

Abstract
The possibility of immobilizing a protein with antigenic properties on a solid support offers significant possibilities in the development of immunosensors and vaccine formulations. For both applications, the orientation of the antigen should ensure ready accessibility of the antibodies to the epitope. However, an experimental assessment of the orientational preferences necessarily proceeds through the preparation/isolation of the antigen, the immobilization on different surfaces and one or more biophysical characterization steps. To predict a priori whether favorable orientations can be achieved or not would allow one to select the most promising experimental routes, partly mitigating the time cost towards the final product. In this manuscript, we apply a simple computational model, based on united-residue modelling, to the prediction of the orientation of the receptor binding domain of the SARS-CoV-2 spike protein on surfaces commonly used in lateral-flow devices. These calculations can account for the experimental observation that direct immobilization on gold gives sufficient exposure of the epitope to obtain a response in immunochemical assays.
Keywords: Immunosensing, Bioconjugation, United residue model, Protein-surface interaction
Graphical abstract
Highlights
-
•
Protein immobilization is a crucial in preparing immunosensors.
-
•
Immobilization must ensure exposition of the epitope.
-
•
A simple computational model can help predicting the exposition.
1. Introduction
The activity and reactivity of an immobilized protein strongly depend on its orientation with respect to the surface of the support in - or on - which it is immobilized. This holds true for enzymes, as well as for antibodies and antigens. Therefore, the possibility to control and manipulate the exposition of the relevant residues and protein surfaces plays an important role in the rational design of devices based on immobilized proteins. Among these devices, immunosensors represent an expanding space for research and market opportunities.
While the path to reach a technologically relevant product must rely upon a strong experimental characterization [1,2], possibly relying upon atomic-level methodologies [[3], [4], [5], [6], [7], [8], [9]], the preparation/isolation of the protein of interest, its immobilization, and the characterization of the resulting composite are complex and time-consuming, therefore it is also true that guidelines for achieving optimal orientations could improve the efficiency of the R&D connected to protein immobilization [10,11]. However, simplified simulation models that would allow for a rapid prediction of the most plausible orientations are not particularly common. In this manuscript we apply a very simple method based on a united-residue modelling of protein-surface interactions, to specifically address the problem of determining the orientation of the SARS-CoV-2 Spike protein Receptor Binding Domain (RBD) on a few prototypical surfaces for biomedical use. United residue modelling of protein-surface interactions is a rather effective model to screen the poses of protein molecules with respect to surfaces [12,13]. The method we apply is based on the works by Jiang, Zhou and del Monte-Martinez [10,[12], [13], [14], [15], [16], [17], [18]], and encompasses van der Waals and electrostatic interactions, as well as covalent immobilization.
The choice of the target protein is motivated by the recent emergence of a new infectious disease (COVID-19) caused by a coronavirus (SARS-CoV-2) [19]. This infectious disease has spread significantly throughout the world, counting 13.841.890 infected people and a death toll of 590.845 as of July 2020 [20]. Models suggests that it will remain circulating and active for several months [21,22], and there is a marked possibility that reinfection is possible [23,24], thus increasing the time of the circulation of the virus. This pandemic outbreak has had a major impact on world economics, with a very long outlook [25]. A capillary control of the diffusion of the infection has proven crucial [26], and serological tests are expected to have a key role in mass screening [27,28].
2. Methods
The structures of the proteins were downloaded from the protein databank (PDB) [29], the pKa values of reactive groups were calculated using PROPKA [30,31], and the interfaces were calculated using the PDBe PISA server [32]. The non-bonded interaction of a residue of type i is represented with a Lennard-Jones (LJ) potential: [12,13].
where r is the nearest distance between the residue and the surface, ε iis the energy at the minimum position, σ iis the equivalent van der Waals radius of each residue and δ i is a size parameter taken from the literature (see tables S2-S7, parameters are taken from [14,16,18,[33], [34], [35], [36], [37]], as indicated in the table captions).
The electrostatic interaction is represented through the Gouy-Chapman potential [12,13,38].
where r is the nearest distance between the i-th residue with charge q i and the surface, σ S is the surface charge density, κ is the inverse Debye Length calculated from the ionic strength I as , and the relative permittivity of the medium is assumed to be distance-dependent (ε r = r) [13,38]. A 1:1 buffer salt concentration of 0.15 mol dm−3 is assumed.
For silica, the surface charge density is estimated to be −0.3C m−2 [37].
For self-assembled charged monolayers (SAM), the charge density is set to +0.02C m−2 for the amino-capped monolayer (SAM-NH2) and to −0.02C m−2 for the carboxyl-capped monolayer (SAM-CO2H) [14,18].
The formation of a covalent bond is treated with the following potential:
where ε B is the bond energy and is set to 600 kJ mol−1 for imino bonds [39] and 100 kJ mol−1 for gold-thiol bonds [[40], [41], [42]], regardless of the starting oxidation state of the thiol [43]. The desolvation energy is already accounted for in the vdW term.
LJ parameters for epoxide-glyoxyl functionalization is assumed to be equal to SAM-CO2H, whereas for gold the parameters have been adapted from reference [36].
The sampling of the relative protein-surface orientations is performed by rotating a plane around the center of mass of the protein. The plane is initially parallel to the z = 0 plane. Only two rotations are necessary, as all the rotations around the normal to the plane will yield the same energy. The first rotation by an angle α ∈ [0, π] is applied around the y-axis, followed by another rotation of an angle β ∈ [0, 2π] around the z-axis, and then a translation is applied to optimize the position, similarly to what is done in the popular PALES software [[44], [45], [46]]. The sampling of the α, β pairs is made uniform by using REPULSION angular sampling [[47], [48], [49]]. The distance of the plane to the protein is then set by minimizing the energy terms described above.
3. Results and discussion
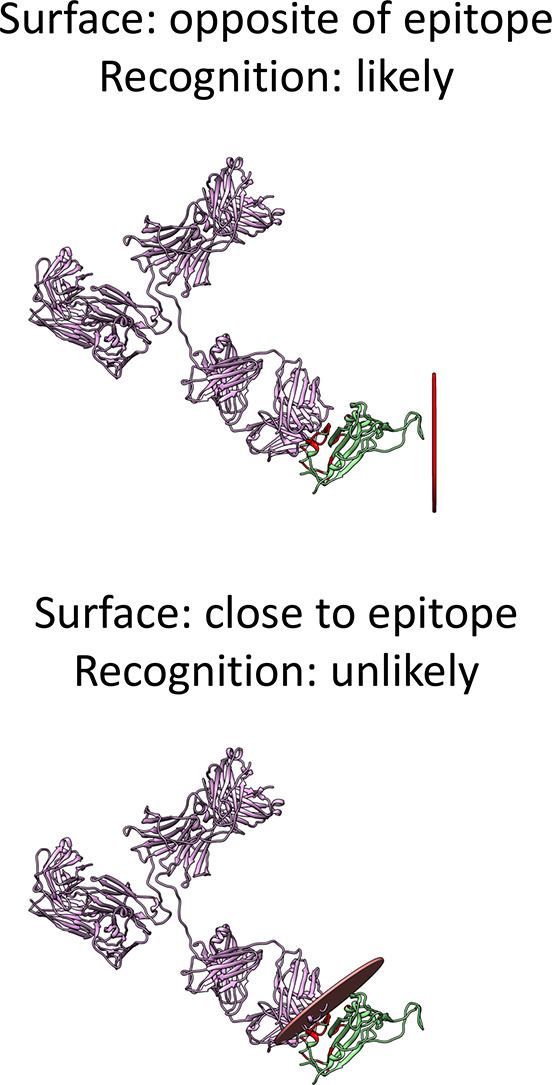
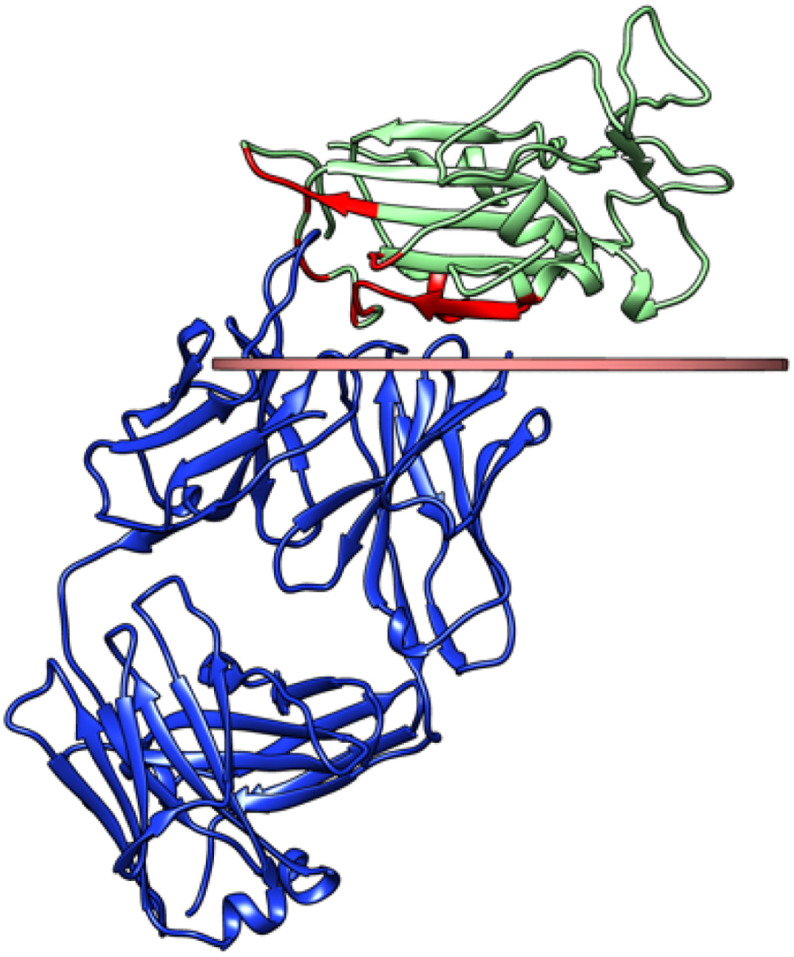
The most important feature of a composite thought for immunochemical applications is that the orientation of the antigen with respect to the surface must ensure the accessibility of the epitope to the antibodies, to guarantee the recognition. Therefore, we have selected the crystallographic structure of RBD in complex with a fragment (FAB) of the human antibody CR3022 (PDB ID: 6W41) [50], and identified the interface residues relevant for the interaction (Fig. 1 and Table S1). In the analysis of the orientations, we assume that the full length antibody will have the same accessibility as the FAB because of the high flexibility of the linkers of the heavy chains (see fig. S1) [[51], [52], [53], [54]]. It is also important to note that, while the spike protein is highly glycosylated at N- and O- positions [55,56], the structured part of the RBD which is recognized by the antibody only carries one glycation at position 343 [55] (pink in Fig. 1), and the glycation site faces away from the antibody binding site. On these grounds, we have not considered glycation (experimentally, this would be done expressing recombinant RBD in prokaryotic cells, whereas glycation would be obtained in human cells [55]).
Fig. 1.

Biological assembly from the crystallographic structure 6 W41. The SARS-CoV-2 receptor binding domain is shown in light green, with the interacting residues highlighted in red and the N-terminus highlighted in green. The glycation site 343 is highlighted in pink. The fragment of the human antibody CR3022 is shown in blue. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.1. Interaction with a hydrophobic surface
Hydrophobic adsorption occurs selectively on hydrophobic carriers at low ionic strength [57]. It is a rather common immobilization protocol, because of its simplicity. The interaction is here represented only through a simple Lennard-Jones (LJ) potential, the parameters of which have been defined according to the hydrophobicity index (table S2) [[33], [34], [35], [36]]. The most probable orientation (5‰ relative population) is shown in Fig. 2 , with the surface represented as a disk. In this, and in the following representations, the interaction is calculated for the antigen alone, and then the complex is shown for examining the interference of the surface with the binding. Of the 2000 considered orientations, 263 are within 10% of the probability of the orientation shown in Fig. 2. Most of those orientations involve contacts between the interface residues and the surface and are therefore expected to be poorly efficient for the recognition. This is not completely unexpected; as hydrophobic carriers mimic the interfaces formed by the naturally occurring interfaces of the proteins.
Fig. 2.
Antigen-FAB complex shown in superposition with the most probable positioning of a hydrophobic surface. The surface is represented as a disk, aligned with the viewer.
3.2. Interaction with charged surfaces
Also this immobilization strategy is rather common because of its simplicity. It is slightly less general, because the outcome strongly depends on the nature of the protein and of the surface. The electrostatic interaction of the i-th residue with the uniformly charged surface with a given charge density is estimated by the Gouy-Chapman potential [12,13], which is added to a LJ term. The parameters defining each system are listed in tables S3-S6.
We have considered the following surfaces:
-
1)
silica - a common chromatographic support with high negative surface charge;
-
2)
positively charged self-assembled monolayer (SAM), with amino capping of the chains [14,18];
-
3)
negatively charged SAM, with carboxylic capping of the chains [14,18].
Supports #2 and #3 imply the possibility of colorimetric detection through gold [58], vide infra.
The most probable orientations are shown in Fig. 3 .
Fig. 3.

Antigen-FAB complex shown in superposition with the most probable positioning with respect to charged surfaces: (a) silica, (b) SAM-CO2H (negative) and (c) SAM-NH2 (positive).
The relative populations of the orientations shown in Fig. 3 are 100% for silica and SAM-NH2. For SAM-CO2H, the orientations in Fig. 3b is populated for about 25%, and there are other 6 orientations out of 2000 that have relative population above 2%, all within a few degrees from the one with highest relative population, except one that is more tilted, yielding a larger accessibility, with a relative population around 4% (Fig. S2).
It is apparent that only negatively charged surfaces allow for the exposition of the epitope, and this is anyway relatively marginal.
These results suggest that it would be nontrivial to achieve a good orientation relying upon adsorption, either based on hydrophobic or on charge interactions. Therefore, we have considered directed approaches based on stronger interactions. In particular, we have considered epoxide-glyoxyl (directed at primary amine moieties) [59] and gold (thiols and disulfide bridges) [58].
The glyoxyl-based approach is quite popular for multipoint orientation-selective immobilization of proteins on surfaces. It involves a two-step mechanism, in the first step, the primary amine groups of the protein are allowed to react with the aldehyde groups to form Schiff base bonds, in the second step the bonds are reduced with sodium borohydride. This kind of immobilization has been simulated in a similar way as described by del Monte-Martìnez et al. [11], assuming a working pH = 7.5, to maximize the reactivity of the N-terminus and at the same time limiting the reactivity of lysine residues (see table S7).
The choice of gold is also extremely popular, because of two reasons: the strong plasmonic response of gold, which causes a purple coloring of the bioconjugate, and because of the relatively easy manipulation required. Current SARS-CoV2 serological tests are indeed based on gold conjugates [60]. The conjugation to the surface is simulated in the same way as the amine-glyoxyl reaction, assuming that all cysteines are equally reactive towards gold (disulfide bridges can interact with gold to a comparable extent as thiols) [43]. The resulting orientation has 100% relative population. Colloidal gold has a net negative surface charge [61], but including the electrostatic term has no impact on the recovered orientation.
In the epoxide-glyoxyl strategy, the conjugation appears to be mostly directed at the N-terminus,1 which is facing away from the recognition interface but is not topologically very remote. Therefore, the epitope will only be partially exposed, whereas for the gold conjugation, ample access to the epitope is possible in the most probable orientation (Figure 4 ).
Fig. 4.

Antigen-FAB complex shown in superposition with the most probable positioning with respect to covalently bound surfaces (a) epoxide-glyoxyl and (b) gold. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Finally, a completely different strategy could be applied for conjugation to (e.g.) gold nanoparticles: the use of a avidin-biotin affinity system [62]. Biotinylation can be achieved through amine-specific reagents [63], and improvement in the selectivity can be achieved with minimal engineering of the sequence [64]. Given that there is a rather substantial difference in the calculated pKas for the different amine sites (see table S7), it can be expected that, for pH values lower than 7, all lysine residues will be protonated and thus less reactive with probability higher than 99%. The N-terminus is not facing the interaction site (see Fig. 1). Therefore, selective biotinylation at the N-terminus is expected to be possible. In this case, the accessibility of the epitope is warranted if the interaction between the antigen and streptavidin, if at all possible, is sufficiently weak.
To explore this possibility we have performed an initial-stage docking using ZDOCK [65], and inspected the first two elements that had a significantly higher ZDOCK score (Fig. S3). The possible interaction between the RBD and streptavidin was investigated also using HADDOCK2.4 [66]. The protein-protein interface residues were predicted with CPORT [67], and then used as “active” and “passive” residues in the HADDOCK calculation. About 10 lowly populated clusters with weak energy were obtained; the most significant three with the lowest HADDOCK-scores are reported in Fig. S3 and their energies in Table S8. Both dockings indicate that, should the interaction occur, it would occur in a position that does not interfere with the antigen-antibody recognition.
4. Conclusions
In this work, we describe the use of united-residue modelling for the prediction of the orientation of the receptor binding domain of the spike protein of the novel coronavirus SARS-CoV-2, a protein of high immunological relevance at the most commonly used surfaces for the preparation of lateral-flow immunochemical devices. With this simple, yet very flexible approach, we find that immobilization on silica, or through glyoxyl reaction of amine residues, or on gold yield orientations compatible with antibody recognition, with gold granting the highest exposition. In this way, we can explain why random conjugation of the RBD to a gold surface yields responsive immunosensors, which are now routinely used. A more detailed experimental verification of the predictions of protein orientation at surfaces represents a significant challenge for the current biophysical methodologies [1]. One can expect that cryo-electron transmission microscopy will be limited by the fact that, in most cases, the surface has higher electron density than that of the protein. Confocal laser scanning microscopy can be used to assess the positioning of the protein with respect to the support, and super-resolution microscopic techniques, such as total internal reflection fluorescence microscopy also allow for the detection of discrete molecular events (e.g., desorption, unfolding, lateral diffusion, …) [2], but the orientation is still a high-hanging fruit by these methodologies. Conversely, the interaction between the protein and the interface can be probed at the atomic level through the application of solid-state NMR [4,[7], [8], [9],[68], [69], [70]], effort which is being started in our lab. Our results suggest that very simple modelling approaches can provide significant hints towards rationally orienting antigens in a way to maximize the exposition of epitopes, and therefore help in the initial moments of the design of conjugates for immunologic applications, when a rapid response to emergency is vital. This is also testified by the emergence of theoretical modelling of several molecular aspects of viral infection and inhibition mechanisms [[71], [72], [73], [74], [75], [76]]. Overall, the expected short-time impact of our work is to provide guidelines to avoid the experimental exploration of immobilization pathways that are less promising.
Declaration of Competing Interest
The authors declare no competing financial interest.
Acknowledgements
Discussion with Luca Sgheri (National Research Council of Italy) and Cesare Bracco (Department of Mathematics and Informatics “Ulisse Dini”, University of Florence) is acknowledged. This work has been supported by the Fondazione Cassa di Risparmio di Firenze, the Italian Ministero della Salute through the grant GR- 2016-02361586, the Italian Ministero dell'Istruzione, dell'Università e della Ricerca through the “Progetto Dipartimenti di Eccellenza 2018-2022” to the Department of Chemistry “Ugo Schiff” of the University of Florence, and the University of Florence through the “Progetti Competitivi per Ricercatori”. The authors acknowledge the support and the use of resources of Instruct-ERIC, a landmark ESFRI project, and specifically the CERM/CIRMMP Italy center. This work is part of the COVID19-NMR project (https://covid19-nmr.de/).
Footnotes
80 orientations out of 2000 account for 99.9% of the relative population. All of these orientations involve binding of the N-terminus.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bpc.2020.106441.
Appendix A. Supplementary data
Supplementary Tables and Figures.
References
- 1.Bolivar J.M., Eisl I., Nidetzky B. Advanced characterization of immobilized enzymes as heterogeneous biocatalysts. Catal. Today. 2016;259:66–80. doi: 10.1016/j.cattod.2015.05.004. [DOI] [Google Scholar]
- 2.Bolivar J.M., Nidetzky B. On the relationship between structure and catalytic effectiveness in solid surface-immobilized enzymes: advances in methodology and the quest for a single-molecule perspective. Biochim. Biophys. Acta BBA - Proteins Proteomics. 1868;2020:140333. doi: 10.1016/j.bbapap.2019.140333. [DOI] [PubMed] [Google Scholar]
- 3.Matlahov I., Geiger Y., Goobes G. Trapping RNase A on MCM41 pores: effects on structure stability, product inhibition and overall enzymatic activity. Phys. Chem. Chem. Phys. 2014;16:9031–9038. doi: 10.1039/c3cp55520h. [DOI] [PubMed] [Google Scholar]
- 4.Mroue K.H., Nishiyama Y., Kumar Pandey M., Gong B., McNerny E., Kohn D.H., Morris M.D., Ramamoorthy A. Proton-detected solid-state NMR spectroscopy of bone with ultrafast magic angle spinning. Sci. Rep. 2015;5:11991. doi: 10.1038/srep11991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Varghese S., Halling P.J., Häussinger D., Wimperis S. High-resolution structural characterization of a heterogeneous biocatalyst using solid-state NMR. J. Phys. Chem. C. 2016;120:28717–28726. doi: 10.1021/acs.jpcc.6b11575. [DOI] [Google Scholar]
- 6.Tian D., Li T., Zhang R., Wu Q., Chen T., Sun P., Ramamoorthy A. Conformations and intermolecular interactions in cellulose/silk fibroin blend films: a solid-state NMR perspective. J. Phys. Chem. B. 2017;121:6108–6116. doi: 10.1021/acs.jpcb.7b02838. [DOI] [PubMed] [Google Scholar]
- 7.Adiram-Filiba N., Schremer A., Ohaion E., Nadav-Tsubery M., Lublin-Tennenbaum T., Keinan-Adamsky K., Goobes G. Ubiquitin immobilized on mesoporous MCM41 silica surfaces - Analysis by solid-state NMR with biophysical and surface characterization. Biointerphases. 2017;12:02D414. doi: 10.1116/1.4983273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Louka A., Matlahov I., Giuntini S., Cerofolini L., Cavallo A., Pillozzi S., Ravera E., Fragai M., Arcangeli A., Ramamoorthy A., Goobes G., Luchinat C. Engineering l-asparaginase for spontaneous formation of calcium phosphate bioinspired microreactors. Phys. Chem. Chem. Phys. PCCP. 2018;20:12719–12726. doi: 10.1039/c8cp00419f. [DOI] [PubMed] [Google Scholar]
- 9.Cerofolini L., Giuntini S., Ravera E., Luchinat C., Berti F., Fragai M. Structural characterization of a protein adsorbed on aluminum hydroxide adjuvant in vaccine formulation. Npj Vaccines. 2019;4:1–5. doi: 10.1038/s41541-019-0115-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cutiño-Avila B., Gil Pradas D., Aragón Abreu C., Fernández Marrero Y., Hernández de la Torre M., Salas Sarduy E., Planes Á. Chávez, Seijas J.M. Guisán, Brito J. Díaz, del Monte-Martínez A. Computer-aided design of bromelain and papain covalent immobilization. Rev. Colomb. Biotecnol. 2014;16:19–28. doi: 10.15446/rev.colomb.biote.v16n1.44184. [DOI] [Google Scholar]
- 11.del Monte-Martínez A., Cutiño-Avila B.V., González-Bacerio J. Rational design strategy as a novel immobilization methodology applied to lipases and phospholipases. In: Sandoval G., editor. Lipases Phospholipases Methods Protoc. Springer; New York, NY: 2018. pp. 243–283. [DOI] [PubMed] [Google Scholar]
- 12.Zhou J., Chen S., Jiang S. Orientation of adsorbed antibodies on charged surfaces by computer simulation based on a united-residue model. Langmuir. 2003;19:3472–3478. doi: 10.1021/la026871z. [DOI] [Google Scholar]
- 13.Xie Y., Zhou J., Jiang S. Parallel tempering Monte Carlo simulations of lysozyme orientation on charged surfaces. J. Chem. Phys. 2010;132 doi: 10.1063/1.3305244. [DOI] [PubMed] [Google Scholar]
- 14.Zhou J., Zheng J., Jiang S. Molecular simulation studies of the orientation and conformation of cytochrome c adsorbed on self-assembled monolayers. J. Phys. Chem. B. 2004;108:17418–17424. doi: 10.1021/jp038048x. [DOI] [Google Scholar]
- 15.Liu J., Liao C., Zhou J. Multiscale simulations of protein G B1 adsorbed on charged self-assembled monolayers. Langmuir. 2013;29:11366–11374. doi: 10.1021/la401171v. [DOI] [PubMed] [Google Scholar]
- 16.Liao C., Xie Y., Zhou J. Computer simulations of fibronectin adsorption on hydroxyapatite surfaces. RSC Adv. 2014;4:15759–15769. doi: 10.1039/C3RA47381C. [DOI] [Google Scholar]
- 17.Zhao D., Peng C., Zhou J. Lipase adsorption on different nanomaterials: a multi-scale simulation study. Phys. Chem. Chem. Phys. 2014;17:840–850. doi: 10.1039/C4CP04696J. [DOI] [PubMed] [Google Scholar]
- 18.Liu J., Yu G., Zhou J. Ribonuclease A adsorption onto charged self-assembled monolayers: a multiscale simulation study. Chem. Eng. Sci. 2015;121:331–339. doi: 10.1016/j.ces.2014.07.021. [DOI] [Google Scholar]
- 19.WHO . 2020. Statement Regarding Cluster of Pneumonia Cases in Wuhan, China.https://www.who.int/china/news/detail/09-01-2020-who-statement-regarding-cluster-of-pneumonia-cases-in-wuhan-china [Google Scholar]
- 20.Dong E., Du H., Gardner L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020;20:533–534. doi: 10.1016/S1473-3099(20)30120-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fanelli D., Piazza F. Analysis and forecast of COVID-19 spreading in China, Italy and France. Chaos Solitons Fractals. 2020;134:109761. doi: 10.1016/j.chaos.2020.109761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carletti T., Fanelli D., Piazza F. COVID-19: the unreasonable effectiveness of simple models, ArXiv200511085 Phys. Q-Bio. 2020 http://arxiv.org/abs/2005.11085 [Google Scholar]
- 23.Kellam P., Barclay W. The dynamics of humoral immune responses following SARS-CoV-2 infection and the potential for reinfection. J. Gen. Virol. 2020 doi: 10.1099/jgv.0.001439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Edridge A.W., Kaczorowska J.M., Hoste A.C., Bakker M., Klein M., Jebbink M.F., Matser A., Kinsella C., Rueda P., Prins M., Sastre P., Deijs M., van der Hoek L. Coronavirus protective immunity is short-lasting. MedRxiv. 2020 doi: 10.1101/2020.05.11.20086439. 2020.05.11.20086439. [DOI] [PubMed] [Google Scholar]
- 25.EU/EA measures to mitigate the economic, financial and social effects of coronavirus, 24 (n.d.).
- 26.Lavezzo E., Franchin E., Ciavarella C., Cuomo-Dannenburg G., Barzon L., Vecchio C.D., Rossi L., Manganelli R., Loregian A., Navarin N., Abate D., Sciro M., Merigliano S., Decanale E., Vanuzzo M.C., Saluzzo F., Onelia F., Pacenti M., Parisi S., Carretta G., Donato D., Flor L., Cocchio S., Masi G., Sperduti A., Cattarino L., Salvador R., Gaythorpe K.A.M., I.C.L.C.-19 R. Team, Brazzale A.R., Toppo S., Trevisan M., Baldo V., Donnelly C.A., Ferguson N.M., Dorigatti I., Crisanti A. Suppression of COVID-19 outbreak in the municipality of Vo, Italy. MedRxiv. 2020 doi: 10.1101/2020.04.17.20053157. 2020.04.17.20053157. [DOI] [Google Scholar]
- 27.Winter A.K., Hegde S.T. The important role of serology for COVID-19 control. Lancet Infect. Dis. 2020;0 doi: 10.1016/S1473-3099(20)30322-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eliaz Y., Danovich M., Gasic G.P. Poolkeh Finds the Optimal Pooling Strategy for a Population-wide COVID-19 Testing (Israel, UK, and US as Test Cases) MedRxiv. 2020 doi: 10.1101/2020.04.25.20079343. 2020.04.25.20079343. [DOI] [Google Scholar]
- 29.Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E., The Protein Data Bank Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H., Robertson A.D., Jensen J.H. Very fast empirical prediction and rationalization of protein pKa values. Proteins. 2005;61:704–721. doi: 10.1002/prot.20660. [DOI] [PubMed] [Google Scholar]
- 31.Søndergaard C.R., Olsson M.H.M., Rostkowski M., Jensen J.H. Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pKa values. J. Chem. Theory Comput. 2011;7:2284–2295. doi: 10.1021/ct200133y. [DOI] [PubMed] [Google Scholar]
- 32.Krissinel E., Henrick K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 33.Miyazawa S., Jernigan R.L. Residue-residue potentials with a favorable contact pair term and an unfavorable high packing density term, for simulation and threading. J. Mol. Biol. 1996;256:623–644. doi: 10.1006/jmbi.1996.0114. [DOI] [PubMed] [Google Scholar]
- 34.Kim Y.C., Hummer G. Coarse-grained models for simulations of multiprotein complexes: application to ubiquitin binding. J. Mol. Biol. 2008;375:1416–1433. doi: 10.1016/j.jmb.2007.11.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bereau T., Deserno M. Generic coarse-grained model for protein folding and aggregation. J. Chem. Phys. 2009;130:235106. doi: 10.1063/1.3152842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brancolini G., Lopez H., Corni S., Tozzini V. Low-resolution models for the interaction dynamics of coated gold nanoparticles with β2-microglobulin. Int. J. Mol. Sci. 2019;20:3866. doi: 10.3390/ijms20163866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu G., Zhou J. Understanding the curvature effect of silica nanoparticles on lysozyme adsorption orientation and conformation: a mesoscopic coarse-grained simulation study. Phys. Chem. Chem. Phys. 2016;18:23500–23507. doi: 10.1039/C6CP01478J. [DOI] [PubMed] [Google Scholar]
- 38.Tsao H.-K. Electrostatic interactions of a string-like particle with a charged plate. J. Colloid Interface Sci. 1998;202:527–540. doi: 10.1006/jcis.1998.5471. [DOI] [Google Scholar]
- 39.Atkins P.W., Paula J.D., Keeler J. Oxford University Press; 2018. Atkins’ Physical Chemistry. [Google Scholar]
- 40.Collard D.M., Anne M. Fox, Use of electroactive thiols to study the formation and exchange of alkanethiol monolayers on gold. Langmuir. 1991;7:1192–1197. doi: 10.1021/la00054a029. [DOI] [Google Scholar]
- 41.Häkkinen H. The gold–sulfur interface at the nanoscale. Nat. Chem. 2012;4:443–455. doi: 10.1038/nchem.1352. [DOI] [PubMed] [Google Scholar]
- 42.Xue Y., Li X., Li H., Zhang W. Quantifying thiol–gold interactions towards the efficient strength control. Nat. Commun. 2014;5:1–9. doi: 10.1038/ncomms5348. [DOI] [PubMed] [Google Scholar]
- 43.Grönbeck H., Curioni A., Andreoni W. Thiols and disulfides on the Au(111) surface: the headgroup−gold interaction. J. Am. Chem. Soc. 2000;122:3839–3842. doi: 10.1021/ja993622x. [DOI] [Google Scholar]
- 44.Zweckstetter M., Bax A. Prediction of sterically induced alignment in a dilute liquid crystalline phase: aid to protein structure determination by NMR. J. Am. Chem. Soc. 2000;122:3791–3792. doi: 10.1021/ja0000908. [DOI] [Google Scholar]
- 45.Zweckstetter M., Hummer G., Bax A. Prediction of charge-induced molecular alignment of biomolecules dissolved in dilute liquid-crystalline phases. Biophys. J. 2004;86:3444–3460. doi: 10.1529/biophysj.103.035790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zweckstetter M. NMR: prediction of molecular alignment from structure using the PALES software. Nat. Protoc. 2008;3:679–690. doi: 10.1038/nprot.2008.36. [DOI] [PubMed] [Google Scholar]
- 47.Bak Null, Nielsen Null. REPULSION, A Novel Approach to Efficient Powder Averaging in Solid-State NMR. J. Magn. Reson. San Diego Calif. 1997;125:132–139. doi: 10.1006/jmre.1996.1087. [DOI] [PubMed] [Google Scholar]
- 48.Bak M., Rasmussen J.T., Nielsen N.C. SIMPSON: a general simulation program for solid-state NMR spectroscopy. J. Magn. Reson. 2000;147:296–330. doi: 10.1006/jmre.2000.2179. [DOI] [PubMed] [Google Scholar]
- 49.Tošner Z., Andersen R., Stevensson B., Edén M., Chr N., Nielsen T. Vosegaard, Computer-intensive simulation of solid-state NMR experiments using SIMPSON. J. Magn. Reson. 2014;246:79–93. doi: 10.1016/j.jmr.2014.07.002. [DOI] [PubMed] [Google Scholar]
- 50.Yuan M., Wu N.C., Zhu X., Lee C.-C.D., So R.T.Y., Lv H., Mok C.K.P., Wilson I.A. 2020. A Highly Conserved Cryptic Epitope in the Receptor-Binding Domains of SARS-CoV-2 and SARS-CoV, Science. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Harris L.J., Larson S.B., Hasel K.W., McPherson A. Refined structure of an intact IgG2a monoclonal antibody. Biochemistry. 1997;36:1581–1597. doi: 10.1021/bi962514+. [DOI] [PubMed] [Google Scholar]
- 52.Harris L.J., Skaletsky E., McPherson A. Crystallographic structure of an intact IgG1 monoclonal antibody11Edited by I. A. Wilson. J. Mol. Biol. 1998;275:861–872. doi: 10.1006/jmbi.1997.1508. [DOI] [PubMed] [Google Scholar]
- 53.Saphire E.O., Parren P.W.H.I., Pantophlet R., Zwick M.B., Morris G.M., Rudd P.M., Dwek R.A., Stanfield R.L., Burton D.R., Wilson I.A. Crystal structure of a neutralizing human IgG against HIV-1: a template for vaccine design. Science. 2001;293:1155–1159. doi: 10.1126/science.1061692. [DOI] [PubMed] [Google Scholar]
- 54.Scapin G., Yang X., Prosise W.W., McCoy M., Reichert P., Johnston J.M., Kashi R.S., Strickland C. Structure of full-length human anti-PD1 therapeutic IgG4 antibody pembrolizumab. Nat. Struct. Mol. Biol. 2015;22:953–958. doi: 10.1038/nsmb.3129. [DOI] [PubMed] [Google Scholar]
- 55.Shajahan A., Supekar N.T., Gleinich A.S., Azadi P. Deducing the N- and O-glycosylation profile of the spike protein of novel coronavirus SARS-CoV-2. Glycobiology. 2020 doi: 10.1093/glycob/cwaa042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Walls A.C., Park Y.-J., Tortorici M.A., Wall A., McGuire A.T., Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;181 doi: 10.1016/j.cell.2020.02.058. 281–292.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Al-Duri B., Robinson E., McNerlan S., Bailie P. Hydrolysis of edible oils by lipases immobilized on hydrophobic supports: effects of internal support structure. J. Am. Oil Chem. Soc. 1995;72:1351–1359. doi: 10.1007/BF02546211. [DOI] [Google Scholar]
- 58.Aubin-Tam M.-E. Conjugation of nanoparticles to proteins. In: Bergese P., Hamad-Schifferli K., editors. Nanomater. Interfaces Biol. Methods Protoc. Humana Press; Totowa, NJ: 2013. pp. 19–27. [DOI] [Google Scholar]
- 59.López-Gallego F., Fernandez-Lorente G., Rocha-Martin J., Bolivar J.M., Mateo C., Guisan J.M. Stabilization of enzymes by multipoint covalent immobilization on supports activated with glyoxyl groups. Methods Mol. Biol. Clifton NJ. 2013;1051:59–71. doi: 10.1007/978-1-62703-550-7_5. [DOI] [PubMed] [Google Scholar]
- 60.Li Z., Yi Y., Luo X., Xiong N., Liu Y., Li S., Sun R., Wang Y., Hu B., Chen W., Zhang Y., Wang J., Huang B., Lin Y., Yang J., Cai W., Wang X., Cheng J., Chen Z., Sun K., Pan W., Zhan Z., Chen L., Ye F. Development and clinical application of a rapid IgM-IgG combined antibody test for SARS-CoV-2 infection diagnosis. J. Med. Virol. 2020 doi: 10.1002/jmv.25727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kumal R.R., Karam T.E., Haber L.H. Determination of the surface charge density of colloidal gold nanoparticles using second harmonic generation. J. Phys. Chem. C. 2015;119:16200–16207. doi: 10.1021/acs.jpcc.5b00568. [DOI] [Google Scholar]
- 62.Oliver C. Colloidal gold/streptavidin methods. Methods Mol. Biol. Clifton NJ. 2010;588:375–380. doi: 10.1007/978-1-59745-324-0_40. [DOI] [PubMed] [Google Scholar]
- 63.Sélo I., Négroni L., Créminon C., Grassi J., Wal J.M. Preferential labeling of α-amino N-terminal groups in peptides by biotin: application to the detection of specific anti-peptide antibodies by enzyme immunoassays. J. Immunol. Methods. 1996;199:127–138. doi: 10.1016/S0022-1759(96)00173-1. [DOI] [PubMed] [Google Scholar]
- 64.Martos-Maldonado M.C., Hjuler C.T., Sørensen K.K., Thygesen M.B., Rasmussen J.E., Villadsen K., Midtgaard S.R., Kol S., Schoffelen S., Jensen K.J. Selective N-terminal acylation of peptides and proteins with a Gly-His tag sequence. Nat. Commun. 2018;9 doi: 10.1038/s41467-018-05695-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pierce B.G., Wiehe K., Hwang H., Kim B.-H., Vreven T., Weng Z. ZDOCK server: interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinforma. Oxf. Engl. 2014;30:1771–1773. doi: 10.1093/bioinformatics/btu097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.van Zundert G.C.P., Rodrigues J.P.G.L.M., Trellet M., Schmitz C., Kastritis P.L., Karaca E., Melquiond A.S.J., van Dijk M., de Vries S.J., Bonvin A.M.J.J. The HADDOCK2.2 web server: user-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 2016;428:720–725. doi: 10.1016/j.jmb.2015.09.014. [DOI] [PubMed] [Google Scholar]
- 67.de Vries S.J., Bonvin A.M.J.J. CPORT: a consensus Interface predictor and its performance in prediction-driven docking with HADDOCK. PLoS One. 2011;6 doi: 10.1371/journal.pone.0017695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Giuntini S., Cerofolini L., Ravera E., Fragai M., Luchinat C. Atomic structural details of a protein grafted onto gold nanoparticles. Sci. Rep. 2017;7:17934. doi: 10.1038/s41598-017-18109-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ravera E., Cerofolini L., Martelli T., Louka A., Fragai M., Luchinat C. 1H-detected solid-state NMR of proteins entrapped in bioinspired silica: a new tool for biomaterials characterization. Sci. Rep. 2016;6:27851. doi: 10.1038/srep27851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huang R., Yamamoto K., Zhang M., Popovych N., Hung I., Im S.-C., Gan Z., Waskell L., Ramamoorthy A. Probing the transmembrane structure and dynamics of microsomal NADPH-cytochrome P450 oxidoreductase by solid-state NMR. Biophys. J. 2014;106:2126–2133. doi: 10.1016/j.bpj.2014.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rao P., Shukla A., Parmar P., Rawal R.M., Patel B., Saraf M., Goswami D. Reckoning a fungal metabolite, Pyranonigrin a as a potential Main protease (Mpro) inhibitor of novel SARS-CoV-2 virus identified using docking and molecular dynamics simulation. Biophys. Chem. 2020;264:106425. doi: 10.1016/j.bpc.2020.106425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Aouidate A., Ghaleb A., Chtita S., Aarjane M., Ousaa A., Maghat H., Sbai A., Choukrad M., Bouachrine M., Lakhlifi T. Identification of a novel dual-target scaffold for 3CLpro and RdRp proteins of SARS-CoV-2 using 3D-similarity search, molecular docking, molecular dynamics and ADMET evaluation. J. Biomol. Struct. Dyn. 2020:1–14. doi: 10.1080/07391102.2020.1779130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Macchiagodena M., Pagliai M., Procacci P. Inhibition of the Main Protease 3CL-pro of the Coronavirus Disease 19 Via Structure-Based Ligand Design and Molecular Modeling, ArXiv200209937 Q-Bio. 2020. http://arxiv.org/abs/2002.09937 [DOI] [PMC free article] [PubMed]
- 74.Macchiagodena M., Pagliai M., Procacci P. Identification of potential binders of the main protease 3CLpro of the COVID-19 via structure-based ligand design and molecular modeling. Chem. Phys. Lett. 2020;750:137489. doi: 10.1016/j.cplett.2020.137489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Procacci P., Macchiagodena M., Pagliai M., Guarnieri G., Iannone F. Interaction of hydroxychloroquine with SARS-CoV2 functional proteins using all-atoms non-equilibrium alchemical simulations. Chem. Commun. 2020 doi: 10.1039/D0CC03558K. [DOI] [PubMed] [Google Scholar]
- 76.Li W. Delving deep into the structural aspects of a furin cleavage site inserted into the spike protein of SARS-CoV-2: a structural biophysical perspective. Biophys. Chem. 2020;264:106420. doi: 10.1016/j.bpc.2020.106420. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Tables and Figures.