

Abstract
Heme oxygenase (HO) enzymes catalyze heme into biliverdin, releasing carbon monoxide (CO) and iron into circulation. These byproducts of heme degradation can have potent cytoprotective effects in the face of stressors such as hypoxia and ischemia-reperfusion events. The potential for exogenous use of CO as a therapeutic agent has received increasing attention throughout the past few decades. Further, HO and CO are noted as putatively adaptive in diving mammals and certain high-altitude human populations that are frequently exposed to hypoxia and/or ischemia-reperfusion events, suggesting that HO and endogenous CO afford an evolutionary advantage for hypoxia tolerance and are critical in cell survival and injury avoidance. Our goal is to describe the importance of examining HO and CO in several systems, the physiological links, and the genetic factors that underlie variation in the HO/CO pathway. Finally, we emphasize the ways in which evolutionary perspectives may enhance our understanding of the HO/CO pathway in the context of diverse clinical settings.
Keywords: carbon monoxide, heme oxygenase, hypoxia, altitude, diving, evolution, cytoprotection
Introduction
In the late 1990s, a concept emerged suggesting that all cells possessed protective genes with the sole purpose to ensure survival (Platt and Nath, 1998). These cytoprotective genes played a role in the production of antioxidants, acute phase proteins, as well as regulating the effects of aging. A few were clearly master regulators, such that when not present, the cell or tissue was sensitive to stress. One of these was Hmox, which encodes the protein heme oxygenase (HO), the rate limiting enzyme responsible for the degradation of heme. HO catabolizes heme into biliverdin, which is subsequently converted to bilirubin by biliverdin reductase, releasing carbon monoxide (CO) and iron as byproducts. Paradoxically, CO was strictly considered a toxic pollutant. This notion stems from CO having a high affinity for the O2-binding site on hemoproteins. Therefore, elevated levels of CO can reduce O2 storage capacity and inhibit O2 transport, resulting in CO toxicity. However, recent studies have revealed the biological effects of the gas, leading to the acceptance of CO having therapeutic and cytoprotective effects in modulating inflammation, cell death, and proliferation (Motterlini and Otterbein, 2010).
Heme oxygenase genes are highly conserved and found in most living organisms (Wilks, 2002; Li and Stocker, 2009). The inducible HO-1 and constitutive HO-2 are ~40% identical in their amino acid sequence in humans (Cruse and Maines, 1988), with HO-1 observed at highest levels in the spleen, lung, and visceral adipose and HO-2 in the testes, heart, brain, and stomach (GTEx, V6). Stressors known to stimulate HO-1 expression include oxidative stress, hypoxia, cytokines, heavy metals, and bacterial endotoxins (Abraham, 2002). HO enzymes function as a crucial mechanism to recycle iron in the body through the degradation of heme (Wilks, 2002). The majority of extant vertebrates are known to utilize iron-containing hemoproteins (e.g., hemoglobin, myoglobin, neuroglobin, cytochromes, catalase, and peroxidase) as a mechanism to transport electrons in the respiratory chain for the production of ATP, to reduce oxidative stress and/or to increase the storage capacity and delivery of oxygen (O2) to cells (Hardison, 1996). Therefore, the ability to recycle iron stores in the body can offer an adaptive advantage if organisms find themselves iron-limited. The regulation of HO enzymes can also influence erythrocyte lifespan. Hmox1−/− mice rarely survive beyond the development and those that do have prolonged erythrocyte lifespans, reduced hemoglobin content, and a reduced erythrocyte size that leads to microcytic anemia (Fraser et al., 2015). Moreover, human patients with reduced erythrocyte lifespans display elevated blood and breath CO levels, suggesting increased HO activity (Strocchi et al., 1992).
Recent studies have shown that species and populations adapted to tolerate chronic hypoxia and/or ischemia/reperfusion (I/R) events have upregulated or modified the HO/CO pathway in a manner that could afford beneficial side effects (Herrera et al., 2008; Simonson et al., 2010; Tift et al., 2014). Here, we highlight some of those model species and populations and discuss how such modifications could provide an evolutionary advantage and how information from these studies may be used to learn more about the applications of the HO/CO pathway in the prevention and treatment of certain diseases (Goebel and Wollborn, 2020).
Evolutionary Insights from Hypoxia-Adapted Populations
The late August Krogh stated that, “for such a large number of problems there will be some animal of choice, or a few such animals, on which it can be most conveniently studied” (Krogh, 1929). Model organisms naturally adapted for life in challenging conditions offer excellent opportunities to understand how extreme phenotypes afford protective advantages that can be directed to improve the treatment and/or the prevention of specific human pathologies (Carey et al., 2012; Carey, 2015). For example, some diving mammals experience dramatic hypoxemia (Meir et al., 2009) and I/R events (Zapol et al., 1979) during their repeated long-duration breath-holds and do not develop injuries from this lifestyle. Species that have adapted to live at high altitude or under other hypoxic conditions (e.g., burrowing) also avoid pathologies associated with exposure to chronic hypoxia (Logan et al., 2020). These natural models offer unique insights into the adaptive mechanisms underlying tolerance to hypoxia and/or I/R injury.
Some deep-diving mammals are known to experience levels of hypoxemia during dives that resemble arterial oxygen saturations of humans breathing ambient air on the summit of Mount Everest (Meir et al., 2009). These deep-divers also appear to be protected from repeated I/R events that occur in a majority of their tissues on a dive-to-dive basis as a result of the “dive response,” characterized by extreme bradycardia and peripheral ischemia to maintain mean arterial blood pressure during breath-holds (Halasz et al., 1974; Zapol et al., 1979; Allen and Vázquez-Medina, 2019). In 1959, Pugh reported levels of CO in the blood of deep-diving Weddell seals from Antarctica that resembled those of cigarette smokers (Pugh, 1959). Since then, Tift et al. (2014) have discovered that deep-diving northern elephant seals also maintain high levels of CO in the blood with carboxyhemoglobin (COHb) levels between 5 and 11% (Tift et al., 2014). While CO was shown to be high in the blood of two deep-diving pinnipeds, the distribution of CO concentrations in the blood of different diving species is still unknown.
The quantity of CO found in the blood of elephant seals is similar to that previously mentioned in laboratory studies, which demonstrate cytoprotective effects from exogenous CO delivery (Nakao et al., 2006). It is hypothesized that the elevated hemoprotein stores seen in deep-diving mammals are the source of the high CO levels, which could be used as a mechanism to reduce the development of tissue injuries from the chronic hypoxemia and I/R events they experience while diving (Tift and Ponganis, 2019). These high CO levels could indicate alterations in the erythrocyte lifespan and HO activity in specific tissues of these animals. Such levels of CO would likely increase the O2-binding affinity of hemoglobin, impacting O2-delivery mechanics. Considering the low arterial pO2 values routinely experienced during breath-holds (<15–20 mmHg), it is possible that a higher hemoglobin-O2 affinity due to moderate COHb levels could preserve blood O2 stores, increasing O2 availability later in the dive (Storz, 2016). Indeed, the Haldane effect suggests that mice exposed to trace amounts of CO will more efficiently acquire O2 and survive longer under conditions of severe hypoxia (Roughton and Darling, 1944). Endogenous CO also impacts mitochondrial O2 consumption and ATP production, which could contribute to hypometabolism during breath-holds and exposure to hypoxia (D’Amico et al., 2006).
Species adapted to live at high altitude face a different hypoxic challenge, in that the ambient environment has a reduced pO2. Certain pathologies may develop due to acute and chronic hypoxia exposure at altitude (e.g., pulmonary and cerebral edema, pulmonary hypertension, and excessive erythrocytosis). Considering hypoxia as a stressor that increases HO-1 activity (Shibahara et al., 2007), the HO/CO pathway is of interest when studying organisms at high altitude. When laboratory rats were brought from an elevation of 1,006 to 3,048 and 4,572 m, their COHb levels increased linearly with altitude from 0.68 to 1.16, and 1.68%, respectively (McGrath, 1992). Similarly, when healthy adult humans were brought from an altitude near sea level to 3,517 m, their COHb levels increased from 0.79 to 0.95% within 20 h (McGrath et al., 1993). When comparing neonatal sheep (lowland species) and llama (highland species) that underwent gestation at high altitude (3,600 m), only sheep developed pulmonary arterial hypertension (Herrera et al., 2008; Llanos et al., 2012). This was associated with reduced soluble guanylate cyclase, HO, and CO production despite increased nitric oxide synthase activity in the lungs of the sheep. In contrast, neonatal llamas avoided pulmonary hypertension and had an increase in pulmonary CO production and HO-1 expression with no change in nitric oxide levels (Herrera et al., 2008). This specific HO/CO protective effect has not been investigated in other species adapted for living with chronic hypoxia.
Humans of Tibetan ancestry exhibit a lower average hemoglobin concentration compared to sojourners and long-term highland residents in the Andes, where individuals exhibit varying degrees of polycythemia (Beall, 2007). Whether relatively lower hemoglobin provides a direct adaptive effect or is the side effect of other traits that provide an advantage remains an important question in the field (Storz, 2010; Simonson 2015). HMOX2 has been detected as a top selection candidate gene in ours and others’ studies of Tibetan adaptation and is hypothesized to play a role in regulating heme catabolism and CO production (Simonson et al., 2010; Yang et al., 2016). A study of 1,250 high-altitude native Tibetans found variants at this locus that are associated with decreased hemoglobin concentration in males, and in vitro analysis indicates that a derived intronic variant (rs4786504) is associated with increased HMOX2 expression (Yang et al., 2016). While it remains to be determined if regulatory variants at this locus lead to increased heme catabolism, such alterations could contribute to the lower hemoglobin concentrations seen in Tibetans living at altitude.
In contrast to Tibetans, adaptive HMOX genetic variants have not yet been identified in Andean or Ethiopian highlanders. However, HO could mitigate various challenges imposed by hypoxia at altitude (e.g., higher Hb-O2 affinity; Storz, 2016). Our recent examination in Andean adult males and females living at 4,350 m shows a positive relationship between endogenous CO in breath and blood with both hemoglobin concentration and altitude (Figure 1). These preliminary findings could reflect elevated erythrocyte destruction, reduced erythrocyte lifespan, and/or increased HO-1 or HO-2 activity. Genetic studies in lowlanders have shown that individuals with increased HO-1 expression are less prone to pathologies, such as diabetes, atherosclerosis, chronic obstructive pulmonary disease (COPD), and arthritis (Motterlini and Otterbein, 2010). Whether these levels contribute to the variability in pathologies associated with excessive erythrocytosis and chronic mountain sickness in Andeans remain to be determined.
Figure 1.

(A) End-tidal carbon monoxide (CO) levels (ppm) in 47 adults from Cerro de Pasco, Peru (high altitude), and 18 adults of mixed ancestry living in San Diego, California, USA (sea level). There was no statistical difference between males (blue) and females (red) from high or low altitude groups (p > 0.8 for both). (B) Relationship between total hemoglobin (g/dl) and the percent carboxyhemoglobin (COHb) in venous blood of adult males (n = 22; blue) and females (n = 21; red) from Cerro de Pasco, Peru (4,330 m).
Clinical Insights
Hypoxemia and I/R events are complications inherent to common disease states that often lead to a suite of downstream problems, including adverse cardiopulmonary effects, inflammation, and tissue death. Induction of a battery of stress response genes, such as HMOX1, respond metabolically to (1) degrade elevated heme released during tissue damage in part due to an increase in intracellular hemoproteins and (2) to generate bioactive products to further enhance cell and tissue recovery (Figure 2). Hypoxia and inflammation often occur in tandem during infection or I/R events, whereby the hypoxia-inducible factor (HIF) pathway interacts with NF-κB signaling to coordinate molecular responses, including regulation of HMOX1 (Lee et al., 1997; Rushworth and O’Connell, 2004).
Figure 2.
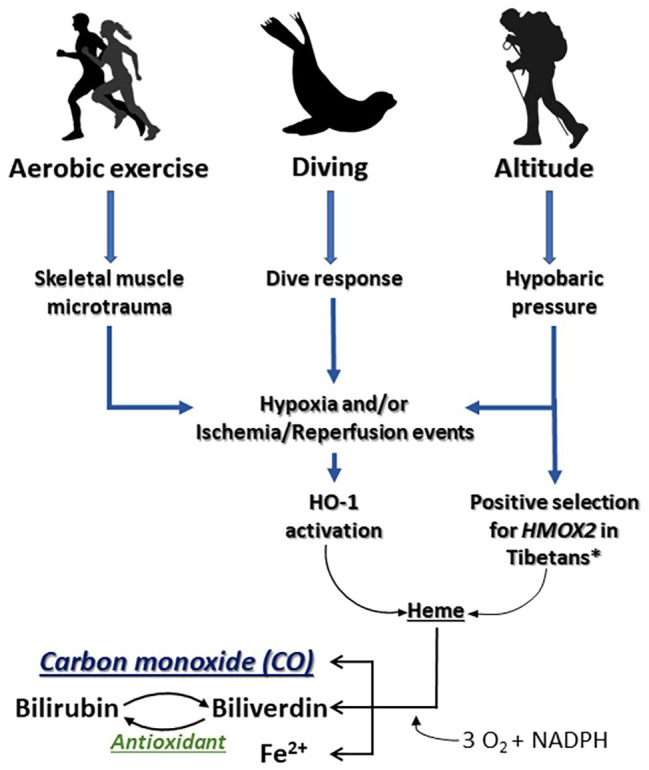
Impacts of aerobic exercise, breath-hold diving, and exposure to altitude on the HO/CO pathway. The stress response protein heme oxygenase-1 (HO-1) responds to intense aerobic exercise and/or skeletal muscle microtrauma by (1) degrading heme that is released during tissue injury as a danger associated molecular pattern and (2) generating bioactive products that contribute to cell and tissue recovery. *Simonson et al., 2010.
Why would evolution result in a system that increases CO in the height of hypoxia or an inflammatory sequelae and ongoing stress response that otherwise decreases tissue O2 availability? Perhaps permissive hypoxia, driven by several selective O2 sensors such as HIF-1α, HIF-2α, and/or mitochondrial oxidases, serves as a mechanism by which cells can dynamically adjust to optimize survival in different environments. In each instance, the availability of O2, which we define as that which is permissive or allowed, dictates specific cellular responses that benefit the needs of the tissue. While O2 is a requisite cofactor for HO-1 activity, HO-1 is also potently induced by hypoxia; in a somewhat paradoxical manner, this results in the generation of CO which then competes with O2 for heme binding sites (Figure 2). This observation has led to speculation that CO creates permissive hypoxia in tissues that serve to modulate cellular energetics and protection (D’Amico et al., 2006).
The effects of HO-1 and CO on tissue protection have been clearly demonstrated in models of I/R injury and many other tissue ischemic pathologies (Wegiel et al., 2014). Due to space constraints, we highlight a few of the seminal findings. Induction of HO-1 or exposure to CO offer dose-dependent anti-inflammatory and cytoprotective effects (Otterbein et al., 2016). The cytoprotective effects of CO were first demonstrated in acute lung injury, and these findings rapidly expanded through the work of numerous laboratories to most models of acute organ injury (Hopper et al., 2018). Administration of exogenous CO or higher HO-1 activity increases the expression of anti-inflammatory cytokines and reduces expression of multiple pro-inflammatory cytokines during acute pulmonary inflammation (Konrad et al., 2016; Zhang et al., 2019). In particular, CO inhibited LPS-induced pro-inflammatory signaling by downregulating NADPH oxidase-dependent reactive oxygen species (ROS) production in macrophages, thereby inhibiting toll-like receptor (TLR) signaling (Nakahira et al., 2006). Additionally, CO can inhibit tumor necrosis factor-induced apoptosis via a p38 MAPK-dependent mechanism (Ryter et al., 2007). These beneficial effects may also result in part from CO stabilization of HIF-1α (Chin et al., 2007). This is important considering the impact of pulmonary function in hypoxia-induced diseases at high altitude (Selland et al., 1993) and the consistent hypoxemia and lung collapse seen during deep-dives in marine mammals (McDonald and Ponganis, 2012).
CO is also hypothesized to play a role in the control of breathing via peripheral O2 sensing in the carotid body, where HO-2 is expressed (Prabhakar, 2012). In the presence of hypoxia, decreased HO-2 activity in glomus cells may reduce CO generation and, since CO inhibits cystathionine-γ-lyase production of the excitatory gasotransmitter H2S, decreased CO generation may result in increased sensory activity of the glomus cells. This is intriguing given that the gene HMOX2 is under evolutionary selection in Tibetan populations, who maintain elevated hypoxic ventilatory responses compared to other high-altitude groups (Beall, 2007).
Organ transplant has been perhaps the most well studied when considering translational potential of the HO/CO pathway. The role of HO-1/CO in the kidney was first described in models of I/R injury, where induction of HO-1 or exposure to CO protected the kidney (Schaaf-Lafontaine and Courtoy, 1986; Blydt-Hansen et al., 2003). This large body of work culminated in the initiation of multiple clinical trials, where exogenous CO has safely been administered to transplant patients1. The mechanism by which CO imparts its salutary effects remain incompletely understood, but likely targets include hemoproteins such as mitochondria oxidases and therein the regulation of bioenergetics, ROS formation, and consumption of O2 (Schallner and Otterbein, 2015; Otterbein et al., 2016). The beneficial effects of HO-1 and CO in organ injury result from preserving mitochondrial health and would dovetail with the observations observed in diving mammals and populations that live at altitude. It is intriguing to speculate that one mechanism for the cytoprotective effects of CO, in instances of tissue hypoxia, may be that CO permits displacement and redistribution of O2 within intracellular stores. The activity of HO-1 is known to increase during hypoxia, increasing CO production, which can alter cellular metabolism; thus, another potential mechanism of CO that plays a role in hypoxia tolerance is slowing or shifting oxidative metabolism to glycolysis through permissive use of O2 (Figure 2).
The HO/CO pathway has also been the focus of many reports in models of cardiovascular disease, including heart failure and cardiac arrest (Shih et al., 2011). Cardiac muscle cells deficient in HO-1 accumulate lethal amounts of ROS, and mice that survive with embryonic HO-1 deletion exhibit many deleterious effects (Kapturczak et al., 2004). Furthermore, Hmox1−/− mice are highly susceptible to I/R injury and, after hypoxia, these animals show evidence of right ventricular infarction (Yoshida et al., 2001; Liu et al., 2005). During reperfusion in the heart, CO administration can decrease infarct size, reduce apoptosis, and increase inotropy. Although those effects were examined mostly in a cardiovascular system, HO-1 also affects other cell types, including skeletal muscle and the physiologic response to exercise (Kozakowska et al., 2018).
Skeletal muscle comprises almost 40% of total body mass in humans, exhibiting major metabolic activity by contributing up to 30% of the resting metabolic rate in adults (Zurlo et al., 1990). The tissue can respond to numerous environmental and physiological challenges (e.g., hypoxia and I/R events) by changing its phenotypic profile and is one commonality across diving animals and individuals living at altitude (Pette and Staron, 2001; Spangenburg et al., 2008; Pabst et al., 2016). Skeletal muscle contraction during exhaustive exercise generates ROS that can promote oxidative damage to myofibers (Reid, 2001), and it has been suggested that HO-1 can protect against exercise-induced injury (Saxena et al., 2010). HO-1 is normally expressed at very low levels in skeletal muscle, but increases dramatically with exhaustive exercise (Pilegaard et al., 2000). The discovery that CO induces mitochondrial biogenesis through specific signaling pathways has raised the possibility that it contributes to the resolution of skeletal muscle injury during and after episodes of oxidative stress, such as exercise or I/R events (Suliman et al., 2007). Intermittent CO breathing after a single exercise test led to an increase in mitochondrial oxidative stress markers and mitochondrial fusion protein expression indicative of mitochondrial biogenesis in skeletal muscle.
From a clinical perspective, based on high disease prevalence and consequences, there is intense interest in both intermittent hypoxemia (as seen in obstructive sleep apnea-OSA) and sustained hypoxemia (as seen in COPD or at high altitude; Soler et al., 2015; Benjafield et al., 2019). The combination of these stimuli (sustained plus intermittent hypoxemia) occurs in some clinical settings, including overlap syndrome (OSA plus COPD), or in OSA patients at high altitude (Marin et al., 2010). The deleterious effects of hypoxemia are well established (Drager et al., 2015; Umeda et al., 2020), although some literature supports a potential protective role of ischemic preconditioning with more mild levels of hypoxemia (Sánchez-de-la-Torre et al., 2018). Regarding CO, some data support its role as a biomarker in OSA (Kobayashi et al., 2008), although its biological impact in these patients has been debated (Owens et al., 2008). Further translational research is certainly encouraged to apply knowledge regarding hypoxia and CO from comparative biology to the clinical setting.
Conclusions
The concept that evolutionary mechanisms have optimized the biology and physiology of HO and CO is relevant in the context of hypoxia adaptation. Examples can be observed across species and under varying physiologic and pathophysiologic conditions ranging from diving mammals to individuals living at altitude, as well as physical exercise and in response to tissue injury. Increasing our understanding of the natural activity and regulation of the HO/CO pathway associated with injury avoidance and maintenance of cellular health will contribute valuable knowledge regarding the benefits of tissue-specific, moderate-level CO exposure.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by UCSD IRB #171772. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
All authors contributed to the article and approved the submitted version.
Conflict of Interest
LO is a Scientific Advisor for Hillhurst Biopharmaceuticals. AM is funded by NHLBI. He reports income from Merck and Livanova related to medical education. ResMed provided a philanthropic donation to UC San Diego.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding. This work was supported by grants from the National Science Foundation (1927616), Department of Defense (W81XWH-16-0464), National Institutes of Health (R01DK119202, R01HL145470), the UCSD Center for Physiological Genomics of Low Oxygen (CPGLO), and American Heart Association (#19CDA34760244).
References
- Abraham N. G. (ed.) (2002). Heme oxygenase in biology and medicine. (New York: Springer Science & Business Media; ). Available at: https://www.springer.com/gp/book/9780306472640#aboutBook [Google Scholar]
- Allen K. N., Vázquez-Medina J. P. (2019). Natural tolerance to ischemia and hypoxemia in diving mammals: a review. Front. Physiol. 10:1199. 10.3389/fphys.2019.01199, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beall C. M. (2007). Two routes to functional adaptation: tibetan and Andean high-altitude natives. Proc. Natl. Acad. Sci. U. S. A. 104, 8655–8660. 10.1073/pnas.0701985104, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjafield A. V., Ayas N. T., Eastwood P. R., Heinzer R., Ip M. S. M., Morrell M. J., et al. (2019). Estimation of the global prevalence and burden of obstructive sleep apnoea: a literature-based analysis. Lancet Respir. Med. 7, 687–698. 10.1016/S2213-2600(19)30198-5, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blydt-Hansen T. D., Katori M., Lassman C., Ke B., Coito A. J., Iyer S., et al. (2003). Gene transfer-induced local heme oxygenase-1 overexpression protects rat kidney transplants from ischemia/reperfusion injury. J. Am. Soc. Nephrol. 14, 745–754. 10.1097/01.asn.0000050760.87113.25, PMID: [DOI] [PubMed] [Google Scholar]
- Carey H. V. (2015). Lessons learned from comparative and evolutionary physiology. Physiology 30, 80–81. 10.1152/physiol.00003.2015, PMID: [DOI] [PubMed] [Google Scholar]
- Carey H. V., Martin S. L., Horwitz B. A., Yan L., Bailey S. M., Podrabsky J., et al. (2012). Elucidating nature’s solutions to heart, lung, and blood diseases and sleep disorders. Circ. Res. 110, 915–921. 10.1161/CIRCRESAHA.111.255398, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin B. Y., Jiang G., Wegiel B., Wang H. J., MacDonald T., Zhang X. C., et al. (2007). Hypoxia-inducible factor 1α stabilization by carbon monoxide results in cytoprotective preconditioning. Proc. Natl. Acad. Sci. U. S. A. 104, 5109–5114. 10.1073/pnas.0609611104, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruse I., Maines M. D. (1988). Evidence suggesting that the two forms of heme oxygenase are products of different genes. J. Biol. Chem. 263, 3348–3353. PMID: [PubMed] [Google Scholar]
- D’Amico G., Lam F., Hagen T., Moncada S. (2006). Inhibition of cellular respiration by endogenously produced carbon monoxide. J. Cell Sci. 119, 2291–2298. 10.1242/jcs.02914, PMID: [DOI] [PubMed] [Google Scholar]
- Drager L. F., Polotsky V. Y., O’Donnell C. P., Cravo S. L., Lorenzi-Filho G., Machado B. H. (2015). Translational approaches to understanding metabolic dysfunction and cardiovascular consequences of obstructive sleep apnea. Am. J. Physiol. Heart Circ. Physiol. 309, H1101–H1111. 10.1152/ajpheart.00094.2015, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser S. T., Midwinter R. G., Coupland L. A., Kong S., Berger B. S., Yeo J. H., et al. (2015). Heme oxygenase-1 deficiency alters erythroblastic island formation, steady-state erythropoiesis and red blood cell lifespan in mice. Haematologica 100, 601–610. 10.3324/haematol.2014.116368, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebel U., Wollborn J. (2020). Carbon monoxide in intensive care medicine-time to start the therapeutic application?! Intensive Care Med. Exp. 8:2. 10.1186/s40635-020-0292-8, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halasz N. A., Elsner R., Garvie R. S., Grotke G. T. (1974). Renal recovery from ischemia: a comparative study of harbor seal and dog kidneys. Am. J. Phys. 227, 1331–1335. 10.1152/ajplegacy.1974.227.6.1331, PMID: [DOI] [PubMed] [Google Scholar]
- Hardison R. C. (1996). A brief history of hemoglobins: plant, animal, protist, and bacteria. Proc. Natl. Acad. Sci. U. S. A. 93, 5675–5679. 10.1073/pnas.93.12.5675, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera E. A., Reyes R. V., Giussani D. A., Riquelme R. A., Sanhueza E. M., Ebensperger G., et al. (2008). Carbon monoxide: a novel pulmonary artery vasodilator in neonatal llamas of the Andean altiplano. Cardiovasc. Res. 77, 197–201. 10.1093/cvr/cvm013, PMID: [DOI] [PubMed] [Google Scholar]
- Hopper C. P., Meinel L., Steiger C., Otterbein L. E. (2018). Where is the clinical breakthrough of heme oxygenase-1/carbon monoxide therapeutics? Curr. Pharm. Des. 24, 2264–2282. 10.2174/1381612824666180723161811, PMID: [DOI] [PubMed] [Google Scholar]
- Kapturczak M. H., Wasserfall C., Brusko T., Campbell-Thompson M., Ellis T. M., Atkinson M. A., et al. (2004). Heme oxygenase-1 modulates early inflammatory responses: evidence from the heme oxygenase-1-deficient mouse. Am. J. Pathol. 165, 1045–1053. 10.1016/S0002-9440(10)63365-2, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M., Miyazawa N., Takeno M., Murakami S., Kirino Y., Okouchi A., et al. (2008). Circulating carbon monoxide level is elevated after sleep in patients with obstructive sleep apnea. Chest 134, 904–910. 10.1378/chest.07-2904, PMID: [DOI] [PubMed] [Google Scholar]
- Konrad F. M., Knausberg U., Höne R., Ngamsri K. -C., Reutershan J. (2016). Tissue heme oxygenase-1 exerts anti-inflammatory effects on LPS-induced pulmonary inflammation. Mucosal Immunol. 9, 98–111. 10.1038/mi.2015.39, PMID: [DOI] [PubMed] [Google Scholar]
- Kozakowska M., Pietraszek-Gremplewicz K., Ciesla M., Seczynska M., Bronisz-Budzynska I., Podkalicka P., et al. (2018). Lack of heme oxygenase-1 induces inflammatory reaction and proliferation of muscle satellite cells after cardiotoxin-induced skeletal muscle injury. Am. J. Pathol. 188, 491–506. 10.1016/j.ajpath.2017.10.017, PMID: [DOI] [PubMed] [Google Scholar]
- Krogh A. (1929). The progress of physiology. Science 70, 200–204. 10.1126/science.70.1809.200, PMID: [DOI] [PubMed] [Google Scholar]
- Lee P. J., Jiang B. H., Chin B. Y., Iyer N. V., Alam J., Semenza G. L., et al. (1997). Hypoxia-inducible factor-1 mediates transcriptional activation of the heme oxygenase-1 gene in response to hypoxia. J. Biol. Chem. 272, 5375–5381. 10.1074/jbc.272.9.5375, PMID: [DOI] [PubMed] [Google Scholar]
- Li C., Stocker R. (2009). Heme oxygenase and iron: from bacteria to humans. Redox Rep. 14, 95–101. 10.1179/135100009x392584, PMID: [DOI] [PubMed] [Google Scholar]
- Liu X., Wei J., Peng D. H., Layne M. D., Yet S. -F. (2005). Absence of heme oxygenase-1 exacerbates myocardial ischemia/reperfusion injury in diabetic mice. Diabetes 54, 778–784. 10.2337/diabetes.54.3.778, PMID: [DOI] [PubMed] [Google Scholar]
- Llanos A. J., Ebensperger G., Herrera E. A., Reyes R. V., Cabello G., Díaz M., et al. (2012). The heme oxygenase-carbon monoxide system in the regulation of cardiorespiratory function at high altitude. Respir. Physiol. Neurobiol. 184, 186–191. 10.1016/j.resp.2012.05.003, PMID: [DOI] [PubMed] [Google Scholar]
- Logan S. M., Szereszewski K. E., Bennett N. C., Hart D. W., van Jaarsveld B., Pamenter M. E., et al. (2020). The brains of six African mole-rat species show divergent responses to hypoxia. J. Exp. Biol. 223:jeb215905. 10.1242/jeb.215905, PMID: [DOI] [PubMed] [Google Scholar]
- Marin J. M., Soriano J. B., Carrizo S. J., Boldova A., Celli B. R. (2010). Outcomes in patients with chronic obstructive pulmonary disease and obstructive sleep apnea: the overlap syndrome. Am. J. Respir. Crit. Care Med. 182, 325–331. 10.1164/rccm.200912-1869OC, PMID: [DOI] [PubMed] [Google Scholar]
- McDonald B. I., Ponganis P. J. (2012). Lung collapse in the diving sea lion: hold the nitrogen and save the oxygen. Biol. Lett. 8, 1047–1049. 10.1098/rsbl.2012.0743, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J. J. (1992). Effects of altitude on endogenous carboxyhemoglobin levels. J. Toxicol. Environ. Health 35, 127–133. 10.1080/15287399209531601, PMID: [DOI] [PubMed] [Google Scholar]
- McGrath J. J., Schreck R. M., Lee P. S. (1993). Carboxyhemoglobin levels in humans: effects of altitude. Inhal. Toxicol. 5, 241–249. 10.3109/08958379309034504 [DOI] [Google Scholar]
- Meir J. U., Champagne C. D., Costa D. P., Williams C. L., Ponganis P. J. (2009). Extreme hypoxemic tolerance and blood oxygen depletion in diving elephant seals. Am. J. Phys. Regul. Integr. Comp. Phys. 297, R927–R939. 10.1152/ajpregu.00247.2009, PMID: [DOI] [PubMed] [Google Scholar]
- Motterlini R., Otterbein L. E. (2010). The therapeutic potential of carbon monoxide. Nat. Rev. Drug Discov. 9, 728–743. 10.1038/nrd3228, PMID: [DOI] [PubMed] [Google Scholar]
- Nakahira K., Kim H. P., Geng X. H., Nakao A., Wang X., Murase N., et al. (2006). Carbon monoxide differentially inhibits TLR signaling pathways by regulating ROS induced trafficking of TLRs to lipid rafts. J. Exp. Med. 203, 2377–2389. 10.1084/jem.20060845, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakao A., Toyokawa H., Tsung A., Nalesnik M. A., Stolz D. B., Kohmoto J., et al. (2006). Ex vivo application of carbon monoxide in University of Wisconsin solution to prevent intestinal cold ischemia/reperfusion injury. Am. J. Transplant. 6, 2243–2255. 10.1111/j.1600-6143.2006.01465.x, PMID: [DOI] [PubMed] [Google Scholar]
- Otterbein L. E., Foresti R., Motterlini R. (2016). Heme oxygenase-1 and carbon monoxide in the heart: the balancing act between danger signaling and pro-survival. Circ. Res. 118, 1940–1959. 10.1161/CIRCRESAHA.116.306588, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens R. L., Yim-Yeh S., Malhotra A. (2008). Carbon monoxide poisoning, or carbon monoxide protection? Chest 134, 895–896. 10.1378/chest.08-1728, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabst D. A., McLellan W. A., Rommel S. A. (2016). How to build a deep diver: the extreme morphology of mesoplodonts. Integr. Comp. Biol. 56, 1337–1348. 10.1093/icb/icw126, PMID: [DOI] [PubMed] [Google Scholar]
- Pette D., Staron R. S. (2001). Transitions of muscle fiber phenotypic profiles. Histochem. Cell Biol. 115, 359–372. 10.1007/s004180100268, PMID: [DOI] [PubMed] [Google Scholar]
- Pilegaard H., Ordway G. A., Saltin B., Darrell Neufer P. (2000). Transcriptional regulation of gene expression in human skeletal muscle during recovery from exercise. Am. J. Physiol. Endocrinol. Metab. 279, E806–E814. 10.1152/ajpendo.2000.279.4.E806, PMID: [DOI] [PubMed] [Google Scholar]
- Platt J. L., Nath K. A. (1998). Heme oxygenase: protective gene or Trojan horse. Nat. Med. 4, 1364–1365. 10.1038/3947, PMID: [DOI] [PubMed] [Google Scholar]
- Prabhakar N. R. (2012). Carbon monoxide (CO) and hydrogen sulfide (H(2)S) in hypoxic sensing by the carotid body. Respir. Physiol. Neurobiol. 184, 165–169. 10.1016/j.resp.2012.05.022, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh L. G. (1959). Carbon monoxide content of the blood and other observations on Weddell seals. Nature 183, 74–76. 10.1038/183074a0, PMID: [DOI] [PubMed] [Google Scholar]
- Reid M. B. (2001). Nitric oxide, reactive oxygen species, and skeletal muscle contraction. Med. Sci. Sports Exerc. 33, 371–376. 10.1097/00005768-200103000-00006, PMID: [DOI] [PubMed] [Google Scholar]
- Roughton F. J. W., Darling R. C. (1944). The effect of carbon monoxide on the oxyhemoglobin dissociation curve. Am. J. Phys. 141, 737–737. 10.1152/ajplegacy.1944.141.5.737-s [DOI] [Google Scholar]
- Rushworth S. A., O’Connell M. A. (2004). Haem oxygenase-1 in inflammation. Biochem. Soc. Trans. 32, 1093–1094. 10.1042/BST0321093, PMID: [DOI] [PubMed] [Google Scholar]
- Ryter S. W., Kim H. P., Nakahira K., Zuckerbraun B. S., Morse D., Choi A. M. K. (2007). Protective functions of heme oxygenase-1 and carbon monoxide in the respiratory system. Antioxid. Redox Signal. 9, 2157–2173. 10.1089/ars.2007.1811, PMID: [DOI] [PubMed] [Google Scholar]
- Sánchez-de-la-Torre A., Soler X., Barbé F., Florés M., Maisel A., Malhotra A., et al. (2018). Cardiac troponin values in patients with acute coronary syndrome and sleep apnea: a pilot study. Chest 153, 329–338. 10.1016/j.chest.2017.06.046, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena S., Shukla D., Saxena S., Khan Y. A., Singh M., Bansal A., et al. (2010). Hypoxia preconditioning by cobalt chloride enhances endurance performance and protects skeletal muscles from exercise-induced oxidative damage in rats. Acta Physiol. 200, 249–263. 10.1111/j.1748-1716.2010.02136.x, PMID: [DOI] [PubMed] [Google Scholar]
- Schaaf-Lafontaine N., Courtoy R. (1986). Transfer into the mouse of mast cells differentiated in vitro. C. R. Seances Soc. Biol. Fil. 180, 234–242. PMID: [PubMed] [Google Scholar]
- Schallner N., Otterbein L. E. (2015). Friend or foe? Carbon monoxide and the mitochondria. Front. Physiol. 6:17. 10.3389/fphys.2015.00017, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selland M. A., Stelzner T. J., Stevens T., Mazzeo R. S., McCullough R. E., Reeves J. T. (1993). Pulmonary function and hypoxic ventilatory response in subjects susceptible to high-altitude pulmonary edema. Chest 103, 111–116. 10.1378/chest.103.1.111, PMID: [DOI] [PubMed] [Google Scholar]
- Shibahara S., Han F., Li B., Takeda K. (2007). Hypoxia and heme oxygenases: oxygen sensing and regulation of expression. Antioxid. Redox Signal. 9, 2209–2225. 10.1089/ars.2007.1784, PMID: [DOI] [PubMed] [Google Scholar]
- Shih R. -H., Cheng S. -E., Hsiao L. -D., Kou Y. R., Yang C. -M. (2011). Cigarette smoke extract upregulates heme oxygenase-1 via PKC/NADPH oxidase/ROS/PDGFR/PI3K/Akt pathway in mouse brain endothelial cells. J. Neuroinflammation 8:104. 10.1186/1742-2094-8-104, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonson T. S. (2015). Altitude adaptation: a glimpse through various lenses. High Alt. Med. Biol. 16, 125–137. 10.1089/ham.2015.0033, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonson T. S., Yang Y., Huff C. D., Yun H., Qin G., Witherspoon D. J., et al. (2010). Genetic evidence for high-altitude adaptation in Tibet. Science 329, 72–75. 10.1126/science.1189406, PMID: [DOI] [PubMed] [Google Scholar]
- Soler X., Gaio E., Powell F. L., Ramsdell J. W., Loredo J. S., Malhotra A., et al. (2015). High prevalence of obstructive sleep apnea in patients with moderate to severe chronic obstructive pulmonary disease. Ann. Am. Thorac. Soc. 12, 1219–1225. 10.1513/AnnalsATS.201407-336OC, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangenburg E. E., Le Roith D., Ward C. W., Bodine S. C. (2008). A functional insulin-like growth factor receptor is not necessary for load-induced skeletal muscle hypertrophy. J. Physiol. 586, 283–291. 10.1113/jphysiol.2007.141507, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storz J. F. (2010). Evolution. Genes for high altitudes. Science 329, 40–41. 10.1126/science.1192481, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storz J. F. (2016). Hemoglobin-oxygen affinity in high-altitude vertebrates: is there evidence for an adaptive trend? J. Exp. Biol. 219, 3190–3203. 10.1242/jeb.127134, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strocchi A., Schwartz S., Ellefson M., Engel R. R., Medina A., Levitt M. D. (1992). A simple carbon monoxide breath test to estimate erythrocyte turnover. J. Lab. Clin. Med. 120, 392–399. PMID: [PubMed] [Google Scholar]
- Suliman H. B., Carraway M. S., Ali A. S., Reynolds C. M., Welty-Wolf K. E., Piantadosi C. A. (2007). The CO/HO system reverses inhibition of mitochondrial biogenesis and prevents murine doxorubicin cardiomyopathy. J. Clin. Invest. 117, 3730–3741. 10.1172/JCI32967, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tift M. S., Ponganis P. J. (2019). Time domains of hypoxia adaptation-elephant seals stand out among divers. Front. Physiol. 10:677. 10.3389/fphys.2019.00677, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tift M. S., Ponganis P. J., Crocker D. E. (2014). Elevated carboxyhemoglobin in a marine mammal, the northern elephant seal. J. Exp. Biol. 217, 1752–1757. 10.1242/jeb.100677, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umeda A., Miyagawa K., Mochida A., Takeda H., Takeda K., Okada Y., et al. (2020). Intermittent hypoxia, energy expenditure, and visceral adipocyte recovery. Respir. Physiol. Neurobiol. 273:103332. 10.1016/j.resp.2019.103332, PMID: [DOI] [PubMed] [Google Scholar]
- Wegiel B., Nemeth Z., Correa-Costa M., Bulmer A. C., Otterbein L. E. (2014). Heme oxygenase-1: a metabolic Nike. Antioxid. Redox Signal. 20, 1709–1722. 10.1089/ars.2013.5667, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilks A. (2002). Heme oxygenase: evolution, structure, and mechanism. Antioxid. Redox Signal. 4, 603–614. 10.1089/15230860260220102, PMID: [DOI] [PubMed] [Google Scholar]
- Yang D., Peng Y., Ouzhuluobu, Bianbazhuoma, Cui C., Bianba, et al. (2016). HMOX2 functions as a modifier gene for high-altitude adaptation in Tibetans. Hum. Mutat. 37, 216–223. 10.1002/humu.22935, PMID: [DOI] [PubMed] [Google Scholar]
- Yoshida T., Maulik N., Ho Y. S., Alam J., Das D. K. (2001). H(mox-1) constitutes an adaptive response to effect antioxidant cardioprotection: a study with transgenic mice heterozygous for targeted disruption of the heme oxygenase-1 gene. Circulation 103, 1695–1701. 10.1161/01.CIR.103.12.1695, PMID: [DOI] [PubMed] [Google Scholar]
- Zapol W. M., Liggins G. C., Schneider R. C., Qvist J., Snider M. T., Creasy R. K., et al. (1979). Regional blood flow during simulated diving in the conscious Weddell seal. J. Appl. Physiol. 47, 968–973. 10.1152/jappl.1979.47.5.968, PMID: [DOI] [PubMed] [Google Scholar]
- Zhang R., Pan K., Hao Y., Yip C., Ko W. (2019). Anti-inflammatory action of HO-1/CO in human bronchial epithelium in response to cationic polypeptide challenge. Mol. Immunol. 105, 205–212. 10.1016/j.molimm.2018.12.002, PMID: [DOI] [PubMed] [Google Scholar]
- Zurlo F., Larson K., Bogardus C., Ravussin E. (1990). Skeletal muscle metabolism is a major determinant of resting energy expenditure. J. Clin. Invest. 86, 1423–1427. 10.1172/JCI114857, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.