

Abstract
Background:
The introduction of biological disease-modifying anti-rheumatic drugs (bDMARDs) into clinical practice has dramatically improve the clinical outcomes of individuals with rheumatoid arthritis (RA). However, bDMARDs are associated with high costs, which has resulted in restricted treatment access and a burden on medical insurance finances. Although biosimilars offer cost-saving, their effectiveness and safety must be established in Post-Marketing Surveillance (PMS). Infliximab (IFX), a chimeric monoclonal antibody to TNF-alpha, is the first bDMARD; its biosimilar, CT-P13, is the first biosimilar DMARD approved for RA treatment in Japan. We will evaluate whether switching from originator IFX to CT-P13 is not inferior for maintaining non-clinical relapse to continued treatment with originator IFX in RA patients achieving clinical remission.
Methods/design:
This study is an interventional, multicenter, open-label, single-arm against historical control and noninferiority clinical trial with a 24-week follow-up. Eighty RA patients who are treated by originator IFX for ≥24 weeks and are achieving clinical remission will be included. Patients will be switched to CT-P13 with the unchanged dosing regimen. We will evaluate disease activity by measuring clinical disease activity indices and by using musculoskeletal ultrasound (MSUS). The primary endpoint is the ratio of patients who experience a nonclinical relapse during the study period. Important secondary endpoints are the changes from the baseline of the MSUS scores. We will also comprehensively analyze the serum levels of many biomarkers such as cytokines and chemokines.
Discussion:
The study results are expected to show the noninferiority of switching to CT-P13 over the continuation of originator IFX. The strength of this study is its prospective evaluation of therapeutic efficacy using not only clinical disease activity indices but also MSUS to accurately and objectively evaluate disease activity at the joint level among patients drawn from multiple centers with a standardized evaluation by MSUS. We will explore whether parameters at baseline can predict a nonclinical relapse after switching from originator IFX to CT-P13 by integrating multilateral assessments, i.e., clinical disease activity indices, MSUS findings, and serum biomarkers.
Trial registration:
This study was registered in the Japan Registry of Clinical Trials (https://jrct.niph.go.jp) on October 11, 2019 as jRCTs071190030.
Keywords: biomarker, biosimilar, CT-P13, infliximab, musculoskeletal ultrasound, originator, rheumatoid arthritis
1. Introduction
Rheumatoid arthritis (RA) is characterized by persistent synovitis, systemic inflammation, and autoantibodies.[1] Uncontrolled active RA causes joint damage, disability, decreased quality of life, and comorbidities. The tight control of the disease activity of RA following the treat-to-target (T2T) strategy is thus recommended.[2] Advances in the treatment of RA, such as the use of biological disease-modifying anti-rheumatic drugs (bDMARDs), have provided better clinical outcomes, including the achievement of clinical remission for patients with RA. Clinicians also aim to achieve not only clinical remission but also imaging remission and immunological remission.[3]
The pathophysiology of RA is associated with several inflammation cascades. One key inflammation cascade includes the overproduction and overexpression of tumor necrosis factor (TNF). This pathway drives both synovial inflammation and joint destruction.[1] Infliximab, a chimeric monoclonal antibody to TNF-alpha, was the first bDMARD to demonstrate a dramatic change in the treatment of RA. IFX is extremely effective in suppressing disease activity and the progression of joint destruction.[4–6] However, although bDMARDs are highly effective, they are costly.
CT-P13 is a biosimilar of originator IFX, developed by Celltrion (Incheon, South Korea). A “biosimilar” is a biotherapeutic product that is similar in terms of quality, safety, and efficacy to an already licensed reference biotherapeutic product (i.e., originator).[7] CT-P13 was approved in 2014 as the first biosimilar DMARD (bsDMARD) for RA treatment in Japan.[8] The introduction of bsDMARDs is expected to reduce the patients economic burden and improve medical insurance finances. The biosimilar CT-P13 and the originator IFX have been shown to be pharmacokinetically equivalent and comparable in efficacy and safety.[8] Switching from originator IFX to CT-P13 was reported to be not clinically inferior to continued treatment with originator IFX[9] or CT-P13.[10] However, in all of these previous studies, the endpoints of efficacy were based on clinical disease activity indices, and the evaluation of disease activity based on high-sensitivity imaging modalities such as joint musculoskeletal ultrasound (MSUS) was not performed.
MSUS is usually used to evaluate the disease activity of RA,[11,12] and MSUS experts have stated that RA patients treated with DMARDs should undergo assessments with MSUS since MSUS better shows the activity of synovial inflammation compared to a clinical examination,[11,12] indicating that the use of MSUS to assess the therapeutic response can be of great help in clinical practice.[11–14] MSUS is a noninvasive, objective, relatively inexpensive, and repeatable imaging modality that is suitable for treatment monitoring.[11,12]
As mentioned above, clinical remission can be achieved in a relatively large number of RA patients by introducing bDMARD therapy, but residual synovitis detected by MSUS is known to remain at a certain frequency even in patients who achieve clinical remission.[15,16] The residual synovitis is an important finding that predicts joint destruction and clinical relapse. It is therefore important to accurately evaluate disease activity at the joint level by using MSUS as well as clinical disease activity indices including subjective parameters. We will therefore also use MSUS assessments to determine whether switching from originator IFX to CT-P13 is not inferior to continued treatment with originator IFX in RA patients achieving clinical remission. A multicenter collaborative study that prospectively evaluates disease activity using MSUS standardized at a high level is rare, even worldwide.
We will evaluate the changes of disease activity after RA patients are switched from originator IFX to CT-P13 by using MSUS as well as clinical disease activity indices so that the patients disease activity will be more accurately assessed. We will also comprehensively analyze the serum level of many biomarkers such as cytokines and chemokines. We will explore whether the patients parameters at baseline can be used to predict a nonclinical relapse after switching from originator IFX to CT-P13 by integrating multilateral assessments including clinical disease activity indices, MSUS findings, and biomarkers.
We named this clinical trial Infliximab Similarity Investigation by Ultrasound Study I (IFX-SIRIUS STUDY I). Herein, we describe the final protocol (version 2.0; December 15, 2019) for the study. We are also planning a study (IFX-SIRIUS STUDY II) to evaluate the changes in disease activity by MSUS as well as clinical disease activity indices after the discontinuation of CT-P13 in patients who have not experienced a clinical relapse following this study.
2. Objectives
2.1. Primary objective
The principal objective of the study is to determine whether switching from originator IFX to CT-P13 is not inferior in maintaining nonclinical relapse to continued treatment with originator IFX in RA patients achieving clinical remission.
2.2. Secondary objectives
We will assess disease activity by MSUS after patients are switched from originator IFX to CT-P13. We will explore whether the clinical and/or MSUS findings and/or biomarkers at baseline can be used to predict a nonclinical relapse after switching from originator IFX to CT-P13.
3. Methods/design
3.1. Study design
The study design is in accordance with the Standard Protocol Items: Recommendations for Interventional Trials and Consolidated Standards of Reporting Trials 2010 guidelines.[17,18] (Additional File 1). The study is a prospective, open-label, interventional and noninferiority clinical trial. This is a single-arm against historical control using a previous report[19] and a multicenter RA ultrasound prospective cohort we are investigating.[20] The study will be conducted at the following 17 centers: Nagasaki University Hospital, Asahi General Hospital, National Hospital Organization Chiba-East Hospital, Eiraku Clinic, Hamanomachi Hospital, Japanese Red Cross Nagasaki Genbaku Hospital, Kagawa University Hospital, University of Miyazaki Hospital, Nagasaki Kita Hospital, Osaka City University Hospital, Osaka Medical College Hospital, Sagawa Akira Rheumatology Clinic, Sasebo Chuo Hospital, Sasebo City General Hospital, Shiminnomori Hospital, Utazu Hospital, and Yoshitama Clinic for Rheumatic Diseases. In total, 80 patients with RA will be assigned to switch from originator IFX to CT-P13. The duration of the intervention is 24 weeks. The study design is summarized in Figure 1.
Figure 1.
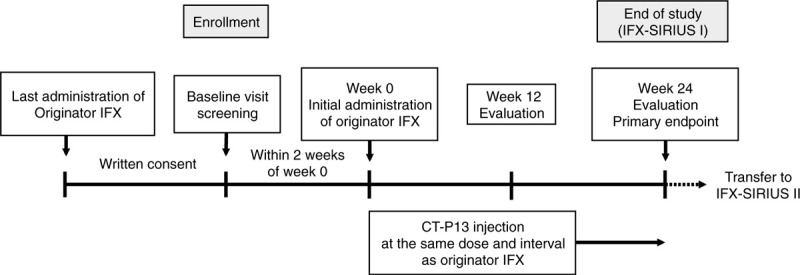
Study design. IFX = infliximab.
3.2. Approvals
The study was approved by the certified review board (CRB) of Nagasaki University (CRB approval no.: CRB19-010). The study is registered in the Japan Registry of Clinical Trials (https://jrct.niph.go.jp) as jRCTs071190030. We will conduct the study in accordance with the principles of the Declaration of Helsinki and the Clinical Trials Act (Act No. 16 of April 14, 2017), the Act on the Protection of Personal Information and related regulatory notifications, and this clinical study protocol. Participants will be provided with an explanation regarding the study by their treating rheumatologist and will be asked to voluntarily sign an informed consent form before their participation.
3.3. Participants
3.3.1. Inclusion criteria
Patients must meet all of the following requirements to be considered for entry into the study:
-
1.
≥20 years old,
-
2.
with the diagnosis of RA based on the American College of Rheumatology (ACR) /European League Against Rheumatism (EULAR) 2010 RA Classification Criteria,[21]
-
3.
treated with originator infliximab for ≥24 weeks and achieving clinical remission defined as a Disease Activity Score 28 (DAS28)-erythrocyte sedimentation rate (ESR) <2.6 at the last administration of originator IFX and at baseline,
-
4.
able and willing to give written informed consent and comply with the requirements of the study protocol.
3.3.2. Exclusion criteria
The exclusion criteria are as follows:
-
1.
the concurrent use of a corticosteroid equivalent to >10 mg/day of prednisolone,
-
2.
a previous use of a biosimilar for IFX,
-
3.
treated with a biologic DMARD other than originator IFX or a bsDMARD,
-
4.
a history of infusion reaction due to originator IFX that required medication,
-
5.
treated with a corticosteroid or antirheumatic drug and changed the dose within 8 weeks prior to the baseline visit,
-
6.
the use of a prohibited drug or therapy within 8 weeks prior to the baseline visit,
-
7.
current pregnancy, breastfeeding, or noncompliant with a medically approved contraceptive regimen during and 6 months after the study period, or
-
8.
being considered unsuitable for this study by the investigator.
3.3.3. Sample size
The sample size with a statistical power of 0.8, in a test for noninferiority with a significance level of 0.05, was estimated based on the results from the test for noninferiority on 2000 simulation datasets. Each simulation dataset was generated by random sampling from a binomial distribution with a probability of 0.16. The reason for the probability (0.16) and the procedures in the test for noninferiority are detailed below in Section 3.8.
3.3.4. Intervention
Patients will receive intravenous CT-P13 with an unchanged dosing regimen throughout the study period. CT-P13 will be administered at the same dose (dose per kg) and the same interval as the originator IFX before switching.
All of the patients must continue to take the same doses of methotrexate (MTX) and oral corticosteroid that they were taking before the switch, throughout the study period. During the study period, the following treatments are prohibited: the administration of a bDMARD or JAK inhibitor, the concomitant use of an immunosuppressant (azathioprine, cyclophosphamide, cyclosporine) or oral corticosteroids equivalent to >10 mg/day of prednisolone, intra-articular corticosteroid injections at joints, and nonsteroidal anti-inflammatory drug (NSAID) suppositories. During the study period, the dosage of any NSAID can be modified within the range of its approved doses in Japan.
3.3.5. Patient discontinuation criteria
A patient may be discontinued prematurely for the following reasons (patients who discontinue before completing the trial will not be replaced):
Patient experiences a clinical relapse.
CT-P13 must be reduced or discontinued for some reason(s).
Continuing participation is inadvisable due to adverse event(s).
The patient asks to leave the trial.
In the Principal Investigator's opinion, continuation in the trial would be detrimental to the patient's well-being.
3.4. Outcome measurements
Study visits will take place at baseline and after 12 and 24 weeks of treatment. The assessments are presented in Table 1. Clinical assessors will be blinded to joint assessments by MSUS.
Table 1.
Treatment schedule and outcome measures.
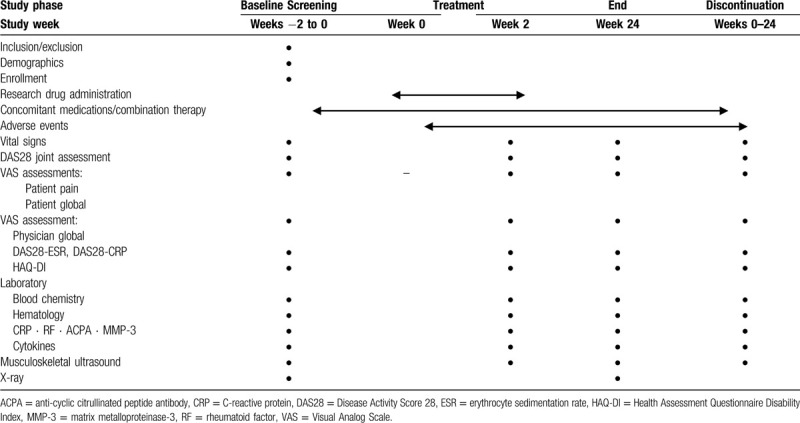
3.4.1. Clinical disease activity
Clinical disease activity was evaluated by each of the attending physicians (Japan College of Rheumatology [JCR]-certified rheumatologists) based on the values of the DAS28-ESR and the DAS28-C reactive protein (CRP) level.[22] At each visit, 28 joints including the bilateral glenohumeral, elbow, wrist, metacarpophalangeal (MCP), interphalangeal (IP), proximal interphalangeal (PIP) of the hand, and knee joints were assessed for tenderness and swelling. Each patients global assessment (PtGA) and evaluators global assessment (EGA) will be established on a 0–100 mm visual analog scale. The patients’ functional assessment will be evaluated by the Health Assessment Questionnaire-Disability Index (HAQ-DI).[23]
3.4.2. MSUS assessments
Participants will undergo imaging by MSUS at baseline, week 12 and week 24. The MSUS examination of each patient will be performed by 1 of the JCR-certified sonographers. A systematic multiplanar grayscale (GS) and power Doppler (PD) examination of each patient's joints will be performed using a multifrequency linear transducer (12–24 MHz). PD will be used depending on which Doppler modality is the most sensitive on the individual machines. The Doppler settings will be adjusted at each hospital according to published recommendations.[24] There will be no change in the MSUS settings during the study and no upgrading of software.
Articular synovitis will be assessed by MSUS at dorsal views of 22 joints: bilateral wrist joints, 1st–5th MCP joints, the IP joints, and the 2nd–5th PIP joints. Each joint is scored for GS as well as PD on a scale from 0 to 3 in a semiquantitative manner. The sum of the GS scores or the PD scores is considered the total GS score or total PD score, respectively. We will also assess the Outcome Measures in Rheumatology (OMERACT)-EULAR combined PDUS score (i.e., the combined PD score) and the Global OMERACT-EULAR Synovitis Score (GLOESS).[25,26] The combined PD score is combined with synovial hypertrophy shown by GS and PD.[25,26]
3.4.3. X-ray imaging
X-ray images of bilateral hands (posteroanterior view) and feet (anteroposterior view) will be conducted. Joint damage progression will be evaluated based on the van der Heijde-modified total Sharp score (vdH-mTSS) method including 16 areas in each hand for erosions and 15 for joint-space narrowing.[27]
3.4.4. Biomarker measurements
The patients serum concentrations of the following biomarkers will be measured. Rheumatoid factor (RF) will be measured by a latex agglutination turbidimetric immunoassay (LATIA) (LZ test “Eiken” RF). Anti-cyclic citrullinated peptide antibodies (ACPA) will be measured by a chemiluminescent immunoassay (CLEIA) (STACIA MEBLux test CCP). Matrix metalloproteinase-3 (MMP-3) will be measured by a latex turbidimetric immunoassay (LTIA) (Panaclear MMP-3 ’Latex’). Multiplex cytokine/chemokine bead assays will be performed using diluted serum supernatants and a MILLIPLEX MAP Human Cytokine/Chemokine Magnetic Bead Panel (Merck Millipore) – Bio-Plex Pro Human Cytokine Assays (Bio-Rad) analyzed with a Bio-Plex MAGPIX Multiplex Reader (Bio-Rad) according to the manufacturer's instructions.
The cytokines/chemokines that are measured by the bead panel include interleukin (IL)-1α, IL-1β, IL-1 receptor antagonist, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL-12 (p40), IL-12 (p70), IL-13, IL-15, IL-17, IL-18, interferon-gamma (IFN-γ), IFN-α2, CXCL1 (growth-related oncogene [GRO]), granulocyte-macrophage colony-stimulating factor (GM-CSF), granulocyte colony-stimulating factor (G-CSF), CX3CL1 (fractalkine), flt-3 ligand, fibroblast growth factor (FGF)-2, eotaxin, epidermal growth factor (EGF), soluble CD40 ligand (sCD40L), vascular endothelial growth factor (VEGF), TNF-β, TNF-α, transforming growth factor (TGF)-α, CCL4 (macrophage inflammatory protein [MIP]-1β), CCL3 (MIP-1α), CCL22 (macrophage-derived chemokine [MDC]), CCL7 (monocyte chemotactic protein [MCP]-3), CCL2 (MCP-1), CXCL10 (IFN-γ-inducible protein [IP]-10), vascular cell adhesion molecule-1 (VCAM-1), and intercellular adhesion molecule-1 (ICAM-1). The serum levels of IL-6 and TNFα will be measured with specific enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems).
3.5. Study endpoints
3.5.1. Primary endpoint
The primary endpoint is the proportion of patients who experience a clinical relapse during the period from baseline to week 24. Clinical relapse is defined as
-
1.
a change from the baseline value in the DAS28-ESR (ΔDAS28-ESR) ≥1.2 or in the DAS28-ESR ≥3.2, and
-
2.
an increase in the DAS28-ESR value due to elevated disease activity of RA.
3.5.2. Secondary endpoints
The secondary endpoints of this study are the following:
-
1.
the changes of the total PD and GS scores and the combined PD score from baseline to weeks 12 and 24, and
-
2.
the changes in the DAS28-ESR and DAS28-CRP values from baseline to weeks 12 and 24.
3.5.3. Exploratory endpoints
For further research we will assess the following:
-
1.
the change in vdH-mTSS from baseline to week 24
-
2.
the change in the HAQ-DI data from baseline to weeks 12 and 24
-
3.
the changes in the serum levels of biomarkers from baseline to weeks 12 and 24
-
4.
the DAS28-ESR and DAS28-CRP values at baseline and weeks 12 and 24
-
5.
the total PD and GS scores and the combined PD score at baseline, week 12, and week 24
-
6.
the vdH-mTSS at baseline and week 24
-
7.
the HAQ-DI at baseline, week 12, and week 24
-
8.
the serum levels of biomarkers at baseline, week 12, and week 24
3.6. Adverse events
All adverse events (AEs) that occur between the administration of CT-P13 and the end of week 24 will be recorded. If necessary, the investigators will administer treatments. A serious AE (SAE) is defined as any adverse reaction resulting in any of the following outcomes: a life-threatening condition or death; a condition that requires inpatient hospitalization or the prolongation of existing hospitalization; threatening to cause disability or disability, a congenital anomaly, or a birth defect. All SAEs will be documented in the medical records and reported to the CRB by the responsible investigator in accordance with Japanese regulations.
3.7. Data collection and management
Appropriate and authorized persons (investigators, clinical trial physicians, and clinical trial collaborators) will prepare a case report form (CRF) for each patient. All data recorded in the CRF must be consistent with the original material unless the data recorded directly in the CRF are used as the source material. According to the schedule presented in Table 1, the investigator will collect data at each patient visit during the study. The investigators will be provided access to an online, web-based, electronic data-capture system. Only the investigator will be able to enter and modify data in the electronic CRF (e-CRF). All study findings and documents will be regarded as confidential. Patients will be identified on the e-CRF by their patient number and/or birth date, not by name. The confidentiality of the documents that identify the patient must be maintained by the investigator so that the anonymity of the participants is ensured. During the study, a sponsor-investigator will perform regular site visits to review the protocol compliance, conduct source data verification, assess drug accountability and management, and ensure that the study is being conducted according to pertinent regulatory and protocol requirements.
3.8. Statistical analysis method
This study was designed to test the noninferiority of CT-P13 to the originator IFX. The non-inferiority margin was decided based on the results of 2 clinical studies[19,20]; the members of the present studys research group agreed with the validity of the studies design to assume the ’historical evidence of sensitivity to drug effects (HESDE) detailed in the U.S. Food and Drug Administration (FDA) guidance[28] among these 2 studies and IFX-SIRIUS STUDY I (i.e., the present study) and to use them as a historical control for this study. Using the results of these 2 prior studies, we examined the proportions of patients with a clinical relapse during the observation period of 24 weeks among those who had achieved clinical remissions, at the time of enrollment, with TNF inhibitors. The difference between these 2 studies is in the treatment administered during the observation period. For the patients in the 1 study,[20] the treatment with TNF inhibitors was continued during the observation period; in the other study,[19] these medications were stopped during the observation period. The inferiority margin, 11.2%, was decided as the lower boundary of the 95% confidence interval of the difference between these 2 proportions. Regarding the expected proportion of patients with a clinical relapse in the IFX-SIRIUS STUDY I, we assume that it will be approximately the same as the results of study,[20] which was16.0%.
The null hypothesis for noninferiority is rejected when the upper boundary of the confidence interval of the difference in the proportion of patients with experience of a clinical relapse during the study period is not larger than the value of 27.2% (i.e., the summation of the noninferiority margin, 11.2%, and the expectation of the proportion of patients with experience of a clinical relapse during the study period, 16.0%). All of the confidence intervals of proportions will be obtained using the Wilson score interval.
The analysis from which the main results are obtained is that on the Per Protocol Set—which consists of the patients evaluated for the primary endpoint in (at least) 2 scheduled visits; 1 at baseline and the other at the visit at week 24 or at which a clinical relapse is observed—in order to maintain the validity with the assumption of the HESDE[28] with patients in the 2 historical control studies.
Other statistical analyses are planned to be exploratively conducted on the relationships between results obtained as the exploratory outcomes, which are detailed above in Section 3.5.3.
4. Discussion
The introduction of biologics into clinical practice has dramatically improved the management of a number of immune-mediated inflammatory diseases, including RA.[29] However, the currently available biologics are expensive, which has led to restricted treatment access for patients with RA.[30,31] Biosimilars offer cost-savings and health gains for patients and will play an important role in treating rheumatic diseases.[9,31,32] A drug's price is one of the key factors for drug selection, and the cost-saving for CT-P13 was 48%(in terms of average NHI price as of February, 2020) compared with originator IFX in Japan. Since the introduction of biosimilars could reduce financial burdens on healthcare budgets, the Japanese government is promoting the use of biosimilars. However, some physicians are cautious about the application of biosimilars in clinical settings because they have questions about the efficacy and safety of biosimilars. The use of biosimilars will be promoted if patients on stable treatment with an originator biologic can safely be switched to the biosimilar.
The efficacy and safety equivalence of CT-P13 to originator IFX in patients with RA has been demonstrated.[8,33,34] It was also reported that switching from originator IFX to CT-P13 is not inferior to continued treatment with originator IFX.[9,10] However, it is difficult to produce generic drugs that have the same molecular structure as the originator biologic because biologics are high-molecular-weight compounds, and thus biosimilars are fundamentally different from generic drugs. It will therefore be most important to establish pharmacovigilance databases across countries that are adequate to monitor the biosimilars efficacy and safety after marketing approval.
The principal objective of the study is to evaluate whether switching from originator infliximab to CT-P13 is not inferior in maintaining a nonclinical relapse to continued treatment with originator IFX in RA patients achieving clinical remission. The strength of this study is its prospective evaluation of therapeutic efficacy using not only clinical disease activity indices but also MSUS to accurately and objectively evaluate disease activity at the joint level in a patient series drawn from multiple centers with a standardized evaluation by MSUS. We will also comprehensively analyze the serum levels of many biomarkers such as cytokines and chemokines. We will explore whether parameters at baseline can be used to predict non-relapse after switching from originator IFX to CT-P13 by integrating multilateral assessments (i.e., the clinical disease activity indices, MSUS findings, and biomarkers). We are also planning to evaluate the non-relapse status after the discontinuation of CT-P13 in patients who have not experienced a clinical relapse in the IFX-SIRIUS STUDY II following the IFX-SIRIUS STUDY I. It would be cost-effective if we could predict from the results of these 2 consecutive studies that patients on stable treatment with originator IFX could switch to CT-P13 without a relapse and could then discontinue CT-P13 without a relapse before switching.
5. Trial status
The IFX-SIRIUS I study received ethical approval on September 17, 2019. Recruitment started in December 2019 and is expected to finish June 30, 2022.
Acknowledgments
We thank our colleagues and staff at the Rheumatology Department of Nagasaki University Hospital for their support.
Author contributions
All authors have given their final approval of the manuscript to be published as presented.
Conceptualization: S. Kawashiri, T. Shimizu, H. Yamamoto, A. Kawakami
Formal analysis: S. Kawashiri, T. Shimizu, S. Sato, S. Morimoto
Investigation: S. Kawashiri, T. Shimizu, R. Sumiyoshi, A. Kawakami
Methodology: S. Kawashiri, T. Shimizu, S. Sato, S. Morimoto, N. Hosogaya, C. Fukushima, H. Yamamoto, A. Kawakami
Project administration: S. Kawashiri, T. Shimizu, Y. Kawazoe
Writing – original draft: S. Kawashiri, T. Shimizu
Writing – review & editing: S. Kawashiri, T. Shimizu, S. Sato, S. Morimoto, Y. Kawazoe, A. Kawakami.
Footnotes
Abbreviations: ACPA = anti-cyclic citrullinated peptide antibodies, ACR = American College of Rheumatology, bDMARD = biological disease-modifying anti-rheumatic drug, CRB = certified review board, CRF = case report form, CRP = C-reactive protein, DAS28 = Disease Activity Score-28, DMARD = disease-modifying antirheumatic drug, e-CRF = electronic case report form, EGA = evaluator's global assessment, ESR = erythrocyte sedimentation rate, EULAR = European League Against Rheumatism, GS = gray scale, HAQ-DI = Health Assessment Questionnaire-Disability Index, HESDE = ’historical evidence of sensitivity to drug effects, IP = interphalangeal, JCR = Japan College of Rheumatology, MCP = metacarpophalangeal, MMP-3 = matrix metalloproteinase-3, MSUS = musculoskeletal ultrasound, MTX = methotrexate, OMERACT = Outcome Measures in Rheumatology, PD = power Doppler, PIP = proximal interphalangeal, PtGA = patient's global assessment, RA = rheumatoid arthritis, TNF = tumor necrosis factor, vdH-mTSS = van der Heijde-modified total Sharp score.
How to cite this article: Kawashiri Sy, Shimizu T, Sato S, Morimoto S, Kawazoe Y, Sumiyoshi R, Hosogaya N, Fukushima C, Yamamoto H, Kawakami A. Switching from originator infliximab to biosimilar infliximab in Japanese patients with rheumatoid arthritis achieving clinical remission (the IFX-SIRIUS study I): study protocol for an interventional, multicenter, open-label, single-arm and noninferiority clinical trial with clinical, ultrasound, and biomarker assessments. Medicine. 2020;99:30(e21151).
This study was funded by Celltrion Healthcare Co., Ltd. (Inchon, South Korea).
The study protocol has been approved by the certified review board of Nagasaki University. The reference number is CRB19-010. Written informed consent will be provided by patients before enrollment.
Results obtained in this trial will be published in an international journal and may be presented at international scientific meetings. This information will be included in the patient consent form.
AK received research funding from ONO Pharmaceutical Co., Sanofi K.K., Asahi Kasei Pharma Corporation, Taisho Toyama Pharmaceutical Co., Teijin Pharma Co., Sumitomo Dainippon Pharma Co., Ltd., Chugai Pharmaceutical Co., MSD Co., Kowa Pharmaceutical Co. Ltd., Taisho Toyama Pharmaceutical Co., Pfizer Japan Inc., Kyowa Hakko Kirin Co., Boehringer Ingelheim Japan, Abbvie GK, Astellas Pharma Inc., Eli Lilly Japan, Daiichi Sankyo Co., Takeda Pharmaceutical Co., Kissei Pharmaceutical Co., Mitsubishi Tanabe Pharma Co., Otsuka Pharmaceutical Co., Cosmic Corporation Co. Ltd., Nihon Medi-Physics Co., Ltd., Daiichi Sankyo Co., Bristol-Myers Squibb, Janssen Pharmaceutical K.K., Mochida Pharmaceutical Co., Ltd, AstraZeneca Co., YL Biologics Co., Alexion GK, and Eisai Co. The other authors declare that they have no competing interests.
The trial is sponsored by Nagasaki University Graduate School of Biomedical Sciences. The sponsor representative is Dr. A. Kawakami.
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
- [1].Scott DL, Wolfe F, Huizinga TW. Rheumatoid arthritis. Lancet 2010;376:1094–108. [DOI] [PubMed] [Google Scholar]
- [2].Smolen JS, Breedveld FC, Burmester GR, et al. Treating rheumatoid arthritis to target: 2014 update of the recommendations of an international task force. Ann Rheum Dis 2016;75:3–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Schett G, Emery P, Tanaka Y, et al. Tapering biologic and conventional DMARD therapy in rheumatoid arthritis: Current evidence and future directions. Ann Rheum Dis 2016;75:1428–37. [DOI] [PubMed] [Google Scholar]
- [4].St Clair EW, van der Heijde DM, Smolen JS, et al. Combination of infliximab and methotrexate therapy for early rheumatoid arthritis: A randomized, controlled trial. Arthritis Rheum 2004;50:3432–43. [DOI] [PubMed] [Google Scholar]
- [5].Lipsky PE, van der Heijde DM, St Clair EW, et al. Infliximab and methotrexate in the treatment of rheumatoid arthritis. Anti-tumor necrosis factor trial in rheumatoid arthritis with concomitant therapy study group. N Engl J Med 2000;343:1594–602. [DOI] [PubMed] [Google Scholar]
- [6].Yamanaka H, Tanaka Y, Sekiguchi N, et al. Retrospective clinical study on the notable efficacy and related factors of infliximab therapy in a rheumatoid arthritis management group in Japan (RECONFIRM). Mod Rheumatol 2007;17:28–32. [DOI] [PubMed] [Google Scholar]
- [7].World Health Organization, Expert Committee on Biological Standardization. Guidelines on evaluation of similar biotherapeutic products (SBPs). XXX 2009;http://www.who.int/biologicals/areas/biological_therapeutics/BIOTHERAPEUTICS_FOR_WEB_22APRIL2010.pdfhttp://www.who.int/biologicals/areas/biological_therapeutics/BIOTHERAPEUTICS_FOR_WEB_22APRIL2010.pdf (accessed 4 Apr 2016). [Google Scholar]
- [8].Takeuchi T, Yamanaka H, Tanaka Y, et al. Evaluation of the pharmacokinetic equivalence and 54-week efficacy and safety of CT-P13 and innovator infliximab in Japanese patients with rheumatoid arthritis. Mod Rheumatol 2015;25:817–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Jørgensen KK, Olsen IC, Goll GL, et al. NOR-SWITCH study group. Switching from originator infliximab to biosimilar CT-P13 compared with maintained treatment with originator infliximab (NOR-SWITCH): a 52-week, randomised, double-blind, non-inferiority trial. Lancet 2017;389:2304–16. [DOI] [PubMed] [Google Scholar]
- [10].Tanaka Y, Yamanaka H, Takeuchi T, et al. Safety and efficacy of CT-P13 in Japanese patients with rheumatoid arthritis in an extension phase or after switching from infliximab. Mod Rheumatol 2017;27:237–45. [DOI] [PubMed] [Google Scholar]
- [11].Colebatch AN, Edwards CJ, Ostergaard M, et al. EULAR recommendations for the use of imaging of the joints in the clinical management of rheumatoid arthritis. Ann Rheum Dis 2013;72:804–14. [DOI] [PubMed] [Google Scholar]
- [12].D’Agostino MA, Terslev L, Wakefield R, et al. Novel algorithms for the pragmatic use of ultrasound in the management of patients with rheumatoid arthritis: from diagnosis to remission. Ann Rheum Dis 2016;75:1902–8. [DOI] [PubMed] [Google Scholar]
- [13].Naredo E, Moller I, Cruz A, et al. Power Doppler ultrasonographic monitoring of response to anti-tumor necrosis factor therapy in patients with rheumatoid arthritis. Arthritis Rheum 2008;58:2248–56. [DOI] [PubMed] [Google Scholar]
- [14].Kawashiri SY, Nishino A, Shimizu T, et al. Ultrasound disease activity of bilateral wrist and finger joints at three months reflects the clinical response at six months of patients with rheumatoid arthritis treated with biologic disease-modifying anti-rheumatic drugs. Mod Rheumatol 2017;27:252–6. [DOI] [PubMed] [Google Scholar]
- [15].Nguyen H, Ruyssen-Witrand A, Gandjbakhch F, et al. Prevalence of ultrasound-detected residual synovitis and risk of relapse and structural progression in rheumatoid arthritis patients in clinical remission: a systematic review and meta-analysis. Rheumatology (Oxford) 2014;53:2110–8. [DOI] [PubMed] [Google Scholar]
- [16].Kawashiri SY, Suzuki T, Nakashima Y, et al. Ultrasonographic examination of rheumatoid arthritis patients who are free of physical synovitis: Power Doppler subclinical synovitis is associated with bone erosion. Rheumatology (Oxford) 2014;53:562–9. [DOI] [PubMed] [Google Scholar]
- [17].Chan AW, Tetzlaff JM, Altman DG, et al. SPIRIT 2013 statement: defining standard protocol items for clinical trials. Ann Intern Med 2013;158:200–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Schulz KF, Altman DG, Moher D, et al. CONSORT 2010 statement: updated guidelines for reporting parallel group randomised trials. BMJ 2010;340:c332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Brocq O, Millasseau E, Albert C, et al. Effect of discontinuing TNFalpha antagonist therapy in patients with remission of rheumatoid arthritis. Joint Bone Spine 2009;76:350–5. [DOI] [PubMed] [Google Scholar]
- [20].Nishino A, Kawashiri SY, Koga T, et al. Ultrasonographic efficacy of biologic and targeted synthetic disease-modifying antirheumatic drug therapy in rheumatoid arthritis from a multicenter rheumatoid arthritis ultrasound prospective cohort in Japan. Arthritis Care Res (Hoboken) 2018;70:1719–26. [DOI] [PubMed] [Google Scholar]
- [21].Aletaha D, Neogi T, Silman AJ, et al. 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League against rheumatism collaborative initiative. Arthritis Rheum 2010;62:2569–81. [DOI] [PubMed] [Google Scholar]
- [22].Prevoo ML, van’t Hof MA, Kuper HH, et al. Modified disease activity scores that include twenty-eight-joint counts. Development and validation in a prospective longitudinal study of patients with rheumatoid arthritis. Arthritis Rheum 1995;38:44–8. [DOI] [PubMed] [Google Scholar]
- [23].Fries JF, Spitz P, Kraines RG, et al. Measurement of patient outcome in arthritis. Arthritis Rheum 1980;23:137–45. [DOI] [PubMed] [Google Scholar]
- [24].Torp-Pedersen ST, Terslev L. Settings and artefacts relevant in colour/power Doppler ultrasound in rheumatology. Ann Rheum Dis 2008;67:143–9. [DOI] [PubMed] [Google Scholar]
- [25].D’Agostino MA, Boers M, Wakefield RJ, et al. Exploring a new ultrasound score as a clinical predictive tool in patients with rheumatoid arthritis starting abatacept: Results from the APPRAISE study. RMD Open 2016;2:e000237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Terslev L, Naredo E, Aegerter P, et al. Scoring ultrasound synovitis in rheumatoid arthritis: A EULAR-OMERACT ultrasound taskforce-Part 2: reliability and application to multiple joints of a standardised consensus-based scoring system. RMD Open 2017;3:e000427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].van der Heijde D. How to read radiographs according to the Sharp/van der Heijde method. J Rheumatol 2000;27:261–3. [PubMed] [Google Scholar]
- [28].U.S. Food and Drug Administration. Non-Inferiority Clinical Trials to Establish Effectiveness Guidance for Industry. 2016;https://www.fda.gov/media/78504/download [Google Scholar]
- [29].Kuek A, Hazleman BL, Ostör AJ. Immune-mediated inflammatory diseases (IMIDs) and biologic therapy: A medical revolution. Postgrad Med J 2007;83:251–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Modena V, Bianchi G, Roccatello D. Cost-effectiveness of biologic treatment for rheumatoid arthritis in clinical practice: An achievable target? Autoimmun Rev 2013;12:835–8. [DOI] [PubMed] [Google Scholar]
- [31].Yoo DH, Prodanovic N, Jaworski J, et al. Efficacy and safety of CT-P13 (biosimilar infliximab) in patients with rheumatoid arthritis: Comparison between switching from reference infliximab to CT-P13 and continuing CT-P13 in the PLANETRA extension study. Ann Rheum Dis 2017;76:355–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Dorner T, Strand V, Cornes P, et al. The changing landscape of biosimilars in rheumatology. Ann Rheum Dis 2016;75:974–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Yoo DH, Hrycaj P, Miranda P, et al. A randomised, double-blind, parallel-group study to demonstrate equivalence in efficacy and safety of CT-P13 compared with innovator infliximab when coadministered with methotrexate in patients with active rheumatoid arthritis: The PLANETRA study. Ann Rheum Dis 2013;72:1613–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Yoo DH, Racewicz A, Brzezicki J, et al. A phase III randomized study to evaluate the efficacy and safety of CT-P13 compared with reference infliximab in patients with active rheumatoid arthritis: 54-week results from the PLANETRA study. Arthritis Res Ther 2016;18:82. [DOI] [PMC free article] [PubMed] [Google Scholar]