

Abstract
Heparins are the most pharmaceutically important polysaccharides. These heparin-based anticoagulant/antithrombotic agents include unfractionated heparins, low molecular weight heparins (LMWHs) and ultralow molecular weight heparins (ULMWHs). Heparins exhibit their pharmacological and biological activities through interaction with heparin-binding proteins. The prototypical heparin-binding protein is antithrombin III (AT), responsible for heparin’s anticoagulant/antithrombotic activity. This study describes a filter-trapping method to isolate the chains in enoxaparin, a LMWH, which bind to AT. We demonstrate this method using the ULMWH, fondaparinux, which consists of a single well defined AT binding site. The interacting chains of enoxaparin are then characterized by activity assays, top-down liquid chromatography-mass spectrometry, and capillary zone electrophoresis mass spectrometry. This filter-trapping assay is an improvement over affinity chromatography for isolating heparin chains interacting with heparin binding proteins.
Keywords: heparin-binding proteins, filter trapping, capillary electrophoresis, mass spectrometry, heparin, antithrombin III
1. Introduction
Glycosaminoglycans (GAGs) are linear polysaccharides that are ubiquitously found at the cell surface and in the extracellular matrix of animal cells (Listik et al., 2019). The structural diversity of GAGs not only comes from various disaccharide-repeating units but also from differential sulfation of the individual monosaccharides (DeAngelis, Liu, & Linhardt, 2013). The diverse structural motifs, their polydispersity and their non-template biosynthesis makes structural analysis of GAGs quite difficult (Mende et al., 2016). GAGs exhibit numerous biological activities through their interaction with a diverse collection of proteins (Capila & Linhardt, 2002). The structure-function relationships of GAGs are still poorly understood due to their structural complexity, primarily the slow development of methods for structural characterization. Profiling disease-associated GAGs is essential for the understanding of their functions at a molecular level, while also facilitating the identification of diagnostic biomarkers and the better design of therapeutic drugs. Developing a method that can selectively assess the activities of GAGs is an attractive approach. One that examines the interaction between heparin and various proteins could play an important role in the regulation of normal physiological and pathophysiological processes leading to the application of heparin outside its normal role as an anticoagulant /antithrombotic agent (Liu, Wang, Yu, Chen, & He, 2017; Yan et al., 2017).
Low molecular weight heparins (LMWHs) are anticoagulant drugs derived from unfractionated heparin (UFH) through controlled chemical or enzymatic depolymerization. LMWHs have a reduced incidence of side effects such as heparin-induced thrombocytopenia (HIT) (Junqueira, Perini, Penholati, & Carvalho, 2012). In addition, LMWHs exhibit prolonged half-lives and a more predictable pharmacological response (Garcia, Baglin, Weitz, & Samama, 2012). Enoxaparin (Lovenox®) the most widely used LMWH is prepared through benzyl esterification and alkaline depolymerization of UFH. Due to this manufacturing process, enoxaparin has unsaturated uronic acid residues (ΔUA, 4-deoxy-α-L-threo-hex-4-eno-pyranosyl uronic acid) at most of its non-reducing ends and 1,6-anhydro aminosugars at a small fraction of its reducing ends. LMWH inhibits coagulation by binding to and activating antithrombin III (AT), which then binds to and inhibits coagulation proteases factor IIa and factor Xa (thrombin) (Onishi, St. Ange, Dordick, & Linhardt, 2016). UFH has a sufficiently long chain to bind both AT and thrombin affording a ternary complex, inactivating thrombin and, thus, preventing fibrin clot formation. FXa does not interact directly with heparin but instead is inhibited by heparin-AT binary complex. Unlike UFH, which has a relatively long chain, LMWH has a shorter chain (<15 saccharide units) making it capable of only binding AT and inactivating factor Xa. Thus, LMWHs are considered factor Xa-selective anticoagulant/antithrombotic drugs.
Specific recognition between GAGs and GAG-binding proteins requires both defined GAG sequences and approximate complementary peptide sequences. The general structural requirements for proteins involved in GAG-protein interactions results from motifs having appropriately spaced basic amino acid residues on the protein surface (Cardin & Weintraub, 1989; Hileman, Fromm, Weiler, & Linhardt, 1998). Many GAG-binding proteins have been identified, and their binding sites have been analyzed (Capila & Linhardt, 2002; Gandhi & Mancera, 2008; Kjellén & Lindahl, 2018; Xu & Esko, 2014). The prototypical example of a defined GAG motif for protein binding is a pentasaccharide sequence found in the anticoagulant drug, heparin (Lindahl et al., 1979; Rosenberg, Armand, & Lan, 1978). Since GAGs are highly heterogeneous, it is often difficult to study the specific structural requirements for individual GAG-protein interactions, such as the GAG type, chain length, and sulfation pattern. Pull-down assays are a well-established approach for studying protein-protein and protein-nuclear acid interactions (Jain, Liu, Xiang, & Ha, 2012; Wu, 2006) but these have not been widely applied to GAG-protein interactions (Xu & Esko, 2014). AT has been coupled to CNBr activated Sepharose to obtain an affinity column to fractionate LMWH into high affinity and low affinity pools (Bisio et al., 2009). Free AT was used for first time by Viskov and coworkers to bind active oligosaccharides to assess the interacting species by LC-MS (Viskov et al., 2013). Surface non-covalent affinity mass spectrometry (SNA-MS) has been used to directly isolate, enrich, and sequence GAGs binding to specific proteins (Keiser, Venkataraman, Shriver, & Sasisekharan, 2001). Surface plasmon resonance imaging (SPRi) has recently been coupled with MALDI-TOF MS for the structural determination of heparin and heparin sulfate interaction with various cytokines (Przybylski, Gonnet, Saesen, Lortat-Jacob, & Daneil, 2020). Microarrays of oligosaccharides probed with fluorescently labeled heparin binding proteins are now widely used in the study of GAG-protein interactions (Jose, Noti, & Seeberger, 2006; Shipp & Hsieh-Wilson, 2007; Yang et al., 2017).
In this work we explore the use of a filter-entrapment enrichment method to prepare GAGs and GAG oligosaccharides with low and high affinity for AT and to examine their structure using capillary zone electrophoresis (CZE)-tandem mass spectrometry (MS/MS). This study focuses on differences in anticoagulant activity, AT-binding, and other structural features of the isolated high and low affinity chains. Finally, we establish an oligosaccharide-protein pull-down assay that can serve as a rapid and moderate-throughput analytical platform for the functional screening of oligosaccharides with affinity for a variety of different GAG-binding proteins.
2. Materials and methods
2.1. Materials
Enoxaparin LMWH standard was obtained from the United States Phaemacopeia (USP, Rockville, MD). Arixtra® (fondaparinux, sodium) was purchased from AuroMedics Pharma LLC (East Windsor, NJ). Human antithrombin III (AT) and platelet factor 4 (PF4) were purchased from Hyphen BioMed (Neuville-sur-Oise, France). Molecular weight cut membrane (50 kDa) was purchased from GE healthcare (Pittsburgh, PA). Recombinant Flavobacterium heparinum heparin lyase I, II, III (EC Nots. 4.2.2.7, 4.2.2.X, and 4.2.2.8, respectively) were expressed in Escherichia coli and purified in our laboratory as previously described (Su et al., 1996). Unsaturated heparin disaccharide standards were purchased from Iduron (Manchester, UK). Biophen heparin anti-Xa (2 stages) and anti-IIa (2 stages) kits were purchased from Aniara (West Chester, OH, USA). Bare fused silica (BFS) Capillary Zone Electrophoresis (CZE) (360 mm o.d. × 50 mm i.d.) was purchased from PolyMicro Technologies (Phoenix, AZ).
2.2. Filter-entrapment enrichment for AT and fondaparinux/enoxaparin binding
The strategy of pull-down assay for the high AT affinity binding LMWH as accomplished using filter-entrapment enrichment method (Fig. 1) AT (375 μg) was reconstituted by dissolving in 0.1 mL of distilled water and mixed well with (100 μg) fondaparinux or enoxaparin in (1 mL) of distilled water, and loaded into the top chamber of a centrifugal spin tube with a 50 kDa molecular weight cut-off (MWCO). The mixture was placed in an ice-bath and chilled for 10 min. The tube was centrifuged at 1500 × g for 10 min and washed 3-times with 1 mL of distilled water at 4 °C. Next, 1 mL of 0.2 M NaCl was added to the top chamber and centrifuged under the same conditions 3-times and the permeate (low affinity fraction) was collected. Finally, the high affinity fraction was eluted by 3 washes (1 mL each) of 2 M NaCl using the same centrifuge conditions. The low and high affinity fractions were both desalted by dialysis using a 1 kDa membrane and lyophilized for further analysis.
Fig. 1.
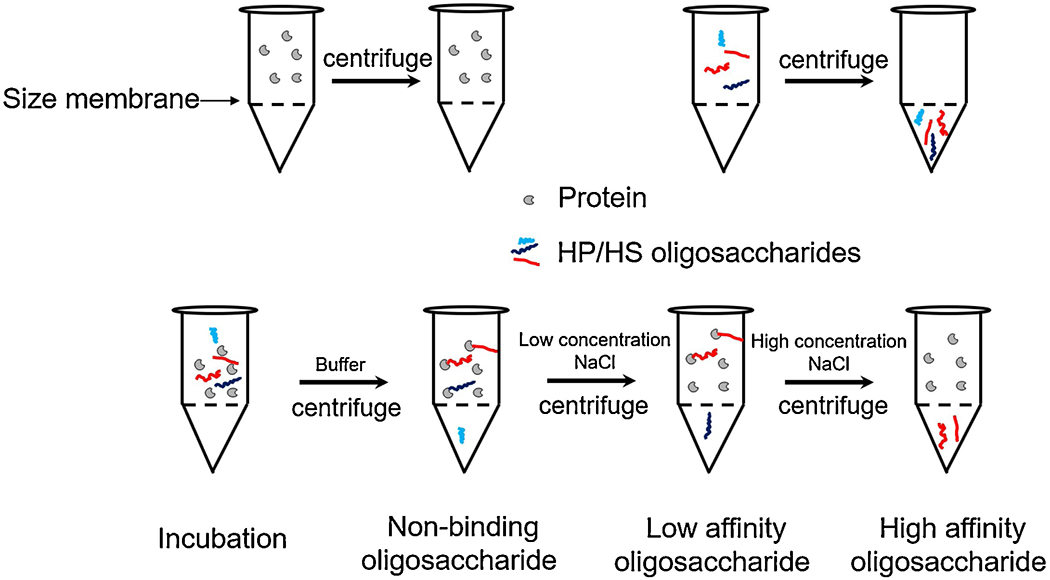
Scheme for filter-entrapment enrichment pull-down assay.
We detected AT protein by two methods. A NanoDrop UV-Vis spectrophotometer was used to detect protein at A280. The second, sensitive and semi-quantitative method used to detect protein was SDS-PAGE. We used different concentration of BSA as a standard and visualized the intensity with Image J software to calculate the concentration of AT.
2.3. Anticoagulant activity
The anti-Xa and anti-IIa activities of the high and low affinity fractions of LMWH were determined using BIOPHEN Heparin Anti-Xa (2 stages) and Anti-IIa (2 stages) kits following the protocols provided by the manufacturer. Briefly, AT (reagent 1), factor Xa (reagent 2Xa) and factor Xa specific chromogenic substrate (reagent 3Xa) were used for determining anti-Xa activity, and AT (regent 1), human thrombin (reagent 2IIa) and factor Xa (thrombin) specific chromogenic substrate (reagent 3IIa) were used for determining anti-IIa activity. Each reagent was reconstituted with 1 mL of distilled water and shaken until fully dissolved. After a 1/5 dilution in the appropriate buffer (Tris-EDTA-NaCl-PEG, pH 8.4) for reagent 1 and reagent 2Xa or 2IIa and distilled water for reagent 3Xa or 3IIa to restore the reagents immediately before use. Reference standard and dilute samples of LMWHs were prepared at the appropriate concentrations. LMWH samples (40 μL) were added into a 96-well plate and incubated for 5 min at 37 °C, 40 μL reagent 1 was added and mixed well and incubated for 2 min, 40 μL reagent 2Xa or 2IIa was next added and incubated for 2 min, 40 μL reagent 3Xa or 3IIa was added last and incubated for another 2 min. The reactions were stopped with adding 80 μL of 50 mM acetic acid. The absorbance was then determined at 405 nm. The anti-Xa and anti-IIa activities were calculated using a standard curve of different concentration of enoxaparin standards.
2.4. Surface plasmon resonance analysis
Biotinylated heparin prepared by conjugating the reducing end of heparin to amine-PEG3-Biotin (Pierce, Rockford, IL) (Kim et al., 2017) was immobilized to a streptavidin (SA)-coated chip based on the manufacturer’s protocol. Surface plasmon resonance (SPR) measurements were performed on a BIAcore 3000 instrument (GE, Uppsala, Sweden) operated using BIAcore 3000 control and BIAevaluation software (version 4.0.1). Solution competition studies between surface heparin and LMWH to measure IC50 was performed using SPR. AT (250 nM) or PF4 (125 nM) mixed with different concentration of high-affinity or low-affinity LMWHs in HBS-EP buffer (0.01 M HEPES, 0.15 M NaCl, 3 mM EDTA, 0.005% surfactant P20, pH 7.4) were injected over the heparin chip at a flow rate of 30 μL/min, respectively. After each run, dissociation and regeneration were performed using sequential injecting with 10 mM glycine-HCl (pH 2.5) and 2 M NaCl to obtain fully regenerated surface. For each set of competition experiments, a control experiment (only protein without LMWH) was performed to make certain that the surface was completely regenerated and that the results obtained between runs were comparable.
2.5. Disaccharides composition analysis
Disaccharides composition was determined by on-line reversed phase ion-pairing (RPIP) liquid chromatography mass spectrometry (LC-MS) performed on an Agilent 1200 LC/MSD instrument (Agilent Technologies, Inc., Wilmington, DE). Enoxaparin fractions (10 μg) were exhaustive digested using a mixture of heparin lyase I, II and III (10 mU each) in digestion buffer (50 mM ammonium acetate containing 2 mM calcium chloride, pH 7.0) at 37 °C overnight. The reaction was terminated by boiling for 10 min and the denatured enzymes were removed by centrifugation at 13,400 × g for 10 min. The supernatants were lyophilized and re-dissolved in water at 1 μg/μL for LC-MS analysis. LC was performed on an Agilent 1200 LC system at 45 °C using an Agilent Poroshell 120 EC-C18 (2.7 μm, 2.1 × 100 mm) column. Mobile phase A was 38 mM ammonium acetate and 12 mM tributylammonium acetate in 15% acetonitrile aqueous solution (pH 6.5), mobile phase B was 38 mM ammonium acetate and 12 mM tributylammonium acetate in 65% acetonitrile aqueous solution (pH 6.5). The mobile phase was passed through the column at a flow rate of 100 μL/min and gradient from 2% to 40% mobile phase B in 25 min, then rose to 60% in following 0.2 min, and a 5 min flow was applied to elute all compounds. MS was equipped with a 6300 ion trap, the parameters in negative-ion mode were set as follows: scan range 300-800 m/z, nebulizer 40 psi, dry gas 8L/min, dry temperature 350 °C.
2.6. Tetrasaccharide analysis using online RPIP LC-MS
Enoxaparin (50 μg) was digested using heparinase II (10 mU) in digestion buffer at 37 °C for 2 h to obtain resistant tetrasaccharides. The LC-MS method parameters and conditions were same as used in the disaccharide analysis described except the flow rate was 120 μL/min and gradient was from 2% to 30% of mobile phase B in 40 min, then rose to 60% for the following 15 min.
2.7. Tetrasaccharide analysis by CZE-MS analysis
Bare fused silica was etched with concentrated hydrofluoric acid (HF) at one end to reduce the other diameter of the capillary for use in the sheath flow CE interface described below. For the etching process, the outlet of the capillary was placed in concentrated HF for 45-60 min. The capillary tip was then washed profusely with water. Then, the etched capillary was coated with Aminomethyltriethoxysilane (AHS) to render a cation coated capillary (Sanderson et al., 2018). Coating solutions were prepared in toluene with 1% concentration of either AHS. To clean and prepare for coating, the capillary was rinsed with 0.1 M NaOH, water, methanol, dry acetone, and dry toluene, respectively, for 30 min each. The capillary was then coated by 1% AHS for 1 h. The capillary was consecutively flushed with dry toluene, dry acetone, and methanol for 30 min to remove excess coating solution. Finally, the capillary was equilibrated with background electrolyte buffer (BGE, 25 mM ammonium acetate in 70% methanol) for 1 h. Once degradation becomes apparent, BFS capillary can be easily cleaned by flushing sodium hydroxide for a short time; however, the coatings are stripped in basic conditions and must be reapplied by repeating the coating procedure. In some experiments. 0.1-1% formic acid (FA) or 0.02–0.1% diethylamine (DEA) was added to the BGE.
CZE separations were conducted with an Agilent HP 3D capillary electrophoresis instrument (Wilmington, DE). A base fused silica capillary, as described above, was used for CZE of GAG analyte. The total length of the capillary ranged from 52 to 60 cm, and its inner diameter was 50 μm with a volume of approximately 1 μL. The aqueous GAG sample was injected for 3 s at 950 mbar followed by a BGE injection for 10 s at 10 mbar. The injected volume was 0.1 nL. The ionic strength of the injected sample plug is 2-3 orders of magnitude less than that of the background electrolyte, and samples tacking is expected under these conditions which provides as sharp sample front. The capillary was then placed into a BGE vial for separation. For most experiments, a separation voltage of −30 kV was applied to the capillary. An EMASS-II (CMP Scientific, Brooklyn, NY) CE-MS interface was employed to couple the CE with a Thermo Scientific Velos Orbitrap Elite mass spectrometer (Bremen, Germany). The etched capillary outlet was inserted inside a cation-coated glass emitter tip with a 30 μm orifice (CMP Scientific, Brooklyn, NY). The etched capillary was positioned 0.3-0.5 mm from the end of the emitter orifice to create a mixing volume of approximately 15 nL, and the end of the emitter was filled with sheath liquid (SL, 25 mM ammonium acetate 70% MeOH). An external power supply provided the transmitter with a nanoelectrospray voltage (nESI) ranging from −1.7 to −1.85 kV. MS detection was performed in negative ion mode. Prior to CZE-MS experiments, a semi-automatic optimization of source parameters was performed using sucrose octasulfate to improve sensitivity of sulfated GAGs and reduce sulfate decomposition during ion transfer prior to MS analysis. The Orbitrap was scanned from m/z 150-2000 to detect oligosaccharides in the GAGs with a specified resolution of 120,000.
Tandem mass spectrometry was performed using negative electron transfer dissociation (NETD), as described previously (Leach, Riley, Westphall, Coon, & Amster, 2017). Fluoranthene (Sigma-Aldrich, St Louis, MO) served as the reagent ion. Fluoranthene radical cations were produced by electron ionization, and allowed to react with GAG anions in the dual linear ion trap.
2.8. Enoxaparin oligosaccharide mapping
Online hydrophilic interaction chromatography (HILIC) ESI-LTQ-Orbitrap-FTMS (ThermoFisher Scientific) was performed to analyze enoxaparin oligosaccharides as previously described (Li, Zhang, Zaia, & Linhardt, 2012). Briefly, a Luna HILIC column (2.0 mm × 150 mm, 200 Å, Phenomenex, Torrance, CA) was used for separation. Mobile phase A (MPA) was 5 mM ammonium acetate with HPLC water. Mobile phase B (MPB) was 5 mM ammonium acetate prepared in 98% acetonitrile with 2% of HPCL water. HPLC binary pump was used to deliver the gradient form 10% MPA to 35% in 40 min at a flow rate of 150 μL/min. Samples were prepared at 1 mg/mL using 10% MPA and the injection volume was 8 μL. Data was processed by a series of software. Charge deconvolution was autoprocessed by DeconTools software. Enoxaparin oligosaccharide structural assignments were performed by automatic processing using GlycReSoft 1.0 software (Maxwell et al., 2012). The compositional data were further compared using GlycCompSoft (Wang et al., 2016) after matching with the hypothesis generated in GlycReSoft.
3. Results and discussion
3.1. Filter-enrichment entrapment of fondaparinux and AT
GAGs have important biological activities mediated by their interaction with a diverse collection of GAG-binding proteins. Unfortunately, structural requirements for most GAG-protein interactions are not well-characterized. We set out to build a medium throughput, generally applicable method for screening the functional binding of oligosaccharides to different proteins. Our approach relies on a simple filter-entrapment enrichment pull-down method. A molecular weight cut-off membrane was selected through which oligosaccharides could pass but in which protein would be retained for use as an enrichment filter. No AT was lost in these studies due to adsorption to the membrane as all of the AT added could be detected in the solution above the membrane even after multiple uses. GAG interaction with GAG-binding proteins is primarily driven through ionic interactions (Capila & Linhardt, 2002; Hileman, Jennings, & Linhardt, 1998). Oligosaccharides with high binding affinity should be retained by the membrane together with protein through ionic interactions. Non-binding and excess oligosaccharides are first washed through the membrane, then the low affinity oligosaccharides are eluted using a low concentration sodium chloride and, finally, high affinity oligosaccharide are recovered by elution with a high concentration sodium chloride (Fig. 1).
We first tested the feasibility of this method by examining fondaparinux (Arixtra®) AT binding. (Fondaparinux is a well-studied homogeneous pentasaccharide having a molecular weight 1507 Da and the sequence α-D-GlcNAc6S(1→4)-β-D-GlcA(1→4)-α-D-GlcNS3S6S(1→4)-α-L-IdoA2S(1→4)-α-D-GlcNS6S-O-methyl glycoside (where GlcA is glucuronic acid, GlcNAc is N-acetyl glucosamine, IdoA is iduronic acid, and S is sulfo) and widely used in clinical as an anticoagulant drug due to its tight binding to AT (Chang et al., 2014). A molecular weight cut-off of 50 kDa membrane was chosen since AT has a molecular weight of 58 kDa. AT was retained in the top chamber of the centrifugal spin tube and remain active for up to 7 days at low temperature (4 °C) under low centrifugal force (1500 × g). More than 99.9% percent of fondaparinux could be eluted through the membrane of the centrifugal spin tube at the same conditions. In filter enrichment fondaparinux and AT were mixed and incubated for 10 min, no fondaparinux was detected by LC-MS in wash, but it was eluted in 2 M NaCl elution step (Fig. 2). When thermally inactivated AT was used with fondaparinux as a negative control, the results showed that all of the fondaparinux eluted in the wash and no fondaparinux was detected in 2 M NaCl elution step.
Fig. 2.
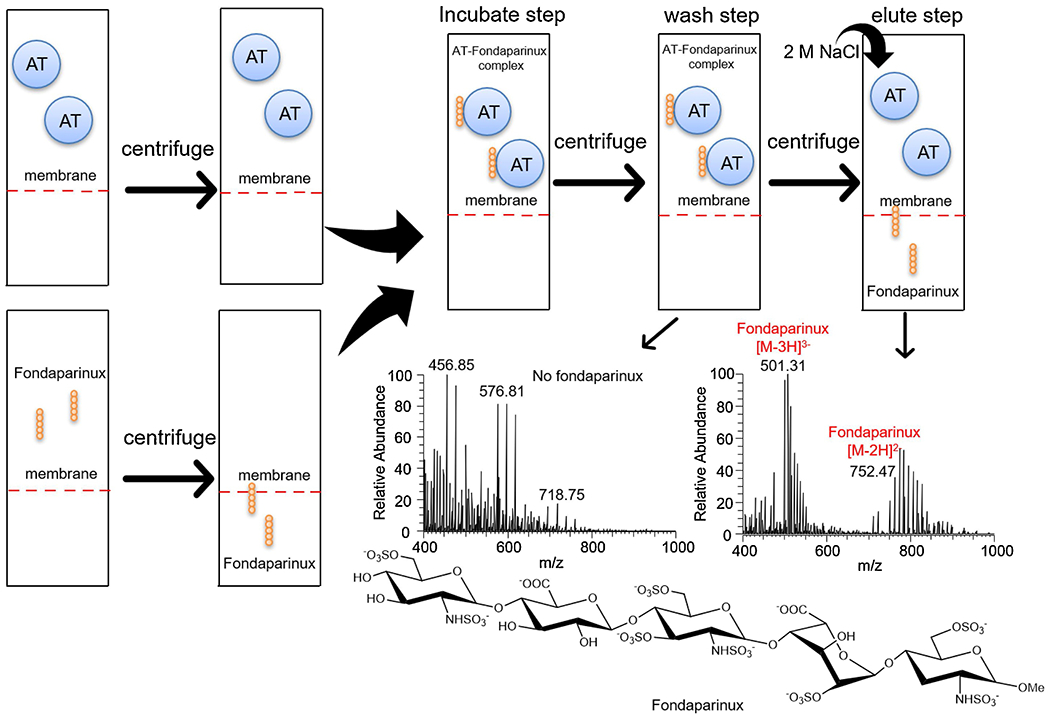
Pull-down method for fondaparinux and AT binding. The monoisotopic peak of fondaparinux was determined to be 1506.95 Da showed as 501.31 (z = 3) and 752.47 (z = 2), sodium adducts and desulfation fractions were also observed in the cluster of peaks.
3.2. Low molecular weight heparin and AT binding
A similar procedure of filter-entrapment enrichment pull-down assay was used to fractionate the LMWH, enoxaparin with AT except for the addition of another step, elution with 0.2 M NaCl in order to obtain a low-affinity fraction prior to the 2 M NaCl elution. The results showed that after 10-times cycles of pull-down assay using 375 μg AT protein we obtained 430 μg of a low affinity enoxaparin fraction and 138 μg of a high affinity fraction as determined using a micro-carbazole colorimetric heparin assay. These two fractions were desalted using a 1 kDa dialysis membrane and lyophilized for activity analysis and structural characterization. Previous studies showed that enoxaparin contained only about 12% high affinity chains (Lin, Sinha, & Betz, 2001).
Difficulties in structural characterization of GAGs and evaluating their biological role is partly ascribed to an incomplete understanding of carbohydrate-protein interactions (Capila & Linhardt, 2002). Progress in preparation of pure, structurally defined oligosaccharide (Zhang et al., 2017) and the rapid development of mass spectrometry (Persson, Vorontsov, Larson, & Nilsson, 2020; Solakyildirim, 2019) has led to an improved understanding of the precise structural requirements for GAG interaction with proteins. We initially considered using AT immobilized to magnetic nanoparticles in our pull-down assay. Proteins are generally much less stable than GAGs and most research has focused on the identification of GAG-binding proteins. Protein can lose most of their activity during immobilization to resins or nanoparticles. Based on our research, the immobilization of AT on magnetic nanoparticles generally retains only 5~10% of AT activity and the capacity of magnetic nanoparticles for AT relatively low resulting in only 10 μg of active immobilized AT per 1 mg magnetic nanoparticles. Only one heparin-binding site is present in each 58 kDa AT. Thus, 10 μg of AT immobilized on 1 mg of magnetic nanoparticles would bind to less than 1 μg of a 4 kDa LMWH chain. We next considered using preparative SPR to enrich high affinity chains of LMWH. Preparative SPR has been reported to be useful in enriching proteins in protein-protein interaction and was compatible with MALDI-TOF MS for protein identification (Przybylski et al., 2020). Unfortunately, in the case of GAG-protein interactions, the immobilization capacity of protein in all four channels of an SPR chip was < 10 μg. Thus, the maximum amount of high affinity LMWH chains that could be isolated in each cycle was < 20 ng. Although this approach requires a very small amount of protein, the high affinity LMWH obtained would be insufficient for structural analysis even using many purification cycles. In our filter entrapment method, no protein immobilization is required and, therefore, all the protein used should be active, resulting in a 10-fold enhancement of recovery. Moreover, free (non-immobilized) AT was found to be very stable under our filter entrapment conditions allowing at least after 10 cycles. The recovered oligosaccharides were next used for activity and structural analysis. The filter entrapment method is general and can be used with other proteins and other oligosaccharides and low molecular weight polysaccharides. This method does not allow the elution with a linear NaCl gradient, which can be useful to separate species with increasing affinity towards AT (Bisio et al., 2001).
3.3. Anticoagulant activity
The anticoagulant activity of the LMWH fractions prepared by AT filter-entrapment enrichment were next measured using USP enoxaparin monograph activity assays. Enoxaparin fraction eluting at 2 M NaCl and showing high affinity binding to AT had an anti-Xa activity of 240 U/mg and anti-IIa activity of 101 U/mg, 4.2-fold and 2.5-fold higher, respectively, that the enoxaparin starting material (Table 1). The fraction showing low AT affinity, eluting at 0.2 M NaCl, had an anti-Xa activity of 43 U/mg and no measurable anti-IIa activity. These results were consistent with previously published activity studies on enoxaparin (Brufatto, Ward, & Nesheim, 2003).
Table 1.
Summary of anticoagulant activity and IC50 values of LMWHs
| Anticoagulant activity (U/mg) | IC50 (μg/mL) | |||
|---|---|---|---|---|
| Anti-Xa | Anti-IIa | AT | PF4 | |
| Enoxaparin | 97 | 24 | 11.1 | 2.9 |
| Low affinity Enoxaparin | 43 | 0 | 5.4 | 5.0 |
| High affinity Enoxaparin | 240 | 101 | 16.8 | 1.0 |
3.4. Surface plasmon resonance analysis
High AT binding affinity and low PF4 binding affinity are critical in the clinical use of LMWHs. A LMWH with high AT affinity and, thus, high anticoagulant activity (Onishi, St. Ange, Dordick, & Linhardt, 2016), can be used in a lower gravimetric dose resulting in lower HIT. HIT is an immunological disorder caused by antibodies to PF4-heparin complexes (Rauova et al., 2006), thus, a reduction in PF4 affinity should also result in reduced HIT. Recently, our group developed a competitive SPR method to measure the binding of heparin and LMWHs with PF4 (Zhao et al., 2017; Zhang, Datta, Dordick, & Linhardt, 2020). In this competitive binding method, AT (250 nM) or PF4 (125 nM) is mixed with different concentrations of an enoxaparin sample in HBS-EP buffer and injected over and SPR chip having heparin immobilized. When the heparin binding sites on AT or PF4 are occupied with enoxaparin, the ability of AT or PF4 to bind to surface-immobilized heparin is prevented, resulting in a reduction in SPR signal (Fig. 3). IC50 values could be calculated from the plots. The fraction with high-affinity for AT showed a lower IC50 value of 5.4 μg/mL than enoxaparin, with an IC50 of 11.1 μg/mL. The fraction with a low-affinity for AT showed a higher IC50 value of 16.8 μg/mL. Competitive binding studies on PF4 showed that fractions with high-affinity for AT showed a higher IC50 value of 5.0 μg/mL than enoxaparin, with an IC50 of 2.9 μg/mL, and fraction with a low-affinity for AT showed a lower IC50 value of 1.0 μg/mL. These results suggest that fractions with a high-affinity for AT will reduce the risk of HIT.
Fig. 3.
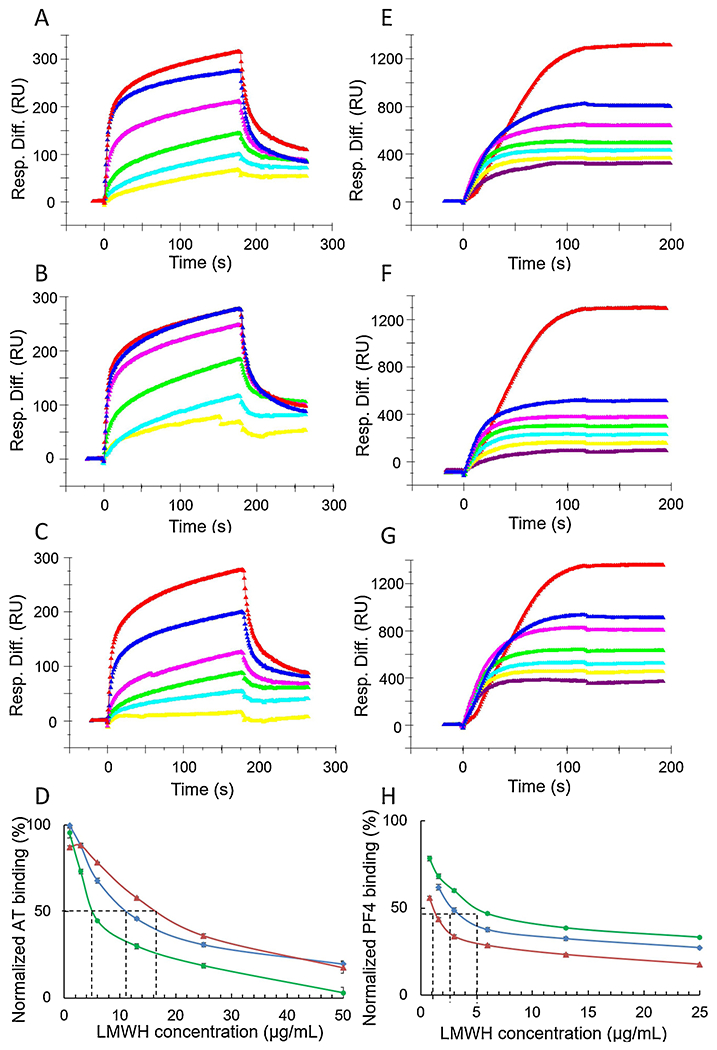
Surface plasmon resonance sensorgrams and IC50 measurement of Enoxaparin, low affinity enoxaparin and high affinity enoxaparin using surface competition SPR. Left pane (A-D) was AT and enoxaparin competition. Control and concentration of enoxaparin in A, B and C (from top bottom): 250 nM AT control, 50, 25, 13, 6, 3 μg/mL Enoxaparin. (A) Competition SPR sensorgrams of AT-heparin interaction inhibiting by original enoxaparin; (B) Competition SPR sensorgrams of AT-heparin interaction inhibiting by low affinity enoxaparin; (C) Competition SPR sensorgrams of AT-heparin interaction inhibiting by high affinity enoxaparin; (D) IC50 measurement for AT binding, red (low affinity) IC50=16.8 μg/mL; blue (original) IC50=11.1 μg/mL; green (high affinity) IC50=5.4 μg/mL. Right pane (E-H) was PF4 and enoxaparin competition. Control and concentration of enoxaparin in E, F and G (from top bottom): 125 nM PF4 control, 50, 25, 13, 6, 3, 1 μg/mL Enoxaparin. (E) Competition SPR sensorgrams of PF4-heparin interaction inhibiting by original enoxaparin; (F) Competition SPR sensorgrams of PF4-heparin interaction inhibiting by high affinity enoxaparin; (G) Competition SPR sensorgrams of PF4-heparin interaction inhibiting by high affinity enoxaparin; (H) IC50 measurement for PF4 binding, red (low affinity) IC50=1.0 μg/mL; blue (original) IC50=2.9 μg/mL; green (high affinity) IC50=5.0 μg/mL.
3.5. Disaccharide analysis
Compositional analysis of disaccharide building blocks was achieved through the exhaustive digestion of enoxaparin with a combination of heparin lyase I, II and III followed by quantitative analysis using ion-pairing reverse-phased (IPRP) HPLC with detection by ion-trap mass spectrometry. Disaccharide analysis provides basic information on the sulfation pattern present in a LMWH. It is noteworthy that sites downstream from 3-O-sulfated glucosamine residue, the central residue present in the AT pentasaccharide binding site are resistant to heparin lyase cleavage (Fig. 4A, Fig. 5A). Thus, the 3-O-sulfated residues require tetrasaccharide analysis, described in the next section. The results of disaccharide analysis showed that there were no significant differences in the disaccharide compositions of enoxaparin, low AT affinity enoxaparin and high AT affinity
Fig. 4.
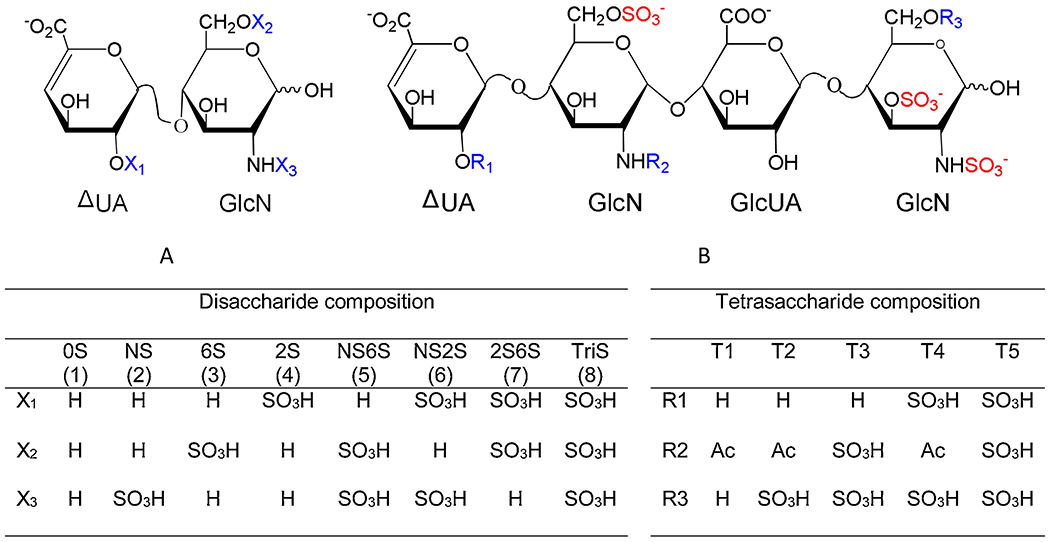
Disaccharide and tetrasaccharide structures. A. Disaccharide structures, (1) to (8) in left table was corresponding to peak 1 to peak 8 in Fig. 5A; B. Five common 3-O-sulfate tetrasaccharide structures.
Fig. 5.
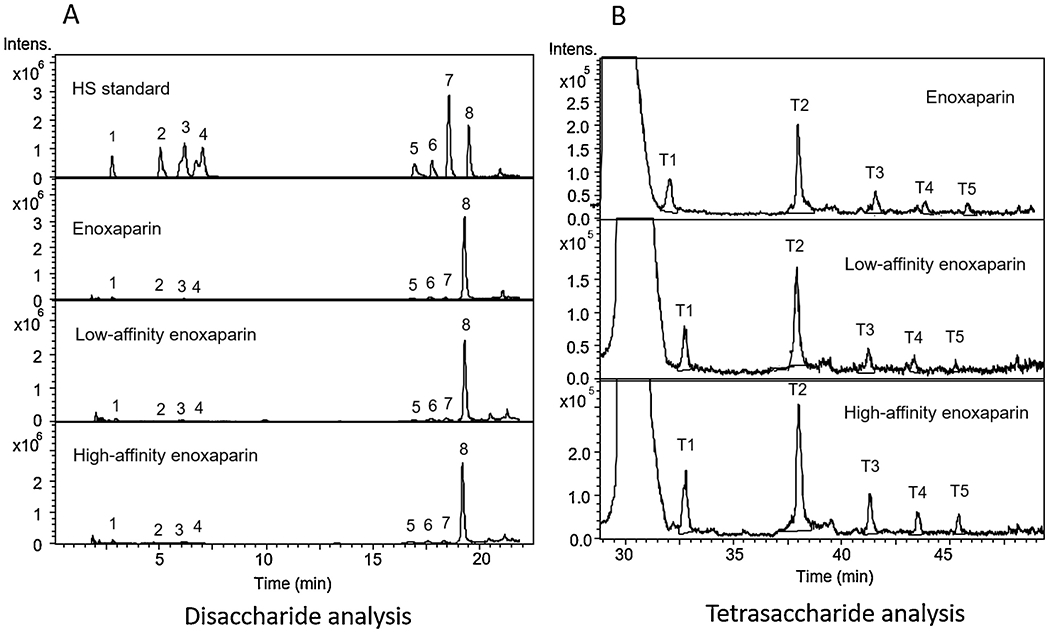
Extracted ion chromatogram (EIC) of disaccharide and tetrasaccharide analysis by LC-MS. A. Disaccharide EIC. 1. 0S; 2. NS; 3. 6S; 4. 2S; 5. NS6S; 6. NS2S; 7. 2S6S; 8. TriS. B. Tetrasaccharide EIC, T1 (m/z = 477, z = 2); T2 (m/z = 517, z=2); T3 (m/z = 536, z = 2); T4 (m/z = 557, z = 2); T4 (m/z = 576, z = 2).
3.6. Tetrasaccharide analysis
LMWH was exhaustively treated with heparin lyase II resulting lyase-resistant tetrasaccharides in addition to the disaccharides. These tetrasaccharides, containing 3-O-sulfo groups, originate from AT binding pentasaccharide sequences. Five common 3-O-sulfo containing tetrasaccharides have been structural characterized (Chen et al., 2017). Their structures are ΔUA-GlcNAc6S-GlcUA-GlcNS3S (T1), ΔUA-GlcNAc6S-GlcUA-GlcNS3S6S (T2), ΔUA-GlcNS6S-GlcUA-GlcNS3S (T3), ΔUA2S-GlcNAc6S-GlcUA-GlcNS3S6S (T4), and ΔUA2S-GlcNS6S-GlcUA-GlcNS3S6S (T5) as shown in (Fig. 4B). The extracted ion chromatography (EIC) of 477, 517, 536, 557 and 576 are shown in Fig. 5B. The results of tetrasaccharide analysis showed that the total contents of these five 3-O-sulfated tetrasaccharides was 162% in high AT affinity enoxaparin fraction compared to the original enoxaparin. In contrast, the total content of tetrasaccharides for low AT affinity enoxaparin was only 72% compared to the original enoxaparin. The amount of the individual 3-O-sulfated tetrasaccharides and their relative percentages are showed in Table 2.
Table 2.
Tetrasaccharide peak areas analyzed by LC-MS
| Peak areas | T1-T5 | ||||||
|---|---|---|---|---|---|---|---|
| T1 | T2 | T3 | T4 | T5 | T1-T5 | Relative content | |
| Enoxaparin | 1.2 × 106 | 3.3 × 106 | 9.4 × 105 | 3.5 × 105 | 3.0 × 105 | 6.1 × 106 | 100% |
| Low affinity enoxaparin | 9.4 × 105 | 2.4 × 106 | 6.4 × 105 | 3.0 × 105 | 1.3 × 105 | 4.4 × 106 | 72% |
| High affinity enoxaparin | 2.1 × 106 | 5.2 × 106 | 1.3 × 106 | 7.7 × 105 | 5.5 × 105 | 9.9 × 106 | 162% |
3.7. CZE-MS MS analysis
The mixture of tetrasaccharides produced by enzymatic digestion of the low-affinity pull-down enriched components of enoxaparin shown in Figure 2 were further analyzed using capillary zone electrophoresis (CZE) and tandem mass spectrometry (CZE-MS/MS). Previous work has established that CZE can separate isomeric GAG oligomers, which can then be analyze on-line by mass spectrometry and tandem mass spectrometry (Sanderson et al., 2018; Stickney et al., 2019). The buffer required for good CZE separation produced lower charge states than we observe by direct infusion of GAGs, and this makes subsequent tandem mass spectrometry more challenging for highly sulfated oligomers such as those examined here. Figure 6a shows the base-peak electropherogram for the separation of the mixture of tetrasaccharides. The molecular weights of the components in this separation correspond to the compositions of compounds T1, T2, T3, and T5.
Fig. 6.
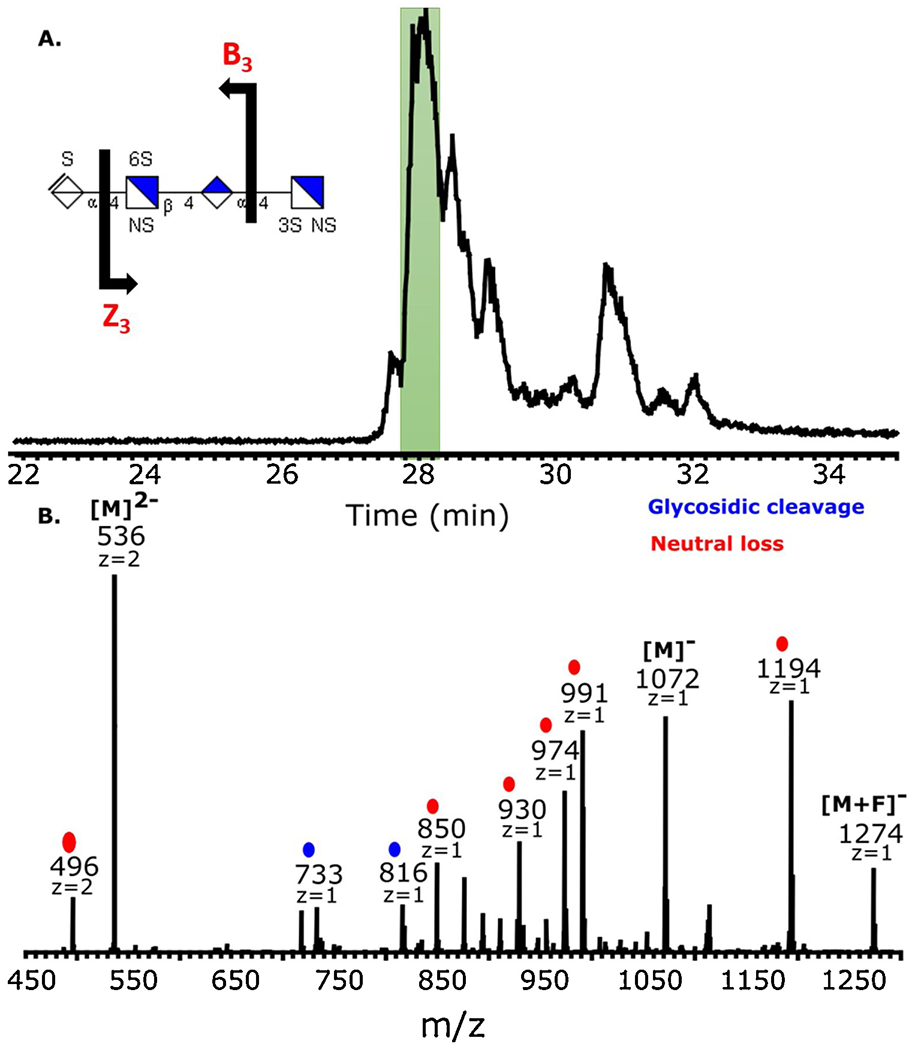
Capillary zone electrophoresis negative electron transfer dissociation tandem mass spectrometry (CZE-MS) data of tetrasaccharides from digestion of a pull-down enrichment fraction of AT binders in enoxaparin. (a) Base peak electropherogram for CZE separation of the tetrasaccharide mixture. (b) NETD activation of m/z 535.992−, the most intense peak at 28 min. The inset in panel (a) shows a possible structure based on the glycosidic fragments shown. The assignments of 3S versus 6S sulfation is not possible without cross-ring fragmentation.
Figure 6b shows the MS/MS spectrum of a precursor ion of m/z 536, with a charge state of (2−), corresponding to the expected molecular weight of T3, using negative electron transfer dissociation (NETD). The two fragments marked in blue are the result of glycosidic cleavage and those marked in red represent a neutral loss, such as SO3, CO2, H2O, H, or some combination of these. The majority of ions in this MS/MS spectrum are from neutral losses from the charge-reduced precursor ion at m/z 1072, or its fluoranthene adduct at m/z 1274. This is expected when the precursor charge is much lower than the number of sulfo modifications, which are 2 and 5, respectively. However, the two glycosidic cleavages that are observed provide enough information to assign the number of sulfo modifications in each monosaccharide residue. The fragments m/z = 733 (B3) and m/z = 816 (Z3) reveal the presence of one sulfate on the unsaturated uronic acid and two sulfates on the glucosamine in the reducing position. This result was not expected as the tetrasaccharide extracted by EIC and thought to be analyzed has a different structure (ΔUA-GlcNS6S-GlcUA-GlcNS3S6S). The tetrasaccharide analyzed in CE-MS appears to be an isomer of the tetrasaccharide extracted by EIC. The isomer is most likely to be ΔUA2S-GlcNS-GlcUA-GlcNS3S6S and ΔUA2S-GlcNS6S-GlcUA-GlcNS3S as previously reported (Alekseeva, Urso, Mazzini, & Naggi, 2019). It is also likely that the sample being analyzed is a mixture of these two structures, and we only collected MS/MS data for one of the two. Overall, the CE-NETD MS/MS method offers a promising platform for the analysis of complex mixtures of GAG. It allows both chemical composition and structural information to be deduced from GAG mixtures.
3.8. Oligosaccharides mapping
Intact enoxaparin chains of low and high-affinity were next examined by top-down analysis using HILIC-FT-MS. The analysis of variation (ANOVA) was performed to illustrate the differences and similarities between low AT affinity enoxaparin, high AT affinity enoxaparin and enoxaparin chains. There was no difference in the number or type of oligosaccharide species. We did observe significant differences of normalized abundance of oligosaccharides in different affinity fractions. In oligosaccharide chains with saturated non-reducing ends (Fig. 7A), both of low and high AT affinity enoxaparin showed decreased amounts of chains shorter than pentasaccharides. The low AT affinity chains showed an increase in saturated chains from hexasaccharide to decasaccharides. Chains having the composition [0, 3, 3, 0, 9], [0, 4, 3, 0, 10], where these numbers represent [ΔHexA, HexA, HexN, Ac, SO3−], were the most representative chains in the low AT affinity enoxaparin fraction. The normalized abundance of these two chains showed a significant increase (P<0.01) in low-affinity fractions and significant decrease (P<0.01) in high-affinity fractions. From dp12 to dp16, the majority of chains showed an increase in the high-affinity fraction. AT affinity was also reflected in sulfation levels, generally the more sulfation the higher the AT binding affinity for a given chain length. These results were similar for fractions in which the non-reducing end was unsaturated (Fig. 7B). In the fractions in which the reducing end was a 1,6-anhydro residue the differences were not as significant as in fractions without a 1,6-anhydro residue, suggesting that the presence of this residue has no relationship with AT binding affinity (Fig. 7C).
Fig. 7.
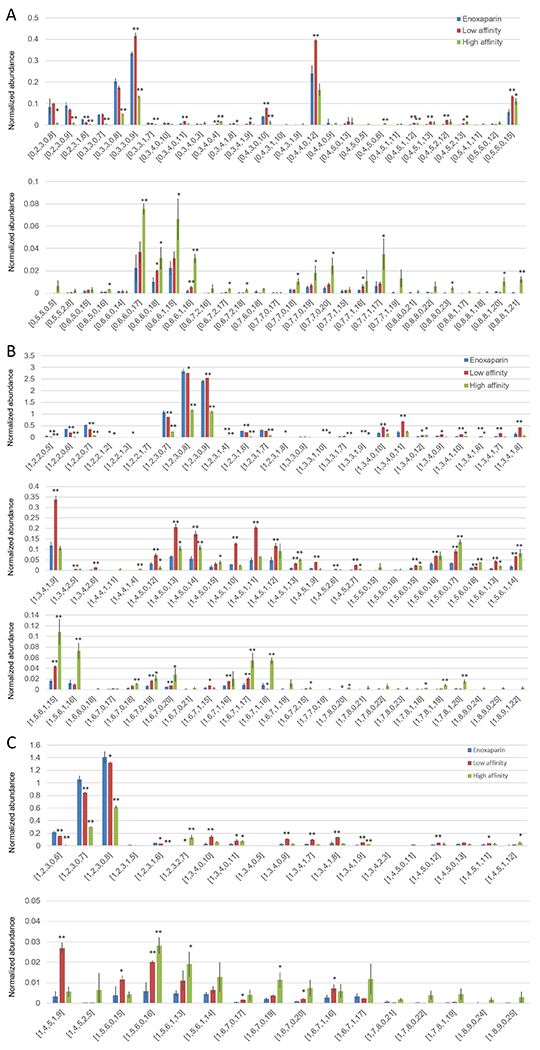
HILIC-FT-MS oligosaccharides mapping of enoxaparin. ANOVA was used for comparison of high and low affinity enoxaparin to original enoxaparin, * for 0.01<p<0.05, ** for p<0.01. A. Enoxaparin whose non-reducing end was saturated, B. Enoxaparin whose non-reducing end was unsaturated, C. Enoxaparin whose reducing end was 1,6-anhydro.
4. Conclusion
A strategy for combining filter-entrapment enrichment with comprehensive analytical methods was used to study the AT affinity domain of enoxaparin chains. This pull-down assay provided a powerful approach for the analysis of heparin oligosaccharide capture and should be applicable for the study of heparin and heparan sulfate interactions with diverse proteins. Such studies should facilitate discovery of GAG structures associated with diseases and lead to a better understanding of the biological functions of GAGs.
Highlights.
Low molecular weight heparins were fractionated using a filter binding assay
The polysaccharide chains binding to antithrombin III showed enhanced activity
Interacting and non-interacting chains were subjected to top-down analysis
Analysis was performed using LC- and CZE- mass spectrometry
Acknowledgements
This study was supported by grant # DK111958 and CA231074 from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interest.
The authors declare no conflicts of interest.
References
- Alekseeva A, Urso E, Mazzini G, & Naggi A (2019). Heparanase as an additional tool for detecting structural peculiarities of heparin oligosaccharides. Molecules, 24, 4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisio A, Ambrosi LD, Gonella S, Guerrini M, Guglieri S, Maggia G, et al. (2001). Preserving the original heparin structure of a novel low molecular weight heparin by γ-irradiation. Arzneimittelforschung Drag Research, 51(II), 806–813. [DOI] [PubMed] [Google Scholar]
- Bisio A, Vecchietti D, Citterio L, Guerrini M, Raman R, Bertini S, et al. (2009). Structural features of low-molecular-weight heparins affecting their affinity to antithrombin. Thrombosis Haemostasis, 102, 865–873. [DOI] [PubMed] [Google Scholar]
- Brufatto N, Ward A, & Nesheim ME (2003). Factor Xa is highly protected from antithrombin-fondaparinux and antithrombin-enoxaparin when incorporated into the prothrombinase complex. Journal of Thrombosis and Haemostasis, 1, 1258–1263. [DOI] [PubMed] [Google Scholar]
- Capila I, & Linhardt RJ (2002). Heparin-protein interactions. Angewandle Chemie International Edition, 41(3), 391–412. [DOI] [PubMed] [Google Scholar]
- Cardin AD, & Weintraub HJR (1989). Molecular modeling of protein-glycosaminoglycan interactions. Arteriosclerosis, 9, 21–32. [DOI] [PubMed] [Google Scholar]
- Chang CH, Lico LS, Huang TY, Lin SY, Chang CL, Arco SD, et al. (2014). Synthesis of the heparin-based anticoagulant drug fondaparinux. Angewandle Chemie International Edition, 41(37), 9876–9879. [DOI] [PubMed] [Google Scholar]
- Chen Y, Lin L, Agyekum I, Zhang X St. Ange K, Yu Y, et al. (2017). Structural analysis of heparin-derived 3-O-sulfated tetrasaccharides: antithrombin binding site variants. Journal of Pharmaceutical Sciences, 106, 973–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeAngelis PL, Liu J, & Linhardt RJ (2013). Chemoenzymatic synthesis of glycosaminoglycans: Re-creating, re-modeling and re-designing nature’s longest or most complex carbohydrate chains. Glycobiology, 23, 764–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi NS, & Mancera RL (2008). The structure of glycosaminoglycans and their interactions with proteins. Chemical Biological Drug Design, 72, 455–482. [DOI] [PubMed] [Google Scholar]
- Garcia DA, Baglin TP, Weitz JI, & Samama MM (2012). Parenteral anticoagulants. Antithrombotic therapy and prevention of thrombosis. (9th ed.) American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest, 141, e24S–e43S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hileman RE, Fromm JR, Weiler JM, & Linhardt RJ (1998). Glycosaminoglycan protein interaction: Definition of consensus sites in glycosaminoglycan binding proteins. Bioassays, 20, 156–167. [DOI] [PubMed] [Google Scholar]
- Hileman RE, Jennings RN, & Linhardt RJ (1998). Thermodynamic analysis of heparin interaction with a basic cyclic protein using isothermal filtration calorimetry. Biochemistry, 37, 15231–15237. [DOI] [PubMed] [Google Scholar]
- Jain A, Liu R, Xiang YK, & Ha T (2012). Single-molecule pull-down for studying protein interactions. Nature Protocol, 7, 445–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jose L, Noti C, & Seeberger PH (2006). Microarrays of synthetic heparin oligosaccharides. Journal of the American Chemical Society, 128, 2766–2767. [DOI] [PubMed] [Google Scholar]
- Junqueira DR, Perini E, Penholati RR, & Carvalho MG (2012). Unfractionated heparin versus low molecular weight heparin for avoiding heparin-induced thrombocytopenia in postoperative patients. Cochrane Database of Systems Review, 12, CD007557. [DOI] [PubMed] [Google Scholar]
- Keiser N, Venkataraman G, Shriver Z, & Sasisekharan R (2001). Direct isolation and sequencing of specific protein-binding glycosaminoglycans. Nature Medicine, 7, 123–128. [DOI] [PubMed] [Google Scholar]
- Kim SY, Zhao J, Liu X, Fraser K, Lin L, Zhang F, et al. (2017). Interaction of Zika virus with glycosaminoglycans. Biochemistry, 56, 1151–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjellén L, & Lindahl U (2018). Specificity of glycosaminoglycan-protein interactions. Current Opinions in Structural Biology, 50, 101–108. [DOI] [PubMed] [Google Scholar]
- Leach FE, Riley NM, Westphall MS, Coon JJ, & Amster IJ (2017). Negative electron transfer dissociation sequencing of increasingly sulfated glycosaminoglycan oligosaccharides on an Orbitrap mass spectrometry. Journal of the American Society for Mass Spectrometry, 28, 1844–1854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Zhang F, Zaia J, & Linhardt RJ (2012). Top-down approach for the direct characterization of low molecular weight heparins using LC-FT-MS. Analytical Chemistry, 84, 8822–8829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin PH, Sinha U, & Betz A (2001). Antithrombin binding of low molecular weight heparins and inhibition of factor Xa. Biochimica et Biophysica Acta, 1526, 105–113. [DOI] [PubMed] [Google Scholar]
- Listik E, Azevedo Margues Gaschler J, Matias M, Neuppmann Feres MF, Toma L, & Raphaelli Nahás-Scocate AC (2019). Proteoglycans and dental biology: the first review. Carbohydrate Polymer, 225, 115199. [DOI] [PubMed] [Google Scholar]
- Lindahl U, Bäckström G, Höök M, Thunberg L, Fransson LA, & Linker A (1979). Structure of the antithrombin-binding site in heparin. Proceedings of the National Academy of Sciences of the United States of America, 76, 3198–3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JR, Wang HF, Yu DF, Chen XY, & He SY (2017). Modulation of binding to vascular endothelial growth factor and receptor by heparin derived oligosaccharide. Carbohydrate Polymer, 174, 558–564. [DOI] [PubMed] [Google Scholar]
- Maxwell E, Tan Y, Tan Y, Hu H, Benson G, Aizikov K, et al. (2012). GlycReSoft: a software package for automated recognition of glycans from LC/MS data. PloS One, 7, e45474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mende M, Bednarek C, Wawryszyn M, Sauter P, Biskup MB, Schepers U, et al. (2016). Chemical synthesis of glycosaminoglycan. Chemical Review, 116, 8193–8255. [DOI] [PubMed] [Google Scholar]
- Onishi A St. Ange K, Dordick JS, & Linhardt RJ (2016). Heparin and anticoagulation. Frontiers in Bioscience, 21, 1372–1392. [DOI] [PubMed] [Google Scholar]
- Persson A, Vorontsov E, Larson G, & Nilsson J (2020). Glycosaminoglycan domain mapping of cellular chondroitin/dermatan sulfates. Scientific Reports, 10, 3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przybylski C, Gonnet F, Saesen E, Lortat-Jacob H, & Daneil R (2020). Surface plasmon resonance imaging coupled to on-chip mass spectrometry: a new tool to probe protein-GAG interactions. Analytical Bioanalytical Chemistry, 412, 507–519. [DOI] [PubMed] [Google Scholar]
- Rauova L, Zhai L, Kowalska MA, Arepally GM, Cines DB, & Poncz M (2006). Role of platelet surface PF4 antigenic complexes in heparin-induced thrombocytopenia pathogenesis: diagnostic and therapeutic implications. Blood, 107, 2346–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg RD, Armand G, & Lan L (1978). Structure-function relationships of heparin species. Proceeding of the National Academy of Sciences USA, 75, 3065–3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson P, Stickney M, Leach FE, Xia J, Yu Y, Zhang F, et al. (2018). Heparin/heparan sulfate analysis by covalently modified reverse polarity capillary zone electrophoresis-mass spectrometry. Journal of Chromatography A, 1545, 75–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipp EL, & Hsieh-Wilson LC (2007). Profiling the sulfation specificities of glycosaminoglycan interactions with growth factors and chemotactic proteins using microarrays. Chemistry & Biology, 14, 195–208. [DOI] [PubMed] [Google Scholar]
- Solakyildirim K (2019). Recent advances in glycosaminoglycan analysis by various mass spectrometry techniques. Analytical and Bioanalytical Chemistry, 411, 3731–3741. [DOI] [PubMed] [Google Scholar]
- Stickney M, Sanderson P, Leach FE, Zhang F, Linhardt RJ, & Amster IJ (2019). Online capillary zone electrophoresis negative electron transfer dissociation tandem mass spectrometry of glycosaminoglycan mixtures. International Journal of Mass Spectrometry, 445, 116209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su H, Blain F, Musil RA, Zimmermann J, Gu K, & Bennett DC (1996). Isolation and expression in Escherichia coli of hepB and hepC, genes coding for the glycosaminoglycan-degrading enzymes heparinase II and heparinase III, respectively, from Flavobacterium heparinum. Applied Environmental Microbiology, 62(8), 2723–2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viskov C, Elli S, Urso E, Gaydesi D, Mourier P, Herman F, et al. (2013). Heparin dodecasaccharide containing two antithrombin-binding pentasaccharides. Journal of Biological Chemistry, 288(36), 25895–25907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Liu X, Li L, Zhang F, Hu M, Ren F, et al. (2016). GlycCompSoft: software for automated comparison of low molecular weight heparins using top-down LC/MS data. PloS One, 12, e0167727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu KK (2006). Analysis of protein-DNA binding by streptavidin-agarose pulldown. Methods in Molecular Biology, 338, 281–290. [DOI] [PubMed] [Google Scholar]
- Xu D, & Esko JD (2014). Demystifying heparan sulfate-protein interactions. Annual Reviews in Biochemistry, 83, 129–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y, Du S, Ji Y, Su N, Wang Y, Mei X, et al. (2017). Discovery of enzymatically depolymerized heparins capable of treating Bleomycin-induced pulmonary injury and fibrosis in mice. Carbohydrate Polymer, 174, 82–88. [DOI] [PubMed] [Google Scholar]
- Yang J, Hsieh PH, Liu X, Zhou W, Zhang X, Zhou J, et al. (2017). Construction and characterization of a heparan sulphate heptasaccharide microarray. Chemical Communications, 53, 1743–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Datta P, Dordick JS, & Linhardt RJ (2020). Evaluating heparin products for heparin-induced thrombocytopenia using surface plasmon resonance. Journal of Pharmaceutical Sciences, 109, 975–980. [DOI] [PubMed] [Google Scholar]
- Zhang X, Pagadala V, Jester HM, Lim AM, Pham TQ, Goulas AMP, et al. (2017). Chemoenzymatic synthesis of heparan sulfate and heparin oligosaccharides and NMR analysis: paving the way to a diverse library for glycobiologists. Chemical Science, 8, 7932–7940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Liu X, Malhotra A, Li Q, Zhang F, & Linhardt RJ (2017). Novel method for measurement of heparin anticoagulant activity using SPR. Analytical Biochemistry, 526, 39–42. [DOI] [PMC free article] [PubMed] [Google Scholar]