

Abstract
Background & Aims:
We investigated whether ABL proto-oncogene 1, non-receptor tyrosine kinase (ABL1) is involved in development of hepatocellular carcinoma (HCC).
Methods:
We analyzed clinical and gene expression data from The Cancer Genome Atlas. Albumin-Cre (HepWT) mice and mice with hepatocyte-specific disruption of Abl1 (HepAbl−/− mice) were given hydrodynamic injections of plasmids encoding the sleeping beauty transposase and transposons with the MET gene and a catenin beta 1 gene with an N-terminal truncation, which induces development of liver tumors. Some mice were then gavaged with the ABL1 inhibitor nilotinib or vehicle (control), daily for 4 weeks. We knocked down ABL1 with short hairpin RNAs in Hep3B and Huh7 HCC cells and analyzed their proliferation and growth as xenograft tumors in mice. We performed RNA sequencing and gene set enrichment analysis of tumors. We knocked down or overexpressed NOTCH1 and MYC in HCC cells and analyzed proliferation. We measured levels of phosphorylated ABL1, MYC, and NOTCH1 by immunohistochemical analysis of an HCC tissue microarray.
Results:
HCC tissues had higher levels of ABL1 than non-tumor liver tissues, which correlated with shorter survival times of patients. HepWT mice with the MET and catenin beta 1 transposons developed liver tumors and survived a median 64 days; HepAbl−/− mice with these transposons developed tumors that were 50% smaller and survived a median 81 days. Knockdown of ABL1 in human HCC cells reduced proliferation, growth as xenograft tumors in mice, and expression of MYC, which reduced expression of NOTCH1. . Knockdown of NOTCH1 or MYC in HCC cells significantly reduced cell growth. NOTCH1 or MYC overexpression in human HCC cells promoted proliferation and rescued the phenotype caused by ABL1 knockdown. The level of phosphorylated (activated) ABL1 correlated with levels of MYC and NOTCH1 in human HCC specimens. Nilotinib decreased the expression of MYC and NOTCH1 in HCC cell lines, reduced their growth of xenograft tumors in mice, and slowed growth of liver tumors in mice with MET and catenin beta 1 transposons, reducing tumor levels of MYC and NOTCH1.
Conclusions:
HCC samples have increased levels of ABL1 compared with non-tumor liver tissues, and increased levels of ABL1 correlate with shorter survival times of patients. Loss or inhibition of ABL1 reduces proliferation of HCC cells and slows growth of liver tumors in mice. Inhibitors of ABL1 might be used for treatment of HCC.
Keywords: hepatocarcinogenesis, mouse model, signal transduction, oncogene
Lay Summary:
We identified a protein that is upregulated in human HCC samples. When we deleted or inhibited this protein in mice, liver tumors grew more slowly and the mice had longer survival times.
Introduction
Hepatocellular carcinoma (HCC) is the major form of liver cancer. It is the sixth most common malignancy globally and ranks fourth in total cancer-related deaths annually.1 The five-year overall survival of patients with a new diagnosis of HCC is less than 18%, and a majority of HCC patients present with advanced disease so that treatment options are limited.2 Currently, first-line therapeutic agents for advanced HCC, either sorafenib or lenvatinib, increase survival by only approximately 3 months.3 Recently a number of drugs, including regorafenib, cabozantinib, and nivolumab, have been approved by the FDA for second-line treatment of HCC.3 However, these drugs only offer a further increase in overall survival of 3–5 months. Therefore, it is imperative to develop new and more effective therapeutic strategies and agents to treat HCC, but achieving this goal requires a better understanding of the molecular signaling pathways that drive or mediate the development of the disease.
Abelson tyrosine-protein kinase 1 (ABL1) is a non-receptor tyrosine kinase of the Abelson-like family. It is mostly known for its involvement in leukemias harboring the Philadelphia chromosome, which results from the translocation of the short arms of chromosomes 9 and 22, creating a fusion of the BCR gene to the second exon of the ABL1 gene, resulting in the production of the BCR-ABL fusion protein.4 Recent evidence has shown that ABL1 also plays an important role in the development of solid tumors, such as melanoma, breast cancer, ovarian cancer and lung cancer, by an independent mechanism not involving any fusion oncoproteins.4 We previously reported that ABL1 is overexpressed and activated in human HCC specimens.5 However, it remains to understand the role of ABL1 in hepatocarcinogenesis, since this is critical for determining whether ABL1 is a suitablea candidate target in the treatment of HCC.
We report here that overexpression of ABL1 correlates with a poor prognosis in HCC. We investigated the role of ABL1 in HCC growth using in vitro and mouse models. We found that ABL1 inhibition impaired HCC growth and extended overall survival of mice with HCC. Mechanistically, we found that inhibition of ABL1 suppresses HCC cell growth by decreasing NOTCH1 expression through the regulation of c-MYC. Collectively, our data strongly suggest that ABL1 is involved in the pathogenesis of HCC and that its inhibition could be a promising novel strategy to treat this disease.
Methods
Cells and treatments
Huh7 cells were purchased from JCRB Cell Bank. Hep3B, Skep1, SNU423, SNU449, SNU475, PLC/PRF, SNU387 and 293T cells were purchased from ATCC. All cells were cultured as described previously.6
For knockdown experiments, Huh7 and Hep3B cells were infected with lentiviral pLKO.1 particles which contain ABL1, NOTCH1, c-MYC or scrambled shRNA and selected with 2μg/mL puromycin for 5 days. Lentiviral pLKO.1 plasmids for shABL1 (Supporting Table 1), shNOTCH1 (Supporting Table 1), shc-MYC7 or scrambled shRNA (SHC002, Sigma-Aldrich) were packaged with pCMV-dr8.2 (Addgene) and pCMV-VSVG (Addgene) in 293T cells to produce lentiviral particles as previously described.7
For NOTCH1 overexpression experiments, the NOTCH1 (NOTCH intercellular domain (NICD)) expression plasmid (EF.hICN1.CMV.GFP), purchased from Addgene (#17623), was packaged with CMV-dr8.2 and pCMV-VSVG in 293T cells to produce lentiviral particles. Six days after infection with the lentiviral particles, GFP-positive scrambled and ABL1 KD Huh7 cells expressing NOTCH1 were sorted by flow cytometry (FACSAria Cell Sorter). The proliferation of these cells was then analyzed using alamarBlue assay as previously described.6
For experiments involving the overexpression of c-MYC, the pBpuro c-MycER™ retroviral plasmid (gifted from Dr. Gerard Evan)8 and control pBpuro retroviral plasmids (Addgene, #1764) were packaged with pMD.MLV and pMD.G/pVSV.G in 293T cells to produce retroviral particles. Six days after infection with the retroviral particles, scrambled and ABL1 KD Huh7 cells were treated with 100nM 4-hydroxytamoxifen. The proliferation of these cells was then analyzed using alamarBlue assay.
For ABL inhibitor experiments, HCC cells were seeded into 96 well plates. After 24h. in culture, the cells were treated with nilotinib (LC lab, Cat#N-8207) (1–20μM) or GNF-5 (Selleckchem, Cat#S7526) (1–20μM); cell proliferation was then analyzed using either SRB9 or alamarBlue assay after 48 or 72 hours.
Mice and treatments
All animals received humane care according to the “Guide for the Care and Use of Laboratory Animals”. The procedures for all animal experiments detailed below were approved by the Institutional Animal Care and Use Committee of Loyola University Chicago. All mice were housed in micro-isolator cages in a room illuminated from 7:00 AM to 7:00 PM (12:12-hr. light-dark cycle) and were given access to water and chow ad libitum.
To generate mice with hepatocyte-specific Abl1-deficiency, Abl1flox/flox mice (JAX, Cat # 013224) were backcrossed to C57BL/6J mice for five generations and then were mated with Albumin-Cre mice (JAX, Cat # 003574). The resulting offspring Alb-Cre; Abl1flox/+ mice were then mated to generate the Albumin-Cre and Alb-Cre; Abl1flox/flox littermates. The age (6–8 weeks old) - and gender-matched Albumin-Cre and Alb-Cre; Abl1flox/flox mice were injected with plasmids, encoding the Sleeping Beauty transposase (HSB2) and transposons with GFP (pT3-GFP) or MET gene and catenin beta 1 gene with the N terminal truncation (referred to here as MET/CAT) as described previously.10
Four weeks after MET/CAT injection, C57BL/6J wildtype mice were given with vehicle (30% captisol), nilotinib (20mg/kg), or Sorafenib (25mg/kg) by oral gavage daily for 4 weeks or prior to being sacrificed (some mice treated with vehicle had to be euthanized earlier due to tumor burdens). Age- and gender-matched mice were allocated to different treatment groups. Both male and female mice were used in the experiments. Six-to-eight week old mice were used for hydrodynamic injections.
Xenograft Model: Huh7 cells (5×106 in 100μL serum-free medium) were injected into the left or right flanks of the 8–12-week-old SCID-bg mice. Three weeks post-injection, some mice were given with vehicle (30% captisol) or nilotinib (20mg/kg) by oral gavage daily for 10 days. Tumor volumes were measured daily using a caliper until the day of sacrifice.
Western blotting
Western blotting was performed as previously described.6 Information on primary antibodies is shown in Supporting Table 2.
Quantitative real-time polymerase chain reaction
Cellular or tissue mRNA was extracted using Zymo mini-columns and quantitative real-time polymerase chain reactions were performed as previously described.6 Primers used for RT-PCR are listed in Supporting Table 3.
Immunohistochemical (IHC) staining
IHC was performed as previously described.10 Human tissue microarrays (TMAs) LV801, LV807, and LV8012 were purchased from US Biomax (Rockville, MD, USA). LV801 and LV807 contain a total of 66 cases of HCC and 50 cases of HCC adjacent normal or normal liver tissues. LV8012 contains 80 cases of HCC (TNM stage II-IV). The IHC signals were quantified visually. The staining was scored as − (0, negative), + (1, weak signal), + + (2, moderate signal), + + + (3, strong signal) by two independent observers including a pathologist from Loyola University Chicago; a sample was rated as positive if it showed at least 1% of cells with a staining score ≥1+. For IHC on mouse samples, cells with positive staining were scored in at least 5 fields at 400× or 200× magnification and reported as mean ± SD. Information on primary antibodies for IHC is listed in Supporting Table 2.
TUNEL staining
TUNEL staining was performed as previously described.6 The TUNEL-positive cell number was scored in at least five fields at 400× magnification/mouse and reported as means ±SD. Three or more mice were used in each group.
RNA sequencing (RNA-seq) and analysis
RNA from scrambled control and ABL1-KD Huh7 cells was extracted using RNeasy Plus Micro Kit (Qiagen, Germantown, MD). RNA-seq was performed by Novogene Corporation. Gene set enrichment analysis was performed using the 3.0 GSEA software. The RNA sequencing data were deposited into the NCBI’s Gene Expression Omnibus database (GEO GSE133294. URL: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE133294).
Chromatin immunoprecipitation assay
Huh7 cell were cultured as described above and chromatin immunoprecipitation (ChIP) assays were performed as previously described11 using a c-MYC antibody (#5605, Cell Signaling). The primers used are listed in Supporting Table S3.
Proximity ligation assay
Proximity ligation assay (PLA) was performed using the Duolink® In Situ Red Starter Kit Mouse/Rabbit (Millipore Sigma) according to the manufacturer’s instructions. Details are provided in supplemental materials.
Human sample analysis
Alterations of ABL1, NOTCH1, and c-MYC mRNA were analyzed from publicly available TCGA data.12 Analysis of gene expression, Kaplan-Meier survival analyses and correlations were performed using R 3.6.0, Python 3.0 and GraphPad Prism 8.0 software. Details are provided in supplemental materials.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 8.0 software. Variation is indicated using standard error presented as means ± standard deviations (SD). Statistical significance was calculated using the two-tailed Student’s t- test, except for the experiments involving repeated measures, which were analyzed using 2-way ANOVA. P < 0.05 was considered significant. The means ± SDs are shown in the Figures where applicable.
Results
High expression of ABL1 in human HCCs is positively correlated with shorter survival times of patients
We previously reported that ABL1 is overexpressed in human HCC specimens with a small sample size.5 To confirm the results with a larger sample size, we analyzed the TCGA database and confirmed that ABL1 was expressed at higher levels in HCCs as compared to normal liver tissues (Fig. 1A). In addition, we found that higher ABL1 expression is positively correlated with poorer prognosis in human HCC patients from the TCGA database (Fig. 1B and Fig. S1), which suggests that it could be a good prognostic factor. We also performed two additional HCC tissue microarrays, which contain 66 cases of HCC and 50 cases of HCC adjacent normal or normal liver tissues. Consistent with previous results,5 we found that ABL1 protein levels were significantly higher in tumors compared to normal liver tissues (Fig. 1, C&D). Kinase activity is critical for the functions of ABL1.13 Phosphorylation of Tyr412, which is located in the kinase activation loop of ABL1, is required for its kinase activity.14, 15 We found that the level of p-ABL1 (p-Tyr412) is largely absent in normal liver tissues but is abundant in HCC specimens (Fig. 1, C&E). It is notable that 86% of HCC specimens with high p-ABL1 (p-Tyr412) staining also express high levels of ABL1 (Fig. 1, D&E). In general, these data indicate that ABL1 is overexpressed and activated in human HCCs, and that these factors correlate with shorter survival times of patients.
Figure 1. High expression of ABL1 in human HCCs is positively correlated with shorter survival times of patients.
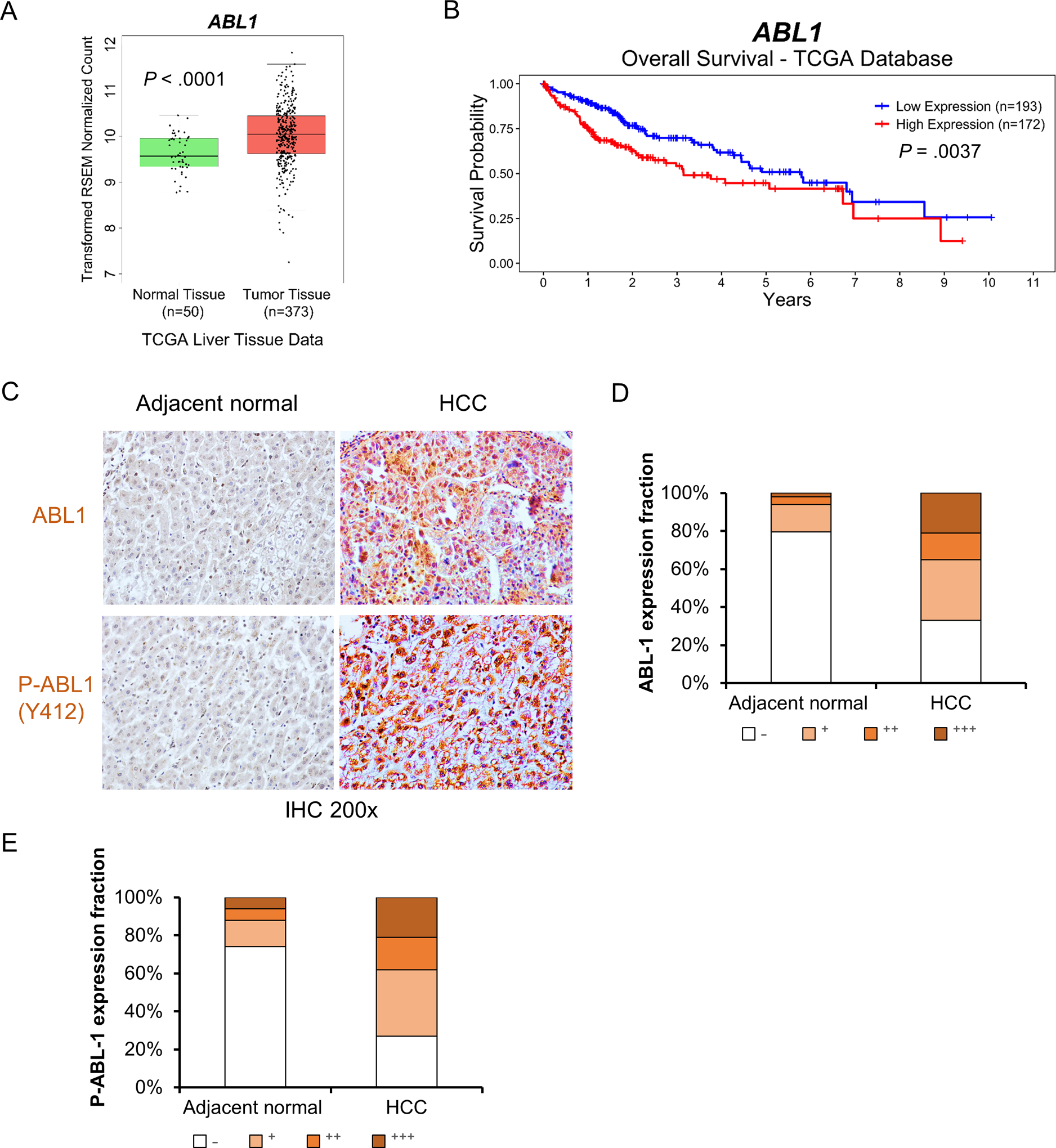
(A) Relative expression of ABL1 mRNA in normal liver and HCC specimens from the TCGA database. (B) ABL1 mRNA expression is correlated with shorter survival times of HCC patients. (C) Representative photos of ABL1 and p-ABL1 IHC staining in adjacent normal liver and HCC specimens from tissue microarrays. (D&E) Quantification of ABL1 and p-ABL1 IHC staining from tissue microarrays (66 cases of HCC and 50 normal tissue specimens).
Deletion of Abl1 in hepatocytes does not affect morphology, histology, proliferation, nor apoptosis in mouse livers
To investigate the role of ABL1 in liver tumorigenesis, we generated mice with hepatocyte-specific deletion of Abl1 (Albumin-Cre; Abl1flox/flox). Albumin-Cre; Abl1flox/flox mice (referred to here as HepAbl1−/−) express Cre recombinase from the albumin promoter, which is specifically expressed in hepatocytes. HepAbl1−/− mice are viable, fertile and visually indistinguishable from wild-type (Albumin-Cre, referred to here as HepWT) mice, suggesting that Abl1 is not required for normal liver development. We confirmed that Cre recombinase expression in hepatocytes removes the Abl1 allele between two loxP sites (Fig. S2A) and showed that ABL1 expression was decreased in both whole liver tissue and hepatocytes (Fig. S2B). There was no significant difference in morphology or histology of livers between HepWT and HepAbl1−/− mice (Fig. S2, C&D). Furthermore, Abl1 deficiency did not affect cell proliferation or apoptosis in mouse livers (Fig. S2, E–H). These results suggest that deletion of Abl1 in hepatocytes does not affect mouse liver homeostasis.
Deficiency of Abl1 in hepatocytes suppresses MET/CAT-induced HCC development
MET can bind directly to ABL1 and activate it in mouse mammary tumors and breast cancer cells.16 To determine the role of ABL1 in HCC development, we used the MET (MET)/β-catenin (CAT)-driven HCC model, which is useful for studying the functions of genes in hepatocarcinogenesis because of its clinical relevance and efficiency of HCC induction.10, 17 We found that levels of phosphorylation of both ABL1 on Tyr412 and CRKL, a direct target of ABL kinases18 commonly used to assess ABL kinase activity, was increased in MET/CAT-induced liver tumors, suggesting that ABL1 is activated in MET/CAT-induced HCC (Fig. 2A). Expression of ABL1 was also increased in MET/CAT-induced liver tumors (Fig. 2A). In contrast, expression or phosphorylation of ABL1 was not altered in the diethylnitrosamine (DEN)-induced HCC model (Fig. S3). These observations suggest that the MET/CAT model is suitable for studying the role of ABL1 in hepatocarcinogenesis. We hydrodynamically injected age- and gender-matched HepWT and HepAbl1−/− mice with plasmids encoding the Sleeping Beauty transposase (HSB2) and transposons with the MET/CAT oncogenes. Comparable transfection efficiency was observed in WT and Abl1 KO mouse livers (Fig. S4). Intriguingly, we found that the overall tumor load and tumor sizes in HepAbl1−/− mice was significantly decreased compared to HepWT mice (Fig. 2B). The relative liver weight, including tumor versus body weight, in HepAbl1−/− mice was decreased by 50% compared to HepWT mice (Fig. 2C). Importantly, the HepWT mice with a liver tumor burden died at the age of 55–70 days (median survival 64 days), compared to a median survival of 81 days in HepAbl1−/− mice (Fig. 2D). These data indicate that Abl1 deficiency in hepatocytes suppresses MET/CAT-induced HCC growth and prolongs survival of mice with HCC.
Figure 2. Deletion of Abl1 suppresses tumor development and prolongs survival in the MET/CAT-induced HCC mouse model.
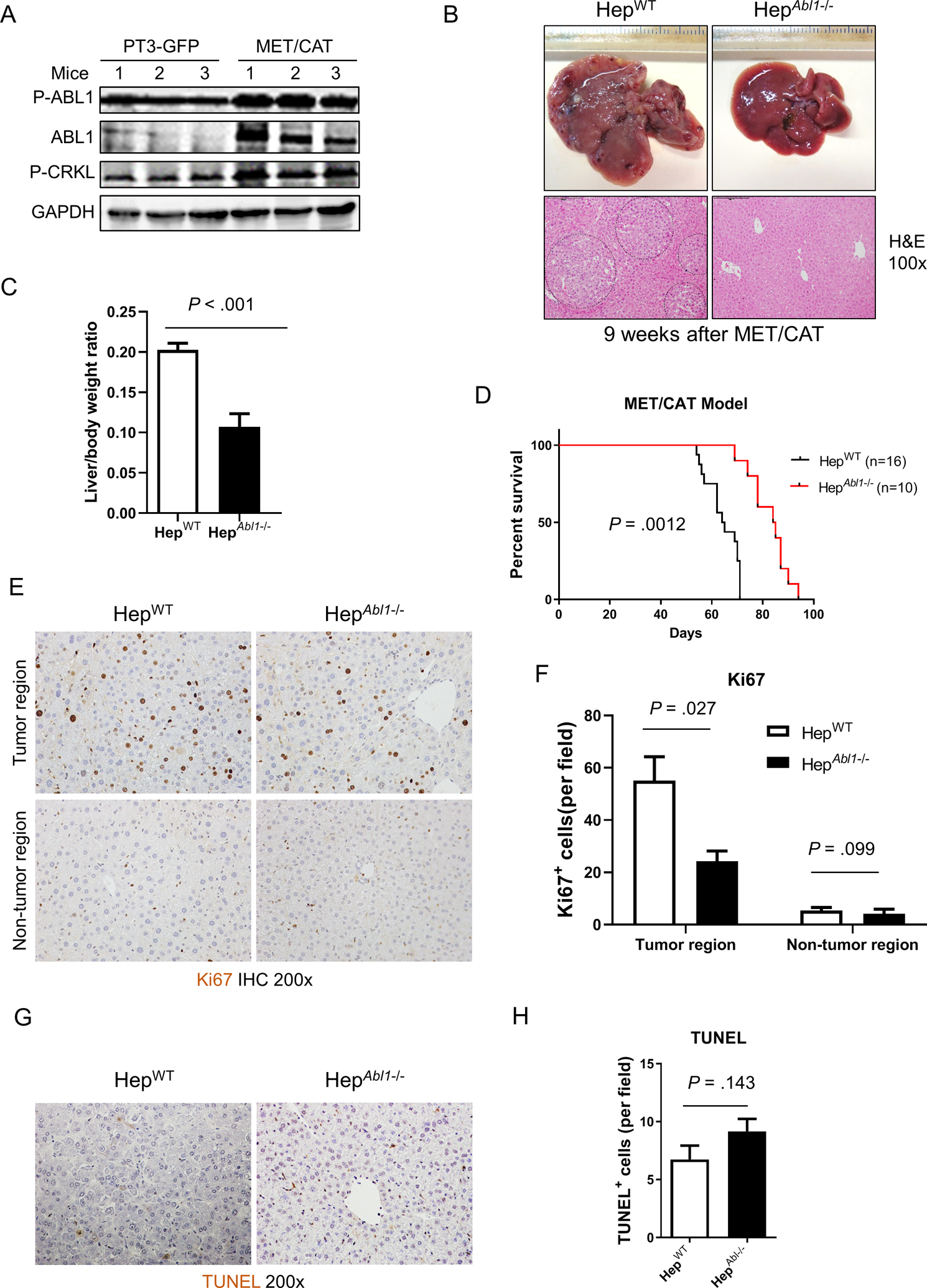
(A) Levels of p-ABL1 (p-Y412), ABL1, p-CRKL and GAPDH proteins in the livers of HepWT mice 9 weeks after hydrodynamic injection of MET/CAT or pT3-GFP. (B) Photographs and H&E staining of livers of HepWT and HepAbl1−/− mice 9 weeks after injection of MET/CAT. (C) Liver body/weight ratios were analyzed in the mice from (B) (n=6). (D) Survival curves of HepWT and HepAbl1−/− mice after injection of MET/CAT. (E) Hepatocyte proliferation of HepWT and HepAbl1−/− mice 9 weeks after injection of MET/CAT was examined by immunohistochemistry for Ki67. (F) Quantification of Ki67 staining for (E) (n=4). (G) Apoptosis in the livers of HepWT and HepAbl1−/− mice 9 weeks after injection of MET/CAT was examined by TUNEL staining. (H) Quantification of TUNEL staining for (G) (n=4).
ABL1 regulates cell survival and proliferation.4 Suppression of HCC development by the deletion of Abl1 could be due to increased apoptosis or decreased proliferation of tumor cells. We first analyzed proliferation in the MET/CAT-injected livers from HepWT and HepAbl1−/− mice by Ki67 staining. The number of Ki67-positive cells was significantly decreased in tumors but not in tumor-free areas in Abl1-deficient livers compared to WT livers (Fig. 2E and 2F). We did not find significant differences in apoptosis in livers of HepWT mice compared to those of HepAbl1−/− mice (Fig. 2G and 2H). These results demonstrate that Abl1 deficiency in hepatocytes decreases tumor cell proliferation but not cell survival in MET/CAT-induced HCC.
Knockdown of ABL1 reduces human HCC cell proliferation and suppresses tumor growth
We further investigated the role of ABL1 in human HCC. To achieve this goal, we used short hairpin RNA (shRNA) to knock down ABL1 expression in two HCC cell lines that show high levels of ABL1 mRNA, Hep3B and Huh7, (Fig. S5). Western blotting showed that ABL1 was successfully knocked down by two shRNAs in both cell lines (Fig. 3A). Importantly, knockdown of ABL1 significantly reduced cell growth in both Hep3B and Huh7 cells (Fig. 3B). Consistently, ABL1 knockdown decreased growth as xenograft tumors in mice (Fig. 3, C–E). Tumors grown from ABL1-knockdown (KD) cells also displayed less cell proliferation compared to those grown from scrambled-RNA control cells (Fig. 3, F&G). However, there was no significant difference in apoptosis in tumors from the two groups (Fig. 3, H&I). Collectively, these results indicate that knockdown of ABL1 reduces human HCC cell proliferation and suppresses tumor growth.
Figure 3. ABL1 Knockdown reduces human HCC cell proliferation and suppresses tumor growth.
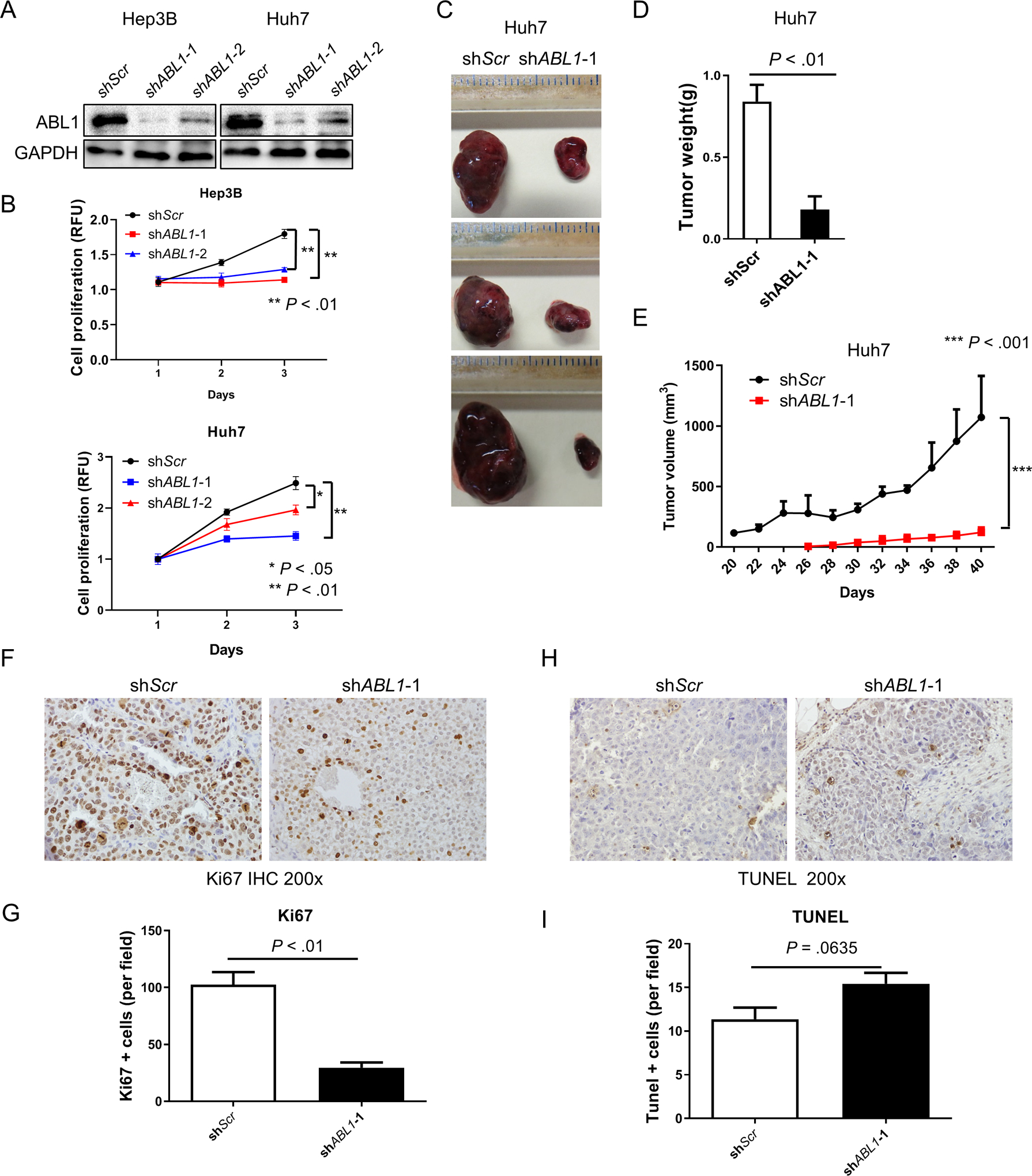
(A) ABL1 and GAPDH protein expression in scrambled-RNA and ABL1-KD Hep3B and Huh7 cells was determined by Western blotting. (B) Quantification of cell proliferation from scrambled-RNA and ABL1-KD Hep3B and Huh7 cells at different time points after seeding. (C) SCID-bg mice were injected on their flanks with scrambled-RNA (left) and ABL1-KD Huh7 cells (right); after 40 days, gross images of tumors are shown. (D) Tumor weight from scrambled-RNA and ABL1-KD Huh7 cell-injected mice (n=5). (E) Tumor volumes from scrambled-RNA and ABL1-KD Huh7 cell-injected mice (n=5/group). (F) Ki67 staining of the tumors was examined by IHC. (G) Quantification of Ki67 staining. (H) Apoptosis of the tumors was examined by TUNEL staining. (I) Quantification of TUNEL staining.
Knockdown of ABL1 inhibits HCC cell proliferation by decreasing NOTCH1 expression
To determine the molecular mechanisms by which ABL1 promotes HCC cell proliferation, we performed RNA-seq analysis using scrambled-control and ABL1-KD cells and proceeded to do Gene Set Enrichment Analysis (GSEA). NOTCH signaling is one of the most significantly downregulated gene pathways that results from ABL1 knockdown (Fig. 4, A&B). NOTCH signaling has been shown to play an important role in tumor cell growth in many types of cancer,19 including HCC. We therefore hypothesized that knockdown of ABL1 inhibits HCC cell proliferation by inhibiting signaling along the NOTCH pathway. We used real-time PCR assays to confirm that ABL1 knockdown decreased mRNA expression for a number of NOTCH signaling pathway genes, including NOTCH1, NOTCH3, JAG1, LFENG and DTX1 (Fig. 4C). We also found that expression of NOTCH downstream targets, including CyclinD1, NRARP, HES1 and HES2, were reduced by knockdown of ABL1 in HCC cells (Fig. 4D), suggesting that NOTCH activity was suppressed in ABL1-KD cells.
Figure 4. ABL1 Knockdown inhibits HCC cell proliferation by decreasing NOTCH1 expression.
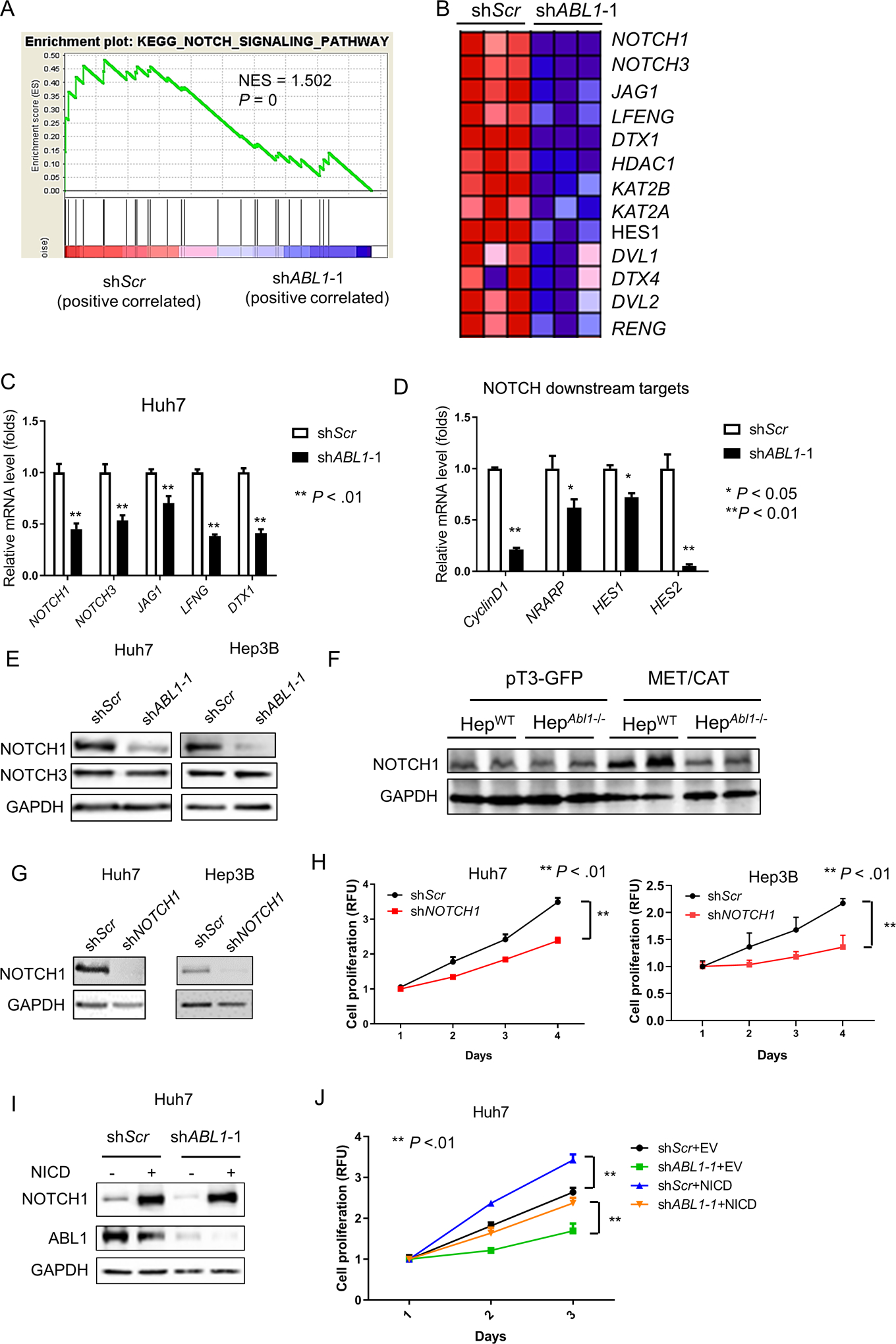
(A) GSEA shows that the NOTCH signaling pathway is enriched in ABL1-KD Huh7 cells. (B) A heat-map indicates the expression of genes in the NOTCH signaling pathway is decreased by ABL1 knockdown in Huh7 cells. (C) Relative mRNA levels of genes in the NOTCH signaling pathway in scrambled-RNA and ABL1-KD Huh7 cells. (D) Relative mRNA levels of NOTCH downstream targets in the scrambled-RNA and ABL1-KD Huh7 cells. (E) NOTCH1, NOTCH3 and GAPDH protein expression in scrambled-RNA and ABL1-KD HCC cells was determined by Western blotting. (F) Expression of NOTCH1 and GAPDH proteins in whole livers of HepWT and HepAbl1−/− mice injected with pT3-GFP or MET/CAT for 9 weeks was determined by Western blotting. (G) NOTCH1 and GAPDH protein expression in scrambled-RNA and NOTCH1-KD Hep3B and Huh7 cells was determined by Western blotting. (H) Quantification of cell proliferation from scrambled-RNA and NOTCH1-KD HCC cells at different time points after seeding. (I) Expression of NOTCH1, ABL1 and GAPDH proteins in control (infected with EF.CMV.GFP) and NOTCH1-overexpressed (infected with EF.hICN1.CMV.GFP) Huh7 scrambled and ABL1-KD cells was determined by Western blotting. (J) Quantification of cell proliferation from control and NOTCH1-overexpressed Huh7 scrambled and ABL1-KD cells at different time points after seeding.
Because NOTCH signaling receptors NOTCH1 and NOTCH3 play critical roles in HCC cell growth,19–21 we focused on testing whether knockdown of ABL1 inhibits cell proliferation by decreasing the expression of either NOTCH1 or NOTCH3 in HCC cells. We first performed Western blotting to determine whether the expression of NOTCH1 and/or NOTCH3 proteins were also decreased by knockdown of ABL1 in HCC cells. Intriguingly, expression of NOTCH1 but not NOTCH3 protein was significantly lower in ABL1-KD cells compared to scrambled-RNA control cells (Fig. 4E), suggesting a possible post-transcriptional, translational or post-translational mechanism by which NOTCH3 might be regulated in ABL1 KD cells. We therefore focused on NOTCH1 and hypothesized that knockdown of ABL1 inhibits cell proliferation by decreasing NOTCH1 in HCC cells. We found that expression of NOTCH1 protein was higher in MET/CAT-induced liver tumors compared to control mouse liver, and was reduced in liver tumors when Abl1 was deleted in hepatocytes (Fig. 4F). Consistently, expression of NOTCH1-targeted genes was also downregulated in Abl1-KO mouse HCCs (Fig. S6, A–C). To further determine if decreased NOTCH1 expression could inhibit HCC cell proliferation, we knocked down NOTCH1 using shRNA in Huh7 and Hep3B cells (Fig. 4G). We tested five distinct shRNAs (data not shown) and only one shRNA efficiently decreased NOTCH1 expression in HCC cells (Fig. 4G). We found that knockdown of NOTCH1 significantly reduced HCC cell growth (Fig. 4H). In addition, NOTCH1 overexpression promoted cell proliferation and rescued the phenotype caused by ABL1 knockdown in HCC cells (Fig. 4, I&J). Overall, our data demonstrate that knockdown of ABL1 inhibits HCC cell growth by decreasing NOTCH1 expression.
Knockdown of ABL1 decreases NOTCH1 expression through regulation of c-MYC in HCC cells
We next investigated the molecular mechanisms by which ABL1 knockdown decreases NOTCH1 expression in HCC cells. As NOTCH1 mRNA levels were reduced by ABL1 knockdown, we reasoned that this knockdown affected the transcription and/or post-transcriptional processing of NOTCH1 mRNA. MicroRNAs have been shown to directly target NOTCH1 and reduce NOTCH1 mRNA levels.22 We therefore hypothesized that ABL1 knockdown might decrease NOTCH1 expression by increasing the expression of some microRNA(s). Using TargetScan, we identified several microRNAs, including miR-150–5p, miR-34–5p, and miRNA-146b-5p, which can potentially directly target NOTCH1. However, we found that only the expression of miR-146b-5p was increased when ABL1 was knocked down in HCC cells (Fig. S7, A–C). To determine if ABL1 knockdown decreases NOTCH1 expression through increasing miR-146b-5p expression in HCC cells, we used miR146b-5p mimics or inhibitors. We confirmed that the miR146b-5p mimic effectively induced expression of miR146b-5p and inhibitors of miR146b-5p sufficiently decreased expression of miR146b-5p (Fig. S7, D&E). However, while we had expected that overexpression of miR146b-5p by the mimic miRNA would decrease expression of NOTCH1 in HCC cells; instead we found that NOTCH1 expression was slightly increased (Fig. S7F). Consistent with this observation, we found that miR146b-5p inhibition decreased the expression of NOTCH1 in HCC cells (Fig. S7G). These data would appear to suggest that ABL1 knockdown might not decrease NOTCH1 expression by increasing the expression of microRNAs.
We therefore tested whether ABL1 knockdown reduces the transcription of NOTCH1 by regulating transcription factors. ABL1 can regulate phosphorylation of the transcription factor c-MYC and its transcriptional activity.23 c-MYC plays a critical role in HCC development.24 Interestingly, GSEA indicated that MYC’s target gene set was downregulated in ABL1 knockdown cells (Fig. 5A). Consistently, c-MYC protein level was much lower in ABL1-KD HCC cells compared to scrambled-RNA cells (Fig. 5B). Expression of c-MYC protein was higher in MET/CAT-induced liver tumors compared to control mouse liver, and was reduced in liver tumors when Abl1 was deleted in hepatocytes (Fig. S8). Phosphorylation of c-MYC on Ser62, which is regulated by ABL123 and is critical for c-MYC stabilization,25 was also decreased by ABL1 knockdown (Fig. 5B). In addition, using proximity ligation assay,26 we found a strong interaction between ABL1 and c-MYC in HCC cells (Figs. 5C, S9). Although c-MYC has been shown to be a direct transcriptional target downstream of NOTCH1 in T-cell acute lymphoblastic leukemia,27 we found that c-MYC protein was not significantly affected by either knockdown or overexpression of NOTCH1 in HCC cells (Fig. S10). On the other hand, enhanced c-MYC expression may increase the level of NOTCH1 mRNA through regulation of NRF2.28, 29 We therefore hypothesized that knockdown of ABL1 decreases NOTCH1 expression through regulation of c-MYC in HCC cells. We found that both mRNA and protein expression of NOTCH1 was decreased when c-MYC was knocked down in HCC cells (Fig. 5, D&E). In line with this, c-MYC knockdown inhibited HCC cell growth (Fig. 5F). We analyzed the promoter of human NOTCH1 and identified a putative binding site for c-MYC (Fig.S11 A). The ChIP assay revealed that c-MYC directly binds to the promoter of NOTCH1 in HCC cells (Fig.S11, B&C). Moreover, overexpression of activated c-MYC promoted cell proliferation and restored decreased NOTCH1 expression and cell growth caused by ABL1 knockdown in HCC cells (Fig. 5, G&H). Considered altogether, these data indicate that knockdown of ABL1 decreases NOTCH1 expression through regulation of c-MYC in HCC cells.
Figure 5. Knockdown of ABL1 decreases NOTCH1 expression through regulation of c-MYC in HCC cells.
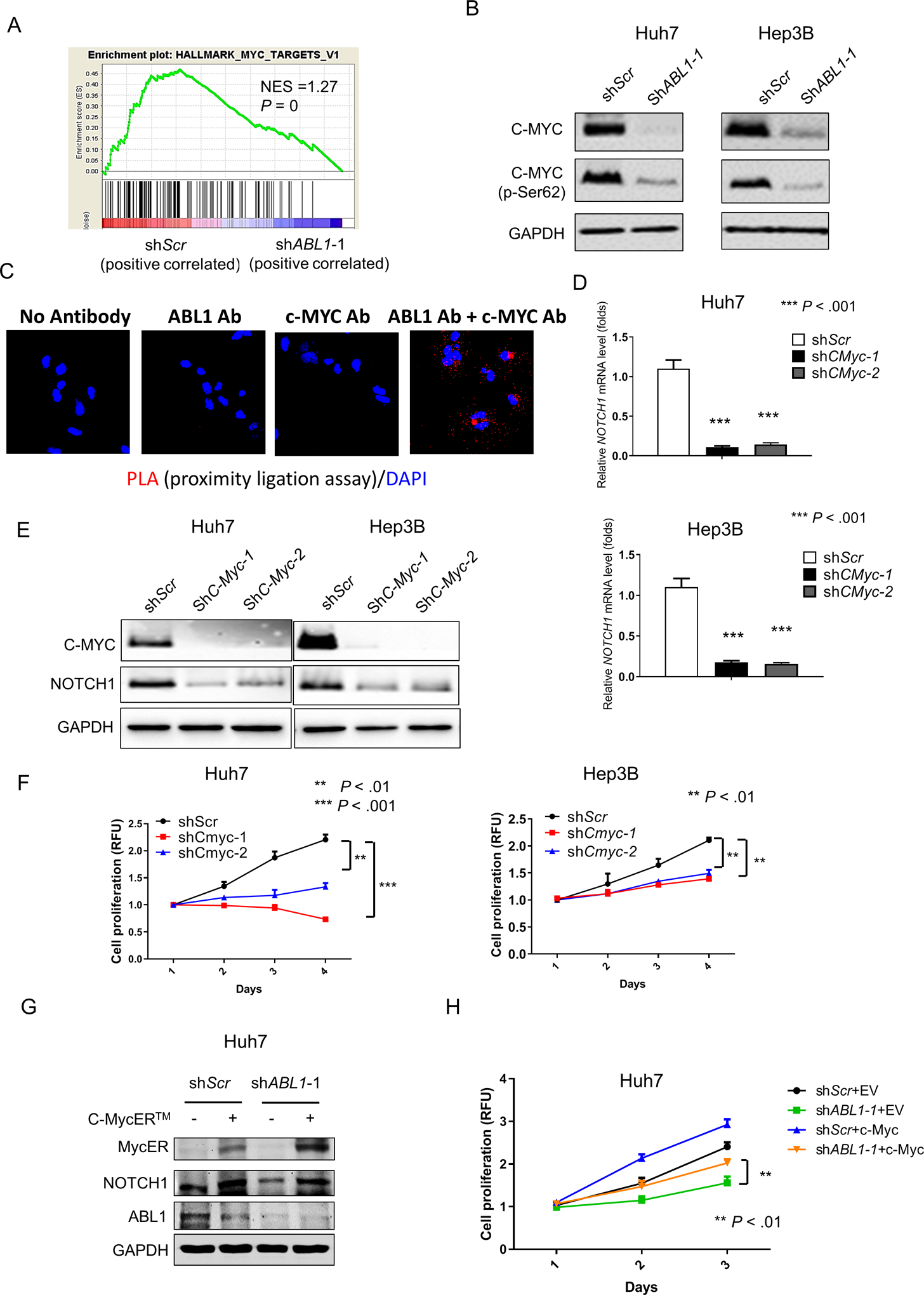
(A) GSEA shows that the MYC targets are enriched in ABL1-KD Huh7 cells. (B) Levels of c-MYC, p-c-MYC (Ser62) and GAPDH proteins in scrambled-RNA and ABL1-KD HCC cells was determined by Western blotting. (C) The interaction of ABL1 and c-MYC was examined using PLA in Huh7 cells. (D) Relative mRNA levels of NOTCH1 in scrambled-RNA control and c-MYC-KD HCC cells. (E) Expression of c-MYC, NOTCH1 and GAPDH proteins in scrambled-RNA and c-MYC-KD HCC cells was determined by Western blotting. (F) Quantification of cell proliferation from scrambled-RNA and c-MYC-KD HCC cells. (G) Expression of MycER (activated fusion c-MYC), NOTCH1, ABL1 and GAPDH proteins in control (infected with the pBpuro retroviral particle) and c-MYC-overexpressed (infected with the pBpuro c-MYCER™ retroviral particle) Huh7 scrambled and ABL1-KD cells was determined by Western blotting. (H) Quantification of cell proliferation from in control and c-MYC-overexpressed Huh7 scrambled and ABL1-KD cells.
Expression of p-ABL1, c-MYC and NOTCH1 is positively correlated in human HCC
Our data indicate that ABL1 regulates the phosphorylation of c-MYC leading to increased c-MYC protein levels, which results in enhanced NOTCH1 mRNA expression and promotes HCC cell growth (Fig. 6A). To determine whether the ABL1/c-MYC/NOTCH1 axis is also relevant in human HCC, we examined the levels of p-ABL1 (Y412) (an indicator of ABL1 activity), c-MYC and NOTCH1 in human HCC specimens using tissue microarrays. The expression of both c-MYC and NOTCH1 proteins was significantly correlated with levels of p-ABL1 in human HCC specimens (Fig. 6, B–D). In addition, NOTCH1 protein expression correlated with c-MYC in human HCC (Fig. 6E). We further analyzed the TCGA database and found that NOTCH1 mRNA level positively correlated with ABL1 mRNA level (Fig. 6F). However, c-MYC mRNA level had no correlation with either ABL1 or NOTCH1 mRNA level in the TCGA HCC samples (Fig. S12), suggesting that ABL1 may not regulate c-MYC at transcript level in human HCC. Overall, these data indicate that the ABL1/c-MYC/NOTCH1 axis is important in human HCC.
Figure 6. Levels of p-ABL1, c-MYC or NOTCH1 are positively correlated in human HCC.
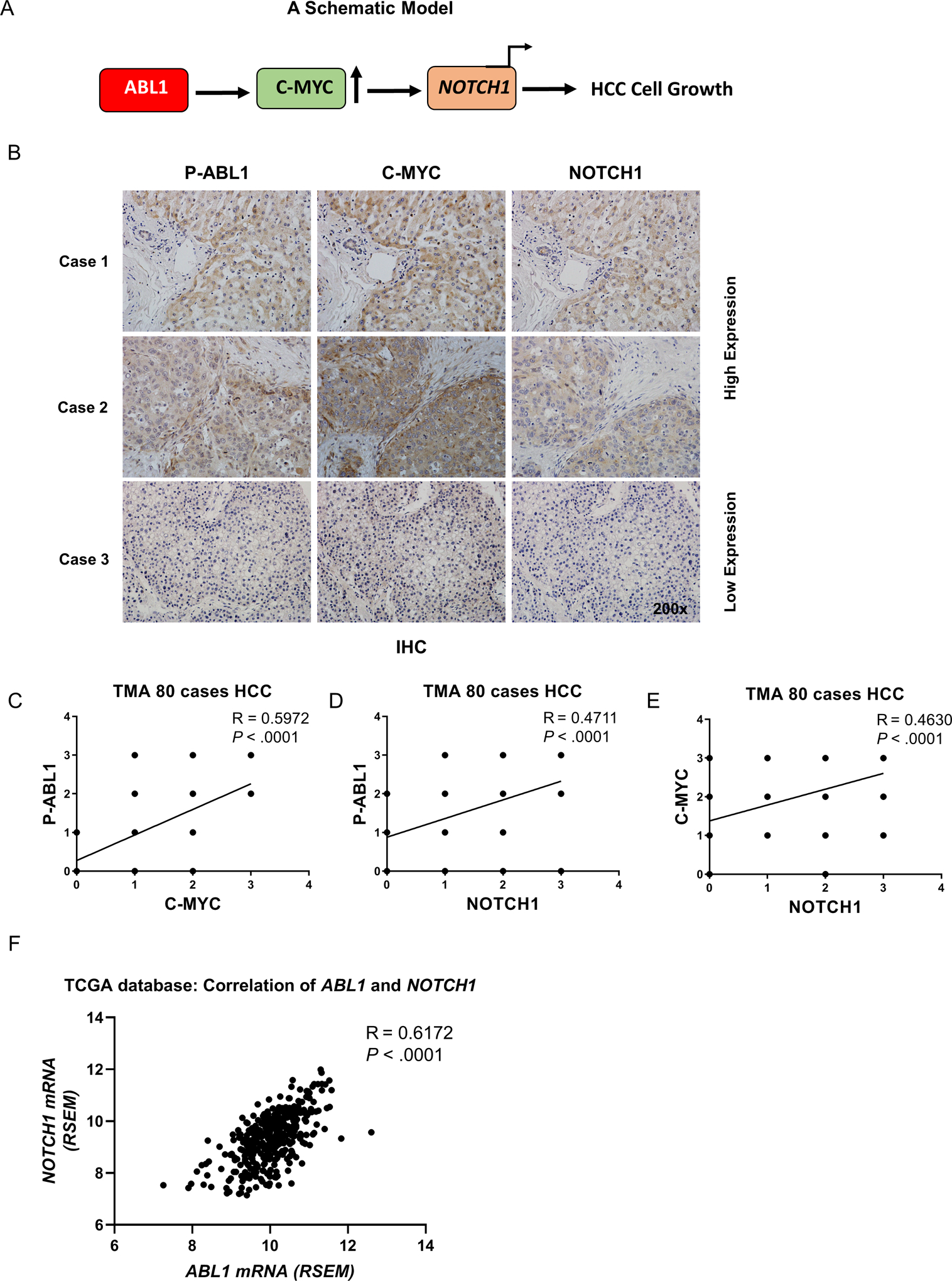
(A) A schematic model: ABL1 activation (due to ABL1 overexpression or other reasons) leads to an increase of c-MYC protein expression and its transcriptional activity with respect to NOTCH1, thereby promoting HCC cell proliferation. (B) Levels of p-ABL1 (p-Y412), c-MYC and NOTCH1 in human HCC specimens (TMA) were determined by IHC staining. (C) Correlation of p-ABL1 and c-MYC levels in human HCC specimens was analyzed. (D) Correlation of p-ABL1 and NOTCH1 levels in human HCC specimens was analyzed. (E) Correlation of c-MYC and NOTCH1 expression in human HCC specimens was analyzed. (F) Correlation of ABL1 and NOTCH1 mRNAs in HCC samples from the TCGA database was analyzed.
ABL1 inhibitors suppress HCC growth in preclinical models
The above results encouraged us to examine whether ABL1 inhibitors could be useful to treat HCC in preclinical models, which would provide a translational basis for the potential clinical use of ABL1 inhibitors in the treatment of HCC patients. Several ABL kinase inhibitors are already used clinically or are under investigation in clinical trials for treating chronic myelogenous leukemia and other cancers,4 but their efficacy in HCC remains unknown. Nilotinib, an ATP-competitive inhibitor of ABL kinases, has been approved by the FDA to treat Philadelphia chromosome-positive chronic myelogenous leukemia.30 GNF-5, a newer allosteric inhibitor that targets the myristate pocket of ABL kinases, has been tested in preclinical leukemia models.31 Because ABL1 knockdown inhibits HCC cell growth (Fig. 3), we hypothesized that ABL inhibitors would be efficacious in treating HCC. First, we confirmed that both nilotinib and GNF-5 effectively inhibit ABL kinase activity, as indicated by phosphorylation of CRKL in HCC cells (Fig. S13, A&B). Further, we found that both nilotinib and GNF5 inhibited HCC cell growth in vitro (Figs. 7A, S13C, and S14A). Importantly, the sensitivity of nilotinib positively correlated with the expression of ABL1 (Figs. 7B and S14B). We also found there is a positive correlation between ABL1 and c-MYC expression in human HCC cell lines (Figs. S14B&C). However, there is no significant correlation between ABL1 and NOTCH1 in these HCC cell lines (Fig. S14B&C), suggesting other mechanisms may also regulate NOTCH1 expression. It is notable that NOTCH1 expression is positively correlated with ABL1 in HCC cells without p53 mutations (Hep3B, Huh7, SK-Hep1 and SNU-423), but there is no correlation in p53-mutant HCC cells (SNU-449, SNU-475, PLC/PRF/5, and SNU-387), suggesting that p53 mutations may affect NOTCH1 expression. Consistent with the functional inhibition of ABL1 using shRNAs, nilotinib effectively decreased the expression of c-MYC and NOTCH1 (Fig. 7C). Moreover, nilotinib significantly suppressed tumor growth (Fig. 7, D–G) and proliferation (Fig. S15, A–D) in an HCC xenograft model. To further test the efficacy of ABL inhibitors in mouse models with intact immune responses, we injected wild-type C57BL/6 mice with MET/CAT to induce HCC and gavaged these mice with nilotinib or vehicle solution by oral gavage daily for 4 weeks. We found that nilotinib treatment dramatically inhibited MET/CAT-induced tumor growth (Fig. 7 H&I) and proliferation (Fig. S16), decreased the expression of MYC and NOTCH1 (Fig. 7J), and prolonged the survival of animals with HCC (Fig. 7K). In contrast, sorafenib was not effective in inhibiting MET/CAT-induced HCC growth (Fig. S17). Taken together, these data indicate that ABL1 inhibitors effectively suppress HCC growth in preclinical models.
Figure 7. ABL1 inhibitors suppress human HCC growth in vitro and in vivo.
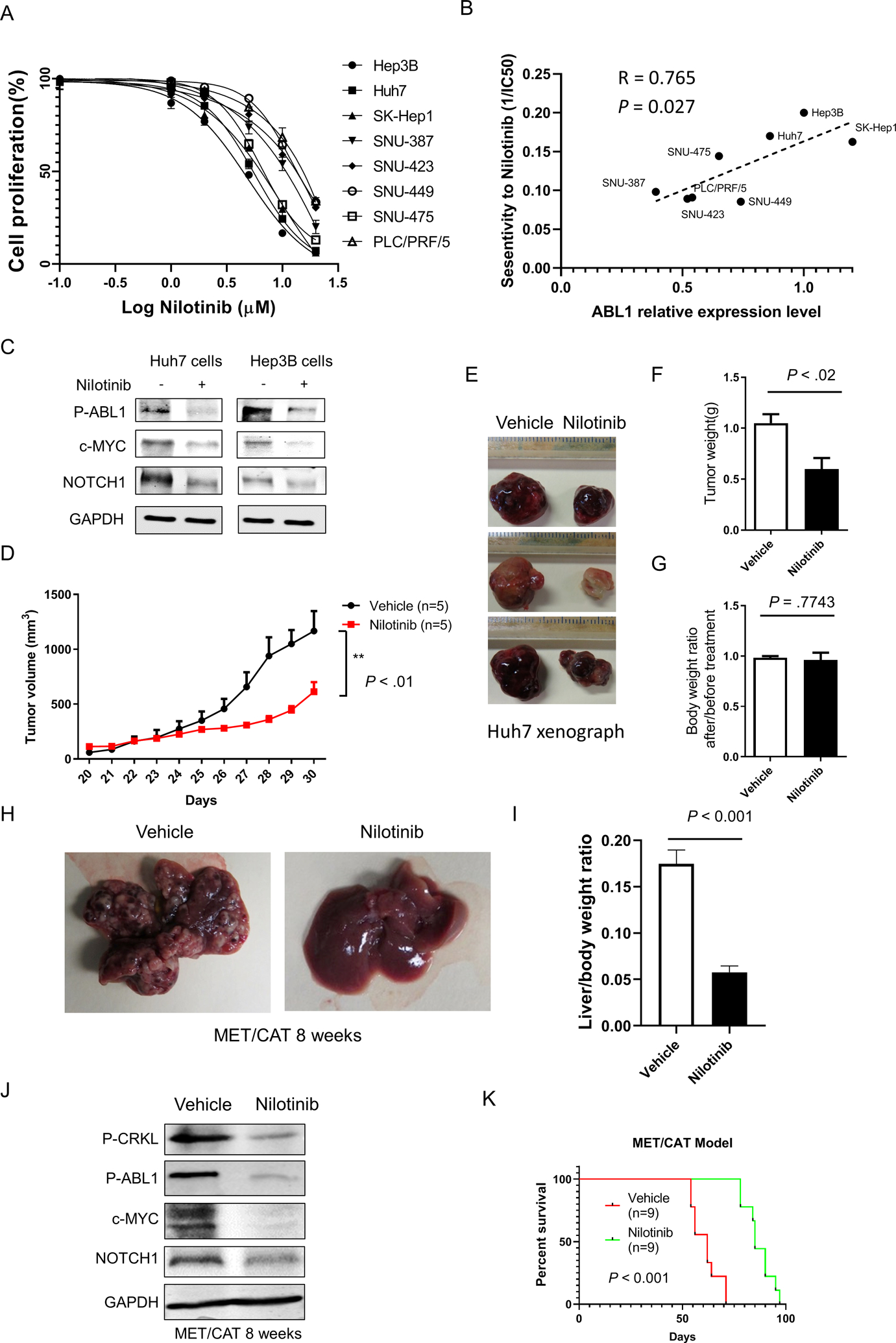
(A) Quantification of cell proliferation of HCC cells treated with vehicle or nilotinib at different dose after 48 hours treatment. (B) Correlation between ABL1 expression and sensitivity to nilotinib in HCC cells described in A. (C) Levels of p-ABL, c-MYC, NOTCH1, and GAPDH proteins in HCC cells treated with vehicle or 5μM Nilotinib for 24h. (D) SCID-bg mice injected with Huh7 cells were gavaged with vehicle or nilotinib for 10 days and Tumor volumes from mice (n=5/group) were measured. (E) Gross images of tumors from vehicle- or nilotinib-gavaged animals are shown. (F) Tumor weights from mice from D. (G) Body weight ratios from mice after/before nilotinib treatment. (H) C57BL/6 mice were injected with MET/CAT to induce HCC. Four weeks after such injection, the mice were gavaged with vehicle or nilotinib for 4 weeks (some mice treated with vehicle had to be euthanized earlier due to tumor burdens), and gross images of livers from vehicle- and nilotinib-gavaged animals at 8 weeks after MET/CAT injection are shown. (I) Liver/body weight ratios from the mice as (I) (n=6/group). (J) Levels of p-CRKL, p-ABL, c-MYC, NOTCH1, and GAPDH proteins in the mouse livers from H. (K) Survival graphs for mice gavaged with vehicle or nilotinib (n=9/group).
Discussion
Tyrosine kinases have been shown to be good targets for treating cancer, including HCC. Currently, four out of every five drugs approved to treat advanced HCC are tyrosine kinase inhibitors.2 Despite the success of TKIs in treating HCC, many patients do not respond. There is significant genetic and biological heterogeneity in HCC. A better understanding of tyrosine kinases in the development HCC is necessary in order to predict response to therapy and to develop new therapies. Only a few tyrosine kinases have been shown to play key roles in HCC initiation and progression.32 In this study, we found that ABL1 is overexpressed and activated in human HCCs, and ABL1 inhibition effectively suppresses HCC growth in human and mouse preclinical models, suggesting that inhibition of ABL1 may be a promising new strategy to treat HCC. We showed that ABL1 inhibitors such as nilotinib significantly inhibit HCC growth in vitro and in vivo. Our findings provide translational support for the development of a clinical trial to assess the safety and efficacy of nilotinib in treating HCC.
How ABL1 is activated in HCC remains unclear. MET can bind directly to ABL1 and activate it in breast cancer cells.16 We found that ABL1 is activated in MET/CAT-induced HCC tumors, suggesting that ABL1 might be activated by MET in human HCC. In addition, we found that ABL1 expression was also increased in MET/CAT-induced liver tumors. Consistently, ABL1 mRNA expression is highly correlated with the gene signature of activated MET in human HCC (Fig. S18).17 Therefore, it is possible that ABL1 mRNA expression can be upregulated by activated MET in HCC. SP1 can modulate ABL1 expression at the transcription level,33 and MET can induce the phosphorylation of SP1 and enhance its transcriptional activity.34 Thus, MET activation may increase ABL1 mRNA expression through the regulation of SP1. This hypothesis will be tested in our future studies.
For the first time, we demonstrated that the NOTCH signaling pathway is suppressed by ABL1 knockdown in HCC cells. NOTCH signaling is a crucial determinant of tumor cell growth in many types of cancer, including HCC.19 However, NOTCH inhibitors such as GSI cause gastrointestinal toxicity due to its off-target effects.35 Our data suggest a possible new strategy to inhibit the NOTCH pathway, which would be to inhibit ABL1. As functional inhibition of ABL1 showed low and tolerable toxicity, ABL1 inhibitors might be useful to treat HCC cases that exhibit upregulated NOTCH signaling. It remains to be determined whether the regulation of the NOTCH signaling pathway by ABL1 is tissue- or cell-context-dependent. Our findings might provide a broader application for using ABL1 inhibitors in other type of cancers with activated NOTCH. For example, NOTCH signaling is known to play a critical role in intrahepatic cholangiocarcinoma (ICC).36 Currently, there are no targeted therapy options available for ICC. It would be intriguing to test whether ABL1 inhibitors might be useful in inhibiting ICC growth in future studies.
ABL kinases function by regulating more than 100 different targets in a cell-context-specific manner, making it difficult to select appropriate downstream targets of ABL1 for study.4 ABL1 has been reported to phosphorylate AKT and ERK to regulate cancer cell proliferation and survival.37 However, ABL1 knockdown did not affect phosphorylation of either AKT or ERK in HCC cells (Fig. S19), which supports the concept that ABL1’s regulation of different targets is cell-context-dependent. We found that ABL1 knockdown decreases c-MYC protein expression and further demonstrated that decreased c-MYC results in reduced NOTCH1 expression in HCC cells. These data reveal a novel mechanism by which ABL1 promotes cell growth in HCC. c-MYC is a well-known proto-oncogene which plays a critical role in many cancers including liver cancer.38 However, it is still currently “untargetable” clinically. Our results suggest that inhibition of ABL1 might be useful to treat HCC cases demonstrating high expression of c-MYC. Although c-MYC is a direct transcriptional target downstream of NOTCH1 in T-ALL,27 our data suggest that c-MYC may not be regulated by NOTCH1 in HCC cells. This is not particularly surprising, as NOTCH1 regulates different targets in tissue- and cell-context-dependent manners.39 It remains unknown whether c-MYC regulates NOTCH1 in other contexts, as this mechanism has not been previously reported. It will be instructive to examine whether this regulation occurs in other cancers. Besides the c-MYC/NOTCH1 axis, we also identified other signaling pathways regulated by ABL1 in HCC cells through RNA-seq analysis. The gene sets that are most significantly downregulated by ABL1 knockdown include hypoxia, the p53 pathway, glycolysis and androgen response (Fig. S20), which have been shown to regulate tumor growth and progression.40–43 It is possible that ABL1 promotes HCC development through regulation of these signaling pathways, and we plan to investigate this question in our future studies.
In conclusion, our study shows that ABL1 plays a critical role in the development of HCC by regulating the c-MYC/NOTCH1 axis. Inhibition of ABL1 represents a promising new strategy to treat HCC in patients who overexpress this protein-tyrosine kinase. A number of ABL kinase inhibitors have been developed and are being used clinically for leukemia and gastrointestinal stromal tumors. These inhibitors might also prove to useful in treating HCC, especially in patients showing overexpression and/or activation of ABL1.
Supplementary Material
What you need to know:
BACKGROUND AND CONTEXT: The oncogene ABL1 might be involved in the pathogenesis of hepatocellular carcinoma (HCC).
NEW FINDINGS: HCC samples have increased levels of ABL1 compared with non-tumor liver tissues, and loss or inhibition of ABL1 reduces proliferation of HCC cells and slows growth of tumors in mice.
LIMITATIONS: This study was performed using human tissue samples, cell lines, and mice. Further studies in humans are needed.
IMPACT: Inhibitors of ABL1 might be developed for treatment of HCC
Acknowledgements
The authors thank Drs. Clodia Osipo, Nancy Zeleznik-Le, and Jiwang Zhang of Loyola University Chicago and Dr. Ann Marie Pendergast of Duke University for their helpful discussions and advice.
Grant Support: This work was supported in part by ACS RSG-18-107 (W. Qiu), NIH R01NS105640 (A. Koleske), NIH R01MH115939 (A. Koleske), NIH R03CA195183 (W. Qiu), R03CA184652 (W. Qiu), and R01CA197128 (W. Qiu).
Abbreviations:
- HCC
hepatocellular carcinoma
- ABL1
Abelson tyrosine-protein kinase 1
- CAT
constitutively active β-catenin
- WT
wild type
- H&E
hematoxylin and eosin
- GSEA
gene set enrichment analysis
- TCGA
The Cancer Genome Atlas
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: The authors have no financial or other conflicts to disclose. All authors agreed on the submission.
References
- 1.Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68:394–424. [DOI] [PubMed] [Google Scholar]
- 2.Heimbach JK, Kulik LM, Finn RS, et al. AASLD guidelines for the treatment of hepatocellular carcinoma. Hepatology 2018;67:358–380. [DOI] [PubMed] [Google Scholar]
- 3.Marrero JA, Kulik LM, Sirlin CB, et al. Diagnosis, Staging, and Management of Hepatocellular Carcinoma: 2018 Practice Guidance by the American Association for the Study of Liver Diseases. Hepatology 2018;68:723–750. [DOI] [PubMed] [Google Scholar]
- 4.Greuber EK, Smith-Pearson P, Wang J, et al. Role of ABL family kinases in cancer: from leukaemia to solid tumours. Nat Rev Cancer 2013;13:559–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chitsike Lennox D X, Breslin Peter, Qiu Wei. ABL1 is Overexpressed and Activated in Hepatocellular Carcinoma. Journal of Cancer and Tumor International 2017;6:8. [Google Scholar]
- 6.Wang F, Bank T, Malnassy G, et al. Inhibition of insulin-like growth factor 1 receptor enhances the efficacy of sorafenib in inhibiting hepatocellular carcinoma cell growth and survival. Hepatol Commun 2018;2:732–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arteaga M, Shang N, Ding X, et al. Inhibition of SIRT2 suppresses hepatic fibrosis. Am J Physiol Gastrointest Liver Physiol 2016;310:G1155–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Littlewood TD, Hancock DC, Danielian PS, et al. A modified oestrogen receptor ligand-binding domain as an improved switch for the regulation of heterologous proteins. Nucleic Acids Res 1995;23:1686–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc 2006;1:1112–6. [DOI] [PubMed] [Google Scholar]
- 10.Shang N, Arteaga M, Zaidi A, et al. FAK is required for c-Met/beta-catenin-driven hepatocarcinogenesis. Hepatology 2015;61:214–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shang N, Wang H, Bank T, et al. Focal Adhesion Kinase and beta-Catenin Cooperate to Induce Hepatocellular Carcinoma. Hepatology 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cancer Genome Atlas Research Network. Electronic address wbe, Cancer Genome Atlas Research N. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017;169:1327–1341 e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Etten RA. Cycling, stressed-out and nervous: cellular functions of c-Abl. Trends Cell Biol 1999;9:179–86. [DOI] [PubMed] [Google Scholar]
- 14.Dorey K, Engen JR, Kretzschmar J, et al. Phosphorylation and structure-based functional studies reveal a positive and a negative role for the activation loop of the c-Abl tyrosine kinase. Oncogene 2001;20:8075–84. [DOI] [PubMed] [Google Scholar]
- 15.Brasher BB, Van Etten RA. c-Abl has high intrinsic tyrosine kinase activity that is stimulated by mutation of the Src homology 3 domain and by autophosphorylation at two distinct regulatory tyrosines. J Biol Chem 2000;275:35631–7. [DOI] [PubMed] [Google Scholar]
- 16.Li R, Knight JF, Park M, et al. Abl Kinases Regulate HGF/Met Signaling Required for Epithelial Cell Scattering, Tubulogenesis and Motility. PLoS One 2015;10:e0124960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tao J, Xu E, Zhao Y, et al. Modeling a human hepatocellular carcinoma subset in mice through coexpression of met and point-mutant beta-catenin. Hepatology 2016;64:1587–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Jong R, ten Hoeve J, Heisterkamp N, et al. Tyrosine 207 in CRKL is the BCR/ABL phosphorylation site. Oncogene 1997;14:507–13. [DOI] [PubMed] [Google Scholar]
- 19.Yuan X, Wu H, Xu H, et al. Notch signaling: an emerging therapeutic target for cancer treatment. Cancer Lett 2015;369:20–7. [DOI] [PubMed] [Google Scholar]
- 20.Wu CX, Xu A, Zhang CC, et al. Notch Inhibitor PF-03084014 Inhibits Hepatocellular Carcinoma Growth and Metastasis via Suppression of Cancer Stemness due to Reduced Activation of Notch1-Stat3. Mol Cancer Ther 2017;16:1531–1543. [DOI] [PubMed] [Google Scholar]
- 21.Zhou L, Zhang N, Song W, et al. The significance of Notch1 compared with Notch3 in high metastasis and poor overall survival in hepatocellular carcinoma. PLoS One 2013;8:e57382. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22.Mei J, Bachoo R, Zhang CL. MicroRNA-146a inhibits glioma development by targeting Notch1. Mol Cell Biol 2011;31:3584–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sanchez-Arevalo Lobo VJ, Doni M, Verrecchia A, et al. Dual regulation of Myc by Abl. Oncogene 2013;32:5261–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dang H, Takai A, Forgues M, et al. Oncogenic Activation of the RNA Binding Protein NELFE and MYC Signaling in Hepatocellular Carcinoma. Cancer Cell 2017;32:101–114 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sears R, Nuckolls F, Haura E, et al. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev 2000;14:2501–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soderberg O, Gullberg M, Jarvius M, et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods 2006;3:995–1000. [DOI] [PubMed] [Google Scholar]
- 27.Palomero T, Lim WK, Odom DT, et al. NOTCH1 directly regulates c-MYC and activates a feedforward-loop transcriptional network promoting leukemic cell growth. Proc Natl Acad Sci U S A 2006;103:18261–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.DeNicola GM, Karreth FA, Humpton TJ, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011;475:106–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wakabayashi N, Shin S, Slocum SL, et al. Regulation of notch1 signaling by nrf2: implications for tissue regeneration. Sci Signal 2010;3:ra52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weisberg E, Catley L, Wright RD, et al. Beneficial effects of combining nilotinib and imatinib in preclinical models of BCR-ABL+ leukemias. Blood 2007;109:2112–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang J, Adrian FJ, Jahnke W, et al. Targeting Bcr-Abl by combining allosteric with ATP-binding-site inhibitors. Nature 2010;463:501–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Regad T Targeting RTK Signaling Pathways in Cancer. Cancers (Basel) 2015;7:1758–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Long J, Liao G, Wang Y, et al. Specific protein 1, c-Abl and ERK1/2 form a regulatory loop. J Cell Sci 2019;132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reisinger K, Kaufmann R, Gille J. Increased Sp1 phosphorylation as a mechanism of hepatocyte growth factor (HGF/SF)-induced vascular endothelial growth factor (VEGF/VPF) transcription. J Cell Sci 2003;116:225–38. [DOI] [PubMed] [Google Scholar]
- 35.Venkatesh V, Nataraj R, Thangaraj GS, et al. Targeting Notch signalling pathway of cancer stem cells. Stem Cell Investig 2018;5:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Geisler F, Strazzabosco M. Emerging roles of Notch signaling in liver disease. Hepatology 2015;61:382–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Structure Hantschel O., regulation, signaling, and targeting of abl kinases in cancer. Genes Cancer 2012;3:436–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gabay M, Li Y, Felsher DW. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb Perspect Med 2014;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Andersson ER, Sandberg R, Lendahl U. Notch signaling: simplicity in design, versatility in function. Development 2011;138:3593–612. [DOI] [PubMed] [Google Scholar]
- 40.Nath B, Szabo G. Hypoxia and hypoxia inducible factors: diverse roles in liver diseases. Hepatology 2012;55:622–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meng X, Franklin DA, Dong J, et al. MDM2-p53 pathway in hepatocellular carcinoma. Cancer Res 2014;74:7161–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alves AP, Mamede AC, Alves MG, et al. Glycolysis Inhibition as a Strategy for Hepatocellular Carcinoma Treatment? Curr Cancer Drug Targets 2019;19:26–40. [DOI] [PubMed] [Google Scholar]
- 43.Ma WL, Lai HC, Yeh S, et al. Androgen receptor roles in hepatocellular carcinoma, fatty liver, cirrhosis and hepatitis. Endocr Relat Cancer 2014;21:R165–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.