

Abstract
Background & Aims:
Advanced pancreatic ductal adenocarcinoma (PDAC) is resistant to therapy, including immune checkpoint inhibitors. We evaluated the effects of a neutralizing antibody against programmed cell death 1 (PDCD1, also called PD1) and an agonist of OX40 (provides a survival signal to activated T cells) in mice with pancreatic tumors.
Methods:
We performed studies in C57BL/6 mice (controls), KrasG12D/+;Trp53R172H/+;Pdx-1-Cre (KPC) mice, and mice with orthotopic tumors grown from Panc02 cells, KrasG12D;P53flox/flox;PDX-1-Cre;Luciferase (KPC-Luc) cells, or mT4 cells. After tumors developed, mice were given injections of control antibody or anti-OX40 and/or anti-PD1 antibody. Some mice were then given injections of antibodies against CD8, CD4, or NK1.1 to deplete immune cells, and IL4 or IL7RA to block cytokine signaling. Bioluminescence imaging was used to monitor tumor growth. Tumor tissues collected and single-cell suspensions were analyzed by time of flight mass spectrometry analysis. Mice that were tumor-free 100 days after implantation of orthotopic tumors were rechallenged with PDAC cells (KPC-Luc or mT4) and survival was measured. Median levels of PD1 and OX40 mRNAs in PDACs were determined from the Cancer Genome Atlas and compared with patient survival times.
Results:
In mice with orthotopic tumors, all those given control antibody or anti-PD1 died within 50 days, whereas 43% of mice given anti-OX40 survived for 225 days; almost 100% of mice given the combination of anti-PD1 and anti-OX40 survived for 225 days, and tumors were no longer detected. KPC mice given control antibody, anti-PD1, or anti-OX40 had median survival times of 50 days or less, whereas mice given the combination of anti-PD1 and anti-OX40 survived for a median 88 days. Mice with orthotopic tumors that were given the combination of anti-PD1 and anti-OX40 and survived 100 days were rechallenged with a second tumor; those re-challenged with mT4 cells survived an additional median 70 days and those re-challenged with KPC-Luc cells survived long term, tumor free. The combination of anti-PD1 and anti-OX40 did not slow tumor growth in mice with antibody-mediated depletion of CD4+ T cells. Mice with orthotopic tumors given the combination of anti-PD1 and anti-OX40 that survived after complete tumor rejection were re-challenged with KPC-Luc cells; those with depletion of CD4+ T cells before the re-challenge had uncontrolled tumor growth. Furthermore, KPC orthotopic tumors from mice given the combination contained an increased number of CD4+ T cells that expressed CD127, compared with mice given control antibody. The combination of agents reduced the proportion of T-regulatory and exhausted T cells and decreased T-cell expression of GATA3; tumor size was negatively associated with numbers of infiltrating CD4+ T cells, CD4+CD127+ T cells, and CD8+CD127+ T cells and positively associated with numbers of CD4+PD1+ T cells, CD4+CD25+ T cells, and CD8+PD1+ T cells. PDACs with high levels of OX40 and low levels of PD1 were associated with longer survival times of patients.
Conclusions:
Pancreatic tumors appear to evade the immune response by inducing development of immune-suppressive T cells. In mice, the combination of anti-PD1 inhibitory and anti-OX40 agonist antibodies reduces the proportion of T-regulatory and exhausted T cells in pancreatic tumors and increases numbers of memory CD4+ and CD8+ T cells, eradicating all detectable tumor. This information might be used in development of immune-based combination therapies for PDAC.
Keywords: CyTOF, immune checkpoint inhibitor, immune-based therapy, mouse model
Graphical Abstract
Lay Summary:
In studies of mice, the authors identified mechanisms by which pancreatic ductal adenocarcinomas avoid attack by the immune system and agents that might increase the anti-tumor immune response.
Introduction
Pancreatic ductal adenocarcinoma (PDAC), comprising 95% of pancreatic cancers, is notorious for its resistance to virtually all therapeutic modalities. Recent advances in cancer immunotherapy using antibodies that block programmed cell death protein 1 (PD-1), its ligand (PD-L1), and cytotoxic T-lymphocyte antigen 4 (CTLA-4) pathways1 can lead to remarkable clinical responses in patients with tumors such as melanoma that are otherwise refractory to conventional treatment. However, as a cancer type with relatively modest tumor mutational burden2 and few infiltrating effector T cells, PDAC has been recognized as an immune-privileged3 or “cold” tumor, and the use of immune checkpoint monotherapy has failed to induce meaningful clinical responses2. This immune privilege has been attributed to multiple factors4, including intratumoral heterogeneity5 of PDAC stroma; oncogenic KRAS signaling, believed to mediate immune evasion6; the presence of T-cell subsets (Th2 cells7 and regulatory T [Treg] cells8) with protumor activity; the absence of infiltrating immune effectors9; and the local effect of tumor microenvironment (TME)–associated immunosuppressive cytokines10, 11.
To address some of these obstacles, we postulated that a combination of immune costimulation–based strategies would substantially increase the tumoricidal effect against PDAC. The two treatment antibodies we used are fundamentally different in their mechanisms of action: anti–PD-1 is an antagonist antibody that blocks PD-1, and anti-OX40 is an agonist antibody that stimulates OX40, which engages intracellular mediators in the tumor necrosis factor receptor superfamily and initiates a survival signal in T cells in the context of T-cell receptor activation12.
To evaluate these combinations in an in vivo model system, we chose to use both a syngeneic orthotopic PDAC model and a genetically engineered autochthonous mouse model (GEMM) since these models represent a rigorous challenge by nonimmunogenic cancers and mimic the human pancreatic tumor microenvironment with high fidelity13. Spontaneous tumor developing in GEMM14 histologically resembles human PDAC tumors and, like locally advanced or metastatic PDAC in patients, is largely resistant to almost all therapies.
In both PDAC models, we evaluated different regimens comprised of immune costimulatory or co-inhibitory antibodies that were pharmaceutically available at the time, including anti–CTLA-4, anti–PD-1, anti–PD-L1, anti–4–1BB, and anti-OX40, and found the combination of antibodies against OX40 and PD-1 to be uniquely effective in inducing durable tumor regression. This long-lasting effect on tumor control was underlined by the models’ resilience to rechallenge with a second, independently derived, histologically aggressive, syngeneic PDAC line. Exploration of the immune mechanisms of this combination approach revealed a unique confluence of events leading to tumor eradication and maintenance of robust immunologic memory.
Materials and Methods
Orthotopic tumor model and treatment with antibodies against immune costimulatory and coinhibitory molecules.
All animal procedures, along with justification of the selected strains for experimental arms and the statistical justification of the number of mice to be used and randomized, were reviewed and approved by the MD Anderson Cancer Center Animal Care and Use Committee.
Male C57BL/6, or albino, Rag2 knockout mice were obtained from The Jackson Laboratory. As a congenic strain of C57BL/6 mice, C57BL/6 albino mice (B6(Cg)-Tyrc−2J/J), with homozygous Tyrc−2J, have an albino coat that improves the transmission of light and allows detection at the level of 10 cells15 in IVIS bioluminescence imaging, facilitating in vivo noninvasive, longitudinal monitoring of tumor growth and quantitation of microscopic tumor burden. On day 1, mouse PDAC cells (Panc02, KrasG12D;P53flox/flox;PDX-1-Cre;Luciferase [KPC-Luc]10, and mT416) were surgically implanted into the pancreas as described previously17.
One week after tumors were established in vivo, treatment was begun with antibodies of the polyclonal rat immunoglobulin G isotype control, anti-OX40 monotherapy, anti–PD-1 monotherapy, or the combination of anti–PD-1 and anti-OX40. IVIS bioluminescence imaging was used to monitor tumor growth weekly. On days 8–9 after the treatments started, we collected tumor tissues and prepared single-cell suspensions for time-of-flight mass spectrometry (CyTOF) analysis.
Mice were treated with 4 mg/kg body weight of anti–PD-1 or anti-OX40 on days 7, 10, 13, 19, 22, and 25, intraperitoneally. Antibody sources are listed in Table S1.
GEMM.
The KrasG12D/+;Trp53R172H/+;Pdx-1-Cre genetically engineered mouse model (KPCGEMM)13, 18 was used for treatment evaluation using the following breeding strategy: LSLKrasG12D (strain #01XJ6) mice were crossed with mutated LSL-p53R172H mice (strain #01XM2) and pancreas-specific Cre mice (PDX-1-Cre, strain #01XL5) to yield mice that possessed a conditional p53 mutation and endogenous levels of mutant KrasG12D expressed by pancreatic tissue.
Spontanously developed PDAC was diagnosed by a palpable solid tumor in the left peritoneal cavity, which was verified by MRI scan. When the mice presented with hunched posture, hyperpnea or dyspnea, ascites, or lethargy, they were euthanized, and the survival date was recorded.
The Cancer Genome Atlas RNA expression and survival analysis.
The median PD-1 and OX40 expression values of 179 RNA samples from The Cancer Genome Atlas were used as cutoffs to classify the samples into low- and high-expression groups for survival analysis.
Immune memory assay.
After more than 100 days following primary orthotopic tumor implantation (KPC, KPC-Luc, or Panc02-Luc), mice that had survived treatment with anti-OX40, with or without anti–PD-1, were confirmed to be tumor free; they were then rechallenged with a mouse PDAC line (KPC-Luc or mT4). These rechallenged mice received no further treatment. Naïve age-matched mice (de novo mice), which had not received primary tumor implantation, were injected with the same number of rechallenge cells, then divided into four groups and treated. All tumor-bearing mice were monitored for survival.
Antibody-mediated assay of CD4, CD8, and natural killer cell depletion; IL-4 neutralization; and IL-7Rα blocking.
Antibodies against CD8, CD4 antibody, NK1.1, IL-4, IL-7Rα, or control isotype were injected into the mice (250 μg in 100 μl phosphate-buffered saline, intraperitoneally) twice weekly for 2 weeks beginning on day 6 (one day before treatment). Antibody sources are listed in Table S1.
CyTOF19.
A single-cell suspension from each sample was counted, and 1.5 M cells per sample were stained in a cocktail of antibodies against surface markers, followed by staining with cisplatin. After barcoding with a Cell-ID 20-Plex barcoding kit, 20 samples were mixed and stained with a cocktail of antibodies against intracellular or transcription markers. Finally, the samples were stained with Ir-intercalator and resuspended in 0.1% bovine serum albumin in Milli-Q water for a Helios mass cytometer analysis.
MATLAB for t-distributed stochastic neighbor embedding (tSNE) plot, heat map, and plot cluster tSNE.
To visualize the CyTOF data, we ran Cyt in MATLAB to transform high-dimensional cytometry data onto a two-dimensional tSNE scatter plot. We then ran PhenoGraph to meta-cluster the gates and channels and visualized the clusters using heat maps.
Statistical analysis.
Statistical differences between the control and experimental groups were determined using the unpaired two-tailed Student t-test; a one-way analysis of variance, followed by Fisher least significant difference multiple comparison tests; or a two-way analysis of variance. Normality tests, along with variations within and between groups, were performed before the significance of differences was evaluated. The relationship between gene expression and tumor size was determined by Pearson correlation (GraphPad Prism V6 and SPSS for Mac; IBM). Survival curves were constructed using the Kaplan-Meier method, and statistical significance was determined using the log-rank test. P values less than 0.05 were considered statistically significant. Representative experiments are shown; each experiment was repeated independently at least three times.
Results
Combination therapy with anti–PD-1 and anti-OX40 eradicates tumor and extends overall survival.
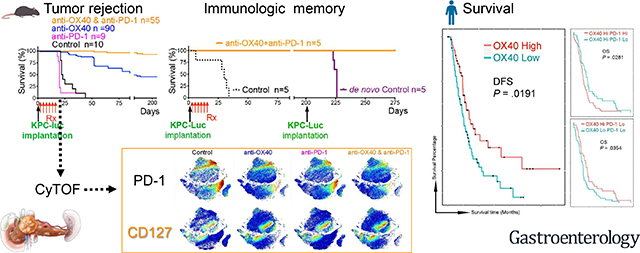
To explore the anti-tumor potential of combination immunotherapy in PDAC, we first used a KPC-Luc syngeneic orthotopic model (Fig. 1A) to test the efficacy of the agonist costimulatory antibody anti-OX40 and the checkpoint inhibitory antibody anti–PD-1 individually and in combination. We observed distinct patterns of efficacy (Fig. 1B–E): a partial response to anti-OX40 monotherapy (3 responders and 2 non-responders in Fig. 1C), a complete response to the combination of anti–PD-1 and anti-OX40 (all 5 were responders in Fig. 1E), and for control and anti-PD-1 monotherapy groups, progressive disease with death and significant tumor burden requiring euthanization within 50 days after tumor implantation (Fig. 1B and D).
Figure 1. Combination of OX40 agonist and PD-1 antagonist eradicates PDAC in tumor-bearing mice.
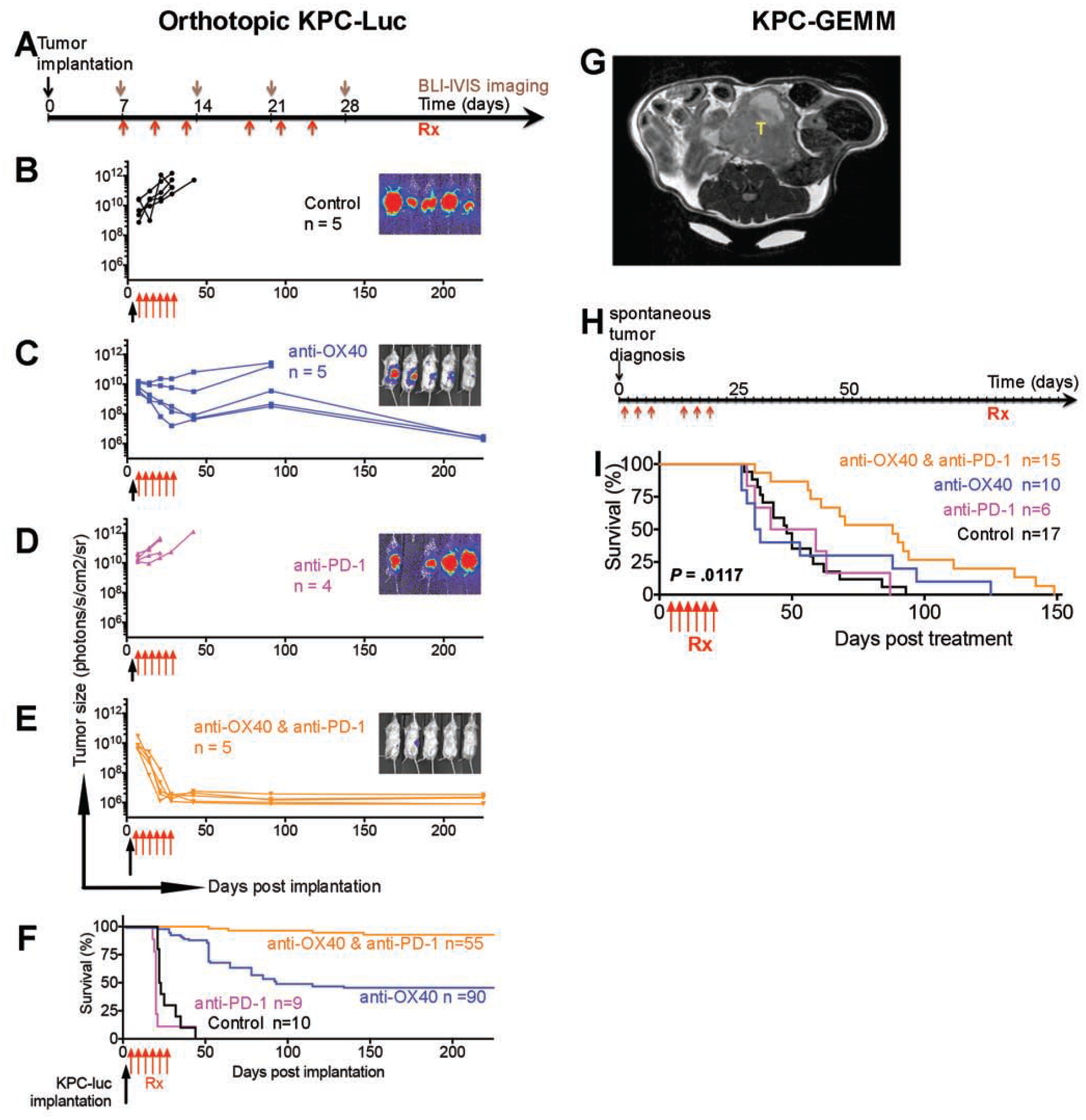
Tumor size was measured by photons/sec/cm2/sr in the IVIS bioluminescence imaging; the normalized image at day 28 is on the right of each growth curve. Color-matched curves were applied to groups on all panels.
(A) Tumor implantation and treatment schedule for KPC-Luc syngeneic orthotopic tumor models.
(B-D) Tumor growth in mice treated with isotype control (B, black), anti-OX40 (C, blue), and anti–PD-1 (D, magenta).
(E) Tumor shrinkage in mice treated with a combination of anti–PD-1 and anti-OX40 (orange). The eliminated tumors were visualized by trace luciferase signals. P values from two-way analysis of variance (ANOVA) were 0.0001 for anti-OX40 vs. control, 0.9542 for anti–PD-1 vs. control, <0.0001 for combination vs. control, 0.0006 for anti–PD-1 vs. anti-OX40, <0.0001 for combination vs. anti-OX40, and <0.0001 for combination vs. anti–PD-1.
(F) Survival curve of tumor-bearing mice after orthotopic tumor implantation. Median survival durations of mice treated with control, anti-OX40, anti–PD-1, and combinaton were 22.5 days, 93 days, 20 days, and more than 225 days after treatment, respectively (P < 0.0001).
(G) MRI diagnosis of a representative spontaneous PDAC tumors (T).
(H) Treatment schedule of spontaneous PDAC tumors in KPC-GEMM mice.
(I) Survival curve of tumor-bearing GEMM mice. Treatments were started when the solid tumor was palpable. Median survival times of mice treated with control, anti-OX40, anti–PD-1, and combination were 48 days, 37 days, 50.5 days, and 88 days after treatment, respectively (log-rank test, P = 0.0117). Significant P values between groups were 0.0011 for combination vs. control, 0.0102 for combination vs. anti–PD-1, and 0.0494 for combination vs. anti-OX40.
The long-term (225 days) tumor-free survival rate was 43% for mice given anti-OX40 monotherapy (Fig. 1C and F) in the KPC-Luc orthotopic model and almost 100% for mice given the combination of anti–PD-1 and anti-OX40 (Fig. 1E and F). The same experiments performed in a Panc02 syngeneic orthotopic model yielded similar results (Fig. S1). By 225 days of follow-up, responses after anti-OX40 monotherapy or after the combination of anti–PD-1 and anti-OX40 were also durable; these mice survived as long as healthy mice would (Fig. 1F).
Next, we validated the survival rate of mice receiving anti-OX40 monotherapy and the combination of anti–PD-1 and anti-OX40 in the spontaneous KPC-GEMM (LSLKrasG12D/+;LSL-p53R172H/+;PDX-1-Cre)18, which recapitulates the complexity of the TME in human PDAC18. The tumor-bearing KPC-GEMM (Fig. 1G) were treated at the same dosage and interval (Fig. 1H) as were used in the orthotopic models (Fig. 1A). As expected, the combination of anti–PD-1 and anti-OX40 significantly increased median survival compared with the control, anti–PD-1 monotherapy, or anti-OX40 monotherapy (88 days vs. 48, 37, or 50.5 days, respectively, P = 0.0117, Fig. 1H–I). No KPC-GEMM experienced tumor-free survival, which underscores the implication of TME in affecting the efficacy of antibody-mediated immunotherapy.
Combination therapy with anti–PD-1 and anti-OX40 results in potent anti-tumor immune memory and secondary prevention.
We were able to produce long-term tumor-free survival in the orthotopic models treated with anti-OX40 alone or in combination with anti–PD-1. We therefore hypothesized that anti-OX40, with or without anti–PD-1, can produce a durable anti-tumor immune response and potent anti-tumor immune memory.
For the orthotopic models that experienced a complete response to anti-OX40 (with or without anti–PD-1), we confirmed the disappearance of tumor using IVIS bioluminescence imaging at >100 days after the first tumor implantation. We then rechallenged these mice with a second implantation. The rechallenged mice received no additional therapy (Fig. 2 and Fig. S2). The survival curves of these mice were compared with those of age-matched C57BL/6 mice that received de novo tumor cell implantation and antibody treatment (isotype control, anti-OX40 monotherapy, anti–PD-1 monotherapy, or combination therapy with anti–PD-1 and anti-OX40; Fig. 2A–B).
Figure 2: Surviving mice after anti-OX40 and anti-PD-1 immunotherapy reject rechallenged tumors.
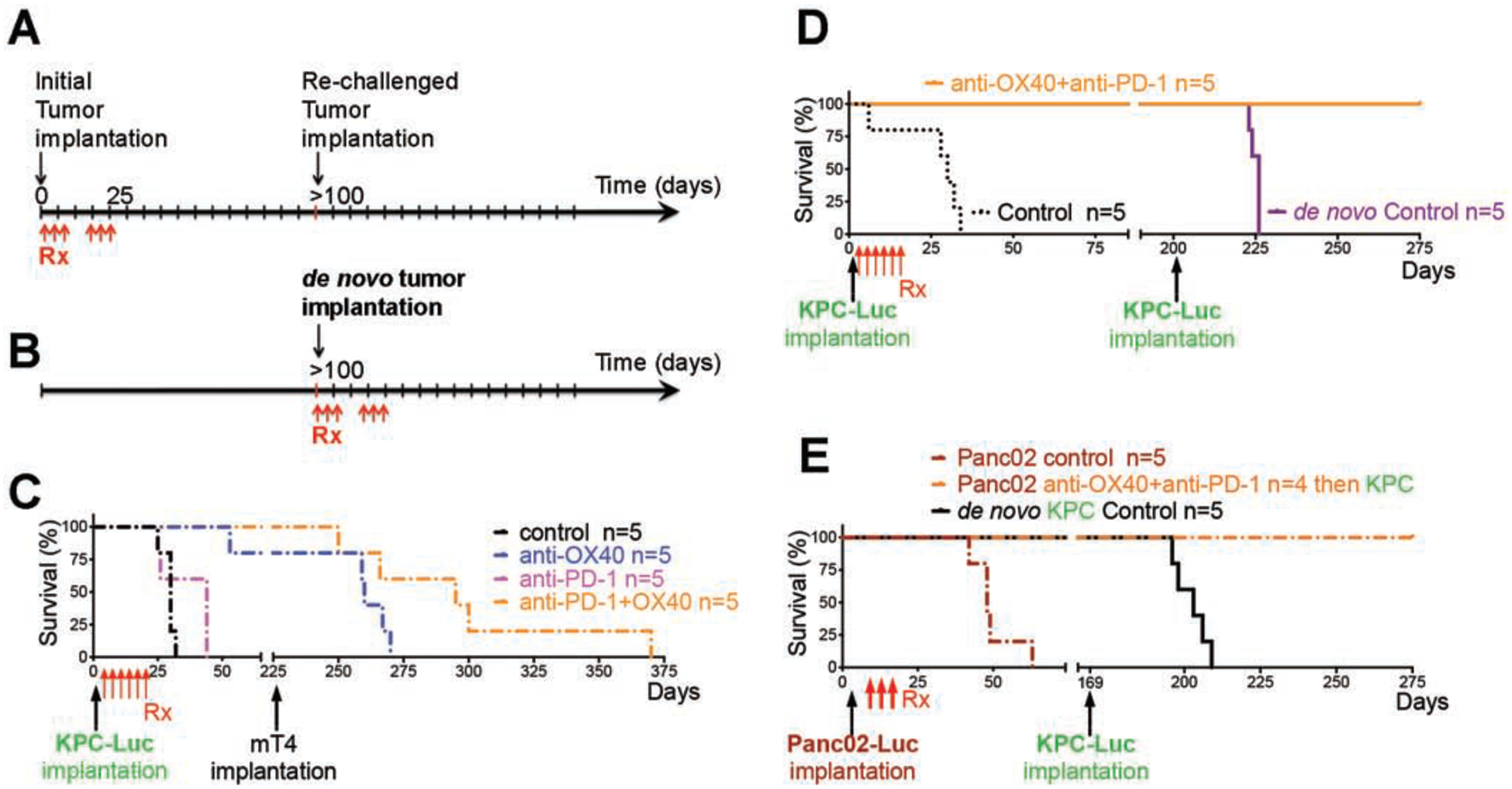
(A) Initial PDAC cells were orthotopically implanted followed by immunotherapy. The surviving mice given anti-OX40 or the combination of anti-OX40 and anti-PD-1 immunotherapies were rechallenged with a second implantation of syngeneic PDAC cells from either the same cell line as the first or a different cell line.
(B) Age-matched C57BL/6 albino mice received de novo mT4 cell implantation.
(C) Survival of mT4-rechallenged mice in the absence of further immunotherapy. The control and anti–PD-1–treated mice, which died before possible mT4 rechallenge, are included for comparison.
(D-E) Survival of KPC-Luc–rechallenged mice in the absence of further immunotherapy. KPC-Luc (D) or Panc02-Luc (E) cells were used as the initial tumor. Control mice (dash-dotted line), which died before possible KPC-Luc rechallenge, and de novo KPC-Luc mice (solid line), which died within 50 days after implantation, are included for comparison.
mT4 is an independently derived, syngeneic KPC PDAC cell line that is highly metastatic and aggressive16. The mT4-rechallenged mice that had received anti-OX40 monotherapy (Fig. 2C) survived as long as the de novo mice did after combination therapy (Fig. 2D). The rechallenged mice that had received combination therapy with anti–PD-1 and anti-OX40 experienced exceptionally long survival following secondary implantation with mT4, with a median survival of 295 days after initial implantation or 70 days after rechallenge (Fig. 2C), in contrast with 263.5 days after initial implantation or 38.5 days after rechallenge in the rechallenged mice that had received anti-OX40 monotherapy (Fig. 2C), 16 days in the control-treated de novo mice, and 42 days in the combination-treated de novo mice (Fig. S2D).
Of note, the rechallenge KPC-Luc cells were incapable of establishing a tumor mass in mice previously cured of either KPC-Luc (Fig. 2E and Fig. S2A and C–D) or Panc02-Luc (Fig. 2F and Fig. S2B) tumors. Furthermore, T cells from KPC-Luc tumors yielded higher T-cell cross-reactivity to shared pancreatic tumor antigens in KPC or Panc02 than to mT4 (Fig. S2A–B and E–F). Taken together, these results indicate that rechallenge tumors can be eliminated by rapid immunologic recall from memory T cells elicited by previous anti–PD-1 and anti-OX40 therapy.
Antitumor immune response derived from anti-OX40 agonist and PD-1 blockade is wholly CD4 T cell dependent, partially CD8 T cell dependent, and natural killer cell independent.
To distinguish the essential immune phenotype from the non-essential inflammatory infiltration associated with anti-OX40 and anti–PD-1 treatment, we systematically depleted CD4, CD8, or natural killer cell populations after administering treatment regimens in the KPC-Luc model (Fig. 3A–E and Fig. S3). In the settings of anti-OX40 monotherapy (Fig. 3F) and the combination of anti-OX40 and anti–PD-1 (Fig. 3G), depletion of CD4 (Fig. 3C) completely abolished the antitumor effect, which indicates that CD4 cells dictate the efficacy of the antitumor immune response. Compared with the control, depletion of CD8 (Fig. 3D) partially interfered with the antitumor response to the treatments, suggesting that CD8 T cells play a role that is contributory but clearly more limited than that of CD4 cells. As expected, an mT4-Luc model confirmed that CD4 T cells were more important than CD8 T cells in the antitumor immune response elicited by anti-OX40 and anti–PD-1 (Fig. S4). Moreover, NK1.1 antibody depletion had no effect on tumor growth following treatment, suggesting that NK1.1-expressing cells are not necessary for the antitumor immune response in these regimens (Fig. S3), a finding that is consistent with the uniformly low frequency of NK1.1-expressing cell populations in the tumor-infiltrating heat map (cf. cluster 22 in Fig. 4F).
Figure 3. CD4 cells are required for maximal tumor elimination and immunologic memory after the treatment with anti-OX40 and anti–PD-1.
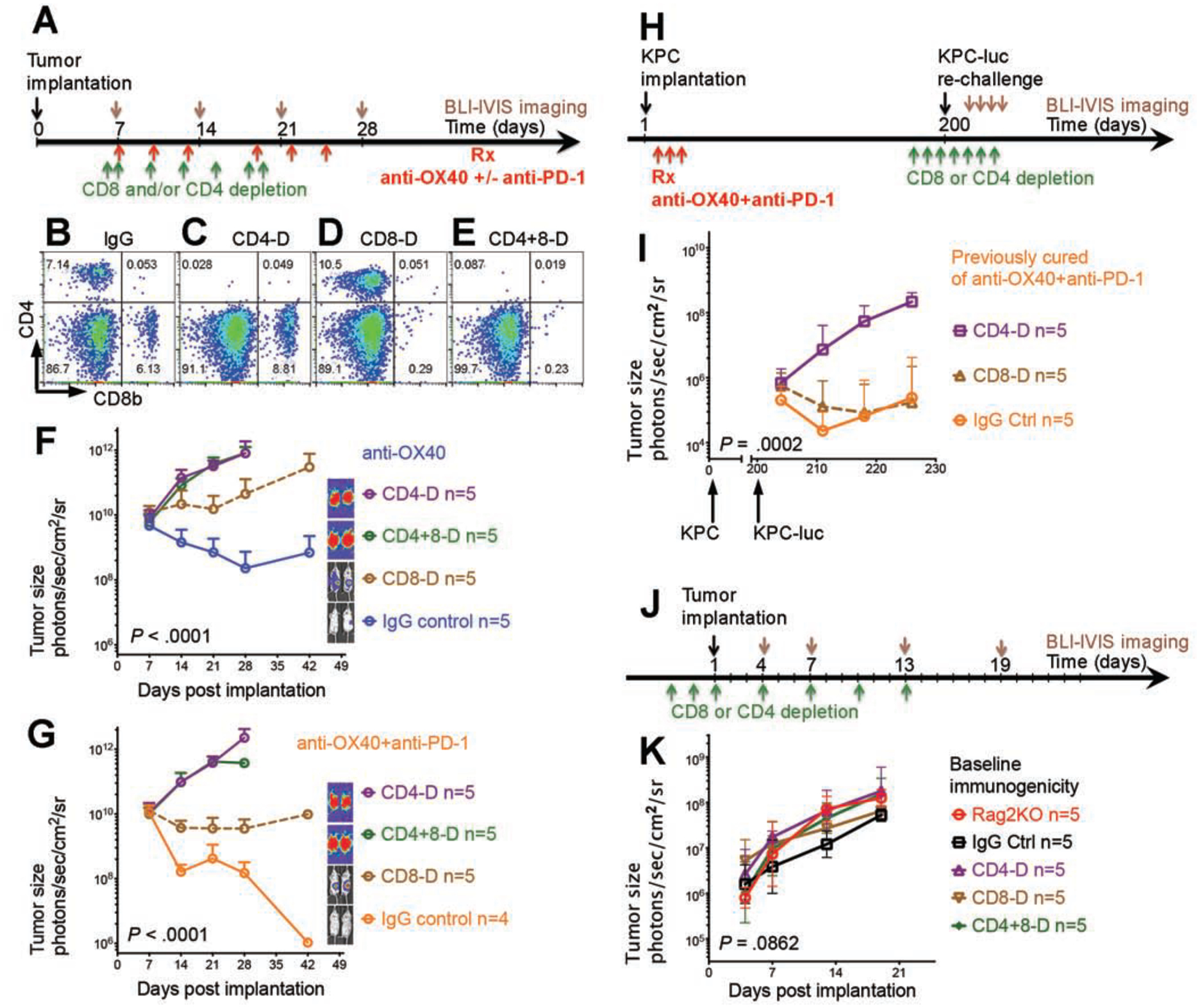
(A, H, J) Schedule of tumor implantation and injection of antibodies for cell depletion in mice receiving treatment only (A), in mice rechallenged after treatment (H), and in mice monitored for the baseline immunogenicity of KPC-Luc tumor (J).
(B–E) Depletion of CD4 (CD4-D) (C), CD8 (CD8-D) (D), or double depletion of CD4 and CD8 (CD4+8-D) (E) was confirmed by flow cytometry, and no cells were depleted after control immunoglobulin G (IgG) antibody injection (B).
(F–G) The antitumor effect was completely abolished after depletion of CD4 cells with or without depletion of CD8 cells; however, the antitumor effect was only partially reduced by depletion of CD8 cells. IVIS bioluminescence images of tumor size in mice are shown on the right in the setting of anti-OX40 monotherapy (F) and combination therapy with anti-OX40 and anti–PD-1 (G).
(I) In mice previously cured by combination therapy with anti-OX40 and anti–PD-1, the immunologic memory effect was completely abolished after depletion of CD4 cells; however, the immunologic memory effect was intact after depletion of CD8 cells.
(K) KPC-Luc tumor exhibited similar growth curves in Rag2 knockout, CD4- and/or CD8-depleted, and immunocompetent control mice.
Figure 4. Identification of tumor-infiltrating T lymphocytes by anti-OX40 or anti–PD-1 by CyTOF analysis.
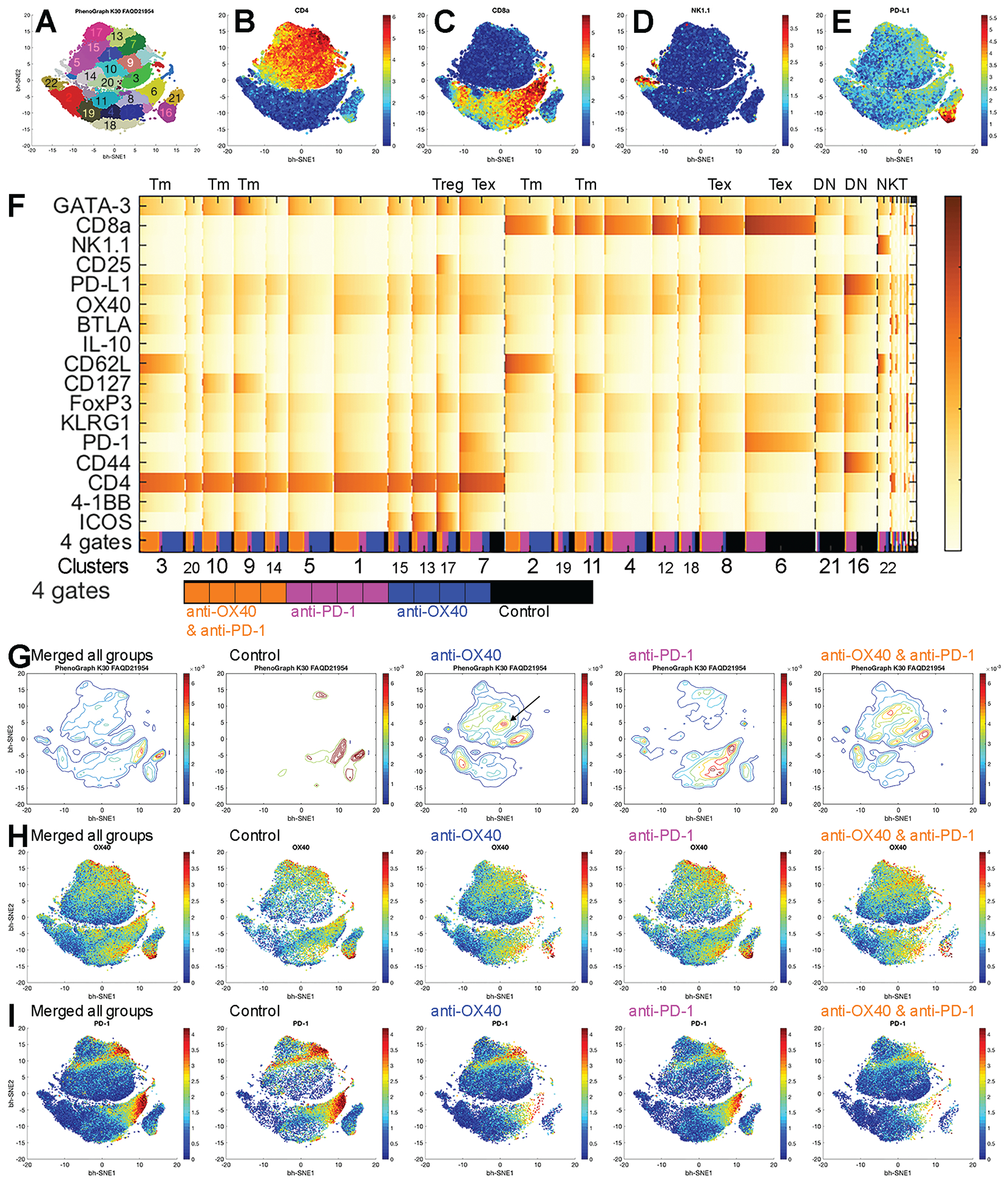
(A-E) tSNE plots of all T cells (A) with expression levels of CD4 (B), CD8 (C), NK1.1 (D), and PD-L1 (E) from all samples with number- and color-coded clusters.
(F) Heat map of normalized marker expression of T-cell clusters with the fractions of four gates. Gate source shows the pooled samples in each group at the bottom line of the heat map. Black: control, blue: anti-OX40 monotherapy, magenta: anti–PD-1 monotherapy, and orange: combination of anti–PD-1 and anti-OX40. Tm: memory T cells, Tex: PD-1 exhausted T cells, and DN: CD4 and CD8 double negative T cells.
(G-I) tSNE plots of all samples and control, anti-OX40, anti–PD-1, and anti–PD-1 and anti-OX40 groups.
(G) Density plots. Arrow in the anti-OX40 plot indicates GATA-3 CD4 cells.
(H) Plots of OX40 expression.
(I) Plots of PD-1 expression.
To determine whether immunologic memory was conveyed by CD4 cells or by CD8 cells, we depleted CD4 or CD8 cells in mice previously cured of a KPC tumor challenge by anti-OX40 and anti–PD-1 therapy (Fig. 3H), and then we rechallenged the mice with the a KPC-Luc tumor 200 days later. Remarkably, we found that CD4 depletion again abolished the antitumor effect while CD8 depletion did not interfered, suggesting that CD4 cells were required to mediate immunologic memory, but CD8 cells were redundant in the memory recall response (Fig. 3I and Fig. S5) to this poorly immunogenic tumor (Fig. 3J–K and Fig. S6). These data indicate that the CD4 population is essential in orchestrating tumor elimination and antitumor memory after treatment with anti-OX40, alone or with anti–PD-1.
Anti-OX40 agonist or PD-1 blockade transforms the immune landscape of the TME.
To characterize the influence of various treatment regimens on the tumor immune landscape, we we resected tumors from syngeneic KPC tumor–bearing mice after treatment (Fig. S7) and performed CyTOF analysis on the tumor-infiltrating lymphocytes therein (Figs. 4, 5, and S8).
Figure 5. Frequency of identified subsets changed after treatments and was correlated with tumor size.
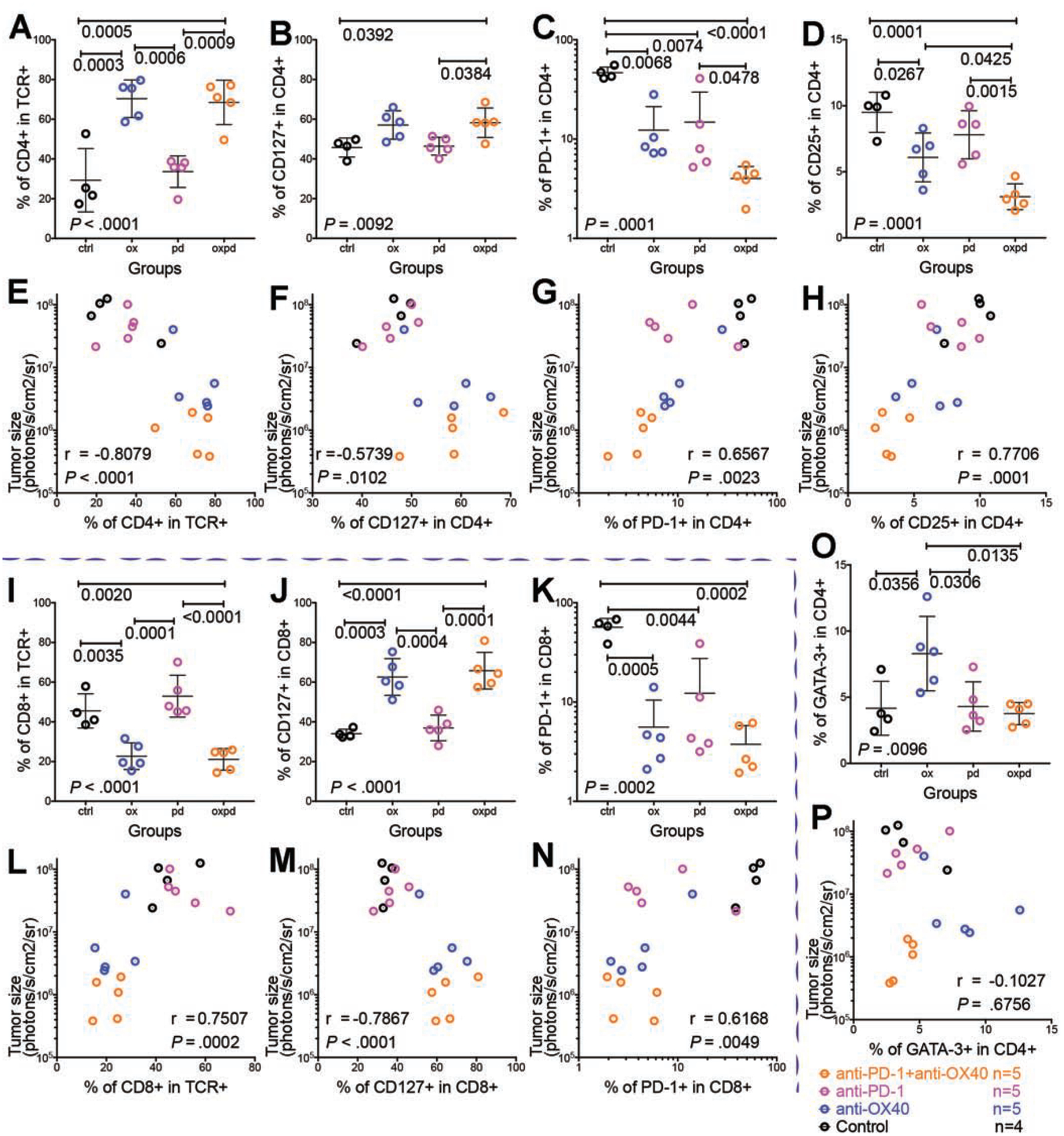
Shown are groups given isotype control (black, ctrl), anti-OX40 agonist monotherapy (blue, ox), anti–PD-1 blockade monotherapy (magenta, pd), and the combination of anti–PD-1 blockade and anti-OX40 agonist (orange, oxpd).
(A–H, O, P) Subsets in the CD4 population
(I-N) Subsets in the CD8 population.
(A) Increased fraction of CD4 T cells after anti-OX40 and combination treatment. TCR: T-cell receptor
(B) Increased CD127 memory subset in CD4 cells after combination treatment.
(C) Decreased PD-1 exhausted subset in CD4 cells after all treatments.
(D) Decreased CD25 Treg cell subsets in CD4 cells after anti-OX40 and combination treatment. (E, F, G, H) Correlations of CD4 T cell subsets and tumor size: CD4 (E), CD127 (F), PD-1 (G), and CD25 (H).
(I) Decreased fraction of CD8 T cells after anti-OX40 and combination treatment.
(J) Increased CD127 memory subset in CD8 cells after anti-OX40 and combination treatment. (K) Decreased PD-1 exhausted subset in CD8 cells after all treatments.
(L, M, N) Correlations of CD8 T cell subsets and tumor size: CD8 (L), CD127 (M), and PD-1 (N).
(O) Increased GATA-3 cell phenotype in CD4 cells after anti-OX40; this reversed to baseline after the addition of anti–PD-1.
(P) No correlation between GATA-3 cell phenotype and tumor size. P values are on the plots. Pearson r correlation was applied. Scattered dot plots are displayed with mean ± standard deviation.
We analyzed the immune signatures of OX40, OX40L, PD-1, and PD-L1 on T cells to generate plot cluster tSNE output (Fig. S9A–C) in individual tumor-bearing hosts on a convergent molecular landscape (Fig. S9D–O) with differential expression of the selected T-cell markers. Hosts in the control and anti–PD-1 monotherapy groups were segregated from those in the anti-OX40 monotherapy and combination anti-OX40 and anti–PD-1 groups (Fig. S9A). Expression level is presented on the basis of the intensity of the T-cell markers in each host. Compared with controls, hosts receiving anti-OX40, with or without anti–PD-1, exhibited low levels of CD8, NK1.1, OX40, OX40L, PD-1, PD-L1, CD44, Foxp3, CD25, and KLRG1 but high levels of CD4 and CD127. The only significant finding in hosts receiving anti–PD-1 monotherapy was lower PD-1 expression than that in the control group. These features suggest that anti-OX40 treatment alters the immune landscape in responders by expanding the T-cell memory population as defined by expression of CD127 in both CD4 and CD8 subsets.
tSNE plots (Fig. S8) were used to define alterations in population structure, and the PhenoGraph method was used to analyze cellular abundance and diversity with meta-clustering according to immune signatures and Euclidean distance matrix in the four treatment groups of mice. Of the 40 total clusters in the tSNE plot, clusters 1 to 22 (Fig. 4A) encompassed 96.4% of the total population (visualized in the heat map [Fig. 4F]); clusters 23 to 40 represented very low-frequency events. Positional clustering of T-cell subsets appeared as expected in tSNE plots: CD4 T cells (Fig. 4B) on the top, CD8 T cells (Fig. 4C) on the bottom, NK1.1-expressing cells (Fig. 4D) on the bottom left, and double-negative T cells on the bottom right (Fig. 4E), with a high PD-L1 phenotype. Heat maps (four conditions merged, Fig. 4F; individual conditions, Fig. S10) show the unsupervised classification of these immune sub-populations.
In mice receiving isotype control (Fig. S10A and Fig. 4F), the tumor-infiltrating immune cells uniformly presented with a PD-1 exhausted phenotype (Fig. 4G; Tex clusters, Fig. 4F). A terminally differentiated, immunosuppressive phenotype (PD-L1hi/CD44hi/IL-10hi) in T cells negative for both CD4 and CD8 (DN clusters, Fig. 4F) presented in significantly larger numbers in this control cohort than in any of the treated cohorts.
In mice receiving anti-OX40 monotherapy (Fig. S10B and Fig. 4F), the tumor-infiltrating lymphocyte population included higher levels of memory T cells than the control group did (CD62L with or without CD127) (Tm clusters, Fig. 4F). Only a small population of PD-1 exhausted cells was observed, suggesting that treatment with anti-OX40 agonist alone may not be sufficient to attain a complete response.
Mice receiving anti–PD-1 monotherapy (Fig. S10C and Fig. 4F) showed an expanded population of PD-1lo CD8 cells (compared with the control group) and a minor CD4 T cell population (Fig. 4G). PD-1 exhausted cells were abundant, but fewer than those in the control group (Fig. 4H). A subpopulation of ICOS, PD-1lo, and CD4 T cells were also present; although phenotypically consistent with an anti-tumor effector cell type20, their role in tumor rejection in this setting is unclear.
In mice receiving both anti-OX40 and anti–PD-1 (Fig. S10D and Fig. 4F), the tumor-infiltrating lymphocytes presented a similar phenotypic profile to those of the anti-OX40 monotherapy group when visualized by tSNE analysis and heat map. As with anti-OX40 monotherapy, CD4 T cells predominated, consistent with the significant contribution of CD4 T cells to the antitumor effect observed above (Fig. 3). Notable differences from the OX40 monotherapy group were an increase in OX40 cells and a near absence of PD-1 exhausted cells (Fig. 4H).
Altogether, this analysis of the immune landscape reveals that anti-OX40 or anti–PD-1 treatment transforms the immune-privileged PDAC TME into a favorable anti-tumor microenvironment characterized by CD4 dominance, increased T-cell memory, and decreased T-cell exhaustion.
PD-1 blockade augments the efficacy of anti-OX40 agonist through expansion of memory T cell population and elimination of Treg cells, GATA-3 cells, and exhausted T cells.
To better understand differences in the rates of response to anti-OX40 monotherapy vs. the combination of anti-OX40 and anti–PD-1 therapy (Fig. 1C and E), we performed an in-depth correlation analysis of tumor size and the subset frequency after each of the four treatments to identify the determinant subsets of the immune cell population associated with an anti-tumor immune response (Fig. 5).
Compared with the control, anti-OX40 monotherapy significantly increased the fraction of CD4 T cells (Fig. 5A), CD8 memory cells (Fig. 5J), and GATA-3 cells (Fig. 5O) and decreased the fraction of CD8 T cells (Fig. 5I), PD-1 exhausted cells (Fig. 5C and 5K), and CD25 Treg cells (Fig. 5D). Compared with anti-OX40 monotherapy, the addition of anti–PD-1 significantly promoted the anti-tumor effect of anti-OX40 by increasing the fraction of total CD4 T cells and memory CD8 cells and by further decreasing the fraction of Treg cells and exhausted T cells. Moreover, the combination therapy significantly increased the CD4 memory population (Fig. 5B) compared with that seen with anti-OX40 monotherapy. Surprisingly, the addition of anti–PD-1 also suppressed the growth of a population of GATA-3 cells that was augmented by anti-OX40 monotherapy (represented by a focused tSNE region in the anti-OX40 plot [arrow] in Fig. 4G and by a clear signal of GATA-3+CD4 cells in Fig. 5O).
Tumor size after treatment was negatively associated with infiltrating CD4 cells (Fig. 5E), CD4+CD127 cells (Fig. 5F), and CD8+CD127 cells (Fig. 5M) and positively associated with CD4+PD-1 cells (Fig. 5G), CD4+CD25 cells (Fig. 5H), CD8 cells (Fig. 5L), and CD8+PD-1 cells (Fig. 5N). In these correlation plots, the non-responders after anti-OX40 monotherapy (blue dots in Fig. 5E–H and L–N, but not 5P) were clustered with all the progressors in the control group and anti–PD-1 monotherapy group, but the responders after anti-OX40 monotherapy were clustered with all the responders in the combination group. Thus, treatment-induced anti-tumor response is associated with a distinct immunophenotypic TME profile.
In addition, comparisons of surface marker expression among individual cohors demonstrated a pattern of mutually exclusive expression of CD127 and PD-1 (Fig. S12), regardless of therapy. As a result of this dichotomous differentiation, CD127 T cells (Fig. S9I) were found primarily in the combination group, contributing to tumor elimination in responders, while PD-1 T cells (Fig. S9J) were detected mostly in the control group, contributing to the uncontrolled tumor growth in progressors.
CD127 (IL-7Rα) plays an important role in T-cell memory and homeostasis21. We were surprised to find dramatic expansion of CD127 cells commensurate with exceptional treatment response (Fig. 5F and M). Next, to find out whether CD127 was a determinant signal or an outcome of anti-tumor efficacy, we injected CD127 antibody in the KPC-Luc model (Fig. S11), blocking the binding of IL-721. In anti-OX40 monotherapy and the combination of anti-OX40 and anti–PD-1, blocking CD127 did not affect the initial anti-tumor response, but did impair tumor elimination at later stages (week 5, Fig. S11), which indicated that anti-OX40– and anti– PD-1–derived CD127 signaling is required for immune memory, sustaining the anti-tumor immune response and long-term tumor-free survival.
Overall, anti-OX40 agonist monotherapy showed a double-edged effect on immune response by inducing T-cell memory expansion, preventing T-cell exhaustion, and depleting Treg cells to improve anti-tumor cytotoxicity but also promoting a GATA-3+ cell subset that favors Th2-mediated tumor progression, leading to a 60% response rate. In contrast, the anti–PD-1 antagonist synergized with anti-OX40 in inducing T-cell memory expansion, preventing T-cell exhaustion, and depleting Treg cells but reversed the effect of anti-OX40 on GATA-3 signaling, leading to a 100% response rate. Therefore, co-administration of an anti–PD-1 antagonist, although ineffective as monotherapy, played an indispensable role in driving the anti-OX40– induced immune landscape towards optimal tumor elimination.
Upregulation of OX40 combined with downregulation of PD-1 in untreated PDAC patients are associated with a more favorable prognosis.
We postulated that the prognosis of PDAC patients exhibiting an endogenous baseline TME profile characterized by decreased PD-1 expression and increased OX40 expression would resemble the favorable milieu seen with the combination of anti–PD-1 and anti-OX40 treatment.
To test this hypothesis, we assessed the survival of PDAC patients according to mRNA expression levels of PD-1 and OX40 extracted from the Cancer Genome Atlas RNA sequencing database. The assumption that PD-1 and OX40 expression in these samples was derived from T cells, and not tumor cells present in the sample was validated using publicly available datasets. PD-1 and OX40 were not found to be expressed by tumor cells among samples collected in the Human Protein Atlas22; and in the BioGPS23 (Dataset: GeneAtlas MOE430, gcrma), PD-1 and OX40 mRNA expression was detectable only in lymphocytes and lymphoid tissues but not non-immune cells. Thus, regardless of the data source, mRNA expression levels of OX40 and PD-1 among tumor samples collected from pancreatic cancer patient reflects their states in the immune compartment and can be used to correlate T-cell phenotype with the disease progression. Patients with high OX40 expression had significantly longer disease-free survival than did those with low OX40 expression (Fig. 6B), but not longer overall survival (Fig. 6A). PD-1 level did not contribute to a difference in overall survival (Fig. 6C) or disease-free survival (Fig. 1D). Most notably, among patients with high OX40 expression, those with low PD-1 displayed statistically significantly longer overall survival than did those with high PD-1 (Fig. 6E), as well as elevated tails on the disease-free survival curve (Fig. 6F), although the median disease-free survival rates did not improve. Likewise, in patients with low PD-1 expression, those with high OX40 had significantly longer overall survival than did those with low OX40 (Fig. 6G), as well as elevated tails on the disease-free survival curve (Fig. 6H), but no significant improvement in median disease-free survival rates. These significant curve shifts and elevated survival tails provide prognostic (albeit circumstantial) validation of our results seen in multiple preclinical models and prospects for selecting patients in future clinical trials.
Figure 6. Patients’ prognosis is associated with the expression levels of OX40 and PD-1 in PDAC tumors in The Cancer Genome Atlas dataset.
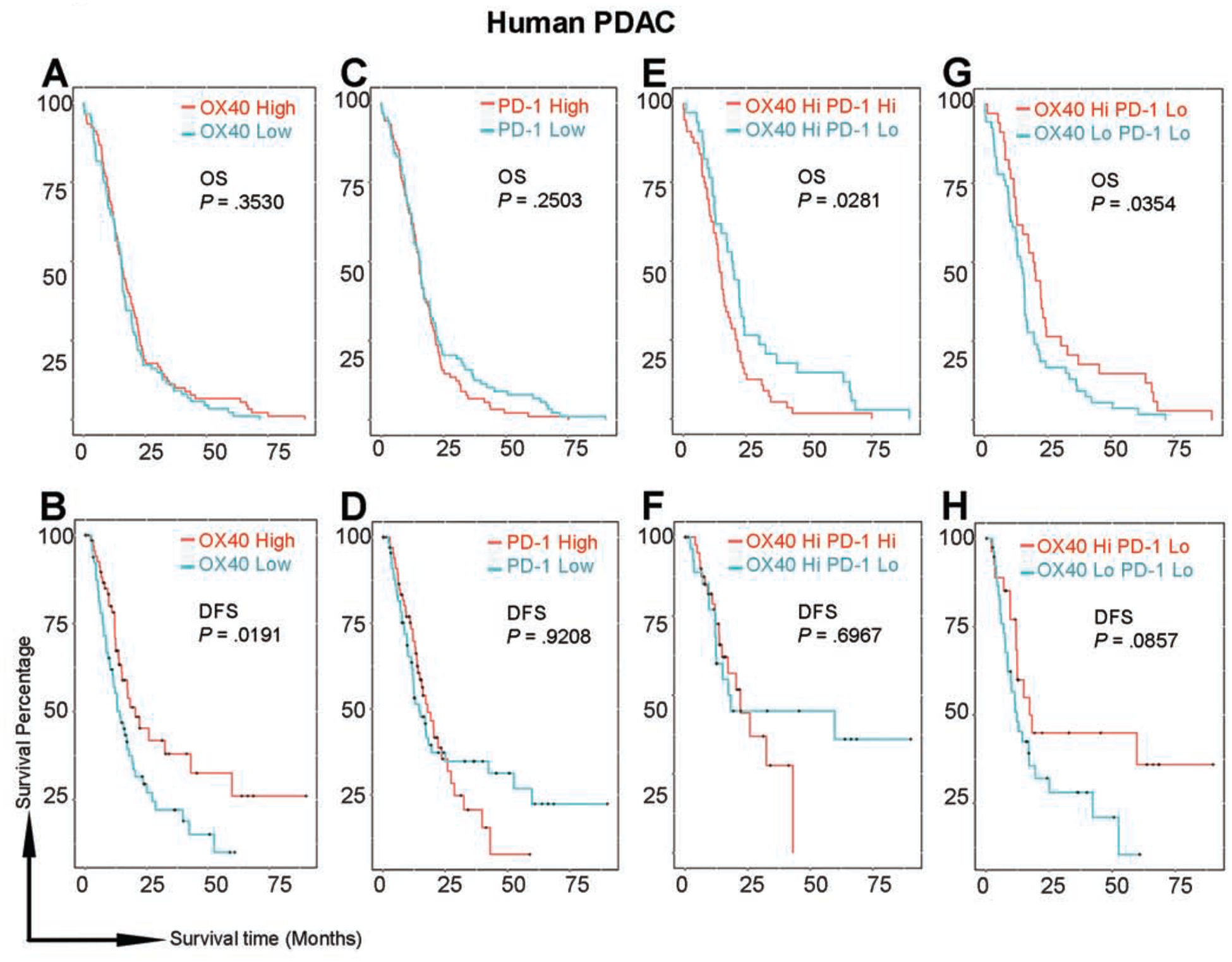
(A, C, E, G) Overall survival (OS) did not significantly differ between patients grouped by OX40 (A) or PD-1 (C) levels only. Among patients with high OX40 expression (E), patients with low PD-1 lived significantly longer than did those with high PD-1. Among patients with low PD-1 expression (G), patients with high OX40 lived significantly longer than did those with low OX40.
(B, D, F, G) Disease-free survival (DFS) was significantly longer in patients with high OX40 than in those with low OX40 (B). There were no differences in DFS in other comparisons; however, tails on the survival curves were elevated in patients with low PD-1 compared with patients with high PD-1 in both the whole population (D) and in the subpopulation with high OX40 (F). Among patients with low PD-1 (H), the tail was elevated in patients with high OX40.
Discussion
Monotherapy with an immune checkpoint inhibitor can lead to complete responses in immunogenic tumors, but challenges remain in identifying a strategy to treat non-immunogenic tumors such as pancreatic cancer. In our study, the tumoricidal activity of monotherapy was inadequate, but the combination of an anti–PD-1 antagonist and anti-OX40 agonist was exceptionally effective in a highly lethal pancreatic tumor model, completely eradicating disease, producing long-term tumor-free survival in PDAC-bearing hosts, and rejecting rechallenge with an aggressive tumor variant. The combined strategy of anti–PD-1 blockade and anti-OX40 agonist was confirmed in repeated experiments using different orthotopically implanted models, as well as GEMM model of pancreatic cancer. Supporting these results was the finding that endogenous RNA expression of high OX40 and low PD-1 among tumor biopsies in patients with PDAC was associated with a favorable prognosis. Together, these findings demonstrate a translational rationale for clinical trials of the combination of anti-OX40 agonist and PD-1 blockade against human PDAC.
To delineate possible mechanisms of this synergy, we performed comprehensive analysis of tumor samples from different treatment cohorts. In contrast to the predominant CD8 T cell– mediated responses often observed in immunogenic tumors, a significant infiltration of CD4 T cells was detected in PDAC tumors of mice receiving anti-OX40 alone or in combination with PD-1 blockade; although depletion of either the CD8 or the CD4 T-cell subset impacted survival, selective depletion of CD4 T cells in these mice had the greatest effect and completely abolished tumor eradication, subverted the development of immunologic memory, and led to decreased host survival. This requirement for CD4 T cells may be one reason why strategies directed at augmenting CD8 T cells alone have been insufficient24, 25, and such a requirement is consistent with a retrospective study26 demonstrating that the presence of tumor-infiltrating CD4 T cells was associated with tumor differentiation, lower recurrence, and favorable survival among PDAC patients treated with neoadjuvant chemotherapy. In our studies, augmentation of the CD4 T cell population using OX40 monotherapy demonstrated an antitumor effect, but this response was suboptimal. While OX40 treatment enhanced CD127+CD8 T cell memory, attenuated T-cell exhaustion, and depleted regulatory cells, it also enriched the population of GATA3 cells among CD4 cells. The addition of PD-1 blockade, while contributing further to T-cell memory expansion and mitigation of T-cell exhaustion, also, significantly reversed the potentially negative influence of OX40-mediated GATA3 cell polarization, maximizing the antitumor effect.
However, OX40 is a pleiotropic costimulatory molecule with diverse effects that appear to depend on antigen dose27, and its specific mechanism of action in reshaping the tumor milieu, its role in tumor surveillance and tumor rechallenge, and its effect on T-cell subsets both direct and indirect, warrant further investigation.
Our findings in PDAC support a new treatment approach for immune-privileged tumors and contrast previous reports in immunogenic tumor models using the combination of OX40 costimulation and PD-1/PD-L1 blockade. While this combination has seen efficacy in other studies28, it has been reported that concurrent25, 29 administration of anti–PD-1 and anti-OX40 abolished the tumoricidal effect of anti-OX40 alone. These differences were attributed to T-cell apoptosis and tissue/organ-specific disease responses induced by anti–PD-1 in the context of “high antigen” exposure among immunogenic tumors. In contradistinction to these studies where responses were predominantly CD8 T cell dependent, we observed an absolute requirement for CD4 T cells in PDAC tumor–bearing animals experiencing long-term disease-free survival. Recently, in a pancreatic cancer model, treatment with anti-OX40 and anti–PD-1 after vaccine priming24 reduced the growth of subcutaneous Panc02 tumors. Although significant in its use of a nonimmunogenic tumor, that study had design limitations of clinical significance for human disease, because subcutaneous implantation does not recapitulate the pancreatic tumor microenvironment (as would be seen in orthotopic or KPC-GEMM models), and Panc02 cells lack the driver mutation Kras, seen in over 90% of PDAC patients, factors that may account for discrepant immunobiologic findings.
In conclusion, our work uncovers the basis for an effective immunotherapy regimen against pancreatic cancer, a prototypic immune-privileged tumor. In addition to reinvigorating exhausted T cells, anti–PD-1 incorporation improves anti-OX40–induced memory differentiation and Treg cell elimination and mitigates anti-OX40–induced GATA-3 cell signaling. The combination of anti–PD-1 and anti-OX40 boosts CD4 T cell–based tumor eradication and generates robust memory recall, a strategy that merits consideration for clinical studies in the treatment of patients with advanced pancreatic cancer.
Supplementary Material
What you need to know:
BACKGROUND AND CONTEXT:
Advanced pancreatic ductal adenocarcinoma is resistant to immune checkpoint inhibitors.
NEW FINDINGS:
In mice, pancreatic tumors appear to evade the immune response by inducing development of immune-suppressive T cells. The combination of anti-PD1 inhibitory and anti-OX40 agonist antibodies reduces the proportion of T-regulatory and exhausted T cells in pancreatic tumors and increases numbers of memory CD4+ and CD8+ T cells to eradicate tumors.
LIMITATIONS:
This study was performed in mice and using data from patient tissues. Studies are needed in humans.
IMPACT:
This information might be used in development of immune-based combination therapies for PDAC.
Acknowledgments
We thank Drs. Stephen E. Ullrich, Lu Huang, Tihui Fu, Xuejun Zhang, Stephen Mok, Todd Bartkowiak, Ashvin R. Jaiswal, Zuliang Jie, Naveen Sharma, Yu Zheng, Hu Jiemiao, Qiuming He, Shumin Li, Renata S. Collazo, and Luis M. Vence, for their input and consultation; Mr. Nasser Kazimi for animal maintenance; and the general support of all of Dr. Cassian Yee’s laboratory staff members. We also thank Mr. Duncan Mak and Dr. Jared Burks for expert advice and technical assistance with the mass cytometry analysis. We thank Ms. Ann M. Sutton, Ms. Sarah J. Bronson, and Scientific Publications, Research Medical Library at The University of Texas MD Anderson Cancer Center for editing this manuscript. We thank Dr. David A. Tuveson at Cold Spring Harbor Laboratory for sharing the mT4 pancreatic cancer cell line.
Grant Support: This work was supported by an MD Anderson Pancreatic Cancer Moon Shot to C.Y., A.M., and Y.M.; MD Anderson Start-up Fund to C.Y.; MD Anderson MRP Fund to C.Y., A.M., and Y.M.; and a Hirshberg Foundation for Pancreatic Cancer Research seed grant to Y.M. The Animal Facilities, the Cytogenetics and Cell Authentication Core, the Sequencing Core, and the Flow Cytometry and Cellular Imaging Core Facility at MD Anderson are partly funded by National Cancer Institute Cancer Center Support Grant P30CA016672. Partial funding support to A.M. is also provided by U24CA224020.
This work was also supported by Parker Institute for Cancer Immunotherapy
Abbreviations:
- CyTOF
time-of-flight mass spectrometry
- IgG
immunoglobulin G
- GEMM
genetically engineered autochthonous mouse model
- KPC-GEMM
KrasG12D/+;Trp53R172H/+;Pdx-1-Cre genetically engineered mouse model
- KPC-Luc
KrasG12D;P53flox/flox;PDX-1-Cre;Luciferase
- PD-1
programmed cell death protein 1
- PDAC
pancreatic ductal adenocarcinoma
- Treg cell
regulatory T cell
- tSNE
t-distributed stochastic neighbor embedding
- TCGA
The Cancer Genome Atlas
- TME
tumor microenvironment
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: The authors declare no potential conflicts of interest.
References
- 1.Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science 2018;359:1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ying H, Dey P, Yao W, et al. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev 2016;30:355–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Katlinski KV, Gui J, Katlinskaya YV, et al. Inactivation of Interferon Receptor Promotes the Establishment of Immune Privileged Tumor Microenvironment. Cancer Cell 2017;31:194–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feig C, Jones JO, Kraman M, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A 2013;110:20212–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clark CE, Hingorani SR, Mick R, et al. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res 2007;67:9518–27. [DOI] [PubMed] [Google Scholar]
- 6.Balli D, Rech AJ, Stanger BZ, et al. Immune cytolytic activity stratifies molecular subsets of human pancreatic cancer. Clin Cancer Res 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Monte L, Reni M, Tassi E, et al. Intratumor T helper type 2 cell infiltrate correlates with cancer-associated fibroblast thymic stromal lymphopoietin production and reduced survival in pancreatic cancer. J Exp Med 2011;208:469–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nummer D, Suri-Payer E, Schmitz-Winnenthal H, et al. Role of tumor endothelium in CD4+ CD25+ regulatory T cell infiltration of human pancreatic carcinoma. J Natl Cancer Inst 2007;99:1188–99. [DOI] [PubMed] [Google Scholar]
- 9.Sousa CM, Kimmelman AC. The complex landscape of pancreatic cancer metabolism. Carcinogenesis 2014;35:1441–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma Y, Hwang RF, Logsdon CD, et al. Dynamic mast cell-stromal cell interactions promote growth of pancreatic cancer. Cancer Res 2013;73:3927–3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ma Y, Ullrich SE. Intratumoral mast cells promote the growth of pancreatic cancer. Oncoimmunology 2013;2:e25964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Croft M Control of immunity by the TNFR-related molecule OX40 (CD134). Annu Rev Immunol 2010;28:57–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hingorani SR, Wang L, Multani AS, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005;7:469–83. [DOI] [PubMed] [Google Scholar]
- 14.Evans RA, Diamond MS, Rech AJ, et al. Lack of immunoediting in murine pancreatic cancer reversed with neoantigen. JCI Insight 2016;1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rabinovich BA, Ye Y, Etto T, et al. Visualizing fewer than 10 mouse T cells with an enhanced firefly luciferase in immunocompetent mouse models of cancer. Proc Natl Acad Sci U S A 2008;105:14342–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boj SF, Hwang CI, Baker LA, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015;160:324–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chang DZ, Ma Y, Ji B, et al. Mast Cells in Tumor Microenvironment Promotes the in vivo Growth of Pancreatic Ductal Adenocarcinoma. Clin Cancer Res 2011;17:7015–7023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hingorani SR, Petricoin EF, Maitra A, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003;4:437–50. [DOI] [PubMed] [Google Scholar]
- 19.Kimball AK, Oko LM, Bullock BL, et al. A Beginner’s Guide to Analyzing and Visualizing Mass Cytometry Data. J Immunol 2018;200:3–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Metzger TC, Long H, Potluri S, et al. ICOS Promotes the Function of CD4+ Effector T Cells during Anti-OX40-Mediated Tumor Rejection. Cancer Res 2016;76:3684–9. [DOI] [PubMed] [Google Scholar]
- 21.Kaech SM, Tan JT, Wherry EJ, et al. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol 2003;4:1191–8. [DOI] [PubMed] [Google Scholar]
- 22.Uhlen M, Fagerberg L, Hallstrom BM, et al. Tissue-based map of the human proteome. Science 2015;347. [DOI] [PubMed] [Google Scholar]
- 23.Wu C, Orozco C, Boyer J, et al. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol 2009;10:R130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kinkead HL, Hopkins A, Lutz E, et al. Combining STING-based neoantigen-targeted vaccine with checkpoint modulators enhances antitumor immunity in murine pancreatic cancer. JCI Insight 2018;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shrimali RK, Ahmad S, Verma V, et al. Concurrent PD-1 Blockade Negates the Effects of OX40 Agonist Antibody in Combination Immunotherapy through Inducing T-cell Apoptosis. Cancer Immunol Res 2017;5:755–766. [DOI] [PubMed] [Google Scholar]
- 26.Nejati R, Goldstein JB, Halperin DM, et al. Prognostic Significance of Tumor-Infiltrating Lymphocytes in Patients With Pancreatic Ductal Adenocarcinoma Treated With Neoadjuvant Chemotherapy. Pancreas 2017;46:1180–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rogers PR, Croft M. CD28, Ox-40, LFA-1, and CD4 modulation of Th1/Th2 differentiation is directly dependent on the dose of antigen. J Immunol 2000;164:2955–63. [DOI] [PubMed] [Google Scholar]
- 28.Polesso F, Weinberg AD, Moran AE. Late-Stage Tumor Regression after PD-L1 Blockade Plus a Concurrent OX40 Agonist. Cancer Immunol Res 2019;7:269–281. [DOI] [PubMed] [Google Scholar]
- 29.Messenheimer DJ, Jensen SM, Afentoulis ME, et al. Timing of PD-1 Blockade Is Critical to Effective Combination Immunotherapy with Anti-OX40. Clin Cancer Res 2017;23:6165–6177. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.