

Abstract
We present solid-state NMR measurements of β-strand secondary structure and inter-strand organization within a 150 kDa oligomeric aggregate of the 42-residue variant of the Alzheimer’s amyloid-β peptide (Aβ(1–42)). We build upon our previous report of a β-strand spanned by residues 30–42, which arranges into an antiparallel β-sheet. New results presented here indicate that there is a second β-strand formed by residues 11–24. Contrary to expectations, NMR data indicate that this second β-strand is organized into a parallel β-sheet despite the co-existence of an antiparallel β-sheet in the same structure. In addition, the in-register parallel β-sheet commonly observed for amyloid fibril structure does not apply to residues 11–24 in the 150 kDa oligomer. Rather, we present evidence for an inter-strand registry shift of 3 residues that likely alternates in direction between adjacent molecules along the β-sheet. We corroborated this unexpected scheme for β-strand organization using multiple 2-dimensional NMR and 13C-13C dipolar recoupling experiments. Our findings indicate a previously unknown assembly pathway and inspire a suggestion as to why this aggregate does not grow to larger sizes.
Keywords: Amyloid-β oligomer, Alzheimer’s disease, peptide aggregate pathways, solid state NMR, out-of-register parallel β-sheet
Introduction
Research on the role of amyloid-β (Aβ) peptide assembly in Alzheimer’s disease (AD) can be described in terms of two presently unanswered questions: 1) what aggregated Aβ structures are possible under different aggregation pathways; and 2) what roles do different aggregated Aβ structures play in cellular pathology in the brain? The evidence for the central role played by Aβ aggregation in AD is reviewed elsewhere [1–3]. Aggregation pathways are complex, producing multiple possible structures broadly classified as oligomers (dimers through 50-mers), protofibrils (hundreds of molecules), and amyloid fibrils (thousands to millions of molecules). The documented ability of the 40-residue variant of Aβ (Aβ(1–40)) to form multiple distinct fibril structures [4–6] highlights the complexity of Aβ aggregation pathways and suggests that there may also be considerable diversity in possible oligomer and protofibril structures. Recently reported structures of fibrils of the 42-residue variant of Aβ (Aβ(1–42)) [7–11] and mutant Aβ(1–40) [12, 13] differ from any previously reported structures of Aβ(1–40) fibrils, indicating that a small difference in primary structure could dramatically affect aggregation pathways. Furthermore, the toxicity of Aβ aggregates has been shown to be structure dependent: investigations of Aβ toxicity in cell culture, animal models, and human brain tissue has highlighted the special toxicity of oligomers over toxicity levels associated with protofibrils and fibrils [2]. In vivo, oligomers could diffuse over larger length scales than the larger Aβ aggregates and may interact in specific ways with cellular membranes and receptors. Furthermore, Aβ oligomers comprise an extremely diverse group [14] and include endogenous [15] as well as synthetic species prepared in vitro [16].
It is not known why Aβ would assemble into any structure within the range of reported oligomer masses (10–200 kDa or 2–50 molecules) without undergoing further assembly into amyloid fibrils. Specifically, the 150 kDa (~32 molecules) oligomers investigated in this study do not elongate in the presence of Aβ monomers to form amyloid fibrils, seed assembly of fibrils from soluble Aβ, or show enhanced fluorescence with thioflavin T (ThT) [17]. In contrast, protofibrils do undergo further assembly to fibrils and show enhanced fluorescence with ThT [18, 19]. Measurements based on methods such as Fourier transform infrared spectroscopy (FTIR) and circular dichroism (CD) establish β-strand secondary structure for 2–4mers and 150 kDa Aβ oligomers in our studies, as with some studies on other oligomer preparations [17, 20, 21]. A planar β-sheet of any size would have dangling hydrogen bond donors and acceptors at its ends, and these ends would be expected to be capable of recruiting additional Aβ molecules into the β-sheet. However, without more structural knowledge it is unclear whether this reasoning would apply to oligomers in the 150 kDa range. Data on Aβ(1–42) oligomers in the smaller mass range under 50 kDa (2–10 molecules) offer some clues, with structures that may or may not contain β-strand secondary structure. In some studies of smaller oligomers, conversion from non-β-strand to β-strand secondary structure is associated with growth to larger sizes and conversion to fibrils [22, 23]. Other studies propose low-molecular-weight oligomer structures that are geometrically incompatible with the approximately planar structure of high-molecular-weight β-sheets and may be incapable of undergoing further assembly without conformational rearrangement [24]. Laganowsky et. al. [25] observed a barrel-shaped oligomer formed by a fragment of αB crystallin and suggest that Aβ could form a similar non-planar structure.
The preparation and characteristics of 150 kDa oligomers share many features with those of globulomers, including their cellular toxicity [17, 26, 27]. The assembly pathway that produces globulomers and the 150 kDa oligomer in our investigation is driven by interaction with the anionic detergent sodium dodecyl sulfate (SDS) near its critical micelle concentration [17]. Initially 2–4mer structures are formed. Dialysis to remove SDS, which is necessary for our solid-state NMR measurements, results in an increase in aggregate size to 150 kDa (~32-mers), as determined by multi-angle light scattering [27]. Globulomers completely inhibited long-term potentiation in rat hippocampal slices and directly modulated recombinant P/Q-type and N-type calcium channels in HEK293 cells [28, 29]. In addition, 150 kDa oligomers but neither monomers nor fibrils induced senescence in brain endothelial cells [30]. Both polyclonal and specific monoclonal antibodies (mAbs) were raised against globulomers [26, 31], and one mAb (A-887755) prevented synapse loss in a mouse model of AD [32].
In this report, we present experimental evidence that there are two β-strands within the secondary structure of Aβ(1–42) when assembled into 150 kDa oligomers. The amino acid sequence for this peptide is DAEFRHDSGY10 EVHHQKLVFF20 AEDVGSNKGA30 IIGLMVGGVV40 IA. Previously, we reported that residues 30–42 form a β-strand (the C-strand) that is organized into an antiparallel β-sheet centered at residue V36 [33, 34]. New results in this report reveal the presence of a second β-strand spanning residues 11–24, which we call the N-strand. The antiparallel inter-molecular organization of the C-strand would motivate the hypothesis that the N-strand must also be arranged into an antiparallel β-sheet. However, NMR experiments designed here to test this hypothesis indicate otherwise. Therefore, the 150 kDa oligomer structure is incompatible with any previously proposed Aβ aggregate structure known to us.
Results
Imaging of oligomers with electron microscopy reveals sizes consistent with previously reported results
Figure 1 shows a negative-stain transmission electron microscopy (TEM) image of 150 kDa oligomers. Images contain structures that appear approximately circular (globular) with approximately uniform diameters under 10 nm. This size is consistent with our previous measurements using light scattering and electrophoresis [17, 27, 33]. Furthermore, oligomer size can be estimated using the Aβ(1–42) fibril structure produced by Colvin et. al. [8], assuming that oligomer and fibrils have similar densities. The fibril structure has a β-strand spacing of 0.45 nm within each β-sheet and consists of 2 molecules in the cross section perpendicular to the β-sheet axis. Therefore, a fibril segment with a mass of 150 kDa would have a length of 7.5 nm. Considering the uncertainty in particle size estimation with negative-stain TEM, the image in Figure 1 is consistent with our previous measurements of oligomer size by light scattering. These results are also consistent with our previously reported imaging by atomic force microscopy [33].
Figure 1:

A) A negative-stain TEM image of 150 kDa oligomers. B) The region from Panel A indicated by the dashed square, enlarged to more clearly show oligomer size. The white gridlines are separated by 10 nm.
2D 13C NMR spectra reveal two β-strands and evidence for multi-site occupancy
Spectral assignments, correspondences between 13C NMR peaks and isotopically labeled sites, were determined by collecting 2-dimensional (2D) 13C-13C NMR spectra with a short mixing time for dipolar recoupling on samples that were uniformly 13C-labeled (with 13C at every C site) within selected amino acids. For example, Figure 2 shows a 2D-fpRFDR spectrum [35] from a 150 kDa oligomer sample (Sample 1) that was 13C-labeled uniformly at K16, F20, V24, and G37. The pattern of off-diagonal peaks between directly bonded 13C atoms makes it possible to determine spectral assignments by analyzing crosspeak patterns that are unique to each uniformly labeled amino acid (see colored lines in Figure 2). Table 1 reports the isotopic labeling employed for the full series of 150 kDa oligomer samples examined in the study. Figures S1 and S2 show the 2D-fpRFDR spectra for samples, with labels chosen so that structure could be assessed for the whole peptide. Table S1 tabulates all 13C NMR peak positions (chemical shifts) and peak widths we have measured from spectra of Samples 1–13 (Figures 2, S1, and S2) as well as Sample 14 (published in [33]).
Figure 2:
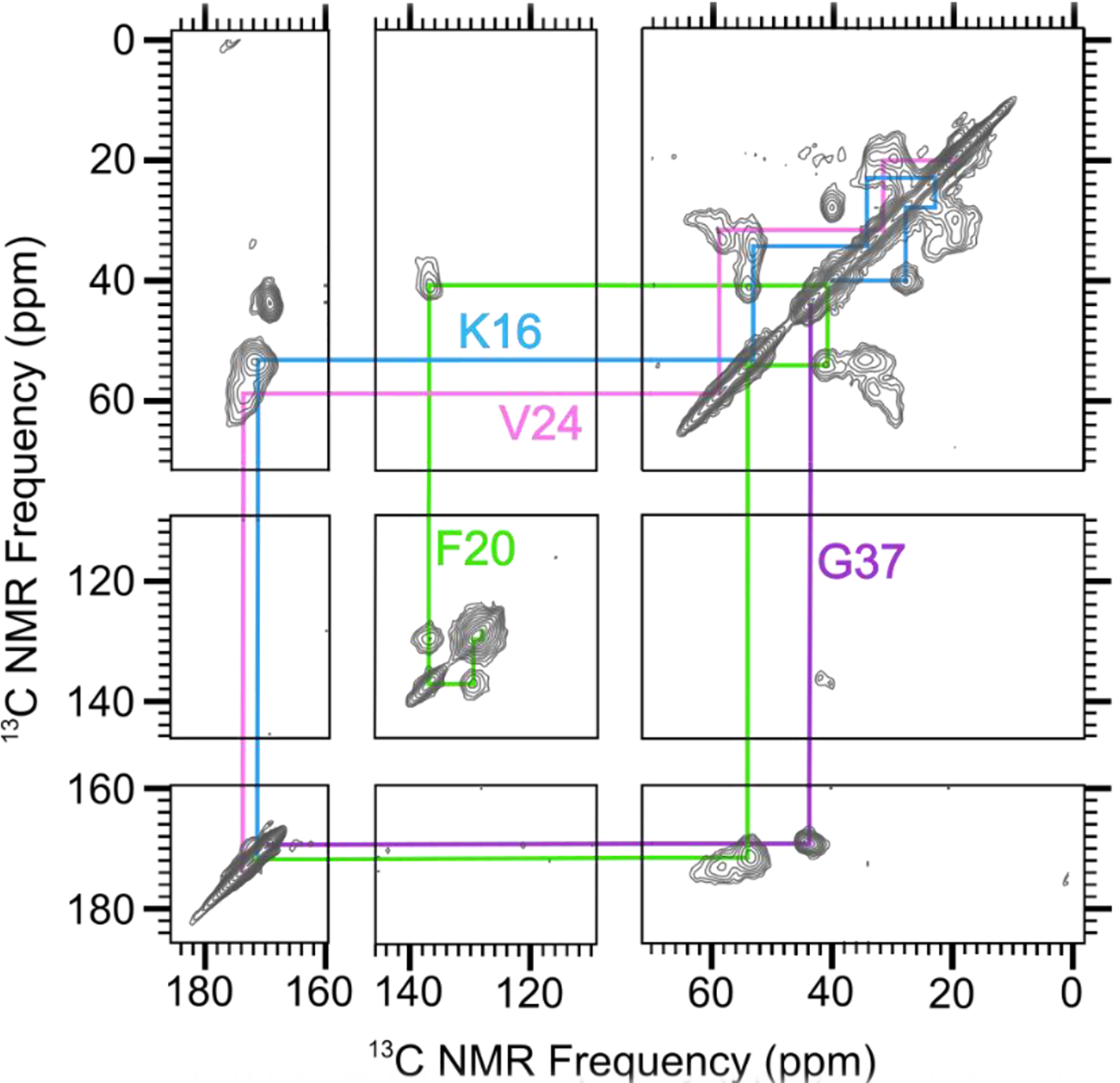
A 2D-fpRFDR spectrum of 150 kDa Aβ(1–42) oligomer sample that was uniformly labeled with 13C at K16, F20, V24, and G37 (Sample 1 in Table 1). Colored lines indicate spectral assignments based on crosspeaks between directly bonded 13C atoms.
Table 1.
Isotopic labeling employed for the 150 kDa oligomer samples analyzed in this study.
| Sample | Isotopic Labeling |
|---|---|
| Uniform 13C-labeling and 15N- labeling at the indicated residues: | |
| 1 | K16, F20, V24, G37 |
| 2 | D7, G9, E11.L17, F19, A21 |
| 3 | E11, F19, 131, V36 |
| 4 | E11.L17.A21, M35, G38 |
| 5 | I32, M35, G37, V40 |
| 6 | Q15, V18, A21 |
| 7 | S8, Y10, V12, L34, G38, 141 |
| 8 | V12, E22, S26, N27, G33 |
| 9 | V12, F20, D23, K28, G29 |
| 10 | E11, H13, Q15, L17 |
| 11 | E11, K16, F19, V36 |
| 12 | A2, E3, F4, G9, V39 |
| 13 | H14, K16, L34, A42 |
| 14 | F19, V24, G25, A30, 131, L34, M35 [33] |
| Selective 13C-labeling at the indicated sites: | |
| A | L17 CO |
| B | V18 CO |
| C | F19 CO |
| D | F20 CO |
| E | A21 CO |
| F | L17 CO and F19 CO |
| G | V18COandA21 CO |
| H | L17 CO and A21 CO |
| I | V18 CO and A21 CO (50% isotope-diluted) |
We assessed peptide secondary structure through analysis of 13C NMR frequencies (chemical shifts) for backbone C sites. Figure 3A reports secondary 13C chemical shifts for all CO, Cα, and Cβ for most of the residues in the oligomer and indicates the presence of 2 β-strand regions. Secondary chemical shifts are measured NMR peak frequencies relative to reported values for corresponding atoms in the same amino acids within model random-coil peptides in solution [36]. Secondary structure is known to correlate with 13C chemical shift when CO, Cα, and Cβ chemical shifts deviate in systematic ways from corresponding random-coil values for contiguous sequences of amino acids within the primary structure [37]. In particular, β-strand secondary structure typically observed in Aβ aggregates corresponds to negative CO and Cα secondary shifts and positive Cβ secondary shifts. To be more precise, we fed the assigned chemical shifts and peptide sequence into a computer program called TALOS-N [38] to predict the backbone torsion angles (ϕ/ψ). For the 150 kDa oligomer, TALOS-N predicts the presence of two β-strands as shown in Figure 3A. The β-strands span residues 11–24 and 30–42; we refer to them as the N-strand and C-strand, respectively. The regions spanned by residues 1–10 and 25–29 are predicted to be an unstructured segment and a turn, respectively. The detailed output of the TALOS program, which includes estimated uncertainty in torsion angle predictions, is shown in Table S2.
Figure 3.
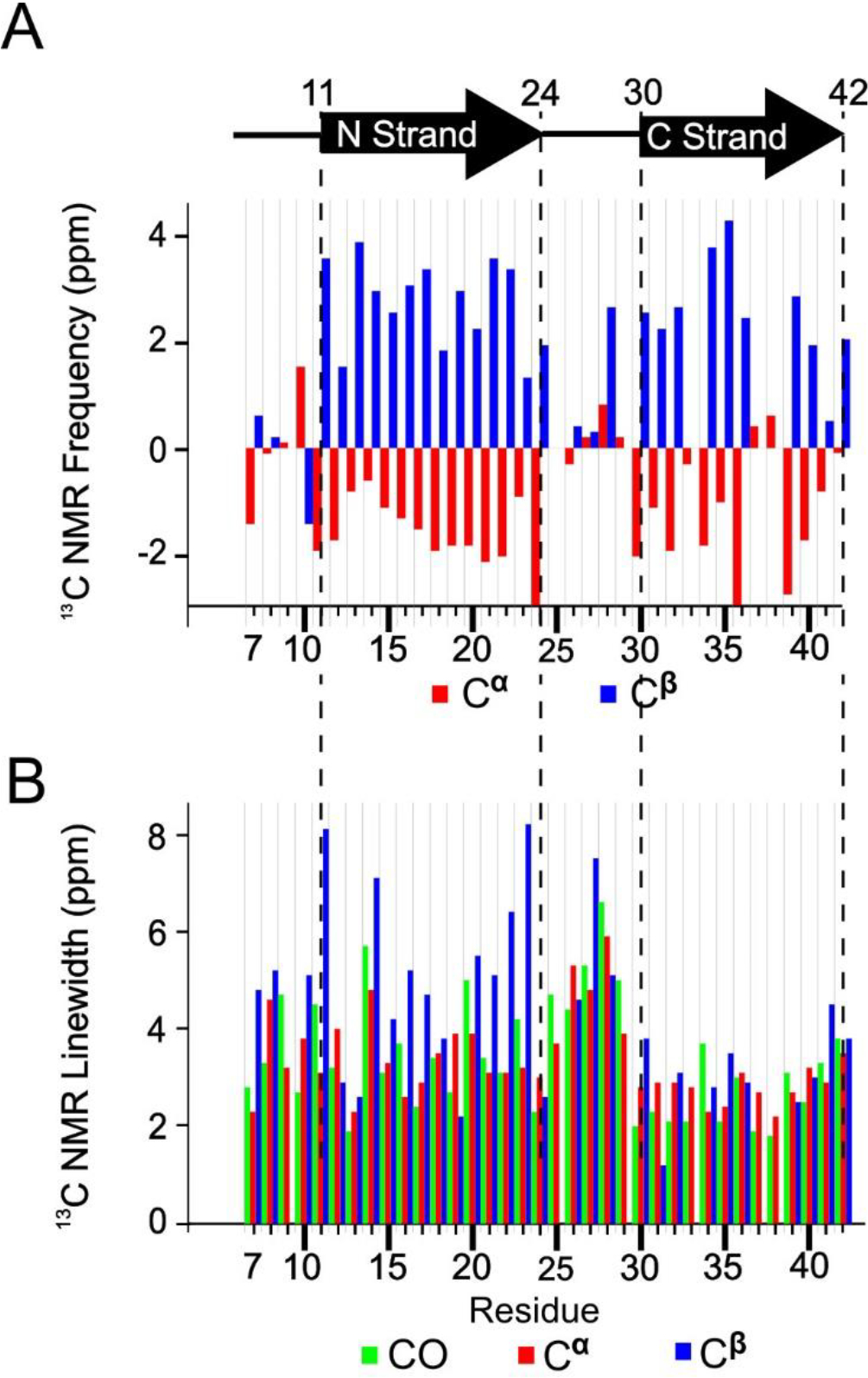
A) Secondary structure of peptide residues in the 150 kDa Aβ(1–42) oligomer, predicted by TALOS-N software, and the measured secondary 13C NMR backbone chemical shifts upon which the TALOS-N prediction is based. B) The NMR linewidths of CO (green), Cα (red), and Cβ (blue) reported as full widths at half maximum measured by nonlinear regression of cross peaks in the 2D-fpRFDR 13C-13C NMR spectra to Gaussian functions.
An interesting observation is that the linewidths were larger for N-strand 13C signals than the C-strand counterparts, especially for the residues near the ends of the N-strand (Figure 3B). On average, the CO, Cα, and Cβ linewidths were 3.3 ± 0.5, 3.2 ± 0.5, and 4.0 ± 1.1 ppm (95% confidence region for full width half maximum), respectively, for the N-strand residues. The corresponding average linewidths for the C-strand were 2.5 ± 0.4, 2.7 ± 0.2, and 3.0 ± 0.7 ppm. Figure S3 shows that, based on a t-test, the CO and Cα linewidths differ significantly between the N- and C-strands. Figure S4 further illustrates this point, showing crosspeaks in 2D-fpRFDR spectra from the 6 valine residues in the peptide (3 within each strand). The NMR signals from the N-strand valine residues (V12, V18, and V24) exhibit more evidence of disorder via asymmetric 2D-NMR crosspeaks than the C-strand valine residues (V36, V39, and V40). In addition, although crosspeaks from the valines in the N-strand have broader linewidths, the chemical shifts of the main Cα-Cβ crosspeaks of every valine are still consistent with β-strand conformation (Figure S4B). In terms of molecular structure, these spectra suggest that N-strand residues occupy multiple magnetically inequivalent sites while C-strand residues do not. This inequivalence will be discussed further as more results are presented.
Most of our subsequent analysis of β-strand arrangements was based on the interpretation that N-strands assemble into a β-sheet that contains only N-strands, and that C-strands assemble into distinct β-sheets in the oligomer. This assumption is supported by the aforementioned difference between the N- and C-strand 13C peak linewidths. Nevertheless, after presenting results that constrain relative orientations of adjacent N-strands, we do consider below a β-sheet model in which N-strands and C-strands co-assemble into the same β-sheet.
PITHIRDS-CT data and 2D-DARR negative results do not support in-register parallel or antiparallel N-strand β-sheet
13C-13C dipolar couplings obtained via the PITHIRDS-CT NMR experiment [39] (Figure 4A) indicate that N-strands are not assembled into an in-register parallel β-sheet. To reach this conclusion, PITHIRDS-CT decays, or 13C NMR peak intensity as a function of the effective duration of 13C-13C dipolar recoupling, were measured for a series of oligomer samples that were each selectively 13C-labeled at a single backbone CO site within the N-strand (Samples A-E, Table 1). With only one 13C-labeled site per molecule, this measurement is expected to result in a decay curve that is bounded by the simulated (dashed) curves in Figure 4A if 13C atoms on adjacent molecules are separated by distances of approximately 0.5 nm. The measured decays are all considerably weaker than the simulated curves, indicating that we did not detect the influence of inter-molecular dipolar couplings. As detailed under Materials and Methods, the theoretical curves were obtained by simulating spin dynamics for eight coupled 13C atoms arranged in straight lines with constant 13C-13C spacings of 0.5 or 0.6 nm under the influence of the PITHIRDS-CT pulse sequence. The linear geometry for 13C atoms was inspired by the in-register parallel β-sheet structure that is common to amyloid fibrils. In this configuration, a 13C atom placed at any backbone CO site or an alanine Cβ site would be arranged into a linear configuration with a nearest-neighbor spacing of 0.48 nm. Empirically, we have observed that a 13C PITHIRDS-CT curve that is bounded by the simulated curves indicates that each 13C atom in the sample is within approximately 0.5 nm from at least one other 13C atom1. Figure 4B demonstrates this observation for previously published data on Aβ(1–42) amyloid fibrils [33], in which β-strands are known to form in-register parallel β-sheets [42]. The accompanying schematic illustrates the expected relative positions of 13C-labeled sites within the C-strands. Figure S5 shows that, although the decays corresponding to 13C labeling at CO sites appear closer to the theoretical curve for 0.6nm inter-nuclear spacing in Figure 4B, this discrepancy influenced by the effect of chemical shift anisotropy on PITHIRDS-CT decays when the 13C-labeled site is a CO. We include more atomic details in a model of the parallel β-sheet in Figure S6A.
Figure 4.
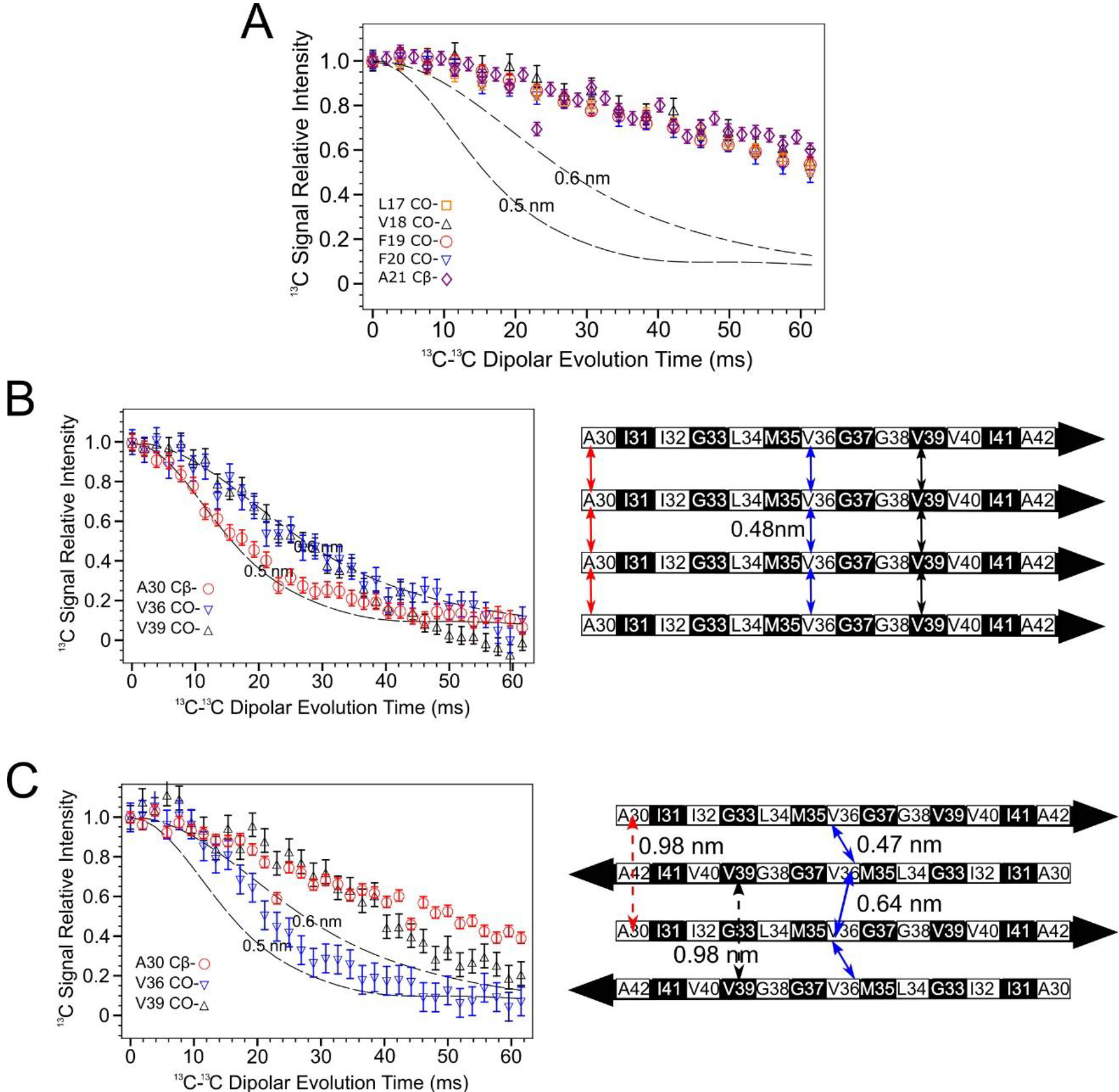
PITHIRDS-CT 13C-13C dipolar recoupling curves rule out an antiparallel alignment of N-strands. A) Curves measured for 150 kDa Aβ(1–42) oligomer samples 13C-labeled at one backbone CO position per molecule within the N-strand (Samples A-E). B) Curves from Aβ(1–42) fibril samples 13C labeled at single near-backbone positions on the C-strand (left) [33] and a schematic of an in-register parallel β-sheet formed by C-strand (right). The black and white coloring for each residue in the β-strand schematic indicate whether each sidechain is above (black) or below (white) the plane of the β-sheet. The double-headed colored arrows indicate the distances between equivalent backbone CO or Cβ sites on adjacent molecules that are short enough (0.6 nm or less) to measurably affect PITHIRDS-CT decays. C) Curves from 150 kDa oligomer samples 13C-labeled at single near-backbone positions on the C-strand (left) [33] and a schematic of an antiparallel C-strand β-sheet (right). Black and white shading on the β-strand schematic is as in Panel B. Since this β-sheet is centered at V36, the CO site of this residue would be the only backbone CO site that would yield a strong PITHIRDS-CT decay if selectively 13C-labeled (double headed arrows with solid line). Distances for other backbone CO sites are too great (e.g., double headed arrows with dashed line). Dashed lines in PITHIRDS-CT panels indicate simulated 13C inter-atomic distances that were calculated for a linear array of eight 13C spins separated by the indicated identical constant distances as outlined in the Methods.
The PITHIRDS-CT decays in Figure 4A as noted are weak and exhibit no dependence on the residue position of the 13C-labeled backbone site, also ruling out an antiparallel β-sheet for N-strands. To reinforce this interpretation, the expected pattern of PITHIRDS-CT decays for an antiparallel β-sheet are illustrated with previously published data on the C-strands [33, 34], which were found to form an antiparallel β-sheet structure (Figure 4C; see also the all-atom β-sheet model in Figure S6A). Since V36 is at the center of the C-strand antiparallel β-sheet, 13C isotopic labeling of the CO site of V36 results in an expected nearest-neighbor 13C-13C distance of less than 0.5 nm and a PITHIRDS-CT decay that is similar to that observed for in-register parallel β-sheets. Although arrangement of 13C atoms in this case would not be linear, we expect the simulated curves to bound experimental decays observed for 13C spin systems in which nearest-neighbor 13C-13C distances are near 0.5 nm. For 13C-labeling at other backbone sites within the C-strand, the antiparallel configuration corresponds to significantly larger 13C-13C distances (above 0.7 nm). In fact, weaker PITHIRDS-CT decays were observed with 13C labeling at the A30 Cβ or the V39 CO site. Figure S7 illustrates that this behavior would be expected for any antiparallel β-sheet composed of β-strands with identical amino acid sequences on different molecules: there is a single amino acid at the “center” with short inter-molecular distance between backbone carbonyls. However, several N-strand antiparallel β-sheet models with different center residues and the sidechain orientations (Figure S8 and S9) failed to support the experimental data. Based on the secondary structure determination for the 150 kDa oligomer (Figure 3A) and the knowledge that C-strands form an antiparallel β-sheet, we assumed that the N-strands would also arrange into an antiparallel β-sheet with L17 or V18 at the center. However, the data in Figure 4A indicates that no residue between L17 and A21 (inclusive) corresponds to the center of an antiparallel β-sheet.
In addition to PITHIRDS-CT experiments, we also used 2D-dipolar assisted rotational resonance (DARR) [43, 44] measurements to probe for a possible antiparallel N-strand β-sheet. This technique, when employed with a 500 ms mixing time for 13C-13C dipolar recoupling (500 ms 2D-DARR), produces spectra illustrated in Figure 5A and is capable of detecting 2D NMR crosspeaks corresponding to 13C atoms on distinct residues that are in dipolar “contact”. That is, we expected to observe inter-residue crosspeaks in 500 ms 2D-DARR spectra when both residues are 13C uniformly labeled and at least one inter-residue pair of 13C atoms is within 0.6 nm [45, 46]. We use contact charts like Figure 5B to facilitate a direct comparison between 2D-DARR NMR data and molecular modeling predictions. The predictions are based on a model of a Type-I antiparallel β-sheet centered at L17 (see Figures 5C, S7 and S8B). In the contact chart, each row and column correspond to the stretch of N-strand residues, and a grid square is shaded gray or red if the all-atom model predicts a corresponding inter-residue contact. Gray shading is for residue pairs that, by virtue of being close in sequence, would exhibit intramolecular contacts if 13C-labeled (see Figure S10) and cannot be used to report on inter-molecular arrangements with the employed labeling scheme. In Figure 5A, the 2D-DARR contacts detected between L17 and F19 and between F19 and A21 provide examples of such intramolecular contacts and serve as positive controls. Red shading in the contact chart is for residue pairs that are predicted by the β-sheet molecular model to only form inter-molecular contacts. Circles in Figure 5B and dashed arrows in Figure 5C indicate residue pairs which were uniformly 13C-labeled in the same sample but failed to produce detected inter-residue 2D-DARR crosspeaks. The spectrum in Figure 5A is marked to show where crosspeaks between E11 and A21, between E11 and F19, and between E11 and L17 would have been expected. More details of molecular modeling are shown in Figures S8 and S9; these figures describe a series of antiparallel β-sheet models centered at different residues within the N-strand. We show a contact chart for each model, indicating that we were unable to detect predicted 2D-DARR inter-residue contacts. The observed 2D-DARR negative results contradict every antiparallel β-sheet model with the center located between residues 14 and 18 (inclusive). Figure S11 exhibits selected slices taken from 2D-DARR spectra, with dashed rectangles indicating frequencies at which inter-residue crosspeaks were predicted but not observed. Although the absences of expected 2D-DARR crosspeaks may not be considered conclusive on their own (crosspeaks could be missing because of limited sensitivity), these negative results complement the PITHIRDS-CT data in Figure 4A as well as data showing observed inter-molecular 2D-DARR crosspeaks within the N-strand presented in the next section.
Figure 5.
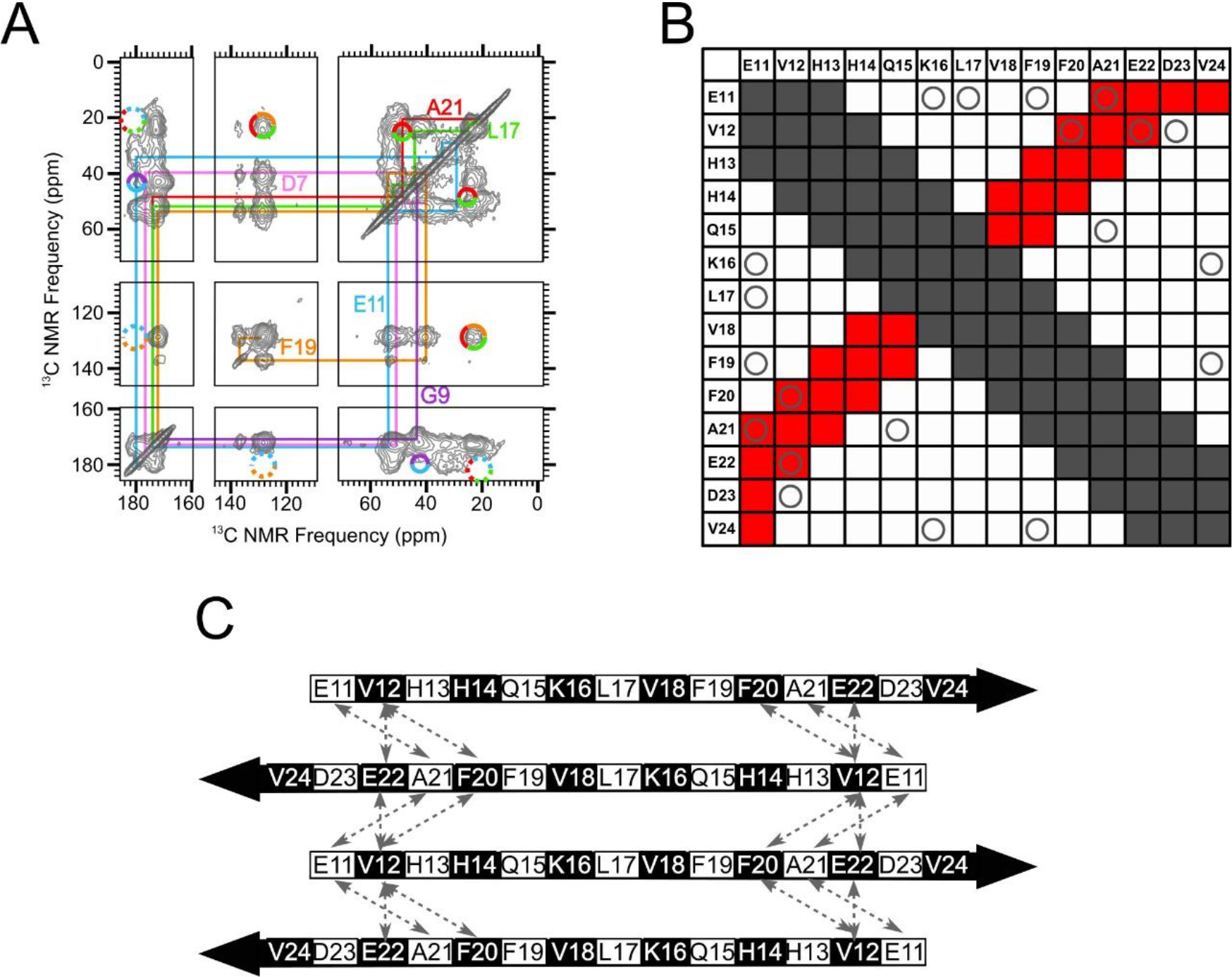
An in-register antiparallel β-sheet centered at L17 was not supported experimentally. A) A 500 ms 2D-DARR NMR spectrum of Sample 2, which was uniformly 13C-labeled at residues D7, G9, E11, L17, F19 and A21. To clarify spectral assignments, the colored lines indicate cross-peaks between directly bonded 13C atoms within each amino acid (intra-residue cross-peaks). The multi-colored circles drawn with solid lines indicate observed cross-peaks between 13C atoms on different labeled residues (inter-residue cross-peaks). The multicolored circles drawn with dotted lines indicate where inter-residue cross-peaks would be expected, based on an all-atom model of a N-strand antiparallel β-sheet centered at L17, but were not observed. (B) A contact chart that summarizes pairs of residues within the N-strand that are expected to exhibit inter-residue cross-peaks in 500 ms 2D-DARR spectra for an antiparallel β-sheet centered at L17. Squares colored gray indicate pairs with intra-residue cross-peaks or inter-residue cross-peaks that are uninformative because they occur through the primary sequence (atoms within a 0.5 nm distance). The squares colored red indicate pairs with predicted cross-peaks based on the model of an N-strand antiparallel β-sheet centered at L17. The “O” symbols represent the tested pairs from all 13C-labeled peptides that we examined which did not show cross-peaks. C) A schematic of an N-strand antiparallel β-sheet centered at L17. The dashed double-headed arrows indicate pairs of residues that correspond to circles in red squares in Panel B. Black and white shading on the β-strand schematic indicate whether an amino acid sidechain is above or below the plane of the diagram, respectively.
2D-DARR and PITHIRDS-CT results support out-of-register parallel β-sheet models for the N-strand
Our inability to detect evidence for either an antiparallel or an in-register parallel N-strand β-sheet compelled us to consider out-of-register parallel β-sheet models. Representations of all-atom parallel β-sheet models are shown in Figure S12 and S13. Schematic diagrams of these models will be used in the main text to guide the presentation of experimental results. We identify each parallel β-sheet model by the registry shift between adjacent strands: Models 2, 3, and 4 incorporate parallel N-strand β-sheets with registry shifts 2, 3, and 4, respectively. As will be discussed shortly, we also considered models in which the registry shift alternates in direction between adjacent molecules in the β-sheet. Parallel β-sheet models with alternating registry shifts will be designated as ±2, ±3, and ±4.
Figure 6A shows a 500 ms 2D-DARR spectrum of Sample 1. This sample was uniformly 13C-labeled at K16, F20, V24, and G37. With this spectrum, we observed inter-residue contacts between K16 and F20 and between F20 and V24. Since these contacts each correspond to a pair of residues that are 4 residues apart within the N-strand, they must arise from inter-molecular 13C-13C dipolar couplings because intramolecular distances would be beyond the detectable range (see Figure S10). Additional 2D-DARR spectra showing detected inter-residue crosspeaks are presented in Figures S14–S17. A total of 9 inter-residue contacts, for N-strand residue pairs that are separated by 3 or 4 residues in sequence, were observed and shown as stars in Figure 6B. These positive results (namely, detected 2D-DARR contacts), as well as the negative results reported above (contacts anticipated but not observed), are consistent with the red shading in Figure 5B, predicted using Model 3 (Figure 6C and Figure S12B). Model ±3 (Figure 6D and Figure S13B) predicts a similar pattern of 2D-DARR contacts. Figures S12 and S13 further compare the pattern of observed and unobserved 2D-DARR contacts with Models 2, ±2, 4, and ±4; these models could also be consistent our 2D-DARR data. Thus, while our 2D-DARR data support the presence of registry-shifted parallel β-sheets, they do not precisely constrain the magnitude of the registry shift. It is also notable that the pattern of predicted 500 ms 2D-DARR contacts is not sensitive to alternation of registry shift direction. Nevertheless, there is an important difference between models with constant compared to alternating registry shifts: comparison of Figures 6C and 6D illustrates that alternation introduces magnetic inequivalence to the β-sheet structure. Taking F19 and its proximate residues for example, Figure 6C shows that, in Model 3, every F19 residue is adjacent to K16 on one neighboring strand and A21 on the other. In Model ±3 depicted in Figure 6D, half of the F19 residues are adjacent to two K16 residues on neighboring strands and half are adjacent to two E22 residues. Thus, an alternating registry shift would predict that isotopically labeled F19 residues would have two distinct sets of NMR peaks. Since we may lack the resolution to clearly distinguish between distinct sets of F19 peaks, this type of magnetic inequivalence could explain why we observed broader and more asymmetric NMR peaks for the N-strand than for the C-strand (see Figure 3B and Figure S4).
Figure 6.
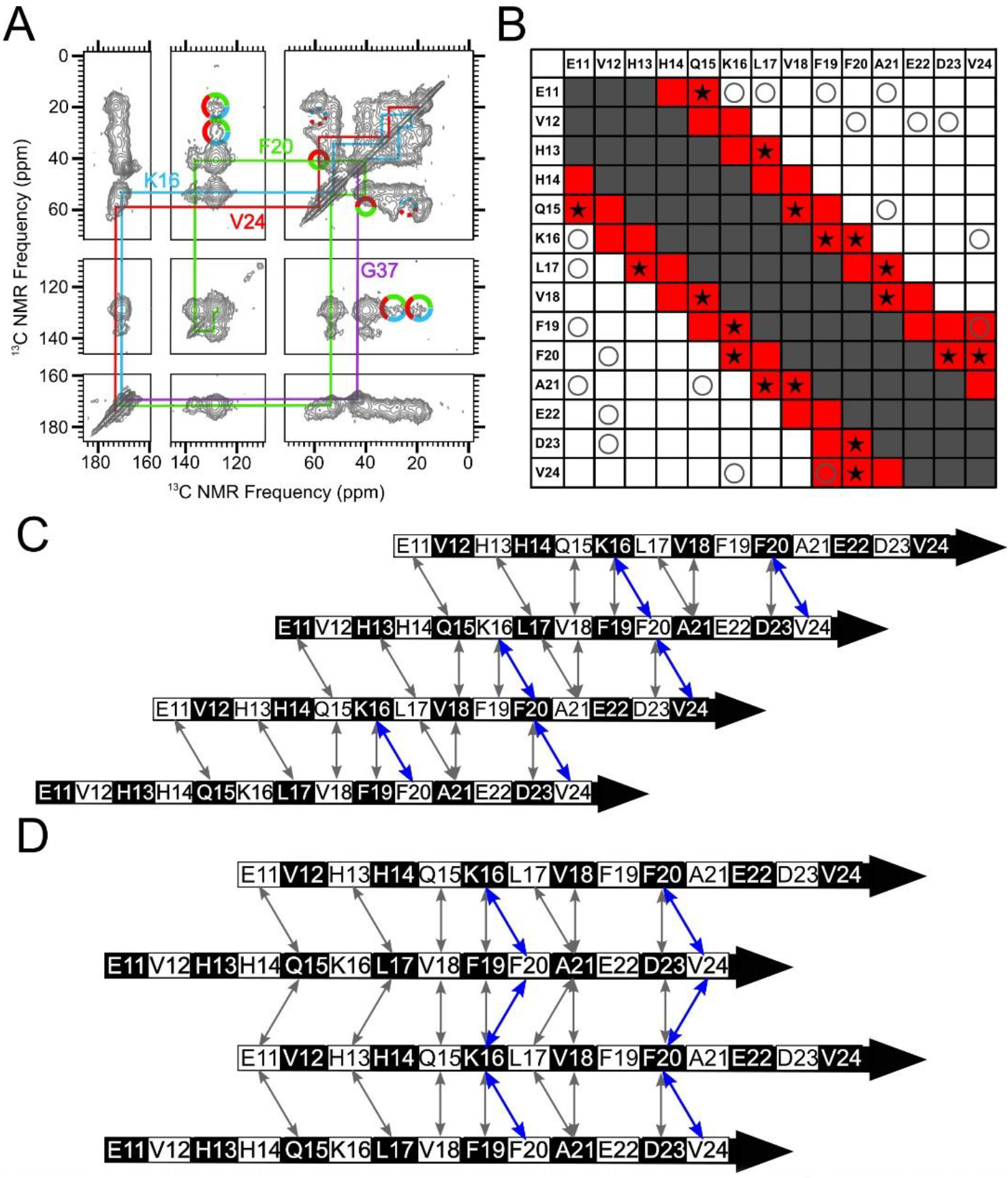
A parallel N-strand β-sheet shifted three residues out of register was consistent with experimental data. A) A 500 ms 2D-DARR spectrum of Sample 1, which was uniformly 13C-labeled at residues K16, F20, V24 and G37. The colored lines indicate intra-residue cross-peaks, and the multi-colored solid circles indicate observed inter-residue cross-peaks. The multi-colored dotted circles indicate where inter-residue cross-peaks may be anticipated but were not observed (See Figure S10 for slices). B) A contact chart similar to Figure 4B, but with red-colored squares corresponding to predicted 500 ms 2DDARR contacts for Model 3. The star symbols (★) indicate test pairs whose cross-peaks were observed experimentally in 500 ms 2D-DARR spectra collected on samples in which the corresponding residues were both uniformly 13C-labeled. The “O” symbols and gray-shaded squares match their designations in Figure 4B. C) A schematic of Model 3. Black and white shading on the β-strand schematics indicate whether an amino acid sidechain is above or below the plane of the diagram, respectively. D) A schematic of Model ±3. The double-headed arrows in Panels C and D convey the same information as the star symbols in Panel B, and the residue pairs whose cross-peaks are observed in panel A (K16/F20, F20/V24) are highlighted in blue.
To validate our interpretation of the data in terms of the out-of-register parallel N-strand β-sheet models, we considered two explanations of our experimental observations. The first possibility is that 2D-DARR experiments are more sensitive than we had expected, and the technique can detect crosspeaks between labeled residues that are separated by distances larger than 0.6 nm. If this alternative hypothesis were true, 2D-DARR crosspeaks detected between residues that are separated by 3 or 4 residues in sequence could be due to intramolecular 13C-13C dipolar couplings. To rule out this possibility, we considered a published 2D-DARR spectrum from an Aβ(1–40) fibril sample that was uniformly 13C-labeled at residues K16, F19, A21, E22, I32 and V36 [4]. It was shown previously that residues K16 and F19 in this structure are both within a β-strand that is organized into an in-register parallel β-sheet [4, 47]. We did not detect crosspeaks between K16 and F19 in the spectrum from this fibril, even though it exhibited a similar signal-to-noise ratio and narrower peaks compared to the spectrum in Figure 6A. For an in-register parallel β-sheet, the closest distance between K16 and F19 in the same strand would be 1 nm. This result supports the expectation that the 2D-DARR experiments would not detect crosspeaks between 13C-labeled residues separated by distances of 1 nm (3 residues) or longer. To further confirm the inter-molecular nature of the K16/F19 contact observed in 150 kDa oligomers, we compared the 2D-DARR spectrum for oligomer Sample 11 (which contains uniform 13C-labels at residues E11, K16, F19, and V36) with that for Sample 11 diluted to 30% with unlabeled peptide (Figure S18). This comparison showed attenuation of the crosspeaks between K16 and F19 as a result of isotopic dilution, indicating that the crosspeaks between K16 and F19 are due to 13C-13C dipolar couplings between labeled residues on different molecules.
The second alternative explanation is that that the observed inter-residue 2D-DARR crosspeaks are due to stacking of distinct N-strand β-sheets rather than contacts between neighboring β-strands in the same β-sheet. It is well known that amyloid fibrils are often composed of β-sheets stacked such that sidechains form steric zipper interfaces [48, 49]. To examine this explanation and obtain more specific constraints on N-strand backbone alignment within the β-sheet, we performed PITHIRDS-CT measurements on samples that were each selectively 13C-labeled at two backbone CO positions in the N-strand. With 13C-labeling only at backbone sites, PITHIRDS-CT results report specifically on organization of adjacent β-strands within the same β-sheet: peptide backbone atoms on different stacked β-sheets would be separated by distances that are too large (> 0.7 nm) to affect PITHIRDS-CT decays. We previously employed a similar strategy to detect out-of-register parallel β-sheet structure within nanofibers of the RADA16-I designer peptide (amino acid sequence RADA4RADA8RADA12RADA16 with acetylated and amidated N- and C- termini, respectively) [50]. 13C-Labeling at a single A8 CO site on the β-strand backbone of RADA16-I fibrils yielded a weak PITHIRDS-CT decay that is not consistent with an in-register parallel β-sheet. With a different RADA16-I sample that was labeled with 13C at two CO sites separated by 2 residues (A4 and A6), we were able to detect a PITHIRDS-CT decay that is consistent with a parallel β-sheet and a registry shift of 2 (Figure S19A and B). A similar PITHIRDS-CT decay analysis was conducted on the series of 150 kDa oligomer Samples F, G, and H that were 13C-labeled at two CO sites within the N-strand (Table 1). In comparison to the decay observed with 13C-labeling of the oligomer at only one site (Sample A), samples with pairs of 13C-labeled CO sites exhibited measurably stronger PITHIRDS-CT decays (Figure 7A). For Samples F, G, and H, the 13C-labeled sites were either 2 residues apart (Sample F: L17 and F19), 3 residues apart (Sample G: V18 and A21), or 4 residues apart (Sample H: L17 and A21). Stronger PITHIRDS-CT for Samples F, G, and H relative to Sample A supports the presence of an out-of-register parallel N-strand β-sheet. The registry shifts introduce inter-molecular 13C-13C couplings. It should be noted that the PITHIRDS-CT curve for labeling of RADA16-I at only a single CO site (A8) per molecule (Figure S19A) resulted in a stronger PITHIRDS-CT than the analogous two CO-site labels applied to Aβ(1–42) 150 kDa oligomers. Unlike the oligomers, RADA16-I forms a single β-strand per molecule, and β-sheets stack along compact interfaces composed only of alanine methyl sidechains. Consequently, the distance between A8 CO sites on different β-sheets is in the detectable range (0.63 nm) and shorter than would be possible with oligomers. Further analysis, presented in the next section, is necessary to explain why observed decays are weaker than the theoretical curves.
Figure 7.
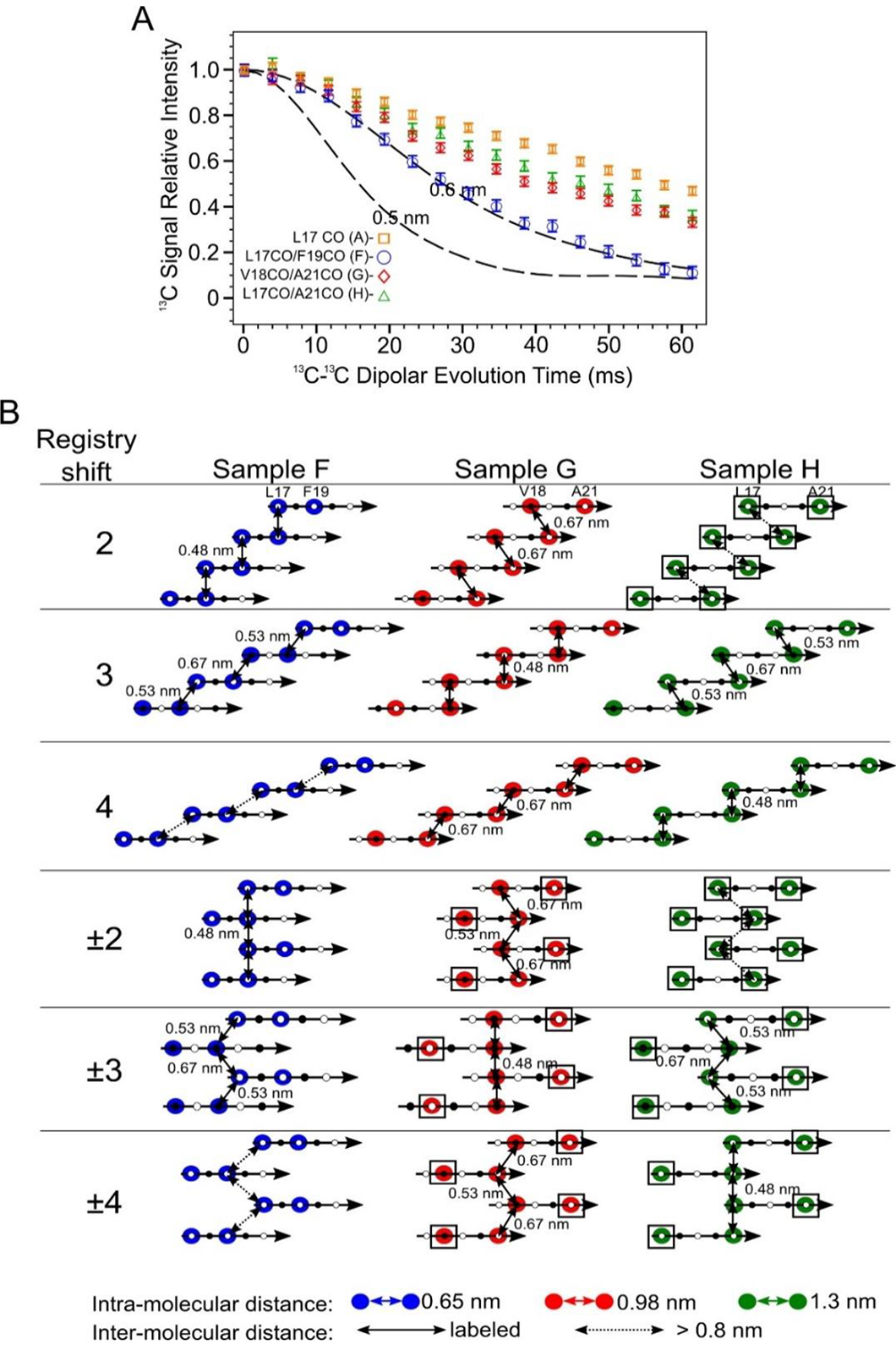
Doubly 13CO labeled PITHIRDS-CT data indicates out-of-register alignments of the N-strands. A) PITHIRDS-CT data for 150 kDa oligomers 13C-labeled at two backbone CO positions within the N-strand (Samples F, G and H). For comparison, the PITHIRDS-CT curves for Sample A (13C at L17 CO) is also plotted. Dashed lines in Panel A were the same simulated curves as in Figure 3. B) Diagrams illustrating the relative positions of 13C-labeled CO sites for Samples F, G and H, predicted by Models 2, 3, 4, ±2, ±3 and ±4. Colored circles indicate residues in which CO sites are 13C-labeled. Doubled headed arrows indicate 13C-13C distances between the labeled sites. Boxes around circles indicate positions of uncoupled spins.
Registry shift within the N-strand β-sheet is likely to be 3 residues and alternate in direction (±3)
The PITHIRDS-CT decays in Figure 7A can be rationalized in terms of relative atomic positions predicted by molecular models of out-of-register parallel β-sheets. Figure 7B illustrates the relative positions of the 13C-labeled sites for Samples F, G, and H within candidate models with registry-shifted parallel N-strand β-sheets. More detailed depictions, including representations of all-atom β-sheet models, are shown in Figure S12 and S13. Figure 7B also shows how relative atomic positions change between models with different registry shifts.
Two limiting cases for PITHIRDS-CT decays illustrate the incompatibility of measured PITHIRDS-CT decays with most of the registry-shifted parallel N-strand β-sheet models considered here. The simulated curves in Figure 7A correspond to one limiting case: these curves bracket the expected PITHIRDS-CT decays for systems in which every 13C-labeled site experience a dipolar coupling with at least one other 13C-labeled site within 0.48 nm. The result would be a “fully coupled” 13C spin system of isotopically labeled sites with the strongest possible 13C-13C couplings for the structures being considered. The single 13C-labeled Aβ(1–42) fibrils (Figure 4B) and the doubly 13C-labeled RADA16-I nanofiber (Figure S19B) are two examples of fully coupled spin systems. At the other extreme, we call a 13C spin system “fully uncoupled” if every 13C-labeled site is at least 0.7 nm from every other 13C-labeled site. For a fully uncoupled 13C-spin system, we would expect to observe a PITHIRDS-CT decay that is indistinguishable from the decay observed with only one backbone CO 13C site per molecule (Samples A-E, Figure 4A). Even when a spin system is not fully uncoupled, we refer to 13C atoms as “uncoupled spins” when they are at least 0.7 nm away from all other 13C-labeled sites (marked by black boxes in Figure 7B). Note that fully uncoupled 13C spin systems do exhibit measurable PITHIRDS-CT decays, most likely due to nuclear spin relaxation effects, chemical shift anisotoropy, and dipolar couplings to the 1% natural abundance 13C background on adjacent C sites. The PITHIRDS-CT decays measured for Samples F, G, and H are inconsistent with both the fully coupled and fully uncoupled extreme cases. The decays for these samples are all weaker than the theoretical curves that correspond to fully coupled spin systems, but they are measurably stronger than the decays measured for samples 13C-labeled at one backbone CO site per molecule. In addition, we verified the presence of inter-molecular 13C-13C dipolar couplings in Sample G with a PITHIRDS-CT experiment on an isotopically diluted sample (Figure S20). Models 2, 3, and 4 each predict a fully coupled 13C spin system for one sample among Samples F, G, and H, while Models 2 and ±2 predict the fully uncoupled 13C spin systems for Sample H (Figure 7B). Since these predictions of spin systems are not confirmed in experiments, only two registry-shifted parallel β-sheet models can be consistent with the data because they predict PITHIRDS-CT decays for Samples F, G, and H that are in between the limiting cases; these are Models ±3 and ±4.
Figure 8 compares measured PITHIRDS-CT decays from Samples F, G, and H to simulated curves that are revised using 13C atom coordinates in Models ±3 and ±4. This comparison, along with trends predicted by relative atomic positions (Figure 7B), points to Model ±3 as the most likely model to fit the data. The clearest distinction between Model ±3 and Model ±4 is given by the prediction of the PITHIRDS-CT decay for Sample F. Model ±3 predicts a curve for this sample that more closely resembles the experimental curve. The observation that the measured Sample F PITHIRDS-CT decay is stronger than the prediction based on Model ±4 indicates the influence of inter-molecular 13C-13C dipolar couplings, as predicted by Model ±3. Models ±3 and ±4 predict similar PITHIRDS-CT decays for Sample G (13C-labeled backbone CO sites 3 residues apart) and Sample H (13C-labeled sites 4 residues apart), with both models predicting that 50% of the 13C spins are uncoupled. Model ±3 does predict slightly shorter 13C-13C distances for Sample G than for Sample H, but the difference between these two measured curves is not large enough to overcome the uncertainty in the analysis.
Figure 8.
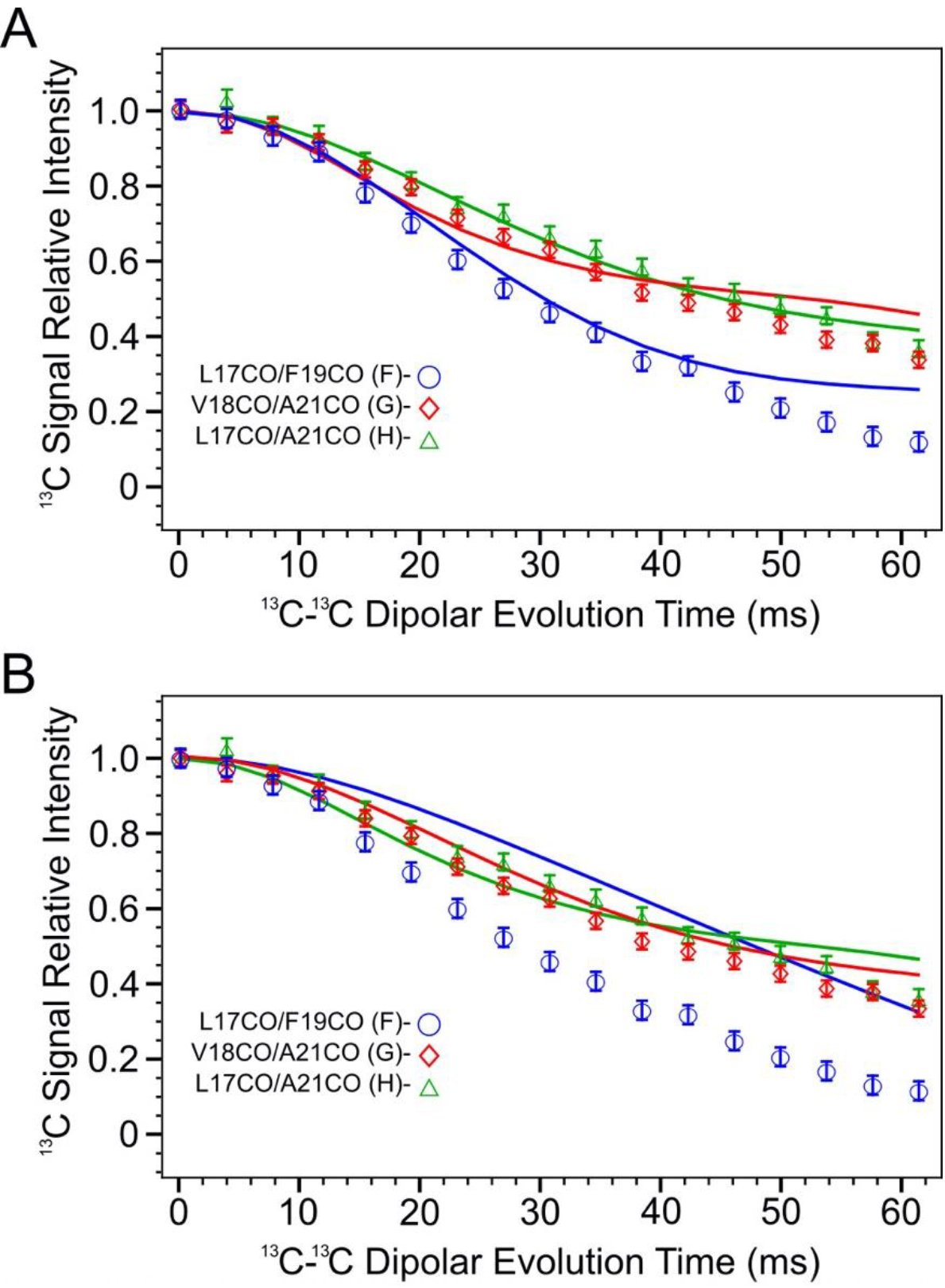
Simulated PITHIRDS-CT curves and measured data for samples F, G and H according to 13CO atom coordinates in model ±3 (Panel A) and ±4 (Panel B). Each simulated curve has the same color as the corresponding data series (blue: sample F, red: sample G, and green: sample H).
Discussion
The most significant result of the present analysis is that the 150 kDa Aβ(1–42) oligomer structure includes both antiparallel and parallel organization of neighboring β-strands. Our previously published work reported 2D NMR contacts and 13C-13C dipolar recoupling data that are consistent with antiparallel arrangement of neighboring C-strands centered at V36 [33, 34]. In the present study, similar experimental techniques applied to the N-strand did not reveal the expected antiparallel N-strand arrangements; inter-residue 2D-DARR contacts anticipated by this arrangement were not observed. Instead, we detected inter-molecular 2D-DARR contacts between N-strand residues that are consistent with a registry-shifted parallel β-sheet. If one assumes that β-sheets containing N-strands include no C-strands, Figure 9A is the N-strand β-sheet model that agrees best with the 2D NMR and 13C-13C PITHIRDS-CT data presented thus far. To summarize the evidence for this figure, the β-strand secondary structure spanning residues 11–24 is based on TALOS-N predictions from CO, Cα, and Cβ NMR 13C chemical shifts (Figure 3). The registry shift of 3 amino acids between adjacent strands is our interpretation of the pattern of 500 ms 2D-DARR inter-residue contacts between residues within the N-strand (Figure 6) and the weakly coupled PITHIRDS-CT decays for Samples F, G, and H (Figure 7). The alternating pattern of registry shifts (±3) yields the best agreement between experimentally measured decays of 13C NMR signal intensity of PITHRIDS-CT experiments on samples that were 13C-labeled at two backbone carbonyl sites within the N-strand (Figure 8).
Figure 9.
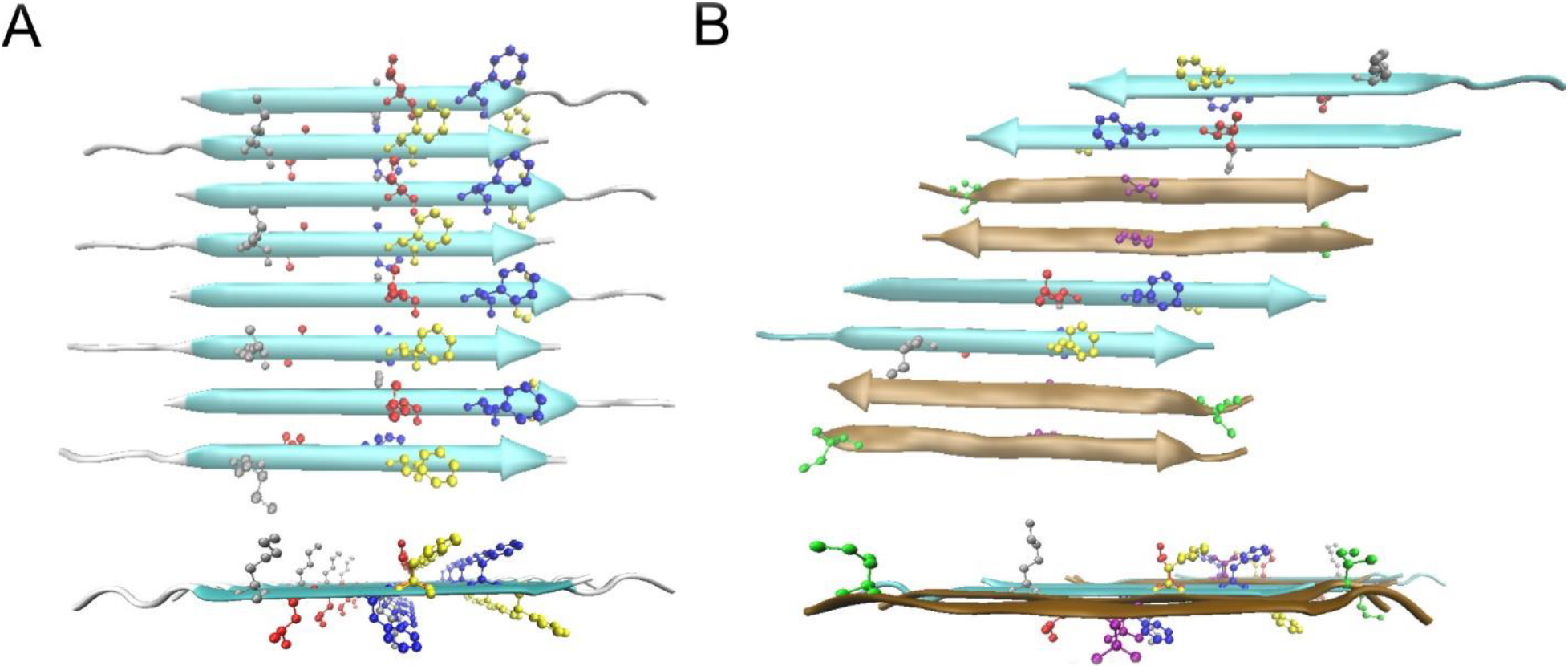
Possible β-sheet structures in the Aβ(1–42) 150kDa oligomers. A) The top and side views of the all-atom structural model of Model ±3 of N-strands. The color code for residues are: Grey K16, Red L17, Blue F19, Yellow F20. B) The top and side view of one structural model of a mixed β-sheet formed by both N-strands and C-strands. The N-strands are colored in cyan and the C-strands are in ochre. The color code for residues are: Grey K16, Red L17, Blue F19, Yellow F20, Green I31, Purple V36.
To simplify our presentation of results, we made the assumption that the N-strands assemble into a β-sheet that is composed only of N-strands. This assumption is supported by the observation that 13C NMR linewidths are measurably broader for the N-strand than the C-strand (Figures 3 and S3), which suggests that the N-strand and C-strand are organized into different β-sheets. Nevertheless, it is worth noting that our observed 13C-13C PITHIRDS-CT decays (Figures 4, 7, and 8) and 2D-DARR contacts between nonadjacent N-strand residues (Figures 5 and 6) are also harmonious with a minimal structural unit composed of 2 N-strands arranged in parallel with a registry shift of 3. One model that includes N-strands associated in registry-shifted parallel pairs is shown in Figure 9B: in this model, N-strands and C-strands and C-strands co-assemble into the same β-sheet. Such a configuration could result from Aβ(1–42) molecules in β-hairpin conformations, as has been proposed by Hoyer et. al. [51] and Doi et. al. [52], or from domain swapping as has been proposed by Stroud et. al. [20]. The model in Figure 9B includes pairs of C-strands organized antiparallel, as would be predicted by our previously published NMR constraints [34]. Further examination of the model in Figure 9B, given in Figure S21 and S22, indicates that this model would predict the observed 500 ms 2D-DARR contacts (Figure 6B) and the 13C-13C PITHIRDS-CT data (Figures 4A and 7A) . Figure S23 illustrates a possible mechanistic argument for why N-strands could form pairs of hydrogen bonded β-strands while being constrained to avoid formation of a larger continuous β-sheet: the N-strands could be forced into this configuration if C-strands self-assemble into an antiparallel β-sheet before N-strands can form structure.
Figure 10 shows that further work is necessary to fully understand the structure of the 150 kDa oligomer with a chart of 2D-DARR contacts between N-strand and C-strand residues. The observation of 11 inter-strand 2D-DARR contacts indicates that the N-strand and C-strand must be closely associated, most likely through sidechain stacking interfaces. The model in Figure 9B does predict some close proximities between residues in the N- and C-strands without sidechain stacking, raising the possibility that the N-strand and C-strand co-assemble into the same β-sheets. However, this model is insufficient to explain all the observed (and unobserved) 2D-DARR contacts in Figure 10 (see Figure S21C). Therefore, none of the structural possibilities presented fully explain the organization of N-strands with C-strands. We suggest that constraints on oligomer nanoscale dimensions are necessary in order to fully understand the oligomer molecular structure. We will subsequently compare these structural constraints with previously published structural data on Aβ fibril, protofibrils, and oligomers. However, we are aware of no previously proposed molecular structural model that simultaneously includes both parallel and antiparallel inter-molecular alignments of β-strands.
Figure 10.
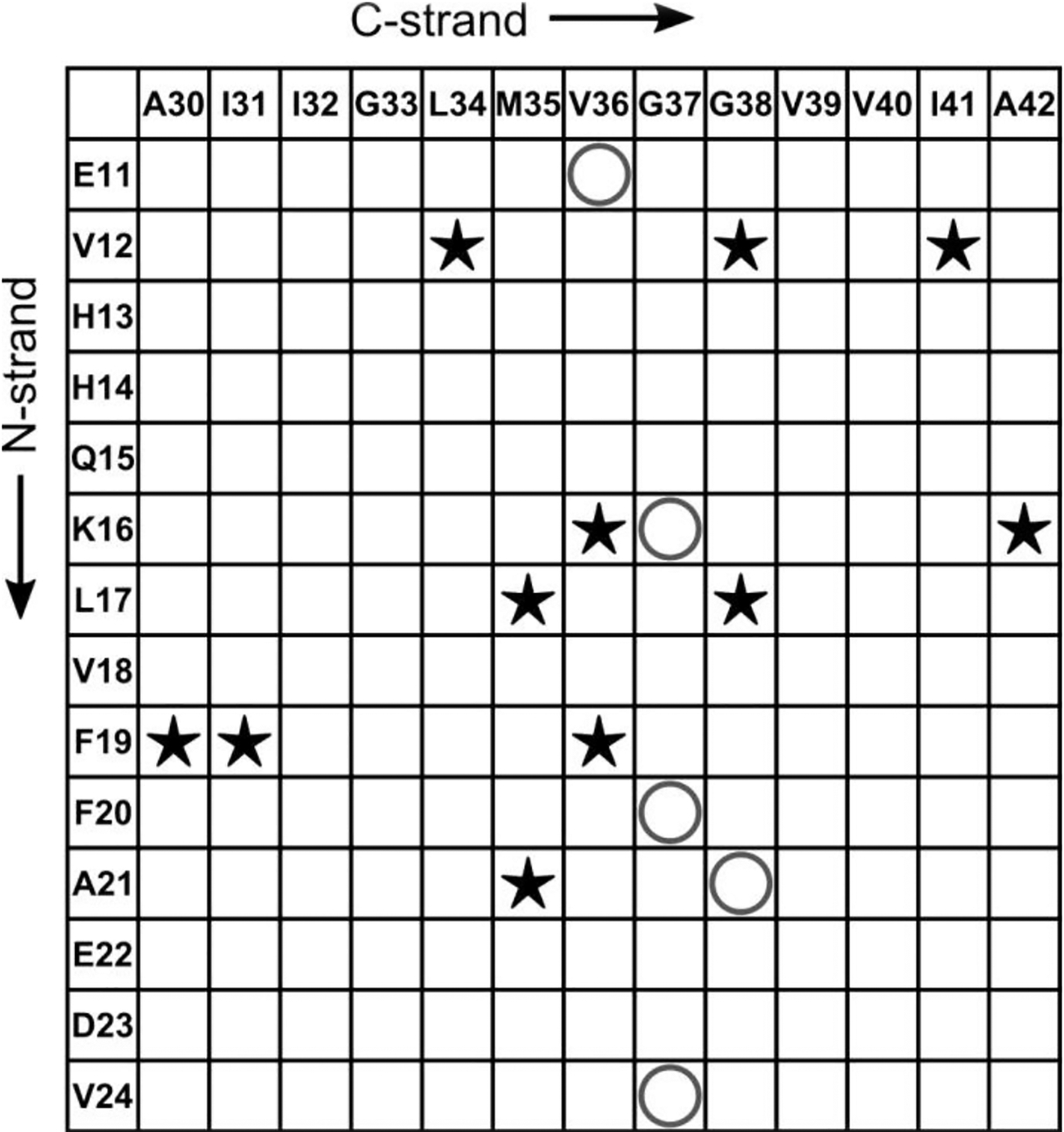
A contact chart that summarizes the experimentally detected and non-detected interactions between the residues in N-strand and that in C-strand. The star symbols (★) indicate pairs of residues whose cross-peaks were observed in 500 ms 2D-DARR spectra, while the “O” symbols represent the pairs from all 13C-labeled peptides that did not show cross-peaks in spectra.
The best-studied Aβ aggregate structures are amyloid fibrils – the main component in amyloid plaques. These fibrils are nanofibers each composed of thousands of Aβ molecules, with fibril widths between 5 and 10 nm and lengths ranging from 100 nm to above 1 μm [53]. In the literature, there are some detailed molecular structures of an Aβ(1–42) amyloid fibril [7–11] and a number of Aβ(1–40) fibril structural models [4, 54] based on well-defined experimental constraints on β-strand secondary structure and inter-strand organization. In each published model, every molecule adopts the same conformation within the amyloid fibril, with 2 or 3 β-strand domains per molecule arranging into “U-shaped” or “S-shaped” conformations (Figure S24A and B). In these fibril structures, β-strands organize into β-sheets such that each β-sheet is composed of equivalent β-strand segments (e.g., a β-strand formed by residues 12 to 23). The result is that β-sheets are close to planar (some twist can be observed along the fibril) and can extend to include thousands of β-strands. Most amyloid fibrils are composed of in-register parallel β-sheets, but Qiang et. al. [12] showed that the Iowa mutant (D23N) of Aβ(1–40) can form a fibril composed of either antiparallel β-sheets or in-register parallel β-sheets depending on the assembly conditions (Figure S24C). It is notable that the planar nature of an amyloid fibril appears to necessitate either that all β-sheets in a fibril be composed of parallel β-strands or that all β-sheets be composed of antiparallel β-strands. We are aware of no instance of co-existence of parallel and antiparallel β-sheets in one fibril structure. The presence of parallel and anti-parallel β-sheet in one structure, just as in our 150kDa Aβ oligomers, could possibly suppress amyloid fibril development.
Other conditions of Aβ aggregation generate protofibrils, which can undergo further assembly to fibrils and show enhanced fluorescence with ThT. The few NMR structural studies that have been conducted on Aβ protofibrils have not been very informative, but some of them have proposed that the protofibrils contain a β-hairpin structure (an intra-molecular antiparallel β-strand) [52, 55] and that they would convert into mature amyloid fibrils by conformation changes. A more recent NMR study revealed a hexameric barrel as the building block of a protofibril sample [56] with an engineered disulfide-containing Aβ(1–42) that locks into a peptide β-hairpin (Aβcc) [57, 58]. These hexameric barrels can interact to form elongated protofibrils that resemble wild type Aβ(1–42) protofibrils, but they cannot proceed to fibrils. Proposed β-hairpin secondary structures that are not locked by covalent crosslinks and their hydrophobic interactions in protofibrils could in principle rearrange to the configuration of mature fibrils [52], but there is as yet no direct experimental evidence of this rearrangement. β-Hairpins also could in principle arrange parallel and anti-parallel β-strands into mixed β-sheets with some similarity to those in Figure 9B, and we do not have enough experimental data to completely exclude all such β-hairpin structures in 150 kDa oligomers. However, such β-hairpins may not be building blocks that are stable enough to resist conversion to fibrils.
The “Aβ oligomer” is the general term for smaller Aβ aggregates, and it involves a wide range of sizes from dimers to protofibrils. Conformation-selective antibodies distinguished two types of endogenous oligomers [59] and surveyed their distribution in the brain. Both types have analogues among synthetic oligomers produced in vitro, but the synthetic oligomers can be produced in larger amounts that allow their structural characterization at the molecular level. One type is closely related to the amyloid fibril. This type may act as intermediates in the fibril aggregation process [60] or be the product of fibril-induced Aβ assembly [61]. Studies on some oligomers of this “intermediate” type found they usually have in-register parallel β-sheets like fibrils [59, 60]. Potapov et. al. [62] used solid-state NMR to show that the fibril-like “U-shape” conformation of the Aβ peptide was already formed in the early oligomer stage. Based on solid-state NMR data, Tycko has suggested that early Aβ oligomers contain antiparallel β-sheets that are “off-pathway” for fibril formation and that these oligomers dissociate before reaggregating as “on-pathway” oligomers with parallel β-sheet structure [63]. Off-pathway oligomers correspond to the second type of oligomer noted above, with assembly pathways and secondary structures different from those of amyloid fibrils, making them “fibril-irrelevant.” Aggregation conditions play a significant role in determining whether this type of Aβ oligomer can be detected in vitro. Initial treatment of synthetic Aβ(1–40) or Aβ(1–42) at high pH and monomer isolation by a technique like size exclusion chromatography (SEC) are necessary to insure that residual aggregates are eliminated [64]. Subsequent aggregation in dilute salt buffers like buffered saline lead to fibril formation through protofibril intermediates [19]. No other oligomers are detected by biophysical methods during such aggregation unless sensitive techniques like microfluidic modulation spectroscopy (MMS) are employed to detect minor components [23]. To obtain in vitro preparations that are largely off-pathway oligomers, it is necessary to treat monomeric Aβ(1–42) with agents that promote aggregation. One such agent is DMSO2, which generates oligomers called Aβ-derived diffusible ligands (ADDLs) [66]. A second is SDS at a concentration just below its critical micelle concentration, which leads to globulomers and the closely related 150 kDa oligomers described here [17, 26]. It is noteworthy that only Aβ(1–42) and not Aβ(1–40) forms these off-pathway oligomers [16, 17, 26]. ADDL preparations contain heterogenous Aβ(1–42) off-pathway oligomers and display two SEC peaks corresponding to large aggregates (65–80 kDa) and monomers [2, 67]. No ADDL characterizations by NMR have been reported.
The 150kDa Aβ oligomers are no doubt fibril-irrelevant oligomers (or off-pathway for fibril formation). First, the oligomer samples are very stable and cannot spontaneously convert into fibrils even in the presence of Aβ(1–42) monomers. Second, Aβ(1–42) peptide molecules first form dimers to tetramers during preparation and then further assemble into 150 kDa oligomers [17]. The final oligomer sample shows little fluorescence response to ThT, which indicates no fibril is involved in the formation of the oligomer. Third, the secondary structures deduced from NMR in our oligomer samples are fundamentally distinct from those in the fibrils. For metastable Aβ oligomers with protein-like size (50~200 kDa), the 150 kDa structure may demand more compact folding rather than growth capability, and thus the difference in secondary structure is expected.
Remarkably, the out-of-register parallel β-sheet structure, with alternating registry shifts of +3 and −3 in the N-strand region that we deduce for 150 kDa oligomers, is a very unusual arrangement. Although there are few examples of structures with such shifts, one is the out-of-register anti-parallel β-sheets formed by Aβ11–25 fibrils formed at pH 2.4 [68]. The β-strands in these fibrils correspond to the N-strand region in 150 kDa oligomers but are anti-parallel rather than parallel. A second example is the model peptide ccβ-p, which has pH-dependent registry shift numbers in anti-parallel β-sheets that form fibrils [69]. One registry shift is +3, and this odd number registry shift would create a flip-over between neighboring β-strands. The flip-over would require that sidechains from the same residue on adjacent molecules alternately point up and down within one β-sheet (see Figure S13B). The N-strand region of Aβ(1–42) and the ccβ-p model peptide (Ac-SIREL EARIR ELELR IG-NH2) both contain several charged sidechains, and thus the pH dependence indicates that sidechain charges may motivate the registry shifts. Further tests are needed to confirm this assumption. It is also worth noting that secondary structures involving antiparallel and out-of-register parallel β-sheets have been reported in other oligomers. Raussens and co-workers [70] observed a conversion of anti-parallel β-sheets in Aβ(1–42) oligomers to parallel β-sheets in Aβ(1–42) fibrils by FTIR. Eisenberg and co-workers crystallized oligomers produced from peptide fragments of several disease-related amyloid proteins including Aβ and found out-of-register anti-parallel β-sheets in oligomers and fibrils [25, 71]. A recent solid state NMR study by Ishii and co-workers [72] of an Aβ(1–42) oligomer called an SPA revealed a structure with some similarities to our 150 kDa oligomer, including aggregate dimensions, predicted β-strand regions, and 13C linewidths (3–4 ppm) on NMR spectra. However, fpRFDR-CT NMR measurements of SPA selectively labeled at 13CO of A30, L34, or V39 indicated out-of-register parallel β-sheets, in contrast to the C-strand structure in our 150 kDa oligomers. These studies either didn’t have enough data to reveal all the secondary structures in the oligomers or only focused on segments of Aβ peptides. However, they did provide evidence for the existence of diverse assembly pathways for different secondary structures that lead to oligomer, an idea which had also been proposed by some simulation studies [73]. Our study is the first to demonstrate the coexistence of out-of-register parallel β-sheets and anti-parallel β-sheets within a single oligomer formed by non-modified Aβ(1–42) peptide.
The coexistence of two different β-strand alignments in 150 kDa oligomers is unexpected but also sheds more light on the full structure of the oligomers. For example, an arrangement alternative to that in Figure 9B could involve the stacking of two separate β-sheets, namely a fully anti-parallel C-strand sheet (Figure S5A, left) and an out-of-register parallel N-strand sheet (Figure 9A). Domain swapping of N- and C-strands could be further introduced to expand the possible arrangements of the two β-sheets. Finally, a possible 150 kDa oligomer structure with different alignments of β-sheets and domain swapping might suggests that the N-strand and the C-strand form β-sheets in different stages of oligomer aggregation.
Materials and Methods
Chemicals and Aβ(1–42) peptide synthesis
Chemical reagents used in the preparation of oligomer samples were purchased from Sigma-Aldrich (St. Louis, MO). Aβ(1–42) peptides (sequence DAEFR HDSGY EVHHQ KLVFF AEDVG SNKGA IIGLM VGGVV IA) with or without 13C and 15N labels were synthesized by New England Peptide (Gardner, MA) and by the Proteomics Core at the Mayo Clinic (Rochester, MN), both equipped Liberty Blue solid-phase peptide synthesizers from CEM (Matthews, NC). The isotope-labeled compounds used in syntheses were all purchased from Cambridge Isotope Laboratories (Tewksbury, MA).
Aβ(1–42) 150kDa oligomer preparations for solid-state NMR
The crude product of Aβ(1–42) peptide synthesis was dried from hexafluoroisopropanol, dissolved with 0.1 M NaOH, and subjected to size-exclusion chromatography (SEC) on a Superdex 75HR 10/30 column (Amersham Pharmacia, Piscataway, NJ) equilibrated with 20 mM sodium phosphate, pH 7.5 (NaP buffer) at a flow rate of 0.5 mL/min.) to isolate Aβ(1–42) monomers as previously described [33, 34]. Aliquots of SEC-purified Aβ monomer were incubated overnight at room temperature with 50 mM sodium chloride and 4 mM SDS to give initial small oligomers called 2–4mers [33]. The solution of 2–4mers was then dialyzed against 20 mM NaP for 48–72 h with at least five buffer changes and then against 10 mM NaP for 3–4 h to remove SDS and reduce the concentration of salt. The quality of oligomer samples was tested by CD and ThT fluorescence at each step of preparation. Finally, residual or unassembled monomers were removed by filtering with an Amicon Ultra 4 centrifugal concentration/filtration device, which has a molecular mass cutoff of 50 kDa. More details in the preparation of these 150 kDa oligomers can be found in our previous reports [33, 34].
For solid-state NMR experiments, at least five preparations were performed for each sample to provide sufficient amounts of oligomers (5–9 mg). The preparations for one sample were combined, flash-frozen, and immediately lyophilized. The lyophilized oligomer samples were stored at −80 °C until use. The isotope-diluted samples, e.g. Sample I, were prepared from isotope-labeled and unlabeled Aβ(1–42) monomers mixed in the desired ratio.
Solid-state NMR experiments
All the solid-state NMR experiments were performed on a Bruker narrow-bore 11.7 Tesla magnet (1H frequency of 500 MHz), equipped with a 3.2-mm HCN MAS probe and a 3.2-mm double-resonance magic angle spinning (MAS) probe. The 2D fpRFDR [35] and 2D DARR [43, 44] spectra are 2D 13C-13C exchange experiments with different mechanisms to reintroduce dipolar coupling between 13C and thus providing cross-peaks. Proton decoupling with a 1H radiofrequency field of 100 kHz was used in fpRFDR recoupling periods and acquisitions, and two-pulse-phase modulation (TPPM) [74] was selected to be the decoupling method. In 2D DARR experiments, continuous irradiation with power corresponding to 11 kHz nutation frequencies (same as the MAS spinning rate) in the 1H channel was applied during the exchange periods. The lengths of exchange periods were set to 50 ms or 500 ms for verifying intra-residue contacts or detecting inter-residue long-distance contacts, respectively. For 2D fpRFDR experiment, the power of the π pulse on 13C channel was adjusted to 33 kHz to match the duration (15.2 μs) of one-third of rotor period at 22 kHz MAS. The signal averaging of 2D fpRFDR and 2D DARR required 36 to 48 h to produce spectra in Figure S1, S2 and S14 to S17. For isotope-diluted samples, the signal averaging was increased to 72 h due to less 13C in the sample. To determine the positions and the linewidths of crosspeaks on 2D fpRFDR spectra, non-linear fitting with a 3D gaussian function was performed for each crosspeak. We treated board crosspeaks as single 3D gaussian peak, although it might consist of several overlapping peaks from the same residue in different conformations.
PITHIRDS-CT experiments [39] were performed on sample A to I with a MAS spinning rate of 12.5 kHz. The dipolar recoupling time was adjusted by number of blocks of pulses (k1, k2 and k3 defined by Tycko [39]), and it was fixed to be between 0 and 61.4 ms in our measurements. 100 kHz proton decoupling was conducted by continuous wave decoupling during PIRHIRDS recoupling and acquisition. PITHIRDS curves in Figure 4A and 7A were from signal averaging of about 24 hours. All the peak intensities in PITHIRDS data sets were corrected by subtracting signals contributed by natural abundance 13C in Aβ(1–42) molecule. We estimate natural abundance signal by multiplying the number of similar 13C sites with the isotopic abundance of 13C, and we assume the correction intensity is constant for all evolution time. For backbone 13CO labels in Aβ(1–42), there are 35 similar CO sites per molecule (excluding all glycines and the C-terminus). For alanine 13Cβ labels, there are 22 similar methyl sites per molecule.
Transmission Electron Microscopy
Imaging by TEM was performed on 150 kDa oligomers prepared with the same protocol we use for solid-state NMR, but without the lyophilization step. The samples were imaged through negative stain TEM (2% Uranyl acetate) on a CM120 BioTwin. Dilution series were conducted to determine optimal concentration for particle dispersion. Grids were prepared within 72 hours of sample preparation, as aggregation would become an issue after this window.
Molecular modeling
Anti-parallel and parallel β-sheets were built by assembling β-strands with VMD (Visual Molecular Dynamics) scripts [75]. A single β-strand was generated by Ambertools [76] using standard backbone torsion angles (anti-parallel β-sheets: ϕ=−139°, ψ=135°; parallel β-sheets: ϕ=−119°, ψ=113°). Anti-parallel β-sheets were built by replicating a two-stranded sheet, in which two replicate β-strands ran in opposite directions and were at an inter-strand distance of 0.48 nm. In-register parallel β-sheets were built by replicating a single β-strand with an inter-strand distance of 0.48 nm. Out-of-register parallel β-sheets were built upon in-register ones in two steps. First, an in-register parallel β-sheet consisting of Alanine residues was built in the abovementioned way. Second, an out-of-register β-sheet in Aβ sequence was constructed by replacing Alanine residues with the corresponding Aβ residues. For parallel β-sheets with n Aβ residues per strand and a registry shift of i, residues 1 to n in the first strand were replaced, residues i to i+n in the second strand were replaced, and so on. Residue replacements were carried out with the Mutagenesis Wizard in PyMOL [77].
NMR-related spin simulations
Simulated PITHIRDS-CT curves were generated using SPINEVOLUTION [78] with the use of parameters that matched the experimental conditions. Briefly, all 13C atoms were treated as identical spins and their positions were fixed by atomic coordinates. All the initial spin vectors were in +x direction, and they evolved according to the pulse sequence of PITHIRDS. The intensities of detected signal at different time points were stored and were used to plot the simulation curves. The REPULSION powder averaging scheme (376 pairs of α and β Euler angles and 36 γ angles) was used for the simulations [79].
Simulations for singly labeled samples (Figure 4) were based on a linear 8-spin system, which is a linear array of eight 13C spins separated by identical constant distances. For doubly labeled samples, the simulated curves (Figure 8) were generated from a 16-spin system, which used the coordinates of sixteen 13CO sites from eight strands in the idealized models of out-of-register β-sheets. For the 4-spin simulation in Figure S21B, only four 13CO sites from two neighboring strands were involved. In addition, two sets of parameters of chemical shift anisotropy were used for spins in each simulation. One set is isotropic chemical shift (δaniso= 0 ppm, ηΩ = 0, αΩ = 0°, βΩ = 0°, γΩ = 0°), and the other is the anisotropic parameters measured from L17 CO (δaniso= −75 ppm, ηΩ = 0.75, αΩ = 0°, βΩ = 0°, γΩ = 0°). These parameters are required input for SPINEVOLUTION [78].
Supplementary Material
Research highlights.
The assembly pathways and structural details of Aβ oligomers are not understood.
Two β-strand regions were located on the Aβ(1–42) sequence in 150 kDa oligomers.
The N-strand (E11-V24) was characterized to arrange into out-of-register parallel β-sheets.
The out-of-register parallel β-sheets and the antiparallel β-sheets coexist in the oligomer structure.
The co-existence of β-sheets with distinct organizations may differentiate oligomer and fibril assembly pathways.
Acknowledgements
This work was supported by the Alzheimer’s Association (grant NIRG-10–173755 to A.K.P.), the National High Magnetic Field Laboratory User Collaboration Research Grant Program, and the National Institute on Aging of the National Institutes of Health (award number R01AG045703). We gratefully acknowledge Evan K. Roberts for helping us analyze 2D NMR spectra and for proposing possible interpretations of data. The authors acknowledge the use of instruments at the NMR center at the Georgia Institute of Technology.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Discrepancies between PITHIRDS-CT experiments and simulations are known to occur [40, 41]. The reason(s) for the discrepancies are not fully understood, but possibly include deviations of atomic positions from idealized geometries, thermal fluctuations in positions of 13C atoms within the molecular ensemble, and chemical shift anisotropy or spin relaxation for which the PITHIRDS-CT pulse sequence does not fully compensate.
Although DMSO was introduced to insure that initial synthetic Aβ(1–42) solutions remain disaggregated [65], it actually induces aggregate formation. Treatment of Aβ(1–42) films with DMSO and dilution into aqueous buffer [66] followed by SEC showed an increase in the amount of the aggregate peak near the void volume relative to that of Aβ(1–42) samples which had not been exposed to DMSO (T. L. Rosenberry, unpublished observations).
References
- [1].Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med 2016;8:595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Cline EN, Bicca MA, Viola KL, Klein WL. The Amyloid-β oligomer hypothesis: beginning of the third decade. J. Alzheimers Dis 2018;64:S567–S610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Mroczko B, Groblewska M, Litman-Zawadzka A, Kornhuber J, Lewczuk P. Amyloid β oligomers (AβOs) in Alzheimer’s disease. J. Neural Transm 2018;125:177–91. [DOI] [PubMed] [Google Scholar]
- [4].Paravastu AK, Leapman RD, Yau WM, Tycko R. Molecular structural basis for polymorphism in Alzheimer’s beta-amyloid fibrils. Proc. Natl. Acad. Sci. U. S. A 2008;105:18349–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Qiang W, Yau W-M, Lu J-X, Collinge J, Tycko R. Structural variation in amyloid-β fibrils from Alzheimer’s disease clinical subtypes. Nature. 2017;541:217–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kollmer M, Close W, Funk L, Rasmussen J, Bsoul A, Schierhorn A, et al. Cryo-EM structure and polymorphism of Abeta amyloid fibrils purified from Alzheimer’s brain tissue. Nat. Commun 2019;10:4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Xiao Y, Ma B, McElheny D, Parthasarathy S, Long F, Hoshi M, et al. Aβ(1–42) fibril structure illuminates self-recognition and replication of Amyloid in Alzheimer’s. Nat. Struc. Mole. Biol 2015;22:499–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Colvin MT, Silvers R, Ni QZ, Can TV, Sergeyev I, Rosay M, et al. Atomic resolution structure of monomorphic Aβ42 amyloid fibrils. J. Am. Chem. Soc 2016;138:9663–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wälti MA, Ravotti F, Arai H, Glabe CG, Wall JS, Böckmann A, et al. Atomic-resolution structure of a disease-relevant Aβ(1–42) amyloid fibril. Proc. Natl. Acad. Sci. U. S. A 2016;113:E4976–E84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gu L, Tran J, Jiang L, Guo Z. A new structural model of Alzheimer’s Abeta42 fibrils based on electron paramagnetic resonance data and Rosetta modeling. J. Struct. Biol 2016;194:61–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gremer L, Schölzel D, Schenk C, Reinartz E, Labahn J, Ravelli RBG, et al. Fibril structure of amyloid-β(1–42) by cryo–electron microscopy. Science. 2017;358:116–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Qiang W, Yau W-M, Luo Y, Mattson MP, Tycko R. Antiparallel β-sheet architecture in Iowa-mutant β-amyloid fibrils. Proc. Natl. Acad. Sci. U. S. A 2012;109:4443–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Schutz AK, Vagt T, Huber M, Ovchinnikova OY, Cadalbert R, Wall J, et al. Atomic-resolution three-dimensional structure of amyloid beta fibrils bearing the Osaka mutation. Angew. Chem. Int. Ed 2015;54:331–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hayden EY, Teplow DB. Amyloid beta-protein oligomers and Alzheimer’s disease. Alzheimers Res. Ther 2013;5:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–9. [DOI] [PubMed] [Google Scholar]
- [16].Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, et al. Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA 1998;95:6448–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Rangachari V, Moore BD, Reed DK, Sonoda LK, Bridges AW, Conboy E, et al. Amyloid-β(1−42) rapidly forms protofibrils and oligomers by distinct pathways in low concentrations of sodium dodecylsulfate. Biochemistry. 2007;46:12451–62. [DOI] [PubMed] [Google Scholar]
- [18].Walsh DM, Lomakin A, Benedek GB, Condron MM, Teplow DB. Amyloid beta-protein fibrillogenesis. Detection of a protofibrillar intermediate. J. Biol. Chem 1997;272:22364–72. [DOI] [PubMed] [Google Scholar]
- [19].Nichols MR, Moss MA, Reed DK, Lin WL, Mukhopadhyay R, Hoh JH, et al. Growth of beta-amyloid(1–40) protofibrils by monomer elongation and lateral association. Characterization of distinct products by light scattering and atomic force microscopy. Biochemistry. 2002;41:6115–27. [DOI] [PubMed] [Google Scholar]
- [20].Stroud JC, Liu C, Teng PK, Eisenberg D. Toxic fibrillar oligomers of amyloid-β have cross-β structure. Proc. Natl. Acad. Sci. U. S. A 2012;109:7717–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sarroukh R, Goormaghtigh E, Ruysschaert JM, Raussens V. ATR-FTIR: a “rejuvenated” tool to investigate amyloid proteins. Biochim. Biophys. Acta 2013;1828:2328–38. [DOI] [PubMed] [Google Scholar]
- [22].Ahmed M, Davis J, Aucoin D, Sato T, Ahuja S, Aimoto S, et al. Structural conversion of neurotoxic amyloid-β1–42 oligomers to fibrils. Nat. Struct. Mol. Biol 2010;17:561–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Shea D, Hsu C-C, Bi TM, Paranjapye N, Childers MC, Cochran J, et al. α-Sheet secondary structure in amyloid β-peptide drives aggregation and toxicity in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2019;116:8895–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Yu L, Edalji R, Harlan JE, Holzman TF, Lopez AP, Labkovsky B, et al. Structural characterization of a soluble Amyloid β-peptide oligomer. Biochemistry. 2009;48:1870–7. [DOI] [PubMed] [Google Scholar]
- [25].Laganowsky A, Liu C, Sawaya MR, Whitelegge JP, Park J, Zhao M, et al. Atomic view of a toxic amyloid small oligomer. Science. 2012;335:1228–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Barghorn S, Nimmrich V, Striebinger A, Krantz C, Keller P, Janson B, et al. Globular amyloid β-peptide1−42 oligomer − a homogenous and stable neuropathological protein in Alzheimer’s disease. J. Neurochem 2005;95:834–47. [DOI] [PubMed] [Google Scholar]
- [27].Moore BD, Rangachari V, Tay WM, Milkovic NM, Rosenberry TL. Biophysical analyses of synthetic Amyloid-β(1−42) aggregates before and after covalent cross-linking. Implications for deducing the structure of endogenous Amyloid-β oligomers. Biochemistry. 2009;48:11796–806. [DOI] [PubMed] [Google Scholar]
- [28].Mezler M, Barghorn S, Schoemaker H, Gross G, Nimmrich V. A beta-amyloid oligomer directly modulates P/Q-type calcium currents in Xenopus oocytes. Br.J.Pharmacol 2012;165:1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hermann D, Mezler M, Müller MK, Wicke K, Gross G, Draguhn A, et al. Synthetic Aβ oligomers (Aβ1–42 globulomer) modulate presynaptic calcium currents: Prevention of Aβ-induced synaptic deficits by calcium channel blockers. Eur. J. Pharmacol 2013;702:44–55. [DOI] [PubMed] [Google Scholar]
- [30].Singh Angom R, Wang Y, Wang E, Pal K, Bhattacharya S, Watzlawik JO, et al. VEGF receptor-1 modulates amyloid beta 1–42 oligomer-induced senescence in brain endothelial cells. FASEB J. 2019;33:4626–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Hillen H, Barghorn S, Striebinger A, Labkovsky B, Muller R, Nimmrich V, et al. Generation and therapeutic efficacy of highly oligomer-specific beta-amyloid antibodies. J. Neurosci 2010;30:10369–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Dorostkar MM, Burgold S, Filser S, Barghorn S, Schmidt B, Anumala UR, et al. Immunotherapy alleviates amyloid-associated synaptic pathology in an Alzheimer’s disease mouse model. Brain. 2014;137:3319–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Tay WM, Huang D, Rosenberry TL, Paravastu AK. The Alzheimer’s amyloid-beta(1–42) peptide forms off-pathway oligomers and fibrils that are distinguished structurally by intermolecular organization. J. Mol. Biol 2013;425:2494–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Huang D, Zimmerman MI, Martin PK, Nix AJ, Rosenberry TL, Paravastu AK. Antiparallel beta-sheet structure within the C-terminal region of 42-residue Alzheimer’s Amyloid-beta peptides when they form 150-kDa oligomers. J. Mol. Biol 2015;427:2319–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ishii Y 13C–13C dipolar recoupling under very fast magic angle spinning in solid-state nuclear magnetic resonance: Applications to distance measurements, spectral assignments, and high-throughput secondary-structure determination. J. Chem. Phys 2001;114:8473–83. [Google Scholar]
- [36].Wishart DS, Bigam CG, Holm A, Hodges RS, Sykes BD. 1H, 13C and 15N random coil NMR chemical shifts of the common amino acids. I. Investigations of nearest-neighbor effects. J. Biomol. NMR 1995;5:67–81. [DOI] [PubMed] [Google Scholar]
- [37].Fritzsching KJ, Yang Y, Schmidt-Rohr K, Hong M. Practical use of chemical shift databases for protein solid-state NMR: 2D chemical shift maps and amino-acid assignment with secondary-structure information. J. Biomol. NMR 2013;56:155–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Shen Y, Bax A. Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J. Biomol. NMR 2013;56:227–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Tycko R Symmetry-based constant-time homonuclear dipolar recoupling in solid state NMR. J. Chem. Phys 2007;126:064506. [DOI] [PubMed] [Google Scholar]
- [40].Gorkovskiy A, Thurber KR, Tycko R, Wickner RB. Locating folds of the in-register parallel β-sheet of the Sup35p prion domain infectious amyloid. Proc. Natl. Acad. Sci. U. S. A 2014;111:E4615–E22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Tycko R, Sciarretta KL, Orgel JPRO, Meredith SC. Evidence for novel β-sheet structures in Iowa mutant β-Amyloid fibrils. Biochemistry. 2009;48:6072–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Gregory DM, Benzinger TLS, Burkoth TS, Miller-Auer H, Lynn DG, Meredith SC, et al. Dipolar recoupling NMR of biomolecular self-assemblies: determining inter- and intrastrand distances in fibrilized Alzheimer’s β-amyloid peptide. Solid State Nucl. Magn. Reson 1998;13:149–66. [DOI] [PubMed] [Google Scholar]
- [43].Takegoshi K, Nakamura S, Terao T. 13C–1H dipolar-assisted rotational resonance in magic-angle spinning NMR. Chem. Phys. Lett 2001;344:631–7. [Google Scholar]
- [44].Takegoshi K, Nakamura S, Terao T. 13C–1H dipolar-driven 13C–13C recoupling without 13C rf irradiation in nuclear magnetic resonance of rotating solids. J. Chem. Phys 2003;118:2325–41. [Google Scholar]
- [45].Crocker E, Patel AB, Eilers M, Jayaraman S, Getmanova E, Reeves PJ, et al. Dipolar assisted rotational resonance NMR of tryptophan and tyrosine in rhodopsin. J. Biomol. NMR 2004;29:11–20. [DOI] [PubMed] [Google Scholar]
- [46].Naito A, Okushita K, Nishimura K, Boutis GS, Aoki A, Asakura T. Quantitative analysis of solid-state homonuclear correlation spectra of antiparallel β-sheet alanine tetramers. J. Chem. Phys. B 2018;122:2715–24. [DOI] [PubMed] [Google Scholar]
- [47].Petkova AT, Ishii Y, Balbach JJ, Antzutkin ON, Leapman RD, Delaglio F, et al. A structural model for Alzheimer’s beta -amyloid fibrils based on experimental constraints from solid state NMR. Proc. Natl. Acad. Sci. U. S. A 2002;99:16742–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Sawaya MR, Sambashivan S, Nelson R, Ivanova MI, Sievers SA, Apostol MI, et al. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature. 2007;447:453–7. [DOI] [PubMed] [Google Scholar]
- [49].Schmidt M, Rohou A, Lasker K, Yadav JK, Schiene-Fischer C, Fandrich M, et al. Peptide dimer structure in an Abeta(1–42) fibril visualized with cryo-EM. Proc. Natl. Acad. Sci. U. S. A 2015;112:11858–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Cormier AR, Pang X, Zimmerman MI, Zhou HX, Paravastu AK. Molecular structure of RADA16-I designer self-assembling peptide nanofibers. ACS Nano. 2013;7:7562–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Hoyer W, Gronwall C, Jonsson A, Stahl S, Hard T. Stabilization of a beta-hairpin in monomeric Alzheimer’s amyloid-beta peptide inhibits amyloid formation. Proc. Natl. Acad. Sci. U. S. A 2008;105:5099–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Doi T, Masuda Y, Irie K, Akagi K-i, Monobe Y, Imazawa T, et al. Solid-state NMR analysis of the β-strand orientation of the protofibrils of amyloid β-protein. Biochem. Biophys. Res. Commun 2012;428:458–62. [DOI] [PubMed] [Google Scholar]
- [53].Tycko R Molecular structure of aggregated Amyloid-β: insights from solid-state Nuclear Magnetic Resonance. Cold Spring Harb. Perspect. Med 2016;6:a024083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Lu J-X, Qiang W, Yau W-M, Schwieters Charles D, Meredith Stephen C, Tycko R. Molecular structure of β-Amyloid fibrils in Alzheimer’s disease brain tissue. Cell. 2013;154:1257–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Scheidt HA, Morgado I, Huster D. Solid-state NMR reveals a close structural relationship between amyloid-beta protofibrils and oligomers. J. Biol. Chem 2012;287:22822–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Lendel C, Bjerring M, Dubnovitsky A, Kelly RT, Filippov A, Antzutkin ON, et al. A hexameric peptide barrel as building block of amyloid-β protofibrils. Angew. Chem. Int. Ed 2014;53:12756–60. [DOI] [PubMed] [Google Scholar]
- [57].Sandberg A, Luheshi LM, Sollvander S, Pereira de Barros T, Macao B, Knowles TP, et al. Stabilization of neurotoxic Alzheimer amyloid-beta oligomers by protein engineering. Proc. Natl. Acad. Sci. U. S. A 2010;107:15595–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Dubnovitsky A, Sandberg A, Rahman MM, Benilova I, Lendel C, Hard T. Amyloid-beta protofibrils: size, morphology and synaptotoxicity of an engineered mimic. PLoS One. 2013;8:e66101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Liu P, Reed Miranda N, Kotilinek Linda A, Grant Marianne KO, Forster Colleen L, Qiang W, et al. Quaternary structure defines a large class of Amyloid-β oligomers neutralized by sequestration. Cell Rep. 2015;11:1760–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Chimon S, Shaibat MA, Jones CR, Calero DC, Aizezi B, Ishii Y. Evidence of fibril-like β-sheet structures in a neurotoxic amyloid intermediate of Alzheimer’s β-amyloid. Nat. Struct. Mol. Biol 2007;14:1157–64. [DOI] [PubMed] [Google Scholar]
- [61].Cohen SI, Linse S, Luheshi LM, Hellstrand E, White DA, Rajah L, et al. Proliferation of amyloid-beta42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. U. S. A 2013;110:9758–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Potapov A, Yau W-M, Ghirlando R, Thurber KR, Tycko R. Successive stages of Amyloid-β self-assembly characterized by solid-state Nuclear Magnetic Resonance with dynamic nuclear polarization. J. Am. Chem. Soc 2015;137:8294–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Tycko R Amyloid polymorphism: structural basis and neurobiological relevance. Neuron. 2015;86:632–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Teplow DB. Preparation of Amyloid β-protein for structural and functional studies Methods in Enzymology: Academic Press; 2006. p. 20–33. [DOI] [PubMed] [Google Scholar]
- [65].Stine WB Jr., Dahlgren KN, Krafft GA, LaDu MJ. In vitro characterization of conditions for amyloid-beta peptide oligomerization and fibrillogenesis. J. Biol. Chem 2003;278:11612–22. [DOI] [PubMed] [Google Scholar]
- [66].Chromy BA, Nowak RJ, Lambert MP, Viola KL, Chang L, Velasco PT, et al. Self-assembly of Abeta(1–42) into globular neurotoxins. Biochemistry. 2003;42:12749–60. [DOI] [PubMed] [Google Scholar]
- [67].Hepler RW, Grimm KM, Nahas DD, Breese R, Dodson EC, Acton P, et al. Solution state characterization of amyloid beta-derived diffusible ligands. Biochemistry. 2006;45:15157–67. [DOI] [PubMed] [Google Scholar]
- [68].Petkova AT, Buntkowsky G, Dyda F, Leapman RD, Yau WM, Tycko R. Solid state NMR reveals a pH-dependent antiparallel beta-sheet registry in fibrils formed by a beta-amyloid peptide. J. Mol. Biol 2004;335:247–60. [DOI] [PubMed] [Google Scholar]
- [69].Verel R, Tomka IT, Bertozzi C, Cadalbert R, Kammerer RA, Steinmetz MO, et al. Polymorphism in an amyloid-like fibril-forming model peptide. Angew. Chem. Int. Ed 2008;47:5842–5. [DOI] [PubMed] [Google Scholar]
- [70].Cerf E, Sarroukh R, Tamamizu-Kato S, Breydo L, Derclaye S, Dufrêne YF, et al. Antiparallel β-sheet: a signature structure of the oligomeric amyloid β-peptide. Biochem. J 2009;421:415–23. [DOI] [PubMed] [Google Scholar]
- [71].Liu C, Zhao M, Jiang L, Cheng P-N, Park J, Sawaya MR, et al. Out-of-register β-sheets suggest a pathway to toxic amyloid aggregates. Proc. Natl. Acad. Sci. U. S. A 2012;109:20913–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Xiao Y, Matsuda I, Inoue M, Sasahara T, Hoshi M, Ishii Y. NMR-based site-resolved profiling of beta-amyloid misfolding reveals structural transitions from pathologically relevant spherical oligomer to fibril. J. Biol. Chem 2020;295:458–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Urbanc B, Cruz L, Teplow DB, Stanley HE. Computer simulations of Alzheimer’s amyloid beta-protein folding and assembly. Curr. Alzheimer Res 2006;3:493–504. [DOI] [PubMed] [Google Scholar]
- [74].Bennett AE, Rienstra CM, Auger M, Lakshmi KV, Griffin RG. Heteronuclear decoupling in rotating solids. J. Chem. Phys 1995;103:6951–8. [Google Scholar]
- [75].Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J. Mol. Graph 1996;14:33–8, 27–8. [DOI] [PubMed] [Google Scholar]
- [76].Salomon-Ferrer R, Case DA, Walker RC. An overview of the Amber biomolecular simulation package. Wires Comput Mol Sci. 2013;3:198–210. [Google Scholar]
- [77].Schrodinger LLC. The PyMOL molecular graphics system, Version 1.8 2015.
- [78].Veshtort M, Griffin RG. SPINEVOLUTION: a powerful tool for the simulation of solid and liquid state NMR experiments. J. Magn. Reson 2006;178:248–82. [DOI] [PubMed] [Google Scholar]
- [79].Bak M, Nielsen NC. REPULSION, A novel approach to efficient powder averaging in solid-state NMR. J. Magn. Reson 1997;125:132–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.