

Abstract
Oral administration of docetaxel in combination with the CYP3A4 inhibitor ritonavir is used in clinical trials to improve oral bioavailability of docetaxel. Diarrhea was the most commonly observed and dose‐limiting toxicity. This study combined preclinical and clinical data and investigated incidence, severity and cause of oral docetaxel‐induced diarrhea. In this study, incidence and severity of diarrhea in patients were compared to exposure to orally administered docetaxel. Intestinal toxicity after oral or intraperitoneal administration of docetaxel was further explored in mice lacking Cyp3a and mice lacking both Cyp3a and P‐glycoprotein. In patients, severity of diarrhea increased significantly with an increase in AUC and C max (P = .035 and P = .025, respectively), but not with an increase in the orally administered dose (P = .11). Furthermore, incidence of grade 3/4 diarrhea after oral docetaxel administration was similar as reported after intravenous docetaxel administration. Intestinal toxicity in mice was only observed at high systemic exposure to docetaxel and was similar after oral and intraperitoneal administration of docetaxel. In conclusion, our data show that the onset of severe diarrhea after oral administration of docetaxel in humans is similar after oral and intravenous administration of docetaxel and is caused by the concentration of docetaxel in the systemic blood circulation. Mouse experiments confirmed that intestinal toxicity is caused by a high systemic exposure and not by local intestinal exposure. Severe diarrhea in patients after oral docetaxel is reversible and is not related to the route of administration of docetaxel.
Keywords: diarrhea, oral docetaxel, toxicity
Abbreviations
- AUCinf
the area under the plasma concentration‐time curve extrapolated to infinity
- AUCs
the area under the plasma concentration‐time curves
- Cmax
maximum plasma concentration
- Cyp3a
murine Cytochrome P450 3a
- Cyp3a‐/‐ mice
Cyp3a knockout mice
- Cyp3a/Mdr1a/b‐/‐ mice
Cyp3a and Mdr1a/b P‐gp knockout mice
- CYP3A4
Cytochrome P450 3A4
- Mdr1a/b P‐gp
murine P‐glycoprotein
- NCI‐CTCAE
National Cancer Institute's Common Terminology Criteria for AEs criteria
- NSCLC
non–small cell lung cancer
- P‐gp
P‐glycoprotein
- PK
pharmacokinetic
- SD
standard deviation
1. INTRODUCTION
Docetaxel is currently widely used as an intravenously administered anticancer agent for solid malignancies, such as non–small cell lung cancer (NSCLC), breast, gastric, prostate, and head‐and‐neck cancer. 1 In the last years, we investigated the oral administration of docetaxel to increase patient convenience, to reduce treatment costs and to circumvent the use of polysorbate 80, a pharmaceutical excipient well known for causing hypersensitivity reactions. 2 , 3 The poor bioavailability of docetaxel after oral administration could be increased by using a solid dispersion pharmaceutical formulation and by co‐administration of the Cytochrome P450 3A4 (CYP3A4) inhibitor ritonavir, making oral administration of docetaxel feasible. 4 , 5 , 6
These results encouraged us to perform two phase I dose escalation studies to determine safety and preliminary efficacy of this concept with oral docetaxel in combination with ritonavir. However, oral docetaxel in combination with either 100 mg or 200 mg ritonavir resulted in a modified toxicity profile of docetaxel compared to its intravenous administration. Within both studies, diarrhea was the most commonly observed toxicity and it was also dose limiting. 7 , 8 In contrast, the most common treatment‐related adverse events reported after intravenous administration of docetaxel were alopecia, anemia, leukocytopenia, and neutropenia. 9 Similarly, in preclinical experiments degeneration and necrosis of the intestinal mucosa was observed 3 days after oral administration of 10 mg kg−1 docetaxel in mice lacking murine P‐glycoprotein (Mdr1a/b P‐gp) and Cytochrome P450 3a (Cyp3a). 10
In the case of the applied oral docetaxel formulation, both clinical and preclinical data suggest intestinal toxicity by oral docetaxel as major cause for the observed diarrhea. Therefore, it is important to understand the mechanism behind the development of diarrhea as this may help to develop possible measures to prevent it. Damage to the intestinal mucosa can lead to an imbalance between absorption and secretion of fluids leading to diarrhea. This damage could be an effect of mitotic arrest of intestinal crypt cells caused by exposure to chemotherapeutic agents in the systemic circulation as observed after administration of 5‐flourouracil, 11 or it could be a direct local effect of intestinal luminal drug on the intestinal or colonic epithelium as is believed to be the case after irinotecan administration. 12 , 13
In this study, we examined mice lacking Cyp3a and mice lacking both Cyp3a and Mdr1a/b P‐gp, in order to mimic the clinical conditions wherein Cyp3a and possibly P‐glycoprotein (P‐gp) are inhibited, as is the case when oral docetaxel formulations (eg, drinking solution, Modradoc001 capsule, or Modradoc006 tablet) are administered with ritonavir. Clinical data of phase I trials with oral docetaxel as drinking solution, ModraDoc001 capsule or Modradoc006 tablet were analyzed to investigate the severity and the duration of intestinal toxicity after oral administration of the drug. 7 , 8 Our study aimed to elucidate whether the intestinal toxicity is caused by (a) a direct local effect, and thus related to the amount of docetaxel present in the lumen of the gastrointestinal tract, or (b) a systemic effect and therefore related to the docetaxel concentration in the systemic circulation. The data obtained from the mouse experiments were compared to the data derived from clinical studies with orally administered docetaxel. Furthermore, incidence and severity of diarrhea after orally administered docetaxel was compared to previously reported incidence and severity after intravenously administered docetaxel.
2. MATERIALS AND METHODS
2.1. Animal studies
2.1.1. General design
In a previously published study, severe intestinal toxicity in mice was observed after oral administration of 10 mg kg−1 docetaxel to Cyp3a and Mdr1a/b P‐gp knockout (Cyp3a/Mdr1a/b−/−) mice. 10 However, after administration of the same dose in Cyp3a knockout (Cyp3a−/−) mice, no toxicity was observed. Therefore, a pharmacokinetic dose‐finding study and a toxicity study were designed (Figure 1).
FIGURE 1.

Study design of preclinical studies in mice. Panel A shows the dose levels tested in the pharmacokinetic dose‐finding study. Panel B shows the design of the toxicity study. In the toxicity study, bodyweight of the mice was measured daily at day 1‐3. AUC0‐inf and C max values represent the mean ± SD. Abbreviations: AUC0‐inf, area under the plasma‐concentration curve in µg mL−1 h−1; C max: maximum observed plasmaconcentrations in ng mL‐1; Cyp3a‐/‐, Cyp3a knockout mice ; Cyp3a/Mdr1a/b‐/‐, Cyp3a; ip, intraperitoneal; Mdr1a/b P‐gp knockout mice; N, number of animals. #Dose not tolerated due to high amount of ethanol needed for dissolution of docetaxel. Also high variability in exposure between individuals. $Due to technical problems during administration blood samples of only four animals were obtained
In the dose‐finding study (Figure 1, Panel A), five mice per treatment group were used. As the highest dose level in Cyp3a4−/− mice was not tolerable, less mice were used for this dose level. For the intraperitoneal administration eight mice were used at the first dose level tested, as a higher variation after i.p. administration than after oral administration was expected. Since no higher variation was observed, five mice in subsequent dose levels were used. The sample size for the dose‐finding study (n = 5) was based on a power analysis that assumed an α of 0.2 and a β of 0.05. The sample size was calculated using the spreadsheet of Boston University. 14 Mean plasma AUC and standard deviations from previous experiments using 10 mg/kg dosing of docetaxel in Cyp3a/Mdr1a/b−/− were used for estimation of the effect size (16 466 ± 2020 ng h/mL). 10 The sample size was powered to pick up a 20% difference in the AUC.
The toxicity study (Figure 1, panel B) was initially started with a cohort for oral administration in which six mice per group were used. The oral doses were selected to obtain a comparable systemic exposure in both strains. In a second cohort, the intraperitoneal administration was started. The intraperitoneal doses were selected to obtain a similar exposure as the highest administered oral dose for that strain. To validate the results between the first and last cohort, three additional mice were added to the oral group with 10 mg/kg and compared to the six mice in the first cohort. No differences in the results between the cohorts were observed and data were pooled for the analyses. Three days after oral or intraperitoneal administration of docetaxel to mice, total body necropsy was performed and tissues and organs were fixed. The sections were reviewed by an animal pathologist, who was blinded to the dose level, route of administration and strain.
2.1.2. Drugs and chemicals
Docetaxel and ritonavir for mice studies were purchased from Sequoia Research Products. Drug‐free lithium‐heparinized human plasma was obtained from Bioreclamation LLC. All other chemicals were of analytical grade and obtained from commercial sources.
2.1.3. Animals
All mouse experiments were approved by the Animal Experiments Review Board of the Netherlands Cancer Institute (Amsterdam), complying with Dutch legislation and in accordance with European Directive 86/609/EEC. Mice were housed and handled according to institutional guidelines complying with Dutch legislation. Mice were kept in a temperature‐controlled environment with a 12‐hour light/ 12‐hour dark cycle and received a standard diet (AM‐II, Hope Farms, Woerden, The Netherlands) and acidified water ad libitum. Strains used in this study were Cyp3a knockout (Cyp3a−/−) 15 and combined Cyp3a and Mdr1a/b P‐gp knockout mice (Cyp3a/Mdr1a/b−/−) 10 . All strains had a > 99% FVB genetic background. In all experiments, male mice of 8‐14 weeks of age were used.
2.1.4. Docetaxel administration in mice
Prior to the experiments, stock solutions containing 1, 3, 6, 9, and 36 mg mL−1 docetaxel in ethanol:polysorbate 80 (1:1, v/v) were prepared and stored at −20°C. On the day of the experiments stock solutions were diluted with water to obtain solutions containing various concentrations of docetaxel in ethanol:polysorbate 80:water (1:1:10, v/v). For the 180 mg kg−1 dose level in Cyp3a−/− mice, stock solutions were diluted with ethanol:polysorbate 80 (1:1) instead of water to avoid precipitation of docetaxel. Animals were fasted 2 hours before oral drug administration to minimize variation in absorption. Docetaxel was administered orally or intraperitoneally at various doses using a total volume of 10 µL per kg of body weight. Oral administration was performed by gavage into the stomach using a blunt‐ended needle. Intraperitoneal administration was performed by injection into the peritoneal cavity.
2.1.5. Sample collection and analysis for dose‐finding study in mice
Multiple blood samples (~50 µL) were collected from the tail vein at 15 and 30 minutes and 1, 2, 4, 8, and 24 hours after administration, using heparinized capillary tubes (Oxford Labware). Blood samples were centrifuged at ambient temperature at 8000 g for 5 minutes and subsequently plasma was collected. All samples were stored at −20°C until analysis. A previously developed LC‐MS/MS assay was used to quantify docetaxel in plasma samples of mice. 16 D9‐labeled docetaxel was used as internal standard for docetaxel. Mouse plasma samples of 20 µL were diluted with 180 µL of drug‐free human plasma prior to sample pretreatment. Human plasma was used for dilution of the samples as the concentrations in the undiluted mouse plasma were outside the calibration range and also to mimic the calibration standards that were prepared in human plasma. Sample pretreatment was started by adding a small volume of internal standard working solution to the samples. Subsequently, the samples were mixed briefly, tertiary‐butyl methyl ether was added and the samples were shaken for 10 minutes at 1250 rpm. The samples were centrifuged at 23 000 g, snap‐frozen, and the organic layer was collected. After evaporation of the organic layer, the samples were reconstituted with 100 µL reconstitution solvent and an aliquot was injected into the LC‐MS/MS system.
2.1.6. Histological analysis for toxicity study in mice
Three days after oral or intraperitoneal administration of docetaxel to mice, total body necropsy was performed and tissues and organs were fixed in EAF fixative (ethanol/acetic acid/formaldehyde/saline at 40:5:10:45 v/v) and embedded in paraffin. Sections were cut at 2 µm from the paraffin blocks and stained with hematoxylin and eosin (HE) according to standard procedures. The sections were reviewed with a Zeiss Axioskop2 Plus microscope (Carl Zeiss Microscopy) equipped with Plan‐Apochroma and Plan‐Neofluar objectives. The reviewing animal pathologist was blinded to the dose level, route of administration and strain. Images were captured with a Zeiss AxioCam HRc digital camera and processed with AxioVision 4 software (both from Carl Zeiss Vision, Munich, Germany).
2.2. Clinical trials
2.2.1. General design
Two previously performed trials with orally administered docetaxel (as ModraDoc001 capsule and ModraDoc006 tablet) were included in our analysis. 7 , 8 The following data were derived for each patient in these studies: administered dose, measured plasma concentrations, time of sampling, onset and grade of diarrhea, duration of diarrhea, and patient characteristics for demographic evaluation. Per patient, PK parameters of docetaxel were related to events of diarrhea by coupling the highest grade of the diarrhea to the corresponding the area under the plasma concentration‐time curve extrapolated to infinity (AUCinf), maximum plasma concentration (C max), and daily dose. For the diarrhea events without corresponding PK parameter, the maximum observed AUCinf and the corresponding C max of that patient were used. In total, 112 patients were included. The demographic and baseline characteristics of the patients and treatment schedules of the different studies and cohorts are described in Table 1.
TABLE 1.
Patient demographics and study details of two clinical studies used for PK data in humans
| QD dose escalation | BID dose escalation | Total | ||||
|---|---|---|---|---|---|---|
| Character | N | % | N | % | N | % |
| Number of patients | 67 | 45 | 112 | |||
| Sex | ||||||
| Male – female | 37‐30 | 26‐19 | 63‐49 | |||
| Age | ||||||
| Median (range) | 58 (36‐79) | 58 (41‐77) | 58 (36‐79) | |||
| WHO status | ||||||
| 0 | 33 | 49% | 21 | 47% | 54 | 48% |
| 1 | 29 | 43% | 22 | 49% | 51 | 46% |
| 2 | 5 | 7% | 2 | 4% | 7 | 6% |
| Tumor characteristics | ||||||
| NSCLC | 30 | 45% | 21 | 47% | 51 | 46% |
| UCC | 5 | 7% | 4 | 9% | 9 | 8% |
| Ovary | 4 | 6% | 2 | 4% | 6 | 5% |
| Primary unknown | 4 | 6% | 0 | 0% | 4 | 4% |
| Other | 24 | 36% | 18 | 40% | 42 | 38% |
| Dosage form |
Drinking solution (n = 5) ModraDoc001/r (n = 43) ModraDoc001/r (n = 19) |
ModraDoc001 (n = 17), ModraDoc006/r (n = 28) | ||||
| Daily docetaxel dose | 30, 40, 50, 60, 80 mg | 40, 50,60, 80 mg | ||||
| Daily ritonavir dose | 100, 200 mg | 200 mg | ||||
| Schedule | QD | BID | ||||
| PK assessments | Week 1 and 2 | Week 1 and 3 | ||||
| PK schedule | Predose, 0.25, 0.5, 0.75, 1, 1.5, 2, 4, 7, 10, 24, and 48 h | Predose, 0.5, 1, 1.5, 2, 3, 4, 6, 7, 7.5, 8, 8.5, 9, 10, 11, 13, 24 and 48 h | ||||
| References | [8] | [7] | ||||
The first study was a phase I dose escalation study with weekly once daily (QD) oral docetaxel in combination with ritonavir. The second study was a dose escalation study with weekly bi‐daily (BID) oral docetaxel in combination with ritonavir.
Abbreviations: BID, Weekly bi‐daily; N, number; QD, weekly once daily.
2.2.2. Clinical studies included
Briefly, the first study was a phase I study with weekly once daily oral docetaxel in combination with ritonavir. 8 This study included 67 patients in several cohorts in a dose escalation design. In this dose escalating study docetaxel was administered as drinking solution in the first dose level (30 mg docetaxel, n = 5) and as ModraDoc001 capsules (n = 43) or ModraDoc006 tablets (n = 19) in the other dose levels. The once weekly doses of the other dose levels were 30, 40, 50, 60, and 80 mg docetaxel in combination with 100 mg or 200 mg ritonavir. Patients received the treatment until progressive disease or until unacceptable toxicity despite dose reduction.
The second study 7 was a dose escalation study with oral docetaxel (as ModraDoc001 capsules) in combination with ritonavir administered weekly according to a bi‐daily schedule. The study design was comparable to the first study. This study included 17 patients treated with docetaxel as ModraDoc001 capsules at three dose levels and 28 patients treated with docetaxel as ModraDoc006 tablets. The weekly doses were 40, 60, and 80 mg docetaxel as ModraDoc001 capsules or 40 mg, 50 mg, and 60mg docetaxel as ModraDoc006 tablets and 200 mg ritonavir.
The clinical studies had similar enrollment criteria, which were in line with general exclusion criteria for Phase I studies in oncology. Patients were eligible if they had a histological or cytological proof of cancer, no standard treatment options available and adequate bone marrow, renal, and hepatic function. 7 , 8 Patients with known alcoholism, drug addiction and/or psychotic disorders were considered not suitable for adequate follow‐up, and thus excluded. Patients were not allowed to concomitantly use P‐gp and CYP3A modulating drugs, H2‐receptor antagonists or proton pump inhibitors. Other exclusion criteria included bowel obstructions that might influence drug absorption and previous anticancer therapy within 4 weeks prior to the first dose of oral docetaxel. The adverse events were determined using the National Cancer Institute's Common Terminology Criteria for AEs criteria (NCI‐CTCAE v3.0).
2.2.3. Docetaxel administration in patients
In the clinical studies, docetaxel was administered to patients as drinking solution (iv formulation, Taxotere®, Rhone‐Poulenc Rorer/Aventis), capsules (ModraDoc001 10 mg capsules, Department of Pharmacy & Pharmacology, The Netherlands Cancer Institute), or tablets (ModraDoc006 10 mg capsules, Department of Pharmacy & Pharmacology, The Netherlands Cancer Institute). Ritonavir was administered as tablets (Norvir®; Abbott). Patients received orally 30 to 80 mg of docetaxel coadministered with ritonavir at a dose of 100 or 200 mg according to a weekly schema. Patients were fasted 2 hours before and 1 hour after oral drug administration to minimize variation in absorption.
2.2.4. Sample collection and analysis
The PK of docetaxel were monitored according to various schedules in the different cohorts and studies during the first 24 or 48 hours (Table 1). Blood samples were collected in heparinized tubes and centrifuged at 4°C at 1500 g for 10 minutes. Subsequently, plasma was collected and stored at −20°C until the time of analysis. A previously developed LC‐MS/MS assays was used to quantify docetaxel in plasma samples. 17 D9‐labeled docetaxel was used as internal standard for docetaxel. Sample pretreatment of human plasma was as for diluted mouse plasma (see above).
2.3. Pharmacokinetic calculations and statistical analysis
PK parameters in mice, including the AUCs, were calculated using the software package PK Solutions 2.0.2 (SUMMIT, Research Services). The individual PK parameters of the patients were analyzed using descriptive noncompartmental PK methods and validated R scripts (R version 2.13.1). The AUCs were estimated by the linear trapezoidal (absorption phase) and logarithmic trapezoidal rule (elimination phase). The AUCinf was calculated by extrapolation. All PK data of the animal and human studies are presented as mean ± standard deviation (SD).
For statistical testing in animal experiments, one‐way ANOVA was used when multiple groups were compared and the Bonferroni post hoc correction was used to accommodate multiple testing. The two‐sided unpaired Student's t test was used when treatments or differences between two groups were compared. Data that did not show normal distribution were log‐transformed to normalize the distribution of the datasets for statistical comparison. During all statistical analyses in animal experiments, differences in group sizes were considered in the calculations. The human data were analyzed using a proportional odds model for testing of the relation between increase in severity of diarrhea and increase in AUCinf, C max, or dose. Complete statistical reports are in Data S1.
2.4. Study approval
All animal experiments were performed according to EU and Dutch national legislation. All experimental protocols were assessed and approved by the institutional animal care and use committee. Two previously performed trials with orally administered docetaxel were included in our analysis. 7 , 8 The included clinical studies were approved by the Medical Ethics Committee of the Netherlands Cancer Institute and were conducted in accordance with the Declaration of Helsinki. Written informed consent was obtained from all patients prior to study entry. The studies were registered under identifier NCT01173913 (NIH register) and ISRCTN32770468 (ISRCTN register), respectively.
3. RESULTS
3.1. Dose finding of docetaxel in mice
A dose‐finding study was performed to select dose levels for the toxicity experiment. Previously, severe toxicity (including intestinal toxicity) was observed 3 days after single oral administration of 10 mg kg−1 docetaxel to combined Cyp3a/Mdr1a/b−/− mice, but no toxicity was observed after administration of the same dose to Cyp3a−/− mice. 10 Although the same dose was administered to both mouse strains, the AUCinf of docetaxel was significantly higher as a result of additional P‐gp knock‐out in the Cyp3a/Mdr1a/b−/− strain. 10 With this dose‐finding study, we aimed to select a low and a high dose for each strain to compare intestinal toxicity in a subsequent experiment. At each dose level (low and high), we aimed for a comparable plasma exposure for both strains. Moreover, we aimed for selection of an intraperitoneal dose for each strain that results in comparable exposure as the highest selected dose level for that strain.
We started to determine the plasma AUCinf in Cyp3a−/− mice and Cyp3a/Mdr1a/b−/− mice after oral administration of 10 mg kg−1 docetaxel. We observed a 27‐fold higher plasma AUCinf in Cyp3a/Mdr1a/b−/− mice than in Cyp3a−/− mice (Figure 2, panel A). Subsequently, we increased the oral dose for Cyp3a−/− mice in several steps to try to reach similar plasma exposure as observed in Cyp3a/Mdr1a/b−/− mice after oral administration of 10 mg kg−1 docetaxel (Figure 1). However, a similarly high exposure in Cyp3a−/− mice could not be reached as it was observed that an oral dose higher than 60 mg kg−1 docetaxel in Cyp3a−/− mice did not result in a further increase in AUCinf compared to a dose of 60 mg kg−1 (Figure 1). This is most likely due to the limited water solubility of docetaxel. Because of the limited volume in the intestinal tract, docetaxel could precipitate and therefore not be absorbed efficiently from the intestinal lumen.
FIGURE 2.

Plasma concentration‐time curves obtained after administration of docetaxel to Cyp3a−/− and Cyp3a/Mdr1a/b−/− mice. Panel A shows plasma concentration‐time curves of oral administration of different doses of docetaxel to Cyp3a−/− and Cyp3a/Mdr1a/b−/− mice. Panel B and C show plasma‐concentration‐time curves after oral or intraperitoneal administration of docetaxel to Cyp3a/Mdr1a/b−/− (panel B) and Cyp3a−/− (panel C) mice. Inset in panel A, B and C show the AUCinf. All AUCsinf differ mutually significantly (P < .001) as calculated with ANOVA of the Log‐transformed data with Bonferroni's post hoc test (panel A) or as calculated with a two‐sided unpaired Student's t test (panel B and C), unless otherwise specified (NS). For all groups n = 5 animals were used (n = 4 for 12.5 mg kg−1 in Cyp3a/Mdr1a/b−/− mice due to technical problems during administration). Inserts show mean ± standard deviations. Abbreviations: AUCinf, area under the plasma concentration‐time curves extrapolated from zero to infinity; C max, maximum plasma concentration; Cyp3a−/−, Cyp3a knock‐out; Cyp3a/Mdr1a/b−/−, Cyp3a and P‐glycoprotein knock‐out; NS, not significant
After increasing the oral docetaxel dose in Cyp3a−/− mice, we decreased the oral docetaxel dose in Cyp3a/Mdr1a/b−/− mice to try to reach a similar plasma exposure as observed in Cyp3a−/− mice after oral administration of 10 mg kg−1 docetaxel (Figure 1). At a dose of 1.67 mg kg−1, the C max observed in Cyp3a/Mdr1a/b−/− mice was similar to the C max observed in Cyp3a−/− mice after a dose of 10 mg kg−1. Moreover, the AUCinf in Cyp3a/Mdr1a/b−/− after administration of 1.67 mg kg−1 was comparable to the AUCinf after administration of 60 mg kg−1 in Cyp3a−/− mice. The difference in shape of the plasma concentration‐time curves between Cyp3a−/− and Cyp3a/Mdr1a/b−/− mice is caused by the Mdr1a/b P‐gp effect on the elimination of docetaxel. 18
Based on the observed results, we selected an oral dose level of 1.67 and 10 mg kg−1 for the Cyp3a/Mdr1a/b−/− strain and an oral dose level of 10 and 60 mg kg−1 for the Cyp3a−/− strain for future toxicity studies. After testing various doses of intraperitoneally administered docetaxel (Figure 1), it was observed that an intraperitoneal dose of 12 mg kg−1 docetaxel used for Cyp3a/Mdr1a/b−/− mice and an intraperitoneal dose of 5 mg kg−1 docetaxel used for Cyp3a−/− mice resulted in a similar AUCinf as obtained after oral administration of the highest selected dose level for that strain (Figure 2, panel B and C). Therefore, both intraperitoneal doses were also selected for the toxicity experiment.
3.2. Toxicity after oral and intraperitoneal administration of docetaxel in mice
For toxicity experiments, docetaxel was administered orally once at doses of 1.67 and 10 mg kg−1 in Cyp3a/Mdr1a/b−/− mice and at doses of 10 and 60 mg kg−1 in Cyp3a−/− mice. Docetaxel was also administered intraperitoneally once at doses of 5 and 12 mg kg−1 in Cyp3a−/− and Cyp3a/Mdr1a/b−/− mice, respectively. The different dosages and administration routes were used to allow comparison of docetaxel toxicity between the strains at similar plasma exposure levels (Figure 2A, inset and Table 2). Pathological examination performed 72 hours after oral administration of 10 mg kg−1 docetaxel to Cyp3a/Mdr1a/b−/− mice revealed a significant reduction in hematopoietic cells in spleen and bone marrow (see Table 2 and Figure 3), which did not occur after a low dose of docetaxel (1.67 mg kg−1). The intestinal toxicity observed at 10 mg kg−1 consisted of severe degeneration of the large and small intestinal mucosa with depletion of the crypts and inflammatory infiltrations in the lamina propria. This toxicity was found in all mice in this group and was similar to previously observed toxicity after administration of the same dose of oral docetaxel in this strain. 10 After oral administration of the same dose (10 mg kg−1 docetaxel) to Cyp3a−/− mice no signs of severe toxicity were observed, but the mean AUC in these mice was almost 28‐fold lower than in Cyp3a/Mdr1a/b−/− mice after the same dose. Even at the maximum achievable AUCinf in Cyp3a−/− mice after an oral dose of 60 mg kg−1 docetaxel, only mild toxicity in the intestinal mucosa and spermatogenic cells was observed (Table 2). However, the maximum AUCinf in Cyp3a−/− mice was still 10.7‐fold lower than the AUCinf in Cyp3a/Mdr1a/b−/− mice after oral administration of 10 mg kg−1. The observed toxicity in Cyp3a−/− mice after a dose of 60 mg kg−1 docetaxel was characterized as increased mitosis and apoptosis of cells in the mucosa of the small intestine in four of nine mice and testicular degeneration in three of nine mice. Administration of a low oral dose of 1.67 mg kg−1 docetaxel in Cyp3a/Mdr1a/b−/− mice did not result in signs of toxicity. The mean AUCinf in these mice was comparable to the mean AUCinf after a dose of 10 mg kg−1 docetaxel in Cyp3a−/− mice.
TABLE 2.
Overview of toxicity observed after various doses of docetaxel, administered orally or intraperitoneally to Cyp3a/Mdr1a/b−/− and Cyp3a−/− mice
| Cyp3a/Mdr1a/b−/− mice | Cyp3a −/− mice | |
|---|---|---|
| IP dose |
Dose: 12 mg kg−1 AUCinf: 54.9 ± 7.1 µg mL−1 h−1 Cmax : 3880 ± 356 ng mL−1 Toxicity: severe toxicity. Observations: Depletion of crypts in mucosa of intestine and colon, intestinal inflammation, edema in mucosa of colon, depletion of hematopoietic cells in bone marrow, reduced hematopoietic activity in spleen |
Dose: 5 mg kg−1 AUCinf: 5.5 ± 1.0 µg mL−1 h− Cmax : 817 ± 137 ng mL−1 Toxicity: little toxicity. Observations: Increase in mitosis and apoptosis in intestinal mucosa, incidental depletion of hematopoietic cells in bone marrow, incidentally reduced hematopoietic activity in spleen and incidental testicular degeneration. |
| High oral dose |
Dose: 10 mg kg−1 AUCinf: 48.3 ± 13.8 µg mL−1 h− Cmax : 3766 ± 572 ng mL−1 Toxicity: severe toxicity. Observations: Depletion of crypts in mucosa of intestine and colon, intestinal inflammation, edema in mucosa of colon, depletion of hematopoietic cells in bone marrow, reduced hematopoietic activity in spleen |
Dose: 60 mg kg−1 AUCinf: 4.5 ± 0.8 µg mL−1 h− Cmax : 1234 ± 281 ng mL−1 Toxicity: little toxicity. Observations: Increase in mitosis and apoptosis in intestinal mucosa and necrosis of spermatogenetic cells. |
| Low oral dose |
Dose: 1.67 mg kg−1 AUCinf: 3.6 ± 0.5 µg mL−1 h− Cmax : 400 ± 80 ng mL−1 Toxicity: no toxicity. Observations: Lesions in testis and testicular degeneration incidentally observed. |
Dose: 10 mg kg−1 AUCinf: 1.7 ± 0.4 µg mL−1 h− Cmax : 391 ± 196 ng mL−1 Toxicity: no toxicity. Observations: No abnormalities detected |
Abbreviations: AUCinf, area under the plasma concentration‐time curves extrapolated from zero to infinity; C max, maximum plasma concentration; Cyp3a−/−, Cyp3a knock‐out; Cyp3a/Mdr1a/b−/−, Cyp3a and P‐glycoprotein knock‐out; IP, intraperitoneal.
FIGURE 3.

Microphotograph of a typical HE section of the ileum (upper panels, original magnification 20×) and bone marrow (lower panels, original magnification 10×) of Cyp3a/Mdr1a/b−/− and Cyp3a−/− mice after single oral or intraperitoneal administration of docetaxel. Mice were sacrificed for pathological examination 72 hours after docetaxel administration. The Cyp3a/Mdr1a/b−/− mice showed no toxicity after oral administration of 1.67 mg kg−1 (panel A and E), but showed severe toxicity after oral administration of 10 mg kg−1 (B and F) or intraperitoneal administration of 12 mg kg−1 (C and G). The severe toxicity observed in Cyp3a/Mdr1a/b−/− mice was depletion of crypts in small intestine (B and C) and depletion of hematopoietic cells in bone marrows (F and G). Cyp3a−/− mice showed an increase in mitosis and apoptosis in intestinal mucosa, but no changes in bone marrow after oral administration of 60 mg kg−1 (D and H). Abbreviations: Cyp3a−/−, Cyp3a knock‐out; Cyp3a/Mdr1a/b−/−, Cyp3a and P‐glycoprotein knock‐out; HE, hematoxylin and eosin stain; L, intestinal lumen; M, mucosa; LP, lamina propria. Arrows indicate deep crypts
In none of the mice of both strains diarrhea was observed after oral administration of docetaxel. In Cyp3a/Mdr1a/b−/− mice, a loss of body weight was observed after administration of 10 mg kg−1 docetaxel. The average body weight of these mice was decreased to 88% of the initial body weight in 3 days. The body weight after 3 days of all other mouse groups was 99%‐105% of the initial body weight.
After intraperitoneal administration of 5 mg kg−1 docetaxel in Cyp3a−/− mice, similar mild toxicity was observed as after oral administration of 60 mg kg−1 docetaxel to these mice (see Table 2). Plasma AUCs and C max values were similar under these conditions (Figure 2, panel C). For both intraperitoneal (5 mg kg−1) and oral (60 mg kg−1) administration of docetaxel in Cyp3a−/− mice, changes in the mucosa of the small intestine were observed in four of six mice. Incidentally (one of six mice), depletion of hematopoietic cells in bone marrow and reduced hematopoietic activity in spleen or testicular degeneration were observed after intraperitoneal administration. Strikingly, in Cyp3a/Mdr1a/b−/− mice, toxicity was also similar after intraperitoneal administration of 12 mg kg−1 docetaxel and oral administration of 10 mg kg−1 (Table 2). Again, plasma AUCs and C max values, whereas much higher than in the other tested strains, were similar under these two conditions (Figure 2, panel B). The toxicity after both administration routes included severe degeneration of intestinal mucosa and depletion of the crypts combined with inflammatory infiltrations. In all mice of both strains, no diarrhea was observed after intraperitoneal administration of docetaxel. The mean body weight after 3 days was 87% and 95% of the initial body weight in Cyp3a/Mdr1a/b−/− and Cyp3a−/− mice, respectively. These data suggest that in mice, intestinal docetaxel toxicity is more related to the systemic exposure level than to the drug administration route.
3.3. Toxicity after oral administration of docetaxel in humans
To quantify the intestinal toxicity in humans, data from 112 subjects derived from two phase I studies involving oral docetaxel administration were combined. 7 , 8 Two subjects were excluded from the analyses, since these patients never started the treatment after enrollment. In both studies the range of the daily doses of docetaxel was 30‐80 mg administered according to a weekly schedule, and the PK parameters of docetaxel were determined in all patients.
Of the 110 evaluable patients, 83 patients (75%) had suffered from diarrhea, of which 71 patients (65% of total) had diarrhea considered related to study drug. Patients with an event of diarrhea which was considered unrelated or unlikely related to oral docetaxel treatment were excluded from the analyses (eg, in case of pre‐existing diarrhea or disease‐related chronic diarrhea). Life‐threatening or disabling diarrhea (grade 4) was not seen during the studies. For the majority of the patients (51 of 71 patients with study drug‐related diarrhea, 72%) the worst treatment‐related event of diarrhea started within 3 weeks after the start of treatment and for most patients (54 patients, 76%) the related event of diarrhea resolved within 2 weeks after start of loperamide treatment.
Treatment‐related diarrhea was of grade 1 severity in 43 patients (39% of total) and of grade 2 severity in 18 patients (16% of total). In case of grade 1‐2 diarrhea, patients were advised to use loperamide, except on the day of docetaxel administration since loperamide is metabolized via CYP3A4. 19 , 20 Neither dose interruption nor reduction was implemented. Of all patients with treatment‐related grade 1‐2 diarrhea, 66% recovered within 1 week after start of loperamide treatment, whereas for 8% of the patients recovery took 2 weeks (1 patient recovered after 7 weeks and for 23% of the patients the exact duration remains unknown).
Grade 3 diarrhea was observed in 10 patients (9% of total). In 8 of these patients, grade 3 diarrhea started during the first 2 weeks of treatment. Diarrhea was labeled as serious adverse event for eight patients suffering from grade 3 diarrhea since hospitalization was needed. During an event of grade 3 diarrhea, loperamide was given and docetaxel was withheld according to protocol until recovery to ≤ grade 1. Afterward, docetaxel was restarted at a lower dose. The mean duration of grade 3 diarrhea was short (range, 1‐12 days) and in most cases patients recovered fully from diarrhea after start of rescue treatment. After recovery, a dose reduction for docetaxel was successfully applied (n = 7) or docetaxel treatment was discontinued (n = 3).
In Figure 4 the severity of the diarrhea is plotted against the AUCinf, the corresponding C max and the daily dose as boxplot. Severity of the diarrhea increases significantly with increasing AUCinf and C max (P = .035 and P = .025, respectively). An increasing orally administered dose did not result in significantly increasing severity of diarrhea (P = .107).
FIGURE 4.
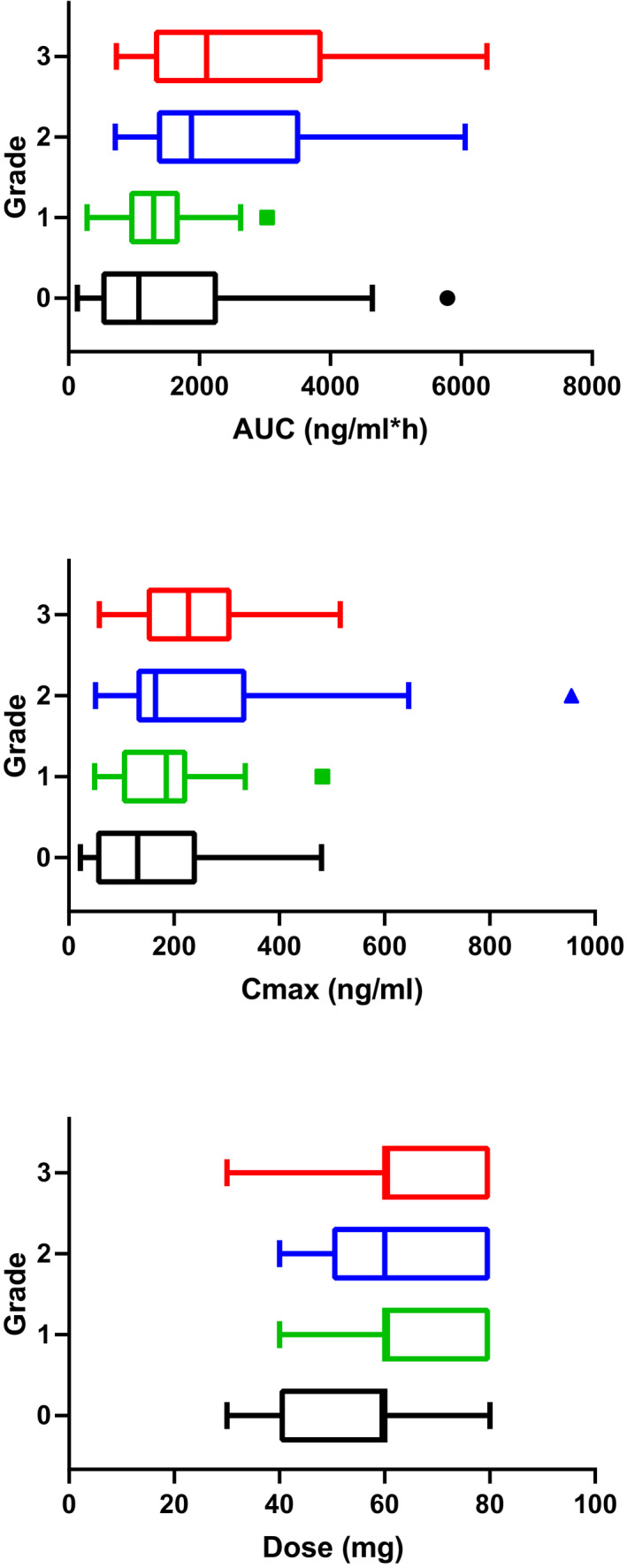
Relationship between the severity of diarrhea (Grade 0‐3) after oral administration of docetaxel and AUCinf of docetaxel, C max and the daily dose given boxplot. Grade 4 diarrhea is not seen in the clinical studies. In total data of 110 evaluable subjects were plotted (grade 0: n = 39, grade 1: n = 43, grade 2: n = 18, grade 3: n = 10). Abbreviations: AUC, area under the plasma concentration‐time curves; C max, maximum plasma concentration; n, number of patients
4. DISCUSSION
A limitation in the treatment with most oral anticancer drugs is the development of gastrointestinal adverse effects. Development of diarrhea can be the major cause for treatment discontinuation and its severity proves to be a dose‐limiting toxic event. 21 During development of novel oral formulations of docetaxel (ModraDoc001 capsules and ModraDoc006 tablets) significant diarrhea was encountered. This led to the execution of preclinical studies to unravel the mechanism of this toxicity.
We used mice lacking Cyp3a with and without intact Mdr1a/b P‐gp expression to better understand the cause of the intestinal toxicity as observed in patients after orally administered of docetaxel. The Cyp3a‐deficient mice are used to reflect the co‐administration of docetaxel with the CYP3A inhibitor ritonavir in humans. Although human CYP3A has no clear direct murine orthologues, 22 there is a broad functional overlap between human CYP3A and murine Cyp3a for the metabolism of docetaxel. 15 , 23 Human MDR1 function is covered by murine Mdr1a and Mdr1b. 24 Despite the limitations associated with extrapolation of preclinical data, mice lacking Cyp3a with and without functional Mdr1a/b expression might be used as a model for oral co‐administration of docetaxel and ritonavir in humans. 25
In our study, Cyp3a/Mdr1a/b−/− mice and Cyp3a−/− mice received high and low doses of oral docetaxel. Each strain also received an intraperitoneal dose which resulted in comparable plasma exposures as the high oral doses. This enabled us to discriminate between local (intestinal) vs systemic exposure in relation to toxicity. Severe intestinal toxicity was observed after the high oral dose in Cyp3a/Mdr1a/b−/− mice (10 mg kg−1 docetaxel). However, also after intraperitoneal administration of a dose of 12 mg kg−1, similarly severe intestinal toxicity was observed in this strain. Using intraperitoneal administration of docetaxel, the initial intestinal uptake step (of docetaxel from the gut lumen) is circumvented while a distribution phase is maintained (and thus a high peak concentration as observed after intravenous administration is circumvented). Since oral and intraperitoneal administration of docetaxel resulted in comparable plasma AUCs, it is likely that the observed intestinal toxicity is caused by docetaxel in the systemic circulation rather than by a direct effect on the intestinal mucosal cells of docetaxel during absorption from the gut lumen. After both routes of administration, depletion of cells in the deep crypts of the intestine was observed upon histological investigation. These crypt cells are not directly exposed to the intestinal content nor involved in drug absorption, but are stem cells and progenitor cells (that finally proliferate to intestinal epithelial cells). 26 The depletion of the deep crypt cells supports the hypothesis that intestinal toxicity is caused by systemic exposure to docetaxel, since similar findings (mitotic arrest and apoptosis in crypts of the mucosa) have been reported in the literature with other systemically applied anticancer drugs. For instance, after intraperitoneal administration of cisplatin, decreased crypt cell production rates were observed, 27 leading to reduced height of the villi and loss of mucosal function. The reduced size of villi was also observed in duodenal mucosal biopsies taken 14 days after administration of oral tegafur/gimeracil/oteracil (S‐1) combined with intravenous administration of cisplatin and docetaxel to patients with metastatic gastric cancer. 28 Patients with a more severe grade of diarrhea showed a greater decrease in villus seize, indicating loss of mucosal function due to reduced villus size.
The toxicity data in our clinical studies with oral docetaxel showed that patients with a higher AUCinf suffered from more severe diarrhea. Although the differences in study desing, investigators, and patient management, an indirect comparison with other docetaxel trials in literature (Table 3) shows that the overall incidence of treatment‐related diarrhea (grade 1‐4) at the highest dose levels of oral docetaxel in our dose escalation studies was two‐fold higher than after weekly iv treatment. 29 , 30 , 31 , 32 , 33 , 34 , 35 , 36 , 37 However, this was mainly due to the high incidence of grade 1/2 diarrhea after oral docetaxel administration. The incidence of grade 3/4 diarrhea after oral docetaxel was in the same range as after iv treatment. The plasma AUCs in patients after oral administration of these doses of docetaxel were comparable to those after iv administration of standard doses of docetaxel. 7 , 8 , 38 , 39 This indicates that severe diarrhea is most probably caused by docetaxel exposure in the systemic circulation rather than by local exposure in the intestinal tract.
TABLE 3.
Incidence and severity of diarrhea in various published trials in humans after intravenous administration of docetaxel compared to incidence and severity of diarrhea in humans after the highest dose‐levels of oral administration of docetaxel
| Regime | Dose | Tumor | N | Overall | Gr 1 −2 | Gr 3 ‐ 4 | References |
|---|---|---|---|---|---|---|---|
| Intravenous docetaxel, every 3 wk: | 100 mg m−2 | Breast | 25 | 72% | 68% | 4% | [34] |
| 100 mg m−2 | Breast | 256 | 45% | 40% | 5% | [33] | |
| 80 mg m−2 | Breast | 56 | 30% | 30% | 0% | [35] | |
| 75 mg m−2 | NSCLC | 110 | 21% | 18% | 3% | [29] | |
| 75 mg m−2 | Breast | 54 | 37% | 26% | 11% | [37] | |
| 75 mg m−2 | Prostate | 176 | 46% | 44% | 2% | [30] | |
| Intravenous docetaxel, every 2 wks: | 50 mg m−2 | Prostate | 170 | 37% | 36% | 1% | [30] |
| Intravenous docetaxel, every week: | 33.3 mg m−2 | NSCLC | 110 | 26% | 23% | 3% | [29] |
| 30 mg m−2 | Breast | 48 | 45% | 27% | 8% | [37] | |
| 40 mg m−2 | Breast | 20 | >30% a | >25% a | 5% | [32] | |
| 36 mg m−2 | NSCLC | 30 | >24% a | >10% a | 14% | [31] | |
| 35 mg m−2 | NSCLC | 36 | 9% | 3% | 6% | [36] | |
| Oral docetaxel, every week: | b | c | 112 | 65% | 55% | 9% | [7, 8] |
Abbreviations: Gr, grade refers to the severity of diarrhea; NSCLC, non–small cell lung cancer; Ref, reference.
Grade 1 toxicity was not reported.
Daily doses of 30/100, 40/100, 60/100, 80/100, 40/200, 50/200, 60/200, and 80/200 mg docetaxel and ritonavir, respectively (every week).
Different tumor types, patients were eligible if they were diagnosed with a histological or cytological proof of cancer, if there were no standard curative or palliative treatment options available and if docetaxel treatment was appropriate for further treatment.
The lack of intestinal toxicity after oral administration of 60 mg kg−1 docetaxel to Cyp3a−/− mice shows that the absolute amount of docetaxel present in the intestinal lumen is not directly related to the development of toxicity. In Cyp3a/Mdr1a/b−/− mice, an oral dose of only 10 mg kg−1 docetaxel already resulted in severe toxicity including intestinal toxicity indicating that docetaxel must be absorbed to cause intestinal toxicity. In Cyp3a−/− mice this absorption is blocked by Mdr1a/b P‐gp at the enterocyte apical membrane. In patients, the severity of diarrhea does not appear to be related to the orally administered dose (ie, amount of docetaxel present in the gastrointestinal tract). Therefore, incomplete absorption of an oral formulation of docetaxel most likely does not increase the risk of severe diarrhea, or of other types of intestinal toxicity.
Our mice data show that a high AUC of docetaxel in the systemic blood circulation is responsible for degeneration of the intestinal mucosa and depletion of the crypts combined with inflammatory infiltrations. Despite the severe changes in the intestinal mucosa of the mice, no diarrhea was observed. Based on body weight loss in mice with severe toxicity, it is possible that the mice did not develop diarrhea, because they stopped eating and drinking early after the development of toxicities in the gastrointestinal tract. It is also possible that diarrhea would have developed after the 3 days used in our preclinical study as observed for 5‐FU treatment by Wu et al 40 In humans, death of colonic crypt cells can result in a cascade of effects whereby immature crypt cells release more secretory compounds and thereby cause diarrhea. 41 The damaged colonic crypts are also not able to absorb chloride, the driving force of water absorption in the colon. Degeneration of intestinal mucosa and inflammatory infiltrations can also lead to inflammatory diarrhea. 42 , 43 This is also seen during colonoscopy and in colon biopsies of patients who had developed intravenous docetaxel‐induced pseudo‐membranous colitis and in duodenal mucosal biopsies after an intravenous docetaxel‐containing chemotherapy regimen. 28 , 44 , 45 Therefore, it is likely that the onset of severe diarrhea in humans after docetaxel treatment is caused by malfunction of the intestinal tract due to similar structural changes of the intestinal mucosa as observed in mice.
Although these results suggest that severe toxicity in the intestine is caused by docetaxel exposure in the systemic circulation and not by a direct local effect, the increase in mild and moderate diarrhea (grade 1 and 2) after oral administration of docetaxel remains unexplained. Most events of mild and moderate diarrhea after oral administration occurred in the evening after treatment, but also some days later. We observed no difference in incidence of mild to moderate diarrhea between weekly once daily and weekly twice daily administration. In all cases, the mild and moderate diarrhea could be successfully treated with loperamide. Since oral docetaxel administration has only been explored in a few clinical studies, limited data are available regarding the pathophysiology of these mild and moderate toxicities. Short‐term locally high docetaxel concentrations in the human enterocyte might cause apoptosis of part of the intestinal epithelial cells, although apoptosis of epithelial cells was not observed in our mouse experiments.
All patients in our study received ritonavir, which can also induce apoptosis in human intestinal epithelial cells and thereby decrease barrier function of the epithelial layer. 46 Cell death in the epithelial cells can cause synthesis of inflammatory cytokines, which eventually can cause mucositis. 42 , 43 Loss of epithelial cells might cause the observed onset of diarrhea, which is also seen after ritonavir treatment of HIV patients (100‐400 mg a day). 47 Since ritonavir is necessary to reach systemic exposure of orally administered docetaxel, we could not distinguish between the contribution of docetaxel and ritonavir in the onset of mild and moderate diarrhea. After single oral administration of both docetaxel and ritonavir as monotherapy, the intestinal villi could be damaged as well. This damage could lead to a reduced surface area for absorption resulting in diarrhea via secretory mechanisms. 41 , 47 The higher incidence of mild and moderate diarrhea after oral co‐administration of docetaxel and ritonavir than after iv administration of docetaxel can therefore be caused by a local effect of both ritonavir and docetaxel. These events can be treated with loperamide, but should be carefully monitored by the treating physicians. 41
In conclusion, our data indicate that diarrhea upon oral docetaxel administration with ritonavir is not directly related to the amount of docetaxel present in the lumen of the gastrointestinal tract. Moreover, the onset of severe diarrhea after oral co‐administration of docetaxel and ritonavir in humans appears to be caused by the exposure of docetaxel in the systemic blood circulation, is reversible and is not related to the route of administration of docetaxel.
CONFLICTS OF INTEREST
The research group of AH Schinkel receives revenue from commercial distribution of some of the mouse strains used in this study. JH Beijnen and JHM Schellens have received a grant for translational research (ZonMW code 40‐41200‐98‐004), hold a patent on oral taxane formulations, own stocks and are (part‐time) employees of Modra Pharmaceuticals, a spin out company developing oral taxane formulations. The other authors declare that they have no conflict of interest.
AUTHOR CONTRIBUTIONS
JH, FS, JB, JHS, and SM designed the study. JH carried out animal experiments. JYS reviewed animal sections. FS and VW collected clinical trial data. JH and HR performed bioanalysis of samples. JH, FS, JYS, JL, and AS analyzed animal data. JH, FS, VW, JB, JHS, and SM analyzed clinical trial data. JH and FS drafted the manuscript. All authors had final approval of the submitted and published versions.
JH and FS share the position as first author, since this study combines animal and human data to support each other and to come to an overall conclusion. The order of the authors is based on alphabetical order of their last name.
Supporting information
Data S1
ACKNOWLEDGMENTS
We thank Dr Erik van Werkhoven (Biometrics Department, The Netherlands Cancer Institute, Amsterdam, The Netherlands) for his help with the statistical analysis of the clinical data. We acknowledge the workers of the animal facility of the Netherlands Cancer Institute for the daily care of animals used for this study and the workers of Central Data Management of the Netherlands Cancer Institute for their assistance with registration of clinical trial data.
Hendrikx JJMA, Stuurman FE, Song J‐Y, et al. No relation between docetaxel administration route and high-grade diarrhea incidence. Pharmacol Res Perspect. 2020;8:e00633 10.1002/prp2.633
Jeroen J. M. A. Hendrikx and Frederik E. Stuurman share the position as first author.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Gligorov J. Preclinical pharmacology of the taxanes: Implications of the differences. Oncologist. 2004;9(suppl_2):3‐8. 10.1634/theoncologist.9-suppl_2-3 [DOI] [PubMed] [Google Scholar]
- 2. Jibodh RA, Lagas JS, Nuijen B, Beijnen JH, Schellens JHM. Taxanes: old drugs, new oral formulations. Eur J Pharmacol. 2013;717(1–3):40‐46. 10.1016/j.ejphar.2013.02.058 [DOI] [PubMed] [Google Scholar]
- 3. Koolen SLW, Beijnen JH, Schellens JHM. Intravenous‐to‐oral switch in anticancer chemotherapy: a focus on docetaxel and paclitaxel. Clin Pharmacol Ther. 2010;87(1):126‐129. 10.1038/clpt.2009.233 [DOI] [PubMed] [Google Scholar]
- 4. Moes JJ, Koolen SLW, Huitema ADR, Schellens JHM, Beijnen JH, Nuijen, B . Pharmaceutical development and preliminary clinical testing of an oral solid dispersion formulation of docetaxel (ModraDoc001). Int J Pharm. 2011;420(2):244‐250. 10.1016/j.ijpharm.2011.08.041 [DOI] [PubMed] [Google Scholar]
- 5. Oostendorp RL, Huitema ADR, Rosing H, et al. Coadministration of ritonavir strongly enhances the apparent oral bioavailability of docetaxel in patients with solid tumors. Clin Cancer Res. 2009;15(12):4228‐4233. 10.1158/1078-0432.CCR-08-2944 [DOI] [PubMed] [Google Scholar]
- 6. Sawicki E, Beijnen JH, Schellens JHM, Nuijen B. Pharmaceutical development of an oral tablet formulation containing a spray dried amorphous solid dispersion of docetaxel or paclitaxel. Int J Pharm. 2016;511(2):765‐773. 10.1016/j.ijpharm.2016.07.068 [DOI] [PubMed] [Google Scholar]
- 7. de Weger VA, Stuurman FE, Hendrikx JJMA, et al. A dose‐escalation study of bi‐daily once weekly oral docetaxel either as ModraDoc001 or ModraDoc006 combined with ritonavir. Eur J Cancer. 2017;86:217‐225. 10.1016/j.ejca.2017.09.010 [DOI] [PubMed] [Google Scholar]
- 8. de Weger VA, Stuurman FE, Koolen SLW, et al. A phase I dose escalation study of once‐weekly oral administration of docetaxel as ModraDoc001 capsule or ModraDoc006 tablet in combination with ritonavir. Clin Cancer Res. 2019;25(18):5466‐5474. 10.1158/1078-0432.CCR-17-2299 [DOI] [PubMed] [Google Scholar]
- 9. European medicines agency (EMA): Taxotere ‐ Summary of Product Characteristics (SmPC). Available at http://www.ema.europa.eu. Accessed March 15, 2020.
- 10. van Waterschoot RAB, Lagas JS, Wagenaar E, et al. Absence of both cytochrome P450 3A and P‐glycoprotein dramatically increases docetaxel oral bioavailability and risk of intestinal toxicity. Can Res. 2009;69(23):8996‐9002. 10.1158/0008-5472.CAN-09-2915 [DOI] [PubMed] [Google Scholar]
- 11. Inomata A, Horii I, Suzuki K. 5‐Fluorouracil‐induced intestinal toxicity: what determines the severity of damage to murine intestinal crypt epithelia? Toxicol Lett. 2002;133(2–3):231‐240. [DOI] [PubMed] [Google Scholar]
- 12. Ikuno N, Soda H, Watanabe M, Olca M. Irinotecan (CPT‐11) and characteristic mucosal changes in the mouse ileum and cecum. J Nat Cancer Inst. 1995;87(24):1876‐1883. [DOI] [PubMed] [Google Scholar]
- 13. Sandmeier D, Chaubert P, Bouzourene H. Irinotecan‐induced colitis. Int J Surg Path. 2005;13(2):215‐218. [DOI] [PubMed] [Google Scholar]
- 14. IACUC (no date) Statistical Explanation Sample Spreadsheet. Available at: https://www.bu.edu/researchsupport/compliance/animal‐care/working‐with‐animals/research/sample‐size‐calculations‐iacuc/
- 15. van Herwaarden AE, Wagenaar E, van der Kruijssen CMM, et al. Knockout of cytochrome P450 3A yields new mouse models for understanding xenobiotic metabolism. J Clin Invest. 2007;117(11):3583‐3592. 10.1172/JCI33435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kuppens IELM, van Maanen MJ, Rosing H, Schellens JHM, Beijnen JH. Quantitative analysis of docetaxel in human plasma using liquid chromatography coupled with tandem mass spectrometry. Biomed Chromatogr. 2005;19(5):355‐361. 10.1002/bmc.457 [DOI] [PubMed] [Google Scholar]
- 17. Hendrikx JJMA, Hillebrand MJX, Thijssen B, et al. A sensitive combined assay for the quantification of paclitaxel, docetaxel and ritonavir in human plasma using liquid chromatography coupled with tandem mass spectrometry. J Chromatogr B. 2011;879(28):2984‐2990. 10.1016/j.jchromb.2011.08.034 [DOI] [PubMed] [Google Scholar]
- 18. van Waterschoot RAB, Lagas JS, Wagenaar E, Rosing H, Beijnen JH, Schinkel AH. Individual and combined roles of CYP3A, P‐glycoprotein (MDR1/ABCB1) and MRP2 (ABCC2) in the pharmacokinetics of docetaxel. Int J Cancer. 2010;127(12):2959‐2964. 10.1002/ijc.25279 [DOI] [PubMed] [Google Scholar]
- 19. Kim K‐A, Chung J, Jung H‐D, Park J‐Y. Identification of cytochrome P450 isoforms involved in the metabolism of loperamide in human liver microsomes. Eur J Clin Pharmacol. 2004;60(8):575‐581. 10.1007/s00228-004-0815-3 [DOI] [PubMed] [Google Scholar]
- 20. Marechal JD, Yu J, Brown S, et al. In silico and in vitro screening for inhibition of cytochrome P450 CYP3A4by comedications commonly used by patients with cancer. Drug Metab Dispos. 2006;34(4):534‐538. 10.1124/dmd.105.007625 [DOI] [PubMed] [Google Scholar]
- 21. Loriot Y, Perlemuter G, Malka D, et al. Drug Insight: gastrointestinal and hepatic adverse effects of molecular‐targeted agents in cancer therapy. Nat Clin Pract Onco. 2008;5(5):268‐278. 10.1038/ncponc1087 [DOI] [PubMed] [Google Scholar]
- 22. Nelson DR, Zeldin DC, Hoffman SMG, Maltais LJ, Wain HM, Nebert, DW . Comparison of cytochrome P450 (CYP) genes from the mouse and human genomes, including nomenclature recommendations for genes, pseudogenes and alternative‐splice variants. Pharmacogenetics. 2004;14(1):1‐18. 10.1097/00008571-200401000-00001 [DOI] [PubMed] [Google Scholar]
- 23. van Waterschoot RAB, Schinkel AH. A critical analysis of the interplay between cytochrome P450 3A and P‐glycoprotein: recent insights from knockout and transgenic mice. Pharmacol Rev. 2011;63(2):390‐410. 10.1124/pr.110.002584 [DOI] [PubMed] [Google Scholar]
- 24. Schinkel AH, Jonker JW. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: an overview. Adv Drug Deliv Rev. 2003;55(1):3‐29. [DOI] [PubMed] [Google Scholar]
- 25. Koolen SLW, van Waterschoot RAB, van Tellingen O, et al. From mouse to man: predictions of human pharmacokinetics of orally administered docetaxel from preclinical studies. J Clin Pharmacol. 2012;52(3):370‐380. 10.1177/0091270010397051 [DOI] [PubMed] [Google Scholar]
- 26. Gehart H, Clevers H. Tales from the crypt: new insights into intestinal stem cells. Nat Rev Gastroenterol Hepatol. 2019;16(1):19‐34. 10.1038/s41575-018-0081-y [DOI] [PubMed] [Google Scholar]
- 27. Allan SG, Smyth JF. Small intestinal mucosal toxicity of cis‐platinum–comparison of toxicity with platinum analogues and dexamethasone. Br J Cancer. 1986;53(3):355‐360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Miyoshi J, Miyamoto H, Goji T, et al. Serum diamine oxidase activity as a predictor of gastrointestinal toxicity and malnutrition due to anticancer drugs. J Gastroenterol Hepatol. 2015;30(11):1582‐1590. 10.1111/jgh.13004 [DOI] [PubMed] [Google Scholar]
- 29. Gridelli C, Gallo C, Di Maio M, et al. A randomised clinical trial of two docetaxel regimens (weekly vs 3 week) in the second‐line treatment of non‐small‐cell lung cancer. The DISTAL 01 study. Br J Cancer. 2004;91(12):1996‐2004. 10.1038/sj.bjc.6602241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kellokumpu‐Lehtinen P‐L, Harmenberg U, Joensuu T. 2‐Weekly versus 3‐weekly docetaxel to treat castration‐resistant advanced prostate cancer: a randomised, phase 3 trial. Lancet Oncol. 2013;2045(12):1‐8. 10.1016/S1470-2045(12)70537-5 [DOI] [PubMed] [Google Scholar]
- 31. Lilenbaum RC, Schwartz MA, Seigel L, et al. Phase II trial of weekly docetaxel in second‐line therapy for nonsmall cell lung carcinoma. Cancer. 2001;92(8):2158‐2163. [DOI] [PubMed] [Google Scholar]
- 32. Mey U, Gorschluter M, Ziske C, Kleinschmidt R, Glasmacher A, Schmidt‐Wolf, IGH . Weekly docetaxel in patients with pretreated metastatic breast cancer: a phase II trial. Anticancer Drugs. 2003;14(3):233‐238. 10.1097/01.cad.0000060631.27490.fe [DOI] [PubMed] [Google Scholar]
- 33. O’Shaughnessy J, Miles D, Vukelja S, et al. Superior survival with capecitabine plus docetaxel combination therapy in anthracycline‐pretreated patients with advanced breast cancer: phase III trial results. J Clin Oncol. 2002;20(12):2812‐2823. 10.1200/JCO.2002.09.002 [DOI] [PubMed] [Google Scholar]
- 34. Pacilio CM, Morabito A, Nuzzo A, et al. Is epirubicin effective in first‐line chemotherapy of metastatic breast cancer (MBC) after an epirubicin‐containing adjuvant treatment? A single centre phase III trial. Br J Cancer. 2006;94(9):1233‐1236. 10.1038/sj.bjc.6603096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rugo HS, Topeck AT, Joy AA, et al. Randomized, placebo‐controlled, double‐blind, phase II study of axitinib plus docetaxel versus docetaxel plus placebo in patients with metastatic breast cancer. J Clin Oncol. 2011;29(18):2459‐2465. 10.1200/JCO.2010.31.2975 [DOI] [PubMed] [Google Scholar]
- 36. Serke M, Schoenfeld N, Loddenkemper R. Weekly docetaxel as second‐line chemotherapy in advanced non‐small cell lung cancer: phase II trial. Anticancer Res. 2004;24(2C):1211‐1216. [PubMed] [Google Scholar]
- 37. Stemmler HJ, Harbeck N, Groll de Rivera I, et al. Prospective multicenter randomized phase III study of weekly versus standard docetaxel (D2) for first‐line treatment of metastatic breast cancer. Oncology. 2011;79(3–4):197‐203. 10.1159/000320640 [DOI] [PubMed] [Google Scholar]
- 38. Baker SD, Zhao M, Lee CKK, et al. Comparative pharmacokinetics of weekly and every‐three‐weeks docetaxel. Clin Cancer Res. 2004;10(6):1976‐1983. 10.1158/1078-0432.CCR-0842-03 [DOI] [PubMed] [Google Scholar]
- 39. Bruno R, Hille D, Riva, A , et al. Population pharmacokinetics/pharmacodynamics of docetaxel in phase II studies in patients with cancer. J Clin Oncol. 1998;16(1):187‐196. [DOI] [PubMed] [Google Scholar]
- 40. Wu Z, Han X, Qin S, et al. Interleukin 1 receptor antagonist reduces lethality and intestinal toxicity of 5‐Fluorouracil in a mouse mucositis model. Biomed Pharmacother. 2011;65(5):339‐344. 10.1016/j.biopha.2011.04.013 [DOI] [PubMed] [Google Scholar]
- 41. Gibson RJ, Keefe DMK. Cancer chemotherapy‐induced diarrhoea and constipation: mechanisms of damage and prevention strategies. Support Care Cancer. 2006;14(9):890‐900. 10.1007/s00520-006-0040-y [DOI] [PubMed] [Google Scholar]
- 42. Sonis ST. The pathobiology of mucositis. Nat Rev Cancer. 2004;4(4):277‐284. 10.1038/nrc1318 [DOI] [PubMed] [Google Scholar]
- 43. Sultani M, Stringer AM, Bowen JM, Gibson RJ. Anti‐inflammatory cytokines: important immunoregulatory factors contributing to chemotherapy‐induced gastrointestinal mucositis. Chemother Res Pract. 2012;2012:490804 10.1155/2012/490804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ibrahim NK, Sahin AA, Dubrow RA, et al. Colitis associated with docetaxel‐based chemotherapy in patients with metastatic breast cancer. Lancet. 2000;355(9200):281‐283. 10.1016/S0140-6736(99)06195-4 [DOI] [PubMed] [Google Scholar]
- 45. Stemmler HJ, Kenngott S, Diepolder H, Heinemann V. Gastrointestinal toxicity associated with weekly docetaxel treatment. Ann Oncol. 2002;13(6):978‐981. 10.1093/annonc/mdf084 [DOI] [PubMed] [Google Scholar]
- 46. Bode H, Lenzner L, Kraemer OH, et al. The HIV protease inhibitors saquinavir, ritonavir, and nelfinavir induce apoptosis and decrease barrier function in human intestinal epithelial cells. Antiviral Ther. 2005;10(5):645‐655. [PubMed] [Google Scholar]
- 47. MacArthur RD, DuPont HL. Etiology and pharmacologic management of noninfectious diarrhea in HIV‐infected individuals in the highly active antiretroviral therapy era. Clin Infect Dis. 2012;55(6):860‐867. 10.1093/cid/cis544 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.