

Abstract
Background
Traumatic brain injury (TBI) is a leading cause of death and disability worldwide. Cerebral oedema, the accumulation of fluid within the brain, is believed to be an important contributor to the secondary brain damage that occurs following injury. The release of kinins is thought to be an important factor in the development of cerebral vasogenic oedema and the use of bradykinin beta‐2 receptor antagonists, which prevent the release of these kinins, has been proposed as a potential therapeutic intervention.
Objectives
The objective was to assess the safety and effectiveness of bradykinin beta‐2 receptor antagonists for TBI.
Search methods
We searched the Cochrane Injuries Group Specialised Register, Cochrane Central Register of Controlled Trials (The Cochrane Library 2010 Issue 2), MEDLINE (Ovid SP), EMBASE (Ovid SP), ISI Web of Science: Science Citation Index Expanded (SCI‐EXPANDED), ISI Web of Science: Conference Proceedings Citation Index‐Science (CPCI‐S), Zetoc: British Library's table of contents of journal articles and conference proceedings, PubMed, and Current Controlled Trials covering all available years up to 20th May 2010. We also searched the Internet and checked the reference lists of relevant papers and other reviews to identify any further studies.
Selection criteria
Randomised controlled trials of beta‐2 receptor antagonists versus placebo for TBI.
Data collection and analysis
Two authors independently screened search results and assessed the full texts of potentially relevant studies for inclusion. Data were extracted and the risks of bias assessed. Relative risks (RR) and 95% confidence intervals (CIs) were calculated and data were pooled using a fixed‐effect model.
Main results
Four studies involving 406 participants were included. All four studies reported the effects of beta‐2 receptor antagonists on mortality. The pooled RR for mortality was 0.84 (95% CI 0.55 to 1.29). Two studies measured disability and the RR of death or severe disability with beta‐2 receptor antagonists was 0.81 (95% CI 0.59 to 1.09). One trial reported data on adverse events, the RR of at least one serious adverse event was 1.37 (95% CI 0.76 to 2.46) and the RR of local skin reactions was 13.79 (95% CI 0.85 to 224.81). Two studies measured the effect on intracranial pressure (ICP), only in one study did this finding reach statistical significance. There was no evidence for the presence of heterogeneity.
Authors' conclusions
There is no reliable evidence that bradykinin beta‐2 receptor antagonists are safe or effective for use in TBI patients, and they should not be used outside the context of well conducted trials. Further adequately powered and well conducted randomised controlled trials are required.
Keywords: Humans, Adrenergic beta-2 Receptor Antagonists, Acute Disease, Brain Edema, Brain Edema/drug therapy, Brain Injuries, Brain Injuries/drug therapy, Brain Injuries/mortality, Intracranial Pressure, Peptides, Peptides/therapeutic use, Quinolines, Quinolines/therapeutic use, Randomized Controlled Trials as Topic
Plain language summary
Bradykinin beta‐2 receptor antagonists for traumatic brain injury
Traumatic brain injury (TBI) is a leading cause of death and disability worldwide. Not all damage to the brain occurs at the moment of injury. The injury sustained at the moment of impact (primary brain injury) initiates a sequence of mechanisms which cause further brain damage (secondary brain injury).
One consequence of the mechanisms triggered by primary injury is the accumulation of fluid within the brain. This condition is known as cerebral oedema and leads to raised intracranial pressure (pressure within the skull), which contributes to secondary brain injury.
The use of a group of drugs known as bradykinin beta‐2 receptor antagonists is being investigated as a potential treatment for TBI. It is proposed that they can inhibit the mechanism which causes cerebral oedema and therefore prevent the elevation of intracranial pressure with subsequent brain damage.
The authors of this review searched for all randomised controlled trials investigating the effects of bradykinin beta‐2 receptor antagonists in traumatically brain injured patients. The authors found four trials involving 406 patients. Whilst the overall effect estimates suggest that bradykinin beta‐2 receptor antagonists may reduce mortality and disability, they also suggest that they may increase the number of serious adverse events. However, all of these results are consistent with the play of chance. The findings indicate that there is no evidence to support the use of bradykinin beta‐2 receptor antagonists for TBI.
Bradykinin beta‐2 receptor antagonists are not presently registered for use in patients. Because the safety and effectiveness of bradykinin beta‐2 receptor antagonists have not been reliably ascertained, they should not be used outside the context of well conducted trials.
Background
Description of the condition
Traumatic brain injury (TBI), defined as a blow or jolt to the head or penetrating head injury that disrupts the function of the brain (CDC 2006), is a leading cause of death and disability worldwide. The majority of TBI occurs in the 15 to 45 years age group, with the highest rate amongst males (Bruns 2003; Tagliaferri 2006). Road traffic crashes (RTCs) are the leading cause of TBI accounting for 40% to 50% of serious injuries, followed by falls and violence (WHO 2006). A review of European epidemiological data by Tagliaferri 2006 et al estimated a TBI (hospitalised and fatal) incidence of 235 per 100,000 per year and a case fatality rate of 11 per 100. Each year in the USA, a total of 1.4 million people suffer a TBI, of whom an estimated 50,000 die and 235,000 are hospitalised (CDC 2006). Almost half of all TBI patients suffer long‐term disability (Thornhill 2000; Whitnall 2006). This coupled with the fact that most TBI occurs in young adults further exacerbates the medical, social and financial burden. Furthermore, this burden is expected to grow, particularly in low and middle income countries, largely due to the predicted increase in RTCs and the associated TBIs.
There are two distinct phases of injury associated with TBI. Primary injury describes the irreversible, direct injury sustained at the moment of impact. Pathophysiological studies indicate that this primary injury initiates a sequence of mechanisms which cause further brain damage, known as secondary brain injury. The impact of secondary injury has been demonstrated by research in which it was found that approximately 40% of TBI patients who were able to speak or follow commands shortly after the event later died from their injury (Reilly 1975).
How the intervention might work
The focus in minimising primary brain injury is placed on injury prevention interventions, whilst secondary brain injury requires effective therapeutic interventions to interrupt the mechanisms that can lead to further neural damage.
Secondary brain damage is caused by physiological assaults that result from a cascade of mechanisms which are initiated immediately after the time of the primary injury. Cerebral oedema is one of the physiological assaults contributing to secondary brain injury. There are two types of cerebral oedema, vasogenic and cytotoxic. Vasogenic oedema involves the accumulation of fluid within the brain that results from the opening of the blood‐brain barrier (BBB), while cytotoxic oedema is due to sustained intracellular water collection (Unterberg 1991; Vincent 2005).
Vasogenic cerebral oedema is thought to be, in part, caused by a sequence of events that begins with the release of kinins, products of the kinin‐kallikrein system (Francel 1992; Mayhan 1996; Unterberg 1991). These kinins stimulate a number of neurochemical disturbances which are thought to initiate and potentiate cerebral oedema (Marmarou 2005). In particular, bradykinin (BK) is believed to contribute to the breakdown of the endothelial junctions that comprise the BBB, which enables the movement of substances into the brain and leads to an accumulation of fluid, that is causing vasogenic cerebral oedema (Unterberg 1991; Unterberg 2004). This is thought to further increase intracranial pressure, which can lead to cerebral ischaemia and additional structural neural damage (Vincent 2005).
In consideration of the role of kinins in vasogenic oedema and their contribution to secondary brain injury, the use of beta‐2 receptor antagonists has been proposed (Francel 1992; Marmarou 1999). These agents act to block the activity of the kinin‐kallikrein system, thus preventing the production of kinins and their impact on the BBB. In this way it is hypothesised that the administration of bradykinin beta‐2 antagonists can prevent elevation of intracranial pressure. There are promising results for the effects of beta‐2 receptor antagonists on neurological function and brain oedema from studies in animal models of TBI (Ivashkova 2006; Kaplanski 2002; Stover 2000).
Why it is important to do this review
Beta‐2 receptor antagonists are not registered for use in traumatically brain injured patients. The evidence for the effectiveness and safety of the use of bradykinin beta‐2 antagonists in TBI has yet to be ascertained. Hence we conducted a systematic review of randomised controlled trials.
Objectives
To assess the evidence on the effects of bradykinin beta‐2 receptor antagonists for the treatment of acute traumatic brain injury.
Methods
Criteria for considering studies for this review
Types of studies
Randomised controlled trials.
Types of participants
Patients of any age with acute traumatic brain injury of any severity.
Types of interventions
Bradykinin beta‐2 receptor antagonists versus placebo.
For studies in which different doses of the intervention drug were compared with placebo, the intervention groups were combined and compared with the control group.
Types of outcome measures
Primary outcomes
Mortality
Disability as measured on the Glasgow Outcome Score (GOS)
Secondary outcomes
Intracranial pressure (ICP)
S100B levels
Side effects, including local skin reactions
Search methods for identification of studies
Searches were not restricted by date, language or publication status.
Electronic searches
We searched the following electronic databases:
Cochrane Injuries Group's Specialised Register (searched 20 May 2010)
Cochrane Central Register of Controlled Trials (The Cochrane Library 2010 Issue 2)
MEDLINE (Ovid SP) 1950 to May (week 1) 2010
EMBASE (Ovid SP) 1980 to (week 19) May 2010
ISI Web of Science: Science Citation Index Expanded (SCI‐EXPANDED) 1970 to May 2010
ISI Web of Science: Conference Proceedings Citation Index‐ Science (CPCI‐S) 1990 to May 2010
Zetoc: British Library's table of contents of journal articles and conference proceedings (http://zetoc.mimas.ac.uk/) (searched 20 May 2010)
PubMed [www.ncbi.nlm.nih.gov/sites/entrez/] (searched 20 May 2010 (limit: added in the last 90 days)
LILACS (to May 2010)
Current Controlled Trials (to May 2010),
The full search strategies are presented in Appendix 1.
Searching other resources
We searched the reference lists of the eligible studies and any review articles for further potentially eligible articles. We searched the Internet using the Google (www.google.com) search engine, with selected terms from the above strategy, for any further unpublished or grey literature.
Data collection and analysis
Selection of studies
Two authors independently examined titles, abstracts, and keywords of citations from electronic databases for eligibility of studies. We obtained the full text of all potentially relevant records and two authors independently assessed whether each met the pre‐defined inclusion criteria. We resolved any disagreements by discussion.
Data extraction and management
Two authors extracted data from each eligible study using a standard form that was developed specifically for this review. We extracted data on the following:
study design;
participant characteristics;
intervention characteristics;
outcome measures;
statistical analysis.
Assessment of risk of bias in included studies
This was assessed using The Cochrane Collaboration's tool for assessing risk of bias presented in Higgins 2008.
The following domains were assessed for each study:
sequence generation,
allocation concealment,
blinding,
incomplete outcome data,
other potential sources of bias.
We completed a risk of bias table for each study, incorporating a description of the study's performance against each of the above domains and our overall judgment of the risk of bias for each entry, as follows:
'Yes' indicates low risk of bias,
'Unclear' indicates unclear or unknown risk of bias,
'No' indicates high risk of bias.
Measures of treatment effect
We calculated relative risks (RR) and 95% confidence intervals (CI) for mortality for each trial. RR and 95% CIs were also calculated for GOS data, which we dichotomised into favourable (moderate disability, good recovery; GOS 4 and 5) and unfavourable (death, vegetative state, severe disability; GOS 1 to 3) outcomes.
Dealing with missing data
We contacted the original investigators to obtain missing data.
Assessment of heterogeneity
We examined trial characteristics in terms of participants, interventions and outcomes for evidence of clinical heterogeneity. Statistical heterogeneity was examined by both the I2 and Chi2 statistics. The I2 statistic describes the percentage of total variation across studies due to heterogeneity rather than chance. A value of 0% indicates no observed heterogeneity, and larger values show increasing heterogeneity; substantial heterogeneity is considered to exist with I2 > 50% (Higgins 2008). For the Chi2 statistic, a P value of < 0.10 was used to indicate the presence of statistically significant heterogeneity.
Possible sources of heterogeneity were assessed by subgroup and sensitivity analyses, as described below.
Assessment of reporting biases
We planned to investigate the presence of reporting (publication) bias using funnel plots, however there were too few included studies to enable meaningful analysis.
Data synthesis
We judged that the trials were sufficiently homogenous, both clinically and statistically, and pooled data using the fixed‐effect model.
Subgroup analysis and investigation of heterogeneity
We planned to conduct a subgroup analysis according to the severity of TBI (mild and moderate; GCS 9 to 15) versus severe (GCS 3 to 8) assuming that there was at least one study in each category. However there were insufficient data for such an analysis.
Sensitivity analysis
We planned to conduct a sensitivity analysis according to allocation concealment (adequate versus inadequate) assuming that there was at least one study in each category. However there were insufficient data for such an analysis.
Results
Description of studies
Results of the search
The combined search strategy identified 254 citations, of which 23 were judged to be potentially eligible based on title or abstract, or both, and the full texts were obtained. After a full text review, four completed studies (BRAIN 2009; Marmarou 1999; Marmarou 2005; Narotam 1998) were judged to be eligible and were included in this review.
Included studies
Two studies were conducted in the USA, one in South Africa and one was an international, multi‐centre trial. Sample sizes were n = 228, n = 133, n = 25 and n = 20 for BRAIN 2009, Marmarou 1999, Marmarou 2005 and Narotam 1998, respectively.
The BRAIN 2009 trial recruited patients aged 16 to 65 years with a GCS ≤ 12 and a computerised tomography (CT) scan indicating the presence of an intracranial abnormality consistent with trauma who presented within eight hours of injury. Marmarou 1999 and Marmarou 2005 involved patients aged 16 to 70 years with a severe TBI (GCS 3 to 8), while Narotam 1998 recruited patients aged 12 years and over with moderate TBI (GCS 9 to 12).
Patients in the BRAIN 2009 trial were allocated to one of four groups: low dose (10 mg loading dose + 5 mg/day), medium dose (20 mg loading dose + 10 mg/day), high dose (30 loading dose + 15 mg/day) Anatibant®, or placebo.
Marmarou 1999 compared a group receiving continuous intravenous infusion of 3 μg/kg/min of Bradycor™ with a control group receiving placebo (reconstituted with lactated Ringer's solution).
Patients were randomised to three treatment arms in Marmarou 2005: one group received a single dose of 3.75 mg Anatibant®, the second group received a single dose of 22.5 mg Anatibant®, and the final group was the control and received placebo.
The intervention group in Narotam 1998 received 3 μg/kg/min of Bradycor™ (CP‐0127) over a seven‐day period and the control group received placebo (lactated Ringer's solution).
All four studies measured mortality. In addition, BRAIN 2009 measured serious adverse events and in‐hospital morbidity (GCS, Disability Rating Scale, Oxford Handicap Scale); Marmarou 1999 and Marmarou 2005 also measured ICP and long‐term disability according to the GOS.
Further details are presented in the 'Characteristics of included studies' table.
Risk of bias in included studies
For further details regarding the performance of the studies against each domain, please see the 'Risk of bias' tables and Figure 1 and Figure 2.
1.

Methodological quality graph: review authors' judgements about each methodological quality item presented as percentages across all included studies. Please note that four trials are included in this review.
2.

Methodological quality summary: review authors' judgements about each methodological quality item for each included study.
Allocation
Adequate sequence generation
A computer‐generated randomisation list was used in all four trials, thus the risk of bias for this item was judged to be low.
Allocation concealment
Allocation concealment was judged to be adequate in BRAIN 2009 and Marmarou 1999. There was insufficient information presented to determine the adequacy of allocation concealment in Marmarou 2005 and Narotam 1998.
The risk of bias for this item was judged as unclear for Marmarou 2005 and Narotam 1998, and low for BRAIN 2009 and Marmarou 1999.
Blinding
All three trials reported that the treating clinicians were blind to allocation status, although in Narotam 1998 two physicians were not blinded for safety reasons.
For Marmarou 1999 and Marmarou 2005 it is unclear if outcome assessors were blind to allocation status. In Narotam 1998 they are reported as being blind to allocation. In BRAIN 2009 all study personnel and participants were blind to allocation status.
The risk of bias for this item was judged as unclear for Marmarou 1999 and Marmarou 2005, and low for BRAIN 2009 and Narotam 1998.
Incomplete outcome data
Reasons for the attrition or exclusion of participants were not reported in Marmarou 1999 or Marmarou 2005. In Narotam 1998 there were no withdrawals, drop‐outs, attrition differences between groups or losses to follow up. In BRAIN 2009 there was a small amount of missing data for certain outcomes, although reasons for this were unlikely to be related to true outcomes and analyses were performed on an intention‐to‐treat basis.
The risk of bias for this item was judged as unclear for Marmarou 1999 and Marmarou 2005, and low for BRAIN 2009 and Narotam 1998.
Selective reporting
We were able to access the protocol for one (BRAIN 2009) of the included trials.
In the methods of Marmarou 2005 it was stated that ICP outcome data were collected, however such data were not presented in the results.
The risk of bias for this item was judged as low for BRAIN 2009, unclear for Marmarou 1999 and Narotam 1998, and high for Marmarou 2005.
Other potential sources of bias
In Marmarou 2005 dilution errors resulted in an incorrect dosage of the active drug being administered to some intervention patients. Additionally there were between‐group differences in terms of TBI severity of the participants.
The source of funding of Marmarou 2005 was not reported. BRAIN 2009, Marmarou 1999 and Narotam 1998 were all funded by pharmaceutical companies involved in the development of the intervention drug.
The risk of bias for this item was judged as low for BRAIN 2009, Marmarou 1999 and Narotam 1998; and high for Marmarou 2005.
Effects of interventions
Mortality
All four included studies reported mortality data. The pooled relative risk (RR) of death at end of follow up was RR 0.84 (95% CI 0.55 to 1.29). There was no evidence of heterogeneity (Chi2 = 3.34, df = 2, P = 0.19, I2 = 40%).
Disability
Two studies reported disability data measured on the GOS. The pooled RR for death or severe disability (GOS 1 to 3) was 0.81 (95% CI 0.59 to 1.09). There was no evidence of heterogeneity (Chi2 = 0.02, df = 1, P = 0.89, I2 = 0%).
Intracranial pressure (ICP)
Three studies reported collecting ICP data as outcome measures, however data were only presented in two (Marmarou 1999; Narotam 1998). A pooled analysis was not appropriate and the data are presented narratively below. Both studies detected a beneficial effect of beta‐2 antagonists on controlling ICP, although the effect was statistically significant for just one study. Marmarou 1999 Mean per cent time above 20 mm Hg for patients in the intervention group was consistently lower than the control group for the first five days post‐injury, although the differences in the daily means were not statistically significant.
Narotam 1998 Elevated ICP (defined as sustained elevations of > 20 mm Hg) occurred in one of the 11 patients in the intervention group compared to seven of the nine control patients (P = 0.005). The mean peak ICP in the control group was 33.56 (± 4.47) mm Hg which occurred at a mean of 2.78 (± 0.47) days after injury; and 20.0 (± 1.92) mm Hg in the intervention group, occurring at a mean 4.16 (± 0.52) days after injury. The mean change in peak ICP from baseline in the intervention group was 9.45 (± 2.01) which was significantly lower than that in the control group (21.89 ± 4.69), P = 0.0018.
S100B levels
None of the included studies reported data on S100B levels.
Adverse events
Local skin reactions
Two studies (BRAIN 2009; Marmarou 2005) collected data on local skin reactions as a potential adverse effect of the intervention, although only the BRAIN 2009 trial presented the figures. The RR for local skin reaction was 13.79 (95% CI 0.85 to 224.81).
The actual outcome data were not reported in Marmarou 2005, although it was stated that "all the local injection sites reactions (erythema and subcutaneous nodules), the incidence of which tended to increase with the concentration of Anatibant, reversed spontaneously".
Serious adverse events
The BRAIN 2009 trial collected data on serious adverse events. These were defined as any untoward medical occurrence that: 1) was fatal; 2) life threatening; 3) required or prolonged hospitalisation; 4) resulted in persistent or significant disability or incapacity; 5) was medically significant in that it may jeopardise the patient and may require medical or surgical interventions to prevent one of the outcomes listed above; or 6) congential anomalies.
The number of patients with at least one serious adverse event was higher in the intervention group. The RR for at least one serious adverse event was 1.37 (95% CI 0.76 to 2.46).
Subgroup analysis
There were insufficient data to perform subgroup analyses.
Discussion
Summary of main results
There is no reliable evidence that bradykinin beta‐2 receptor antagonists are effective or safe for use in patients with traumatic brain injury (TBI). Whilst the pooled effect estimates show non‐significant reductions in mortality and severe disability with beta‐2 receptor antagonists, they also indicate a non‐significant increase in serious adverse events. However, all of these findings are consistent with the play of chance.
These findings are based on four RCTs involving 406 TBI patients.
Quality of the evidence
Two of the included studies were judged to have adequate allocation concealment, for the remaining two studies this was rated as unclear. Blinding of outcome assessors was unclear in the two trials that collected long‐term GOS disability data.
Marmarou 1999 originally aimed to recruit 160 patients, 80 in each study arm, however the sponsor (SmithKline Beecham) stopped recruitment at 139 patients. The reason for this action was reported as being "because of results of animal toxicology studies conducted during the course of the trial. These results, although largely inconclusive after repeat studies were performed, were sufficient to prompt SmithKline Beecham to terminate the trial". Although 139 patients were enrolled, data were only presented for 133 and the reason for the exclusion of six patients was not given. For outcomes at six months, data were presented for 116 participants (58 in each group), thus a total of 17 (13%) patients were not included in the analysis. The reasons for these exclusions were not explicit in the trial report.
BRAIN 2009 was a well conducted trial, judged to be at low risk of bias for all domains. The trialists originally planned to recruit 400 patients however recruitment was stopped by the sponsor at 228 participants, consequently the trial's power to detect an effect was reduced.
The small sample size is likely to be the reason for the uneven distribution in the severity of TBI noted in Marmarou 2005. Based on GCS at baseline, patients allocated to the intervention group were more severely injured than patients in the control group. There were also dosage errors in administration of the study drug, which meant that the exact allocated dose was not given in some patients.
The length of time between injury and administration of the drug in the trials may be considered a potential weakness. The inflammatory process is understood to be initiated immediately after injury. It therefore may be reasonable to believe that a treatment aimed at interrupting this process needs to be administered as early as possible to maximise any beneficial effect. The shortest eligible time period adopted in the included trials was within eight hours of injury, used in both BRAIN 2009 and Marmarou 2005, which still may have meant that patients were treated with beta‐2 receptor antagonists relatively late in the inflammatory process. Future trials might consider adopting a more restrictive eligibility criterion in terms of time since injury, in an attempt to administer the treatment as early as possible in the inflammatory process. However, any time period has to also be clinically feasible.
Potential biases in the review process
This systematic review addresses a focused research question and uses pre‐defined inclusion criteria and methodology to select and appraise eligible studies.
As with all systematic reviews, the possibility of publication bias should be considered as a potential threat to validity. However, in light of our extensive and sensitive searching we believe that the risk of such a bias affecting the results is minimal.
For the mortality outcome we pooled the number of deaths at the end of the follow‐up period in each trial. For Marmarou 1999 and Marmarou 2005 this was six months, for BRAIN 2009 and Narotam 1998 it was 15 and 14 days, respectively. Pooling data from such different lengths of follow up may be considered a potential weakness.
There were a number of points on which we sought further clarification from the trial authors. Unfortunately we did not receive a response to our queries regarding either Marmarou 1999 or Marmarou 2005.
Authors' conclusions
Implications for practice.
Bradykinin beta‐2 receptor antagonists are not licensed for use in TBI patients.
Because the safety and effectiveness of beta‐2 receptor antagonists have yet to be reliably ascertained, these agents should not be used outside the context of well conducted RCTs.
Implications for research.
Further adequately powered well designed RCTs are required to determine the the safety and effectiveness of beta‐2 receptor antagonists in TBI patients.
What's new
| Date | Event | Description |
|---|---|---|
| 21 May 2010 | New search has been performed | The search has been updated to 19 May 2010. No new trials were identified; the conclusions remain the same. |
History
Protocol first published: Issue 3, 2007 Review first published: Issue 1, 2008
| Date | Event | Description |
|---|---|---|
| 19 January 2010 | New search has been performed | The results of the BRAIN trial have been incorporated. The trial had been an ongoing study in previous versions of the review. The conclusions remain unchanged. The title has been changed to specify the type of beta‐2 receptor antagonist (bradykinin). |
| 20 May 2008 | Amended | The assessment of methodological quality sections in the 'Methods' and 'Results' section have been revised in light of updated Cochrane guidance presented in Higgins 2008. Risk of bias tables have been completed. |
| 11 April 2008 | New search has been performed | Searches have been updated; no new studies were identified. |
| 26 March 2008 | Amended | Converted to new review format. |
Acknowledgements
Thanks to Pablo Perel and Ian Roberts for their help and advice in preparing this systematic review.
Thanks also to Pradeep Narotam and Haleema Shakur for providing further trial details.
Appendices
Appendix 1. Search strategies
Cochrane Injuries Group Specialised Register (searched 20 May 2010) (beta‐2 or B2 or bradycor or anatibant or icatibant or cereport or bradykinin) Cochrane Central Register of Controlled Trials (The Cochrane Library 2010, Issue 2) #1 MeSH descriptor Craniocerebral Trauma explode all trees #2 MeSH descriptor Cerebrovascular Trauma explode all trees #3 MeSH descriptor Brain Edema explode all trees #4 (brain or cerebral or intracranial) near3 (oedema or edema or swell*) #5 MeSH descriptor Glasgow Coma Scale explode all trees #6 MeSH descriptor Glasgow Outcome Scale explode all trees #7 MeSH descriptor Unconsciousness explode all trees #8 glasgow near3 (coma or outcome) near3 (score or scale) #9 Unconscious* or coma* or concuss* or 'persistent vegetative state' #10 "Rancho Los Amigos Scale" #11 (head or crani* or cerebr* or capitis or brain* or forebrain* or skull* or hemispher* or intra‐cran* or inter‐cran*) near3 (injur* or trauma* or damag* or wound* or fracture* or contusion*) #12 Diffuse near3 axonal near3 injur* #13 (head or crani* or cerebr* or brain* or intra‐cran* or inter‐cran*) near3 (haematoma* or hematoma* or haemorrhag* or hemorrhag* or bleed* or pressure) #14 (#1 OR #2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8 OR #8 OR #9 OR #10 OR #11 OR #12 OR #13) #15 MeSH descriptor Bradykinin explode all trees #16 MeSH descriptor Receptor, Bradykinin B2 explode all trees #17 bradykinin* #18 MeSH descriptor Receptors, Adrenergic, beta‐2 explode all trees #19 (adrenergic* or antagonist*) near3 (B2 or Beta‐2) #20 Anatibant or LF16‐0687 or XY2405 #21(Bradycor or CP‐0127 or Icatibant or CP‐0597 or Cereport) #22 (#15 OR #16 OR #17 OR #18 OR #19 OR #20 OR #21) #23 (#14 AND #22) MEDLINE (Ovid SP) 1950 to May (week 1) 2010 1. exp CRANIOCEREBRAL TRAUMA/ 2. exp Cerebrovascular Trauma/ 3. exp BRAIN EDEMA/ 4. ((brain or cerebral or intracranial) adj3 (oedema or edema or swell*)).ab,ti. 5. exp GLASGOW COMA SCALE/ 6. exp GLASGOW OUTCOME SCALE/ 7. exp UNCONSCIOUSNESS/ 8. (Glasgow adj3 (coma or outcome) adj3 (scale* or score*)).ab,ti. 9. (Unconscious* or coma* or concuss* or 'persistent vegetative state').ab,ti. 10. "Rancho Los Amigos Scale".ab,ti. 11. ((head or crani* or cerebr* or capitis or brain* or forebrain* or skull* or hemispher* or intra‐cran* or inter‐cran*) adj3 (injur* or trauma* or damag* or wound* or fracture* or contusion*)).ab,ti. 12. "Diffuse axonal injur*".ab,ti. 13. ((head or crani* or cerebr* or brain* or intra‐cran* or inter‐cran*) adj3 (haematoma* or hematoma* or haemorrhag* or hemorrhag* or bleed* or pressure)).ab,ti. 14. or/1‐13 15. exp Receptors, Bradykinin/ 16. exp Bradykinin/ 17. bradykinin*.ab,ti. 18. exp Receptors, Adrenergic, beta‐2/ 19. ((adrenergic* or antagonist*) adj3 (B2 or Beta‐2)).ab,ti. 20. (Anatibant or LF16‐0687 or XY2405).ab,ti. 21. (Bradycor or CP‐0127 or Icatibant or CP‐0597 or Cereport).ab,ti. 22. or/15‐21 23. 14 and 22 EMBASE (Ovid SP) 1980 to (week 19) 2010 1. exp Brain Injury/ 2. exp Brain Edema/ 3. exp Glasgow Coma Scale/ 4. exp Glasgow Outcome Scale/ 5. exp Rancho Los Amigos Scale/ 6. exp Unconsciousness/ 7. ((brain or cerebral or intracranial) adj3 (oedema or edema or swell*)).ab,ti. 8. ((head or crani* or cerebr* or capitis or brain* or forebrain* or skull* or hemispher* or intra‐cran* or inter‐cran*) adj3 (injur* or trauma* or damag* or wound* or fracture* or contusion*)).ab,ti. 9. (Glasgow adj3 (coma or outcome) adj3 (scale* or score*)).ab,ti. 10. Rancho Los Amigos Scale.ab,ti. 11. (Unconscious* or coma* or concuss* or 'persistent vegetative state').ab,ti. 12. Diffuse axonal injur*.ab,ti. 13. ((head or crani* or cerebr* or brain* or intra‐cran* or inter‐cran*) adj3 (haematoma* or hematoma* or haemorrhag* or hemorrhag* or bleed* or pressure)).ab,ti. 14. or/1‐13 15. exp Bradykinin Receptor/ 16. exp Bradykinin/ 17. bradykinin*.ab,ti. 18. exp Beta 2 Adrenergic Receptor/ 19. ((adrenergic* or antagonist*) adj3 (B2 or Beta‐2)).ab,ti. 20. (Anatibant or LF16‐0687 or XY2405).ab,ti. 21. (Bradycor or CP‐0127 or Icatibant or CP‐0597 or Cereport).ab,ti. 22. or/15‐21 23. 14 and 22 ISI Web of Science: Science Citation Index Expanded (SCI‐EXPANDED) 1970 to 19 May 2010 and ISI Web of Science: Conference Proceedings Citation Index‐ Science (CPCI‐S) 1990 to May 2010 Topic=((brain or head) SAME (injur* or trauma* or damag* or wound* or fracture* or contusion* or haematoma* or hematoma* or haemorrhag* or hemorrhag* or bleed* or pressure)) AND Topic=(beta‐2 or B2 or bradycor or anatibant or icatibant or cereport or bradykinin) AND Topic=(random* or trial*) PubMed [www.ncbi.nlm.nih.gov/sites/entrez/] searched 29th May 2010 (limit: added in the last 90 days) Craniocerebral Trauma [mh] OR Brain Edema [mh] OR Glasgow Coma Scale [mh] OR Glasgow Outcome Scale [mh] OR Unconsciousness [mh] OR Cerebrovascular Trauma [mh] OR ((head OR cranial OR cerebral OR brain* OR intra‐cranial OR inter‐cranial) AND (haematoma* OR hematoma* OR haemorrhag* OR hemorrhage* OR bleed* OR pressure)) OR (Glasgow AND scale) OR ("diffuse axonal injury" OR "diffuse axonal injuries") OR ("persistent vegetative state") OR ((unconscious* OR coma* OR concuss*) AND (injury* OR injuries OR trauma OR damage OR damaged OR wound* OR fracture* OR contusion* OR haematoma* OR hematoma* OR haemorrhag* OR hemorrhag* OR bleed* OR pressure)) AND (Bradycor OR CP‐0127 OR Anatibant OR LF16‐0687 OR XY2405 OR B2 OR Beta‐2 OR Bradycor OR CP‐0127 OR Anatibant OR LF16‐0687 OR XY2405 AND (randomized controlled trial[pt] OR controlled clinical trial[pt]) OR (randomized OR randomised OR randomly OR placebo[tiab]) OR (trial[ti]) OR ("Clinical Trials as Topic"[MeSH Major Topic])) NOT (("Animals"[Mesh]) NOT ("Humans"[Mesh] AND "Animals"[Mesh])) Zetoc searched (20 May 2009) Beta 2 + head Beta 2 + brain B2 + head B2 + brain
Zetoc (11 April 08) (Brain [and] injur* [and] (B2 or Beta‐2 or Bradykinin* or Anatibant or LF16‐0687 or XY2405 or Bradycor or CP‐0127)) or (Head [and] injur* [and] (B2 or Beta‐2 or Bradykinin* or Anatibant or LF16‐0687 or XY2405 or Bradycor or CP‐0127))
LILACS (May 2010) (head OR brain OR cranial OR cerebral OR brain$ OR intra‐cranial OR inter‐cranial [Palavras] AND (haematoma$ OR hematoma$ OR haemorrhag$ OR hemorrhage$ OR bleed$ OR pressure OR injury$ OR injuries OR trauma OR damage OR damaged OR wound$ OR fracture$) [Palavras] AND (bradykinin* OR B2 OR Beta2 OR B‐2 OR Beta‐2 OR Anatibant OR LF16‐0687 OR XY2405 OR Bradycor OR CP‐0127 OR icatibant OR cereport OR CP‐0597 OR (adrenergic* and antagonist*)) [Palavras]
Data and analyses
Comparison 1. Beta‐2 receptor antagonist versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mortality at end of follow‐up period | 4 | 379 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.84 [0.55, 1.29] |
| 2 Death or severe disability (GOS 1 to 3) at end of follow‐up period | 2 | 139 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.81 [0.59, 1.09] |
| 3 Adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3.1 Skin reactions at injection site | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 Serious adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
1.1. Analysis.
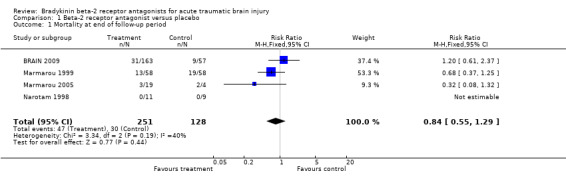
Comparison 1 Beta‐2 receptor antagonist versus placebo, Outcome 1 Mortality at end of follow‐up period.
1.2. Analysis.

Comparison 1 Beta‐2 receptor antagonist versus placebo, Outcome 2 Death or severe disability (GOS 1 to 3) at end of follow‐up period.
1.3. Analysis.

Comparison 1 Beta‐2 receptor antagonist versus placebo, Outcome 3 Adverse events.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
BRAIN 2009.
| Methods | Trial location: International, multi‐centre. | |
| Participants | n = 228 TBI patients aged 16 to 65 years with a GCS of 12 or less, with a CT scan showing an intracranial abnormality consistent with trauma and presenting within 8 hours of injury. | |
| Interventions | Four groups:
Data from intervention groups were combined for the review's analyses. |
|
| Outcomes | Mortality. In‐hospital morbidity (assessed by GCS, DRS and HIREOS). 15‐day follow up. | |
| Notes | Funded by Xytis Pharmaceuticals. Planned sample size of 400, sponsor stopped trial after recruitment of 228 patients. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Adequate sequence generation? | Low risk | Computer randomisation. |
| Allocation concealment? | Low risk | Central allocation. |
| Blinding? All outcomes | Low risk | "All study personnel and participants were to be blinded to treatment assignment for the duration of the study". |
| Incomplete outcome data addressed? All outcomes | Low risk | Reasons for missing outcome data unlikely to be related to true outcome. Intention‐to‐treat analysis performed. |
| Free of selective reporting? | Low risk | Data for all outcomes pre‐specified in the trial protocol are presented in the final report. |
| Free of other bias? | Low risk | The study appears to be free of other sources of bias. |
Marmarou 1999.
| Methods | Trial location: USA. | |
| Participants | Patients aged 16 to 70 years with severe traumatic brain injury (GCS 3 to 8). 139 patients recruited, 133 patients were randomised and received allocated treatment, reasons for exclusion of the six patients were not reported. | |
| Interventions | Two groups. Intervention: n = 66, 3ug/kg/min Bradycor ‐ continuous iv infusion over five days. Infusion beginning within 12 hours of injury. Control: n = 67, placebo (reconstituted with lactated Ringer's solution). | |
| Outcomes | Mortality.
GOS at three and six months.
ICP measurements.
Blood pressure.
Heart rate.
Therapeutic intervention.
GCS.
Neuropsychological tests.
Adverse events. Length of follow up: first 14 days, long‐term outcome assessed at three and six months after injury. |
|
| Notes | Trial sponsored by SmithKline Beecham Pharmaceuticals. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Adequate sequence generation? | Low risk | A computer‐generated randomisation list with sequential blocks of four randomisation numbers assigned to each site. |
| Allocation concealment? | Low risk | The study drug and placebo were identical in appearance and packaging. |
| Blinding? All outcomes | Unclear risk | Study personnel were blinded to allocation status. It is unclear whether long‐term outcome assessors of disability at three and six months were blinded to allocation status. |
| Incomplete outcome data addressed? All outcomes | Unclear risk | The report states that the sponsor stopped the trial at an accrual of 139 patients, 133 of these were randomised and received the allocated dose, subsequent analyses are based on these 133 patients. The reason for the exclusion of the six remaining participants is not stated. 13% (n = 17) of patients were lost (or excluded) at six months. |
| Free of selective reporting? | Unclear risk | We were unable to locate the protocol for this trial. |
| Free of other bias? | Low risk | The study appears to be free of other sources of bias. |
Marmarou 2005.
| Methods | Trial location: USA. | |
| Participants | Patients aged 16 to 70 years with severe traumatic brain injury (GCS 3 to 8 or if intubated a motor score of 2 to 5). 25 patients were randomised. | |
| Interventions | Intervention 1: n = 10 received single dose of 3.75mg Anatibant.
Intervention 2: n = 10 received single dose of 22.5mg Anatibant.
Control: n = 5 received placebo. Administered within eight hours of injury, or 12 hours if surgery was required. |
|
| Outcomes | GOS at one, three and six months after injury. Pharmacokinetic assessments. ICP recorded for the first five days. Therapeutic interventions. Vital signs and body temperature. Assessment of GCS ‐ daily. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Adequate sequence generation? | Low risk | A computer‐generated randomisation was used to allocate patients. |
| Allocation concealment? | Unclear risk | Patients were sequentially assigned to a randomisation number and to the matching treatment. |
| Blinding? All outcomes | Unclear risk | Clinicians were blinded to allocation status. It is unclear whether long‐term outcome assessors were blind to allocation status. |
| Incomplete outcome data addressed? All outcomes | Unclear risk | Twenty‐five patients were randomised; one patient in the control group died before treatment and was excluded from the trial's analyses (NB this death has been included in this review's analyses). 8% of patients were lost to follow up at six months. |
| Free of selective reporting? | High risk | We were unable to locate the protocol for this trial. In the trial's methods section it stated that outcome data on intracranial pressure were collected, however these data were not reported in the results section. |
| Free of other bias? | High risk | Due to dilution errors the exact doses of 3.75mg and 22.5mg were not administered in some patients. Comparison of baseline characteristics indicated that patients randomised to intervention group were more severely injured, and amongst intervention groups those in 3.75mg group were more severely injured than the 22.5mg group. |
Narotam 1998.
| Methods | Trial location: Durban, South Africa. | |
| Participants | Patients aged 12 years and over with focal closed traumatic brain injury (GCS 9 to 14), presenting within 24 and 96 hours of injury. 20 patients were randomised. | |
| Interventions | Two groups. Intervention: n = 11 received Bradycor CP‐0127 (3ug/kg/min) over a seven day period. Control: n = 9 received placebo (lactated Ringer's solution). | |
| Outcomes | Control of ICP, improvement in neurological function (measured by GCS), need for surgical interventions to excise the contusion. Monitoring of vital signs and laboratory parameters. Maintenance of adequate CPP. Adverse events. 14‐day all‐cause mortality. | |
| Notes | Trial funded by Cortech Inc ‐ inventor and owner of CP‐0127. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Adequate sequence generation? | Low risk | Patients were randomised according to a computer‐generated randomisation code. |
| Allocation concealment? | Unclear risk | No information reported. |
| Blinding? All outcomes | Low risk | This was an open label study. Two physicians in the study group were aware of allocation status for safety reasons, although actively treating physicians were blinded. Outcome assessors were blind to allocation status. |
| Incomplete outcome data addressed? All outcomes | Low risk | No withdrawals, drop‐outs, attrition between groups or losses to follow up reported. |
| Free of selective reporting? | Unclear risk | We were unable to locate the protocol for this trial. |
| Free of other bias? | Low risk | The study appears to be free of other sources of bias. |
Differences between protocol and review
The assessment of methodological quality used in earlier versions of this review, and for the protocol, has been replaced with an assessment of the risk of bias. This amendment is in response to a change in The Cochrane Collaboration's methodological guidance.
Contributions of authors
KB devised the search strategy and ran the searches. KK and KB screened the search output, assessed papers for eligibility and extracted data. KK entered the data into RevMan and performed the analyses. KK took the lead in writing the review with KB contributing as required.
Sources of support
Internal sources
London School of Hygiene & Tropical Medicine, UK.
External sources
No sources of support supplied
Declarations of interest
None known.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
BRAIN 2009 {published data only}
- Shakur H, Andrews P, Asser T, Balica L, Boeriu C, Diego Ciro Quintero J, et al. The BRAIN TRIAL: a randomised, placebo controlled trial of a Bradykinin B2 receptor antagonist (Anatibant) in patients with traumatic brain injury. Trials 2009;10:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Marmarou 1999 {published data only}
- Marmarou A, Nichols J, Burgess J, Newell D, Troha J, Burnham D, et al. Effects of the bradykinin antagonist Bradycor (deltibant, CP‐1027) in severe traumatic brain injury: results of a multi‐center, randomized, placebo‐controlled trial. American Brain Injury Consortium Study Group. Journal of Neurotrauma 1999;16(6):431‐44. [DOI] [PubMed] [Google Scholar]
Marmarou 2005 {published data only}
- Marmarou A, Guy M, Murphey L, Roy F, Layani L, Combal JP, et al. A single dose, three‐arm, placebo‐controlled, phase I study of the bradykinin B2 receptor antagonist Anatibant (LF16‐0687Ms) in patients with severe traumatic brain injury. Journal of Neurotrauma 2005;22(12):1444‐55. [DOI] [PubMed] [Google Scholar]
Narotam 1998 {published data only}
- Narotam PK, Rodell TC, Nadvi SS, Bhoola KD, Troha JM, Parbhoosingh R, et al. Traumatic brain contusions: a clinical role for the kinin antagonist CP‐0127. Acta Neurochirurgica 1998;140(8):793‐802. [DOI] [PubMed] [Google Scholar]
Additional references
Bruns 2003
- Bruns J, Hauser A. The epidemiology of traumatic brain injury: a review. Epilepsia 2003;10:2‐10. [DOI] [PubMed] [Google Scholar]
CDC 2006
- National Center for Injury Prevention and Control. Traumatic brain injury ‐ factsheet. CDC ‐ www.cdc.gov/ncipc/tbi 2006.
Francel 1992
- Francel PC. Bradykinin and neuronal injury. Journal of Neurotrauma 1992;9 Suppl 1:27‐45. [PubMed] [Google Scholar]
Higgins 2008
- Higgins JPT, Green S, editors. Cochrane Handbook for Systematic Reviews of Interventions Version 5.0.0 [updated February 2008]. The Cochrane Collaboration, 2008. Available from www.cochrane‐handbook.org.
Ivashkova 2006
- Ivashkova Y, Svetnitsky A, Mayzler O, Pruneau D, Benifla M, Fuxman Y, et al. Bradykinin B2 receptor antagonism with LF 18‐1505T reduces brain edema and improves neurological outcome after closed head trauma in rats. Journal of Trauma 2006;61(4):879‐85. [DOI] [PubMed] [Google Scholar]
Kaplanski 2002
- Kaplanski J, Pruneau D, Asa I, Artru A, Azez A, Ivashkova Y, et al. LF 16‐0687 Ms, a bradykinin B2 receptor antagonist, reduces brain edema and improves long‐term neurological function recovery after closed head trauma in rats. Journal of Neurotrauma 2002;19(8):953‐64. [DOI] [PubMed] [Google Scholar]
Mayhan 1996
- Mayhan WG. Role of activation of bradykinin B2 receptors in disruption of the blood‐brain barrier during acute hypertension. Brain Research 1996;738:337‐41. [DOI] [PubMed] [Google Scholar]
Reilly 1975
- Reilly PL, Graham DI, Adams JH, Jennett B. Patients with head injury who talk and die. Lancet 1975;2(7931):375‐7. [DOI] [PubMed] [Google Scholar]
Stover 2000
- Stover JF, Dohse NK, Unterberg AW. Significant reduction in brain swelling by administration of nonpeptide kinin B2 receptor antagonist LF 16‐0687Ms after controlled cortical impact injury in rats. Journal of Neurosurgery 2000;92(5):853‐9. [DOI] [PubMed] [Google Scholar]
Tagliaferri 2006
- Tagliaferri F, Compagnone C, Korsic M, Servadei F, Kraus J. A systematic review of brain injury epidemiology in Europe. Acta Neurochirurgica 2006;148(3):255‐68. [DOI] [PubMed] [Google Scholar]
Thornhill 2000
- Thornhill S, Teasdale GM, Murray GD, McEwen J, Roy CW, Penny KI. Disability in young people and adults one year after head injury: prospective cohort study. BMJ 2000;320(7250):1631‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Unterberg 1991
- Unterberg A, Baethmann A, Lanksch W. Prevention and treatment of secondary brain damage. Clinical aspects. Chest 1991;100:200‐2. [PubMed] [Google Scholar]
Unterberg 2004
- Unterberg AW, Stover J, Kress B, Kiening KL. Edema and brain trauma. Neuroscience 2004;129:1021‐9. [DOI] [PubMed] [Google Scholar]
Vincent 2005
- Vincent JL, Berré J. Primer on medical management of severe brain injury. Critical Care Medicine 2005;33(6):1392‐9. [DOI] [PubMed] [Google Scholar]
Whitnall 2006
- Whitnall L, McMillan TM, Murray GD, Teasdale GM. Disability in young people and adults after head injury: 5 ‐ 7 year follow up of a prospective cohort. Journal of Neurology, Neurosurgery and Psychiatry 2006;77:640‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
WHO 2006
- Neurological Disorders ‐ Public Health Challenges. WHO 2006.