

Abstract
miRNA-135a-5p upregulation has been identified in renal fibrosis in diabetic nephropathy (DN) with an incompletely known mechanism. Previous data showed that Sirtuin 1 (SIRT1) serves as a novel therapeutic target for DN and interact with the transforming growth factor-β/mothers against decapentaplegic homolog (TGF-β/Smad) signaling pathway. The aim of this study was to investigate the regulatory relationship between miR-135a-5p and SIRT1. The expression of miR-135a-5p and SIRT1 was detected using reverse transcription-quantitative PCR and western blotting. The renal fibrosis and Smad3 signaling pathway were assessed by western blotting, by analyzing protein expression of collegen1A1, α-smooth muscle actin (α-SMA), fibronectin (FN), epithelial-cadherin, Smad3 and phosphorylated Smad3 (p-Smad3). The target binding between miR-135a-5p and SIRT1 was predicted on TargetScan Human software, and confirmed by dual-luciferase reporter assay and RNA immu-noprecipitation. The results demonstrated miR-135a-5p is upregulated and SIRT1 was downregulated in the serum and renal tissue of DN patients, and TGFβ1-induced DN cell models in human HK-2 and HMCs. Knockdown of miR-135a-5p and overexpression of SIRT1 could inhibit TGFβ1-induced renal fibrosis in vitro. Moreover, SIRT1 was a downstream target for miR-135a-5p. Silencing of SIRT1 could abolish the suppressive role of miR-135a-5p knockdown in TGFβ1-induced HK-2 and HMCs. The TGFβ1 induced p-Smad3 expression in HK-2 and HMCs, which could be attenuated by miR-135a-5p knockdown via SIRT1. In conclusion, knockdown of miR-135a-5p inhibits TGFβ1-induced renal fibrosis by targeting SIRT1 and inactivating Smad3 signaling, providing a novel insight into miR-135a-5p as a potential therapeutic approach for DN.
Keywords: miR-135a-5p, sirtuin 1, mothers against decapentaplegic homolog 3, renal fibrosis, diabetic nephropathy
Introduction
Diabetic nephropathy (DN) is a hard microvascular complication of diabetes (1). Histologically, mesangial cell proliferation and podocyte damage are the major pathological features in the early stage of DN (2). DN is structurally characterized by the thickening of glomerular and tubular basement membranes, which have been attributed to extracellular matrix (ECM) synthesis (3-5). Deposition of ECM proteins such as collagens and fibronectin in the tubulointerstitium and glomerular mesangium contribute to tubulointerstitial fibrosis and glomerulosclerosis, eventually resulting in renal fibrosis (6,7). Pathologically, the progression of renal fibrosis is one of the hallmarks of DN and further predicts the deterioration of renal function (8,9). Considering that tubulointerstitial fibrosis is the key underlying pathology, understanding the regulatory mechanism underlying fibrogenesis in the interstitium is key to developing therapeutic targets for DN (10). However, to date, the option to target renal fibrosis is still not available in the clinic. Thus, more molecular pathways should be identified for new therapeutic strategies for DN.
Transforming growth factor-β (TGFβ) is a vital cytokine that promotes sclerosis and has been well documented in fibrosis development in DN (11). Highly expressed TGFβ occurs universally in chronic kidney diseases in both animal models and humans. For example, animal models of spontaneous diabetes have demonstrated increased TGFβ1 mRNA expression at the onset of hyperglycemia (12,13); in addition, TGFβ receptors have been described to be upregulated in renal disease models including DN. A previous study revealed that TGFβ1 was increased in several types of cells in the diabetic kidney, including mesangial cells, thus contributing to fibrotic events and cell survival (10). Therefore, TGFβ has been evaluated as a major target for DN treatment. In addition, evidence indicates a primary role for TGFβ and its downstream signaling cascades in the progression of renal diseases (14). TGFβ regulates numerous cell behaviors including cell proliferation, differentiation, adhesion and apoptosis (15,16). In addition, several intracellular signaling cascades have been identified for TGFβ-induced renal fibrosis, such as the mothers against decapentaplegic homologs (Smads), mitogen activated protein kinase (MAPK) and Jagged/Notch signaling pathway (10). Thus, it is important to further understand the molecular mechanisms of TGFβ-induced fibrotic events, which may lead to more effective approaches for DN treatment.
MicroRNAs (miRNAs/miRs) are highly conserved small non-coding RNAs (~22 nucleotides) with no protein coding capacity. Generally, miRNAs act as a mediator of functional gene expression by interacting with the 3′ untranslated region (3′UTR) of the target gene. It has been proposed that miRNAs modulate the effects of TGFβ1 in renal fibrosis, such as miRNA (miR)-433 (17). Cumulative evidence has demonstrated the close association between dysregulation of miRNAs and the progression of diabetes and diabetic complications, including DN (18,19). A number of miRNAs have been identified as early biomarkers in various chronic kidney diseases due to their consistency and reproducibility in human peripheral blood (20). In addition, integrated serum miRNAs expression profiling may be used in DN for identification of novel miRNAs (21). Among them, miR-135a-5p was significantly upregulated in the serum and renal tissues from patients with DN compared with healthy controls (21). However, the role and mechanism of miR-135a-5p in DN hasn't been fully illuminated.
Sirtuin 1 (SIRT1) belongs to a highly conserved family of NAD+-dependent deacetylase and serves as a therapeutic target for DN (22). The present study aimed to investigate the expression of miR-135a-5p in peripheral blood of patients with DN, as well as the role and mechanism of miR-135a-5p in TGFβ1-induced cell model of renal fibrosis.
Materials and methods
Clinical specimens
Patients were diagnosed with DN confirmed by renal biopsy, with nodular sclerosis (Kinnelstiel-Wilson lesion) in the glomerulus between January 2015 and December 2017. Peripheral venous blood samples were collected from 30 patients with DN (age, 43-73 years; male, 46.7%) and 30 patients with diabetes without DN (age, 38-68 years; male, 50.0%) after informed consent was provided by each individual. The clinical characteristics of the participants are presented in Table I. The blood samples were snap-frozen in liquid nitrogen (-79°C). All protocols involving human subjects were approved by the Ethics Committee of the Zhongnan Hospital of Wuhan University. The renal tissue sections were obtained from 10 of 30 renal biopsy specimens, and the control tissues were adjacent normal renal tissue sections from patients with renal carcinoma with normal kidney function (data not shown).
Table I.
Clinical characteristics of patients with DN or healthy controls (Control).
| Clinical features | DN (n=30) | Control (n=30) |
|---|---|---|
| Age, mean years | 60.23 | 62.45 |
| Sex, male/female | 16/14 | 15/15 |
| UAER, µg/min | 248.75 | 14.35 |
| Scr, µmol/l | 120.35 | 63.27 |
| BUN, mmol/l | 12.87 | 5.35 |
UAER, urine albumin excretion rate; BUN, blood urea nitrogen; Scr, serum creatinine; DN, diabetic nephropathy.
Cells and cell culture
Human proximal tubule cell lines (HK-2, cat. no. CRL-2190) were purchased from the American Type Culture Collection, and a human renal mesangial cell line (HMC, cat. no. 4200) was obtained from ScienCell Research Laboratories, Inc. HK-2 and HMCs were cultured and passaged in growth culture medium containing 90% Dulbecco's modified Eagle medium (DMEM; Gibco; Thermo Fisher Scientific, Inc.), 10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.) and 1% antibiotics (100 U/ml penicillin and 100 µg/ml streptomycin; Invitrogen; Thermo Fisher Scientific, Inc.) at 37°C in a humidified environment containing 5% CO2 for indicated times.
TGFβ1 treatment and high glucose treatments
HK-2 and HMCs at 60% confluency were seeded into 6-well plates (Corning, Inc.) overnight. For TGFβ1 treatment, the cells were made quiescent by incubation with serum-free medium for 16 h when grown to 80-90% confluence. A recombinant human TGFβ1 (cat. no. P01137; R&D Systems, Inc.) at a concentration of 10 ng/ml was added to the cell growth culture for 24 h to detect changes in the expression of miR-135a-5p and SIRT1, fibrosis response and TGFβ1/Smad3 signaling. For high glucose treatment, different concentrations of D-glucose (5-30 mmol/l) were added into DMEM, and HMC and HK-2 cells were incubated for 48 h before detection of miR-135a-5p expression.
Cell transfection
HK-2 and HMCs at 60% confluency were seeded into 6-well plates (Corning, Inc.) and incubated overnight. miR-135a-5p mimic (5′-UAU GGC UUU UUA UUC CUA UGU GA-3′), miR-135a-5p inhibitor (anti-miR-135a-5p; 5′-UCA CAU AGG AAU AAA AAG CCA UA-3′), specific small interfering (si)RNA against SIRT1 (siSIRT1; sense, 5′-GAU GAA GUU GAC CUC CUC ATT-3′ and antisense, 5′-UGA GGA GGU CAA CUU CAU CTT-3′) and the corresponding negative controls miR-NC mimic (5′-GUC CAG UGA AUU CCC AG-3′), anti-NC (5′-TCA CAA CCT CCT AGA AAG AGT AGA-3′) and siNC (sense, 5′-TTC TCC GAA CGT GTC ACG T-3′ and anti-sense, 5′-ACG UGA CAC GUU CGG AGA A-3′) were acquired from Shanghai GenePharma Co., Ltd. The overexpression of SIRT1 was performed using a pcDNA3.1 plasmid (Invitrogen; Thermo Fisher Scientific, Inc.). All transfections of oligo-nucleotides (30 nM) or vectors (2 µg) were performed using Lipofectamine® 2000 (Invitrogen; Thermo Fisher Scientific, Inc.), and the transfected cells were collected for further analysis after transfection for 36 h.
Reverse transcription-quantitative PCR (RT-qPCR)
For examination of the expression of SIRT1 mRNA and miR-135a-5p, total RNA was extracted from blood and treated cells using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.). The first-strand cDNA was synthesized at 42°C for 15 min using Fastking RT kit (Tiangen Biotech Co., Ltd.) and miRNA First-Strand cDNA Synthesis kit (Tiangen Biotech Co., Ltd.). qPCR was performed with SuperReal PreMix Plus (SYBR Green; Tiangen Biotech Co., Ltd.) on the ABI PRISM 7500 Fast Real-time PCR System (Applied Biosystems; Thermo Fisher Scientific, Inc.). The thermocycling conditions were 40 cycles of 95°C for 15 sec, 60°C for 60 sec and 95°C for 15 sec. Relative gene expression was normalized to GAPDH (for mRNAs) and U6 small nuclear RNA (U6; for miR-135a-5p). The reactions were performed in at least three independent runs using the following primers: SIRT1 isoform 2 forward, 5′-TGT GTC ATA GGT TAG GTG GTG A-3′ and reverse, 5′-AGC CAA TTC TTT TTG TGT TCG TG-3′; GAPDH forward, 5′-CTG GGC TAC ACT GAG CAC C-3′ and reverse, 5′-AAG TGG TCG TTG AGG GCA ATG-3′; miR-135a-5p forward, 5′-TTG GTC TTG TTT CCC GGT CC-3′ and reverse, 5′-TCA CAG CTC CAC AGG CT A AC-3′; U6 forward, 5′-CTC GCT TCG GCA GCA CA-3′ and reverse, 5′-AAC GCT TCA CGA ATT TGC GT-3′. Expression levels were normalized to the respective internal controls and calculated using the 2−ΔΔCq method (23).
Western blotting
To examine the expression of collagen I, α-smooth muscle actin (α-SMA), fibronectin (FN) and epithelial (E)-cadherin, total protein was extracted from blood and treated cells using RIPA lysis buffer (Beyotime Institute of Biotechnology) supplemented with the protease inhibitor phenylmethanesulfonyl fluoride (MedChemExpress). A total of 20 µg of protein determined by Bradford protein assay (Bio-Rad Laboratories, Inc.) was loaded for the standard procedures of western blotting. The proteins were transferred to PVDF membranes and incubated in a blocking buffer (3% BSA; R&D Systems, Inc.) for 1 h at 25°C and with primary antibodies overnight at 4°C. The primary antibodies against SIRT1 (cat. no. 8469; 1:1,000), collagen1A1 (cat. no. 84336; 1:1,000), α-SMA (cat. no. 14968; 1:1,000), E-cadherin (cat. no. 14472; 1:1,000), Smad3 (cat. no. 9513; 1:1,000), phosphorylated (p)-Smad3 (cat. no. 9520; 1:1,000) and GAPDH (cat. no. 97166; 1:1,000) were purchased from Cell Signaling Technology, Inc. The antibody against FN (cat. no. 8422; 1:200) was provided by Santa Cruz Biotechnology, Inc. After that, the membranes were washed with Tris-buffered saline containing 0.1% Tween-20, and then incubated with horseradish peroxidase-conjugated secondary mouse (cat. no. 7076; 1:2,500) or rabbit (cat. no. 7074; 1:2,500) antibody from Cell Signaling Technology, Inc. at 25°C for 1.5 h. Protein bands were finally detected by using an enhanced chemiluminescent substrate (Pierce; Thermo Fisher Scientific, Inc.). The relative protein expression was normalized to GAPDH according to the gray intensity determined on Image-Pro Plus 6.0 software (Media Cybernetics, Inc.).
Luciferase reporter assay and RNA immunoprecipitation (RIP)
According to bioinformatics algorithms, human SIRT1 3′ untranslated region (UTR) contained a potential target site of hsa-miR-135a-5p. Then, the wild type of SIRT1 3′ UTR fragment (SIRT1-wt) containing AAA AAG CCA U was cloned by PCR methods into pGL4 vector (Promega Corporation), as well as the mutated SIRT1 3′ UTR sequence (SIRT1-mut) containing UUU UUC GGU A. HK-2 and HMCs were transfected according to the following groups: SIRT1-wt + miR-NC mimic, SIRT1-wt + miR-135a-5p mimic, SIRT1-wut + miR-NC mimic, SIRT1-wut + miR-135a-5p mimic. After incubation for 24 h, cells were collected to measure Firefly and Renilla luciferase activities using the Dual-Luciferase Reporter assay system (Promega Corporation).
Magna RIP™ RNA-binding protein immunoprecipitation kit (EMD Millipore) was used according to the manufacturer's instructions. Briefly, HK-2 and HMC cells transfected with miR-135a-5p mimic or miR-NC mimic were extracted in RIP lysis buffer. Then, the cell extract was incubated with protein A/G magnetic beads pre-coated with argonaute 2 or IgG antibody and diluted with Salt solution II. The immunoprecipitated contents were treated with Proteinase K and incubated in TRIzol® reagent (Invitrogen) to detect the expression of SIRT1 mRNA with RT-qPCR.
Statistical analysis
Data are presented as the mean ± standard error of the mean from three independent experiments and were analyzed using SPSS 19.0 software (SPSS, Inc.). The P-values were evaluated using one-way analysis of variance followed by Tukey's post hoc test. Spearman's rank correlation analysis was performed to confirm the correlation between miR-135a-5p and SIRT1 in DN patients. P<0.05 was considered to indicate a statistically significant difference.
Results
miR-135a-5p is upregulated and SIRT1 is downregulated in patients with DN
Previous studies suggested the enhancement of miR-135a-5p expression in renal fibrosis and the important role of SIRT1 in mesangial cells and renal fibrosis (12,18). Consistently, the present study observed a significantly increased expression of miR-135a-5p (Fig. 1A) and a lower expression of SIRT1 (Fig. 1B and C) in the serum of patients with DN (n=30, Table I) as measured by RT-qPCR and western blotting. Moreover, the expression of miR-135a-5p and SIRT1 in the renal tissues was also detected. As presented in Fig. S1A and B, miR-135a-5p was upregulated, whereas SIRT1 was downregulated in the 10/30 renal biopsy specimens compared with normal renal tissues. In addition, there was a negative correlation between miR-135a-5p and SIRT1 expression in renal tissues of DN patients, according to Spearman's rank correlation analysis (Fig. S1C). These data suggested that miR-135a-5p and SIRT1 were involved in renal fibrosis.
Figure 1.

Expression of miR-135a-5p and SIRT1 in patients with DN. (A) Serum miR-135a-5p and (B) SIRT1 mRNA levels were detected by reverse transcription-quantitative PCR in control patients with diabetes without DN and those with DN. (C) Western blotting was used to detected SIRT1 protein levels. Data were plotted as the mean ± standard error of mean and performed in triplicate. *P<0.05 vs. normal. DN, diabetic nephropathy; Normal, patients with diabetes without DN; SIRT1, sirtuin 1; miR, microRNA.
miR-135a-5p expression is increased during TGFβ1-induced renal fibrosis in vitro
To examine the relevance of miR-135a-5p in renal fibrosis in DN, a cell model of renal fibrosis in HMC and HK-2 cells was constructed. First, miR-135a-5p expression was monitored in various glucose concentration stimulation in HMC and HK-2 cells. As a result, 15-30 mmol/l of D-glucose induced an increase in miR-135a-5p expression at 48 h (Fig. S2). Subsequently, HMC and HK-2 cells were exposed to 10 ng/ml TGFβ1 for 24 h for renal fibrosis analysis. As presented in Fig. 2A, miR-135a-5p was highly expressed in TGFβ1-induced HMC and HK-2 cells. The levels of collagen 1A1, α-SMA and FN were significantly promoted, whereas E-cadherin was inhibited under TGFβ1 stimulation (Fig. 2B). These data suggested that TGFβ1 treatment induced renal fibrosis in HMC and HK-2 cells, accompanied with upregulation of miR-135a-5p.
Figure 2.

Role of miR-135a-5p in TGFβ1-induced renal fibrosis in HMC and HK-2 cells. HMC and HK-2 cells were treated with 10 ng/ml TGFβ1 for 24 h. (A) miR-135a-5p expression was detected by reverse transcription-quantitative PCR. (B) Expression of collagen 1A1, α-SMA, FN and E-cadherin was measured by western blotting. *P<0.05 vs. the control cells (without TGFβ1 treatment). FN, fibronectin; SMA, smooth muscle actin; TGF, transforming growth factor; HMC, human mesangial cells; miR, microRNA; E, epithelial.
Knockdown of miR-135a-5p inhibits TGFβ1-induced renal fibrosis in HMC and HK-2 cells
Considering the upregulation of miR-135a-5p during renal fibrosis, miR-135a-5p was knocked down in HMC and HK-2 cells by transient transfection of anti-miR-135a-5p. During TGFβ1 exposure, anti-miR-135a-5p-transfected cells exhibited lower expression levels of miR-135a-5p compared with that of anti-NC-transfected cells (Fig. 3A). In addition, the levels of collagen 1A1, α-SMA and FN were decreased under TGFβ1 stimulation when miR-135a-5p was knocked down, whereas that of E-cadherin was elevated compared with anti-NC-transfected cells (Fig. 3B). These results demonstrated that miR-135a-5p knockdown may alleviate TGFβ1-induced renal fibrosis in HMC and HK-2 cells.
Figure 3.

Effects of the miR-135a-5p inhibitor on TGFβ1-induced HMC and HK-2 cells. miR-135a-5p expression in HMC and HK-2 cells was silenced by transient transfection of anti-miR-135a-5p. (A) Levels of miR-135a-5p were detected by reverse transcription-quantitative PCR. (B) Levels of collagen 1A1, α-SMA, FN and E-cadherin were measured by western blotting. *P<0.05 vs. anti-NC cells. FN, fibronectin; SMA, smooth muscle actin; TGF, transforming growth factor; HMC, human mesangial cells; miR, microRNA; E, epithelial; NC, negative control.
SIRT1 is downregulated, and overexpression of SIRT1 exerts a suppressive role in TGFβ1-induced renal fibrosis in vitro
To examine the effects of SIRT1 in renal fibrosis in DN, the present study determined its expression in a cell model of renal fibrosis in HMC and HK-2 cells. As presented in Fig. 4A, SIRT1 was expressed at a low level in TGFβ1-induced HMC and HK-2 cells. Thus, SIRT1 was overexpressed in HMC and HK-2 cells using ectopic expression of a recombinant vector pcDNA-SIRT1 (Fig. 4B). Of note, during TGFβ1 stimulation, levels of collagen 1A1, α-SMA and FN were significantly decreased, whereas E-cadherin was enhanced by SIRT1 upregulation compared with the negative control (Fig. 4C). These results demonstrated that overexpression of SIRT1 high expression may relieve TGFβ1-induced renal fibrosis in HMC and HK-2 cells.
Figure 4.
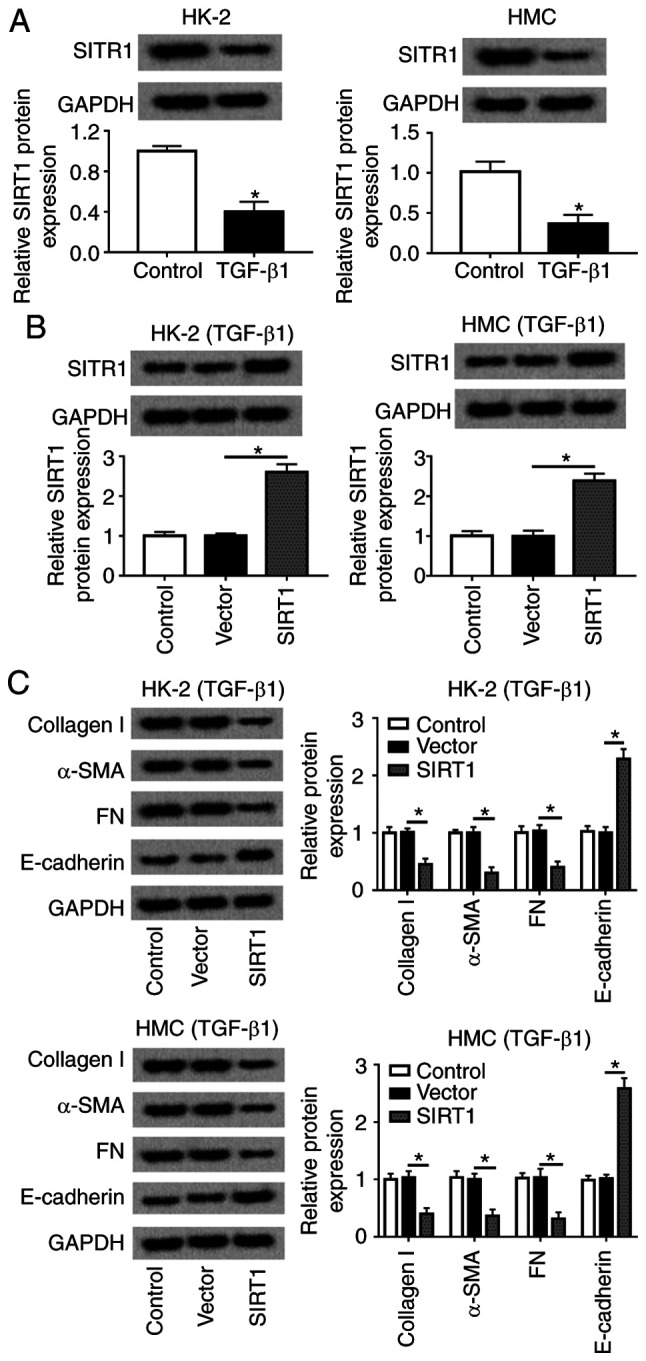
Role of SIRT1 in TGFβ1-induced HMC and HK-2 cells. HMC and HK-2 cells were treated with 10 ng/ml TGFβ1 for 24 h. (A) SIRT1 protein expression was detected. (B) Levels of SIRT1 were identified when HMC and HK-2 cells transfected pcDNA-SIRT1 (SIRT1). (C) Expression of collagen 1A1, α-SMA, FN and E-cadherin was measured by western blotting in SIRT1-overexpressed HMC and HK-2 cells. *P<0.05 vs. control cells (vector). FN, fibronectin; SMA, smooth muscle actin; TGF, transforming growth factor; HMC, human mesangial cells; miR, microRNA; E, epithelial; NC, negative control; SIRT1, sirtuin 1.
miR-135a-5p regulates SIRT1 expression via target binding
The regulatory relationship between miR-135a-5p and SIRT1 was further investigated. Algorithms analysis by TargetScan Human database (http://www.targetscan.org/vert_72/) identified the targets of miR-135a-5p, and the 3′UTR of human SIRT1 exhibited a highly conserved binding site for miR-135a-5p (Fig. 5A). To verify this, a luciferase reporter assay was performed. The luciferase reporter vectors integrating wt or mut SIRT1 3′UTR fragment were constructed, and HMC and HK-2 cells were co-transfected with SIRT1-wt/mut and either miR-135a-5p or miR-NC mimic. First, RT-qPCR analysis was used to confirm the high miR-135a-5p expression level in the mimic-transfected HMC and HK-2 cells (Fig. S3A). The luciferase activity was significantly reduced in cells transfected with the miR-135a-5p mimic and SIRT1-wt; however, no differences were observed in the SIRT1-mut groups (Fig. 5B). RIP assay further identified the target binding of miR-135a-5p and SIRT1 (Fig. 5C). A western blot assay demonstrated that SIRT1 expression was inhibited by the miR-135a-5p mimic but promoted by anti-miR-135a-5p in HMC and HK-2 cells compared with the corresponding NCs (Fig. 5D). These data supported the hypothesis that SIRT1 was a direct target of miR-135a-5p.
Figure 5.
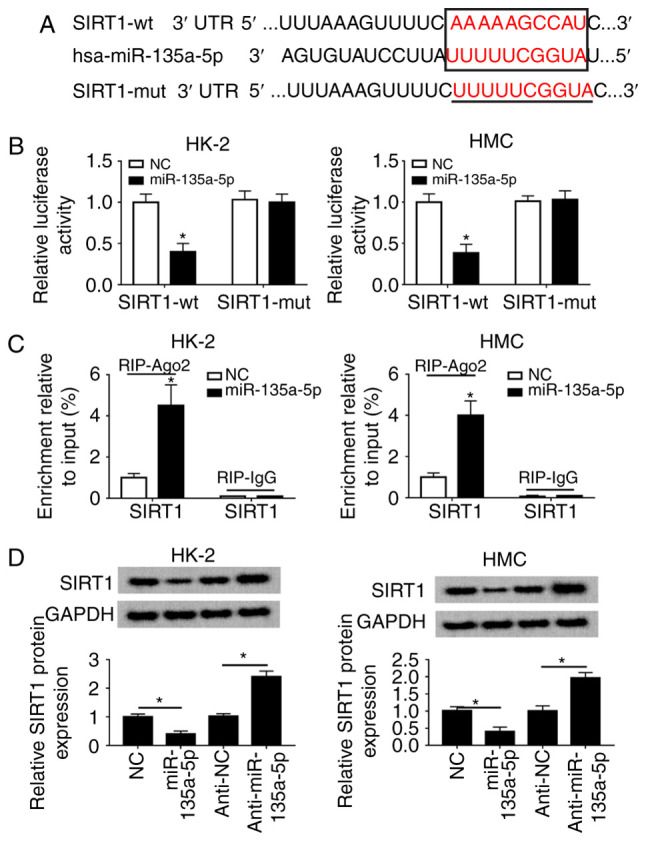
miR-135a-5p directly targets SIRT1 3′UTR. (A) TargetScan Human algorithm predicted the target sequence of hsa-miR-135a-5p in the SIRT1 3′ UTR. (B) Luciferase reporter assay was used to validate the relative luciferase activity of vectors containing the SIRT1-wt/mut in HMC and HK-2 cells when co-transfected with miR-135a-5p mimic or NC. (C) RNA immunoprecipitation assay was performed to further identify the level of SIRT1 mRNA in HMC and HK-2 cells co-transfected with miR-135a-5p or NC. (D) Western blotting was used to analyze the SIRT1 protein levels in HMC and HK-2 cells transfected with anti-miR-135a-5p, miR-135a-5p and the corresponding controls. Data were plotted as the mean ± standard error of the mean and performed in triplicate. *P<0.05 vs. control cells (NC or anti-NC). FN, fibronectin; SMA, smooth muscle actin; TGF, transforming growth factor; HMC, human mesangial cells; miR, microRNA; E, epithelial; NC, negative control; UTR, untranslated region; mut, mutant; wt, wild-type; miR-135a-5p, miR-135a-5p mimic; anti-miR-135a-5p, miR-135a-5p inhibitor.
SIRT1 mediates the role of miR-135a-5p knockdown in TGFβ1-induced renal fibrosis in vitro
Rescue experiments were performed to clarify the effects of SIRT1 dysregulation on the role of miR-135a-5p in HK-2 and HMC cells. As presented in Fig. 6A, HMC and HK-2 cells were divided into four transfection groups: Anti-NC, anti-miR-135a-5p, anti-miR-135a-5p + scramble and anti-miR-135a-5p + siSIRT1. The upregulation of SIRT1 induced by anti-miR-135a-5p was impaired by siSIRT1 (Fig. 6A), and western blotting confirmed that siSIRT1 transfection caused a significant decrease of the SIRT1 level in HMC and HK-2 cells compared with the scramble siRNA-transfected cells (Fig. S3B). When miR-135a-5p was inhibited, collagen 1A1, α-SMA and FN synthesis was reduced compared with the NC group, which was blocked by silencing of SIRT1 (Fig. 6B). Knockdown of SIRT1 abolished the effects of anti-miR-135a-5p on E-cadherin expression as indicated in Fig. 6B. These results demonstrated that miR-135a-5p knockdown inhibited TGFβ1-induced renal fibrosis by upregulating SIRT1.
Figure 6.


Influence of SIRT1 silencing on TGFβ1-induced HMC and HK-2 cells. HMC and HK-2 cells were transfected with siRNA against human SIRT1 (siSIRT1), and effect of SIRT1 downregulation in miR-135a-5p-knocked down cell was evaluated using western blotting. (A) Levels of SIRT1 were detected. (B) Expression of collagen1A1, α-SMA, FN and E-cadherin was measured. The quantification was performed on Image J. Data were plotted as mean ± standard error of mean and performed in triplicate. *P<0.05 vs. control cells (anti-NC or anti-miR-135a-5p+scramble). FN, fibronectin; SMA, smooth muscle actin; TGF, transforming growth factor; HMC, human mesangial cells; miR, microRNA; E, epithelial; NC, negative control; si, small interfering.
miR-135a-5p knockdown inactivates the TGFβ1/Smad3 signaling pathway through upregulating SIRT1
To explore the signaling pathway underlying the activity of miR-135a-5p during TGFβ1-induced renal fibrosis, Smad3 activation was measured. The upregulation of p-Smad normalized to total Smad3 was observed in HMC and HK-2 cells under TGFβ1 stimulation compared with that in untreated cells (Fig. 7A). The relative level of p-Smad3 was significantly reduced in the anti-miR-135a-5p group compared with that in the anti-NC group; in addition, the inactivation of Smad3 induced by miR-135a-5p knockdown was reversed by silencing SIRT1 in HMC and HK-2 cells (Fig. 7B). These results indicated that the inhibition of the TGFβ1/Smad3 signaling pathway was involved in the role of miR-135a-5p/SIRT1 in renal fibrosis in vitro.
Figure 7.

Effects of miR-135a-5p on Smad3 activation in TGFβ1-induced HMC and HK-2 cells. (A) Expression of total Smad3 and p-Smad3 in HMC and HK-2 cells under TGFβ1 stimulation was measured by western blotting. *P<0.05 vs. control cells (without TGFβ1 treatment). HMC and HK-2 cells were transfected with anti-miR-135a-5p/NC and siSIRT1/scramble or not. (B) Levels of Smad3 and p-Smad3 in TGFβ1-induced HMC and HK-2 cells. All the quantification of western blotting was performed on Image J. Data were plotted as mean ± standard error of the mean and performed in triplicate. *P<0.05 vs. control cells (anti-NC or anti-miR-135a-5p+scramble). FN, fibronectin; SMA, smooth muscle actin; TGF, transforming growth factor; HMC, human mesangial cells; miR, microRNA; E, epithelial; NC, negative control; Smad3, mothers against decapentaplegic homolog 3; p, phosphorylated.
Discussion
In China, diabetes has become a major public health problem (24), and ~10% of patients with diabetes suffer from DN (25). Progressive renal fibrosis is one of the hallmark pathological characteristics of DN (8). For example, the fibrosis-related genes collagen I, FN, E-cadherin and α-SMA were upregulated in a mouse model of DN (26). Bai et al (27) observed that the levels of Snail, Vimentin, collagen IV and α-SMA were upregulated, and E-cadherin was downregulated in 86 renal biopsies of DN. Putta et al (28) reported that silencing of miR-192 caused downregulation of key profibrotic genes such as collagen 1A2, collagen 4A1 and FN in the glomeruli and cortex of diabetic mice. In addition, it was also suggested that the epithelial-mesenchymal transition (EMT) served as one potential mechanism underlying renal fibrosis in DN (29,30). In the current study, TGFβ1 treatment induced renal fibrosis in HMC and HK-2 cells as demonstrated by the increased synthesis of collagen 1A1, α-SMA and FN, as well as by decreased expression of E-cadherin.
Numerous miRNAs have been reported to be involved in renal fibrosis in DN. For example, Zhao et al (31) demonstrated that miR-23b was expressed at a lower level in the serum of patients with diabetes mellitus and concluded that miR-23b had a protective effect against renal fibrosis in DN. Expression of miR-192 was upregulated by TGF-β1 in cultured glomerular mesangial cells and diabetic glomeruli of mice (28). The specific reduction of renal miR-192 decreased renal fibrosis and improve proteinuria. These results supported the possibility of an anti-miRNA-based translational approach to the treatment of DN. The results of the present study demonstrated that inhibition of miR-135a-5p the upregulation of E-cadherin levels, but reduced collagen 1A1, α-SMA, and FN expression in TGFβ1-induced HMC and HK-2 cells. The present results also revealed that SIRT1 was a target gene of miR-135a-5p, and silencing of SIRT1 abolished the effects of miR-135a-5p on renal fibrosis. In addition, Smad3 activation was altered by miR-135a-5p/SIRT1 in HMC and HK-2 cells.
As an oncogene, miR-135a-5p promotes cell proliferation and metastasis in hepatocellular carcinoma (32,33) and breast cancer (34). However, miR-135a-5p served as an anti-oncogene and targeted HOXA10 to suppress the proliferation of head and neck squamous cell carcinoma (35). Thus, miR-135a-5p may serve a dual role in cancers. In diabetes, expression of this miRNA is considered to be upregulated. For instance, Agarwal et al (36) observed elevated miR-135a-5p levels in human diabetic skeletal muscle. Upregulation of miR-135a-5p was also identified in the serum miRNA expression profile and renal tissues from patients with DN (21). In addition, the biological role and mechanism of miR-135a-5p was preliminarily explored, and the results demonstrated that miR-135a-5p upregulation promoted mesangial cell proliferation by decreasing G1/S arrest and increasing synthesis of ECM proteins such as FN, Vimentin, and collagen I by directly regulating short transient receptor potential channel 1 in a HMC line (21); by contrast, silencing of miR-135a-5p alleviated hyperglycemia and improved glucose tolerance in vivo (36). Thus, miR-135a-5p may be associated with fibrosis and diabetes. The present study focused on the expression of miR-135a-5p in patients with DN, as well as its biological role in TGFβ1-induced renal fibrosis cell models in HMC and HK-2 cells. The results demonstrated that the expression levels of miR-135a-5p were increased in the sera and renal tissues of patients with DN as well as in HMC and HK-2 cells under various glucose concentrations or TGFβ1 stimulation. Functionally, anti-miR-135a-5p attenuated the expression of collagen 1A1, α-SMA and FN, and elevated the levels of E-cadherin under TGFβ1 stimulation in vitro, which was in agreement with the previous findings by He et al (21). In addition, SIRT1 has been demonstrated to mediate the inhibitory activity of miR-135a-5p in the synthesis of fibrosis-related genes. Of note, Wu et al (37) recently investigated the role and possible regulatory mechanism of miR-135a-5p in cardiac fibrosis and reported that cardiac fibroblasts from neonatal rats induced by isoproterenol was inhibited by miR-135a-5p targeting the transient receptor potential melastatin 7. Simultaneously, miR-135a-5p was decreased in ISO-induced cardiac fibrosis in vitro and in vivo (37). These results suggested the complex and vital role of miR-135a-5p in the biological functions of diseases, including cancer and diabetes complications.
SIRT1 has been demonstrated to serve a crucial role in miscellaneous physiological processes through the deacetylation of a number of nuclear proteins such as p53 and NF-κB (22,38,39). Previous studies have demonstrated that SIRT1 reduces apoptosis in TGFβ-treated mesangial cells via acceleration of Smad7 degradation and the TGFβ signaling pathway (40). In addition, SIRT1 regulates fibroblast activation and tissue fibrosis by canonical TGFβ signaling (10). Inhibition of SIRT1 promotes TGFβ1-induced EMT and renal fibrosis in HK-2 cells (26). Accumulating evidence has indicated SIRT1 is affected by miRNAs during fibrogenesis, including that in the kidney. For example, inhibition of miR-133b and miR-199b attenuated TGF-β1-induced EMT and renal fibrosis by targeting SIRT1 (26). miR-34a targeting SIRT1 aggravated high glucose-stimulated tubulointerstitial fibrosis in HK cells (41,42). In the present study, the results demonstrated that SIRT1, which is a vital regulator in the evolution of renal fibrosis in DN (22), was directly suppressed by miR-135a-5p. Furthermore, the current study proposed that the miR-135a-5p/SIRT1 axis may provide a new approach for DN treatment.
Previously, SIRT1 activation had been suggested as a therapeutic strategy in progressive, fibrotic diseases in the kidney, liver, lung and heart (14,43-45). Mechanically, inhibition of the TGFβ1/Smad3 pathway has been attributed to the protective role of SIRT1 activation in organ fibrosis including renal fibrosis (14). For example, co-immunoprecipitation assays have provided direct evidence of an interaction between acetylated Smad3 and SIRT1 (14,46). In the kidney, knockdown of SIRT1 increases the levels of acetylated Smad3, thus substantially enhancing the transcriptional activity of Smad3 following TGF-β1 treatment (14). Additionally, the allosteric modifier of SIRT1 deacetylase ameliorates the TGFβ1-stimulated collagen production, which is accompanied by a reduction of Smad3 reporter activity (47). The present study indicated that the miR-135a-5p/SIRT1/Smad3 pathway was involved in TGFβ1-induced renal fibrosis.
One limitation of the current study was that it did not verify the suppressive activity of miR-135a-5p in diabetic db/db mice by injection of recombinant lentivirus containing miR-135a-5p inhibitor (21,26,28). Furthermore, immunohistochemistry examination of the kidney was not performed (14). TGFβ regulates biological processes by interacting with Smads, MAPK and Jagged/Notch signaling pathways (10). The results of the present study suggested that the miR-135a-5p/SIRT1 axis regulated Smad3 activation; it would be interesting to verify whether the other two signaling pathways may be altered by the functions of the miR-135a-5p/SIRT1 axis. Therefore, the identification of molecular pathways underlying DN would be imperative for development of new therapeutic strategies.
In conclusion, the results of the present study demonstrated that miR-135a-5p knockdown attenuated renal fibrosis in DN by targeting SIRT1 and inactivating the TGFβ1/Smad3 pathway. These results supported the hypothesis that miR-135a-5p may be a novel therapeutic target in suppressing renal fibrosis in DN.
Supplementary Data
Acknowledgments
Not applicable.
Abbreviations
- DN
diabetic nephropathy
- SIRT1
sirtuin 1
- HMC
human mesangial cells
- α-SMA
α-smooth muscle actin
- FN
fibronectin
- ECM
extracellular matrix
- TGFβ
transforming growth factor-β
- Smad
mothers against decapentaplegic homolog
- MAPK
mitogen activated protein kinase
Funding
This study was supported by the Health Commission of Hubei Province scientific research project 'The role and mechanism of interferon regulatory factor 8 in renal tubular injury in diabetic nephropathy' (project no. WJ2019M210) and The Wuhan Science and Technology Bureau (grant no. 2017060201010179).
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Authors' contributions
JZ and LZ conceived and designed the experiments. DZ performed the experiments and acquired funding. XW and JZ contributed the reagents/materials/analysis tools and performed data analysis and interpretation. LZ wrote the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
All protocols involving human subjects were approved by the Ethics Committee of the Zhongnan Hospital of Wuhan University. Informed consent was obtained from all patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Liu ZH. Nephrology in China. Nat Rev Nephrol. 2013;9:523–528. doi: 10.1038/nrneph.2013.146. [DOI] [PubMed] [Google Scholar]
- 2.Coimbra TM, Janssen U, Gröne HJ, Ostendorf T, Kunter U, Schmidt H, Brabant G, Floege J. Early events leading to renal injury in obese Zucker (fatty) rats with type II diabetes. Kidney Int. 2000;57:167–182. doi: 10.1046/j.1523-1755.2000.00836.x. [DOI] [PubMed] [Google Scholar]
- 3.Kanwar YS, Sun L, Xie P, Liu FY, Chen S. A glimpse of various pathogenetic mechanisms of diabetic nephropathy. Annu Rev Pathol. 2011;6:395–423. doi: 10.1146/annurev.pathol.4.110807.092150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gilbert RE, Cooper ME. The tubulointerstitium in progressive diabetic kidney disease: More than an aftermath of glomerular injury? Kidney Int. 1999;56:1627–1637. doi: 10.1046/j.1523-1755.1999.00721.x. [DOI] [PubMed] [Google Scholar]
- 5.Steffes MW, Osterby R, Chavers B, Mauer SM. Mesangial expansion as a central mechanism for loss of kidney function in diabetic patients. Diabetes. 1989;38:1077–1081. doi: 10.2337/diab.38.9.1077. [DOI] [PubMed] [Google Scholar]
- 6.Wang S, Yang Z, Xiong F, Chen C, Chao X, Huang J, Huang H. Betulinic acid ameliorates experimental diabetic-induced renal inflammation and fibrosis via inhibiting the activation of NF-κB signaling pathway. Mol Cell Endocrinol. 2016;434:135–143. doi: 10.1016/j.mce.2016.06.019. [DOI] [PubMed] [Google Scholar]
- 7.Mason RM, Wahab NA. Extracellular matrix metabolism in diabetic nephropathy. J Am Soc Nephrol. 2003;14:1358–1373. doi: 10.1097/01.ASN.0000065640.77499.D7. [DOI] [PubMed] [Google Scholar]
- 8.Song KH, Park J, Park JH, Natarajan R, Ha H. Fractalkine and its receptor mediate extracellular matrix accumulation in diabetic nephropathy in mice. Diabetologia. 2013;56:1661–1669. doi: 10.1007/s00125-013-2907-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wynn TA. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J Clin Invest. 2007;117:524–529. doi: 10.1172/JCI31487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hills CE, Squires PE. The role of TGF-β and epithelial-to mesenchymal transition in diabetic nephropathy. Cytokine Growth Factor Rev. 2011;22:131–139. doi: 10.1016/j.cytogfr.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 11.Sutariya B, Jhonsa D, Saraf MN. TGF-β: The connecting link between nephropathy and fibrosis. Immunopharmacol Immunotoxicol. 2016;38:39–49. doi: 10.3109/08923973.2015.1127382. [DOI] [PubMed] [Google Scholar]
- 12.Chen S, Hong SW, Iglesias-de la Cruz MC, Isono M, Casaretto A, Ziyadeh FN. The key role of the transforming growth factor-beta system in the pathogenesis of diabetic nephropathy. Ren Fail. 2001;23:471–481. doi: 10.1081/JDI-100104730. [DOI] [PubMed] [Google Scholar]
- 13.Sharma K, Jin Y, Guo J, Ziyadeh FN. Neutralization of TGF-beta by anti-TGF-beta antibody attenuates kidney hypertrophy and the enhanced extracellular matrix gene expression in STZ-induced diabetic mice. Diabetes. 1996;45:522–530. doi: 10.2337/diab.45.4.522. [DOI] [PubMed] [Google Scholar]
- 14.Huang XZ, Wen D, Zhang M, Xie Q, Ma L, Guan Y, Ren Y, Chen J, Hao CM. Sirt1 activation ameliorates renal fibrosis by inhibiting the TGF-β/Smad3 pathway. J Cell Biochem. 2014;115:996–1005. doi: 10.1002/jcb.24748. [DOI] [PubMed] [Google Scholar]
- 15.Böttinger EP, Bitzer M. TGF-beta signaling in renal disease. J Am Soc Nephrol. 2002;13:2600–2610. doi: 10.1097/01.ASN.0000033611.79556.AE. [DOI] [PubMed] [Google Scholar]
- 16.Massagué J, Chen YG. Controlling TGF-beta signaling. Genes Dev. 2000;14:627–644. [PubMed] [Google Scholar]
- 17.Kato M. TGF-β-induced signaling circuit loops mediated by microRNAs as new therapeutic targets for renal fibrosis? Kidney Int. 2013;84:1067–1069. doi: 10.1038/ki.2013.297. [DOI] [PubMed] [Google Scholar]
- 18.Lu Z, Wang F. Epigenetic regulations in diabetic nephropathy. J Diabetes Res. 2017;2017:7805058. doi: 10.1155/2017/7805058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cui C, Cui Y, Fu Y, Ma S, Zhang S. Microarray analysis reveals gene and microRNA signatures in diabetic kidney disease. Mol Med Rep. 2018;17:2161–2168. doi: 10.3892/mmr.2017.8177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rysz J, Gluba-Brzózka A, Franczyk B, Jabłonowski Z, Ciałkowska-Rysz A. Novel biomarkers in the diagnosis of chronic kidney disease and the prediction of its outcome. Int J Mol Sci. 2017;18:1702. doi: 10.3390/ijms18081702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He F, Peng F, Xia X, Zhao C, Luo Q, Guan W, Li Z, Yu X, Huang F. MiR-135a promotes renal fibrosis in diabetic nephrop-athy by regulating TRPC1. Diabetologia. 2014;57:1726–1736. doi: 10.1007/s00125-014-3282-0. [DOI] [PubMed] [Google Scholar]
- 22.Kume S, Kitada M, Kanasaki K, Maegawa H, Koya D. Anti-aging molecule, Sirt1: A novel therapeutic target for diabetic nephropathy. Arch Pharm Res. 2013;36:230–236. doi: 10.1007/s12272-013-0019-4. [DOI] [PubMed] [Google Scholar]
- 23.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 24.Yang W, Lu J, Weng J, Jia W, Ji L, Xiao J, Shan Z, Liu J, Tian H, Ji Q, et al. Prevalence of diabetes among men and women in China. N Engl J Med. 2010;362:1090–1101. doi: 10.1056/NEJMoa0908292. [DOI] [PubMed] [Google Scholar]
- 25.Roglic G, Unwin N, Bennett PH, Mathers C, Tuomilehto J, Nag S, Connolly V, King H. The burden of mortality attributable to diabetes: Realistic estimates for the year 2000. Diabetes Care. 2005;28:2130–2135. doi: 10.2337/diacare.28.9.2130. [DOI] [PubMed] [Google Scholar]
- 26.Sun Z, Ma Y, Chen F, Wang S, Chen B, Shi J. miR-133b and miR-199b knockdown attenuate TGF-β1-induced epithelial to mesenchymal transition and renal fibrosis by targeting SIRT1 in diabetic nephropathy. Eur J Pharmacol. 2018;837:96–104. doi: 10.1016/j.ejphar.2018.08.022. [DOI] [PubMed] [Google Scholar]
- 27.Bai X, Geng J, Zhou Z, Tian J, Li X. MicroRNA-130b improves renal tubulointerstitial fibrosis via repression of Snail-induced epithelial-mesenchymal transition in diabetic nephropathy. Sci Rep. 2016;6:20475. doi: 10.1038/srep20475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Putta S, Lanting L, Sun G, Lawson G, Kato M, Natarajan R. Inhibiting microRNA-192 ameliorates renal fibrosis in diabetic nephropathy. J Am Soc Nephrol. 2012;23:458–469. doi: 10.1681/ASN.2011050485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Loeffler I, Wolf G. Epithelial-to-mesenchymal transition in diabetic nephropathy: Fact or fiction? Cells. 2015;4:631–652. doi: 10.3390/cells4040631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Simonson MS. Phenotypic transitions and fibrosis in diabetic nephropathy. Kidney Int. 2007;71:846–854. doi: 10.1038/sj.ki.5002180. [DOI] [PubMed] [Google Scholar]
- 31.Zhao B, Li H, Liu J, Han P, Zhang C, Bai H, Yuan X, Wang X, Li L, Ma H, et al. MicroRNA-23b targets ras GTPase-activating protein SH3 domain-binding protein 2 to alleviate fibrosis and albuminuria in diabetic Nephropathy. J Am Soc Nephrol. 2016;27:2597–2608. doi: 10.1681/ASN.2015030300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zeng YB, Liang XH, Zhang GX, Jiang N, Zhang T, Huang JY, Zhang L, Zeng XC. miRNA-135a promotes hepatocellular carcinoma cell migration and invasion by targeting forkhead box O1. Cancer Cell Int. 2016;16:63. doi: 10.1186/s12935-016-0328-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yao S, Tian C, Ding Y, Ye Q, Gao Y, Yang N, Li Q. Down-regulation of Krüppel-like factor-4 by microRNA-135a-5p promotes proliferation and metastasis in hepatocellular carcinoma by transforming growth factor-β1. Oncotarget. 2016;7:42566–42578. doi: 10.18632/oncotarget.9934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen Y, Zhang J, Wang H, Zhao J, Xu C, Du Y, Luo X, Zheng F, Liu R, Zhang H, Ma D. miRNA-135a promotes breast cancer cell migration and invasion by targeting HOXA10. BMC Cancer. 2012;12:111. doi: 10.1186/1471-2407-12-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo LM, Ding GF, Xu W, Ge H, Jiang Y, Chen XJ, Lu Y. MiR-135a-5p represses proliferation of HNSCC by targeting HOXA10. Cancer Biol Ther. 2018;19:973–983. doi: 10.1080/15384047.2018.1450112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Agarwal P, Srivastava R, Srivastava AK, Ali S, Datta M. miR-135a targets IRS2 and regulates insulin signaling and glucose uptake in the diabetic gastrocnemius skeletal muscle. Biochim Biophys Acta. 2013;1832:1294–1303. doi: 10.1016/j.bbadis.2013.03.021. [DOI] [PubMed] [Google Scholar]
- 37.Wu Y, Liu Y, Pan Y, Lu C, Xu H, Wang X, Liu T, Feng K, Tang Y. MicroRNA-135a inhibits cardiac fibrosis induced by isoproterenol via TRPM7 channel. Biomed Pharmacother. 2018;104:252–260. doi: 10.1016/j.biopha.2018.04.157. [DOI] [PubMed] [Google Scholar]
- 38.Yuan F, Xie Q, Wu J, Bai Y, Mao B, Dong Y, Bi W, Ji G, Tao W, Wang Y, Yuan Z. MST1 promotes apoptosis through regulating Sirt1-dependent p53 deacetylation. J Biol Chem. 2011;286:6940–6945. doi: 10.1074/jbc.M110.182543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Salminen A, Kaarniranta K. NF-kappaB signaling in the aging process. J Clin Immunol. 2009;29:397–405. doi: 10.1007/s10875-009-9296-6. [DOI] [PubMed] [Google Scholar]
- 40.Kume S, Haneda M, Kanasaki K, Sugimoto T, Araki S, Isshiki K, Isono M, Uzu T, Guarente L, Kashiwagi A, Koya D. SIRT1 inhibits transforming growth factor beta-induced apoptosis in glomerular mesangial cells via Smad7 deacetylation. J Biol Chem. 2007;282:151–158. doi: 10.1074/jbc.M605904200. [DOI] [PubMed] [Google Scholar]
- 41.Xue M, Li Y, Hu F, Jia YJ, Zheng ZJ, Wang L, Xue YM. High glucose up-regulates microRNA-34a-5p to aggravate fibrosis by targeting SIRT1 in HK-2 cells. Biochem Biophys Res Commun. 2018;498:38–44. doi: 10.1016/j.bbrc.2017.12.048. [DOI] [PubMed] [Google Scholar]
- 42.Li A, Peng R, Sun Y, Liu H, Peng H, Zhang Z. LincRNA 1700020I14Rik alleviates cell proliferation and fibrosis in diabetic nephropathy via miR-34a-5p/Sirt1/HIF-1α signaling. Cell Death Dis. 2018;9:461. doi: 10.1038/s41419-018-0527-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bugyei-Twum A, Ford C, Civitarese R, Seegobin J, Advani SL, Desjardins JF, Kabir G, Zhang Y, Mitchell M, Switzer J, et al. Sirtuin 1 activation attenuates cardiac fibrosis in a rodent pressure overload model by modifying Smad2/3 transactivation. Cardiovasc Res. 2018;114:1629–1641. doi: 10.1093/cvr/cvy131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang T, Wang J, Pang Y, Dang X, Ren H, Liu Y, Chen M, Shang D. Emodin suppresses silica-induced lung fibrosis by promoting Sirt1 signaling via direct contact. Mol Med Rep. 2016;14:4643–4649. doi: 10.3892/mmr.2016.5838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jiang R, Zhou Y, Wang S, Pang N, Huang Y, Ye M, Wan T, Qiu Y, Pei L, Jiang X, et al. Nicotinamide riboside protects against liver fibrosis induced by CCl4 via regulating the acetylation of Smads signaling pathway. Life Sci. 2019;225:20–28. doi: 10.1016/j.lfs.2019.03.064. [DOI] [PubMed] [Google Scholar]
- 46.Li J, Qu X, Ricardo SD, Bertram JF, Nikolic-Paterson DJ. Resveratrol inhibits renal fibrosis in the obstructed kidney: Potential role in deacetylation of Smad3. Am J Pathol. 2010;177:1065–1071. doi: 10.2353/ajpath.2010.090923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Y, Connelly KA, Thai K, Wu X, Kapus A, Kepecs D, Gilbert RE. Sirtuin 1 activation reduces transforming growth factor-β1-induced fibrogenesis and affords organ protection in a model of progressive, experimental kidney and associated cardiac disease. Am J Pathol. 2017;187:80–90. doi: 10.1016/j.ajpath.2016.09.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article.