrates. In these cases, the reactions proceed faster and with better yields if Cu(OBz)2 is used as a catalyst. This classic method might be interesting to prepare specific structures; however, the requirement of I(III)‐species as electrophiles severely limits its practicability. Attention needs to be paid to the anion accompanying the iodonium moiety. Non‐nucleophilic ones are required to avoid undesired side reactions (Scheme 9a).55
Scheme 9.

Synthetic routes for the preparation of S‐aryl sulfonium salts 51 and scope of the methods.
Alternatively, diaryl(alkoxy)sulfonium salts such as 52 can be easily obtained by alkylation of the corresponding sulfoxide with Meerwein salts. Reaction of these compounds with Grignard reagents takes place smoothly to afford the corresponding triarylsulfonium salts 51 in good to excellent yields (Scheme 9b). While the number of sulfonium salts that have been prepared following this route is broad, the reaction shows the typical limitation associated with the use of Grignard reagents; namely, restricted functional group tolerance.[2a], 56 The initial activation of the sulfoxide with TMSCl or TMSOTf seems to be equally efficient for this synthetic route;57 in addition, isoelectronic sulfiliminium salts 53 show similar reactivity towards Grignards (Scheme 9c).58
From all methods described, probably the most valuable from a synthetic point of view is the one shown in Scheme 9d, which only requires easy available sulfoxides as starting materials. These are activated by an acid anhydride, usually Tf2O, to generate in situ the corresponding S(IV) bistriflate 7. Further reaction with non‐functionalized arenes via electrophilic aromatic substitution affords the desired salts 51a–f. The scope of arenes that satisfactorily get involved in this reaction is relatively broad; it ranges from electron poor rings such as 1,2‐dichlorobenzene to moderately electron rich ones, being benzofuran at the upper limit of nucleophilicity. More electron‐deficient (hetero)arenes are too electron‐poor to react, whereas electron‐richer rings cause problems because of the involvement of radical species by single electron transfer.
The additional advantages of this synthetic method reside in its exquisite regioselectivity, which is governed by the directing effects of the substituents present in the substrate, and the remarkable functional group tolerance. Cyclopropanes, halides, esters, nitriles, ethers, ketones, aldehydes, amides, sulfonamides, thiophenes and pyridines remain untouched during the sulfenylation step (Scheme 9e).[7a], [7b], 59 AlCl3‐activated sulfilimines 54 are able to functionalize C–H bonds from aromatics as well; however, the scope of this transformation has not been studied in detail.57
4.2. Structure of S‐(Aryl) Sulfonium Salts
S‐(aryl)dibenzothiophenium tiflate 51g is showcased as illustrative example of this family of sulfonium salts (Figure 8). As in the other cases already inspected the X‐ray structure of 51g shows a central S1 atom, which adopts a pyramidal geometry (sum of angles around S1 = 300.0°). While an O1–S1 contact between the cationic and anionic elements of the salt is present, the distance separating both atoms (3.251 Å) is longer than in 1a (Figure 3), probably due to the higher steric requirement of the phenyl rest when compared with a linear alkyne substituent. Natural population analysis at the B3LYP/6‐31G* level carried out for the parent S‐(phenyl) structure 51h indicate that the sulfur atom bears a nearly complete positive charge (+0.902e), while the α‐carbon to sulfur, C1, is negatively charged (–0.238e).[59b]
Figure 8.

Molecular diagram of the X‐ray structure of 51g (top), ellipsoids shown at 50 % probability; and representation of the HOMO (left) and LUMO+1 (right) of the parent structure 51h. Partial charges and Wiberg bond orders are calculated at B3LYP/6‐311G* level.
Having three aromatic substituents, the synthetic utility of triaryl‐substituted sulfonium salts can only be harvest if the transfer of only one of these arenes to the desired substrate is chemoselectively achieved. The strategy normally used to attain this goal consists in the embedding of the two rests that should not react into a cyclic scaffold. Taking again 51g as example, any plausible oxidative addition of a metal into the S1–C6 bond will probably be followed by an extremely facile reductive elimination, which regenerates the starting five‐membered thiophenium ring and minimizes the possibility that any further transformation occurs. This effect provided by the chelating template does not operate when the oxidative addition occurs at the S1–C1 bond. As result, further reactivity involving the exocyclic group is not hindered.[59b], 60
Another interesting conclusion can be obtained from the careful inspection of the frontier orbitals. In S‐(phenyl) sulfonium salt 51h the very low‐lying LUMO+1 largely corresponds to the mixture between the π*‐system of the dibenzothiophene platform and the exocyclic σ*(S–Ar) orbital (Figure 8). This distribution of orbital coefficients also suggests that populating this orbital weakens the S1–Ar(exocyclic) bond. Thus, by one electron reduction of 51h, the homolytic cleavage of the S1–Ar(exocyclic) to afford aryl radicals is highly favored (chemoselective) if compared with the cleavage of the other two S–C bonds (S1–C7 and S1–CX), which are constituents of the dibenzothiophenium skeleton.[6f], [7a], 61
4.3. Reactivity of S‐(Aryl) Sulfonium Salts
Being di(aryl/alkyl)sulfides excellent leaving groups, it is not surprising that sulfonium salts of general formula 51 have been recognized as excellent electrophilic partners for C–C and C–X coupling reactions. These transformations normally proceed in the presence of metal catalysts; however, when strong nucleophiles such as alkoxydes, thiolates or even fluoride are employed, the non‐catalyzed reaction still proceeds satisfactorily rendering aryl ethers, thioethers or fluorides.62 Theoretical calculations for the fluoride case indicate that, more than in a typical SNAr mechanism, it is the coordination of fluoride to the S‐center followed by reductive elimination of Ar–F, the pathway that requires lower activation energy. Combined with the site‐selective and functional group tolerant syntheses available for S‐(aryl) sulfonium salts, this reaction allows the late‐stage aromatic fluorination of relatively complex structures under mild conditions (Scheme 10). Slight modifications of the protocol also permit its use for the efficient 18F‐labelling of clinically relevant PET tracers 55–57.[7c], [59a], [62d]
Scheme 10.

Selected examples of metal‐free fluorination of aryl groups.
The use of aryl sulfonium salts 51 as electrophilic partners in Ni‐ and Pd‐catalyzed cross coupling chemistry was also pioneered by the group of Liebeskind in Stille, Negishi and Suzuki–Miyaura reactions. In their experiments they specifically used S‐(aryl) tetrahydrothiophenium salts as a platform that allows the selective S–C(aryl) oxidative addition (Scheme 11a).[5k], 51 The beneficial attributes associated with the use of sulfonium salts are, if compared with aryl halides or triflates, their already mentioned easy and regioselective preparation unfunctionalized arenes and non‐volatility. In addition, they are comparatively more thermally stable than diazonium salts. Moreover, hypervalent I(III) reagents of similar structure are not adequate substrates for coupling reactions because the aryl iodide side products form after the oxidative addition are able to react further under the applied reaction conditions. After the seminal work of Liebeskind, additional variants were developed not only for the Suzuki–Miyaura and Negishi,[5b] but also for the Mizoroki–Heck,[5c], [5h] Sonogashira[5d] and carboxylation reactions63 (Scheme 11). Among other examples it is also worthy to highlight the results of Yorimitsu and co‐workers. These authors have shown that after the initial S–C oxidative addition in substrates of general formula 51o, C–H activation processes may be used to deliver condensed polyarenes 63.[5j], 64
Scheme 11.

Reactivity scope of S‐(aryl) sulfonium salts 51 in typical Pd‐catalyzed coupling reactions.
The employment of aryl sulfonium salts has also been recently extended to the formation of C–X bonds. In concrete Zhang and Tian have reported the Ullmann‐type N‐arylation of anilines using dialkyl(aryl)sulfonium triflates 51p as arylating agents in the presence of Pd[P(tBu)3]2/CuBr as a catalytic system.65 The corresponding diarylamines were prepared in good to high yields (Scheme 11e). In an independent study, Ritter and co‐workers reported an even more versatile approach, which allows extending the scope of this reaction to secondary amines and amides. Depending on the substrate employed, they identified Pd(AlPhos)2COD or [Pd2(dba)3]/RuPhos as the ideal catalysts, in combination with sulfonium salts derived from the thianthrenium unit.[5a] It worth to highlight again that for all the coupling reactions mentioned, di(alkyl/aryl)sulfides are formed as the side products, being these compounds generally inert in the presence of Pd catalysts. This is again an advantage if compared with related I(III)‐species since the latter produce aryl iodides, which may undergo further reaction under the conditions applied.
The reduction of sulfonium salts via single electron transfer to generate alkyl radicals was first introduced by Kellogg and co‐workers employing Ru(bpy)3Cl2 as a photocatalyst and Hantzch esters as reductant.66 That work, which corresponds to the first application reported of photoredox catalysis in organic chemistry, is in the origin of the recent development in the area. Although the specific transformation chosen for their seminal study, namely the desulfuration of phenacyl sulfonium salts to acetophenones, had no strong synthetic relevance; their mechanistic studies paved the way to the further use of sulfonium and other salts, such as iodonium ones, as organic radical promoters under the mild conditions offered by photocatalysis (Scheme 12a). Some years later, Fensterbank, Goddard and Ollivier reported the reduction of triaryl sulfonium salts under analogous conditions, and the subsequent reaction of the in situ generated radicals with olefins to afford the corresponding arylated products 66 (Scheme 12b). The tri(aryl)sulfonium salts 51h used in this study had three identical aryl rests; this obviously limits the reaction scope, but no chemoselectivity problem arises during the key S‐CAr bond cleavage step.[7d]
Scheme 12.

Photoredox‐catalyzed C–C and C–X coupling reactions via selective aryl radical generation.
Making use of photoredox catalysis and the chemoselective S–C(exo) cleavage provided by dibenzothiophenium salts, Procter has been able to develop a metal free coupling between the exo‐aryl group of sulfonium salts and non‐functionalized (hetero)arenes (Scheme 12c).[7a] This strategy pairs in one‐pot the sulfonium salt synthesis and the photocatalytic cross‐coupling steps, allowing a fast construction of biaryl motifs by functionalization of two original C–H bonds. Diazonium salts were known to have very similar reactivity under these photochemical conditions, but their synthesis is not as straightforward (Scheme 12d).67 Finally, by combination of photoredox catalysis with more conventional Cu reactivity, Ritter and co‐workers have been able to develop functional protocols for the late stage fluorination, trifluoromethylation and oxygenation of structurally complex compounds via aryl sulfonium salts (Scheme 12e, f).61, 68 The photochemical borylation of arenes via C–S bond activation has also been recently reported.69 All these methods rely on the crucial regioselective synthesis of sulfonium salts from arenes via C–H sulfenilation.
5. C(sp3)‐Substituted Sulfonium Salts: S‐(Alkyl) Derivatives
S‐(alkyl) sulfonium salts have been classically used as precursors for S‐ylides, which are well‐known reagents for epoxidation, aziridination and cyclopropanation. This chemistry has not only been reviewed,3 but it has also become textbook chemistry.70 Therefore, our discussion will not focus on these transformations. We will only cover reactions in which the alkyl rests are not transformed before their transfer to a given substrate.
5.1. Synthesis of S‐(Alkyl) Sulfonium Salts
A compilation of the most general methods to prepare S‐(alkyl)sulfonium salts is shown in Scheme 13. Dialkyl sulfides are efficiently alkylated to the corresponding sulfonium salts by reaction with alkyl iodides or triflates. The substrate scope of this reaction can be extended to less nucleophilic diaryl sulfides or sterically demanding ones by concomitant employment of alkyl bromides/iodides and AgBF4 or NaClO4 as halogenide scavengers (Scheme 13a).71 Treatment of alkoxy‐substituted sulfonium salts 52 with alkyl Grignard reagents yields the corresponding S‐(alkyl)sulfonium salts 72 as well; however, when softer organocadmium reagents are used as nucleophiles, the chemical yields are improved and racemization processes (in case of enantiopure substrates) are minimized (Scheme 13b).72
Scheme 13.

Synthetic routes for the preparation of S‐alkyl sulfonium salts and scope of the methods.
Finally, the method already described in section 4.1 for the preparation of triarylsulfonium salts from sulfoxides can also be applied for the synthesis of 72‐type structures as well; in this case by employing alkyl, (o‐biaryl)‐substituted sulfoxides as a starting materials. This intramolecular cyclisation is often used for the preparation of S‐(alkyl) salts that cannot be obtained by simple alkylation, such as the Umemoto's reagent 72a, which is arguably the most well‐known example of a sulfonium salt, and an extremely versatile reagent for the introduction of CF3‐groups into elaborated organic substrates.73, 74
5.2. Structure of S‐(Alkyl) Sulfonium Salts
While the only X‐ray structure available of compound 72b is not a well‐defined one;75 still some comparison is with those of 1a, 38h and 51g is possible. In 72b the S1 atom again adopts the expected pyramidal geometry (sum of angles around S1 = 299.1°), which is more acute than in all other cases analysed probably due to the small steric requirements for the methyl rest. Natural population analysis at the B3LYP/6‐31G* level indicates that 72b follows the same tendency already observed: the sulfur atom S1 is highly positively charged (+0.869e); the HOMO can be interpreted as a free electron pair centered at S, and the LUMO largely corresponds to the mixture between the π*‐system of the dibenzothiophene platform and the exocyclic σ*(S–Me) orbital. From this frontier orbital picture can be concluded that nucleophiles will suffer alkylation by reaction with 72b either by attack at the alkyl rest (SN2 mechanism) or at sulfur (followed by ligand coupling), while one electron reduction of 72b should afford alkyl radicals, especially if stabilizing groups are present at the alkyl substituent (Figure 9).
Figure 9.

Calculated structure of cation 72b, representation of its HOMO (left) and LUMO (right) and partial charges and Wiberg bond orders calculated at B3LYP/6‐311G* level.
5.3. Reactivity of S‐(Alkyl) Sulfonium Salts
As predicted from their electronic structure, when combined with nucleophiles, most S‐(alkyl) sulfonium salts 72 serve as soft alkylating reagents. Nucleophiles such as phenols, amine, enolates, and thiolate ions get involved in this reaction, being the operating mechanism proposed strongly depending on their nature. Traditional SN2 reactions are likely to occur for soft nucleophiles (pathway A1, Scheme 14), while sulfuran intermediate formation followed by ligand coupling has been proposed to operate for hard ones (Pathway A2, Scheme 14). Saving the distances in terms of the mechanism, S‐(alkyl) sulfonium salts can be considered softer analogues of trialkyloxonium ones (Scheme 14a, b).76
Scheme 14.

Reactivity outline and synthetic applications of S‐(alkyl)sulfonium salts 72.
S‐(alkyl) sulfonium salts also get involve in metal catalysed coupling reactions. For example, the Umemoto reagent 72a has been intensively used in Pd‐catalyzed trifluoromethylation of aromatic rings after an initial C–H activation step. While the detailed redox chemistry involved in the C–CF3 bond‐forming needs further study, the use of Cu(OAc)2 was found to be essential for enhancing the catalytic turnover. Mechanisms involving Pd(II)/Pd(IV) and/or Pd(0)/Pd(II) pathways have been proposed (Scheme 14c);77 and the extension of the scope of this reaction to non‐fluorinated alkyl rests has been recently reported.78
Finally, as a continuation of the seminal work of Kellogg mentioned in the former section,66 S‐(alkyl) sulfonium salts have also found interesting applications as alkyl radical sources under mild photoredox catalytic conditions.79 An interesting example, which is depicted in Scheme 14d, has been very recently reported by Novák and co‐workers. These authors have developed a decarboxylative Liebeskind‐type benzylation of prolines using dual Ni–Ir catalysts.80 As key steps for this transformation, the authors propose the oxidative addition of a Ni0 catalysts into the C–S bond of the sulfonium salt 72d,followed by reaction of the thus obtained metal complex with a photochemically generated tetrahydropyrroyl radical.
6. Conclusions
Although sulfonium salts have been used since decades in organic synthesis, their chemistry is far from being exhausted, and many exciting transformations are still to be uncovered while exploring this area of the chemical space. Specifically, we envision future applications of sulfonium salts as customized reagents for the late stage functionalization of advanced organic structures, either in combination with traditional coupling catalysis, or with the more trending areas of photoredox catalysis and electrosynthesis. For this specific task the variety of available syntheses for the efficient preparation of sulfonium salts offers the possibility to optimize on demand the structure of the S‐platform independently from the nature of group to be transferred stands. We anticipate that this structural versatility will be fundamental for the discovery and application of new transformations involving these reagents.
Acknowledgements
We thank Dr. Golz (University of Göttingen) for DFT calculations on model substrates. M. Pretor and M. Blaue (University of Göttingen) are also acknowledged for providing graphic material. Support from the European Research Council (ERC CoG 771295) and the Deutsche Forschungsgemeinschaft (INST 186/1237‐1 and INST 186/1324‐1) is gratefully acknowledged. We are also grateful to the University of Göttingen for continued support of our research programs. Open access funding enabled and organized by Projekt DEAL.
Biographies
Sergei I. Kozhushkov was born in 1956 in Kharkov, USSR. He studied chemistry at Lomonosov Moscow State University, where he obtained his doctoral degree in 1983 under the supervision of Professor N. S. Zefirov and performed his Habilitation in 1998. From 1983 to 1991, he worked at Moscow State University and then at Zelinsky Institute of Organic Chemistry. In 1991, he joined the research group of Professor A. de Meijere (Göttingen) as an AvH Research Fellow. Since 1993 he has worked as a Research Associate and since 1996 he has held a position of a Scientific Assistant. In 2001 he was promoted to Senior Scientist at the University of Göttingen. His current research interests focus on the chemistry of highly strained small ring compounds. The results of his scientific activity have been published in over 200 original publications, review articles, book chapters and patents.

Manuel Alcarazo obtained his Ph.D. from the Universidad de Sevilla (Spain) in 2005, having worked under the supervision of Profs. Rosario Fernández and José M. Lassaletta. Following a postdoctoral stay at the MPI für Kohlenforschung in Mülheim an der Ruhr (Germany) working with Professor Alois Fürstner on the synthesis of Ecklonialactones and the design of new C‐based ligands, he began his independent research career in 2009 at the same institution. Since 2015 he is Full Professor for Organic Synthesis at the University of Göttingen. For his research Manuel Alcarazo has been awarded with an ERC StG and an ERC CoG grants. His research interests are focused on the areas of homogeneous (asymmetric) catalysis and the applications of main group chemistry into organic synthesis.

References
- 1.a) Fernández I. and Khiar N., in: Arylsulfonium Salts and Derivatives, in Science of Synthesis, Vol 31a (Ed.: Ramsden C. A.), Thieme, Stuttgart, 2007, pp. 1001–1014; [Google Scholar]; b) Kaiser D., Klose I., Oost R., Neuhaus J. and Maulide N., Chem. Rev, 2019, 119, 8701–8780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Andersen K. K., Cinquini M. and Papanikolaou N. E., J. Org. Chem, 1970, 35, 706–710; [Google Scholar]; b) Baechler R. D. and Mislow K., J. Am. Chem. Soc, 1970, 92, 3090–3093; [Google Scholar]; c) Darwish D. and Scott C. E., Can. J. Chem, 1973, 51, 3647–3648. [Google Scholar]
- 3.a) Oost R., Neuhaus J. D., Merad J. and Maulide N., in: Sulfur Ylides in Organic Synthesis and Transition Metal Catalysis, in Modern Ylide Chemistry. Structure and Bonding, Vol 177, (Ed.: Gessner V.), Springer, Cham, 2017; [Google Scholar]; b) Yanagi Y., Nogi K. and Yorimitsu H., Tetrahedron Lett, 2018, 59, 2951–2959; [Google Scholar]; c) Pulis A. P. and Procter D. J., Angew. Chem. Int. Ed, 2016, 55, 9842–9860; [DOI] [PubMed] [Google Scholar]; Angew. Chem, 2016, 128, 9996–10014. [Google Scholar]
- 4. Zhdankin V. V., in: Hypervalent Iodine Chemistry: Preparation, Structure, and Synthetic Applications of Polyvalent Iodine Compounds, Wiley, Chichester, 2014. [Google Scholar]
- 5.a) Engl P. S., Häring A. P., Berger F., Berger G., Pérez‐Bitrián A. and Ritter T., J. Am. Chem. Soc, 2019, 141, 13346–13351; [DOI] [PubMed] [Google Scholar]; b) Aukland M. H., Talbot F. J. T., Fernández‐Salas J. A., Ball M., Pulis A. P. and Procter D. J., Angew. Chem. Int. Ed, 2018, 57, 9785–9789; [DOI] [PubMed] [Google Scholar]; Angew. Chem, 2018, 130, 9933–9937; [Google Scholar]; c) Uno D., Minami H., Otsuka S., Nogi K. and Yorimitsu H., Chem. Asian J, 2018, 13, 2397–2400; [DOI] [PubMed] [Google Scholar]; d) Tian Z.‐Y., Wang S.‐M., Jia S.‐J., Song H.‐X. and Zhang C.‐P., Org. Lett, 2017, 19, 5454–5457; [DOI] [PubMed] [Google Scholar]; e) Wang S.‐M., Wang X.‐Y., Qin H.‐L. and Zhang C.‐P., Chem. Eur. J, 2016, 22, 6542–6546; [DOI] [PubMed] [Google Scholar]; f) Wang X.‐Y., Song H.‐X., Wang S.‐M., Yang J., Qin H.‐L., Jiang X. and Zhang C.‐P., Tetrahedron, 2016, 72, 7606–7612; [Google Scholar]; g) Cowper P., Jin Y., Turton M. D., Kociok‐Köhn G. and Lewis S. E., Angew. Chem. Int. Ed, 2016, 55, 2564–2568; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem, 2016, 128, 2610–2614; [Google Scholar]; h) Wang S.‐M., Song H.‐X., Wang X.‐Y., Liu N., Qin H.‐L. and Zhang C.‐P., Chem. Commun, 2016, 52, 11893–11896; [DOI] [PubMed] [Google Scholar]; i) Vasu D., Yorimitsu H. and Osuka A., Synthesis, 2015, 47, 3286–3291; [Google Scholar]; j) Vasu D., Yorimitsu H. and Osuka A., Angew. Chem. Int. Ed, 2015, 54, 7162–7166; [DOI] [PubMed] [Google Scholar]; Angew. Chem, 2015, 127, 7268–7272; [Google Scholar]; k) Zhang S., Marshall D. and Liebeskind L. S., J. Org. Chem, 1999, 64, 2796–2804. [DOI] [PubMed] [Google Scholar]
- 6.a) Wang X., Saeva F. D. and Kampmeier J. A., J. Am. Chem. Soc, 1999, 121, 4364–4368; [Google Scholar]; b) Kampmeier J. and Nalli T. W., J. Org. Chem, 1993, 58, 943–949; [Google Scholar]; c) Saeva F. D., Breslin D. T. and Luss H. R., J. Am. Chem. Soc, 1991, 113, 5333–5337; [Google Scholar]; d) Dektar L. and Hacker N. P., J. Am. Chem. Soc, 1990, 112, 6004–6015; [Google Scholar]; e) Saeva F. D., Tetrahedron, 1986, 42, 6123–6129; [Google Scholar]; f) Beak P. and Sullivan T. A., J. Am. Chem. Soc, 1982, 104, 4450–4457; [Google Scholar]; g) Chung S.‐K. and Sasamoto K., J. Chem. Soc., Chem. Commun, 1981, 346–347; [Google Scholar]; h) Péter Á., Perry G. J. P. and Procter D. J., Adv. Synth. Catal. 2020, DOI:‐ 10.1002/adsc.202000220. [DOI] [Google Scholar]
- 7.a) Aukland M. H., Šiaučiulis M., West A., Perry G. J. P. and Procter D. J., Nat. Catal, 2020, 3, 163–169; [Google Scholar]; b) Berger F., Plutschack M. B., Riegger J., Yu W., Spiecher S., Ho M., Frank N. and Ritter T., Nature, 2019, 567, 223–228; [DOI] [PubMed] [Google Scholar]; c) Gendron T., Sander K., Cybulska K., Benhamou L., Sin P. K. B., Khan A., Wood M., Porter M. J. and Årstad E., J. Am. Chem. Soc, 2018, 140, 11125–11132; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Donck S., Baroudi A., Fensterbank L., Goddard J.‐P. and Ollivier C., Adv. Synth. Catal, 2013, 355, 1477–1482. [Google Scholar]
- 8.a) Rangappa P., Shine H. J., Marx J. N., Ould‐Ely T., Kelly A. T. and Whitmire K. H., J. Org. Chem, 2005, 70, 9764–9770; [DOI] [PubMed] [Google Scholar]; b) Ochiai M., Nagoaka T., Sueda T., Yan J., Chen D.‐W. and Miyamoto K., Org. Biomol. Chem, 2003, 1, 1517–1521. [DOI] [PubMed] [Google Scholar]
- 9. Höfer M. and Liskka R., J. Polym. Sci., Part A, 2009, 47, 3419–3430. [Google Scholar]
- 10.a) Waldecker B., Kafuta K. and Alcarazo M., Org. Synth, 2019, 96, 258–276; [Google Scholar]; b) Waldecker B., Kraft F., Golz C. and Alcarazo M., Angew. Chem. Int. Ed, 2018, 57, 12538–12542; [DOI] [PubMed] [Google Scholar]; Angew. Chem, 2018, 130, 12718–12722. [Google Scholar]
- 11. Li X., Golz C. and Alcarazo M., Angew. Chem. Int. Ed, 2019, 58, 9496–9500; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem, 2019, 131, 9596–9600. [Google Scholar]
- 12. Roy A., Maity A., Giri R. and Bisai A., Asian J. Org. Chem, 2020, 9, 226–232. [Google Scholar]
- 13.Special Issue “Hypervalent Iodine Chemistry” (Ed.: Wirth T.), in Topics in Current Chemistry, Vol. 373, Springer, Cham, 2016. [Google Scholar]
- 14.See for example: a) Boelke A., Vlasenko Y. A., Yusubov M. S., Nachtsheim B. J. and Postnikov P. S., Beilstein J. Org. Chem, 2019, 15, 2311–2318; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Alazet S., Preindl J., Simonet‐Davin R., Nicolai S., Nanchen A., Meyer T. and Waser J., J. Org. Chem, 2018, 83, 12334–12356. [DOI] [PubMed] [Google Scholar]
- 15.For a recent review on the synthesis and reactivity of haloacetylenes see: a) Wu W. and Jiang H., Acc. Chem. Res, 2014, 47, 2483–2504; [DOI] [PubMed] [Google Scholar]; b) Liu G., Kong L., Shen J. and Zhu G., Org. Biomol. Chem, 2014, 12, 2310–2321; [DOI] [PubMed] [Google Scholar]; c) Zhang J., Li P. and Wang L., Org. Biomol. Chem, 2014, 12, 2969–2978; [DOI] [PubMed] [Google Scholar]; d) Peng J., Zhao Y., Zhou J., Ding Y. and Chen C., Synthesis, 2014, 46, 2051–2056; [Google Scholar]; e) Ohmura T., Kijima A., Komori Y. and Suginome M., Org. Lett, 2013, 15, 3510–3513. [DOI] [PubMed] [Google Scholar]
- 16. Beringer F. M. and Galton S. A., J. Org. Chem, 1965, 30, 1930–1934. [Google Scholar]
- 17. Ochiai M., Masaki Y. and Shiro M., J. Org. Chem, 1991, 56, 5511–5513. [Google Scholar]
- 18. Zhdankin V. V., Kuehl C. J., Krasutsky A. P., Bolz J. T. and Simonsen A. J., J. Org. Chem, 1996, 61, 6547–6551. [DOI] [PubMed] [Google Scholar]
- 19.a) Ochiai M., Ito T., Takaoka Y., Masaki Y., Kunishima M., Tani S. and Nagao Y., J. Chem. Soc., Chem. Commun, 1990, 118–119; [Google Scholar]; b) Ochiai M., Kunishima M., Nagao Y., Fuji K., Shiro M. and Fujita E., J. Am. Chem. Soc, 1986, 108, 8281–8283. [Google Scholar]
- 20. Bachi M. D., Bar‐Ner N., Crittell C. M., Stang P. J. and Williamson B. L., J. Org. Chem, 1991, 56, 3912–3915. [Google Scholar]
- 21.a) Frei R., Wodrich M. D., Hari D. P., Borin P.‐A., Chauvier C. and Waser J., J. Am. Chem. Soc, 2014, 136, 16563–16573; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chen C. C. and Waser J., Chem. Commun, 2014, 50, 12923–12926; [DOI] [PubMed] [Google Scholar]; c) Fernández‐González D., Brand J. P. and Waser J., Chem. Eur. J, 2010, 16, 9457–9461; [DOI] [PubMed] [Google Scholar]; d) Fernández‐González D., Brand J. P., Mondière R. and Waser J., Adv. Synth. Catal, 2013, 355, 1631–1639; [Google Scholar]; e) Frei R. and Waser J., J. Am. Chem. Soc, 2013, 135, 9620–9623. [DOI] [PubMed] [Google Scholar]
- 22.For selected reviews see: a) Hari D. P., Caramenti P. and Waser J., Acc. Chem. Res, 2018, 51, 3212–3225; [DOI] [PubMed] [Google Scholar]; b) Waser J., Synlett, 2016, 27, 2761–2773; [Google Scholar]; c) Brand J. P. and Waser J., Chem. Soc. Rev, 2012, 41, 4165–4179. [DOI] [PubMed] [Google Scholar]
- 23. Talavera G., Peña J. and Alcarazo M., J. Am. Chem. Soc, 2015, 137, 8704–8707. [DOI] [PubMed] [Google Scholar]
- 24.a) Fatiadi A. J., in: Preparation and Synthetic Applications of Cyano Compounds (Eds.: Patai S. and Rappoport Z.), John Wiley & Sons, New York: 1983, pp. 1057–1303; [Google Scholar]; b) Larock R. C., in: Comprenhensive Organic Transformations: A Guide to Functional Group Preparations, 2nd Ed., Wiley‐VCH, Weinheim, 2010, pp. 819–955. [Google Scholar]
- 25. Fleming F. F., Nat. Prod. Rep, 1999, 16, 597–606. [Google Scholar]
- 26. Fleming F. F., Yao L., Ravikumar P. C., Funk L. and Shook B. C., J. Med. Chem, 2010, 53, 7902–7917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pollack P., in: Fine Chemicals: The industry and the Business, John Wiley & Sons, New Jersey, 2007. [Google Scholar]
- 28. Huang S.‐T., Hsu Y.‐C., Yen Y.‐S., Chou H.‐H., Lin J.‐T., Chang C.‐W., Hsu C.‐P., Tsai C. and Yin D.‐J., J. Phys. Chem. C, 2008, 112, 19739–19747. [Google Scholar]
- 29. Zil'berman E. N., Russ. Chem. Rev, 1986, 55, 39–48. [Google Scholar]
- 30.For a recent review see: Yan G., Zhang Y. and Wang J., Adv. Synth. Catal, 2017, 359, 4068–4105. [Google Scholar]
- 31.a) Nauth A. M. and Opatz T., Org. Biomol. Chem, 2019, 17, 11–23; [DOI] [PubMed] [Google Scholar]; b) Schörgenhumer J. and Waser M., Org. Chem. Front, 2016, 3, 1535–1540; [Google Scholar]; c) Kim J., Kim J. H. and Chang S., Angew. Chem. Int. Ed, 2012, 51, 11948–11959; [DOI] [PubMed] [Google Scholar]; Angew. Chem, 2012, 124, 12114–12125. [Google Scholar]
- 32. Noller C. R., in: Lehrbuch der Organischen Chemie, Springer, Berlin, 2013, p. 1029. [Google Scholar]
- 33.a) Cui J., Song J., Liu Q., Liu H. and Dong Y., Chem. Asian J, 2018, 13, 482–495; [DOI] [PubMed] [Google Scholar]; b) Akula R., Xiong Y. and Ibrahim H., RSC Adv, 2013, 3, 10731–10735; [Google Scholar]; c) Anbarasan P., Neumann H. and Beller M., Chem. Eur. J, 2011, 17, 4217–4222; [DOI] [PubMed] [Google Scholar]; d) Anbarasan P., Neumann H. and Beller M., Chem. Eur. J, 2010, 16, 4725–4728; [DOI] [PubMed] [Google Scholar]; e) Wu Y.‐Q., Limburg D. C., Wilkinson D. E. and Hamilton G. S., Org. Lett, 2000, 2, 795–797; [DOI] [PubMed] [Google Scholar]; f) Hughes T. V., Hammond S. D. and Cava M. P., J. Org. Chem, 1998, 63, 401–402. [Google Scholar]
- 34. Zhdankin V. V., Kuehl C. J., Krasutsky A. P., Bolz J. T., Mismash B., Woodward J. K. and Simonsen A. J., Tetrahedron Lett, 1995, 36, 7975–7978. [Google Scholar]
- 35.a) Declas N., Le Vaillant F. and Waser J., Org. Lett, 2019, 21, 524–528; [DOI] [PubMed] [Google Scholar]; b) Le Vaillant F., Wodrich M. D. and Waser J., Chem. Sci, 2017, 8, 1790–1800; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Frei R., Courant T., Wodrich M. D. and Waser J., Chem. Eur. J, 2015, 21, 2662–2668. [DOI] [PubMed] [Google Scholar]
- 36. Wang X. and Studer A., Angew. Chem. Int. Ed, 2018, 57, 11792–11796; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem, 2018, 130, 11966–11970. [Google Scholar]
- 37.For other applications of this reagent see: Barrado A. G., Zieliński A., Goddard R. and Alcarazo M., Angew. Chem. Int. Ed, 2017, 56, 13401–13405; [DOI] [PubMed] [Google Scholar]; Angew. Chem, 2017, 129, 13586–13590. [Google Scholar]
- 38. Mondal M., Chen S. and Kerrigan N. J., Molecules 2018, 23, No 738, 29 pages. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen J., Li J., Plutschack M. B., Berger F. and Ritter T., Angew. Chem. Int. Ed, 2020, 59, 5616–5620; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem, 2020, 132, 5665–5669. [Google Scholar]
- 40.a) Matsuo J.‐i., Yamanaka H., Kawana A. and Mukaiyama T., Chem. Lett, 2003, 32, 392–393; [Google Scholar]; b) Nenajdenko V. G., Vertelezkij P. V., Gridnev I. D., Shevchenko N. E. and Balenkova E. S., Tetrahedron, 1997, 53, 8173–8180. [Google Scholar]
- 41.a) Boelke A., Caspers L. D. and Nachtsheim B. J., Org. Lett, 2017, 19, 5344–5347; [DOI] [PubMed] [Google Scholar]; b) Stridfeldt E., Seemann A., Bouma M. J., Dey C., Ertan A. and Olofsson B., Chem. Eur. J, 2016, 22, 16066–16070; [DOI] [PubMed] [Google Scholar]; c) Fujiita M., Lee H. J. and Okuyama T., Org. Lett, 2006, 8, 1399–1401; [DOI] [PubMed] [Google Scholar]; d) Ochiai M., Sueda T., Noda R. and Shiro M., J. Org. Chem, 1999, 64, 8563–8567; [Google Scholar]; e) Huang X. and Xu X. H., J. Chem. Soc., Perkin Trans. 1, 1998, 3321–3322; [Google Scholar]; f) Ochiai M., Sumi K., Nagao Y. and Fujita E., Tetrahedron Lett, 1985, 26, 2351–2354. [Google Scholar]
- 42. Yar M., McGarrigle E. M. and Aggarwal V. K., in: e‐EROS Encyclopedia of Reagents for Organic Synthesis; John Wiley & Sons, Hoboken, 2012, pp. 1–6. [Google Scholar]
- 43. Ulbrich J., Bachelorarbeit, University of Göttingen, 2018. [Google Scholar]
- 44. An J., Yang Q.‐Q., Wang Q. and Xiao W.‐J., Tetrahedron Lett, 2013, 54, 3834–3837. [Google Scholar]
- 45.a) Matlock J. V., Svejstrup T. D., Songara P., Overington S., McGarrigle E. M. and Aggarwal V. K., Org. Lett, 2015, 17, 5044–5047; [DOI] [PubMed] [Google Scholar]; b) An J., Chang N.‐J., Song L.‐D., Jin Y.‐Q., Ma Y., Chen J.‐R. and Xiao W.‐J., Chem. Commun, 2011, 47, 1869–1871; [DOI] [PubMed] [Google Scholar]; c) Yar M., McGarrigle E. M. and Aggarwal V. K., Org. Lett, 2009, 11, 257–260; [DOI] [PubMed] [Google Scholar]; d) Yar M., McGarrigle E. M. and Aggarwal V. K., Angew. Chem. Int. Ed, 2008, 47, 3784–3786; [DOI] [PubMed] [Google Scholar]; Angew. Chem, 2008, 120, 3844–3846. [Google Scholar]
- 46.For selected examples, see: a) Ishikawa T., Kasai N., Yamada Y. and Hanamoto T., Tetrahedron, 2015, 71, 1254–1260; [Google Scholar]; b) Mao Z., Qu H., Zhao Y. and Lin X., Chem. Commun, 2012, 48, 9927–9929; [DOI] [PubMed] [Google Scholar]; c) Lin H., Shen Q. and Lu L., J. Org. Chem, 2011, 76, 7359–7369. [DOI] [PubMed] [Google Scholar]
- 47.a) Kurosato F., Ishikawa T., Yamada Y. and Hanamoto T., Synlett, 2015, 26, 1827–1830; [Google Scholar]; b) Maeda R., Ooyama K., Anno R., Shiosaki M., Azema T. and Hanamoto T., Org. Lett, 2010, 12, 2548–2550. [DOI] [PubMed] [Google Scholar]
- 48. Fritz S. P., Moya J. F., Unthank M. G., McGarrigle E. M. and Aggarwal V. K., Synthesis, 2012, 44, 1584–1590. [Google Scholar]
- 49.a) Zhou M., Tan X., Hu Y., Shen H. C. and Qian X., J. Org. Chem, 2018, 83, 8627–8635; [DOI] [PubMed] [Google Scholar]; b) Šiaučiulis M., Ahlsten N., Pulis A. P. and Procter D. J., Angew. Chem. Int. Ed, 2019, 58, 8779–8783; [DOI] [PubMed] [Google Scholar]; Angew. Chem, 2019, 131, 8871–8875. [Google Scholar]
- 50.See for example: a) Watanabe S., Mori E., Nagai H., Iwamura T., Iwama T. and Kataoka T., J. Org. Chem, 2000, 65, 8893–8898; [DOI] [PubMed] [Google Scholar]; b) Watanebe S.‐I., Mori E., Nagai H. and Kataoka T., Synlett, 2000, 49–52; [Google Scholar]; c) Okuyama T., Fujita M., Gronheid R. and Lodder G., Tetrahedron Lett, 2000, 41, 5125–5129; [Google Scholar]; d) Ochiai M., Shu T., Nagaoka T. and Kitagawa Y., J. Org. Chem, 1997, 62, 2130–2138. [DOI] [PubMed] [Google Scholar]
- 51. Srogl J., Allred G. D. and Liebeskind L. S., J. Am. Chem. Soc, 1997, 119, 12376–12377. [Google Scholar]
- 52. Lin H., Dong X., Li Y., Shen Q. and Lu L., Eur. J. Org. Chem, 2012, 4675–4679. [Google Scholar]
- 53. DeVoe R. J., Olofson P. M. and Sahyun M. R., in: Photochemistry and Photophisics of Onium Salts, in Adv. Photochem., Vol. 17 (Eds.: Volman D. H., Hammond G., Neckers D.), John Wiley & Sons, Chichester, 1992, pp. 313–355. [Google Scholar]
- 54. Stephenson C., Yoon T. and MacMillan D. W. C., in: Visible Light Photocatalysis in Organic Chemistry, Wiley‐VCH, Weinheim, 2018. [Google Scholar]
- 55. Crivello J. V. and Lam J. H. W., J. Org. Chem, 1978, 43, 3055–3058. [Google Scholar]
- 56. Andersen K. K. and Papanikolaou N. E., Tetrahedron Lett, 1966, 7, 5445–5449. [Google Scholar]
- 57.a) Imazeki S., Sumio M., Fukuzawa K., Ishihara M. and Akiyama T., Synthesis, 2004, 1648–1654; [Google Scholar]; b) Milller R., Renaldo A. F. and Ito H., J. Org. Chem, 1988, 53, 5571–5573. [Google Scholar]
- 58. Manya P., Sekera A. and Rumpf P., Bull. Soc. Chim. Fr, 1971, 286–294. [Google Scholar]
- 59.a) Xu P., Zhao D., Berger F., Hamad A., Rickmeier J., Petzold R., Kondratiuk M., Bohdan K. and Ritter T., Angew. Chem. Int. Ed, 2020, 59, 1956–1960; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem, 2020, 132, 1972–1976; [Google Scholar]; b) Kafuta K., Korzun A., Böhm M., Golz C. and Alcarazo M., Angew. Chem. Int. Ed, 2020, 59, 1950–1955; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem, 2020, 132, 1966–1971; [Google Scholar]; c) Fernández‐Salas J. A., Pulis A. P. and Procter D. J., Chem. Commun, 2016, 52, 12364–12367; [DOI] [PubMed] [Google Scholar]; d) Shevchenko N. E., Karpov A. S., Zakurdaev E. P., Nenajdenko V. G. and Balenkova E. S., Chem. Heterocycl. Compd, 2000, 36, 137–143. [Google Scholar]
- 60. Kawashima H., Yanagi T., Wu C.‐C., Nogi K. and Yorimitsu H., Org. Lett, 2017, 19, 4552–4555. [DOI] [PubMed] [Google Scholar]
- 61. Li J., Chen J., Sang R., Ham W.‐S., Plutschack M. B., Berger F., Chabbra S., Schnegg A., Genicot C. and Ritter T., Nat. Chem, 2020, 12, 56–62. [DOI] [PubMed] [Google Scholar]
- 62.a) Ming X.‐X., Tian Z.‐Y. and Zang C.‐P., Chem. Asian J, 2019, 14, 3370–3379; [DOI] [PubMed] [Google Scholar]; b) Zhao J.‐N., Kayumov M., Wang D.‐Y. and Zhang A., Org. Lett, 2019, 21, 7303–7306; [DOI] [PubMed] [Google Scholar]; c) Sander K., Galante E., Gendron T., Yiannaki E., Patel N., Kalber T. L., Badar A., Robson M., Johnson S. P., Bauer F., Mairinger S., Stanek J., Wanek T., Kuntner C., Kottke T., Weizel L., Dickens D., Erlandsson K., Hutton B. F., Lythgoe M. F., Stark H., Langer O., Koepp M. and Årstad E. Š., J. Med. Chem, 2015, 58, 6058–6080; [DOI] [PubMed] [Google Scholar]; d) Mu L., Fischer C. R., Holland J. P., Becaud J., Schubiger P. A., Schibli R., Ametamey S. M., Graham K., Stellfeld T., Dinkelborg L. M. and Lehmann L., Eur. J. Org. Chem, 2012, 889–892. [Google Scholar]
- 63. Yanagi T., Somerville R. J., Nogi K., Martín R. and Yorimitsu H., ACS Catal, 2020, 10, 2117–2123. [Google Scholar]
- 64. Nogi K. and Yorimitsu H., Chem. Commun, 2017, 53, 4055–4065. [DOI] [PubMed] [Google Scholar]
- 65. Tian Z.‐Y. and Zhang C.‐P., Chem. Commun, 2019, 55, 11936–11939. [DOI] [PubMed] [Google Scholar]
- 66.a) Van Bergen T. J., Hedstrand D. M., Kruizinga W. H. and Kellogg R. M., J. Org. Chem, 1979, 44, 4953–4962; [Google Scholar]; b) Hedstrand D. M., Kruizinga W. H. and Kellogg R. M., Tetrahedron Lett, 1978, 19, 1255–1258. [Google Scholar]
- 67. Hari D. P., Schroll P. and König B., J. Am. Chem. Soc, 2012, 134, 2958–2961. [DOI] [PubMed] [Google Scholar]
- 68.a) Sang R., Korkis S. E., Su W., Ye F., Engl P. S., Berger F. and Ritter T., Angew. Chem. Int. Ed, 2019, 58, 16161–16166; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem, 2019, 131, 16307–16312; [Google Scholar]; b) Ye F., Berger F., Jia H., Ford J., Worthman A., Börgel J., Genicot C. and Ritter T., Angew. Chem. Int. Ed, 2019, 58, 14615–14619; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem, 2019, 131, 14757–14761. [Google Scholar]
- 69. Huang C., Feng J., Ma R., Fang S., Lu T., Tang W., Du D. and Gao J., Org. Lett, 2019, 21, 9688–9692. [DOI] [PubMed] [Google Scholar]
- 70.a) Neuhaus J. D., Oost R., Merad J. and Maulide N., Top. Curr. Chem. 2018, 376, Article 15; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lu L.‐Q., Li T.‐R., Wang Q. and Xiao W.‐J., Chem. Soc. Rev, 2017, 46, 4135–4149; [DOI] [PubMed] [Google Scholar]; c) Burtoloso A. C. B., Dias R. M. P. and Leonarczyk I. A., Eur. J. Org. Chem, 2013, 5005–5016; [Google Scholar]; d) Zhang Y. and Wang J., Coord. Chem. Rev, 2010, 254, 941–953; [Google Scholar]; e) Li A.‐H., Dai L.‐X. and Aggarwal V. K., Chem. Rev, 1997, 97, 2341–2372. [DOI] [PubMed] [Google Scholar]
- 71.a) Aggarwal V. K., Thompson A. and Jones R. V. H., Tetrahedron Lett, 1994, 46, 8659–8660; [Google Scholar]; b) Kemp D. S. and Vellaccio F., J. Org. Chem, 1981, 46, 1807–1810; [Google Scholar]; c) Trost B. M. and Bogdanowicz M. J., J. Am. Chem. Soc, 1973, 95, 5298–5307. [Google Scholar]
- 72. Andersen K. K., Caret R. L. and Ladd D. L., J. Org. Chem. 1976, 41, 3096‐3100. [Google Scholar]
- 73.a) Shibata N., Matsnev A. and Cahard D., Beilstein J. Org. Chem. 2010, 6, No 65; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Umemoto T., Chem. Rev, 1996, 96, 1757–1778; [DOI] [PubMed] [Google Scholar]; c) Umemoto T. and Ishihara S., J. Am. Chem. Soc, 1993, 115, 2156–2164; [Google Scholar]; d) Umemoto T. and Ishihara S., Tetrahedron Lett, 1990, 31, 3579–3582. [Google Scholar]
- 74. Zhang C., Org. Biomol. Chem, 2014, 12, 6580–6589. [DOI] [PubMed] [Google Scholar]
- 75. Hashmall J. A., Horak V., Khoo L. E., Quicksall C. O. and Sun M. K., J. Am. Chem. Soc, 1981, 103, 289–295. [Google Scholar]
- 76.a) Liu B. and Shine H. J., J. Phys. Org. Chem, 2001, 14, 81–89; [Google Scholar]; b) Umemura K., Matsuyama H. and Kamigata N., Bull. Chem. Soc. Jpn, 1990, 63, 2593–2600. [Google Scholar]
- 77. Wang X., Truesdale L. and Yu J.‐Q., J. Am. Chem. Soc, 2010, 132, 3648–3649. [DOI] [PubMed] [Google Scholar]
- 78. Simkó D. C., Elekes P., Pázmándi V. and Novák Z., Org. Lett, 2018, 20, 676–679. [DOI] [PubMed] [Google Scholar]
- 79. Otsuka S., Nogi K., Rovis T. and Yorimitsu H., Chem. Asian J, 2019, 14, 532–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Varga B., Gonda Z., Tóth B. L., Kotschy A. and Novák Z., Eur. J. Org. Chem. 2019, DOI:‐ 10.1002/ejoc.201900957. [DOI] [Google Scholar]