

Abstract
Backgrounds
The incidence of germline mutations in the newly discovered cryptic exon (E1’) of VHL gene in patients with von Hippel-Lindau (VHL) disease and in patients with paraganglioma or pheochromocytoma (PPGL) is not currently known.
Methods
We studied a large international multicentre cohort of 1167 patients with a previous negative genetic testing. Germline DNA from 75 patients with a single tumour of the VHL spectrum (‘Single VHL tumour’ cohort), 70 patients with multiple tumours of the VHL spectrum (‘Multiple VHL tumours’ cohort), 76 patients with a VHL disease as described in the literature (‘VHL-like’ cohort) and 946 patients with a PPGL were screened for E1’ genetic variants.
Results
Six different genetic variants in E1' were detected in 12 patients. Two were classified as pathogenic, 3 as variants of unknown significance and 1 as benign. The rs139622356 was found in seven unrelated patients but described in only 16 patients out of the 31 390 of the Genome Aggregation Database (p<0.0001) suggesting that this variant might be either a recurrent mutation or a modifier mutation conferring a risk for the development of tumours and cancers of the VHL spectrum.
Conclusions
VHL E1’ cryptic exon mutations contribute to 1.32% (1/76) of ‘VHL-like’ cohort and to 0.11% (1/946) of PPGL cohort and should be screened in patients with clinical suspicion of VHL, and added to panels for Next Generation Sequencing (NGS) diagnostic testing of hereditary PPGL. Our data highlight the importance of studying variants identified in deep intronic sequences, which would have been missed by examining only coding sequences of genes/exomes. These variants will likely be more frequently detected and studied with the upcoming implementation of whole-genome sequencing into clinical practice.
Keywords: von Hippel-Lindau, paraganglioma, cryptic exon
Introduction
Von Hippel-Lindau (VHL) disease is an autosomal-dominant renal cancer predisposition syndrome1 responsible for the development in affected patients of renal cysts or clear cell carcinomas, and other features as retinal or central nervous system haemangioblastomas, pancreatic cysts or neuroendocrine tumours, endolymphatic sac tumours and pheochromocytomas and/or paragangliomas (PPGLs). A germline mutation (including gross deletion) is identified in one of the three exons of VHL in almost all affected patients.2 Nevertheless, some patients with clinically diagnosed VHL disease, but without identified VHL germline mutation, have been reported.3 One of the tumour types of the VHL tumour spectrum, PPGL, are rare neuroendocrine tumours with a great genetic heterogeneity and the highest heritability rate with about 40% of genetically determined forms.4 5 Indeed, to date, approximately 17 susceptibility genes have been reported but two thirds of identified mutations are found in SDHB, SDHD and VHL genes.6 7
Recently, a cryptic exon of VHL gene, named E1’, has been discovered. A germline mutation in the first intronic region which results in creation of a cryptic exon designated E1’ was found in one large family with a typical VHL disease and without any alteration in the other VHL exons.8 VHL gene is one of the major PPGL susceptibility genes but, to date, E1’ exon has not been included in PPGL target gene panels.
Hence, our objective was to assess the prevalence of E1’ germline mutations in two international cohorts of patients: first, in 221 patients with a single or multiple tumours suggesting a VHL disease and then in 946 with a PPGL but without an identified mutation in the three VHL exons or in the main PPGL susceptibility genes, respectively.
Methods
Patient’s selection
A total of 1167 patients were analysed, divided into four different groups:
946 patients with PPGL but without germline mutation in major PPGL susceptibility genes (‘PPGL’ cohort) (table 1 and online supplementary table S1).
76 patients with a VHL disease as defined in the literature,2 9 that is, patients with multiple haemangioblastomas, or a single haemangioblastoma with another tumour of the VHL spectrum, or one tumour of the VHL spectrum (excepted epididymal and renal cysts) and family history of VHL tumour but no germline VHL gene mutation (‘VHL-like’ cohort).
70 patients with multiple tumours of the clinical spectrum of VHL disease but who did not fill the definition of a VHL disease and who had no germline VHL mutation (‘Multiple VHL tumours’ cohort).
75 patients with a single tumour of the VHL spectrum without VHL mutation occurring at a young age (‘Single VHL tumour’ cohort) (table 1 and online supplementary table S2).
Table 1.
Main clinical and tumour characteristics of the different cohorts
| Total patients n=1167 | |
| VHL-like, n=76 patients | |
| Age at first diagnosis mean (min-max) | 45.4 (20–76) |
| Multiple haemangioblastomas | 16 (21%) |
| Haemangioblastoma with another VHL tumour | 28 (37%) |
| One VHL tumour and family history of VHL tumour | 32 (42%) |
| Multiple VHL tumours, n=70 patients | |
| Age at first diagnosis mean, (min-max) | 55 (11–81) |
| Three or more VHL tumours | 3 (4%) |
| Two VHL tumours | 67 (96%) |
| Single VHL tumour, n=75 patients | |
| Age at first diagnosis mean (min-max) | 34.6 (11–78) |
| Clear cell renal cell carcinoma | 3 (4%) |
| Cerebral haemangioblastoma | 27 (36%) |
| Retinal haemangioblastoma | 10 (13.3%) |
| Other tumours | 35 (46.6%) |
| PPGL, n=946 patients | |
| Age at first diagnosis mean (min-max) | 43 (8–94) |
| Benign PPGL | 869 (92%) |
| Single benign PPGL | 771 (82%) |
| Multiple benign PPGL | 98 (10%) |
| Metastatic PPGL | 77 (8%) |
| Single metastatic PPGL | 67 (7%) |
| Multiple metastatic PPGL | 10 (1%) |
| Familial PPGL | 17 (2%) |
PPGL, paraganglioma; VHL, von Hippel-Lindau.
jmedgenet-2019-106519supp001.pdf (55.6KB, pdf)
jmedgenet-2019-106519supp002.pdf (45.8KB, pdf)
Germline DNA from ‘VHL-like’, ‘Multiple VHL tumours’ and ‘Single VHL tumour’ cohorts had been previously tested for VHL gene by Sanger sequencing or Next Generation Sequencing and large rearrangements by MLPA or QMPSF. The procedures used for PPGL diagnosis were in accordance with international guidelines.10 11
Moreover, a control cohort of 198 European subjects without VHL manifestation was analysed in order to determine the frequency of variant in the general population.
Each patient signed a written informed consent for genetic analyses.
Direct sequencing of the E1’ cryptic exon of VHL on germline DNA
Sanger sequencing on germline DNA of E1’ was performed as previously described.8 Variants interpretation was performed by using different criterions: ACMG criteria,12 allele frequency in databases, phenotype of patients and tumour analysis as described below.
VHL gene analysis in tumour
Tumour DNA was extracted from frozen or paraffin embedded tumour by the QIAamp DNA minikit (Qiagen). Loss of heterozygosity (LOH) was evaluated by (1) Sanger sequencing of the E1’ cryptic exon of VHL by mutation-specific primers and (2) microsatellite analysis on D3S1537, D3S1038, D3S1317 D3S3547, D3S3727 as previously described.13 14 VHL gene deletion on tumour DNA was assessed with the SALSA MLPA P016 VHL probemix (MRC-Holland).
CA9 immunochemistry
Immunohistochemistry was performed as previously described on 6 µm slides cut from paraffin-embedded tumours with anti CA9 antibody (1/1500, ab15086, Abcam).15 Antigen retrieval was performed by boiling slides in Tris-EDTA buffer (pH9) for 45 min. Revelation was performed using Histogreen as a chromogen. Images were acquired with a Leica DM400B microscope with Leica Application Suite software V.2.8.1 and a Leica DFC420C camera.
PNMT and VHL RT-qPCR
RNA was extracted from paraffin embedded tumours of six control PPGL (3 NF1-related, 2 RET-related and 1 TMEM127-related PPGL), 5 VHL- related PPGL (all carrying a missense mutation in VHL gene) and patients #3 and #10 PPGL by using the Maxwell 16 LEV RNA FFPE Purification Kit (Promega). RNA was quantified and its purity assessed with a NanoDrop ND-1000 spectrophotometer (Labtech). RT PCR was performed on 1000 ng of RNA with iScript cDNA Synthesis Kit iScript (BioRad). Then, as described in,16 pre amplification of PNMT, VHL, GAPDH and 18S on complementary DNA was performed with SsoAdvanced PreAmp Supermix (BioRad). Because of RNA fragmentation, all primers were designed to amplify amplicons smaller than 100 bp. We used two VHL primer sets. The first set amplified the VHL transcript including exons 1 and 2 (E1-E2) (F: 5'- CATCCACAGCTACCGAGGTC-3' overlapping exons 1 and 2 and R: 5'-GTGTGTCCCTGCATCTCTGA-3' located on exon 2). The second set amplified the VHL transcript with exon 1 and the cryptic exon (E1-E1') (F: 5'-GCATCCACAGCTACCGAGTC-3' overlapping exon 1 and the cryptic exon and R: 5'-AGTCTCCCCAGGAGGAATGT-3' located on the cryptic exon). Quantitative PCR was performed on VHL (E1-E2), PNMT, GAPDH and 18S by SYBR Green Master MixSybrGreen (BioRad) on the C1000 Touch (BioRad) and VHL (E1-E1') was amplified by PCR in parallel with GAPDH. All experiments were performed in duplicate three times.
Statistical analysis
Statistical analysis was carried out with GraphPad software. Differences between allele's frequency in gnomAD and our cohort of patients and relative risk were assessed by χ² tests. A p<0.05 was considered significant.
Results
We analysed the germline DNA of 1167 patients from France, Spain, Canada and the USA. We identified a rare germline genetic variant (minor allele frequency <1%) in the E1’ VHL cryptic exon in 12 patients (1%). One of these patients was classified as ‘VHL-like’ (1 of 76 patients, 1.3%), 2 as ‘Multiple VHL tumours’ (2/70 patients, 2.9%), 1 as ‘Single VHL tumour’ (1/75 patients, 1.3%) and 8 belonged to the ‘PPGL’ cohort (8/946, 0.8%) (table 2). Among these 12 patients, we identified 6 different variants, 4 in the E1’ and 2 at the intron-exon junction and we considered only two variants as pathogenic mutations (figure 1). None of these variants was found in a control cohort of 198 European subjects without VHL manifestations.
Table 2.
Patients with a genetic variant in the E1' cryptic exon of VHL gene
| Patient number | Phenotype (age at first diagnosis) |
Follow-up duration (years) | Germline genetic variant | dbSNP ID | Allele frequency* (%) |
LOH | Somatic VHL mutation | CAIX IHC |
PNMT and VHL (E1-E2) expression | VHL (E1-E1’ expression | Classification of VUS |
| #1 | Carotid body PGL (47) | 5 | c.340+563C>T | NA | No† | ND | Neg | ND | ND | Benign | |
| #2 | Carotid body PGL (34) | 1 | c.340+578C>T | rs139622356 | 0.05 | NA | NA | NA | NA | NA | VUS |
| #3 | PCC (38) |
7 | c.340+578C>T | rs139622356 | 0.05 | No | No | Neg | Normal | Increase | VUS |
| #4 | Carotid body PGL (74) | 1 | c.340+578C>T | rs139622356 | 0.05 | NA | NA | NA | NA | NA | VUS |
| #5 | Carotid body PGL (56) | 1 | c.340+578C>T | rs139622356 | 0.05 | NA | NA | NA | NA | NA | VUS |
| #6 | PGL+ccRCC (81–82) |
2 | c.340+578C>T | rs139622356 | 0.05 | NA | NA | NA | NA | NA | VUS |
| #7 | bilccRCC (49–51) |
NA | c.340+578C>T | rs139622356 | 0.05 | NA | NA | NA | NA | NA | VUS |
| #8 | ccRCC (39) |
NA | c.340+578C>T | rs139622356 | 0.05 | NA | NA | NA | NA | NA | VUS |
| #9 | Multiple retinal HMB (36) | 32 | c.340+617C>G‡ | NA | NA | NA | NA | NA | NA | Pathogenic | |
| #10 | PCC (11) |
15 | c.340+682T>C | NA | No | c.482G>A p.R161Q | Pos | Decrease | Increase | Pathogenic | |
| #11 | Multiple HN PGL (12) | 1 | 340+725A>T | NA | NA | NA | NA | NA | NA | VUS | |
| #12 | PCC (34) |
10 | c.340+866C>A | rs536631685 | 0.02 | NA | NA | NA | NA | NA | VUS |
*Frequency in gnomAD or 1000 Genomes,.
†loss of the mutated allele.
‡Mutation described in Lenglet et al.8
bilccRCC, bilateral clear cell renal cell carcinoma; ccRCC, clear cell renal cell carcinoma; HMB, haemangioblastoma; HN, head and neck; LOH, loss of heterozygosity; NA, none available; ND, not done; Neg, negative immunochemistry; PCC, pheochromocytoma; PGL, paraganglioma; Pos, positive immunochemistry; VHL, von Hippel-Lindau; VUS, variant of uncertain significance.
Figure 1.

VHL E1’ variants identified: mutations identified in patient #10 on the germline DNA and somatic DNA (A); location of germline variants found in VHL E1’(B).
Seven patients (patients #2 to #8) (0.6%) carried the same rare variant of uncertain significance (VUS), c.340+578C>T which is referenced in dbSNP as rs139622356 and has been previously reported in the Genome Aggregation Database (gnomAD). The five remaining patients carried different E1’ variants. One of them (patient #9) harbours the c.340+617C>G mutation previously described in the original paper.8 Patient #12 carried the c.340+866C>A VUS, which is referenced in dbSNP (rs536631685) and 1000 Genomes, but not in the Genome Aggregation Database (gnomAD). Finally, three novel VUS of the E1’ VHL cryptic exon were discovered in the three remaining patients. None of the four patients with a PPGL and an E1’ VUS have developed VHL spectrum tumour(s) during their follow-up and none of them had family history of VHL disease (table 2); however, segregation analysis was only performed in patient #9. The proband’s mother did carry the variant and had a pancreatic cyst and multiple vertebral body haemangiomas which both are evocating of VHL disease.17
Among the remaining 11 patients, 3 tumours were available, 2 as paraffin embedded samples (patients #1, #10) and 1 as a frozen tumour (patient #3). None of them presented a LOH at VHL locus and the mutated allele was lost as determined by Sanger sequencing in tumour #1. In tumour DNA of patient #10, which harbours the c.340+682T>C variant, we identified a second variant in the exon 3 of VHL (c.482G>A; p.Arg161Gln), known to be pathogenic (figure 1). This somatic mutation was previously described in this patient.18 In the absence of LOH, this exon 3 variant may function as the second VHL hit in this tumour. No other mutation of the VHL gene was identified in tumour DNA of patient #3.
To validate and classify these different VUS, we carried out different functional studies on available tumour tissues. We first performed immunohistochemistry to study the expression of CA9, known to be expressed at the membrane of tumour cells in case of VHL inactivation.19 A membranous positive CA9 immunostaining has been previously reported in VHL-related PPGL, haemangioblastoma, endolymphatic tumours and ccRCC.15 20 21 We observed a cluster of tumour cells with a positive membranous CA9 immunostaining only in the PPGL of patient #10 (figure 2A) which can be seen in VHL-related PPGL.15 Then, we assessed the expression of PNMT gene, which is one of the most downregulated genes in VHL-related PPGL,22 23 by RT-qPCR. As expected, the PPGL of patient #10 exhibited a significant low expression of PNMT mRNA, comparable to the VHL-tumours used as controls. On the contrary, the level of PNMT expression was equivalent to control tumours in the PPGL of patient #3, which produced both epinephrine and norepinephrine. Finally, we analysed the expression of VHL gene by RT-qPCR. We assessed the expression of two different VHL mRNA: the mRNA containing the exons 1 and 2 (E1-E2), which will lead with the exon 3 to the expression of the two main VHL proteins (pVHL213 and pVHL160) and VHL mRNA containing the exon 1 and E1' (E1-E1'), which was previously described as increased in tumour or in lymphoblastoid cell lines of patients with E1' mutation. In normal condition, this VHL E1-E1' mRNA is degraded by nonsense-mediated decay (NMD), and in this pathological condition, NMD may be overwhelmed. The PPGL of patients #3 and #10 showed expression of VHL E1-E1' mRNA which was absent in controls, suggesting that the two variants change the VHL mRNA splicing (figure 3). Moreover, the PPGL of patient #10 showed a low expression of VHL E1-E2 mRNA comparable to the VHL-related PPGL used as control (figure 2B). Altogether, these data provide evidence that this VHL E1' mutation (c.340+682T>C) is a pathogenic mutation that combined with the second mutation (c.482G>A; p.Arg161Gln) induce tumorigenesis.
Figure 2.

CA9 immunochemistry in patients tumours with a VHL E1' mutation showed a membranous immunostaining on a cluster of cells in patient #10 PPGL (A). PNMT and VHL (E1-E2 transcript) genes expression by RT-qPCR in patients #3 and #10 showed a low expression of the two genes in patient #10 tumours (B).
Figure 3.
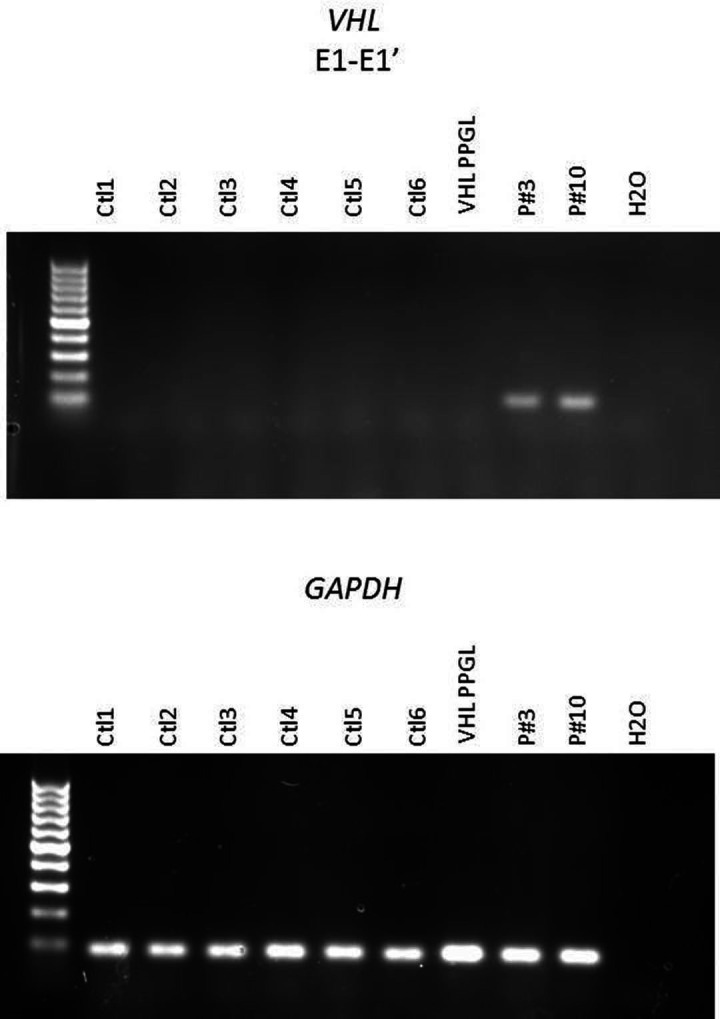
Expression of E1-E1' transcript of VHL gene showed expression only in patients #3 and #10.
Finally, 23 patients carry the c.340+648T>C (rs73024533) variant, previously described in dbSNP, at an heterozygous state. The allele frequency of rs73024533 in our cohort is comparable to that of the gnomAD database and of our control cohort of 198 European subjects (1.9% vs 1.3% and 1.3%, respectively, p=0.0536).
Discussion
E1’ mutations were previously described by Lenglet et al in eight families, either with erythrocytosis or VHL disease. These mutations led to an abnormal VHL mRNA with the insertion of the E1’ in the transcript and to mRNA degradation by NMD and to global defect in VHL protein expression.8
In our large international study, we identified four new germline variants in E1’ VHL gene and we classified two of them as pathogenic, representing 1.3% of ‘VHL-like’ cohort (1/76 patients) and 0.11% of ‘PPGL’ cohort (1/946 patients). Our patients did not have all the manifestation of VHL disease. However, in the single patient in whom a familial genetic screening was performed (patient #9), the proband’s mother had her first screening (cerebral and medullary MRI and abdominal CT scan) at the age of 70 years old, which diagnosed one pancreatic cyst and multiple vertebral body haemangiomas. Interestingly, multiple vertebral body haemangiomas are rare in VHL disease but have been described in patients with Chuvash polycythemia, a disease secondary to a recurrent germline biallelic mutation in VHL gene (c.598C>T, p.Arg200Trp).24 Our data suggest incomplete penetrance of E1’ VHL mutations, as it was previously described for the SDHA gene-another PPGL susceptibility gene- mutations that exhibit a relatively high allele frequency in gnomAD.25
We have identified the same variant c.340+578C>T (rs139622356) in seven patients, but our tumour analyses were not able lead to the classification of this variant in a pathogenic variant. Indeed in one tumour with this variant, we identified the E1-E1' mRNA which suggest that the variant is pathogenic. However, epinephrine secretion and PNMT expression of this tumour are strong indicator against the diagnosis of VHL-related PPGL.26 Moreover, we identified this variant in 0.6% of our cohort, which is 10 times more frequent in our cohort than in reference databases. Indeed, this variant is described in 0.05% of gnomAD subjects (7/1167 vs 16/31 390, p<0.0001). It is noteworthy that in Tuscan and Iberian subjects reported in the 1000 Genomes project, the frequency of this rs139622356 is 0.9%. All these data suggest that this variant could be either a pathogenic variant that is not implicated in the PPGL of our patient because of the lack of LOH/second VHL mutation, or a modifier variant contributing potentially to an 8.5-fold risk (95% CI 4.4 to 14.3, p<0.0001) for development of PPGL or VHL tumours. Hence, more functional analyses and more tumours analyses will be required to achieve a definitive conclusion.
Our study demonstrates that E1’ VHL variants are rare events in ‘VHL-like’ and ‘PPGL’ patients, but nearly as frequent as the VHL mutation rate in exons 1 and 2 in patients with PPGL (in the molecular genetic laboratory of Hôpital Européen Georges Pompidou-Paris-France VHL mutation rate in exon 1 has been reported to be 0.74% (p=0.062), in exon 2: 0.18% (p=0.99) and in exon 3: 0.92% (p=0.0264),27 or as frequent as in exons of other PPGL susceptibility genes (for instance, the mutation rate in exon 1 of SDHD is 0.43%). However, because patients with well-established VHL pathogenic mutations were excluded from our cohort, the current frequency may be an underestimation. As the identification of VHL variants has important implications for management and follow-up of patients and relatives, we suggest that E1’ cryptic exon should be added to NGS diagnostic panels. Considering the genetic heterogeneity of PPGLs and the high rate of detectable driver mutations in these tumours,10 a low frequency of variants in any given new gene/exons is not unexpected. However, the interpretation of these E1' variants might be difficult and more functional analyses has to be designed in order to validate these variants. Finally, our study underlines the importance of variants identified in deep intronic sequences, which would have been missed by examining only coding sequences of genes/exomes. These variants will likely be more frequently detected and studied in the next future with the upcoming implementation of whole-genome sequencing into clinical practice.
Acknowledgments
We thank Catherine Tritscher for administrative assistance and Caroline Travers, Valentin Adamus and Astrid Ramahefasolo for technical assistance. We also thank Pr Pascal Pigny for his help and the PREDIR and TENGEN networks for their help in patient enrolment. The authors would like to express their gratitude to Sophie Devaux for her help.
Footnotes
BC, SF, SG and ML contributed equally.
Contributors: APGR conceived and supervised the study. ABu, APGR, MR, PLD and BG designed the study and analysed the results. JF and NB participated to the data analysis. ABu., BC, SF, SG, ML and ED designed and performed the experiments. PR, JA, IB, BBdP, MC, CD EdM, AE, PH, PK, SL, JLS, ABa, SR, BG, PLD, MR and APGR collected subjects and clinical parameters. ABu and APGR wrote the manuscript. ABu prepared the figures and the tables. All the authors discussed the results and commented the manuscript.
Funding: ABu received a financial support from ITMO Cancer AVIESAN (Alliance Nationale pour les Sciences de la Vie et de la Santé, National Alliance for Life Sciences & Health) within the framework of the Cancer Plan and from la Fondation pour la Recherche Médicale (FDT20170436955). SF is currently supported by an NIH-NIGMS individual predoctoral fellowship grant (F31-GM131634-01) and, previously, by an NIH NRSA Institutional Predoctoral Training Grant T32CA148724 and NRSA F31-GM131634-01. PLD receives funding support from NIH-GM114102, Alex’s Lemonade Stand Cancer Foundation (Innovation Award) and the NCATS- UL1 TR002645. The Genomic Sequencing Facility at the GCCRI is supported by the P30-CA54174 (CTRC at UTHSCSA) and NIH Shared Instrument grant 1S10OD021805-01 (S10 grant). BC is supported by Rafael del Pino Foundation. MR receives funding support from Instituto de Salud Carlos III (ISCIII), through the 'Acción Estratégica en Salud' (AES) (projects PI17/01796), cofounded by the European Regional Development Fund (ERDF), and the Paradifference Foundation.
Disclaimer: The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Competing interests: None declared.
Patient consent for publication: Not required.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data availability statement: All available data are in the article.
References
- 1. Latif F, Tory K, Gnarra J, Yao M, Duh FM, Orcutt ML, Stackhouse T, Kuzmin I, Modi W, Geil L. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 1993;260:1317–20. 10.1126/science.8493574 [DOI] [PubMed] [Google Scholar]
- 2. Lonser RR, Glenn GM, Walther M, Chew EY, Libutti SK, Linehan WM, Oldfield EH, disease vonH-L. Von Hippel-Lindau disease. Lancet 2003;361:2059–67. 10.1016/S0140-6736(03)13643-4 [DOI] [PubMed] [Google Scholar]
- 3. Binderup MLM, Galanakis M, Budtz-Jørgensen E, Kosteljanetz M, Luise Bisgaard M, Prevalence LBM. Prevalence, birth incidence, and penetrance of von Hippel-Lindau disease (vHL) in Denmark. Eur J Hum Genet 2017;25:301–7. 10.1038/ejhg.2016.173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Favier J, Amar L, Gimenez-Roqueplo A-P. Paraganglioma and phaeochromocytoma: from genetics to personalized medicine. Nat Rev Endocrinol 2015;11:101–11. 10.1038/nrendo.2014.188 [DOI] [PubMed] [Google Scholar]
- 5. Gimenez-Roqueplo A-P, Dahia PL, Robledo M. An update on the genetics of paraganglioma, pheochromocytoma, and associated hereditary syndromes. Horm Metab Res 2012;44:328–33. 10.1055/s-0031-1301302 [DOI] [PubMed] [Google Scholar]
- 6. Amar L, Bertherat J, Baudin E, Ajzenberg C, Bressac-de Paillerets B, Chabre O, Chamontin B, Delemer B, Giraud S, Murat A, Niccoli-Sire P, Richard S, Rohmer V, Sadoul J-L, Strompf L, Schlumberger M, Bertagna X, Plouin P-F, Jeunemaitre X, Gimenez-Roqueplo A-P. Genetic testing in pheochromocytoma or functional paraganglioma. J Clin Oncol 2005;23:8812–8. 10.1200/JCO.2005.03.1484 [DOI] [PubMed] [Google Scholar]
- 7. Currás-Freixes M, Piñeiro-Yañez E, Montero-Conde C, Apellániz-Ruiz M, Calsina B, Mancikova V, Remacha L, Richter S, Ercolino T, Rogowski-Lehmann N, Deutschbein T, Calatayud M, Guadalix S, Álvarez-Escolá C, Lamas C, Aller J, Sastre-Marcos J, Lázaro C, Galofré JC, Patiño-García A, Meoro-Avilés A, Balmaña-Gelpi J, De Miguel-Novoa P, Balbín M, Matías-Guiu X, Letón R, Inglada-Pérez L, Torres-Pérez R, Roldán-Romero JM, Rodríguez-Antona C, Fliedner SMJ, Opocher G, Pacak K, Korpershoek E, de Krijger RR, Vroonen L, Mannelli M, Fassnacht M, Beuschlein F, Eisenhofer G, Cascón A, Al-Shahrour F, Robledo M. PheoSeq: a targeted next-generation sequencing assay for pheochromocytoma and paraganglioma diagnostics. J Mol Diagn 2017;19:575–88. 10.1016/j.jmoldx.2017.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lenglet M, Robriquet F, Schwarz K, Camps C, Couturier A, Hoogewijs D, Buffet A, Knight SJL, Gad S, Couvé S, Chesnel F, Pacault M, Lindenbaum P, Job S, Dumont S, Besnard T, Cornec M, Dreau H, Pentony M, Kvikstad E, Deveaux S, Burnichon N, Ferlicot S, Vilaine M, Mazzella J-M, Airaud F, Garrec C, Heidet L, Irtan S, Mantadakis E, Bouchireb K, Debatin K-M, Redon R, Bezieau S, Bressac-de Paillerets B, Teh BT, Girodon F, Randi M-L, Putti MC, Bours V, Van Wijk R, Göthert JR, Kattamis A, Janin N, Bento C, Taylor JC, Arlot-Bonnemains Y, Richard S, Gimenez-Roqueplo A-P, Cario H, Gardie B. Identification of a new VHL exon and complex splicing alterations in familial erythrocytosis or von Hippel-Lindau disease. Blood 2018;132:469–83. 10.1182/blood-2018-03-838235 [DOI] [PubMed] [Google Scholar]
- 9. Maher ER, Neumann HP, Richard S. Von Hippel-Lindau disease: a clinical and scientific review. Eur J Hum Genet 2011;19:617–23. 10.1038/ejhg.2010.175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Toledo RA, Burnichon N, Cascon A, Benn DE, Bayley J-P, Welander J, Tops CM, Firth H, Dwight T, Ercolino T, Mannelli M, Opocher G, Clifton-Bligh R, Gimm O, Maher ER, Robledo M, Gimenez-Roqueplo A-P, Dahia PLM, NGS in PPGL (NGSnPPGL) Study Group . Consensus statement on next-generation-sequencing-based diagnostic testing of hereditary phaeochromocytomas and paragangliomas. Nat Rev Endocrinol 2017;13:233–47. 10.1038/nrendo.2016.185 [DOI] [PubMed] [Google Scholar]
- 11. Lenders JWM, Duh Q-Y, Eisenhofer G, Gimenez-Roqueplo A-P, Grebe SKG, Murad MH, Naruse M, Pacak K, Young WF, Endocrine Society . Pheochromocytoma and paraganglioma: an endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2014;99:1915–42. 10.1210/jc.2014-1498 [DOI] [PubMed] [Google Scholar]
- 12. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, ACMG Laboratory Quality Assurance Committee . Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet Med 2015;17:405–23. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hoefling C, Schmidt H, Meinhardt M, Lohse A, Taubert H, Fuessel S, Schmidt U, Schuster K, Baretton G, Wirth MP, Meye A. Comparative evaluation of microsatellite marker, AP-PCR and CGH studies in primary renal cell carcinoma. Int J Mol Med 2004;13:835–42. 10.3892/ijmm.13.6.835 [DOI] [PubMed] [Google Scholar]
- 14. Girolami F, Passerini I, Gargano D, Frusconi S, Villari D, Nicita G, Torricelli F. Microsatellite analysis of chromosome 3p region in sporadic renal cell carcinomas. Pathol Oncol Res 2002;8:241–4. 10.1007/BF03036738 [DOI] [PubMed] [Google Scholar]
- 15. Favier J, Meatchi T, Robidel E, Badoual C, Sibony M, Nguyen AT, Gimenez-Roqueplo A-P, Burnichon N. Carbonic anhydrase 9 immunohistochemistry as a tool to predict or validate germline and somatic VHL mutations in pheochromocytoma and paraganglioma-a retrospective and prospective study. Mod Pathol 2020;33:57–64. 10.1038/s41379-019-0343-4 [DOI] [PubMed] [Google Scholar]
- 16. Zeka F, Vanderheyden K, De Smet E, Cuvelier CA, Mestdagh P, Vandesompele J. Straightforward and sensitive RT-qPCR based gene expression analysis of FFPE samples. Sci Rep 2016;6:21418 10.1038/srep21418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. van Asselt SJ, de Vries EG, van Dullemen HM, Brouwers AH, Walenkamp AM, Giles RH, Links TP. Pancreatic cyst development: insights from von Hippel-Lindau disease. Cilia 2013;2:3 10.1186/2046-2530-2-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mircescu H, Wilkin F, Paquette J, Oligny LL, Decaluwe H, Gaboury L, Nolet S, Van Vliet G, Deal C. Molecular characterization of a pediatric pheochromocytoma with suspected bilateral disease. J Pediatr 2001;138:269–73. 10.1067/mpd.2001.111316 [DOI] [PubMed] [Google Scholar]
- 19. Ivanov SV, Kuzmin I, Wei MH, Pack S, Geil L, Johnson BE, Stanbridge EJ, Lerman MI. Down-Regulation of transmembrane carbonic anhydrases in renal cell carcinoma cell lines by wild-type von Hippel-Lindau transgenes. Proc Natl Acad Sci U S A 1998;95:12596–601. 10.1073/pnas.95.21.12596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pinato DJ, Ramachandran R, Toussi STK, Vergine M, Ngo N, Sharma R, Lloyd T, Meeran K, Palazzo F, Martin N, Khoo B, Dina R, Tan TM. Immunohistochemical markers of the hypoxic response can identify malignancy in phaeochromocytomas and paragangliomas and optimize the detection of tumours with VHL germline mutations. Br J Cancer 2013;108:429–37. 10.1038/bjc.2012.538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stillebroer AB, Mulders PFA, Boerman OC, Oyen WJG, Oosterwijk E. Carbonic anhydrase IX in renal cell carcinoma: implications for prognosis, diagnosis, and therapy. Eur Urol 2010;58:75–83. 10.1016/j.eururo.2010.03.015 [DOI] [PubMed] [Google Scholar]
- 22. Castro-Vega LJ, Letouzé E, Burnichon N, Buffet A, Disderot P-H, Khalifa E, Loriot C, Elarouci N, Morin A, Menara M, Lepoutre-Lussey C, Badoual C, Sibony M, Dousset B, Libé R, Zinzindohoue F, Plouin PF, Bertherat J, Amar L, de Reyniès A, Favier J, Gimenez-Roqueplo A-P. Multi-Omics analysis defines core genomic alterations in pheochromocytomas and paragangliomas. Nat Commun 2015;6:6044 10.1038/ncomms7044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. López-Jiménez E, Gómez-López G, Leandro-García LJ, Muñoz I, Schiavi F, Montero-Conde C, de Cubas AA, Ramires R, Landa I, Leskelä S, Maliszewska A, Inglada-Pérez L, de la Vega L, Rodríguez-Antona C, Letón R, Bernal C, de Campos JM, Diez-Tascón C, Fraga MF, Boullosa C, Pisano DG, Opocher G, Robledo M, Cascón A. Research resource: transcriptional profiling reveals different pseudohypoxic signatures in SDHB and VHL-related pheochromocytomas. Mol Endocrinol 2010;24:2382–91. 10.1210/me.2010-0256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gordeuk VR, Sergueeva AI, Miasnikova GY, Okhotin D, Voloshin Y, Choyke PL, Butman JA, Jedlickova K, Prchal JT, Polyakova LA. Congenital disorder of oxygen sensing: association of the homozygous Chuvash polycythemia VHL mutation with thrombosis and vascular abnormalities but not tumors. Blood 2004;103:3924–32. 10.1182/blood-2003-07-2535 [DOI] [PubMed] [Google Scholar]
- 25. van der Tuin K, Mensenkamp AR, Tops CMJ, Corssmit EPM, Dinjens WN, van de Horst-Schrivers ANA, Jansen JC, de Jong MM, Kunst HPM, Kusters B, Leter EM, Morreau H, van Nesselrooij BMP, Oldenburg RA, Spruijt L, Hes FJ, Timmers HJLM. Clinical aspects of SDHA-Related pheochromocytoma and paraganglioma: a nationwide study. J Clin Endocrinol Metab 2018;103:438–45. 10.1210/jc.2017-01762 [DOI] [PubMed] [Google Scholar]
- 26. Eisenhofer G, Lenders JWM, Timmers H, Mannelli M, Grebe SK, Hofbauer LC, Bornstein SR, Tiebel O, Adams K, Bratslavsky G, Linehan WM, Pacak K. Measurements of plasma methoxytyramine, normetanephrine, and metanephrine as discriminators of different hereditary forms of pheochromocytoma. Clin Chem 2011;57:411–20. 10.1373/clinchem.2010.153320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ben Aim L, Pigny P, Castro-Vega LJ, Buffet A, Amar L, Bertherat J, Drui D, Guilhem I, Baudin E, Lussey-Lepoutre C, Corsini C, Chabrier G, Briet C, Faivre L, Cardot-Bauters C, Favier J, Gimenez-Roqueplo A-P, Burnichon N. Targeted next-generation sequencing detects rare genetic events in pheochromocytoma and paraganglioma. J Med Genet 2019;56:513–20. 10.1136/jmedgenet-2018-105714 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jmedgenet-2019-106519supp001.pdf (55.6KB, pdf)
jmedgenet-2019-106519supp002.pdf (45.8KB, pdf)