

To the Editor:
Polycythemia vera (PV) is a chronic myeloproliferative neoplasm (cMPN) characterized by stem cell-derived clonal myeloproliferation resulting in panmyelosis with persistently raised hematocrit, increased risk of thrombotic complications, and predisposition to evolve to myelofibrosis or leukemia [1]. Therapy is currently based on phlebotomy to normalize hematocrit, and aspirin. Hydroxyurea is used as first line when cytoreduction is necessary [1], although toxicity can result in inadequate disease management [2]. Recently, ropeginterferon α-2b was approved by European Medicinal Agency as first line for patients without symptomatic splenomegaly [3]. Ruxolitinib is second-line for patients who are refractory and/or intolerant to hydroxyurea [4]; other treatments include busulfan, pipobroman [5], and nonpegylated and pegylated interferons (off-label) [1, 6, 7], but use is limited by side effects and safety concerns. Additional, targeted therapies are therefore needed.
Up to 98% of patients with PV bear the JAK2V617F gene mutation, which activates erythropoietin receptor signaling pathways. Givinostat is a histone-deacetylase (HDAC) inhibitor that selectively targets JAK2V617F cell growth, reducing hematopoietic cell proliferation [8]. The efficacy and safety of givinostat alone or with hydroxyurea has previously been evaluated in two studies in JAK2V617F positive PV [9, 10]. Although these studies confirmed the positive risk-benefit of givinostat, they did not provide comprehensive efficacy evidence for givinostat monotherapy, and did not identify the most appropriate dose. The current study was therefore conducted to support givinostat monotherapy development in PV, aiming to determine the maximum tolerated dose (MTD), and to assess safety and efficacy of this dose.
This multinational, open-label, nonrandomized study was conducted in two parts. Part A (Phase Ib) was dose escalation, with the first 4-week cycle determining the MTD. Part B (Phase II), the proof of concept phase, then evaluated efficacy and safety at this MTD. Full details of the methods are in the supplement. Both parts had 24-week treatment periods, with patients receiving six four-week cycles of givinostat. In Part A, since givinostat 50 mg twice daily (BID) was previously well tolerated, the first cohort of three patients received 100 mg BID, with the dose to be escalated by 50 mg BID in each subsequent cohort according to a 3 + 3 design, adopting a modified Fibonacci escalation scheme, although only after the third patient had been followed for a minimum of one cycle, and tolerability data had been evaluated by the Safety Review Team (Supplementary Table 1). For Part B, patients initially received givinostat at the MTD, with modification permitted to achieve an optimized dose, balancing tolerability, and response.
Eligible patients were aged ≥18 years, with a confirmed PV diagnosis, JAK2V617F positivity assessed by centralized quantitative real-time polymerase chain reaction, and active/not controlled disease, defined as: (1) hematocrit ≥45% or <45% with phlebotomy, and (2) platelet count >400 × 109/l, and (3) white blood cell count >10 × 109/l. Main exclusion criteria were: absolute neutrophil count <1.2 × 109/l; prior JAK2 or HDAC inhibitor treatment; systemic treatment for cMPNs other than aspirin; hydroxyurea, interferon alpha, or anagrelide within 28, 14, or 7 days before enrollment, respectively. All patients provided informed consent. Study registration: ClinicalTrials.gov (NCT01901432).
The primary objectives of Part A were to determine givinostat’s MTD, and to characterize safety and tolerability in terms of treatment-related adverse events (AEs). Secondary endpoints were to evaluate overall response after three and six cycles (using the clinico-hematological European LeukemiaNet (ELN) response criteria [11]), and to characterize pharmacokinetics. For Part B, primary objectives were to evaluate overall response, safety and tolerability after three cycles. Secondary endpoints were to evaluate overall response, safety and tolerability after six cycles, and to characterize pharmacokinetics. Exploratory endpoints are in the supplement.
Twelve patients were studied in Part A, with 35 in Part B (Supplementary Fig. 1, Supplementary Tables 2 and 3). In Part A, during the first cycle one patient receiving givinostat 100 mg BID experienced dose-limiting toxicity: Grade 3 dyspepsia, drug related, resolving with sequelae after treatment. Three additional patients therefore received 100 mg BID, none of whom had dose-limiting toxicity during Cycle 1. Although escalation to higher doses was permitted, the Safety Review Team agreed the MTD was 100 mg BID, given: (a) thrombocytopenia is a known side effect of HDAC inhibitors; (b) a platelet count decrease was observed in subsequent cycles; (c) as givinostat is a chronic treatment, it was preferable to not expose patients to higher doses that could be poorly tolerated during chronic treatment. To more accurately define givinostat’s MTD, three additional patients received an intermediate dose (150 mg daily). Finally, three patients received 50 mg BID, to investigate safety, pharmacokinetics, and pharmacodynamics of this dose. A total of 66.7% of patients experienced at least one drug-related AE, mainly Grade 1 or 2 (Supplementary Table 4), most commonly thrombocytopenia (33.3% of patients). Two patients (16.7%) experienced a serious AE (thrombophlebitis and myocardial infarction), neither drug-related; no patient died. Two patients withdrew due to drug-related AEs (dyspepsia [Grade 3] and thrombocytopenia [Grade 4]), both with 100 mg BID.
The overall response rate in Part A was above 70% (Fig. 1). One patient achieved complete response after three cycles, and one after six cycles. Median givinostat Tmax was 1.5–4 h (Supplementary Table 5), with steady-state reached by day 28 of Cycle 1 (the first repeat-dose pharmacokinetic evaluation). After three cycles, givinostat normalized hematological parameters in 45.5–54.5% of patients (Supplementary Table 6), normalized spleen volume in 54.5%, resolved disease-related symptoms in 63.6% (Supplementary Table 7), and reduced pruritus and JAK2-mutated allele burden (Supplementary Table 8).
Fig. 1. Parts A and B: therapeutic response evaluation (intention-to-treat population).
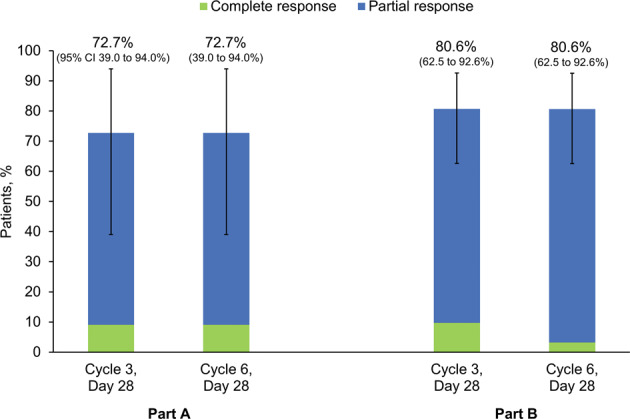
Data are from 11 patients in Part A and 31 patients in Part B.
At the end of Cycles 3 and 6 of Part B, 80.6% of patients were responders (Fig. 1), with three achieving complete response after three cycles and one after six. Overall, 94.3% of patients had at least one drug-related AE, the majority Grade 1 or 2 and none Grade 4 or 5, with most occurring during the first three cycles of treatment (152 out of 190 events). The most common were diarrhea (51.4% of patients), thrombocytopenia (45.7%), and increased blood creatinine (37.1%) (Table 1). Two patients experienced a serious AE, both during the first three cycles, one study drug-related (Grade 3 diarrhea resolving in 7 days without therapy, with study drug temporarily discontinued). Three patients withdrew, one due to study drug-related AEs (Grade 3 neutropenia and Grade 2 thrombocytopenia, both resolving). The other two were withdrawn by their investigators (Supplementary Fig. 1). No patient died, and there were no clinically relevant vital signs or ECG values.
Table 1.
Part B: patients with study drug-related treatment-emergent AEs, overall and by system organ class and preferred term (including only preferred terms reported by one or more patient with Grade 3 events) (safety population).
| System organ class preferred term | Grade 3 | Any grade | ||
|---|---|---|---|---|
| N | % | N | % | |
| Patients with any drug-related AE | 10 | 28.6 | 33 | 94.3 |
| Blood and lymphatic system disorders | 3 | 8.6 | 18 | 51.4 |
| Anemia | 2 | 5.7 | 6 | 17.1 |
| Neutropenia | 1 | 2.9 | 2 | 5.7 |
| Thrombocytopenia | 1 | 2.9 | 16 | 45.7 |
| Cardiac disorders | 0a | 0 | 1 | 2.9 |
| Gastrointestinal disorders | 4 | 11.4 | 26 | 74.3 |
| Diarrhea | 4 | 11.4 | 18 | 51.4 |
| General disorders and administration site conditions | 2 | 5.7 | 9 | 25.7 |
| Asthenia | 2 | 5.7 | 8 | 22.9 |
| Investigations | 0a | 0 | 19 | 54.3 |
| Metabolism and nutrition disorders | 1 | 2.9 | 8 | 22.9 |
| Hypocalcemia | 1 | 2.9 | 4 | 11.4 |
| Nervous system disorders | 0a | 0 | 5 | 14.3 |
| Renal and urinary disorders | 0a | 0 | 2 | 5.7 |
| Respiratory, thoracic and mediastinal disorders | 0a | 0 | 1 | 2.9 |
| Skin and subcutaneous tissue disorders | 1 | 2.9 | 6 | 17.1 |
| Rash | 1 | 2.9 | 1 | 2.9 |
Data are from 35 patients. Grades are based on the National Cancer Institute Common Terminology Criteria for Adverse Events Version 4.03, where Grade 1 are mild events, Grade 2 are moderate, Grade 3 are severe, Grade 4 are life-threatening, and Grade 5 events result in death. There were no Grade 4 or 5 events in Part B of the study.
AE adverse event.
aThere were no Grade 3 events for these system organ classes.
Overall, Part B pharmacokinetics was similar to Part A at comparable doses (Supplementary Tables 9 and 10). Improvements were seen in all individual response criteria (Supplementary Fig. 2; Supplementary Table 11), with white blood cell and platelet counts normalized in 90.3% and 74.2% of patients after three cycles, respectively, and hematocrit in 77.4% and 48.4% after three and six cycles, respectively. Improvements were observed in disease-related symptoms assessed by Myeloproliferative Neoplasm Symptom Assessment Form quality of life (QoL) questionnaire, especially during Cycle 6 (Supplementary Table 12), with a reduction in the proportion with severe pruritus (Score 7–10; Supplementary Fig. 3). Approximately 50% had no headache (Supplementary Table 12), and no patients had severe headache (Supplementary Fig. 4). The proportion of patients without microvascular symptoms improved from baseline (38.7%) to Cycle 6 (51.6%; Supplementary Table 12), with a low proportion having severe symptoms (6.5–12.9%; Supplementary Fig. 5). A total of 19.4% had a spleen volumetric index reduction of at least 35% during treatment, with total spleen normalization in two and three patients after three and six cycles, respectively, and a moderate reduction in JAK2V617F allele burden (Supplementary Table 12). Finally, differential gene expression was observed (Supplementary Fig. 6), with upregulation for GLRX, STAT4 and HDAC3, and downregulation for MYC.
The study aims were achieved, with the MTD, 100 mg BID, determined in Part A, and this dose effective in Part B. In addition to the high overall response rate, givinostat had a positive impact on individual clinico-hematological ELN criteria, both hematological parameters and disease-related symptoms. The three hematological parameters, all abnormal at study entry, were normalized in the majority of patients, and givinostat improved key disease-related symptoms, notably pruritus with complete resolution in many patients, with an associated positive impact on QoL. A reduction in JAK2-mutated allele burden was observed in both parts of the study, and Part B provided clear evidence of differential gene expression with givinostat, consistent with disease pathway regulation. Overall, givinostat was well tolerated with no new safety concerns. Unlike previous studies, the recruited population had active or not controlled disease, and were both high- and low-risk, making comparisons difficult. However, the observed response was greater than for other PV therapies [12–15]. For example, in a study comparing interferon to hydroxyurea, 45% of patients had a hematologic response to either therapy [12], whereas in a second study, 40% of patients had a response of any type to ruxolitinib [13], and in a third the overall response to the HDAC inhibitor vorinostat was 35%, with significant side effects resulting in a high rate of study withdrawal [15].
In conclusion, these data support givinostat monotherapy development in the defined PV target population.
Supplementary information
Acknowledgements
This study was funded by Italfarmaco S.p.A. David Young of Young Medical Communications and Consulting Ltd, a medical writer supported by funding from Italfarmaco S.p.A., provided drafts and editorial assistance to the authors during preparation of this paper. The authors would like to thank the investigators who recruited patients, including Dr Andrzej Hellmann, and the patients at the investigative sites for their support of this study.
Compliance with ethical standards
Conflict of interest
AR has received honoraria for consultancy, and travel support from Italfarmaco, Gilead, Amgen, Novartis, Pfizer, Celgene, Sanofi, Astellas and Roche. AI has received speaker honoraria from Novartis, Pfizer and Incyte. AMV has received honoraria for advisory board participation from Novartis, Celgene, Incyte, CTI and Italfarmaco, and for lectures from Novartis, and CTI. N.v.B. received research funding from Novartis. MFM has received honoraria and has attended advisory boards with Novartis, and has received honoraria from Celgene. RAM has received honoraria as a consultant to Novartis, Sierra Oncology, and La Jolla Oncology, and research support from Incyte, Genentech, Celgene, CTI, and Abbvie. RT has advised and received honoraria from Italfarmaco. PB, SM, and SDT are employees of Italfarmaco SpA, the sponsor of the study. RN, AG, BM, AP, GC, MDM, SL, NC, J-PM, APancrazzi, FG have no conflicts to disclose.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version of this article (10.1038/s41375-020-0735-y) contains supplementary material, which is available to authorized users.
References
- 1.McMullin MF, Harrison CN, Ali S, Cargo C, Chen F, Ewing J, et al. A guideline for the diagnosis and management of polycythaemia vera. A British Society for Haematology Guideline. Br J Haematol. 2019;184:176–91. doi: 10.1111/bjh.15648. [DOI] [PubMed] [Google Scholar]
- 2.Demuynck T, Verhoef G, Delforge M, Vandenberghe P, Devos T. Polycythemia vera and hydroxyurea resistance/intolerance: a monocentric retrospective analysis. Ann Hematol. 2019;98:1421–6. doi: 10.1007/s00277-019-03654-6. [DOI] [PubMed] [Google Scholar]
- 3.Kiladjian J-J, Cassinat B, Soret-Dulphy J, Verger E, Roy L, Rey J, et al. Molecular response to hydroxyurea and ropeginterferon alfa-2B in the PROUD-PV randomized Phase 3 trial. Haematologica. 2017;102:S787. [Google Scholar]
- 4.Mesa RA. New guidelines from the NCCN for polycythemia vera. Clin Adv Hematol Oncol. 2017;15:848–50. [PMC free article] [PubMed] [Google Scholar]
- 5.Kiladjian J-J, Gardin C, Renoux M, Bruno F, Bernard J-F. Long-term outcomes of polycythemia vera patients treated with pipobroman as initial therapy. Hematol J. 2003;4:198–207. doi: 10.1038/sj.thj.6200250. [DOI] [PubMed] [Google Scholar]
- 6.Kiladjian J-J, Chomienne C, Fenaux P. Interferon-alpha therapy in bcr-abl-negative myeloproliferative neoplasms. Leukemia. 2008;22:1990–8. doi: 10.1038/leu.2008.280. [DOI] [PubMed] [Google Scholar]
- 7.Yacoub A, Mascarenhas J, Kosiorek H, Prchal JT, Berenzon D, Baer MR, et al. Pegylated interferon alfa-2a for polycythemia vera or essential thrombocythemia resistant or intolerant to hydroxyurea. Blood. 2019;134:1498–509. doi: 10.1182/blood.2019000428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guerini V, Barbui V, Spinelli O, Salvi A, Dellacasa C, Carobbio A, et al. The histone deacetylase inhibitor ITF2357 selectively targets cells bearing mutated JAK2V617F. Leukemia. 2008;22:740–7. doi: 10.1038/sj.leu.2405049. [DOI] [PubMed] [Google Scholar]
- 9.Rambaldi A, Dellacasa CM, Finazzi G, Carobbio A, Ferrari ML, Guglielmelli P, et al. A pilot study of the histone-deacetylase inhibitor givinostat in patients with JAK2V617F positive chronic myeloproliferative neoplasms. Br J Haematol. 2010;150:446–55. doi: 10.1111/j.1365-2141.2010.08266.x. [DOI] [PubMed] [Google Scholar]
- 10.Finazzi G, Vannucchi AM, Martinelli V, Ruggeri M, Nobile F, Specchia G, et al. A phase II study of givinostat in combination with hydroxycarbamide in patients with polycythaemia vera unresponsive to hydroxycarbamide monotherapy. Br J Haematol. 2013;161:688–94. doi: 10.1111/bjh.12332. [DOI] [PubMed] [Google Scholar]
- 11.Barosi G, Rosti V. Novel strategies for patients with chronic myeloproliferative disorders. Curr Opin Hematol. 2009;16:129–34. doi: 10.1097/MOH.0b013e3283257a9e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gisslinger H, Klade C, Georgiev P, Skotnicki A, Gercheva-Kyuchukova L, Egyed M, et al. Final results from PROUD-PV a randomized controlled Phase 3 trial comparing ropeginterferon alfa-2b to hydroxyurea in polycythemia vera patients. Blood. 2016;128:475. doi: 10.1182/blood.V128.22.475.475. [DOI] [Google Scholar]
- 13.Pugliese N, Giordano C, Nappi D, Luciano L, Cerchione C, Annunziata M, et al. Adding hydroxyurea in combination with ruxolitinib improves clinical responses in hyperproliferative forms of myelofibrosis. Cancer Med. 2019;8:2802–9. doi: 10.1002/cam4.2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kiladjian J-J, Guglielmelli P, Griesshammer M, Saydam G, Masszi T, Durrant S, et al. Efficacy and safety of ruxolitinib after and versus interferon use in the RESPONSE studies. Ann Hematol. 2018;97:617–27. doi: 10.1007/s00277-017-3225-1. [DOI] [PubMed] [Google Scholar]
- 15.Andersen CL, McMullin MF, Ejerblad E, Zweegman S, Harrison C, Fernandes S, et al. A phase II study of vorinostat (MK-0683) in patients with polycythaemia vera and essential thrombocythaemia. Br J Haematol. 2013;162:498–508. doi: 10.1111/bjh.12416. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.