

Abstract
Introduction:
Coronary artery disease (CAD) poses significant morbidity and mortality globally. Despite significant advances in treatment interventions, residual cardiovascular risks remain unchecked. Recent clinical trials have shed light on the potential therapeutic benefits of targeting anti-inflammatory pathways. Myeloperoxidase (MPO) plays an important role in atherosclerotic plaque formation and destabilization of the fibrous cap; both increase the risk of atherosclerotic cardiovascular disease and especially CAD.
Areas covered:
This article examines the role of MPO in the pathogenesis of atherosclerotic CAD and the mechanistic data from several key therapeutic drug targets. There have been numerous interesting studies on prototype compounds that directly or indirectly attenuate the enzymatic activities of MPO, and subsequently exhibit atheroprotective effects; these include aminobenzoic acid hydrazide, ferulic acid derivative (INV-315), thiouracil derivatives (PF-1355 and PF-0628999), 2-thioxanthine derivative (AZM198), triazolopyrimidines, acetaminophen, N-acetyl lysyltyrosylcysteine (KYC), flavonoids, and alternative substrates such as thiocyanate and nitroxide radical.
Expert opinion:
Future investigations must determine if the cardiovascular benefits of direct systemic inhibition of MPO outweigh the risk of immune dysfunction, which may be less likely to arise with alternative substrates or MPO inhibitors that selectively attenuate atherogenic effects of MPO.
Keywords: Myeloperoxidase, Coronary Artery Disease, Inflammation, Oxidative Stress
1. Introduction
Coronary artery disease (CAD) is the leading cause of morbidity and mortality throughout the world, especially in developed countries. Although the treatment of CAD has been significantly improved during the past decades, the mortality rate among patients with CAD is still undeniably high, which accounts for one-third of all deaths in adults 35 years of age or older [1]. The major pathogenesis of CAD is atherosclerosis caused by interaction between cardiovascular risk factors and the immune system [2, 3], resulting in the formation and growth of atherosclerotic plaques obstructing coronary arteries. Those plaques can become vulnerable over time due to persistent inflammatory activation, leading to plaque weakening and subsequent rupture, which results in acute myocardial ischemia and infarction [4]. Therefore, inflammation is apparently one of the main factors for development and progression of CAD, and many studies have demonstrated the ability of inflammatory biomarkers to predict the risk of future CAD, plaque stability, adverse clinical outcomes in CAD patients.
Recently, Ridker et al. studied the therapeutic results of Canakinumab, a monoclonal antibody antagonizing interleukin-1β, in the Canakinumab Anti-inflammatory Thrombosis Outcome Study (CANTOS) [5], in which over 10,000 patients with previous history of CAD and increased serum inflammatory marker (high-sensitivity C-reactive protein) were randomized to receive different doses of Canakinumab or placebo. They demonstrated that during 48 months follow up, Canakinumab treatment was significantly associated with lower incidence of nonfatal myocardial infarction without any alteration of lipid levels. However, higher risk of fatal infection was observed in the treatment group. These findings support that targeting inflammation could be a promising novel therapeutic approach for CAD patients, but the adverse effects on the immune function must be carefully considered.
Among all the inflammatory biomarkers, myeloperoxidase (MPO), an enzyme released from activated leukocytes, has been shown to be mechanistically linked to atherosclerosis, especially CAD, and previous studies suggested that MPO could be useful as a diagnostic marker and prognostic indicator for CAD patients [6–10]. Interestingly, recent research has focused on the hypothesis that MPO might be a potential therapeutic target for novel treatment of CAD. Also, several synthesized MPO inhibitors have been introduced in studies on their atheroprotective effects [11, 12]. This review summarizes current insights into the physiology of MPO, the pathogenic roles of MPO in atherosclerosis, and how it has recently been recognized as a promising therapeutic target for future CAD treatment.
2. Structure and Function of MPO
MPO, one of the heme peroxidase cyclooxygenase enzymes, is a 146 kDa glycosylated homodimeric protein composed of two monomers. Each monomer is formed by a 14.5 kDa light chain and a 58.5 kDa heavy glycosylated chain, which contain a prosthetic heme derivative and a calcium-binding site necessary for enzymatic reactions [13, 14]. MPO is synthesized in human promyelocytic cell line HL-60 and hematopoietic cells within bone marrow during myeloid lineage differentiation and can be found in neutrophils, macrophages, and lymphocyte antigen 6C (Ly-6C)-positive monocytes [15–17]. The synthesis starts with the transcription and translation of the MPO gene located on the long arm of chromosome 17, resulting in a preproMPO, which is then processed in the endoplasmic reticulum to form an active monomeric proMPO by N-glycosylation, transient interaction between the molecular chaperones calnexin and calreticurin, and incorporation with heme from protoporphytin IX. ProMPO is subsequently cleaved by proteolytic enzymes, and dimerized by disulfide bond formation, before being transported to the Golgi and become a mature MPO [18]. This mature MPO is stored in azurophilic granules of neutrophils, mast cells, and tissue macrophages [13, 19, 20] to be used intracellularly and secreted extracellularly [18]. However, some evidence suggests that after differentiation from monocytes, macrophages do not actively synthesize MPO unless they are activated by local cytokines and signaling molecules under inflammatory conditions, especially atherosclerosis, which may initiate transcription of MPO gene in macrophages [21, 22].
The main function of MPO is to produce reactive oxidants for the destruction of ingested microorganisms within phagosomes [23, 24], by utilizing H2O2 and a halide (Cl−, Br−, I−) or the pseudohalide (SCN−) in the catalytic cycle (Figure 1), which starts with the ground state form of MPO (MPO-Fe(III)) [13, 25]. The first step of the catalytic cycle is the formation of compound I (MPO-Fe(IV).π+) by oxidizing the ground state MPO with H2O2. Compound I is then reduced to the ground state MPO by either the ‘halogenation cycle’ or the ‘peroxidase cycle.’ In the halogenation cycle, compound I is reduced by a halide or the pseudohalide to the ground state MPO, resulting in a hypohalous acid (HOX), mostly HOCl under physiologic circumstances [23, 25]. On the other hand, with a high concentration of H2O2, compound I is likely to undergo a 2-step reduction in the peroxidation cycle, and become compound II (MPO-Fe(IV)) as an intermediate before being reduced to the ground state MPO by a variety of substrates such as, O2.-, nitric oxide (NO), nitrite (NO2−), tyrosine, serotonin, catecholamines, ascorbic acid, uric acid, and estrogen [24]. Moreover, the ground state MPO can also be reduced to an enzymatically inactive form MPO-Fe(II) or compound III (MPO-Fe(II)-O2) by a single electron addition, or O2.-, respectively [25]. The redox form of MPO can be transformed to either compound III by binding to O2, or back to the ground state form by single-electron peroxidation. Likewise, compound III can be converted back to the ground state form by O2.-, resulting in H2O2 formation as a byproduct [26].
Figure 1:
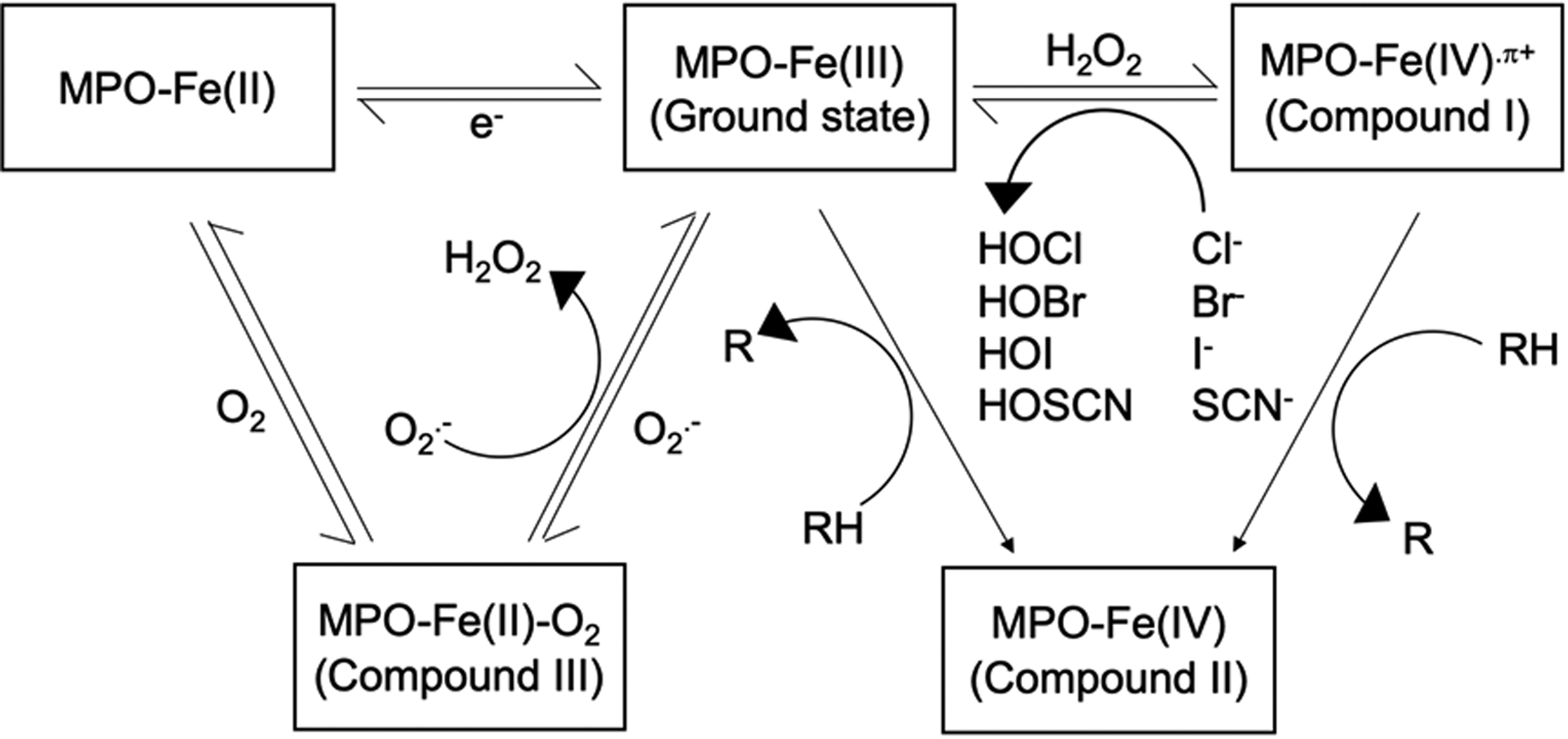
The catalytic cycle of myeloperoxidase. Ground state MPO (MPO-Fe(III)) is oxidized by H2O2 to become Compound I (MPO-Fe(IV).π+), which can be reduced to the ground state MPO by either halogenation cycle, or peroxidation cycle, depending on the concentration of H2O2. In halogenation cycle, halide (Cl−, Br−, or I−) or pseudohalide (SCN−) is used to produce hypohalous acid (HOCl, HOBr, HOI, or HOSCN). In peroxidation cycle, Compound II (MPO-Fe(IV)) is formed as an intermediate. Ground state MPO can also be reduced to MPO-Fe(II) or Compound III (MPO-Fe(II)-O2) by e− or O2.-, respectively. MPO-Fe(II) can be transformed back to the ground state form, or to Compound III by binding to O2. Compound III can also be converted back to the ground state form by O2.-, resulting in the formation of H2O2.
MPO can produce a variety of products in different pathways. Firstly, HOCl is the most abundant physiological product of MPO, which can react with many kinds of protein, lipid, nucleic acids [27], glycosaminoglycans, and some components of extracellular matrix [28, 29]. Interestingly, there are several methods to indirectly measure the enzymatic activities of HOCl which have been linked to atherosclerosis, such as 3-chlorotyrosine [25, 30], 5-chlorouracil [31], and the antibody specific for hypochlorite oxidized protein (HOP-1) [32, 33]. Secondly, MPO directly oxidizes tyrosine and produces tyrosine radicals that give rise to dityrosine, trityrosine, pulcherosine, and isodityrosine, which have been linked to the formation of atherosclerotic plaques [34, 35]. Thirdly, MPO can generate reactive nitrogen species from NO2− either by MPO itself or through HOCl, resulting in .NO2 or NO2Cl, respectively [36, 37]. Interestingly, these nitrogen derivatives also have mechanistic links to atherosclerosis [36, 38]. Lastly, MPO produces reactive cyanate by utilizing urea, H2O2, and SCN−, accounting for up to 50% of overall H2O2 consumption by MPO under physiological conditions [39, 40]. This MPO pathway is more common in patients with chronic kidney disease, due to elevated urea level, and also in patients who smoke, which indirectly increases SCN− [40]. Cyanate has the ability to interact with protein by carbamylation of nucleophilic side chains, such as lysine, resulting in homocitrulline (carbamyl lysine), which subsequently promotes atherosclerosis [40].
3. MPO and Pathogenesis of Atherosclerosis
MPO has been shown to be involved in the development and progression of atherosclerosis [13] since Daugherty et al. reported that MPO was found in atherosclerotic plaques [41]. Subsequently, Sugiyama et al. [42] demonstrated that MPO-containing macrophages were found abundantly within the vulnerable and ruptured plaques rather than earlier stages of atherosclerosis, which contained only a very low amount of MPO or MPO-negative macrophages. A genetic study on subjects with either partial or complete MPO deficiency revealed that the lack of MPO expression provided a significantly lower risk of developing cardiovascular disease [43]. In contrast, genetic polymorphism of MPO has been shown to be associated with increased risk of CAD [44]. Moreover, a study on a cohort of 158 patients with known history of CAD compared with 175 healthy controls, showed that serum MPO levels were significantly associated with CAD [9]. These results shed light on the pathogenic roles and potential clinical utilities of MPO in atherosclerosis and CAD. It is believed that MPO promotes atherosclerosis and CAD through different pathways including chemical modification of lipoproteins, endothelial dysfunction, and weakening of atherosclerotic plaque. Even though there are also few studies contradicting this hypothesis by showing that MPO deficiency is associated with increased atherosclerosis in mouse models [45–47], there is still much more evidence suggesting that MPO contributes to the development and progression of atherosclerosis.
3.1. Effects of MPO on lipoproteins
Among different lipoproteins, low-density lipoprotein (LDL) has been described as one of the key mediators of atherosclerosis, and therapeutic interventions on modifying LDL have provided favorable outcomes for both primary and secondary prevention of atherosclerosis and cardiovascular diseases [48]. MPO-derived HOCl can oxidize LDL by targeting lysine residues on apolipoprotein B-100, the most predominate component of LDL, and it makes LDL become more recognizable by the scavenger receptors on macrophages, leading to the formation of macrophage foam cells within the lipid core of atherosclerotic plaques [49–51]. Reactive nitrogen species produced by MPO are also able to modify LDL to form NO2LDL, another atherogenic form of LDL that macrophages can uptake [36, 52]. Moreover, carbamylation of lysine residues on LDL by MPO-derived cyanate also contributes to foam cell formation and atherosclerosis [40]. Furthermore, oxidized LDL is also able to increase atherosclerotic burden through endothelial dysfunction [53], cellular apoptosis of monocytes [54], and increased inflammatory response [51].
In addition to oxidizing LDL, MPO is capable of modifying high-density lipoprotein (HDL), the only lipoprotein that provides atheroprotective effects by transporting peripheral cholesterol back to the liver. This is also called macrophage reverse cholesterol transport, through the interaction between ATP-binding cassette transporter A1 (ABCA1), G1 (ABCG1) and apolipoprotein A-1 (apoA-1) [48, 55, 56]. MPO binds to helix 8 and oxidizes apoA-1 at tyrosine residues, resulting in impaired ABCA1-dependent cholesterol efflux from macrophages [57, 58]. Interestingly, MPO can also oxidize paraoxonase 1, which is an HDL-associated antioxidant protein that binds to apoA-1, resulting in decreased atheroprotective properties of HDL [59]. Similarly, methionine and tryptophan residues have also been shown to be important targets for MPO-mediated oxidation reactions, which further reduce anti-atherogenic properties of HDL [60–62]. Furthermore, MPO-produced HOCl also causes dysfunctional HDL that competes with native HDL on lipid uptake via scavenger receptors on macrophages [19, 63]. These findings might explain that the presence of dysfunctional HDL is responsible for an increase in cardiovascular risk observed among patients with normal or high serum HDL [6].
3.2. MPO and Endothelial Dysfunction
MPO indirectly causes endothelial dysfunction by interfering with NO metabolism, which is one of the key factors maintaining normal physiology of endothelial cells and the vascular system [6, 19]. Not only does MPO reduce NO bioavailability by consuming NO as a substrate in the catalytic cycle, it has also been shown to be involved in the synthesis of endogenous NO through HOCl [19]. MPO-produced HOCl is capable of chlorinating arginine, which is an important substrate for the formation of the enzyme necessary for the synthesis of NO, nitric oxide synthase (NOS). Interestingly, chlorinated arginine negatively affects the enzymatic activity of NOS [64]. Moreover, NOS can be destabilized by HOCl-mediated direct oxidation, resulting in a further impairment of NO synthesis [65]. Marsche et al. [66] also demonstrated that HOCl-modified HDL is able to downregulate the expression of NOS in endothelial cells.
The association between MPO and endothelial dysfunction has been corroborated in several human studies. Vita et al. [67] studied a cohort of 298 patients which revealed that after adjusting for traditional cardiovascular risk factors, elevated serum MPO was independently associated with endothelial dysfunction assessed by brachial artery flow-mediated dilation. Similarly, another study on patients with symptomatic CAD showed that increased MPO levels in systemic circulation correlated with the degree of microvascular dysfunction measured by the capacity of endothelium-dependent vasodilation after an exposure to acetylcholine [68]. Interestingly, even though CAD patients underwent coronary revascularization, higher MPO levels were independently associated with incomplete ST-segment resolution and lower myocardial blush grade, suggesting that MPO causes impaired myocardial microcirculation in patients with CAD despite coronary revascularization [69].
3.3. MPO and weakening of atherosclerotic plaques
Generally, vulnerability of atherosclerotic plaques is determined by the integrity of the fibrous cap that covers the lipid core created by foam cells [2, 70]. MPO can reduce the thickness of the fibrous cap through three main mechanisms. Firstly, MPO-derived HOCl has been shown to be involved in the apoptosis of endothelial cells by inhibiting Bcl-2 and promoting the expression of cytochrome C within endothelial cells [71]. Secondly, MPO and HOCl activate matrix metalloproteinases (MMPs), enzymes that can degrade the extracellular matrix of the fibrous cap [72, 73]. Thirdly, since MPO is able to bind with some extracellular matrix structural proteins, such as type IV collagen and fibronectin [29], they can be directly oxidized by MPO-derived HOCl, resulting in decreased vascular smooth muscle cell adhesion, which may weaken atherosclerotic plaques and promote plaque rupture [74–76]. Furthermore, some other interesting mechanisms of MPO and weakening of atherosclerotic plaques have also been proposed. Firstly, HOCl has been shown to inhibit one of the isoforms of the enzyme that counteracts MMPs, tissue inhibitor of matrix metalloproteinase-1, by oxidizing cysteine residues at N-terminal, resulting in unopposed MMPs, which further weakens the fibrous cap, and promotes subsequent plaque rupture[77]. Secondly, Oxidized LDL and oxidant stress are also able to weaken atherosclerotic plaques by causing endoplasmic reticulum stress, which subsequently induces macrophage apoptosis within necrotic cores, resulting in more vulnerable plaques [78]. Lastly, MPO has been shown to be associated with the formation of neutrophil extracellular traps, which could activate inflammatory response, and release some atherogenic inflammatory mediators, such as MPO itself, MMPs, and oxidants, resulting in the formation, destabilization, and rupture of atherosclerotic plaques [79–81].
4. Targeting MPO in Coronary Artery Disease
Since MPO has been recognized as an important factor in the development and progression of atherosclerosis, and provides clinical utilities for diagnosis and prognosis in CAD patients [6, 82], recent research has focused on developing MPO inhibitors and alternative substrates as novel therapeutic interventions to improve the standard of care in patients with CAD. There are several pharmacological targets that can inhibit the enzymatic activities of MPO and potentially alter the progression of atherosclerotic plaques and CAD (table 1).
Table 1:
MPO inhibitors with cardioprotective effects shown in studies.
| Drug | Class | Publication | Type of study | Finding |
|---|---|---|---|---|
| ABAH | Benzoic acid hydrazides | Han et al. [86] (2012) | In vitro, human samples from CAD patients and healthy controls | ABAH inhibited MPO activity and neutrophil adhesion with endothelial cell in CAD patients |
| Tiyerili et al. [87] (2016) | In vitro, mouse model of atherosclerosis | High dose ABAH attenuated inflammation, vascular oxidative stress, and improved endothelial function | ||
| INV-315 | Ferulic acid derivative | Liu et al. [90] (2012) | In vivo, mouse model of atherosclerosis | INV-315 was associated with lower inflammation, MPO activity, plaque size, and improved endothelial function |
| PF-1355 | Thiouracil derivative | Ali et al. [11] (2016) | In vivo, mouse model of CAD | PF-1355 reduced inflammation, MPO activity, ischemic reperfusion injury, cardiac remodeling, and improved cardiac function |
| PF-0628999 | Thiouracil derivative | Roth Flach et al. [95] (2019) | In vivo, mouse model of atherosclerosis | PF-0628999 decreased plaque inflammation and necrotic core area |
| AZM198 | 2-thioxanthines derivative | Rashid et al. [99] (2018) | In vivo, mouse model of atherosclerosis | AZM198 increased fibrous cap thickness, and decreased MPO activity |
| Cheng et al. [12] (2019) | In vivo, mouse model of atherosclerosis | AZM198 was associated with improved endothelial function, and lower MPO activity | ||
| Triazolopyrimidines | - | Duclos et al. [101] (2017) | In vitro, human and mouse samples | Triazolopyrimidine inhibited the oxidation of apoA-1 on HDL, and reduced the amount of HOCl produced from neutrophils |
| Acetaminophen | - | Nenseter et al. [105] (1995) | In vitro, human samples | Acetaminophen inhibited MPO-mediated LDL oxidation |
| Chou et al. [106] (2002) | In vitro, human samples | Acetaminophen inhibited MPO-mediated LDL oxidation | ||
| Ozsoy et al. [108] (2007) | In vivo, rabbit model of atherosclerosis | Acetaminophen reduced LDL oxidation | ||
| KYC | - | Zhang et al. [109] (2013) | In vitro, human and bovine samples | KYC inhibited HOCl production, and LDL oxidation mediated by MPO |
| Flavonoids (epicatechin, quercetin, taxifolin, and luteolin) | Flavonoids | Kostyuk et al. [115] (2003) | In vitro, human samples | Flavonoids inhibited MPO-mediated LDL oxidation |
| Epicatechin | Flavonoids | Steffen et al. [117] (2006) | In vitro, human samples | Epicatechin inhibited MPO-mediated LDL oxidation, and attenuated endothelial dysfunction caused by oxidized LDL |
| Quercetin | Flavonoids | Loke et al. [116] (2008) | In vitro, human samples | Quercetin inhibited MPO-mediated LDL oxidation |
| Bhaskar et al. [118] (2013) | In vivo, rabbit model of atherosclerosis | Quercetin reduced MPO activity in the aorta of the rabbits | ||
| Lu et al. [119] (2018) | In vitro, human samples | Quercetin inhibited production of HOCl, and improved endothelial function | ||
| Thiocyanate | Alternative substrates | Morgan et al. [124] (2015) | In vivo, mouse model of atherosclerosis | Thiocyanate reduced plaque size with stable MPO level |
| Zietzer et al. [125] (2019) | In vivo, mouse model of atherosclerosis | Sodium thiocyanate was associated with decreased plaque formation, and improved endothelial function | ||
| Nitroxide radical | Alternative substrates | Kim et al. [126] (2015) | In vivo, mouse model of atherosclerosis | Nitroxide radical stabilized plaques by increasing the collagen and decreasing the lipid content |
ABAH, aminobenzoic acid hydrazide; apoA-1, apolipoprotein A-1; CAD, coronary artery disease; HDL, high density lipoprotein; KYC, N-acetyl lysyltyrosylcysteine; MPO, myeloperoxidase.
4.1. Benzoic Acid Hydrazides
Aminobenzoic acid hydrazide (ABAH) is the first potent MPO inhibitor that has been discovered [83]. ABAH inactivates MPO by undergoing oxidation reaction with MPO itself, resulting in the formation of the radical form of ABAH, which subsequently reduces ground state MPO to MPO-Fe(II) that can react with either O2 or H2O2, leading to enzyme turnover or irreversible inhibition of the enzymatic activities of MPO, respectively [83, 84]. Nonetheless, high O2 and low H2O2 concentration found in extracellular space reduce the potency of ABAH in inhibiting MPO, and under this condition, ABAH can only act as a competitive inhibitor [84, 85].
An in vitro study from Han et al. [86] using the peripheral blood of 20 CAD patients compared with 20 healthy controls, revealed that ABAH significantly reduced the enzymatic activity of MPO and the capability of neutrophils to attach to endothelial cells. The magnitude of these effects also correlated with the dose of ABAH. Tiyerili et al. [87] studied apolipoprotein in E-deficient mice fed a cholesterol-rich diet and two different doses of ABAH. They found that only high-dose ABAH was associated with lower inflammation, vascular oxidative stress, and atherosclerotic plaque area, with improved endothelial function. However, it is believed that to effectively inhibit MPO, ABAH requires an excessive amount of H2O2, which can hardly be found under physiological conditions in humans[85]. Moreover, the inhibitory effect of ABAH on MPO is reversible and would be ineffective against HOCl-mediated atherogenesis [88]. Therefore, ABAH theoretically seems to be an unsuccessful therapeutic intervention for modifying the progression of atherosclerosis and CAD.
4.2. Ferulic Acid Derivative
Ferulic acid, one of the phenolic antioxidants, has been shown to provide inhibitory effects on MPO-mediated tyrosine oxidation[89]. In 2012, Liu et al. [90] synthesized INV-315, a small molecule derived from ferulic acid and investigated its therapeutic effects on the progression of atherosclerotic lesions and endothelial function in a mouse model. They found that INV-315 treatment was associated with lower plaque area, decreased inflammatory cytokine (interleukin-6) levels, reduced MPO enzymatic activity, and improved endothelial function. Interestingly, they demonstrated that INV-315 treatment resulted in enhanced reverse cholesterol transport of HDL from macrophages measured by ex-vivo assays. However, they did not demonstrate the effect of INV-315 on lipoprotein oxidation, which is one of the main atherogenic pathways of MPO.
4.3. Thiouracil Derivatives
Since propylthiouracil, has been used for the treatment of hyperthyroidism by inhibiting thyroid peroxidase (TPO)[91], it has also been shown to have an antagonistic effect against MPO with unclear mechanism [92]. Research focused on discovering a thiouracil derivative that could be more specific for MPO than TPO. In 2015, Ruggeri et al. [93] introduced the synthetic thiouracil derivative, 2-(6-(5-chloro-2-methoxyphenyl)-4-oxo-2-thioxo-3,4-dihydropyrimidin-1(2H)-yl)acetamide (PF-06282999) containing the thiouracil sulfur atom, which can be oxidized by compound I. This results in a thiyl radical that can selectively and irreversibly inhibit enzymatic activity of MPO by forming a covalent bond with the heme moiety within MPO [93, 94].
In 2016, Ali et al. [11] studied the therapeutic effect of another synthetic, thiouracil-derived MPO inhibitor that has similar chemical structure to PF-06282999, 2-(6-(2,5-dimethoxyphenyl)-4-oxo-2-thioxo-3,4-dihydropyrimidin-1(2H)-yl)acetamide (PF-1355). They demonstrated that in myocardial infarcted mice with and without PF-1355 administered orally for 2 days following the ligation of a coronary artery, PF-1355 was associated with significantly lower enzymatic activity of MPO in both intra- and extra-cellular components compared with the control group. By using a magnetic resonance imaging (MRI) agent, MPO-Gd (bis-5hydroxytryptamide-diethylene-triaminepentaacetate-gadolinium), to track extracellular MPO activity in vivo, PF-1355 was associated with less MPO activity, a lower degree of inflammation, and less ischemic reperfusion injury within the infarct lesion. Interestingly, these effects could also be seen on heart tissue obtained at 7 days after ischemic injury, resulting in increased myocardial thickness at the infarct area seen in the treatment group. Furthermore, early initiation of PF-1355 resulted in improved cardiac function and remodeling. Prolonged use of PF-1355 also showed an inverse correlation between the duration of treatment and degree of cardiac remodeling. This was the first study that demonstrated both early and late cardioprotective effects of the thiouracil-derived MPO inhibitor against CAD and ischemic cardiomyopathy.
Recently, another animal study of the potential therapeutic effects of a 4-week supplementation of PF-0628999 on the development of atherosclerotic plaques in the aorta. It revealed that, although PF-0628999 did not reduce the overall plaque size, it was significantly associated with decreased plasma MPO, the necrotic core area of the plaques, and level of inflammation assessed by [18F]-Fluorodeoxyglucose-positron emission tomography (FDG-PET) scan [95]. These results suggested that PF-0628999 could be helpful for plaque stabilization and potentially prevent subsequent plaque rupture.
Nevertheless, there are few animal studies that demonstrate the cardiovascular benefits of MPO inhibitor developed from thiouracil. Although those effects were tested in young mice that could have a different drug metabolism than older mice, the majority of patients with atherosclerotic cardiovascular diseases are older. Hence, the atheroprotective effects of thiouracil-derived MPO inhibitor still need to be vetted by future research in both animal and human models. Unfortunately, the product development of PF-0628999 was discontinued in 2017 without an explanation [96], so PF-1355 is the only thiouracil derivative that can be used for further investigation on cardioprotective effects.
4.4. 2-thioxanthines Derivatives
2-thioxanthines have been described as potent, irreversible, and selective MPO inhibitors [97]. They act as MPO’s suicide substrates after undergoing oxidation by MPO, resulting in radicals that form a covalent bond with the heme prosthetic group in MPO, and subsequently inactivate the enzymatic activity of MPO without affecting the ability of microbe killing by the immune cells [97, 98]. Rashid et al. [99] studied one of the 2-thioxanthine derivatives, AZM198, administered orally for 13 weeks in mice, and demonstrated that AZM198 was significantly associated with lower MPO activity within atherosclerotic plaques measured by MPO-Gd, and liquid chromatography with tandem mass spectrometry (LC-MS/MS), which measures HOCl production from chlorination of dihydroethidium [100]. AZM198 supplementation was also associated with increased fibrous cap thickness without any alteration of inflammatory cells or lipid content in the necrotic cores of the plaques.
Furthermore, another AZM198 study from Cheng et al. [12] showed that mice treated with AZM198 had better endothelial function and decreased enzymatic activity of MPO measured by LC-MS/MS compared with the control group. Notably, there was no significant difference in plasma MPO levels and inflammatory cytokines, suggesting that AZM198 selectively attenuates the effects of MPO on endothelial dysfunction and atherosclerosis. The mechanism by which AZM198 improves endothelial function was described as an increase in cyclic guanosine monophosphate (cGMP) observed in the treatment group, which suggested that there was improved activity of soluble guanylyl cyclase (sGC) as a result of increased NO bioavailability from MPO inhibition. These findings showed the possibility that AZM198 could be an MPO inhibitor to promote plaque stabilization and improve endothelial function, which could subsequently reduce the risk of CAD. However, those results were from young mice as well as the studies on thiouracil derivatives, and further investigation on the atheroprotective effects of 2-thioxanthine derivatives is warranted.
4.5. Triazolopyrimidines
Recently, Duclos et al. [101] discovered a novel reversible MPO inhibitor, triazolopyrimidine (7-(benzyloxy)-3H-[1,2,3]triazolol[4,5-d]pyrimidin-5-amine), which selectively inhibits enzymatic activity of MPO by binding to its active side with matching polar amino acid residues, and interaction between the amine part and heme carboxylic acid. Interestingly, triazolopyrimidine has been shown to be capable of inhibiting the formation of oxidized apoA-1 found on HDL, and the production of HOCl from neutrophils. These proposed mechanisms of triazolopyrimidines on MPO needs to be vetted by future studies both in vitro and in vivo, to provide more details of pharmacokinetics and pharmacodynamics, especially the efficacy of triazolopyrimidines, since it reversibly inhibits MPO.
4.6. Acetaminophen
Acetaminophen (paracetamol) has been one of the most commonly used analgesic and antipyretic drugs, and also play a role in modulating the enzymatic activities of MPO [102, 103]. In 1989, Van Zyl et al. [104] revealed that acetaminophen could attenuated MPO-mediated HOCl production by competing with Cl−. This finding was supported by subsequent studies, which also revealed that this reaction requires high therapeutic concentration of acetaminophen [102, 103], whereas lower concentration of acetaminophen might promote the turnover of compound II and III to ground state MPO [103]. These findings raised the possibility that acetaminophen might be a potential therapeutic intervention for atherosclerosis. However, early preclinical studies showed the contradictory results of the effect of acetaminophen on LDL oxidation. Nenseter et al. [105] and Chou et al. [106] demonstrated that acetaminophen was able to inhibit LDL oxidation, while Kapiotis et al. [107] revealed that acetaminophen catalyzed MPO-mediated LDL oxidation via a phenoxyl radical, which could promote lipid oxidation in LDL. These contradictory results may be attributed to the different experimental strategies and conditions. Despite the controversies of the effect of acetaminophen on LDL oxidation, there is still limited evidence from in vivo studies. Ozsoy et al. [108]studied on hypercholesterolemic rabbits, and demonstrated that administration of acetaminophen at therapeutic dose was associated with reduced MPO-mediated LDL oxidation. Hence, according to the conflicting effects of acetaminophen on LDL oxidation, and limited evidence, acetaminophen might not be a promising candidate for future research on MPO inhibitors and atherosclerosis.
4.7. N-acetyl lysyltyrosylcysteine amide
In 2013, Zhang et al. [109] discovered a tripeptide, N-acetyl lysyltyrosylcysteine amide (KYC), and demonstrated that KYC was able to bind with the active side of MPO and interact with iron-heme site, resulting in decreased MPO-mediated HOCl production, and LDL oxidation. Interestingly, KYC was also found to increase the consumption of H2O2 by MPO, which could reduce oxidative injury to endothelial cells [109]. Although recent studies showed that inhibition of MPO by KYC provided neuroprotective effects in mouse model of stroke [110, 111], there is still limited evidence of the atheroprotective effects of KYC, which need to be investigated by future in vitro and in vivo studies.
4.8. Flavonoids
Flavonoids, substances found fruits, vegetables, and some kinds of tea, have long been shown to be antioxidants and MPO inhibitors, especially quercetin, which is the most potent MPO inhibitor among all flavonoids [112–114]. Early studies demonstrated that flavonoids were able to inhibit LDL oxidation mediated by MPO in vitro [115–117]. Interestingly, Bhaskar et al. [118] studied on rabbit model, and revealed that quercetin was associated with lower MPO activity in the aorta of the rabbits fed with high cholesterol diet. In addition, squercetin was also shown to be able to attenuate the production of HOCl, and endothelial dysfunction in the aorta of mice [119]. These findings suggested potential mechanisms of atheroprotective effects of flavonoids which need to be focused by future research.
4.9. Ceruloplasmin
Ceruloplasmin (Cp), a copper-containing ferroxidase, is one of the acute phase proteins secreted by hepatocytes and macrophages in response to acute stress, and it has been shown to be an endogenous potent inhibitor of MPO [120, 121]. It is believed that Cp interacts with MPO by an electrostatic interaction due to the anionic and cationic nature of Cp and MPO, respectively [121]. However, the detailed mechanism remained largely unclear, and there have been discrepancies regarding the inhibitory efficacy of Cp on MPO, until Chapman et al. [122] demonstrated in vivo interaction between Cp and MPO and significantly reduced enzymatic activity of MPO observed in Cp knock-out (Cp−/−) compared with wild-type mice (Cp+/+). They revealed that Cp prevents the formation of HOCl by reducing compound I to compound II through the peroxidase cycle instead of turning back to the ground state. Ceruloplasmin is also capable of reducing the bioavailability of the ground state MPO by inhibiting the reduction reaction that converts compound II back to the ground state, and driving the formation of compound II from the ground state. These mechanisms may result in the accumulation of compound II and decreased production of HOCl. Preliminary data from Kennedy et al. [123] demonstrated that after coronary artery ligation in both Cp hemizygote (Cp+/−) and wild type mice, the Cp+/− group had increased in vivo MPO activity measured by 24-hour urine. Interestingly, increased myocardial fibrosis was observed in the Cp+/− group compared with the wild type. These findings suggest the cardioprotective effect of Cp and the possibility that Cp can be a novel target for MPO inhibition and atherosclerotic cardiovascular diseases.
4.10. Alternative Substrates
Supplementation of alternative substrates has also been shown to be another interesting approach for reducing the production of MPO-derived HOCl, or the enzymatic activities of MPO, which could attenuate the atherosclerotic burden, and potentially reduce risk of adverse effects of direct inhibition of MPO. Morgan et al. [124] demonstrated in mouse model that thiocyanate (SCN−) supplementation was associated with significantly decreased aortic root plaque areas without any significant changes in MPO levels. Similarly, Zietzer et al. [125] revealed that sodium thiocyanate treatment was associated with reduced atherosclerotic plaque formation and improved endothelial function in mouse model. Furthermore, Kim et al. [126] also showed that supplementation of the stable nitroxide radical, 4-hydroxy-2,2,6,6-tetramethylpiperidine-N-oxyl radical, could stabilize atherosclerotic plaques by increasing the collagen content and decreasing the lipid content within the plaques. Hence, these findings provide insights into the use of alternative substrates as another potential therapeutic target for atherosclerosis, which still needs to be warranted by future research.
5. Conclusion
The role of inflammation in CAD has been increasingly recognized, and recent research focused on discovering novel targets for the treatment of CAD. Since the mechanistic links by which MPO promotes the development and progression of atherosclerosis have been well studied, as well as extensive clinical evidence on MPO and the prognosis of CAD patients, some MPO inhibitors have been developed recently with interesting results shown in both preclinical and animal studies, such as ABAH, INV-315, PF-06282999, PF-1355, AZM198, triazolopyrimidines, acetaminophen, KYC, flavonoids, and alternative substrates. In addition, endogenous Cp also has potential to be an alternative approach to attenuate the atherogenic properties of MPO. Even though some of them were discontinued in research, the findings from previous studies have raised the possibility that MPO may be a promising target for future treatment of CAD. Research needs to verify the therapeutic effects of the current candidates for MPO inhibitor, PF-1355, AZM198, triazolopyrimidines, acetaminophen, KYC, flavonoids, Cp, and alternative substrates, whereas an increase in susceptibility to infection also needs to be considered.
6. Expert Opinion
Among those compounds, findings from recent studies suggested that PF-1355, AZM198, nitroxide radical, and thiocyanate may be the most promising MPO inhibitors for the prevention and treatment of atherosclerotic cardiovascular diseases. Each compound shows different atheroprotective effects. Firstly, PF-1355 is the only MPO inhibitor that could improve cardiac function and prevent ischemic cardiomyopathy [11], which is one of the most common etiologies of heart failure. However, there has been only one study demonstrating cardioprotective effects of PF-1355, and effects of PF-1355 on atherosclerotic burden need to be investigated. Also, since PF-1355 is a thiouracil derivative, there are possible severe adverse effects on the immune system, such as agranulocytosis, which is well known in the use of propylthiouracil for patients with hyperthyroidism [91]. Secondly, AZM198 and nitroxide radical were shown to stabilize atherosclerotic plaque [99, 126], thereby reducing the risk of acute coronary syndrome. Of note, stabilizing atherosclerotic plaque alone may not be able to reduce atherosclerotic burden, improve coronary blood flow, or reduce symptoms for patients with stable CAD. Therefore, AZM198 and nitroxide radical may only be beneficial to patients who have developed significant amount of stable atherosclerotic plaque. Thirdly, thiocyanate has been shown to reduce atherosclerotic plaque formation and progression [124, 125], suggesting that thiocyanate can potentially be used for both primary and secondary prevention of CAD without direct inhibition of MPO.
Nevertheless, there are challenges for the development of MPO inhibitors that need to be considered. It is still unclear whether systemic inhibition of MPO is necessary if mechanisms that directly target regional inflammatory responses can be feasible. The timing and the degree of inhibition may also influence its beneficial cardioprotective effects. Furthermore, whether such approaches can be effective in the subset of patients that have heightened inflammatory milieu would be intriguing. It is important to emphasize, however, that MPO levels tested by clinically available or research-based assays predominantly measure the mass concentration detected in the biospecimens and not their activities. On the other hand, there is a concern that direct inhibition of the inflammatory response may adversely affect the efficacy of the immune system to fight against infection. In addition, there is also relatively small evidence showing that MPO deficiency could be associated with increased atherosclerosis [47]. Hence, further investigations need to corroborate their cardioprotective effects, whereas possible adverse reactions, especially immune dysfunction, need to be monitored closely.
In the near future, based on accumulating evidence on atheroprotective effects of MPO inhibitors, we believe that research will continue to focus on developing different exogenous and endogenous MPO inhibitors, and there will be a variety of compounds discovered and demonstrated as potential novel therapeutic interventions for atherosclerotic cardiovascular diseases in in vitro and animal studies. Moreover, we also believe that the cardioprotective effects of the previous prototype compounds, especially PF-1355, AZM198, nitroxide radical, and thiocyanate will be further demonstrated in animal models, and potentially in clinical studies. However, adverse effects on the immune system may possibly be observed in direct systemic MPO inhibitors, whereas selective MPO inhibitors and alternative substrates, such as nitroxide radical and thiocyanate, may not or minimally affect the ability of immune cells to fight against infection. Therefore, targeting MPO has potential to be one of the most promising therapeutic approaches for CAD and other atherosclerotic cardiovascular diseases.
Article Highlights.
Myeloperoxidase (MPO) has been mechanistically linked to the development and progression of coronary artery disease (CAD) and other atherosclerotic cardiovascular diseases.
MPO plays a role in pathogenesis of atherosclerosis by oxidizing low-density lipoprotein, modifying high-density lipoprotein, promoting endothelial dysfunction, and weakening atherosclerotic plaque.
A variety of MPO inhibitors that provide atheroprotective effects have been developed and investigated in preclinical studies, including benzoic acid hydrazides, ferulic acid derivative, thiouracil derivatives, 2-thioxanthines derivatives, triazolopyrimidines, acetaminophen, N-acetyl lysyltyrosylcysteine amide, flavonoids, ceruloplasmin and alternative substrates.
Promising MPO inhibitors, such as PF-1355, AZM198, and alternative substrates, including nitroxide radical and thiocyanate, have been shown to reduce atherosclerotic burden, stabilize atherosclerotic plaque, and prevent cardiac complications from CAD in in vitro and animal studies.
Direct systemic inhibition of MPO may be associated with immune dysfunction, which needs to be monitored along with potential cardiovascular benefits.
Soon, there will be a variety of novel exogenous and endogenous MPO inhibitors developed and demonstrated in preclinical studies, whereas PF-1355, AZM198, nitroxide radical, and thiocyanate will be further investigated in animal models and potentially clinical studies.
Funding
W Tang is funded by grants from the National Institutes of Health, National Heart, Lung, and Blood Institute (R01HL103931, R01DL106000, R01HL126827) and National Institute of Diabetes and Digestive and Kidney Diseases R01DK106000
Footnotes
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers
- 1.Benjamin EJ, Virani SS, Callaway CW, et al. Heart Disease and Stroke Statistics—2018 Update: A Report From the American Heart Association. Circulation 2018;137(12):e67–e492. [DOI] [PubMed] [Google Scholar]
- 2.Hansson GK. Inflammation, Atherosclerosis, and Coronary Artery Disease. New England Journal of Medicine 2005;352(16):1685–95. [DOI] [PubMed] [Google Scholar]
- 3.Libby P, Loscalzo J, Ridker PM, et al. Inflammation, Immunity, and Infection in Atherothrombosis: JACC Review Topic of the Week. Journal of the American College of Cardiology 2018. 2018/October/23/;72(17):2071–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van der Wal AC, Becker AE, van der Loos CM, et al. Site of intimal rupture or erosion of thrombosed coronary atherosclerotic plaques is characterized by an inflammatory process irrespective of the dominant plaque morphology. Circulation 1994;89(1):36–44. [DOI] [PubMed] [Google Scholar]
- 5.Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. New England Journal of Medicine 2017;377(12):1119–31. [DOI] [PubMed] [Google Scholar]
- 6.Koeth RA, Haselden V, Tang WH. Myeloperoxidase in cardiovascular disease. Adv Clin Chem 2013;62:1–32. [DOI] [PubMed] [Google Scholar]
- 7.Brennan ML, Penn MS, Van Lente F, et al. Prognostic value of myeloperoxidase in patients with chest pain. N Engl J Med 2003;349(17):1595–604. [DOI] [PubMed] [Google Scholar]
- 8.Morrow DA, Sabatine MS, Brennan ML, et al. Concurrent evaluation of novel cardiac biomarkers in acute coronary syndrome: myeloperoxidase and soluble CD40 ligand and the risk of recurrent ischaemic events in TACTICS-TIMI 18. Eur Heart J 2008;29(9):1096–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang R, Brennan ML, Fu X, et al. Association between myeloperoxidase levels and risk of coronary artery disease. Jama 2001;286(17):2136–42. [DOI] [PubMed] [Google Scholar]
- 10.Meuwese MC, Stroes ES, Hazen SL, et al. Serum myeloperoxidase levels are associated with the future risk of coronary artery disease in apparently healthy individuals: the EPIC-Norfolk Prospective Population Study. J Am Coll Cardiol 2007;50(2):159–65. [DOI] [PubMed] [Google Scholar]
- 11.Ali M, Pulli B, Courties G, et al. Myeloperoxidase Inhibition Improves Ventricular Function and Remodeling After Experimental Myocardial Infarction. JACC: Basic to Translational Science 2016;1(7):633–43. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This study demonstrated that PF-1355 attenuated MPO activity, ischemic reperfusion injury, cardiac remodelling, and improved cardiac function.
- 12.Cheng D, Talib J, Stanley Christopher P, et al. Inhibition of MPO (Myeloperoxidase) Attenuates Endothelial Dysfunction in Mouse Models of Vascular Inflammation and Atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology 2019. 2019/July/01;39(7):1448–57. [DOI] [PubMed] [Google Scholar]; • This study demonstrated that AZM198 was associated with reduced MPO activity and improved endothelial function.
- 13.Teng N, Maghzal GJ, Talib J, et al. The roles of myeloperoxidase in coronary artery disease and its potential implication in plaque rupture. Redox report : communications in free radical research 2017;22(2):51–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khan AA, Alsahli MA, Rahmani AH. Myeloperoxidase as an Active Disease Biomarker: Recent Biochemical and Pathological Perspectives. Medical sciences (Basel, Switzerland) 2018;6(2):33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koeffler HP, Ranyard J, Pertcheck M. Myeloperoxidase: its structure and expression during myeloid differentiation. Blood 1985;65(2):484–91. [PubMed] [Google Scholar]
- 16.Swirski FK, Wildgruber M, Ueno T, et al. Myeloperoxidase-rich Ly-6C+ myeloid cells infiltrate allografts and contribute to an imaging signature of organ rejection in mice. J Clin Invest 2010;120(7):2627–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Olsson I, Persson AM, Stromberg K. Biosynthesis, transport and processing of myeloperoxidase in the human leukaemic promyelocytic cell line HL-60 and normal marrow cells. Biochem J 1984;223(3):911–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hansson M, Olsson I, Nauseef WM. Biosynthesis, processing, and sorting of human myeloperoxidase. Arch Biochem Biophys 2006;445(2):214–24. [DOI] [PubMed] [Google Scholar]
- 19.Nicholls SJ, Hazen SL. Myeloperoxidase and Cardiovascular Disease. Arteriosclerosis, Thrombosis, and Vascular Biology 2005;25(6):1102–11. [DOI] [PubMed] [Google Scholar]
- 20.Nussbaum C, Klinke A, Adam M, et al. Myeloperoxidase: A Leukocyte-Derived Protagonist of Inflammation and Cardiovascular Disease. Antioxidants & Redox Signaling 2012. 2013/February/20;18(6):692–713. [DOI] [PubMed] [Google Scholar]
- 21.Nauseef WM. Biosynthesis of human myeloperoxidase. Archives of Biochemistry and Biophysics 2018;642:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumar AP, Piedrafita FJ, Reynolds WF. Peroxisome proliferator-activated receptor gamma ligands regulate myeloperoxidase expression in macrophages by an estrogen-dependent mechanism involving the −463GA promoter polymorphism. J Biol Chem 2004;279(9):8300–15. [DOI] [PubMed] [Google Scholar]
- 23.Kettle AJ, Winterbourn CC. Myeloperoxidase: a key regulator of neutrophil oxidant production. Redox Report 1997. 1997/February/01;3(1):3–15. [DOI] [PubMed] [Google Scholar]
- 24.Klebanoff SJ. Myeloperoxidase: friend and foe. Journal of Leukocyte Biology 2005;77(5):598–625. [DOI] [PubMed] [Google Scholar]
- 25.Podrez EA, Abu-Soud HM, Hazen SL. Myeloperoxidase-generated oxidants and atherosclerosis. Free Radical Biology and Medicine 2000. 2000/June/15/;28(12):1717–25. [DOI] [PubMed] [Google Scholar]
- 26.Winterbourn CC, Hampton MB, Livesey JH, et al. Modeling the reactions of superoxide and myeloperoxidase in the neutrophil phagosome: implications for microbial killing. J Biol Chem 2006;281(52):39860–9. [DOI] [PubMed] [Google Scholar]
- 27.Prütz WA. Hypochlorous Acid Interactions with Thiols, Nucleotides, DNA, and Other Biological Substrates. Archives of Biochemistry and Biophysics 1996. 1996/August/01/;332(1):110–20. [DOI] [PubMed] [Google Scholar]
- 28.Fuchs B, Schiller J. Glycosaminoglycan Degradation by Selected Reactive Oxygen Species. Antioxidants & Redox Signaling 2013. 2014/September/01;21(7):1044–62. [DOI] [PubMed] [Google Scholar]
- 29.Kubala L, Kolářová H, Víteček J, et al. The potentiation of myeloperoxidase activity by the glycosaminoglycan-dependent binding of myeloperoxidase to proteins of the extracellular matrix. Biochimica et Biophysica Acta (BBA) - General Subjects 2013. 2013/October/01/;1830(10):4524–36. [DOI] [PubMed] [Google Scholar]
- 30.Hazen SL, Heinecke JW. 3-Chlorotyrosine, a specific marker of myeloperoxidase-catalyzed oxidation, is markedly elevated in low density lipoprotein isolated from human atherosclerotic intima. J Clin Invest 1997;99(9):2075–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takeshita J, Byun J, Nhan TQ, et al. Myeloperoxidase generates 5-chlorouracil in human atherosclerotic tissue: a potential pathway for somatic mutagenesis by macrophages. The Journal of biological chemistry 2006;281(6):3096–104. [DOI] [PubMed] [Google Scholar]
- 32.Hazell LJ, Arnold L, Flowers D, et al. Presence of hypochlorite-modified proteins in human atherosclerotic lesions. The Journal of clinical investigation 1996;97(6):1535–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hawkins CL, Davies MJ. Detection, identification, and quantification of oxidative protein modifications. The Journal of biological chemistry 2019;294(51):19683–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Savenkova MI, Mueller DM, Heinecke JW. Tyrosyl radical generated by myeloperoxidase is a physiological catalyst for the initiation of lipid peroxidation in low density lipoprotein. Journal of Biological Chemistry 1994;269(32):20394–400. [PubMed] [Google Scholar]
- 35.Jacob JS, Cistola DP, Hsu FF, et al. Human phagocytes employ the myeloperoxidase-hydrogen peroxide system to synthesize dityrosine, trityrosine, pulcherosine, and isodityrosine by a tyrosyl radical-dependent pathway. Journal of Biological Chemistry 1996;271(33):19950–56. [DOI] [PubMed] [Google Scholar]
- 36.Podrez EA, Schmitt D, Hoff HF, et al. Myeloperoxidase-generated reactive nitrogen species convert LDL into an atherogenic form in vitro. Journal of Clinical Investigation 1999;103(11):1547–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eiserich JP, Hristova M, Cross CE, et al. Formation of nitric oxide-derived inflammatory oxidants by myeloperoxidase in neutrophils. Nature 1998;391(6665):393–97. [DOI] [PubMed] [Google Scholar]
- 38.Hazen SL, Zhang R, Shen Z, et al. Formation of nitric oxide-derived oxidants by myeloperoxidase in monocytes pathways for monocyte-mediated protein nitration and lipid peroxidation in vivo. Circulation Research 1999;85(10):950–58. [DOI] [PubMed] [Google Scholar]
- 39.van Dalen CJ, Whitehouse MW, Winterbourn CC, et al. Thiocyanate and chloride as competing substrates for myeloperoxidase. Biochem J 1997;327(Pt 2):487–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Z, Nicholls SJ, Rodriguez ER, et al. Protein carbamylation links inflammation, smoking, uremia and atherogenesis. Nature Medicine 2007;13:1176. [DOI] [PubMed] [Google Scholar]
- 41.Daugherty A, Dunn JL, Rateri DL, et al. Myeloperoxidase, a catalyst for lipoprotein oxidation, is expressed in human atherosclerotic lesions. J Clin Invest 1994;94(1):437–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sugiyama S, Okada Y, Sukhova GK, et al. Macrophage myeloperoxidase regulation by granulocyte macrophage colony-stimulating factor in human atherosclerosis and implications in acute coronary syndromes. Am J Pathol 2001;158(3):879–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kutter D, Devaquet P, Vanderstocken G, et al. Consequences of Total and Subtotal Myeloperoxidase Deficiency: Risk or Benefit ? Acta Haematologica 2000;104(1):10–15. [DOI] [PubMed] [Google Scholar]
- 44.Asselbergs FW, Reynolds WF, Cohen-Tervaert JW, et al. Myeloperoxidase polymorphism related to cardiovascular events in coronary artery disease. The American Journal of Medicine 2004. 2004/March/15/;116(6):429–30. [DOI] [PubMed] [Google Scholar]
- 45.Brennan ML, Anderson MM, Shih DM, et al. Increased atherosclerosis in myeloperoxidase-deficient mice. J Clin Invest 2001;107(4):419–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McMillen TS, Heinecke JW, LeBoeuf RC. Expression of human myeloperoxidase by macrophages promotes atherosclerosis in mice. Circulation 2005;111(21):2798–804. [DOI] [PubMed] [Google Scholar]
- 47.Aratani Y Myeloperoxidase: Its role for host defense, inflammation, and neutrophil function. Archives of Biochemistry and Biophysics 2018;640:47–52. [DOI] [PubMed] [Google Scholar]
- 48.Michos ED, McEvoy JW, Blumenthal RS. Lipid Management for the Prevention of Atherosclerotic Cardiovascular Disease. New England Journal of Medicine 2019;381(16):1557–67. [DOI] [PubMed] [Google Scholar]
- 49.Hazell LJ, Stocker R. Oxidation of low-density lipoprotein with hypochlorite causes transformation of the lipoprotein into a high-uptake form for macrophages. The Biochemical journal 1993;290 (Pt 1)(Pt 1):165–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hazell LJ, van den Berg JJM, Stocker R. Oxidation of low-density lipoprotein by hypochlorite causes aggregation that is mediated by modification of lysine residues rather than lipid oxidation. Biochemical Journal 1994;302(1):297–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Malle E, Marsche G, Arnhold J, et al. Modification of low-density lipoprotein by myeloperoxidase-derived oxidants and reagent hypochlorous acid. Biochimica et biophysica acta 2006;1761(4):392–415. [DOI] [PubMed] [Google Scholar]
- 52.Podrez EA, Febbraio M, Sheibani N, et al. Macrophage scavenger receptor CD36 is the major receptor for LDL modified by monocyte-generated reactive nitrogen species. Journal of Clinical Investigation 2000;105(8):1095–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Steffen Y, Schewe T, Sies H. Epicatechin protects endothelial cells against oxidized LDL and maintains NO synthase. Biochemical and Biophysical Research Communications 2005. 2005/June/17/;331(4):1277–83. [DOI] [PubMed] [Google Scholar]
- 54.Vicca S, Massy ZA, Hennequin C, et al. Apoptotic pathways involved in U937 cells exposed to LDL oxidized by hypochlorous acid. Free Radical Biology and Medicine 2003. 2003/September/15/;35(6):603–15. [DOI] [PubMed] [Google Scholar]
- 55.de la Llera-Moya M, Drazul-Schrader D, Asztalos BF, et al. The ability to promote efflux via ABCA1 determines the capacity of serum specimens with similar high-density lipoprotein cholesterol to remove cholesterol from macrophages. Arterioscler Thromb Vasc Biol 2010;30(4):796–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rosenson RS, Brewer HB Jr., Ansell BJ, et al. Dysfunctional HDL and atherosclerotic cardiovascular disease. Nat Rev Cardiol 2016;13(1):48–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zheng L, Nukuna B, Brennan ML, et al. Apolipoprotein A-I is a selective target for myeloperoxidase-catalyzed oxidation and function impairment in subjects with cardiovascular disease. Journal of Clinical Investigation 2004;114(4):529–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zheng L, Settle M, Brubaker G, et al. Localization of nitration and chlorination sites on apolipoprotein A-I catalysed by myeloperoxidase in human atheroma and associated oxidative impairment in ABCA1-dependent cholesterol efflux from macrophages. Journal of Biological Chemistry 2005;280(1):38–47. [DOI] [PubMed] [Google Scholar]
- 59.Huang Y, Wu Z, Riwanto M, et al. Myeloperoxidase, paraoxonase-1, and HDL form a functional ternary complex. J Clin Invest 2013;123(9):3815–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shao B, Cavigiolio G, Brot N, et al. Methionine oxidation impairs reverse cholesterol transport by apolipoprotein A-I. Proceedings of the National Academy of Sciences 2008;105(34):12224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Smith JD. Myeloperoxidase, inflammation, and dysfunctional high-density lipoprotein. Journal of clinical lipidology 2010. Sep-Oct;4(5):382–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Peng DQ, Brubaker G, Wu Z, et al. Apolipoprotein A-I tryptophan substitution leads to resistance to myeloperoxidase-mediated loss of function. Arterioscler Thromb Vasc Biol 2008;28(11):2063–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Marsche G, Hammer A, Oskolkova O, et al. Hypochlorite-modified high density lipoprotein, a high affinity ligand to scavenger receptor class B, type I, impairs high density lipoprotein-dependent selective lipid uptake and reverse cholesterol transport. J Biol Chem 2002;277(35):32172–9. [DOI] [PubMed] [Google Scholar]
- 64.Zhang C, Reiter C, Eiserich JP, et al. L-arginine chlorination products inhibit endothelial nitric oxide production. J Biol Chem 2001;276(29):27159–65. [DOI] [PubMed] [Google Scholar]
- 65.Stocker R, Huang A, Jeranian E, et al. Hypochlorous Acid Impairs Endothelium-Derived Nitric Oxide Bioactivity Through a Superoxide-Dependent Mechanism. Arteriosclerosis, Thrombosis, and Vascular Biology 2004;24(11):2028–33. [DOI] [PubMed] [Google Scholar]
- 66.Marsche G, Heller R, Fauler G, et al. 2-Chlorohexadecanal derived from hypochlorite-modified high-density lipoprotein - Associated plasmalogen is a natural inhibitor of endothelial nitric oxide biosynthesis. Arteriosclerosis, Thrombosis, and Vascular Biology 2004;24(12):2302–06. [DOI] [PubMed] [Google Scholar]
- 67.Vita JA, Brennan ML, Gokce N, et al. Serum myeloperoxidase levels independently predict endothelial dysfunction in humans. Circulation 2004;110(9):1134–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Baldus S, Heitzer T, Eiserich JP, et al. Myeloperoxidase enhances nitric oxide catabolism during myocardial ischemia and reperfusion. Free Radic Biol Med 2004;37(6):902–11. [DOI] [PubMed] [Google Scholar]
- 69.Yunoki K, Naruko T, Komatsu R, et al. Relation of Elevated Levels of Plasma Myeloperoxidase to Impaired Myocardial Microcirculation After Reperfusion in Patients With Acute Myocardial Infarction. The American Journal of Cardiology 2010. 2010/April/01/;105(7):922–29. [DOI] [PubMed] [Google Scholar]
- 70.Narula J, Nakano M, Virmani R, et al. Histopathologic characteristics of atherosclerotic coronary disease and implications of the findings for the invasive and noninvasive detection of vulnerable plaques. J Am Coll Cardiol 2013;61(10):1041–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sugiyama S, Kugiyama K, Aikawa M, et al. Hypochlorous Acid, a Macrophage Product, Induces Endothelial Apoptosis and Tissue Factor Expression. Arteriosclerosis, Thrombosis, and Vascular Biology 2004;24(7):1309–14. [DOI] [PubMed] [Google Scholar]
- 72.Fu X, Kassim SY, Parks WC, et al. Hypochlorous acid oxygenates the cysteine switch domain of pro-matrilysin (MMP-7). A mechanism for matrix metalloproteinase activation and atherosclerotic plaque rupture by myeloperoxidase. J Biol Chem 2001;276(44):41279–87. [DOI] [PubMed] [Google Scholar]
- 73.Silvestre-Roig C, Winther MPd, Weber C, et al. Atherosclerotic Plaque Destabilization. Circulation Research 2014;114(1):214–26. [DOI] [PubMed] [Google Scholar]
- 74.Bennett MR, Sinha S, Owens GK. Vascular Smooth Muscle Cells in Atherosclerosis. Circulation Research 2016;118(4):692–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cai H, Chuang CY, Vanichkitrungruang S, et al. Hypochlorous acid-modified extracellular matrix contributes to the behavioral switching of human coronary artery smooth muscle cells. Free Radical Biology and Medicine 2019. 2019/April/01/;134:516–26. [DOI] [PubMed] [Google Scholar]
- 76.Vanichkitrungruang S, Chuang CY, Hawkins CL, et al. Oxidation of human plasma fibronectin by inflammatory oxidants perturbs endothelial cell function. Free Radical Biology and Medicine 2019. 2019/May/20/;136:118–34. [DOI] [PubMed] [Google Scholar]
- 77.Wang Y, Rosen H, Madtes DK, et al. Myeloperoxidase inactivates TIMP-1 by oxidizing its N-terminal cysteine residue: an oxidative mechanism for regulating proteolysis during inflammation. J Biol Chem 2007;282(44):31826–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tabas I Macrophage apoptosis in atherosclerosis: consequences on plaque progression and the role of endoplasmic reticulum stress. Antioxidants & Redox Signaling 2009;11(9):2333–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Doring Y, Soehnlein O, Weber C. Neutrophil Extracellular Traps in Atherosclerosis and Atherothrombosis. Circ Res 2017;120(4):736–43. [DOI] [PubMed] [Google Scholar]
- 80.Papayannopoulos V, Metzler KD, Hakkim A, et al. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. The Journal of Cell Biology 2010;191(3):677–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Borissoff JI, Joosen IA, Versteylen MO, et al. Elevated Levels of Circulating DNA and Chromatin Are Independently Associated With Severe Coronary Atherosclerosis and a Prothrombotic State. Arteriosclerosis, Thrombosis, and Vascular Biology 2013;33(8):2032–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Myeloperoxidase Ndrepepa G. - A bridge linking inflammation and oxidative stress with cardiovascular disease. Clin Chim Acta 2019;493:36–51. [DOI] [PubMed] [Google Scholar]
- 83.Kettle AJ, Gedye CA, Hampton MB, et al. Inhibition of myeloperoxidase by benzoic acid hydrazides. Biochem J 1995;308(Pt 2):559–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kettle AJ, Gedye CA, Winterbourn CC. Mechanism of inactivation of myeloperoxidase by 4-aminobenzoic acid hydrazide. Biochem J 1997;321(Pt 2):503–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Galijasevic S The development of myeloperoxidase inhibitors. Bioorg Med Chem Lett 2019;29(1):1–7. [DOI] [PubMed] [Google Scholar]
- 86.Han L, Shen X, Pan L, et al. Aminobenzoic acid hydrazide, a myeloperoxidase inhibitor, alters the adhesive properties of neutrophils isolated from acute myocardial infarction patients. Heart and Vessels 2012. September 01;27(5):468–74. [DOI] [PubMed] [Google Scholar]
- 87.Tiyerili V, Camara B, Becher MU, et al. Neutrophil-derived myeloperoxidase promotes atherogenesis and neointima formation in mice. International Journal of Cardiology 2016. 2016/February/01/;204:29–36. [DOI] [PubMed] [Google Scholar]
- 88.Kettle AJ, Gedye CA, Winterbourn CC. Superoxide is an antagonist of anti-inflammatory drugs that inhibit hypochlorous acid production by myeloperoxidase. Biochemical Pharmacology 1993. 1993/April/25/;45(10):2003–10. [DOI] [PubMed] [Google Scholar]
- 89.Kato Y, Nagao A, Terao J, et al. Inhibition of myeloperoxidase-catalyzed tyrosylation by phenolic antioxidants in vitro. Biosci Biotechnol Biochem 2003;67(5):1136–9. [DOI] [PubMed] [Google Scholar]
- 90.Liu C, Desikan R, Ying Z, et al. Effects of a novel pharmacologic inhibitor of myeloperoxidase in a mouse atherosclerosis model. PLOS ONE 2012;7(12):e50767–e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cooper DS. Antithyroid Drugs. New England Journal of Medicine 2005;352(9):905–17. [DOI] [PubMed] [Google Scholar]
- 92.Lee E, Hirouchi M, Hosokawa M, et al. Inactivation of peroxidases of rat bone marrow by repeated administration of propylthiouracil is accompanied by a change in the heme structure. Biochem Pharmacol 1988;37(11):2151–3. [DOI] [PubMed] [Google Scholar]
- 93.Ruggeri RB, Buckbinder L, Bagley SW, et al. Discovery of 2-(6-(5-Chloro-2-methoxyphenyl)-4-oxo-2-thioxo-3,4-dihydropyrimidin-1(2H)-yl)acet amide (PF-06282999): A Highly Selective Mechanism-Based Myeloperoxidase Inhibitor for the Treatment of Cardiovascular Diseases. J Med Chem 2015;58(21):8513–28. [DOI] [PubMed] [Google Scholar]
- 94.Dong JQ, Varma MV, Wolford A, et al. Pharmacokinetics and Disposition of the Thiouracil Derivative PF-06282999, an Orally Bioavailable, Irreversible Inactivator of Myeloperoxidase Enzyme, Across Animals and Humans. Drug Metabolism and Disposition 2016;44(2):209–19. [DOI] [PubMed] [Google Scholar]
- 95.Roth Flach RJ, Su C, Bollinger E, et al. Myeloperoxidase inhibition in mice alters atherosclerotic lesion composition. PLOS ONE 2019;14(3):e0214150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Li T, Jiang S, Ni B, et al. Discontinued Drugs for the Treatment of Cardiovascular Disease from 2016 to 2018. Int J Mol Sci 2019;20(18). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tiden AK, Sjogren T, Svensson M, et al. 2-thioxanthines are mechanism-based inactivators of myeloperoxidase that block oxidative stress during inflammation. J Biol Chem 2011;286(43):37578–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Björnsdottir H, Welin A, Michaëlsson E, et al. Neutrophil NET formation is regulated from the inside by myeloperoxidase-processed reactive oxygen species. Free Radical Biology and Medicine 2015. 2015/December/01/;89:1024–35. [DOI] [PubMed] [Google Scholar]
- 99.Rashid I, Maghzal GJ, Chen YC, et al. Myeloperoxidase is a potential molecular imaging and therapeutic target for the identification and stabilization of high-risk atherosclerotic plaque. Eur Heart J 2018;39(35):3301–10. [DOI] [PubMed] [Google Scholar]; • This study demonstrated that AZM198 was able to inhibit MPO, and stabilize atherosclerotic plaque by increasing fibrous cap thickness.
- 100.Maghzal GJ, Cergol KM, Shengule SR, et al. Assessment of myeloperoxidase activity by the conversion of hydroethidine to 2-chloroethidium. J Biol Chem 2014;289(9):5580–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Duclos F, Abell LM, Harden DG, et al. Triazolopyrimidines identified as reversible myeloperoxidase inhibitors. MedChemComm 2017;8(11):2093–99. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This study demonstrated that triazolopyrimidines inhibited MPO-mediated HDL oxidation, and HOCl production.
- 102.Koelsch M, Mallak R, Graham GG, et al. Acetaminophen (paracetamol) inhibits myeloperoxidase-catalyzed oxidant production and biological damage at therapeutically achievable concentrations. Biochem Pharmacol 2010;79(8):1156–64. [DOI] [PubMed] [Google Scholar]
- 103.Marquez LA, Dunford HB. Interaction of Acetaminophen with Myeloperoxidase Intermediates: Optimum Stimulation of Enzyme Activity. Archives of Biochemistry and Biophysics 1993. 1993/September/01/;305(2):414–20. [DOI] [PubMed] [Google Scholar]
- 104.Van Zyl JM, Basson K, Van Der Walt BJ. The inhibitory effect of acetaminophen on the myeloperoxidase-induced antimicrobial system of the polymorphonuclear leukocyte. Biochemical Pharmacology 1989. 1989/January/01/;38(1):161–65. [DOI] [PubMed] [Google Scholar]
- 105.Nenseter MS, Halvorsen B, Rosvold O, et al. Paracetamol inhibits copper ion-induced, azo compound-initiated, and mononuclear cell-mediated oxidative modification of LDL. Arterioscler Thromb Vasc Biol 1995;15(9):1338–44. [DOI] [PubMed] [Google Scholar]
- 106.Chou TM, Greenspan P. Effect of acetaminophen on the myeloperoxidase-hydrogen peroxide-nitrite mediated oxidation of LDL. Biochimica et biophysica acta 2002;15:1–2. [DOI] [PubMed] [Google Scholar]
- 107.Kapiotis S, Sengoelge G, Hermann M, et al. Paracetamol Catalyzes Myeloperoxidase-Initiated Lipid Oxidation in LDL. Arteriosclerosis, Thrombosis, and Vascular Biology 1997;17(11):2855–60. [DOI] [PubMed] [Google Scholar]
- 108.Ozsoy MB, Pabuçcuoğlu A. The effect of acetaminophen on oxidative modification of low-density lipoproteins in hypercholesterolemic rabbits. Journal of clinical biochemistry and nutrition 2007;41(1):27–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhang H, Jing X, Shi Y, et al. N-acetyl lysyltyrosylcysteine amide inhibits myeloperoxidase, a novel tripeptide inhibitor. Journal of lipid research 2013;54(11):3016–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yu G, Liang Y, Huang Z, et al. Inhibition of myeloperoxidase oxidant production by N-acetyl lysyltyrosylcysteine amide reduces brain damage in a murine model of stroke. J Neuroinflammation 2016;13(1):016–0583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yu G, Zheng S, Liang Y, et al. Inhibition of myeloperoxidase by N-acetyl lysyltyrosylcysteine amide reduces oxidative stress-mediated inflammation, neuronal damage, and neural stem cell injury in a murine model of stroke. Journal of Pharmacology and Experimental Therapeutics 2017:jpet.117.245688. [DOI] [PubMed] [Google Scholar]
- 112.Shiba Y, Kinoshita T, Chuman H, et al. Flavonoids as Substrates and Inhibitors of Myeloperoxidase: Molecular Actions of Aglycone and Metabolites. Chemical Research in Toxicology 2008. 2008/August/01;21(8):1600–09. [DOI] [PubMed] [Google Scholar]
- 113.Zeka K, Ruparelia K, Arroo RRJ, et al. Flavonoids and Their Metabolites: Prevention in Cardiovascular Diseases and Diabetes. Diseases 2017;5(3):19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hertog MGL, Feskens EJM, Kromhout D, et al. Dietary antioxidant flavonoids and risk of coronary heart disease: the Zutphen Elderly Study. The Lancet 1993. 1993/October/23/;342(8878):1007–11. [DOI] [PubMed] [Google Scholar]
- 115.Kostyuk VA, Kraemer T, Sies H, et al. Myeloperoxidase/nitrite-mediated lipid peroxidation of low-density lipoprotein as modulated by flavonoids. FEBS Letters 2003;537(1–3):146–50. [DOI] [PubMed] [Google Scholar]
- 116.Loke WM, Proudfoot JM, McKinley AJ, et al. Quercetin and Its In Vivo Metabolites Inhibit Neutrophil-Mediated Low-Density Lipoprotein Oxidation. Journal of Agricultural and Food Chemistry 2008. 2008/May/01;56(10):3609–15. [DOI] [PubMed] [Google Scholar]
- 117.Steffen Y, Schewe T, Sies H. Myeloperoxidase-mediated LDL oxidation and endothelial cell toxicity of oxidized LDL: attenuation by (−)-epicatechin. Free radical research 2006. 2006/January/01;40(10):1076–85. [DOI] [PubMed] [Google Scholar]
- 118.Bhaskar S, Kumar KS, Krishnan K, et al. Quercetin alleviates hypercholesterolemic diet induced inflammation during progression and regression of atherosclerosis in rabbits. Nutrition 2013. 2013/January/01/;29(1):219–29. [DOI] [PubMed] [Google Scholar]
- 119.Lu N, Sui Y, Tian R, et al. Inhibitive Effects of Quercetin on Myeloperoxidase-Dependent Hypochlorous Acid Formation and Vascular Endothelial Injury. Journal of Agricultural and Food Chemistry 2018. 2018/May/16;66(19):4933–40. [DOI] [PubMed] [Google Scholar]; •• This study demonstrated that quercetin inhibited MPO-mediated HOCl production, and improved endothelial function.
- 120.Taylor JC, Oey L. Ceruloplasmin: plasma inhibitor of the oxidative inactivation of alpha 1-protease inhibitor. Am Rev Respir Dis 1982;126(3):476–82. [DOI] [PubMed] [Google Scholar]
- 121.Segelmark M, Persson B, Hellmark T, et al. Binding and inhibition of myeloperoxidase (MPO): a major function of ceruloplasmin? Clin Exp Immunol 1997;108(1):167–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chapman ALP, Mocatta TJ, Shiva S, et al. Ceruloplasmin is an endogenous inhibitor of myeloperoxidase. The Journal of biological chemistry 2013;288(9):6465–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kennedy DJ, Weber ME, Wang X, et al. Abstract 19636: Cardioprotective Role of Ceruloplasmin in Heart Failure via Inhibition of Myeloperoxidase Activity. Circulation 2014;130(suppl_2):A19636–A36. [Google Scholar]
- 124.Morgan PE, Laura RP, Maki RA, et al. Thiocyanate supplementation decreases atherosclerotic plaque in mice expressing human myeloperoxidase. Free radical research 2015;49(6):743–49. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This study demonstrated that thiocyanate reduced atherosclerotic plaque size with stable MPO level.
- 125.Zietzer A, Niepmann ST, Camara B, et al. Sodium thiocyanate treatment attenuates atherosclerotic plaque formation and improves endothelial regeneration in mice. PLOS ONE 2019;14(4). [DOI] [PMC free article] [PubMed] [Google Scholar]; • This study demonstrated that sodium thiocyanate was associated with decreased atherosclerotic plaque formation, and improved endothelial function.
- 126.Kim CHJ, Mitchell JB, Bursill CA, et al. The nitroxide radical TEMPOL prevents obesity, hyperlipidaemia, elevation of inflammatory cytokines, and modulates atherosclerotic plaque composition in apoE−/− mice. Atherosclerosis 2015. 2015/May/01/;240(1):234–41. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This study demonstrated that nitroxide radical was able to stabilize atherosclerotic plaque by increasing the collagen and decreasing the lipid content.