

Abstract
Aromatic L-amino acid decarboxylase deficiency (AADCD) is a rare neurotransmitter metabolic disorder caused by DDC gene mutations, which leads to the metabolic disturbance of dopamine and serotonin. Most of the reported cases came from Taiwan China, but patients from mainland China were seldomly reported. The current study was the largest AADCD patient cohort from mainland China. Twenty-three patients with clinical features of AADCD and DDC gene variants were recruited. A total of 16 DDC variants were identified in this study, of which four variants (c.2T>C, c.277A>G, c.1021+1G>A, c.565G>T) were never reported previously. The intronic variant c.714+4A>T was the most common one, with an allele frequency of 45.7%. And patients carried this intronic variant presented with severe clinical manifestations, all of whom were bedridden. In this study, the average onset age was 3.61 ± 1.28 months and the average age of diagnosis was 12.91 ± 5.62 months. Early onset hypotonia, oculogyric crises, and autonomic symptoms such as excessive sweating, nasal congestion and profuse nasal, and oropharyngeal secretions, were common in our patients. Eighteen patients (78.3%) got various degree of improvement after using pyridoxine monotherapy or different combination of pyridoxine, dopamine agonists, and monoamine oxidase (MAO) inhibitors.
Subject terms: Paediatric neurological disorders, Disease genetics
Introduction
Aromatic L-amino acid decarboxylase deficiency (AADCD) is a rare neurotransmitter disorder. AADC is the last enzyme in the biosynthesis of dopamine and serotonin. The deficiency of AADC leads to metabolic disturbance of dopamine, norepinephrine, epinephrine, and serotonin. Most of the clinical manifestations of AADCD could be explained by the deficiency of these neurotransmitters. Dopamine deficiency affects cognitive function, emotion, and voluntary movement. Reduced expression of norepinephrine and epinephrine can impact attention, mood, sleep patterns, cognition, and stress hormone levels. And disturbance of serotonin can affect memory, learning, mood, sleep patterns, cardiovascular function, body temperature, as well as endocrine function [1]. As a result, patients with AADCD clinically characterized by early onset hypotonia, oculogyric crises, dystonia, autonomic dysfunction, impaired development, and some other neurologic dysfunction.
AADCD is an autosomal recessive disorder, which is caused by DDC gene mutation. The DDC gene, located on p12.2-p12.1 of chromosome 7, consists of 15 exons and encodes a 480 amino acid peptide. The AADC enzyme is a homodimeric pyridoxal 5′-phosphate (PLP)-dependent α-decarboxylase, which converts L-dopa and 5-HTP into dopamine and serotonin, respectively. Each monomer consists of a large domain of 309 residues containing the PLP binding site, an 86-residue C-terminal domain, and an N-terminal domain of 85 residues [2]. Previous studies have revealed some residues are important for enzyme catalysis [3, 4], including (1) residues participate in PLP binding: Ser-147, Ser-149, His-192, Asp-271, Asn-300, His-302, Lys-303, and Phe-309; (2) the localization of substrate binding and cofactor stabilization: Trp-71, Tyr-79, Phe-80, Thr-82, Ile-101, Phe-103, and Thr-246. Interestingly, the active sites are different between the apoenzyme form of AADC and the holoenzyme form, and some flexible loop regions play important roles during the apoenzyme to holoenzyme transition [5], including loop1 (residues 66–84), loop2 (residues 100–110), and loop3 (residues 323–357).
In addition to genetic test, AADCD could also be diagnosed by plasma AADC activity or cerebrospinal fluid (CSF) neurotransmitter analysis. Plasma AADC activity would be severely decreased in patients with AADCD. And the typical CSF patterns of AADCD include (1) low levels of 5-hydroxyindoleacetic acid (5-HIAA), homovanillic acid (HVA), and 3-methoxy-4-hydroxyphenylglycol (MHPG), (2) high levels of 3-ortho-methyldopa (3-OMD), L-dopa, and 5-OH tryptophan, (3) normal pterins, including neopterin and biopterin [6]. Highly increased level of 3-OMD in dried blood spot and elevation of vanilla lactic acid (VLA) in urine would be helpful for the diagnosis of AADCD.
Since the first case reported in 1990 [7], over 100 patients have been described, which mainly from Asia (especially Taiwan China) [2, 6]. A newborn screening program of AADCD by testing 3-OMD of dried blood spot was performed in Taiwan, which revealed that the prevalence of AADCD was about 1/32,000 [8]. In this study, 23 patients with AADCD were reported, which was the largest patient cohort from mainland China up to date. In addition, the molecular analysis of DDC mutations identified in our patients were also shown in this study.
Methods
Patients and clinical information
A total of 23 individuals with AADCD were recruited, of which two cases had been reported previously in Chinese [9]. According to the consensus guideline of AADCD, cases could be broadly classified as mild (mild developmental delay, ambulatory without assistance, and mild intellectual disability), severe (without or very limited developmental milestones, and fully dependent), and moderate (in between) [6]. In this group, no mild cases were identified, four patients (17.4%, 4/23) could be classified as moderate, and the other 19 (82.6%, 19/23) patients were classified as severe.
Informed consents were obtained from all patients' parents or guardians. Clinical data, including age of onset, clinical manifestations, auxiliary examination, family history, and treatment were collected.
Molecular analysis
DDC gene mutations were detected by whole exome sequencing. Genomic DNA was extracted from the peripheral blood leukocytes of patients and their parents according to a standard protocol. Library preparation was carried out according to the recommendations of MyGenostics (Beijing, China). Whole-exome regions were captured with the SureSelectXT Human All Exon V6 (Agilent). The enriched libraries were then sequenced on an Illumina NextSeq 500 system (Illumina) using 150 bp paired-end reads. Raw reads were aligned to UCSC hg19 with BWA software. Aligned reads were processed with SAMtools and Picard following the best practice guidelines of the Genome Analysis Tool kit (GATK). Single-nucleotide variants and small insertion-deletions were detected with the GATK Haplotype Caller.
Variants were annotated using ANNOVAR (http://annovar.openbioinformatics.org/en/latest/). Common sites with population allele frequency above 5% according to dbSNP 138, 1000 Genome Project, esp6500si, and ExAC databases were excluded. Pathogenicity of variants was interpreted according to the ACMG Standards and Guidelines of 2015. The 3D models of mutated proteins were generated by using the SWISS-MODEL online server (https://www.swissmodel.expasy.org), except for the two mutations located at splicing sites (c.714+4A>T and c.1021+1G>A).
Results
Characteristics of DDC gene variants
Among our patients, 5 (21.7%) were detected with homozygous DDC variants and 18 (78.3%) with compound heterozygous variants. Sixteen different DDC variants were identified (Table 1). All the DDC variants were inherited from heterozygous parents. The splicing site variant in intron 6 (c.714+4A>T) was the most common variant with an allele frequency of 45.7% (21/46). Seventeen patients (73.9%, 17/23) carried this variant, of which 4 were homozygous and 13 were heterozygous. Other common DDC variants were as following: c.1234C>T (p.R412W, allele frequency 13.0%, 6/46), c.179T>C (p.V60A, allele frequency 6.5%, 3/46), c.175G>A (p.D59N, allele frequency 4.3%, 2/46), c.304G>A (p.G102S, allele frequency 4.3%, 2/46), and c.1297dupA (p.I433fs, allele frequency 4.3%, 2/46). The variants identified just in 1 patient in this study included c.2T>C (p.M1K), c.106G>A (p.G36R), c.170T>C (p.I57T), c.277A>G (p.M93V), c.299G>C (p.C100S), c.478C>G (p.R160G), c.565G>T (p.D189Y), c.1021+1G>A, c.1312T>C (p.C438R), c.1339C>T (p.R447C). Among them, 4 variants (c.2T>C, c.277A>G, c.1021+1G>A, c.565G>T) had not been reported previously. All the novel variants identified in this study were predicted as disease causing or damaging by MutationTaster and SIFT, respectively. Only one variant (c.277A>G) was predicted as benign by Polyphen-2.
Table 1.
DDC gene variants identified in this study
| Nucleotide change | Amino acid change | Allele frequency | MutationTaster | PolyPhen-2 | SIFT |
|---|---|---|---|---|---|
| c.714+4A>T | – | 45.7% | Disease causing | NA | NA |
| c.1234C>T | p.412R>W | 13.0% | Disease causing | Probably damaging with a score of 1.000 | Damaging with a score of 0.000 |
| c.179T>C | p.V60A | 6.5% | Polymorphism | Possibly damaging with a score of 0.802 | Damaging with a score of 0.000 |
| c.175G>A | p.D59N | 4.3% | Disease causing | Probably damaging with a score of 0.999 | Damaging with a score of 0.000 |
| c.304G>A | p.G102S | 4.3% | Disease causing | Probably damaging with a score of 1.000 | Damaging with a score of 0.000 |
| c.1297dupA | p.I433fs | 4.3% | Disease causing | NA | NA |
| *c.2T>C | p.M1K | 2.2% | Disease causing | Probably damaging with a score of 0.964 | Damaging with a score of 0.000 |
| c.106G>A | p.G36R | 2.2% | Disease causing | Probably damaging with a score of 1.000 | Damaging with a score of 0.017 |
| c.170A>G | p.I57T | 2.2% | Disease causing | Benign with a score of 0.139 | Damaging with a score of 0.001 |
| *c.277A>G | p.M93V | 2.2% | Disease causing | Benign with a score of 0.216 | Damaging with a score of 0.001 |
| c.299G>C | p.C100S | 2.2% | Disease causing | Probably damaging with a score of 0.958 | Damaging with a score of 0.016 |
| c.478C>G | p.R160G | 2.2% | Disease causing | Probably damaging with a score of 1.000 | Damaging with a score of 0.025 |
| *c.565G>T | p.D189Y | 2.2% | Disease causing | Probably damaging with a score of 0.997 | Damaging with a score of 0.002 |
| *c.1021+1G>A | – | 2.2% | Disease causing | NA | NA |
| c.1312T>C | p.C438R | 2.2% | Disease causing | Possibly damaging with a score of 0.520 | Damaging with a score of 0.006 |
| c.1339C>T | p.R447C | 2.2% | Disease causing | Probably damaging with a score of 1.000 | Damaging with a score of 0.000 |
NA not applicable
aThese were novel variants identified in this study
Among all the DDC variants found in this study, five were located at exon 2, three were located at exon 3, three were located at exon 14, and two were located at exon 5. The other three variants were located at exon 13, intron 6, and intron 10, respectively (Fig. 1).
Fig. 1.

Genomic organization of DDC gene and variants distribution
Molecular structure
The 3D models of exon mutations were generated by using the SWISS-MODEL online server. The structure of wild-type AADC enzyme is shown in Fig. 2. A total of 14 exon mutations of DDC gene were found in this study, and the structure change of these mutations were further analyzed. Two mutations were located at loop2 (c.299G>C (p.C100S) and c.304G>A (p.G102S)), which was the important structure during the transition of apoenzyme to holoenzyme. Some mutations that affected residues not directly involved in the active sites or the important loop regions, including five mutations (c.2C>T (p.M1K), c.106G>A (p.G36R), c.170T>C (p.I57T), c.175G>A (p.D59N), and c.179T>C (p.V60A)) belonging to the N-terminal domain, three mutations (c.277A>G (p.M93V), c.478C>G (p.R160G), and c.565G>T (p.D189Y)) lying on the large domain, and four mutations (c.1234C>T (p.R412W), c.1297dupA (p.I433fs), c.1312T>C (p.C438R), and c.1339C>T (p.R447C)) locating on the C-terminal domain.
Fig. 2.
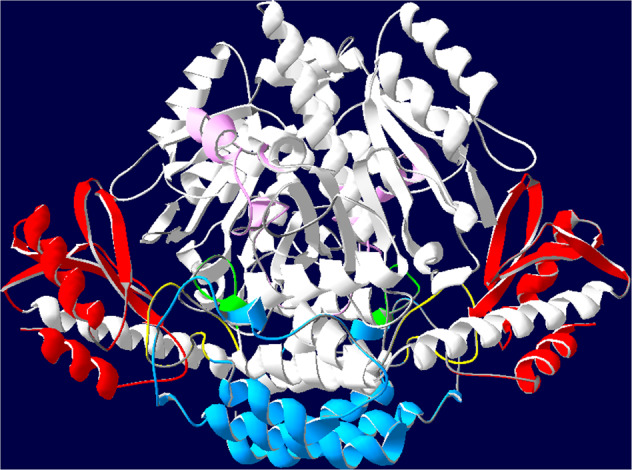
The 3D structure of wild-type AADC. The large domains of both monomers are colored in white except for loops 1, 2, and 3 that are colored yellow, green and pink, respectively. The C-terminal domains are colored in red and the N-terminal domains are colored in blue. The described color code has been used for all the panels of Fig. 3
The first cluster included two mutations located at loop2 (Fig. 3a). Cys100 and Gly102 were located at the C-terminal end of loop2, near the key active sites Ile-101 and Phe-103. However, neither of the mutations p.C100S and p.G102S interrupted the H-bond of Ile-101 and Phe-103. And the p.G102S substitution formed a new H-bond between Ser102 and Thr112.
Fig. 3.

The location of residues subjected to DDC mutations. Residues subjected to mutations (colored in orange) belong to four clusters, including loop 2 (a), N-terminal domain except loop 1 (b), large domain except loop 2 and loop 3 (c), and C-terminal domain (d), respectively
The second cluster included five mutations that affected the residues belonging to N-terminal domain except loop 1 (Fig. 3b). The mutation of the initiation codon p.M1K affected translation, thus the mutated protein lacked an α-helix comprised of 12 amino acids, compared with the wild type. The p.I57T substitution increased two H-bonds in the mutated protein, including one between Thr57 and Glu54, and one between Thr57 and Phe53. There were no significant H-bond changes after substitution of p.G36R, p.D59N, or p.V60A.
The third cluster comprised mutations at the large domain except loop 2 and loop 3 (Fig. 3c). When Met93 mutated to Val93, two H-bonds were newly formed, including one between Val93 and Ala97, and one between Asp92 and Gly96. The p.R106G substitution impaired the H-bond between Arg160 and Leu156, as well as Ala159 and Gln168. And the mutation p.D189Y destroyed the H-bond between Asp189 and His192.
The fourth cluster included four mutations located at the C-terminal domain (Fig. 3d). The mutation p.R412W impaired the H-bond between Arg412 and Asn420. The p.C438R substitution disturbed the H-bond between Asn428 and Ser429. And the p.R447C substitution destroyed the H-bond between Arg447 and Lys443. Moreover, the frameshift mutation c.1297dupA (p.I433fs) resulted in early termination of translation, and the substitution of Ile433 to Asn433 contributed to a new H-bond between Asn417 and Gly442, and impaired the H-bond between Ile433 and Asn420.
Demography
This study included 13 females and 10 males. The 23 patients with AADCD came from 10 different provinces of mainland China, including Fujian, Guangdong, Zhejiang, Chongqing, Shanghai, Hunan, Guizhou, Sichuan, Shandong, and Hebei. It was more common in Southern China, accounting for 87.0% (20/23) in this study, especially from Fujian Province (30.4%, 7/23), Zhejiang Province (17.4%, 4/23), and Guangdong Province (17.4%, 4/23), all of which are coastal provinces near by the Taiwan area. In other hand, 17 patients (73.9%) came from coastal cities while only 6 patients (26.1%) came from inland cities of China.
Clinical features
Clinical manifestation
Of all the patients, the average onset age was 3.61 ± 1.28 months (ranged from 1 month to 7 months), and the average age of diagnosis was 12.91 ± 5.62 months (ranged from 4 months to 25 months). All the patients had developmental delay. Oculogyric crisis and hypotonia as the early symptoms presented in all the patients (100%). Seven patients (30.4%) were misdiagnosed as epilepsy previously because of the oculogyric crisis. Other common symptoms included excessive sweating, nasal congestion, dystonia, and so on. More detailed information could be found in Table 2.
Table 2.
Clinical information of AADC deficiency patients recruited in this study
| No. | Sex | Onset age | Diagnosis age | DDC variants (maternal/paternal) | Severity | Development | EEG | MRI | 3-OMD in dried blood spot (ng/ml) | Urinary VLA (VLA/Crn) | Treatment and effect |
|---|---|---|---|---|---|---|---|---|---|---|---|
| P1 | F | 3 m | 14 m | c.304G>A/c.304G>A | Moderate | Head control at 3 m; turn over at 13 m; now could sit with support and say a few simple words | Normal | Normal | NA | NA | Improved after taking pyridoxine |
| P2 | F | 3 m | 7 m | c.1021+1G>A/c.299G>C | Moderate | Head control at 15 m; turn over at 20 m; now could sit with assistance and grasp small things in a few minutes | δ slow wave | Increased extracerebral space | NA | NA | Improved in head control, hypotonia after taking pyridoxine, selegiline, bromocriptine, rotigotine, and oxymetazoline |
| P3 | F | 5 m | 12 m | c.478C>G/c.565G>T | Moderate | Head control at 10 m; turn over at 19 m; sits without support at 19 m; now could babble | Sharp slow wave | Delayed myelination | 823.017 | NA | Improved in hypotonia, oculogyric crises, and development after taking pyridoxine, selegiline, and rotigotine |
| P4 | F | 6 m | 21 m | c.2T>C/c.277A>G | Moderate | Head control at 12 m; turn over at 14 m; sit without support at 18 m; babbling begins at 8 m; walk independently at 27 m | Normal | Increased extracerebral space | 581.12 | 0.1047 | Improved in hypotonia, sleep disorder, autonomic symptoms, and motor function after taking pyridoxine, selegiline, pramipexole, and trihexyphenidyl |
| P5 | F | 3 m | 17 m | c.714+4A>T/c.714+4A>T | Severe | Bedridden | NA | NA | NA | NA | No improvement after taking pyridoxine and folinic acid |
| P6 | M | 4 m | 12 m | c.714+4A>T/c.714+4A>T | Severe | Bedridden | Normal | Delayed myelination | 873.58 | NA | Improved in autonomic symptoms after taking pyridoxine, pramipexole, selegiline, and trihexyphenidyl |
| P7 | F | 4 m | 8 m | c.714+4A>T/c.714+4A>T | Severe | Bedridden | Normal | Normal | NA | NA | Improved in hypotonia after taking pyridoxine and selegiline |
| P8 | M | 5 m | 20 m | c.714+4A>T/c.714+4A>T | Severe | Bedridden | Normal | Normal | 650 | NA | Improved in hypotonia after taking pyridoxine and rotigotine |
| P9 | F | 3 m | 19 m | c.714+4A>T/c.1234C>T | Severe | Bedridden | NA | NA | NA | NA | Improved in hypotonia and oculogyric crises after taking pyridoxine and selegiline |
| P10 | F | 3 m | 14 m | c.1234C>T/c.174+4A>T | Severe | Bedridden | Normal | NA | NA | NA | Improved in oculogyric crises and mood after taking pyridoxine, folinic acid, and clonazepam |
| P11 | M | 3.5 m | 8 m | c.1234C>T/c.174+4A>T | Severe | Bedridden | NA | Delayed myelination | NA | NA | Pyridoxine was discontinued due to worsening of oculogyric cirses |
| P12 | M | 3 m | 8 m | c.179T>C/c.714+4A>T | Severe | Bedridden | Normal | Delayed myelination | 2110.56 | NA | Improved in hypotonia after taking pyridoxine and selegiline |
| P13 | F | 7 m | 9 m | c.714+4A>T/c.179T>C | Severe | Bedridden | Normal | Increased extracerebral space | 1366.67 | 0.1324 | Improved in autonomic symptoms and dystonia after taking pyridoxine and trihexyphenidyl |
| P14 | F | 3 m | 25 m | c.1297dupA/c.714+4A>T | Severe | Bedridden | Normal | Increased extracerebral space | NA | NA | Improved in hypotonia after taking pyridoxine, selegiline, and rotigotine |
| P15 | M | 4 m | 9 m | c.1297dupA/c.714+4A>T | Severe | Bedridden | Normal | Normal | NA | NA | Improved in autonomic symptoms after taking pyridoxine, L-dopa, ambroxol, biotin, and L-carnitine |
| P16 | M | 2.5 m | 8 m | c.1339C>T/c.714+4A>T | Severe | Bedridden | Normal | Normal | 1428.316 | NA | Limited improvement in excessive sweating after taking pyridoxine, selegiline and trihexyphenidyl; L-dopa was discontinued due to worsening of oculogyric crises |
| P17 | M | 4 m | 18 m | c.714+4A>T/c.106G>A | Severe | Bedridden | NA | NA | 700 | NA | No improvement after taking pyridoxine |
| P18 | M | 4 m | 14 m | c.714+4A>T/c.175G>A | Severe | Bedridden | NA | NA | NA | NA | Improved in ptosis and dystonia after taking pyridoxine, selegiline, pramipexole, and folinic acid |
| P19 | M | 2 m | 5 m | c.1312T>C/c.714+4A>T | Severe | Bedridden | Normal | Normal | NA | NA | No improvement after taking pyridoxine, pramipexole, and trihexyphenidyl |
| P20 | M | 3 m | 4 m | c.714+4A>T/c.1234C>T | Severe | Bedridden | Posterior temporal sharp slow wave | Normal | NA | NA | No improvement after taking levetiracetam |
| P21 | F | 3 m | 11 m | c.714+4A>T/c.175G>A | Severe | Bedridden | Normal | Normal | 1711.79 | 0.3174 | Improved in head control after taking pyridoxine, pramipexole |
| P22* | F | 1 m | 19 m | c.1234C>T/c.170T>C | Severe | Bedridden | Slow background | Normal | NA | NA | Improved in hypotonia and autonomic symptoms after taking pyridoxine and rotigotine |
| P23* | F | 4 m | 15 m | c.1234C>T/c.179T>C | Severe | Bedridden | 2–3.5 Hz slow wave | Normal | NA | NA | Improved in hypotonia after taking pyridoxine and selegiline |
NA not analysis
aThese patients have been reported previously
Four patients (17.4%, 4/23) were classified as moderate, among whom the average onset age was 4.25 ± 1.50 months (ranged from 3 months to 6 months), and the average age of diagnosis was 13.50 ± 5.80 months (ranged from 7 months to 21 months). The developmental was slightly to moderately delayed in these patients, including one patient (P4 with c.2T>C/c.277A>G) could walk independently, one could sit (P3 with c.478C>G/c.565G>T) without support but could not stand, and two (P1 with homozygous variant c.304G>A and P2 with c.1021+1G>A/c.299G>C) could sit with assistance. However, because of hypotonia, patients in this group could only control their heads for a short while. Dystonia was presented in two moderate patients, accounting for 50.0%. Symptoms of autonomic nervous system were common: profuse nasal and oropharyngeal secretions occurred in four patients (100%), excessive sweating in four patients (100%), nasal congestion in four patients (100%), ptosis in two patients (50.0%), and temperature instability in one patient (25.0%). Feeding or swallowing problems were documented in two patients (50.0%). Sleeping disorders were documented in three patients (75.0%), including insomnia and somnolence. All the patients (100%) presented with fatiguability. Irritability was documented in three patients (75.0%). Two patients (50.0%) had small hands and feet. Gastrointestinal problems, such as obstipation, and gastrointestinal reflux, were occurred in two (50.0%), and one (25.0%) patients, respectively.
The other nineteen (82.6%, 19/23) patients were classified as severe, all of whom were bedridden or could only partially control head. The intronic variant c.714+4A>T was more common in severe cases, which was heterozygous in 13 severe patients (68.4%, 13/19) and homozygous in 4 (21.0%, 4/19). That means all patients with c.714+4A>T variant presented with severe clinical manifestation, and none of moderate patients carried heterozygous or homozygous variant of c.714+4A>T. The genotype of the other two severe cases was c.1234C>T/c.170T>C and c.1234C>T/c.179T>C, respectively. Additionally, all the variants detected in the moderate cases were not found in the severe cases. Among the 19 patients with a severe clinical phenotype, the average onset age was 3.47 ± 1.23 months (ranged from 1 month to 7 months), and the diagnostic age was 12.79 ± 5.74 months (ranged from 4 months to 25 months). The developmental milestone was severely delayed in these patients, all of them were bedridden and nobody could control head. Dystonia, which mainly involved limbs, was presented in 12 patients, with a proportion of 63.2%. Symptoms of autonomic nervous system were also common in this group of patients: profuse nasal and oropharyngeal secretions occurred in 18 patients (94.7%), excessive sweating in 16 patients (84.2%), nasal congestion in 15 patients (78.9%), ptosis in 13 patients (68.4%), temperature instability in 9 patients (47.4%), and miosis in 6 patients (31.6%). Feeding or swallowing problems were documented in 9 patients (47.4%). Sleeping disorders were documented in 17 patients (89.5%), including insomnia and somnolence. Fourteen patients (73.7%) presented with irritability. Fatiguability was documented in 10 patients (52.6%). Ten patients (52.6%) had small hands and feet. Gastrointestinal problems, such as obstipation, gastrointestinal reflux, and diarrhea, were occurred in 9 (47.4%), 7 (36.8%), and 4 (21.1%) patients, respectively.
Auxiliary examination
Dried blood spot 3-OMD was tested in nine patients. The average value was 1138.34 ± 538.62 ng/ml, ranged from 581.12 to 2110.56 ng/ml (reference range: 6.33–93.28 ng/ml). Urinary VLA was detected in three patients, with an average value of 0.1848 ± 0.1156, ranged from 0.1047 to 0.3174 (reference range: <0.0157 VLA/Crn). Urinary dopamine level was detected in two patients, which was normal in one patient (879.622 μmol/molCrn, reference ranged from 32 to 2408 μmol/molCrn) and was increased in the other (14320.576 μmol/molCrn). CSF test of neurotransmitter metabolites was performed in only one patient (P16), with significant reduced MHPG (3.55 nmol/L; reference range: 17–114 nmol/L), 5-HIAA (6.73 nmol/L; reference range: 129–410 nmol/L), and HVA (7.87 nmol/L; reference range: 276–1173 nmol/L), together with elevated level of 3-OMD (2095.72 nmol/L, reference range: <60 nmol/L). Hypoglycemia was occurred in two patients (25.0%, 2/8).
Electroencephalogram (EEG) was performed on 18 patients, which was abnormal in 5 patients (27.8%, 5/18), mainly showed slow background. Eighteen patients underwent brain magnetic resonance imaging (MRI) test. Abnormal MRI was reported in eight patients (44.4%, 8/18), including four with delayed myelination and four with increased extracerebral space.
Treatment and therapeutic outcome
Pyridoxine, monoamine oxidase (MAO) inhibitors, and dopamine agonists have been considered as the first line treatment for AADCD patients. In this group, multi drug combined therapy was used in 15 patients, 5 with pyridoxine and MAO inhibitors, 4 with pyridoxine and dopamine agonists, 6 with pyridoxine, MAO inhibitors and dopamine agonists. Three patients had monotherapy of pyridoxine, four patients had pyridoxine combined with other drugs such as trihexyphenidyl and folinic acid. One patient only had levetiracetam treatment, no AADC-related drugs were used at our last follow up. In detail, pyridoxine was used in 22 patients, with a dosage of 10–300 mg/d (1–27.3 mg/kg/d). Selegiline, one of the MAO inhibitors, was used in 11 patients, with a dosage of 1.0–3.3 mg/d (0.08–0.3 mg/kg/d). Dopamine agonists was used in ten patients, including rotigotine in five patients with a dosage of 0.6–1.5 mg/d (0.06–0.1 mg/kg/d), pramipexole in five patients with a dosage of 0.04–0.25 mg/d (0.006–0.03 mg/kg/d), and bromocriptine in one patient with a dosage of 5 mg/d (0.2 mg/kg/d). One patient used rotigotine and bromocriptine at the same time. Trihexyphenidyl, one of the anticholinergic drugs for symptomatic treatment, was used in six patients with a dosage of 2.0–4.0 mg/d (0.2–0.6 mg/kg/d). Other drugs such as folinic acid, clonazepam, and oxymetazoline, were also used in some patients.
Eighteen patients (78.3%) had various degree of improvement (4 moderate cases and 14 severe cases) in oculogyric crisis, hypotonia, and autonomic symptoms. Particularly, all the patients got combination therapy of pyridoxine, dopamine agonists and MAO-inhibitors improved in various degree. However, the motor function was not improved obviously after drug treatment, especially among those with severe phenotype (the therapeutic effect of each patient could be found in Table 2). The other five severe patients (21.7%) had no responses to drug management, including two treated with pyridoxine monotherapy, one with pyridoxine combined with folinic acid, one with pyridoxine combined with pramipexole and trihexyphenidyl, and one with levetiracetam. Two patients were treated with L-dopa firstly, which was discontinued in one because of the worsening of oculogyric crises.
Discussion
AADCD was firstly reported by Hyland et al. [7], and only about 130 cases were published so far [2]. Nearly half of the reported patients were from Asian (especially Taiwan China), though cases from Europe, North America, and Australia had also been reported. However, only five cases from mainland China were reported so far [9–11], of which two individuals were siblings [10]. The two siblings with AADCD died at 12 months and 10 months due to asphyxia and pneumonia, respectively. In this study, 23 AADCD patients from mainland China were reported. Most of these patients came from southern coastal provinces of China, such as Fujian, Zhejiang and Guangdong Province, all of which were near the Taiwan area.
DDC gene, which encodes the AADC enzyme, is the pathogenic gene of this disease. Up to date, approximately eighty variants across the majority of exons of DDC have been identified, with clusters of variants centering on exon 2, 3, and 14 reported in previous study [1]. A total of 16 DDC gene variants were identified in this study, which were mainly clustered in exon 2, 3, 5, and 14. It has been reported previously that the intronic variant c.714+4A>T was the most frequent variant in the Taiwanese patients and individuals with Chinese ancestry [12], accounting for about 75–80% of all DDC variants [13, 14]. In this study, the allele frequency of c.714+4A>T was 45.7%, also the most common one. In addition, the allele frequency of c.714+4A>T of 1171 anonymous randomly selected samples from Taiwan was 0.34%, and the carrier rate was 1:146 [8]. Other common DDC variants identified in this group of patients included c.1234C>T, c.179T>C, c.175G>A, c.304G>A, and c.1297dupA, which was consistent with the reports from Taiwan. The regional distribution study of this cohort showed 87.0% patients were from Southern China, with the similar original background of patients from Taiwan. Four variants (c.2T>C, c.277A>G, c.1021+1G>A, c.565G>T) have not been reported previously. Patients with these novel DDC variants were all came from inland China including Chongqing, Guizhou and Hebei, which may have a different ancestry from those came from southern coastal provinces.
Hypotonia and oculogyric crises are the two most common symptoms in our patient cohort, as well as cases reported in previous studies. Autonomic nervous symptoms, including profuse nasal and oropharyngeal secretions, excessive sweating, nasal congestion, ptosis, and temperature instability, are also common in patients. Other common symptoms include developmental delay, sleeping disorders, irritability, fatiguability, and dystonia. Abnormal EEG was found in 27.8% patients, and abnormal MRI in 44.4% patients. This disease could be diagnosed by the abnormal neurotransmitters′ level in CSF as mentioned above. And diagnosis could be also made based on the severely decreased plasma AADC activity, which may be moderately decreased in heterozygous carriers [15]. As the rapid development of next generation sequencing, a great majority of AADCD patients can be genetically confirmed.
There are some correlations between the DDC genotype and AADCD phenotype. The most frequent variant of c.714+4A>T may lead to a truncated protein and result in severe clinical phenotype [14]. All the patients carried heterozygous or homozygous variant of c.714+4A>T in this study presented with severe clinical manifestation, and none of the patients with moderate severity carry this variant. However, patients with the four novel DDC variants identified in this study presented with less severe clinical manifestations than patients with other variants. The moderate cases could turn over and sit with support at our last follow-up. Particularly, one patient with two novel variants (c.2T>C and c.277A>G) could walk without assistance and got good response after taking pyridoxine, selegiline, pramipexole, and trihexyphenidyl. The clinical phenotype may be also related to the structure and functional changes of the mutated protein. In this study, two mutations (p.C100S and p.G102S) were located at loop2, which may affect the active sites of AADC enzyme. However, patients with these two mutations presented with moderate phenotype. One of the possible reason was that the changes of p.C100S and p.G102S mutations were subtle, and both of them did not impair the vital H-bond between Ile101 and Phe103. Other studies also indicated that the p.G102S substitution only led to a minimal steric hindrance in the cleft between loop 2 and loop 3, and indirectly alter the position of Phe103 [3, 5]. The current knowledge about the correlations between DDC mutations and clinical phenotype remains limited, more detailed information should depend on accumulated patients and more deepened studies.
A consensus treatment guideline of AADCD was published in 2017 [6], which recommended that pyridoxine/pyridoxal phosphate (PLP), dopamine agonists, and MAO-inhibitors should be considered as the first line treatment for AADCD patients. PLP, the active form of pyridoxine, is a cofactor of AADC. So that the use of pyridoxine or PLP might increase the residual activity of AADC enzyme. However, the recommendation of pyridoxine or PLP treatment was mainly based on biochemistry or its cost/availability advantages rather than on clinical evidence [2, 6]. Most of the patients used pyridoxine or PLP combined with other medicines, and only one patients with a mild phenotype got a favorable response among eight patients receiving pyridoxine monotherapy [16]. The dopamine agonists, such as bromocriptine, pramipexole, rotigotine and pergolide, could activate postsynaptic dopamine receptors directly. Improvements of head control, hypotonia, oculogyric crises and autonomic symptoms were reported among AADCD patients treated by dopamine agonists [1, 17]. MAO-inhibitors, such as selegiline and tranylcypromine, inhibit the breakdown of serotonin and dopamine, therefore increase the availability of monoamine. Several studies reported that patients with MAO-inhibitor therapy received improvements in motor function, dystonia, and some autonomic symptoms [18–20]. However, one patient was reported with oculogyric crises deterioration after taking selegiline [20]. Additional symptomatic treatment includes anticholinergic drugs for hypertonia or autonomic symptoms, melatonin for sleeping disorders, oxymetazoline for nasal congestion, and clonidine for irritability [2]. Because of the high levels of L-dopa, L-dopa is not advised as the first line treatment for AADCD patients. However, some patients with L-dopa binding site variants (p.G102S, p.R347Q, p.R160W) were reported with good response to L-dopa [20–23]. In general, the therapeutic improvements were better in moderate patients than those with severe clinical phenotype. Five patients with a severe phenotype failed to respond to drug treatment, including two were treated with pyridoxine monotherapy, one with pyridoxine combined with folinic acid, one with pyridoxine combined with pramipexole and trihexyphenidyl, and one with levetiracetam. Other combination of medicines should be tried in these patients further. And we also found that all the patients treated with combination of pyridoxine, dopamine agonists and MAO-inhibitors got improvement in different degree.
The therapeutic effect was variable among the reported studies [14, 18], which may be due to lack of suitable assessing managements. In a previous survey of 78 patients, only about 19% patients got positive response to drug management [1]. In this study, most of the patients (78.3%) got varying degrees of improvements after taking the first line drugs like pyridoxine, dopamine agonists, and MAO-inhibitors. The good response may be contributed to the earlier medication intervention. In this study, The median diagnostic age was 12 months, ranged from 4 months to 25 months; while the median diagnostic age of the previous study was 3.9 years, ranged from 4 months to 24 years [1]. In addition, Riccardo Montioli et al. indicated that different DDC variants may result in specific enzymatic phenotype and comprehensive understanding of the enzymatic phenotype associated with each pathogenic variant may help to guide therapeutic decision and prevent inappropriate use of medications [5]. For example, patients with mutations that caused a decrease of substrate binding affinity (e.g., p.P47H, p.T69M, p.G102S, p.G123R, p.S250F) may response to L-dopa therapy. And those with mutations did not affect the substrate binding sites may get better response to dopamine agonists and/or MAO-inhibitors theoretically. However, because of the limited reported cases of AADCD and the low frequency of each DDC variant, there is no sufficient evidence to establish a relationship between the mutational impact on the enzyme function and the clinical severity as well as the therapeutic prognosis of this disease. Moreover, the kinetic analysis was only performed for limited AADC mutations, more comprehensive mechanism researches of mutated protein could be benefit for more appropriate therapy and better therapeutic response. Based on our experience, we recommended that patients with DDC mutations beyond the reported substrate binding sites could try pyridoxine, dopamine agonists and MAO-inhibitors combination therapy first, and modify the treatment according to patients’ therapeutic response.
Gene therapy for AADCD have been reported [24–26], which infused the AAV2-hAADC vector directly into the putamen. It was safe and effective. A total of 20 AADCD patients (mean age 6.4 years, range from 1.67 to 19 years old) received gene therapy. They got improvements in motor and cognitive capabilities. However, more studies and long-term follow-up are needed for the safety and effectiveness of the gene therapy for AADCD.
AADCD is a rare neurotransmitter metabolic disorder with limited cases reported worldwide, and only five cases have been reported from mainland China. Patients with infant onset hypotonia, oculogyric crises, excessive sweating and other autonomic symptoms should be considered the diagnosis of AADCD. Further diagnostic tests include CSF neurotransmitter metabolites, plasma AADC activity, and genetic test. Dried blood spot measurement of 3-OMD and urinary measurements of neurotransmitter metabolites are also helpful in diagnosis. The clinical effects of drug treatment have been reported to be limited, and gene therapy would be a promising treatment option for AADCD patients.
Acknowledgements
We would like to thank the reported families for their cooperation in this study. Financial support has been provided by clinical research special fund of Wu Jieping Medical Foundation (grant No. 320.6750.17091, receiptor: Xinhua Bao), the Youth foundation of the National Natural Science Foundation of China (grant No. 81801128, receiptor: Qingping Zhang), and the Peking University First Hospital Scientific Research Seed Fund (grant No. 2018SF013, receiptor: Qingping Zhang).
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Brun L, Ngu LH, Keng WT, Ch’ng GS, Choy YS, Hwu WL, et al. Clinical and biochemical features of aromatic L-amino acid decarboxylase deficiency. Neurology. 2010;75:64–71. doi: 10.1212/WNL.0b013e3181e620ae. [DOI] [PubMed] [Google Scholar]
- 2.Himmelreich N, Montioli R, Bertoldi M, Carducci C, Leuzzi V, Gemperle C, et al. Aromatic amino acid decarboxylase deficiency: Molecular and metabolic basis and therapeutic outlook. Mol Genet Metab. 2019;127:12–22. doi: 10.1016/j.ymgme.2019.03.009. [DOI] [PubMed] [Google Scholar]
- 3.Montioli R, Cellini B, Borri Voltattorni C. Molecular insights into the pathogenicity of variants associated with the aromatic amino acid decarboxylase deficiency. J Inherit Metab Dis. 2011;34:1213–24. doi: 10.1007/s10545-011-9340-6. [DOI] [PubMed] [Google Scholar]
- 4.Guenter J, Lenartowski R. Molecular characteristic and physiological role of DOPA-decarboxylase. Postepy Hig Med Dosw. 2016;70:1424–40. doi: 10.5604/17322693.1227773. [DOI] [PubMed] [Google Scholar]
- 5.Montioli R, Dindo M, Giorgetti A, Piccoli S, Cellini B, Voltattorni CB. A comprehensive picture of the mutations associated with aromatic amino acid decarboxylase deficiency: from molecular mechanisms to therapy implications. Hum Mol Genet. 2014;23:5429–40. doi: 10.1093/hmg/ddu266. [DOI] [PubMed] [Google Scholar]
- 6.Wassenberg T, Molero-Luis M, Jeltsch K, Hoffmann GF, Assmann B, Blau N, et al. Consensus guideline for the diagnosis and treatment of aromatic l-amino acid decarboxylase (AADC) deficiency. Orphanet J Rare Dis. 2017;12:12. doi: 10.1186/s13023-016-0522-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hyland K, Clayton PT. Aromatic amino acid decarboxylase deficiency in twins. J Inherit Metab Dis. 1990;13:301–4. doi: 10.1007/BF01799380. [DOI] [PubMed] [Google Scholar]
- 8.Chien YH, Chen PW, Lee NC, Hsieh WS, Chiu PC, Hwu WL, et al. 3-O-methyldopa levels in newborns: result of newborn screening for aromatic l-amino-acid decarboxylase deficiency. Mol Genet Metab. 2016;118:259–63. doi: 10.1016/j.ymgme.2016.05.011. [DOI] [PubMed] [Google Scholar]
- 9.Chen X, Xu J, Chen L. Aromatic L-amino acid decarboxylase deficiency: a report of two cases and literature review. Linchuang Er Ke Za Zhi. 2019;37:365–8. [Google Scholar]
- 10.Dai L, Ding C, Fang F. A novel DDC gene deletion mutation in two Chinese mainland siblings with aromatic l-amino acid decarboxylase deficiency. Brain Dev. 2019;41:205–9. doi: 10.1016/j.braindev.2018.08.003. [DOI] [PubMed] [Google Scholar]
- 11.Zhu J, Yu F. Feeding difficulty and developmental delay for 8 months and nystagmus for 4 months in an infant. Zhongguo Dang Dai Er Ke Za Zhi. 2017;19:68–72. doi: 10.7499/j.issn.1008-8830.2017.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee NC, Chien YH, Hwu WL. A review of aromatic l-amino acid decarboxylase (AADC) deficiency in Taiwan. Am J Med Genet C Semin Med Genet. 2019;181:226–9. doi: 10.1002/ajmg.c.31670. [DOI] [PubMed] [Google Scholar]
- 13.Hwu WL, Chien YH, Lee NC, Li MH. Natural history of aromatic L-amino acid decarboxylase deficiency in Taiwan. JIMD Rep. 2018;40:1–6. doi: 10.1007/8904_2017_54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee HF, Tsai CR, Chi CS, Chang TM, Lee HJ. Aromatic L-amino acid decarboxylase deficiency in Taiwan. Eur J Paediatr Neurol. 2009;13:135–40. doi: 10.1016/j.ejpn.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 15.Verbeek MM, Geurtz PB, Willemsen MA, Wevers RA. Aromatic L-amino acid decarboxylase enzyme activity in deficient patients and heterozygotes. Mol Genet Metab. 2007;90:363–9. doi: 10.1016/j.ymgme.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 16.Arnoux JB, Damaj L, Napuri S, Serre V, Hubert L, Cadoudal M, et al. Aromatic L-amino acid decarboxylase deficiency is a cause of long-fasting hypoglycemia. J Clin Endocrinol Metab. 2013;98:4279–84. doi: 10.1210/jc.2013-2740. [DOI] [PubMed] [Google Scholar]
- 17.Helman G, Pappa MB, Pearl PL. Widening phenotypic spectrum of AADC deficiency, a disorder of dopamine and serotonin synthesis. JIMD Rep. 2014;17:23–27. doi: 10.1007/8904_2014_327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tay SK, Poh KS, Hyland K, Pang YW, Ong HT, Low PS, et al. Unusually mild phenotype of AADC deficiency in 2 siblings. Mol Genet Metab. 2007;91:374–8. doi: 10.1016/j.ymgme.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 19.Kojima K, Anzai R, Ohba C, Goto T, Miyauchi A, Thony B, et al. A female case of aromatic l-amino acid decarboxylase deficiency responsive to MAO-B inhibition. Brain Dev. 2016;38:959–63. doi: 10.1016/j.braindev.2016.06.002. [DOI] [PubMed] [Google Scholar]
- 20.Manegold C, Hoffmann GF, Degen I, Ikonomidou H, Knust A, Laass MW, et al. Aromatic L-amino acid decarboxylase deficiency: clinical features, drug therapy and follow-up. J Inherit Metab Dis. 2009;32:371–80. doi: 10.1007/s10545-009-1076-1. [DOI] [PubMed] [Google Scholar]
- 21.Chang YT, Sharma R, Marsh JL, McPherson JD, Bedell JA, Knust A, et al. Levodopa-responsive aromatic L-amino acid decarboxylase deficiency. Ann Neurol. 2004;55:435–8. doi: 10.1002/ana.20055. [DOI] [PubMed] [Google Scholar]
- 22.Brautigam C, Wevers RA, Hyland K, Sharma RK, Knust A, Hoffman GF. The influence of L-dopa on methylation capacity in aromatic L-amino acid decarboxylase deficiency: biochemical findings in two patients. J Inherit Metab Dis. 2000;23:321–4. doi: 10.1023/A:1005698223186. [DOI] [PubMed] [Google Scholar]
- 23.Barth M, Serre V, Hubert L, Chaabouni Y, Bahi-Buisson N, Cadoudal M, et al. Kinetic analyses guide the therapeutic decision in a novel form of moderate aromatic acid decarboxylase deficiency. JIMD Rep. 2012;3:25–32. doi: 10.1007/8904_2011_43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chien YH, Lee NC, Tseng SH, Tai CH, Muramatsu SI, Byrne BJ, et al. Efficacy and safety of AAV2 gene therapy in children with aromatic L-amino acid decarboxylase deficiency: an open-label, phase 1/2 trial. Lancet Child Adolesc Health. 2017;1:265–73. doi: 10.1016/S2352-4642(17)30125-6. [DOI] [PubMed] [Google Scholar]
- 25.Kojima K, Nakajima T, Taga N, Miyauchi A, Kato M, Matsumoto A, et al. Gene therapy improves motor and mental function of aromatic l-amino acid decarboxylase deficiency. Brain. 2019;142:322–33. doi: 10.1093/brain/awy331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hwu WL, Muramatsu S, Tseng SH, Tzen KY, Lee NC, Chien YH, et al. Gene therapy for aromatic L-amino acid decarboxylase deficiency. Sci Transl Med. 2012;4:134ra161. doi: 10.1126/scitranslmed.3003640. [DOI] [PubMed] [Google Scholar]