

Abstract
Background
Drug resistance is common in focal epilepsy. In this update, we summarised the current evidence regarding add‐on levetiracetam in treating drug‐resistant focal epilepsy. The original review was published in 2001 and last updated in 2012.
Objectives
To evaluate the effectiveness of levetiracetam when used as an add‐on treatment for people with drug‐resistant focal epilepsy.
Search methods
We searched the Cochrane Register of Studies (CRS Web, which includes the Cochrane Epilepsy Group Specialized Register and CENTRAL), MEDLINE Ovid, ClinicalTrials.gov, and the WHO International Clinical Trials Registry Platform (ICTRP) to November 2018. We contacted the manufacturers of levetiracetam and researchers in the field to seek any ongoing or unpublished trials.
Selection criteria
Randomised, placebo‐controlled trials of add‐on levetiracetam treatment in people with drug‐resistant focal epilepsy.
Data collection and analysis
Two review authors independently selected trials for inclusion, assessed trials for bias, extracted data, and evaluated the overall certainty of the evidence. Outcomes investigated included 50% or greater reduction in focal seizure frequency (response), treatment withdrawal, adverse effects (including a specific analysis of changes in behaviour), cognitive effects, and quality of life (QoL). Primary analysis was intention‐to‐treat. We performed meta‐analysis for all outcomes using a Mantel‐Haenszel approach and calculated risk ratios (RR), with 95% confidence intervals (CI) for all estimates apart from adverse effects (99% CIs). We assessed heterogeneity using a Chi² test and the I² statistic.
Main results
This update included 14 trials (2455 participants), predominantly possessing low risks of bias. Participants were adults in 12 trials (2159 participants) and children in the remaining two (296 participants). The doses of levetiracetam tested were 500 mg/day to 4000 mg/day in adults, and 60 mg/kg/day in children. Treatment ranged from 12 to 24 weeks. When individual doses were examined, levetiracetam at either 500 mg/day or 4000 mg/day did not perform better than placebo for the 50% or greater reduction in seizure frequency outcome (500 mg: RR 1.60, 95% CI 0.71 to 3.62; P = 0.26; 4000 mg: RR 1.64, 95% CI 0.59 to 4.57; P = 0.34). Levetiracetam was significantly better than placebo at all other individual doses (1000 mg to 3000 mg). RR was significantly in favour of levetiracetam compared to placebo when results were pooled across all doses (RR 2.37, 95% CI 2.02 to 2.78; 14 studies, 2455 participants; moderate‐certainty evidence). Dose–response analysis demonstrated that the odds of achieving response (50% or greater reduction in seizure frequency) were increased by nearly 40% (odds ratio (OR) 1.39, 95% CI 1.23 to 1.58) for each 1000 mg increase in dose of levetiracetam. There were important levels of heterogeneity across multiple comparisons.
Participants were not significantly more likely to experience treatment withdrawal with levetiracetam than with placebo (pooled RR 1.11, 95% CI 0.89 to 1.40; 13 studies, 2428 participants; high‐certainty evidence).
Somnolence was the most common adverse effect, affecting 13% of participants, and it was significantly associated with levetiracetam compared to placebo (pooled RR 1.62, 99% CI 1.19 to 2.20; 13 studies, 2423 participants; moderate‐certainty evidence). Changes in behaviour were negligible in adults (1% affected; RR 1.79, 99% CI 0.59 to 5.41), but significant in children (23% affected; RR 1.90, 99% CI 1.16 to 3.11). Levetiracetam had a positive effect on some aspects of cognition and QoL in adults and worsened certain aspects of child behaviour.
Authors' conclusions
Overall, this review update finds that in both adults and children with drug‐resistant focal epilepsy, levetiracetam added on to usual care is more effective than placebo at reducing seizure frequency, it is unlikely to be stopped by patients, and it has minimal adverse effects outside of potential worsening behaviour in children. These findings are unchanged from the previous review update in 2012. This review update contributes two key additional findings: 1. a 500 mg daily dose of levetiracetam is no more effective than placebo at reducing seizures; and 2. the odds of response (50% reduction in seizure frequency) are increased by nearly 40% for each 1000 mg increase in dose of levetiracetam.
It seems reasonable to continue the use of levetiracetam in both adults and children with drug‐resistant focal epilepsy.
Plain language summary
Levetiracetam add‐on for drug‐resistant focal epilepsy
This is an updated version of the Cochrane Review first published in 2001 and last updated in Issue 9, 2012 of the Cochrane Database of Systematic Reviews.
Background and objective
Epilepsy is a one of the most common disorders of the brain, affecting over 70 million people worldwide. Levetiracetam is an antiepileptic drug widely used around the world. In this review, we summarised the current evidence regarding its effectiveness when used as a treatment added on to usual care in people experiencing focal epilepsy that responds poorly to medication.
Methods
The evidence is current to 26 November 2018. Fourteen studies in which people were randomly assigned to either levetiracetam or a dummy tablet (placebo) were included, with a total of 2455 participants (296 of whom were children). Everybody had their usual medications continued, meaning that nobody was left without taking an antiepileptic medicine. Among other things, we looked at how many people had their frequency of seizures reduced by 50% or more when taking levetiracetam versus when taking the placebo tablet. We combined the results of all of these people in order to increase our confidence in how effective levetiracetam is.
Key results
Levetiracetam significantly reduced the frequency of seizures in both adults and children. Just over half of children responded to levetiracetam, and 34% of adults also responded. These responses were much higher than in the placebo groups, indicating that levetiracetam was better than placebo. The most effective doses were those of 1000 mg to 3000 mg levetiracetam. For every 1000 mg increase in dose, the chances that levetiracetam would reduce seizures (fits) appeared to improve by 40%. We noticed that the results were very different in each study we looked at. Therefore, although we could see that levetiracetam works, it was difficult for us to be certain about how large that effect actually is.
Levetiracetam was generally tolerated well by adults and children. Most people managed to complete their course of levetiracetam during the studies. There were very few side effects with levetiracetam. The only side effect that was significantly associated with levetiracetam was somnolence (sleepiness). However, we also noticed that the behaviour of some children taking levetiracetam could worsen significantly.
Overall, it seems reasonable to add levetiracetam to a patient's usual antiepileptic medications if they have focal epilepsy that has responded poorly to other medications.
Summary of findings
Summary of findings 1. Levetiracetam compared to placebo for drug‐resistant focal epilepsy.
| Levetiracetam compared to placebo for drug‐resistant focal epilepsy | ||||||
| Patient or population: drug‐resistant focal epilepsy Setting: outpatients Intervention: levetiracetam Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) |
Relative effect
(95% CI) (99% CI for adverse events) |
№ of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with levetiracetam | |||||
| 50% or greater reduction in focal seizure frequency (responders) intention to treat– all doses | Study population | RR 2.37 (2.02 to 2.78) | 2455 (14 RCTs) | ⊕⊕⊕⊝ Moderatea,b | The odds of response (50% reduction in seizure frequency) was increased by nearly 40% (OR 1.39, 95% CI 1.23 to 1.58) for each 1000 mg increase in dose of levetiracetam. | |
| 172 per 1000 | 404 per 1000 (344 to 473) | |||||
| Treatment withdrawal– all doses | Study population | RR 1.11 (0.89 to 1.40) | 2428 (13 RCTs) | ⊕⊕⊕⊕ Highb | There was no effect on the odds of withdrawal of treatment (OR 0.99, 95% CI 0.85 to 1.15) for each 1000 mg increase in dose of levetiracetam. | |
| 114 per 1000 | 127 per 1000 (101 to 160) | |||||
| Adverse effects: 5 most common adverse effects (any age) – somnolence | Study population | RR 1.62 (1.19 to 2.20) | 2423 (13 RCTs) | ⊕⊕⊕⊝ Moderatec | — | |
| 97 per 1000 | 158 per 1000 (116 to 214) | |||||
| Adverse effects: 5 most common adverse effects (any age) – headache | Study population | RR 0.85 (0.60 to 1.21) | 2423 (13 RCTs) | ⊕⊕⊝⊝ Lowa,c | — | |
| 87 per 1000 | 74 per 1000 (52 to 106) | |||||
| Adverse effects: 5 most common adverse effects (any age) – dizziness | Study population | RR 1.54 (0.99 to 2.42) | 2423 (13 RCTs) | ⊕⊕⊕⊝ Moderatec | — | |
| 49 per 1000 | 76 per 1000 (49 to 119) | |||||
| Adverse effects: 5 most common adverse effects (any age) – fatigue (asthenia) | Study population | RR 1.53 (0.98 to 2.38) | 2423 (13 RCTs) | ⊕⊕⊕⊝ Moderatec | — | |
| 45 per 1000 | 69 per 1000 (44 to 107) | |||||
| Adverse effects: 5 most common adverse effects (any age) – accidental injury | Study population | RR 0.72 (0.49 to 1.06) | 2423 (13 RCTs) | ⊕⊕⊝⊝ Lowa,c | — | |
| 74 per 1000 | 53 per 1000 (36 to 78) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; OR: odds ratio; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded one level due to inconsistency: potentially important heterogeneity present between studies (I² > 45%). bHigh or unclear risk of bias present in 9/14 studies. However, sensitivity analysis restricted to studies of low risk of bias only showed similar numerical results and no changes to conclusions. Therefore, no downgrade for risk of bias. cDowngraded one level due to risk of bias: high or unclear risk of bias present in 9/14 studies (sensitivity analysis not conducted as we deemed it inappropriate to exclude any adverse events).
Background
This is an update to a Cochrane Review that was originally published in 2001 (Chaisewikul 2001), and last updated in 2012 (Mbizvo 2012).
Description of the condition
Epilepsy is a one of the most common disorders of the brain, affecting over 70 million people worldwide. Generally, it is defined as two or more unprovoked seizures occurring more than 24 hours apart, a single unprovoked seizure if recurrence risk is high (a greater than 60% risk over the next 10 years), or the specific diagnosis of an epilepsy syndrome (Fisher 2015). In 2017, the International League Against Epilepsy (ILAE) updated the classification and terminology of seizures and epilepsy to incorporate progress in our understanding of epilepsy since the original 1989 classification and subsequent 2010 revision (Berg 2010; Fisher 2017a; Fisher 2017b; Scheffer 2017). There are now four levels in which a diagnosis of epilepsy can be characterised, and these allow for the varying availability of diagnostic resources worldwide (Fisher 2017a; Fisher 2017b; Scheffer 2017). A level one diagnosis requires a clinician to recognise that a person has had two or more unprovoked seizures, rather than some other types of paroxysmal events. A level two diagnosis subclassifies the epilepsy based on seizure type, where the 'focal' and 'generalised' seizure categories are now supplemented with the two additional categories 'generalised and focal epilepsy' and 'unknown if generalised or focal epilepsy'. These recognise that not all epilepsies can be dichotomised into the simple forms of focal or generalised. A level three diagnosis subclassifies epilepsy by syndrome (e.g. Dravet syndrome, West syndrome, or Lennox‐Gastaut syndrome). A level four diagnosis subclassifies epilepsy by aetiology, and can include complex diagnostic information from neuroimaging, genetics, immunology, and environmental factors (Fisher 2017a; Fisher 2017b; Scheffer 2017).
People with epilepsy not only suffer from the direct consequences of seizures, which include injuries, infections (such as aspiration pneumonia), and death (including a 1% risk of sudden unexpected death in epilepsy), but they also experience multiple neurobiological, cognitive, and psychosocial complications of epilepsy. Low‐ and middle‐income countries contribute nearly 80% of the global burden of epilepsy. This is largely due to the presence of tropical infections such as neurocysticercosis associated with poorer standards of hygiene and health care, and also because there tends to be a large number of children among these populations (Shorvon 1996). The incidence of epilepsy peaks in early childhood before falling to low levels in early adult life and then rising again among the elderly population (Shorvon 1996). Therefore, epilepsy remains a significant burden of disease in high‐income countries not least because of the ageing population structure. The UK National General Practice Study of Epilepsy found that of the 60% of people with epilepsy who have motor seizures, focal‐onset epilepsy is more common than generalised‐onset epilepsy, affecting two‐thirds and one third of the people, respectively (NICE 2012).
The goal of epilepsy treatment is to achieve sustained seizure freedom and to achieve this using a tolerated antiepileptic drug (AED) schedule. Unfortunately, over 75% of people with epilepsy remain untreated and this is mostly resultant from the concentration of epilepsy within low‐ and middle‐income countries. Surprisingly, many people in low‐income countries enter long‐term remission from their epilepsy without AEDs, suggesting that a drug‐independent mechanism to long‐term remission may exist. However, it is doubtless that the use of AEDs is associated with favourable outcomes for people with epilepsy. Various combinations of AEDs can be used to try and achieve those outcomes, with varying success rates. The prognosis in newly diagnosed epilepsy is usually good, with up to 50% of people entering remission (seizure‐freedom for five years on or off treatment) either without treatment or on their first AED (Brodie 2010; Maguire 2011). An additional 10% achieve remission on a second or third drug (Brodie 2010). For the remainder, AEDs may fail to provide remission from seizures. Pharmacoresistance or intolerable drug‐related adverse effects, or both, are major contributors to this. Drug‐resistant epilepsy is defined by the ILAE as that in which there has been failure of adequate trials of two tolerated and appropriately chosen and used AED schedules (whether as monotherapies or in combination) to achieve sustained seizure freedom (Kwan 2010). No seizure frequency requirement is necessary to meet this definition. This allows for those people with infrequent seizures (e.g. occurring once a year) to still be regarded as drug‐resistant, which is relevant to the impact seizures have on lifestyle factors such as driving.
Description of the intervention
Levetiracetam is one of a second generation of AEDs introduced in the late 1990s to early 2000s. It is extensively used worldwide and in all ages for the treatment of drug‐resistant focal epilepsy and other types of epilepsy (Lyseng‐Williamson 2011). A third generation of AEDs has been introduced more recently, since approximately 2010, and these include levetiracetam's newer selective analogue brivaracetam, and others such as eslicarbazepine, perampanel, and vigabatrin (Coppola 2017; Hanaya 2016). However, the second generation is broadly considered the first line of AED therapy in routine clinical practice. This is because there is extensive evidence that these drugs are generally well tolerated (more so than the first generation of AEDs such as phenytoin, carbamazepine, and sodium valproate), and they are largely non‐inferior to both first‐ and third‐generation AEDs (although formal head‐to‐head comparisons remain scarce) (Hanaya 2016). Even since the last update to this review in 2012, levetiracetam has established itself as one of the most popular AED options in routine clinical practice. This is largely because outside of its clinical efficacy in reducing seizures, particularly favourable adverse effects profile, limited interaction with other drugs, and ability to be uptitrated rapidly (Gambardella 2008). These make it an uncomplicated option for clinicians to use. However, it remains important that the evidence base behind its use is kept up‐to‐date, both in terms of efficacy and also in terms of adverse events, so that frontline clinicians are able to help patients come to the most accurately informed decision about their therapy. This information is particularly pressing now given the availability of a host of third‐generation AED options available for patients to choose. Furthermore, in the absence of formal head‐to‐head comparisons of levetiracetam against other AEDs, updated systematic review information on its efficacy is likely to be important for ongoing network meta‐analyses comparing levetiracetam and other AEDs to a common reference AED treatment.
Levetiracetam was first introduced onto the market in April 2000. It has been available as a generic brand in the US since 2008 and in the UK since 2011 (Mbizvo 2012). The drug is available for use by mouth or intravenous infusion. For oral use in adults aged 16 years and over, it is given at an initial dose of 250 mg once daily for one to two weeks, then increased to 250 mg twice daily, then increased in steps of 250 mg twice daily (with the maximum per dose being 1.5 g twice daily), adjusted according to response, with dose to be increased every two weeks (BNF 2019a). Titration can be performed more quickly than this in emergency circumstances. For oral use in children under 16 years of age, it is given at an initial dose of 7 mg/kg to 10 mg/kg once daily, then increased in steps of up to 7 mg/kg to 10 mg/kg twice daily (maximum per dose 30 mg/kg twice daily), with dose to be increased every two weeks (BNF 2019b).
Levetiracetam is only licensed for use in the following circumstances for adults and children by mouth or intravenous infusion: as either monotherapy or add‐on therapy for focal‐onset seizures with or without evolution to bilateral tonic‐clonic seizures, and as add‐on therapy for generalised‐onset motor seizures (including myoclonic seizures and tonic‐clonic seizures) (BNF 2019a). However, in routine clinical practice, it is now popular as an unlicensed monotherapy for generalised‐onset motor seizures also, particularly as a less teratogenic alternative to sodium valproate in women of childbearing age with epilepsy (Tomson 2015). It is also now used frequently as an unlicensed intravenous treatment option in status epilepticus (Cock 2011). A discussion on the unlicensed or intravenous use of levetiracetam is outside of the scope of this current review. This review focuses on updating the evidence base surrounding oral levetiracetam use as add‐on therapy in drug‐resistant focal epilepsy. The review will draw information from randomised, placebo‐controlled trials of add‐on levetiracetam treatment in people (of any age) with drug‐resistant focal epilepsy.
How the intervention might work
Levetiracetam possesses both antiepileptic and anti‐epileptogenic properties (Betts 2000). Its exact mode of action is not completely understood (Xiao 2009). It binds to, and modulates, the synaptic vesicle protein 2A (SV2A); a protein that has some controlling effect on neurotransmitter release from presynaptic vesicles (Gillard 2006; Lynch 2004). It also selectively inhibits N‐type Ca2+ channels and decreases intracellular calcium‐ion increase (both of which negatively impact neurotransmitter release) (Lukyanetz 2002; Niespodziany 2001). There is evidence that it releases γ‐aminobutyric acid (GABA) activity and glycine‐gated currents by acting on their negative allosteric modulators, namely zinc and the beta‐carbolines (Rigo 2002). Neuroprotective effects have also been described (Gibbs 2006). The proposed mechanisms of action of levetiracetam have been largely derived from animal‐model studies, and the results remain to be validated in humans.
With regard to pharmacokinetics, levetiracetam generally demonstrates a favourable profile. Bioavailability is the fraction of a drug's administered dose that reaches the systemic circulation. When a drug is administered orally, bioavailability can be reduced by factors such as the rates of absorption and first‐pass gut and hepatic metabolism. Oral levetiracetam provides close to 100% bioavailability, making it largely bioequivalent to intravenous levetiracetam (Trinka 2011). A drug's susceptibility to oxidative hepatic metabolism and its influence on cytochrome P450 enzyme function in the liver can largely determine the duration and intensity of the pharmacological action of that drug, and its interaction with other drugs. Levetiracetam is advantaged by a lack of oxidative hepatic metabolism or influence on cytochrome P450 enzyme function. Dosing is thus simplified in both adults and children by linear, dose‐proportional kinetics. Plasma concentrations of levetiracetam peak at one hour, and a steady‐state concentration is reached by 48 hours with repeated dosing (usually twice daily). The drug shows no significant pharmacokinetic interactions with other AEDs or with drugs such as warfarin, digoxin, and the oral contraceptive pill; which all interact with the aforementioned hepatic enzyme systems. Clearance of levetiracetam is exclusively renal: 66% unchanged and 24% as an inactive metabolite following hydrolysis of its acetamide group in the blood. Clearance is 30% to 40% higher in children and it is impaired in elderly people or in people with renal impairment (Crepeau 2010; Glauser 2006; Pellock 2001). Dose lowering is required in renal impairment: in adults, at a maximum of 2000 mg daily if the estimated glomerular filtration rate (eGFR) is 50 mL/minute/1.73 m² to 80 mL/minute/1.73 m², 1500 mg if eGFR is 30 mL/minute/1.73 m² to 50 mL/minute/1.73 m², and 1000 mg if eGFR is less than 30 mL/minute/1.73 m² (BNF 2019a). For children, dose is reduced if the eGFR is less than 80 mL/minute/1.73 m² (BNF 2019a).
Why it is important to do this review
It is important that estimates of the overall efficacy, tolerability and safety profiles of AEDs remain updated as new studies are published. This is to help ensure clinicians and patients make informed decisions on treatment using the most accurate and up‐to‐date information available.
Objectives
To evaluate the effectiveness of levetiracetam when used as an add‐on treatment for people with drug‐resistant focal epilepsy.
Methods
Criteria for considering studies for this review
Types of studies
Trials had to meet the following criteria.
Randomised controlled trials (RCTs): included trials were those for which the study author had described the trial as 'a randomised controlled trial' (or words to that effect). A judgement was then made on the risk of selection bias of the included trials, based on the reported methods of random list generation and allocation concealment (see Assessment of risk of bias in included studies for details on which methods conferred a low risk of selection bias).
Placebo‐controlled.
Double, single, or unblinded: a judgement was then made on the risk of performance and detection biases being present in the trial (see Assessment of risk of bias in included studies).
Parallel or cross‐over design: for cross‐over trials, the first treatment period was treated as a parallel trial (i.e. only data from the first treatment period were used).
Consist of a treatment period of at least eight weeks in duration.
Types of participants
Participants had to meet all of the following criteria.
Any age, gender, and ethnic background.
Experiencing drug‐resistant focal epilepsy: that is experiencing focal seizures with or without impairment of consciousness or awareness, with or without evolution to bilateral, convulsive seizures (involving tonic, clonic, or tonic and clonic components). There has been a lack of consensus between studies when defining drug resistance (Mbizvo 2012). Therefore, to allow a fair and inclusive evaluation of all trials that have been said to involve drug‐resistant participants, a specific cut‐off for number of background AEDs and the time period on these was not set. Instead, the requirement was for trials to have described participants on AEDs as having 'failed to respond' or having 'refractory', 'drug‐resistant', or 'uncontrolled' epilepsy (or words to that effect). Information was then collected on the duration of epilepsy, the number of AEDs tried, and the length of time during which seizures had not responded to those AEDs, and the minimum number of seizures required during that time for participants to have been included in the trial. Where relevant, a subgroup analysis was conducted to compare primary outcomes between studies where the mean duration of epilepsy was shorter (less than 12 months) and longer (12 months or greater).
Types of interventions
The active treatment group received treatment with levetiracetam in addition to conventional AED treatment.
The control group received matched placebo in addition to conventional AED treatment.
Types of outcome measures
Primary outcomes
1. 50% or greater reduction in focal seizure frequency
We chose the proportion of people with a 50% or greater reduction in focal seizure frequency in the treatment period compared to the prerandomisation baseline period as it is commonly reported in this type of study, and can be calculated for studies that do not report this outcome provided that baseline seizure data were reported. For the purposes of this review, we termed people who achieved 50% or greater reduction in focal seizure frequency 'responders'.
2. Treatment withdrawal
The proportion of people having treatment withdrawn during the course of the treatment period was used as a measure of global effectiveness. Treatment is likely to be withdrawn due to adverse effects, lack of efficacy, or a combination of both. It is also an outcome to which the participants make a direct contribution. In trials of short duration, it is likely that adverse effects will be the most common reason for withdrawal.
3. Adverse effects
3.1. Five most common adverse effects
The proportion of people experiencing the five most common adverse effects was reported.
3.2. General adverse effects
The proportion of people experiencing the following five adverse effects was also reported (where available and if different from the five most common adverse effects):
ataxia;
dizziness;
fatigue;
nausea;
somnolence.
These adverse effects were chosen as they were considered by the review authors to be common and important adverse effects of AEDs generally.
3.3. Behavioural adverse effects
The proportion of people experiencing adverse effects pertaining to changes in behaviour (e.g. aggression, agitation, anger, anxiety, apathy, depression, hostility, and irritability). Clinicians often consider changes in behaviour to be common adverse effects of levetiracetam (Asconapé 2001; NICE 2012; Penovich 2004).
Secondary outcomes
1. Cognitive effects
At present, there is no consensus as to which instruments should be used to assess the effects of AEDs on cognition, and as a result this has been approached in a heterogeneous way (Cochrane 1998). In view of this difficulty, we intended to tabulate results where a specific instrument had been used to assess the effects of levetiracetam on cognition, but made no attempt to combine the results in a meta‐analysis.
2. Quality of life
There is no consensus as to which instruments should be used to assess quality of life (QoL), and QoL data were also tabulated where a specific instrument had been used to assess the effects of levetiracetam on QoL. However, we made no attempt to combine the results in a meta‐analysis.
Search methods for identification of studies
Electronic searches
The first searches for the original review were in 2000. Subsequent searches were in September 2002, July 2005, January 2010, February 2011, April 2011, August 2012, March 2014, February 2015, March 2017, October 2017, and 26 November 2018. For the latest update, we searched the following databases. There were no language restrictions:
Cochrane Register of Studies (CRS Web, 26 November 2018) using the strategy set out in Appendix 1;
MEDLINE (Ovid, 1946 to November 21, 2018) using the strategy outlined in Appendix 2;
ClinicalTrials.gov (26 November 2018) using the strategy outlined in Appendix 3;
WHO International Clinical Trials Registry Platform (ICTRP; apps.who.int/trialsearch/; 26 November 2018) using the strategy outlined in Appendix 4.
CRS Web includes the Cochrane Epilepsy Group Specialized Register and the Cochrane Central Register of Controlled Trials (CENTRAL).
Searching other resources
References from published studies
We reviewed the reference lists of retrieved studies to search for additional reports of relevant trials.
Other sources
We contacted colleagues in the field for information about any unpublished or ongoing studies.
Data collection and analysis
Selection of studies
Two review authors (GKM and BC or PD) independently assessed trials for inclusion. We first screened titles and abstracts, followed by full‐text reports of potentially eligible trials. A third review author (AGM) resolved any disagreements.
Data extraction and management
Two review authors (GKM and BC or PD) extracted the information shown below from included trials, with any disagreements resolved by similar discussion. We contacted trial authors for any information missing from the published manuscript that was deemed relevant.
Publication details
Year of trial publication.
Methodological/trial design
Method of random sequence generation.
Method of randomisation concealment (allocation concealment).
Method of blinding (of participants, personnel, and investigators).
Whether any randomised participants had been excluded from reported analyses.
Duration of baseline period.
Duration of treatment period (uptitration and maintenance phases).
Dose(s) of levetiracetam tested.
Participant/demographic information
Total number of participants allocated to each treatment group.
Age and sex.
Country or continents from which the majority of participants had been recruited.
Duration of epilepsy.
Number with focal epilepsy.
Seizure classification.
Duration of time in which seizures were drug‐resistant.
Minimum seizure rate required for trial inclusion.
Seizure frequency during the baseline period.
Number of background AEDs.
Outcomes
Number of participants experiencing each outcome (see Types of outcome measures) was recorded per randomised group.
Assessment of risk of bias in included studies
Two review authors (GKM and PD or BC) independently assessed trials for the risks of bias listed below (Higgins 2011). A third review author (AGM) settled any disagreements. Where possible, we used published data, and sought unpublished data when details were unclear or unavailable.
Selection bias: were there adequate methods of random sequence generation and allocation concealment? Methods considered to confer a low risk of selection bias included those using random number tables/electronically generated random numbers for random sequence generation, and those using allocation of sequentially numbered sealed packages of medication, sealed opaque envelopes, or central/telephone randomisation for allocation concealment.
Performance bias: was knowledge of the allocated interventions by participants and personnel adequately prevented during the study? Methods considered to confer a low risk of performance bias included using packaging and tablets that were identical for levetiracetam and placebo.
Detection bias: was knowledge of the allocated interventions by outcome assessors prevented during the study? Studies were regarded at low risk of detection bias when it was specifically described that investigators/outcome assessors were blinded to treatment assignment.
Attrition bias: were incomplete outcome data adequately addressed? Reasons for low risks of this bias included no missing outcome data, reasons for missing outcome data unlikely to be related to true outcome, missing outcome data balanced in numbers across intervention groups, with similar reasons for missing data across groups, or for dichotomous outcome data, the proportion of missing outcomes compared with observed event risk not enough to have a clinically relevant impact on the intervention effect estimate.
Reporting bias: were reports of the study free of suggestion of selective outcome reporting? Risks were regarded as low when the results of all outcomes measured (where the outcome was also relevant to this review) were published.
In addition to providing overall estimates, a sensitivity analysis that excluded trials with unclear or high risks of any of the biases was performed for the primary outcome measure (50% or greater reduction in seizure frequency).
Measures of treatment effect
For dichotomous outcomes, the preferred measure of treatment effect was the Mantel‐Haenszel risk ratio (RR). We reported the outcomes of 50% reduction in seizure frequency and treatment withdrawal with 95% confidence intervals (CI). For individual adverse effects, we used 99% CI to make allowance for multiple testing.
We summarised continuous outcomes (cognitive outcomes and QoL) in tables and in the text rather than enter data into analysis due to our perceived variability of how included studies would assess these outcomes (see Secondary outcomes).
Unit of analysis issues
The unit of allocation and analysis was the participant for all included trials.
We considered a cross‐over trial design as eligible for this review. However, for such studies, we used the first treatment period as a parallel trial (i.e. used only data from the first treatment period).
Dealing with missing data
All analyses included all participants in the treatment groups to which they had been allocated (i.e. an intention‐to‐treat (ITT) approach).
For the efficacy outcome (50% or greater reduction in seizure frequency), we undertook three analyses: primary ITT analysis, worse‐case analysis, and best‐case analysis.
Primary intention‐to‐treat analysis
For this, all randomised participants were analysed in the treatment group to which they had been allocated, irrespective of the treatment that they actually received. Participants randomised but excluded from analysis (e.g. for not completing follow‐up or with inadequate seizure data) were assumed non‐responders.
Worse‐case analysis
Participants randomised but excluded from analysis (e.g. for not completing follow‐up or with inadequate seizure data) were assumed non‐responders in the levetiracetam group and responders in the placebo group.
Best‐case analysis
Participants randomised but excluded from analysis (e.g. for not completing follow‐up or with inadequate seizure data) were assumed responders in the levetiracetam group and non‐responders in the placebo group.
Assessment of heterogeneity
Statistical heterogeneity between trials was checked for each outcome using a Chi² test for heterogeneity and the I² statistic, interpreted as follows (Higgins 2011):
0% to 40%: heterogeneity levels might not be important;
30% to 60%: may represent moderate heterogeneity;
50% to 90%: may represent substantial heterogeneity;
75% to 100%: considerable heterogeneity.
The 2012 update already included regression modelling to assess for trial factors that might explain significant levels of heterogeneity (see Mbizvo 2012). This review update includes a random‐effects model to report results where the levels of heterogeneity were considered important (see Data synthesis). This review also uses updated regression methods to augment the dose–response analysis (see Subgroup analysis and investigation of heterogeneity).
Assessment of reporting biases
To assess selective reporting bias, we assessed the consistency of the measurements and outcomes planned by the original investigators during the trial with those reported within the published paper by comparing the trial protocols (when available) with the information given in the final publication. Where protocols were not available, we compared the 'Methods' and the 'Results' sections of the published papers. We also used our knowledge of the clinical background to identify standard outcome measures usually taken but not reported by the trial investigators.
Where there were 10 or more studies for any comparison or outcome, we investigated the presence of publication bias by inspecting a funnel plot for asymmetry.
Data synthesis
We performed meta‐analysis for all dichotomous outcomes using a Mantel‐Haenszel approach and an RR as the measure of treatment effect.
Provided there was no important heterogeneity (where important heterogeneity was defined as P < 0.05 on Chi² test, I² > 50%, or both), we used a fixed‐effect model for analysis. Where there was important heterogeneity, we included a random‐effects model to report results (see Subgroup analysis and investigation of heterogeneity).
Subgroup analysis and investigation of heterogeneity
We assessed clinical and statistical heterogeneity using the methods outlined in Assessment of heterogeneity. We conducted subgroup analyses separating adult and paediatric trials. We analysed dose–response for the outcomes 50% or greater reduction in focal seizure frequency and treatment withdrawal. We evaluated dose–response in trials with fixed doses (i.e. not doses based on weight) using a generalised linear mixed model (i.e. a model including both fixed and random effects) with the logit link function, as described in Turner 2000, and estimated using the command xtmelogit in STATA SE version 14 (Stata). Study and dose were included as fixed effects within the mixed model while treatment was included as a random‐effect within the mixed model (no random‐effect was included for the constant term of the mixed model). Dose was standardised to dose increases of 1000 mg. This method estimated an odds ratio (OR) as opposed to an RR.
Sensitivity analysis
We performed a sensitivity analysis of the primary outcomes of the review (where possible) based on the methodological quality of the studies, restricting meta‐analysis to only studies with a globally low risk of bias.
Summary of findings and assessment of the certainty of the evidence
We used the GRADE Working Group grades of evidence to provide a 'Summary of findings' table outlining the overall certainty of evidence, the magnitude of effect of the interventions examined, and the sum of available data on most important outcomes (i.e. 50% or greater reduction in seizure frequency, treatment withdrawal, and the five most common adverse effects) (Table 1; Schünemann 2009). Within this, 'assumed risk' (also called baseline risk) was the control event rate and was therefore a measure of the typical burden of these outcomes, and 'corresponding risk' was a measure of the burden of the outcomes after the intervention was applied (i.e. the risk of an outcome in levetiracetam‐treated people based on the relative magnitude of an effect and assumed (baseline) risk). The GRADE system classifies the certainty of evidence into one of four grades:
high: further research is very unlikely to change our confidence in the estimate of effect;
moderate: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate;
low: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate;
very low: any estimate of effect is very uncertain.
A judgement was made on the individual trials used to provide the pooled effect estimates and the certainty of evidence was then downgraded by the presence of bias, inconsistency (heterogeneity), indirectness, imprecision, and publication bias; and upgraded by the presence of a large effect and a dose–response gradient. Only studies with no threats to validity (not downgraded for any reason) can be upgraded.
Two review authors (GKM and SN) independently conducted this process by with any disagreements resolved by discussion with a third review author (AGM).
Results
Description of studies
Results of the search
See Figure 1 for a flow‐diagram summary of the results of database searches and records identified from other sources. The previous update of this review included 11 trials, and four potentially eligible trials remained as awaiting classification pending receipt of further information about the trials (Inoue 2015 (as N01221); Yagi 2010; Zheng 2009; Boon 2002). Inoue 2015, Yagi 2010, and Zheng 2009 are now included in the current review following subsequent receipt of further information. There was no further information for Boon 2002, so this trial has been excluded. The search identified NCT01392768 as an additional potentially eligible trial that awaits classification pending receipt of further information from authors (see Characteristics of studies awaiting classification).
1.
Study flow diagram.
Included studies
See Characteristics of included studies table.
This update included 14 trials (2455 participants), seven of which were published subsequent to the original 2001 review (Glauser 2006; Levisohn 2009; Peltola 2009; Tsai 2006; Wu 2009; Xiao 2009; Zhou 2008). The 2001 review analysed four included studies using both published and unpublished trial information and data (Ben‐Menachem 2000; Betts 2000; Cereghino 2000; Shorvon 2000). The unpublished information was obtained as prepublished study protocols provided by UCB S.A. Pharma sector. These study protocols were also available for use in this current review, in addition to their corresponding published manuscripts. The remaining trials were analysed using published data only (prepublished study protocols were sought, where relevant, but unobtainable). Data from Yagi 2010 and Zheng 2009 were successfully obtained subsequent to the 2012 review using a translator.
Participants were 296 children in two trials (age range four to 16 years) (Glauser 2006; Levisohn 2009). The remaining trials included 2159 adults aged over 16 years. Aside from one cross‐over trial (Shorvon 2000), all trials were parallel design. Trials involving children (Glauser 2006; Levisohn 2009), and trials published earlier (Ben‐Menachem 2000; Betts 2000; Cereghino 2000; Shorvon 2000), recruited from populations within various European countries and the US. Adult trials published later largely recruited from populations within Asian countries (mostly China and Taiwan) (Inoue 2015; Tsai 2006; Wu 2009; Yagi 2010; Zheng 2009; Zhou 2008). One trial recruited from various countries (centres in Finland, India, Mexico, Russia, South Africa, and Ukraine) (Peltola 2009).
Three trials did not report the mean duration of epilepsy (Cereghino 2000; Levisohn 2009; Zheng 2009). For the Cereghino 2000 trial, participants had to have experienced uncontrolled focal epilepsy for at least two years, with a minimum of 12 focal seizures within 12 weeks before study selection and two focal seizures occurring per four weeks during the 12‐week baseline period. This was on a background of at least two AEDs taken simultaneously or consecutively. For the Levisohn 2009 trial, participants had to have experienced uncontrolled focal epilepsy for a minimum of six months, with a minimum of one focal seizure during the four weeks prior to screening. This was on a background of one or two AEDs. Across the remaining trials, the mean duration of epilepsy ranged from seven to 26 years. Within these, the Betts 2000 trial required a minimum of at least four seizures in the six months prior to study entry. Three trials required at least two seizures per four weeks in their 12‐week (Ben‐Menachem 2000; Inoue 2015), and their eight‐week (Peltola 2009), baseline periods. The Zheng 2009 trial required eight seizures during the eight‐week baseline period. Yagi 2010 did not provide information regarding the number of seizures required prior to study entry during the eight‐week baseline period. The remaining six trials required at least four seizures per four weeks in their eight‐ or 12‐week baseline periods (Glauser 2006; Shorvon 2000; Tsai 2006; Wu 2009; Xiao 2009; Zhou 2008). This was on a background of one to four AEDs. The mean duration of epilepsy across all included trials did not range below 12 months.
Treatment periods consisted of the combination of an uptitration and a maintenance phase in all but two trials (Betts 2000 and Peltola 2009 did not involve uptitration). Duration of the treatment periods ranged from 12 to 24 weeks between trials (uptitration range zero to four weeks, maintenance range eight to 24 weeks). The doses of levetiracetam tested were 60 mg/kg/day for children, and a range of 500 mg/day to 4000 mg/day for adults. The Peltola 2009 trial was the only one that tested an extended‐release preparation of levetiracetam (1000 mg dose). The Inoue 2015 trial was the only one that tested levetiracetam 500 mg. The Betts 2000 trial was the only one that tested levetiracetam 4000 mg. We were able to calculate a 50% or greater reduction in seizure frequency for all 14 trials. All trials provided data for treatment withdrawal, while all but two trials (Zheng 2009; Zhou 2008), provided data for adverse effects. Generally, trials published an adverse effect if 5% or more of the participants in any treatment group were affected, but the Betts 2000 and Cereghino 2000 trials used a higher threshold of 10%.
Four trials provided data for cognitive effects and QoL outcomes in adults, but only 619/765 participants randomised to these trials were assessed with the relevant instruments (Betts 2000; Cereghino 2000; Shorvon 2000; Zhou 2008). One trial provided outcome data for cognitive as well as behavioural and emotional effects in children (Levisohn 2009). This trial assessed 73/99 participants randomised with the relevant instruments. The three new trials included in the current update did not contribute any data for the cognitive effects and QoL outcomes.
Overall, there was missing efficacy outcome (50% or greater reduction in seizure frequency) for 97 adults (70 randomised to levetiracetam and 27 to placebo). These participants contributed to the best‐ and worst‐case scenario analyses.
Excluded studies
We excluded one trial from the review (Boon 2002). This was a cross‐over trial and data were not available from the first period to allow this trial to be included in analyses.
Risk of bias in included studies
Figure 2 and Figure 3 summarise the risk of bias of the included trials (see also Characteristics of included studies table).
2.

'Risk of bias' graph: review authors' judgements about each 'Risk of bias' item presented as percentages across all included studies (shown above).
3.

'Risk of bias' summary: review authors' judgements about each 'Risk of bias' item for each included study.
In summary, five RCTs were at overall low risk of bias (selection bias, performance bias, detection bias, attrition bias, and reporting bias; Ben‐Menachem 2000; Cereghino 2000; Peltola 2009; Shorvon 2000; Tsai 2006). For the remaining RCTs, risks were largely unclear although some risks of bias were high for the following trials: Betts 2000; Glauser 2006; Inoue 2015; Yagi 2010; Zheng 2009; Zhou 2008.
Allocation
Eight of the 14 trials described as RCTs provided details of an adequate method of sequence generation and allocation concealment to qualify them at low risk of selection bias (Ben‐Menachem 2000; Betts 2000; Cereghino 2000; Glauser 2006; Peltola 2009; Shorvon 2000; Tsai 2006; Zhou 2008). Five trials generated the random list using random permuted blocks, and concealed allocation by dispensing sequentially numbered sealed packages (Betts 2000; Cereghino 2000; Glauser 2006; Shorvon 2000; Tsai 2006). Ben‐Menachem 2000 achieved randomisation using a minimisation programme, which was concealed by using telephone randomisation. Participants were randomised in a 2:1 ratio to levetiracetam or placebo. Peltola 2009 achieved randomisation and allocation concealment using an interactive voice response system. Participants were randomised in a 1:1 ratio to levetiracetam or placebo. Zhou 2008 used a random numbers table for sequence generation, and participants received an exclusive random number consecutively on entry into the trial, with medication packaged by UCB S.A. Pharma.
The remaining six RCTs were at unclear risk of selection bias, for which full details on the method of random list generation or allocation concealment were not provided (Inoue 2015; Levisohn 2009; Wu 2009; Xiao 2009; Yagi 2010; Zheng 2009). The study sponsor generated randomisation codes for one trial (no further specification given), with participants assigned a randomisation number and given levetiracetam or placebo accordingly (Xiao 2009). This trial described an adequate method of allocation concealment (concealment via the use of numbered containers). Wu 2009 and Levisohn 2009 provided no details on the method of random sequence generation and allocation concealment, although Levisohn 2009 reported that participants were randomised in a 2:1 ratio to levetiracetam or placebo, and that randomisation was stratified for age (four to seven years, eight to 12 years, 13 to 16 years) and number of concomitant AEDs (one or two).
Blinding
All trials were described as double‐blind. Nine trials provided details that packaging and tablets were identical for levetiracetam and placebo and were therefore at low risk of performance bias (blinding of participants and personnel) (Ben‐Menachem 2000; Betts 2000; Cereghino 2000; Glauser 2006; Peltola 2009; Shorvon 2000; Tsai 2006; Wu 2009; Xiao 2009). The remaining five trials did not describe the method used to blind participants and personnel, and were at unclear risk of performance bias (Inoue 2015; Levisohn 2009; Yagi 2010; Zheng 2009; Zhou 2008). The risk of detection bias was low in eight trials that reported that the investigators were blinded to treatment assignment (Ben‐Menachem 2000; Betts 2000; Cereghino 2000; Glauser 2006; Peltola 2009; Shorvon 2000; Tsai 2006; Xiao 2009), and unclear in six trials that did not provide details that the investigators were blinded to treatment assignment (Inoue 2015; Levisohn 2009; Wu 2009; Yagi 2010; Zheng 2009; Zhou 2008).
Incomplete outcome data
Eight trials were at low risk of attrition bias (Ben‐Menachem 2000; Cereghino 2000; Levisohn 2009; Peltola 2009; Shorvon 2000; Tsai 2006; Wu 2009; Xiao 2009). Six trials were at high risk, mainly owing to discrepancies in the denominators compounded by reasons for missing outcome data being potentially related to true outcome, with either imbalance in numbers or reasons for missing data across intervention groups (Betts 2000; Glauser 2006; Inoue 2015; Yagi 2010; Zheng 2009; Zhou 2008, see Characteristics of included studies).
Selective reporting
Twelve trials were at low risk of selective reporting bias (Ben‐Menachem 2000; Cereghino 2000; Glauser 2006; Inoue 2015; Levisohn 2009; Peltola 2009; Shorvon 2000; Tsai 2006; Wu 2009; Xiao 2009; Zheng 2009; Zhou 2008). Betts 2000 was at high risk as there were no uniform baseline seizure data (see Characteristics of included studies table). Yagi 2010 was at unclear risk.
Effects of interventions
See: Table 1
Primary outcome: 50% or greater reduction in focal seizure frequency
Mantel‐Haenszel meta‐analysis
Intention‐to‐treat analysis
All trials (2455 participants) reported results for the 50% or greater reduction in seizure frequency outcome. Considering all doses of levetiracetam evaluated, more participants taking levetiracetam compared to placebo achieved 50% or greater reduction in seizure frequency (pooled RR 2.37, 95% CI 2.02 to 2.78; P < 0.00001; moderate‐certainty evidence; Analysis 1.1; Table 1). Ignoring dose, 36% of participants responded to levetiracetam (95% CI 34% to 38%), with a placebo response of 17% (95% CI 15% to 20%).
1.1. Analysis.

Comparison 1: Levetiracetam versus placebo, Outcome 1: ≥ 50% reduction in focal seizure frequency: intention to treat
The Chi² test for heterogeneity for response to levetiracetam indicated an important level of statistical heterogeneity between trials (Chi² = 24.71, degrees of freedom (df) = 13 (P = 0.03)). This signifies that there was a moderately large (I² = 47%) amount of variation (inconsistency) in the magnitude of the positive effect of levetiracetam overall. This is illustrated by the observations that the proportion of adults responding to levetiracetam varied from 21% to 72%, with a median 42%, and the proportion of children responding was 45% in one trial (Glauser 2006) and 63% in the other trial (Levisohn 2009). Repeating all‐dose analysis using a random‐effects model, levetiracetam was still significantly better than placebo (RR 2.26, 95% CI 1.79 to 2.85; P < 0.00001; Analysis 1.2).
1.2. Analysis.

Comparison 1: Levetiracetam versus placebo, Outcome 2: ≥ 50% reduction in focal seizure frequency: intention to treat (random effects)
There was no clear evidence of publication bias from inspecting asymmetry of a funnel plot (Figure 4).
4.

Funnel plot: 50% responder rate (fixed‐effect analysis, based on Analysis 1.1).
Subgroup analysis: adult trials compared to paediatric trials
The above conclusions remained unchanged when analysis was limited to the trials involving adults (all doses, pooled RR 2.49, 95% CI 2.08 to 2.99; P < 0.00001; Analysis 1.3). Ignoring dose, 34% of adults responded to levetiracetam (95% CI 32% to 37%), with a placebo response of 16% (95% CI 13% to 18%). There remained substantial heterogeneity within the trials involving adults (Chi² = 21.83, df = 11 (P = 0.03); I² = 50%). Repeating analysis using a random‐effects model, levetiracetam was still significantly better than placebo (RR 2.40, 95% CI 1.82 to 3.16; P < 0.00001; Analysis 1.4).
1.3. Analysis.

Comparison 1: Levetiracetam versus placebo, Outcome 3: ≥ 50% reduction in focal seizure frequency: (intention to treat): subgroup analysis by age
1.4. Analysis.

Comparison 1: Levetiracetam versus placebo, Outcome 4: ≥ 50% reduction in focal seizure frequency: (intention to treat): subgroup analysis by age (random effects)
For the two trials that recruited children, the results were sufficiently similar to be combined to give a pooled RR for 50% or greater reduction in seizure frequency of 1.91 (95% CI 1.38 to 2.63; P < 0.0001; Chi² = 1.58, df = 1 (P = 0.21); I² = 37%; Analysis 1.3). 52% of children responded to levetiracetam (95% CI 44% to 59%), with a placebo response of 25% (95% CI 18% to 34%). Overall, there was no statistically significant difference in the results of trials recruiting adults (all levetiracetam doses) and trials recruiting children (test for subgroup differences: Chi² = 2.01, df = 1 (P = 0.16); I² = 50.3%; Analysis 1.3).
Dose–response analysis
When considering individual doses, levetiracetam at either 500 mg or 4000 mg did not perform better than placebo for the 50% or greater reduction in seizure frequency outcome (500 mg: RR 1.60, 95% CI 0.71 to 3.62; P = 0.26; 4000 mg: RR 1.64, 95% CI 0.59 to 4.57; P = 0.3; Analysis 1.1). Levetiracetam was significantly better than placebo at all other individual doses (1000 mg to 3000 mg; Analysis 1.1). We fitted a generalised linear mixed model to the data from Analysis 1.1 to estimate the effect of dose on the 50% or greater reduction in seizure frequency outcome (details in Data synthesis). The odds of response (50% reduction in seizure frequency) were increased by nearly 40% (OR 1.39, 95% CI 1.23 to 1.58) with estimated between‐study standard deviation of 0.49 (standard error (SE) 0.16)) for each 1000 mg increase in dose of levetiracetam. In other words, this translates into an approximate 40% increase in the odds of response with an increase in dose from 1000 mg to 2000 mg of levetiracetam.
Handling missing data: best‐case and worse‐case scenarios
Conclusions remained mostly unchanged in best‐case and worst‐case scenarios compared to the ITT approach (Analysis 1.5; Analysis 1.6). No outcome data was missing in the two trials recruiting children, therefore results and conclusions were identical across all scenarios. An important amount of heterogeneity was present in many of the pooled dose analyses. Results were largely unchanged when analysis was repeated using a random‐effects model for the subgroups with large amounts of heterogeneity (Analysis 1.7; Analysis 1.8).
1.5. Analysis.

Comparison 1: Levetiracetam versus placebo, Outcome 5: ≥ 50% reduction in focal seizure frequency: best case
1.6. Analysis.

Comparison 1: Levetiracetam versus placebo, Outcome 6: ≥ 50% reduction in focal seizure frequency: worst case
1.7. Analysis.

Comparison 1: Levetiracetam versus placebo, Outcome 7: ≥ 50% reduction in focal seizure frequency: best case (random effects)
1.8. Analysis.

Comparison 1: Levetiracetam versus placebo, Outcome 8: ≥ 50% reduction in focal seizure frequency: worst case (random effects)
Overall, the scenario analyses indicated that levetiracetam continued to perform better than placebo for the 50% or greater reduction in seizure frequency outcome even when missing data were taken into account. Significant levels of heterogeneity were compounded by the missing data, meaning that the precise magnitude of positive effect remains unclear in the scenario analyses.
Sensitivity analysis across trials with low risk of bias
When sensitivity analysis was conducted on the five trials possessing a globally low risk of bias (Ben‐Menachem 2000; Cereghino 2000; Peltola 2009; Shorvon 2000; Tsai 2006), the above conclusions were not changed for all three analyses (Analysis 1.9):
1.9. Analysis.

Comparison 1: Levetiracetam versus placebo, Outcome 9: ≥ 50% reduction in focal seizure frequency: sensitivity analysis with trials of low risk of bias only
ITT: pooled RR 2.93 (95% CI 2.25 to 3.81); P < 0.00001; Chi² = 14.10, df = 4 (P = 0.007); I² = 72%;
best case: pooled RR 3.13 (95% CI 2.41 to 4.06); P < 0.00001; Chi² = 12.54, df = 4 (P = 0.01); I² = 68%;
worst case: pooled RR 2.69 (95% CI 2.09 to 3.46); P < 0.00001; Chi² = 14.54, df = 4 (P = 0.006); I² = 72%.
Conclusions were also unchanged when sensitivity analysis was repeated using a random‐effects model (Analysis 1.10).
1.10. Analysis.

Comparison 1: Levetiracetam versus placebo, Outcome 10: ≥ 50% reduction in focal seizure frequency: sensitivity analysis with trials of low risk of bias only (random effects)
Primary outcome: treatment withdrawal
Mantel‐Haenszel meta‐analysis
Intention‐to‐treat analysis
Thirteen trials (2428 participants) reported results for treatment withdrawal (Ben‐Menachem 2000; Betts 2000; Cereghino 2000; Glauser 2006; Inoue 2015; Levisohn 2009; Peltola 2009; Shorvon 2000; Tsai 2006; Wu 2009; Xiao 2009; Yagi 2010; Zhou 2008). Zheng 2009 reported that three participants withdrew from the trial prematurely, one due to adverse reactions and two thought 'treatment was invalid' (translated into Engish from Zheng 2009 published in Chinese). However, it was not stated which treatment groups these individuals withdrew from, therefore these data could not contribute to analysis.
Considering all doses of levetiracetam evaluated, there was no statistically significant difference between levetiracetam and placebo in terms of participants withdrawing from treatment (pooled RR 1.11, 95% CI 0.89 to 1.40; P = 0.36; high‐certainty evidence; Analysis 1.11).
1.11. Analysis.

Comparison 1: Levetiracetam versus placebo, Outcome 11: Treatment withdrawal
A Chi² test for heterogeneity for withdrawal from levetiracetam treatment (all doses) indicated no important statistical heterogeneity overall between trials (Chi² = 12.00, df = 12 (P = 0.45); I² = 0%).
There was no clear evidence of publication bias from inspecting asymmetry of a funnel plot (Figure 5).
5.
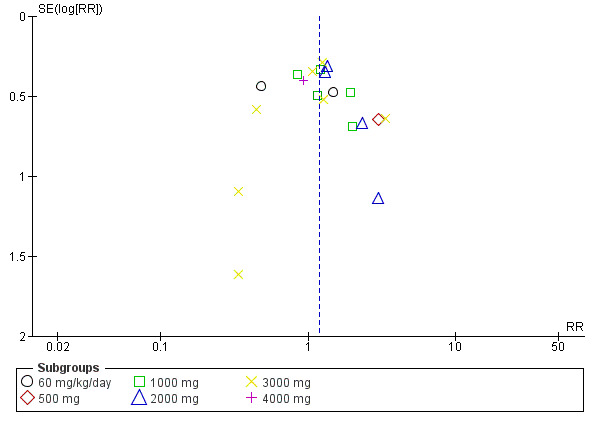
Funnel plot: treatment withdrawal (fixed‐effect analysis, based on Analysis 1.11).
Subgroup analysis: adult trials compared to paediatric trials
There were no statistically significant differences between any dose of levetiracetam and placebo in terms of treatment withdrawal (Analysis 1.11).
These conclusions also remained unchanged when analysis was limited to trials involving children (pooled RR for treatment withdrawal 0.80, 95% CI 0.43 to 1.46; P = 0.46; Chi² = 3.04, df = 1, P = 0.08, I² = 67%) and trials involving adults (pooled RR for treatment withdrawal 1.18, 95% CI 0.92 to 1.51; P = 0.20; Chi² = 7.90, df = 10 (P = 0.64); I² = 0%). Furthermore, there was no statistically significant difference in the results for adults (all doses pooled) and children (test for subgroup differences: Chi² = 1.35, df = 1 (P = 0.24), I² = 26.2%, Analysis 1.12).
1.12. Analysis.

Comparison 1: Levetiracetam versus placebo, Outcome 12: Treatment withdrawal: subgroup analysis by age
Conclusions for treatment withdrawal were unchanged when the analysis was repeated using a random‐effects model for trials recruiting children (Analysis 1.13).
1.13. Analysis.

Comparison 1: Levetiracetam versus placebo, Outcome 13: Treatment withdrawal: subgroup analysis by age (random effects)
Dose–response
We fitted a generalised linear mixed model to the data from Analysis 1.11 to estimate the effect of dose on treatment withdrawal (details in Data synthesis). There was no effect of increasing dose on the odds of withdrawal of treatment (OR 0.99, 95% CI 0.85 to 1.15, with estimated between‐study standard deviation of 0.59 (SE 0.20)) for each 1000 mg increase in dose of levetiracetam).
Sensitivity analysis across trials with low risk of bias
When sensitivity analysis was conducted on the five trials possessing a globally low risk of bias (Ben‐Menachem 2000; Cereghino 2000; Peltola 2009; Shorvon 2000; Tsai 2006), the above conclusions were not changed (pooled RR for treatment withdrawal 1.26, 95% CI 0.90 to 1.76; Chi² = 1.14, df = 4 (P = 0.89); I² = 0%; Analysis 1.14).
1.14. Analysis.

Comparison 1: Levetiracetam versus placebo, Outcome 14: Treatment withdrawal: sensitivity analysis with trials of low risk of bias only
Conclusions were also unchanged when sensitivity analysis was repeated using a random‐effects model (Analysis 1.15).
1.15. Analysis.

Comparison 1: Levetiracetam versus placebo, Outcome 15: Treatment withdrawal: sensitivity analysis with trials of low risk of bias only (random effects)
Primary outcome: adverse effects
Five most common adverse effects
Thirteen trials (2425 participants) reported results for adverse events (Ben‐Menachem 2000; Betts 2000; Cereghino 2000; Glauser 2006; Inoue 2015; Levisohn 2009; Peltola 2009; Shorvon 2000; Tsai 2006; Wu 2009; Xiao 2009; Yagi 2010; Zhou 2008). Zheng 2009 reported that one participant withdrew from the trial due to an adverse reaction but it was not stated which group this participant was in and there was no further information regarding adverse events. Therefore, Zheng 2009 was not included in analysis of adverse events.
Not all trials reported the same adverse effects, which altered the denominators representing number of participants from which RRs were calculated. To give a pooled summary of the five most common adverse effects across trials (2423 safety population), trials where an adverse effect was not reported (i.e. less than 5% or 10% of participants affected: see Included studies) were assigned zero events for that adverse effect. With this analysis, the five most common adverse effects (at any age) were as follows (Analysis 1.16):
1.16. Analysis.

Comparison 1: Levetiracetam versus placebo, Outcome 16: Adverse effects: 5 most common adverse effects (any age)
somnolence: affected 13% of participants (RR 1.62, 99% CI 1.19 to 2.20; P < 0.00001, I² = 0%; moderate‐certainty evidence);
headache: affected 8% of participants (RR 0.85, 99% CI 0.59 to 1.21; P = 0.23, I² = 66%; low‐certainty evidence);
dizziness: affected 7% of participants (RR 1.54, 99% CI 0.98 to 2.41; P = 0.01, I² =15%; moderate‐certainty evidence);
fatigue (asthenia): affected 6% of participants (RR 1.53, 99% CI 0.98 to 2.38; P = 0.01, I² = 0%; moderate‐certainty evidence);
accidental injury: affected 6% of participants (pooled RR 0.72, 99% CI 0.49 to 1.06; P = 0.03, I² = 60%; low‐certainty evidence).
Conclusions were also unchanged when analyses of headache and accidental injury were repeated using a random‐effects model (Analysis 1.17).
1.17. Analysis.

Comparison 1: Levetiracetam versus placebo, Outcome 17: Adverse effects: 5 most common adverse effects (any age, random effects)
The relative commonality of individual adverse effects did not largely alter when analysis was limited to adults (Analysis 1.18), aside from the introduction of infection (RR 1.76, 99% CI 1.03 to 3.02), which was more common than dizziness. Only the RRs for somnolence (RR 1.57, 99% CI 1.13 to 2.20; P = 0.0005) and infection were statistically significant with levetiracetam over placebo. Accidental injury was statistically significantly more associated with placebo than levetiracetam (RR 0.60, 99% CI 0.39 to 0.92; P = 0.002).
1.18. Analysis.

Comparison 1: Levetiracetam versus placebo, Outcome 18: Adverse effects: most common adverse effects in adults
In children, somnolence remained the most common adverse effect, although it was not statistically significant over placebo (RR 1.90, 99% CI 0.88 to 4.09; P = 0.03) and there was a wide CI (Analysis 1.19). The next most common adverse effects in children were vomiting (RR 1.22, 99% CI 0.55 to 2.69; P = 0.52), pharyngitis (RR 1.09, 99% CI 0.47 to 2.50; P = 0.79), aggression (hostility) (RR 1.72, 99% CI 0.64 to 4.63; P = 0.16), and accidental injury (RR 1.63, 99% CI 0.63 to 4.26; P = 0.19). These were no more common than in placebo treatment.
1.19. Analysis.

Comparison 1: Levetiracetam versus placebo, Outcome 19: Adverse effects: most common adverse effects in children
In summary, somnolence was the only adverse effect significantly associated with levetiracetam compared to placebo overall.
General adverse effects
RRs for the general adverse effects (where available) were: ataxia (adults, unpublished data: 1.50, 99% CI 0.43 to 5.26; P = 0.40; Analysis 1.18), nausea (adults: 1.37, 99% CI 0.47 to 4.00; P = 0.44; Analysis 1.18), dizziness (children: 1.52, 99% CI 0.47 to 4.94; P = 0.36; Analysis 1.19), and fatigue ((asthenia), children: 1.82, 99% CI 0.62 to 5.33; P = 0.15; Analysis 1.19).
There were no general adverse effects significantly associated with levetiracetam compared to placebo.
Behavioural adverse effects
There were no individual behavioural adverse effects significantly associated with levetiracetam compared to placebo.
Adverse effects pertaining to changes in behaviour were described as follows (see Analysis 1.20):
1.20. Analysis.

Comparison 1: Levetiracetam versus placebo, Outcome 20: Behavioural changes
hostility: affected 0.98% of participants (RR 1.92, 99% CI 0.56 to 6.60; P = 0.17);
personality disorder: affected 0.82% (RR 1.10, 99% CI 0.30 to 3.95; P = 0.85);
nervousness: affected 0.66% (RR 4.80, 99% CI 0.68 to 34.14; P = 0.04);
depression: affected 0.60% of participants (RR 1.41, 99% CI 0.25 to 7.85; P = 0.61);
aggression: affected 0.60% of participants (RR 1.42, 99% CI 0.27 to 7.42; P = 0.59);
agitation: affected 0.55% of participants (RR 6.17, 99% CI 0.66 to 57.79; P = 0.04);
emotional lability: affected 0.55% of participants (RR 1.44, 99% CI 0.28 to 7.29; P = 0.56);
psychomotor hyperactivity: affected 0.49% of participants (RR 0.42, 99% CI 0.08 to 2.19; P = 0.18);
irritability: affected 0.27% of participants (RR 11.28, 99% CI 0.26 to 495.63; P = 0.10);
abnormal behaviour: affected 0.27% (RR 5.92, 99% CI 0.14 to 255.98; P = 0.22);
altered mood: affected 0.22% of participants (RR 4.85, 99% CI 0.11 to 216.99; P = 0.28);
anxiety: affected 0.22% of participants (RR 4.85, 99% CI 0.11 to 216.99; P = 0.28);
dissociation: affected 0.16% of participants (RR 0.14, 99% CI 0.00 to 6.77; P = 0.19).
When behavioural adverse effects were combined, 4.53% of participants were affected (RR 1.87, 99% CI 1.19 to 2.95; P = 0.0004). In this, 22.64% of children were affected (RR 1.90, 99% CI 1.16 to 3.11; P = 0.0009) and 1.04% of adults were affected (RR 1.79, 99% CI 0.59 to 5.41; P = 0.17).
Secondary outcome: cognitive effects
The additional trials included in this review update did not contribute any data to cognitive effects, meaning that there are no new changes since the previous review for these outcomes.
See Table 2; Table 3; Table 4.
1. Cognitive assessment as mean changes from baseline in variables on neuropsychological tests: Zhou 2008.
| Test | Subscale | Placebo (n = 11) | Levetiracetam 3000 mg/day (n = 13) |
| Verbal Fluency | — | Improved | Improved |
| Trail Making Test | Time on Part A | Improved | Improved |
| Time on Part B | Improved | Improved | |
| Wisconsin Card Sorting Test | Number of correct responses | Improved | Improved |
| Perseverative errors | Improved | Improved | |
| Non‐perseverative errors | Improved | Worsened | |
| Number of categories | Improved | Improved | |
| Performance time | Improved | IMPROVEDa | |
| Digit symbol | — | Worsened | Improved |
| Digit span | — | Worsened | Worsened |
| Stroop Color–Word Interference Task | Reaction time for naming words | Worsened | Improved |
| Correct number of naming words | Worsened | Improved | |
| Reaction time for naming colours | Improved | Improved | |
| Correct number of naming colours | Improved | Worsened | |
| Logic memory | — | Improved | Improved |
| Delayed logical memory | — | Improved | IMPROVEDa |
| Visual memory | — | Improved | Improved |
| Delayed visual memory | — | Worsened | Improved |
| Calculation | — | Worsened | Improved |
n: number of participants. aP < 0.01.
2. Cognitive assessment as least square mean change from baseline (Leiter‐R AM, WRAML‐2, Leiter‐R ERS): Levisohn 2009 and Loge 2010 (children).
| Test | Subscale | Placebo (n = 27) | Levetiracetam 60 mg/kg/day (n = 46) |
| Leiter‐R AM | Composite score | Improved | Improved |
| WRAML‐2 | General memory | Improved | Improved |
| Visual memory | Improved | Improved | |
| Verbal memory | Improved | Improved | |
| Attention/concentration | Improved | Worsened | |
| Leiter‐R ERS | Cognitive/social | Improved | Improved |
| Emotions/regulations | Improved | Improved |
Leiter‐R AM: Leiter International Performance Scale‐Revised Attention and Memory; Leiter‐R ERS: Leiter International Performance Scale‐Revised, Examiner's Rating Scale; n: number of participants; WRAML‐2: Wide Range Assessment of Memory and Learning‐2. aP < 0.1. Note: results were for per‐protocol population
3. Behavioural and emotional functioning assessment as least square mean change from baseline (CBCL and CHQ‐PF50): Levisohn 2009 and Loge 2010 (children).
| Test | Subscale | n | Placebo (n = 27) | n | Levetiracetam 60 mg/kg/day (n = 46) |
| CBCL competence scores | Activities | 22 | WORSENEDa | 41 | Worsened |
| Social | 22 | Worsened | 41 | Worsened | |
| School | 19 | Improved | 35 | Improved | |
| Total competence | 19 | Worsened | 34 | Worsened | |
| CBCL problem scores | Anxious/depressed | 22 | Improved | 43 | Improved |
| Withdrawn/depressed | Improved | Worsened | |||
| Somatic complaints | Improved | Improved | |||
| Social problems | Improved | Worsened | |||
| Thought problems | Improved | Worsened | |||
| Attention problems | Improved | Improved | |||
| Rule‐breaking behaviour | Improved | Worsened | |||
| Aggressive behaviour | IMPROVEDa | WORSENEDa | |||
| Internalising syndromesb | Improved | Improved | |||
| Externalising syndromesc | IMPROVEDa | WORSENEDa | |||
| Total problems | IMPROVEDa | WORSENEDa | |||
| CHQ‐PF50 | Role/social–emotional/behavioural | 27 | Worsened | 45 | Improved |
| Behaviour | 27 | Worsened | 45 | Worsened | |
| Mental health | 27 | Improved | 45 | Improved | |
| Psychosocial summary | 26 | Improved | 44 | Improved |
CBCL: Achenbach Child Behavior Checklist; CHQ‐PF50: Child Health Questionnaire‐Parent Form 50; n: number of participants. aP < 0.05. bInternalising syndromes contain the withdrawn/depressed, anxious/depressed, and somatic complaints scores. cExternalising syndromes contain the aggressive behaviour and rule‐breaking behaviour scores. Note: results were for per‐protocol population.
Table 2 shows results for Zhou 2008. This table shows mean change from baseline for each treatment group, by way of variables within a series of neuropsychological tests. The results indicate that levetiracetam did not lessen/reduce cognitive function (no worsening in variables was statistically significant). Performance time on the Wisconsin Card Sorting Test (WCST) and Delayed Logic Memory significantly improved for participants treated with levetiracetam, but not for those treated with placebo.
Table 3 shows results for Levisohn 2009. This table shows mean change from baseline for each treatment group, by scores within the Leiter International Performance Scale‐Revised Attention and Memory (Leiter‐R AM), Wide Range Assessment of Memory and Learning‐2 (WRAML‐2), and Leiter International Performance Scale‐Revised, Examiner's Rating Scale (Leiter‐R ERS) instruments. The results indicate that levetiracetam did not lessen/reduce/impair cognitive function in children; there were no significant changes in either group of participants.
Table 4 shows more results for the Levisohn 2009 trial. This table shows mean change from baseline for each treatment group, by component of the Achenbach Child Behavior Checklist (CBCL) and Child Health Questionnaire‐Parent Form 50 (CHQ‐PF50). The results demonstrated statistically significant worsening of scores in aggressive behaviour, externalising syndromes (consisting of aggressive behaviour and rule‐breaking behaviour), and total problems in children treated with levetiracetam, but not those treated with placebo.
Secondary outcome: quality of life
The additional trials included in this review update did not contribute any data to QoL assessment, meaning that there are no new changes since the previous review for these outcomes.
See Table 5; Table 6; Table 7; Table 8.
4. Quality of life assessment as mean change from baseline (QOLIE‐31): Cereghino 2000.
| Subscale | Placebo (n = 81) | Levetiracetam | |
| 1000 mg/day (n = 80) | 3000 mg/day (n = 85) | ||
| Overall QoL | Improved | Improved | IMPROVEDa |
| Seizure worry | Worsened | IMPROVEDa | IMPROVEDa |
| Emotional well‐being | Improved | Worsened | Worsened |
| Energy‐fatigue | Worsened | Improved | Worsened |
| Cognitive functioning | WORSENEDa | Improved | Improved |
| Medication effects | Worsened | Improved | Improved |
| Social function | Worsened | Worsened | Improved |
| Health status | Improved | Improved | Improved |
n: number of participants; QoL: quality of life; QOLIE‐31: Quality Of Life in Epilepsy Inventory. aP < 0.05.
5. Quality of life assessment as mean change from baseline (QOLIE‐31): Zhou 2008.
| Subscale | Placebo (n = 11) | Levetiracetam 3000 mg/day (n = 13) |
| Overall QoL | Improved | Improved |
| Seizure worry | Improved | Improved |
| Emotional well‐being | Improved | Improved |
| Energy‐fatigue | Improved | worsened |
| Cognitive functioning | Worsened | IMPROVEDa |
| Medication effects | Worsened | Improved |
| Social function | Improved | IMPROVEDa |
| Health status | Improved | Improved |
n: number of participants; QoL: quality of life; QOLIE‐31: Quality Of Life in Epilepsy Inventory. aP < 0.05.
6. Quality of life assessment as mean change from baseline (ESI‐55): Shorvon 2000.
| QoL domain | Placebo (n = 89) | Levetiracetam | |
| 1000 mg/day (n = 92) | 2000 mg/day (n = 81) | ||
| Health status | IMPROVEDa | IMPROVEDa | IMPROVEDa |
| Role limitation due to memory problems | Improved | IMPROVEDa | Worsened |
| Pain | Worsened | IMPROVEDa | Improved |
| Cognitive functioning | Improved | Improved | Improved |
| Emotional well‐being | Unchanged | Improved | Improved |
| Energy/fatigue | Improved | IMPROVEDa | Improved |
| Social functioning | Improved | IMPROVEDa | Improved |
| Role limitation due to emotional problems | Improved | Improved | Worsened |
| Role limitation due to physical problems | Improved | IMPROVEDa | Improved |
| Physical function | Improved | Worsened | Improved |
| Overall QoL | Improved | Improved | IMPROVEDa |
| Health perceptions | Improved | IMPROVEDa | IMPROVEDa |
ESI‐55: Epilepsy Surgery Inventory Scale; n: number of participants; QoL: quality of life. aP < 0.05. Note: almost all participants provided information for each individual domain.
7. Summary of quality of life: mean change from baseline (ESI‐55 scale): Betts 2000.
| QoL composite score | Period | Placebo | Levetiracetam | ||||
| Number | Mean change | 2000 mg/day: number | 2000 mg/day: mean change | 4000 mg/day: number | 4000 mg/day: mean change | ||
| Mental health | Baseline | 35 | Not applicable | 40 | Not applicable | 37 | Not applicable |
| Mental health | Overall double‐blind | 28 | –1.7 (worsened) | 30 | 1.7 (improved) | 28 | 3.5 (improved) |
| Physical health | Baseline | 29 | Not applicable | 37 | Not applicable | 34 | Not applicable |
| Physical health | Overall double‐blind | 28 | 3.6 (improved) | 30 | 0.8 (improved) | 26 | 2.3 (improved) |
| Role functioning | Baseline | 33 | Not applicable | 38 | Not applicable | 35 | Not applicable |
| Role functioning | Overall double‐blind | 28 | –0.5 (worsened) | 31 | 0.4 (improved) | 27 | 2.3 (improved) |
ESI‐55: Epilepsy Surgery Inventory Scale; n: number of participants; QoL: quality of life.
For adults, two trials used the Quality Of Life in Epilepsy Inventory (QOLIE‐31) as an instrument to measure QoL (Cereghino 2000; Zhou 2008), while two other trials used the Epilepsy Surgery Inventory Scale (ESI‐55) (Betts 2000; Shorvon 2000). One trial assessed cognitive effects using nine tests chosen from the Chinese version of the Wechsler Adult Intelligence Scale‐Revised (WAIS‐RC) and other tests commonly used to assess cognitive function (see Table 2) (Zhou 2008). For children, one trial assessed cognitive effects using the following series of instruments: Leiter‐R AM, WRAML‐2, and Leiter‐R ERS (Levisohn 2009). The same trial used the CBCL and CHQ‐PF50 to assess behavioural and emotional effects.
Table 5 shows results for Cereghino 2000. This table shows mean change from baseline for each treatment group, using the subscale of QOLIE‐31. Results indicate that compared to placebo, participants treated with levetiracetam were significantly less worried about seizures, and participants treated with levetiracetam 3000 mg had a significantly better overall QoL.
Table 6 shows results for Zhou 2008 using the QOLIE‐31. They indicate that compared to placebo, participants treated with levetiracetam had significantly better cognitive functioning and social function.
Table 7 shows results for Shorvon 2000. This table shows mean change from baseline for each treatment group, by domain using the ESI‐55 scale. Results indicate that when compared to placebo, participants treated with levetiracetam scored significantly better for the health perception domain. Participants treated with 1000 mg scored significantly better for the 'role limitation due to memory problems', 'pain', 'energy', 'social functioning', and 'role limitation due to physical problems' domains. Individuals treated with levetiracetam 2000 mg scored better but not statistically significantly for the overall QoL domain.
Table 8 shows results for Betts 2000 using the ESI‐55; however, for this trial we only had aggregate data for the three composite scores of this instrument.
Overall, for adults, results from the Cereghino 2000, Shorvon 2000, and Zhou 2008 trials did indicate that levetiracetam had a positive effect on some aspects of QoL, while results from the Zhou 2008 trial indicated that the drug did not negatively affect and, in a way, improved cognitive function. In children, the results from the Levisohn 2009 trial indicated that levetiracetam did not alter cognitive function but did worsen aspects of child behaviour.
Discussion
Summary of main results
Overall, this review update found that in both adults and children with drug‐resistant focal epilepsy, levetiracetam added on to usual care is more effective than placebo at reducing seizure frequency, it is unlikely to be stopped by patients, and it has minimal adverse effects. These findings are unchanged from the previous review update in 2012 (Mbizvo 2012).
This review update contributes two key additional findings:
a 500 mg daily dose of levetiracetam is no more effective than placebo at reducing seizures; and
the odds of response (50% reduction in seizure frequency) are increased by nearly 40% for each 1000 mg increase in dose of levetiracetam.
In adults, levetiracetam demonstrated statistically significant efficacy over placebo in the outcome 50% or greater reduction in seizure frequency at all doses aside from 500 mg and 4000 mg. Inoue 2015 contributed data to suggest levetiracetam 500 mg lacked efficacy. As this result was only provided by one study, which had a high risk of bias from incomplete outcome data (see Characteristics of included studies), this finding remains to be confirmed in future RCTs. However, if correct, this finding would have important implications given that it is not uncommon, in clinical practice, for patients treated with levetiracetam to remain on a low dose of 250 mg twice daily for prolonged periods of time. It would suggest that there should be a low threshold to escalate the dose beyond 500 mg if well‐tolerated and efficacy aims to be achieved. Betts 2000 contributed data to suggest levetiracetam 4000 mg lacked efficacy. This trial was excluded from the 50% or greater reduction in seizure frequency outcome in both the 2001 (Chaisewikul 2001) and 2012 (Mbizvo 2012) reviews because uniform baseline seizure data were not collected for the trial's participants (see Characteristics of included studies). However, in order to maximise information gathered within the context of few new additional trials published since the 2012 update, we decided to include the Betts 2000 data in the 50% or greater reduction in seizure frequency outcome but also to grade the trial as possessing high risks of bias from incomplete outcome data and selective reporting (see Risk of bias in included studies). The finding that 4000 mg daily of levetiracetam is no more effective than placebo should, therefore, be interpreted with caution and would require confirmation in future RCTs if safe, although we cannot recommend this as the maximum recommended dose of levetiracetam is 3000 mg daily (BNF 2019a). We were able to demonstrate a dose–response relationship for levetiracetam in the order of 40% improvement in response for each 1000 mg increase in dose for adults. This helps supplement previous focused studies on the dose–response of levetiracetam in drug‐resistant focal epilepsy, which managed to use only some of the trials included in the current review update (Snoeck 2007).
For our global outcome of treatment withdrawal, we have insufficient evidence to conclude that levetiracetam is more likely to be withdrawn than placebo in adults. Somnolence and infection were the only adverse effects significantly associated with levetiracetam in adults. Somnolence is a common complaint among people with epilepsy because AEDs and seizures can modify sleep architecture and thereby lead to daytime sleepiness (Manni 2000). It remains possible, therefore, that significant somnolence was a general function of taking an AED rather than a specific marker of levetiracetam. We are unable to explain the apparent correlation between infection and levetiracetam. This adverse effect was apparent in the 2012 review and the three additional studies reported in the current review did not report this adverse effect, meaning no new conclusions are drawn but we note that the CI is wide (99% CI 1.03 to 3.02). This outcome requires further investigation, particularly given that practicing clinicians would not commonly associate levetiracetam with infection. The lack of evidence of treatment withdrawal on levetiracetam would favour that the adverse effects profile of levetiracetam was generally tolerable. Of note, there remained no significant evidence to suggest that behavioural adverse effects were significantly associated with levetiracetam in adults, both individually or as grouped behavioural adverse effects.
There were only two paediatric trials in the 2012 update (Glauser 2006; Levisohn 2009), and the current review identified no more RCTs of levetiracetam published since then for children with drug‐resistant focal epilepsy. The conclusions for children remain unchanged: levetiracetam, at a dose of 60 mg/kg/day, is more effective than placebo at achieving response (50% or greater reduction in seizure frequency), with an RR of 1.91 in favour of levetiracetam. It is also no more likely to be withdrawn by children than placebo. There are no individual adverse effects significantly associated with levetiracetam as compared to placebo in children. However, our focused review of behavioural adverse effects (as a group) suggests that children may be significantly affected by these.
Children remained better responders than adults, with 52% of children responding to levetiracetam compared to 34% of adults. Placebo response was 25% for children and 16% for adults, making the absolute response (levetiracetam minus placebo) 27% for children and 18% for adults. The expected placebo response in drug‐resistant focal epilepsy is 9.3% to 16.6% (Burneo 2002; Guekht 2010). The reasons for larger placebo responses than expected in the current pool of trials are unclear and merit further investigation. However, such a large response to placebo, when taken in the context of levetiracetam demonstrating a significant effect in reducing seizures over placebo, suggests that levetiracetam is a highly effective AED. There were no substantive differences in results between the ITT analysis and the sensitivity best‐case and worst‐case analyses for either adults or children.
As was the case in 2012 (Mbizvo 2012), we have insufficient data and analysis to make firm conclusions about the cognitive effects of levetiracetam and its effect on QoL. Levetiracetam appeared to have a positive effect on cognition and some aspects of QoL in adults. In children, levetiracetam did not appear to alter cognitive function but there was evidence of worsening in certain aspects of child behaviour. The three additional trials included in this update reported no data on these outcomes. We made no attempt at a meta‐analysis across data pertaining to cognitive effects and QoL. It is difficult to be sure of the real‐life impact of any changes in cognitive effects or QoL seen in this analysis and the conclusions remain to be validated in a more detailed investigation on the effects of levetiracetam on cognition and QoL. These outcomes are important because they can place clinical trial evidence of clinical efficacy into the context of meaningful life improvement for patients (Kerr 2011).
Overall completeness and applicability of evidence
The review sought out to evaluate the effectiveness of levetiracetam, added on to usual care, in treating drug‐resistant focal epilepsy. The evidence analysed is highly relevant to the review aims as all RCTs aimed to answer this same question, and pooled results covered adults and children, and a range of low‐, middle‐, and high‐income countries. This is likely to mean that the results of this review are generalisable, with sufficient external validity to suggest levetiracetam is likely to have similar effects in drug‐resistant focal epilepsy populations worldwide. However, the trials analysed in this review treated people with levetiracetam for only 12 to 24 weeks. People with drug‐resistant focal epilepsy need longer‐term treatment than this, yet the conclusions drawn in the current review cannot be applied to periods outside of 12 to 24 weeks of treatment. The conclusions on children are also based on a sample size of fewer than 300 participants. More studies, particularly longer‐term studies, and studies on children will be needed before complete evaluation of the effectiveness of levetiracetam is possible.
Although the results of this review indicate that levetiracetam is an effective add‐on treatment for both adults and children with drug‐resistant focal epilepsy, it cannot tell us how levetiracetam compares with other AEDs in this scenario. This is an extremely important issue for clinicians who are faced with an ever‐increasing number of AEDs to choose from, and head‐to‐head trials are needed to provide the evidence that is needed to enable clinicians to make an evidence‐based choice between AEDs. This review focuses on the use of oral levetiracetam in drug‐resistant focal epilepsy, and the results cannot be generalised to add‐on treatment in people with generalised epilepsy. Likewise, no inference can be made about the efficacy and tolerability of levetiracetam when used as monotherapy or as an intravenous agent.
Certainty of the evidence
The overall certainty of evidence for the response outcome (50% or greater reduction in seizure frequency, all doses) was moderate (see Table 1), indicating that further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. This certainty score was downgraded from high due to the presence of important levels of heterogeneity between trials, and due to high or unclear risks of bias being present in 9/14 studies. Although levetiracetam significantly outperformed placebo for the response outcome, the heterogeneity meant we could not be confident about the magnitude of this effect. The strong performance of levetiracetam in this regard, and its already widespread clinical use for this group of patients, means that there are unlikely to be many future RCTs set up to investigate levetiracetam in this particular population. Therefore, the certainty of evidence for this particular outcome is unlikely to improve in the future. We performed a sensitivity analysis restricting analysis to studies with a low risk of bias (which would act to increase the certainty of evidence). This subgroup analysis did not change the results from the main analysis, suggesting that any downgrade in the certainty of evidence owing to bias would not significantly change our confidence in the primary conclusions drawn.
The overall certainty of evidence for the treatment withdrawal outcome was high (see Table 1), indicating that further research is very unlikely to change our confidence in this estimate of effect.
The overall certainty of evidence for the adverse effects of somnolence, dizziness, and fatigue was moderate (see Table 1). This was downgraded from high due the presence of high or unclear risks of bias in many of the corresponding trials. For headache and accidental injury, the overall certainty of evidence was low because of the presence of high or unclear risks of bias and due to heterogeneity between the corresponding trials. A grading of low indicates that further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change these estimates. This remains to be investigated.
Potential biases in the review process
The influence of a possible information bias cannot be excluded in this review. The original review had unpublished data confidentially made available for inclusion (Chaisewikul 2001), while this update had no such data made available for the trials published since the original review. To illustrate this limitation, the risks of selection, performance, and detection biases were initially regarded as 'unclear' for the Ben‐Menachem 2000 and Shorvon 2000 trials (included in the original review). This judgement was made based on the information available in the published versions of these trials. These trials were regarded as possessing a 'low risk' of these biases only after we had the opportunity to extract further information from the unpublished scripts. It stands to reason that similar discrepancies in information may exist for the other trials regarded as having an 'unclear risk' of certain biases in this review. Most RCTs implement various adequate methods of random sequence generation, allocation concealment, and investigator blinding in their protocols, but not all publish details about these methods. Future trial publications should aim to reduce this discrepancy in information in order to allow a clearer interpretation of the risks of bias. The influence of this possible information bias on the conclusions of this review is likely to be small given that a predominant number of trials had low risks of bias and a subgroup analysis where trials with unclear or high risks of bias were excluded demonstrated negligible changes to the results. Funnel plots suggest that there is unlikely to be existent publication bias in the literature surrounding the subject of this review (Figure 4; Figure 5).
Agreements and disagreements with other studies or reviews
A meta‐analysis of levetiracetam for randomised placebo‐controlled trials in people with refractory epilepsy was published in April 2019 by a group in China (Chen 2019). Although this pooled together results for both focal and generalised epilepsies across 17 trials, 15 trials were in focal epilepsy. We captured all 15 of these same trials in the current review, and after choosing to exclude the Boon 2002 trial (see Excluded studies), we included 14 of them. The Chen 2019 review concluded that levetiracetam is an effective AED for both adults and children with generalised or focal seizures at 1000 mg to 3000 mg or 60 mg/kg/day, with a favourable adverse effects profile. These conclusions echo those of our own within the current review. The agreement of these two reviews on efficacy and tolerability of levetiracetam in focal epilepsy is reassuring. The observation that these two unrelated reviews identified the same trials is also reassuring for quality and completeness of the search methodology and study screening protocols used.
In the current review, behavioural adverse effects, when looked at individually, were not significant in adults or children. Similarly, they were not significant in adults alone. However, when individual behavioural adverse effects were pooled, they became significantly associated with levetiracetam over placebo in children (but not adults). This requires further investigation, as highlighted in more detail in the Mbizvo 2012 update. A systematic review published following that update reported an RR of 2.18 for the total number of behavioural adverse effects for levetiracetam versus placebo in children (Halma 2014). However, in addition to the Glauser 2006 and Levisohn 2009 trials, that systematic review included one RCT of children with generalised epilepsies in the analysis pool. The paucity of further RCTs in children with drug‐resistant focal epilepsy make it difficult for further conclusions to be drawn about behavioural adverse effects in this particular group of children with epilepsy. For both adults and children, it is generally felt, in clinical practice, that changes in behaviour are common when taking levetiracetam. This is resultant from clinical experience although the RCTs do not demonstrate clear evidence for this, as we have demonstrated in the current review. One of the reasons for this discrepancy may be the exclusion, in RCTs, of participants with psychiatric comorbidities (whom clinicians would see routinely). Of note, in one observational study on the adverse effects of 4085 adults treated with AEDs, levetiracetam had the greatest rate of psychiatric and behavioural side effects (PBSE). Furthermore, a history of psychiatric comorbidity was, indeed, one of the risk factors for increased PBSE rate (Chen 2017). It may also be that changes in behaviour manifest soon after starting levetiracetam, meaning that it is withdrawn before the eight‐week minimum treatment period that was set for included trials in this review. Finally, we note that only one of the four early trials from the year 2000 reported behavioural adverse effects (depression in Shorvon 2000). It is possible that the questions or instruments used to pick up adverse effects in RCTs, particularly early ones, were not sensitive for changes in behaviour because these were not the sort of adverse effect expected of an AED.
Authors' conclusions
Implications for practice.
This review provides moderate‐ to high‐certainty evidence that levetiracetam is effective as add‐on treatment in adults and children with drug‐resistant focal epilepsy, although an affective dose is likely to be above 500 mg daily in adults. The maximum effective dose remains unclear, with a suggestion that 4000 mg may not be effective for this particular group of patients. However, this is based on one study with high risk of bias and therefore poor‐quality evidence, meaning that this finding should be interpreted with caution and may not be certain. As the maximum recommended dose of levetiracetam is 3000 mg daily (BNF 2019a), further comment on the implications for clinical practice of levetiracetam 4000 mg is beyond the scope of this review. The review demonstrates that nearly 30% of children may be responsive to adjuvant levetiracetam at a dose of 60 mg/kg/day. Efficacy also remains high in adults, with nearly 20% responding. Each increase of dose by 1000 mg may provide a 40% increase in the odds of achieving response (50% reduction in seizure frequency) in adults. Low‐ to moderate‐certainty evidence provided by this review suggests that all doses appear well tolerated in both adults and children, although there is a possibility of adverse changes in behaviour in children, potentially affecting around 20%. Overall, it appears reasonable to continue the use of adjuvant levetiracetam in clinical practice for treating adults and children with drug‐resistant focal epilepsy. The conclusions cannot be applied to levetiracetam use in generalised epilepsy, as an intravenous agent, or to its use as monotherapy.
Implications for research.
To evaluate further the place of add‐on levetiracetam in drug‐resistant focal epilepsy, further studies are required to address the following:
the efficacy of a 500 mg daily dose of levetiracetam;
the minimum and maximum effective and safe doses of levetiracetam;
the most effective dose of levetiracetam;
the long‐term efficacy and safety of levetiracetam beyond 24 weeks;
the effects of levetiracetam on behaviour;
the effects of levetiracetam on quality of life and cognition;
economic aspects of levetiracetam therapy;
how levetiracetam compares with other add‐on treatments;
placebo response in randomised controlled trials of levetiracetam;
the effectiveness of levetiracetam in children (using larger sample sizes to supplement the currently small body of evidence).
Beyond this group of patients, further investigation will also be needed on how levetiracetam compares with standard AEDs such as: 1. carbamazepine as monotherapy in focal epilepsy, 2. lamotrigine as monotherapy in focal epilepsy, and 3. valproate as monotherapy in generalised epilepsy. The effectiveness of levetiracetam versus standard AEDs will be studied in the SANAD‐II trial.
What's new
| Date | Event | Description |
|---|---|---|
| 27 November 2018 | New citation required but conclusions have not changed | Conclusions are unchanged. |
| 26 November 2018 | New search has been performed | Searches updated 26 November 2018; three new trials have been included (Zheng 2009; Yagi 2010; Inoue 2015). |
History
Protocol first published: Issue 1, 2000 Review first published: Issue 1, 2001
| Date | Event | Description |
|---|---|---|
| 5 October 2017 | New search has been performed | Searches updated. |
| 13 August 2015 | New search has been performed | Added Inoue 2015 to Classification pending references. This supersedes previous reference N01221 and Yagi 2010 in Studies awaiting classification. |
| 24 February 2015 | New citation required but conclusions have not changed | No new relevant studies, conclusions remain unchanged |
| 24 February 2015 | New search has been performed | Searches updated 24 February 2015. |
| 24 March 2014 | New search has been performed | Searches updated. No new studies found. |
| 13 September 2012 | Amended | Missing citation added Yagi 2010 |
| 12 August 2012 | New citation required but conclusions have not changed | Paediatric data has been incorporated into the update. |
| 19 April 2011 | New search has been performed | Addition of seven new trials to the systematic review and meta‐analysis, published after the original 2001 review. |
| 8 November 2009 | Amended | Published notes added. |
| 23 September 2008 | Amended | Converted to new review format. |
| 1 July 2005 | New search has been performed | The date of the latest search for evidence to the review is 1 July 2005, no new studies were identified. In a previous update on 27 September 2002 we found one new study which we included as published data of the study N138 (Ben‐Menachem et al. Efficacy and tolerability of levetiracetam 3000 mg/d in patients with refractory seizures: a multicenter, double‐blind, responder‐selected study evaluating monotherapy. European Levetiracetam Study Group. Epilepsia 2000;41(10):1276‐83). One study was also added to the 'Studies awaiting assessment' section (Boon P et al. Dose‐response effect of levetiracetam 1000 and 2000 mg/day in partial epilepsy. Epilepsy Research 2002;48(1‐2):s77‐89). This will be assessed for inclusion at a later date. |
Acknowledgements
We would like to thank and acknowledge two of the review authors of the original review; Rungsan Chaisewikul and Michael Privitera.
Appendices
Appendix 1. CRS Web search strategy
1. Levetiracetam* or Levitiracetam* or Keppra AND CENTRAL:TARGET
2. MESH DESCRIPTOR Epilepsies, Partial EXPLODE ALL AND CENTRAL:TARGET
3. ((partial or focal) and (seizure* or epilep*)):AB,KW,MC,MH,TI AND CENTRAL:TARGET
4. #2 OR #3 AND CENTRAL:TARGET
5. #1 AND #4
6. (monotherap* NOT (adjunct* OR "add‐on" OR "add on" OR adjuvant* OR combination* OR polytherap*)):TI AND CENTRAL:TARGET
7. #5 NOT #6
8. #7 AND >24/02/2015:CRSCREATED
Appendix 2. MEDLINE search strategy
The following search strategy is based on the Cochrane Highly Sensitive Search Strategy for identifying randomized trials published in Lefebvre 2011.
1. (Lev?tiracetam? or Keppra).tw.
2. exp Epilepsies, Partial/
3. ((partial or focal) and (seizure$ or epilep$)).tw.
4. 2 or 3
5. (randomized controlled trial or controlled clinical trial or pragmatic clinical trial).pt. or (randomi?ed or placebo or randomly).ab.
6. clinical trials as topic.sh.
7. trial.ti.
8. 5 or 6 or 7
9. exp animals/ not humans.sh.
10. 8 not 9
11. 1 and 4 and 10
12. (monotherap$ not (adjunct$ or "add‐on" or "add on" or adjuvant$ or combination$ or polytherap$)).ti.
13. 11 not 12
14. remove duplicates from 13
15. limit 14 to ed=20150224‐20181126
16. 14 not (1$ or 2$).ed.
17. 16 and (2015$ or 2016$ or 2017$ or 2018$).dt.
18. 15 or 17
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
The search strategy below is the original MEDLINE strategy that was used for earlier versions of this review. It is based on the Cochrane Highly Sensitive Search Strategy for MEDLINE as set out in Appendix 4b of the Cochrane Handbook for Systematic Reviews of Interventions (version 4.2.4, updated March 2005) (Higgins 2005).
1. randomized controlled trial.pt.
2. controlled clinical trial.pt.
3. exp Randomized Controlled Trials/
4. exp Random Allocation/
5. exp Double‐Blind Method/
6. exp Single‐Blind Method/
7. clinical trial.pt.
8. Clinical Trial/
9. (clin$ adj trial$).ab,ti.
10. ((singl$ or doubl$ or trebl$ or tripl$) adj (blind$ or mask$)).ab,ti.
11. exp PLACEBOS/
12. placebo$.ab,ti.
13. random$.ab,ti.
14. exp Research Design/
15. or/1‐14
16. (animals not humans).sh.
17. 15 not 16
18. levetiracetam.tw.
19. (epilep$ or seizure$ or convulsion$).tw.
20. exp Seizures/
21. exp Epilepsy/
22. 19 or 20 or 21
23. 17 and 18 and 22
Appendix 3. ClinicalTrials.gov search strategy
Interventional Studies | Epilepsies, Partial | Levetiracetam OR Keppra
Appendix 4. ICTRP search strategy
Condition: partial epilepsy OR focal epilepsy
Intervention: Levetiracetam OR Keppra
Recruitment status: all
Data and analyses
Comparison 1. Levetiracetam versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 ≥ 50% reduction in focal seizure frequency: intention to treat | 14 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1.1 60 mg/kg/day | 2 | 296 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.91 [1.38, 2.63] |
| 1.1.2 500 mg | 1 | 141 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.60 [0.71, 3.62] |
| 1.1.3 1000 mg | 5 | 854 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.30 [1.72, 3.06] |
| 1.1.4 2000 mg | 4 | 533 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.26 [2.16, 4.94] |
| 1.1.5 3000 mg | 8 | 1082 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.59 [2.07, 3.23] |
| 1.1.6 4000 mg | 1 | 77 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.64 [0.59, 4.57] |
| 1.1.7 All doses | 14 | 2455 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.37 [2.02, 2.78] |
| 1.2 ≥ 50% reduction in focal seizure frequency: intention to treat (random effects) | 14 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.2.1 1000 mg | 5 | 854 | Risk Ratio (M‐H, Random, 95% CI) | 2.31 [1.43, 3.75] |
| 1.2.2 2000 mg | 4 | 533 | Risk Ratio (M‐H, Random, 95% CI) | 2.98 [1.59, 5.60] |
| 1.2.3 3000 mg | 8 | 1082 | Risk Ratio (M‐H, Random, 95% CI) | 2.49 [1.80, 3.45] |
| 1.2.4 All doses | 14 | 2455 | Risk Ratio (M‐H, Random, 95% CI) | 2.26 [1.79, 2.85] |
| 1.3 ≥ 50% reduction in focal seizure frequency: (intention to treat): subgroup analysis by age | 14 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.3.1 Children | 2 | 296 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.91 [1.38, 2.63] |
| 1.3.2 Adults (all doses) | 12 | 2159 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.49 [2.08, 2.99] |
| 1.4 ≥ 50% reduction in focal seizure frequency: (intention to treat): subgroup analysis by age (random effects) | 12 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.4.1 Adults (all doses) | 12 | 2159 | Risk Ratio (M‐H, Random, 95% CI) | 2.40 [1.82, 3.16] |
| 1.5 ≥ 50% reduction in focal seizure frequency: best case | 14 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.5.1 60 mg/kg/day | 2 | 296 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.91 [1.38, 2.63] |
| 1.5.2 500 mg | 1 | 141 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.97 [0.90, 4.31] |
| 1.5.3 1000 mg | 5 | 854 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.59 [1.96, 3.43] |
| 1.5.4 2000 mg | 4 | 533 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.00 [2.67, 5.99] |
| 1.5.5 3000 mg | 8 | 1082 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.81 [2.26, 3.50] |
| 1.5.6 4000 mg | 1 | 77 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.69 [1.53, 8.95] |
| 1.5.7 All doses | 14 | 2455 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.63 [2.25, 3.09] |
| 1.6 ≥ 50% reduction in focal seizure frequency: worst case | 14 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.6.1 60 mg/kg/day | 2 | 296 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.91 [1.38, 2.63] |
| 1.6.2 500 mg | 1 | 141 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.42 [0.65, 3.12] |
| 1.6.3 1000 mg | 5 | 854 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.94 [1.48, 2.53] |
| 1.6.4 2000 mg | 4 | 533 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.31 [1.62, 3.30] |
| 1.6.5 3000 mg | 8 | 1082 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.14 [1.75, 2.62] |
| 1.6.6 4000 mg | 1 | 77 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.63 [0.30, 1.35] |
| 1.6.7 All doses | 14 | 2455 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.99 [1.72, 2.30] |
| 1.7 ≥ 50% reduction in focal seizure frequency: best case (random effects) | 14 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.7.1 1000 mg | 5 | 854 | Risk Ratio (M‐H, Random, 95% CI) | 2.63 [1.67, 4.14] |
| 1.7.2 3000 mg | 8 | 1082 | Risk Ratio (M‐H, Random, 95% CI) | 2.74 [1.96, 3.82] |
| 1.7.3 All doses | 14 | 2455 | Risk Ratio (M‐H, Random, 95% CI) | 2.55 [1.99, 3.27] |
| 1.8 ≥ 50% reduction in focal seizure frequency: worst case (random effects) | 14 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.8.1 1000 mg | 5 | 854 | Risk Ratio (M‐H, Random, 95% CI) | 1.94 [1.17, 3.21] |
| 1.8.2 2000 mg | 4 | 533 | Risk Ratio (M‐H, Random, 95% CI) | 2.14 [0.89, 5.11] |
| 1.8.3 3000 mg | 8 | 1082 | Risk Ratio (M‐H, Random, 95% CI) | 2.03 [1.45, 2.84] |
| 1.8.4 All doses | 14 | 2455 | Risk Ratio (M‐H, Random, 95% CI) | 1.87 [1.45, 2.41] |
| 1.9 ≥ 50% reduction in focal seizure frequency: sensitivity analysis with trials of low risk of bias only | 5 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.9.1 Intention to treat | 5 | 1156 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.93 [2.25, 3.81] |
| 1.9.2 Best‐case scenario | 5 | 1156 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.13 [2.41, 4.06] |
| 1.9.3 Worst‐case scenario | 5 | 1156 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.69 [2.09, 3.46] |
| 1.10 ≥ 50% reduction in focal seizure frequency: sensitivity analysis with trials of low risk of bias only (random effects) | 6 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.10.1 Intention to treat | 6 | 1354 | Risk Ratio (M‐H, Random, 95% CI) | 2.81 [1.88, 4.19] |
| 1.10.2 Best‐case scenario | 6 | 1354 | Risk Ratio (M‐H, Random, 95% CI) | 2.95 [2.02, 4.31] |
| 1.10.3 Worst‐case scenario | 6 | 1354 | Risk Ratio (M‐H, Random, 95% CI) | 2.65 [1.79, 3.92] |
| 1.11 Treatment withdrawal | 13 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.11.1 60 mg/kg/day | 2 | 296 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.80 [0.43, 1.46] |
| 1.11.2 500 mg | 1 | 141 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.96 [0.84, 10.47] |
| 1.11.3 1000 mg | 5 | 854 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.23 [0.85, 1.79] |
| 1.11.4 2000 mg | 4 | 533 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.49 [0.97, 2.28] |
| 1.11.5 3000 mg | 7 | 1055 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.11 [0.79, 1.57] |
| 1.11.6 4000 mg | 1 | 77 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.92 [0.42, 2.02] |
| 1.11.7 Any dose | 13 | 2428 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.11 [0.89, 1.40] |
| 1.12 Treatment withdrawal: subgroup analysis by age | 13 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.12.1 Children | 2 | 296 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.80 [0.43, 1.46] |
| 1.12.2 Adults | 11 | 2132 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.18 [0.92, 1.51] |
| 1.13 Treatment withdrawal: subgroup analysis by age (random effects) | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.13.1 Children | 2 | 296 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.28, 2.52] |
| 1.14 Treatment withdrawal: sensitivity analysis with trials of low risk of bias only | 5 | 1156 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.26 [0.90, 1.76] |
| 1.15 Treatment withdrawal: sensitivity analysis with trials of low risk of bias only (random effects) | 5 | 1156 | Risk Ratio (M‐H, Random, 95% CI) | 1.24 [0.89, 1.74] |
| 1.16 Adverse effects: 5 most common adverse effects (any age) | 13 | Risk Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 1.16.1 Somnolence | 13 | 2425 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.62 [1.19, 2.20] |
| 1.16.2 Headache | 13 | 2425 | Risk Ratio (M‐H, Fixed, 99% CI) | 0.85 [0.59, 1.21] |
| 1.16.3 Dizziness | 13 | 2425 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.54 [0.98, 2.41] |
| 1.16.4 Fatigue (asthenia) | 13 | 2425 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.53 [0.98, 2.38] |
| 1.16.5 Accidental injury | 13 | 2425 | Risk Ratio (M‐H, Fixed, 99% CI) | 0.72 [0.49, 1.06] |
| 1.17 Adverse effects: 5 most common adverse effects (any age, random effects) | 13 | Risk Ratio (M‐H, Random, 99% CI) | Subtotals only | |
| 1.17.1 Headache | 13 | 2425 | Risk Ratio (M‐H, Random, 99% CI) | 0.74 [0.38, 1.47] |
| 1.17.2 Accidental injury | 13 | 2425 | Risk Ratio (M‐H, Random, 99% CI) | 0.69 [0.35, 1.36] |
| 1.18 Adverse effects: most common adverse effects in adults | 10 | Risk Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 1.18.1 Accidental injury | 4 | 1023 | Risk Ratio (M‐H, Fixed, 99% CI) | 0.60 [0.39, 0.92] |
| 1.18.2 Ataxia (unpublished data only) | 4 | 1023 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.50 [0.43, 5.26] |
| 1.18.3 Dizziness | 9 | 1813 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.55 [0.95, 2.51] |
| 1.18.4 Fatigue (asthenia) | 4 | 1023 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.47 [0.90, 2.40] |
| 1.18.5 Headache | 7 | 1711 | Risk Ratio (M‐H, Fixed, 99% CI) | 0.78 [0.54, 1.14] |
| 1.18.6 Infection | 4 | 1023 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.76 [1.03, 3.02] |
| 1.18.7 Nausea | 3 | 599 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.37 [0.47, 4.00] |
| 1.18.8 Somnolence | 10 | 2099 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.57 [1.13, 2.20] |
| 1.19 Adverse effects: most common adverse effects in children | 2 | Risk Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 1.19.1 Accidental injury | 1 | 198 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.63 [0.63, 4.26] |
| 1.19.2 Aggression (hostility) | 2 | 296 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.72 [0.64, 4.63] |
| 1.19.3 Cough | 2 | 296 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.39 [0.49, 3.93] |
| 1.19.4 Dizziness | 2 | 296 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.52 [0.47, 4.94] |
| 1.19.5 Fatigue (asthenia) | 2 | 296 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.82 [0.62, 5.33] |
| 1.19.6 Pharyngitis | 2 | 296 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.09 [0.47, 2.50] |
| 1.19.7 Somnolence | 2 | 296 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.90 [0.88, 4.09] |
| 1.19.8 Vomiting | 2 | 296 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.22 [0.55, 2.69] |
| 1.20 Behavioural changes | 6 | Risk Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 1.20.1 Hostility | 1 | 198 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.92 [0.56, 6.60] |
| 1.20.2 Personality disorder | 1 | 198 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.10 [0.30, 3.95] |
| 1.20.3 Nervousness | 1 | 198 | Risk Ratio (M‐H, Fixed, 99% CI) | 4.80 [0.68, 34.14] |
| 1.20.4 Depression | 1 | 324 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.41 [0.25, 7.85] |
| 1.20.5 Aggression | 1 | 98 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.42 [0.27, 7.42] |
| 1.20.6 Agitation | 2 | 254 | Risk Ratio (M‐H, Fixed, 99% CI) | 6.17 [0.66, 57.79] |
| 1.20.7 Emotional lability | 1 | 198 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.44 [0.28, 7.29] |
| 1.20.8 Psychomotor hyperactivity | 1 | 98 | Risk Ratio (M‐H, Fixed, 99% CI) | 0.42 [0.08, 2.19] |
| 1.20.9 Irritability | 1 | 156 | Risk Ratio (M‐H, Fixed, 99% CI) | 11.28 [0.26, 495.63] |
| 1.20.10 Abnormal behaviour | 1 | 98 | Risk Ratio (M‐H, Fixed, 99% CI) | 5.92 [0.14, 255.98] |
| 1.20.11 Altered mood | 1 | 98 | Risk Ratio (M‐H, Fixed, 99% CI) | 4.85 [0.11, 216.99] |
| 1.20.12 Anxiety | 1 | 98 | Risk Ratio (M‐H, Fixed, 99% CI) | 4.85 [0.11, 216.99] |
| 1.20.13 Dissociation | 1 | 94 | Risk Ratio (M‐H, Fixed, 99% CI) | 0.14 [0.00, 6.77] |
| 1.20.14 Combined (regardless of age) | 6 | 926 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.87 [1.19, 2.95] |
| 1.20.15 Combined (children) | 2 | 296 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.90 [1.16, 3.11] |
| 1.20.16 Combined (adults) | 4 | 630 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.79 [0.59, 5.41] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Ben‐Menachem 2000.
| Study characteristics | ||
| Methods | Randomised, double‐blind, PCB‐controlled, parallel trial Multicentre across Europe 2 treatment arms: 1 LEV; 1 PCB Randomisation concealment: telephone randomisation. Random list generation: centralised minimisation procedure of an unbalanced randomisation list (2 LEV:1 PCB) Blinding: identical tablets and packages. Investigators were described as blinded to treatment assignment. If treatment code was broken, the participant had to be removed from the trial Baseline: 12 weeks. Treatment period: 16 weeks (4 weeks' titration, 12 weeks' maintenance) |
|
| Participants | All adults Total randomised 286 adults; all with drug‐resistant focal epilepsy 181 adults to LEV 3000 mg; 105 adults to PCB 48% male Age range: 17–70 years Other AEDs: 1 ≥ 2 focal seizures per 4 weeks during 12‐week baseline ≥ 1‐year history of focal epilepsy Mean duration of epilepsy(years): LEV: 19 (SD 11); PCB: 19 (SD 12); overall: 19 (SD 11) Median baseline seizure frequency per week: 1.7; range: 0.3–1.7 |
|
| Interventions | LEV 3000 mg/day PCB Uptitration dosages: titrated upwards every 2 weeks from 500 mg twice daily to the target dosage of 1500 mg twice daily |
|
| Outcomes | ≥ 50% reduction in seizure frequency Treatment withdrawal Adverse effects |
|
| Notes | 10 participants randomised to levetiracetam were not analysed, with reasons given. Three participants randomised to placebo were not analysed, with reasons also given. These participants contributed to our best‐case and worst‐case sensitivity analyses. In this iteration, we used the published outcome data on 50% or greater reduction in seizure frequency for this trial, having previously used the data obtained in the unpublished trial manuscripts in 2012. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Unbalanced randomisation list (2 LEV:1 PCB) was produced by the central randomisation office. |
| Allocation concealment (selection bias) | Low risk | Investigator confirmed the participants' eligibility, then called the central randomisation office who allocated a treatment number. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Identical LEV and PCB tablets and packaging. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Investigators described as blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants who were randomised were analysed in the groups they were initially assigned to notwithstanding premature terminations. The reasons or outcomes (or both) for premature terminations were stated. There were sufficient data to allow an ITT as well as best‐case and worst‐case sensitivity analyses to be conducted where required. |
| Selective reporting (reporting bias) | Low risk | All outcomes measured (that were also relevant to this review) were published. |
Betts 2000.
| Study characteristics | ||
| Methods | Randomised, double‐blind, PCB‐controlled, parallel trial Multicentre across Europe 3 treatment arms: 2 LEV and 1 PCB Randomisation concealment: allocated sequentially sealed, numbered packages containing either LEV or PCB. Random list generation: computer‐generated random permuted blocks (size 3). Blinding: identical tablets and packages. Investigators were described as blinded to treatment assignment. If treatment code was broken, the participant had to be removed from the trial. Baseline: 4 weeks. No titration period. Treatment period: 24 weeks |
|
| Participants | All adults Total randomised 119 adults 42 adults to LEV 2000 mg; 38 adults to LEV 4000 mg; 39 adults to PCB 61% male Age range: 16–67 years Other AEDs: 1–3 ≥ 4 seizures in 6 months before study entry Mean duration of epilepsy (years): LEV 2000 mg: 21.1 (SD 14.4); LEV 4000 mg: 24.6 (SD 15.6); PCB: 26.0 (SD 13.2) Median of baseline seizure frequency per week: LEV 2000 mg: 1.21; LEV 4000 mg: 1.34; PCB: 1.24 |
|
| Interventions | LEV 2000 mg/day LEV 4000 mg/day PCB add‐on |
|
| Outcomes | Treatment withdrawal Adverse effects QoL and cognitive effects |
|
| Notes | Baseline seizure frequency data were derived from 34 participants in LEV 2000 mg group, 36 participants in LEV 4000 mg group; 36 participants in PCB group In the text for the trial, the number of participants in the inferential ITT population was reported as 27 for LEV 2000 mg, 28 for LEV 4000 mg, and 31 for PCB groups. In a graph for the trial, the number of participants in the inferential ITT population was reported as 26 in LEV 2000 mg, 28 in LEV 4000 mg, and 25 in PCB groups. The number of participants forming the denominator changed without account being provided by the authors on the reasons for this or the participants' response to treatment, resulting in high risk of bias gradings for incomplete outcome data and selective reporting. The missing participants contribute to our best‐case and worst‐case analyses. All participants had drug‐resistant epilepsy and some had generalised‐onset and unclassified seizures. QoL was assessed using the ESI‐55 for 30 or 31 participants in LEV 2000 mg, 26–28 participants in LEV 4000 mg, and 28 participants in PCB groups. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Blocked (restricted) randomisation generated by the RANUNI function of SAS software performed the permutation of the 3 treatment numbers. |
| Allocation concealment (selection bias) | Low risk | Treatment group allocated according to which country the study took place, and the chronological order of their entry into the study. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | 500 mg tablet for LEV and PCB. Identical in taste and appearance. Provided by UCB S.A. Pharma. Participants withdrawn if the double‐blind code was broken. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Investigator described as blinded. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Baseline seizure frequency data were derived from 34 participants in the LEV 2000 mg, 36 in the LEV 4000 mg, and 36 in the PCB groups. In the text for the trial, the number of participants in the inferential ITT population was reported as 27 in the LEV 2000 mg, 28 in the LEV 4000 mg, and 31 in the PCB groups. In a graph for the trial, the number of participants in the inferential ITT population was reported as 26 in the LEV 2000 mg, 28 in the LEV 4000 mg, and 25 in the PCB groups. The number of participants forming the denominator changed without account being provided by the authors on the reasons for this or the participants' response to treatment. |
| Selective reporting (reporting bias) | High risk | Missing data for baseline seizure frequency. |
Cereghino 2000.
| Study characteristics | ||
| Methods | Randomised, double‐blind, PCB‐controlled, parallel trial Multicentre across USA 3 treatment arms: 2 LEV and 1 PCB Randomisation concealment: allocated sequentially sealed, numbered packages containing either LEV or PCB. Random list generation: random permuted blocks Blinding: identical tablets and packages. Investigators were described as blinded to treatment assignment. If treatment code was broken, the participant had to be removed from the trial. Baseline: 12 weeks. Treatment period: 18 weeks (4 weeks' titration, 14 weeks' maintenance) |
|
| Participants | All adults Total randomised 294 adults 98 adults to LEV 1000 mg; 101 adults to LEV 3000 mg; 95 adults to PCB 61% men Age range: 16–70 years Other AEDs ≥ 1 ≥ 2 focal seizures per 4 weeks during 12‐week baseline ≥ 2‐year history of uncontrolled focal epilepsy Mean duration of epilepsy (years): not given Median baseline seizure frequency per week: 2.13; range: 0.15–163.56 |
|
| Interventions | LEV 1000 mg/day LEV 3000 mg/day PCB add‐on Uptitration dosages = LEV dose was escalated at 2‐week intervals during the titration period. Doses of LEV were 333 mg/day for 2 weeks, then 666 mg/day for 2 weeks, and 1000 mg/day started on the first visit of the observation period, or 1000 mg/day, 2000 mg/day, then 3000 mg/day. |
|
| Outcomes | ≥ 50% reduction in seizure frequency Treatment withdrawal Adverse effects QoL and cognitive effects |
|
| Notes | A minority of participants also had generalised or unclassified seizures, or both, in addition to partial‐onset seizures. 1 participant in LEV 1000 mg was excluded from 50% responder analysis. QoL assessed using the QOLIE‐31 for 80 participants in LEV 1000 mg, 85 participants in LEV 3000 mg, and 81 participants in PCB. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was in blocks by study site. Each participant was assigned a unique treatment number, which corresponded to the blinded randomised treatment. |
| Allocation concealment (selection bias) | Low risk | The treatment identity (LEV/PCB) and dosage (1000 mg/day or 3000 mg/day) for each participant were contained in sealed envelopes label with the trial identification and the treatment number. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | All study medication was blinded. The PCB and all strengths of LEV were identical in appearance, shape, size, colour, and smell. Unbroken tablets were equal in taste. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Investigators described as blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants who were randomised (294) were analysed in the groups they were initially assigned. They were included in the analysis regardless of whether they finished the trial. The numbers randomised in each arm were 95 adults to LEV 1000 mg/day, 101 adults to LEV 3000 mg/day, and 95 adults to PBC. There was sufficient data to allow an ITT, as well as best‐case and worst‐case sensitivity analysis to be conducted. |
| Selective reporting (reporting bias) | Low risk | All outcomes measured (that were also relevant to this review) were published. |
Glauser 2006.
| Study characteristics | ||
| Methods | Randomised, double‐blind, PCB‐controlled trial Multicentre (60 centres) across the US and Canada 2 treatment arms: 1 LEV and 1 PCB Randomisation concealment: randomisation schedule was performed by centre and participants were allocated sequentially. Random list generation: computer‐generated schedule with a permuted block (size 4). Blinding: identical tablets and packages. Investigators, site personnel, study personnel from the contract research organisation responsible for the monitoring and conduct of the trial, and study sponsor personnel were described as blinded to treatment assignment. Baseline: 8 weeks. Treatment period: 14 weeks (4 weeks' titration, 10 weeks' maintenance) |
|
| Participants | All children Total randomised 216 children; all with drug‐resistant focal epilepsy 101 children to LEV 60 mg/kg/day and 97 children to PCB 54% boys in LEV; 47% boys in PCB Age range: 3–17 years Other AEDs 1 or 2 ≥ 4 focal seizures per 4 weeks during 8‐week baseline ≥ 4 focal seizures during 4 weeks before screening Diagnosis of uncontrolled focal epilepsy made ≥ 6 months before screening Mean duration of epilepsy (years): LEV: 7.4; PCB: 6.8 Median baseline seizure frequency per week: LEV: 4.7 (range 0–696); PCB: 5.3 (range 0–467) |
|
| Interventions | LEV 60 mg/kg/day PCB add‐on Uptitration dosages 20 mg/kg/day, increasing every 2 weeks |
|
| Outcomes | ≥ 50% reduction in seizure frequency Treatment withdrawal Adverse effects |
|
| Notes | Before breaking the blinding, 18 participants were excluded, including all 16 participants at 1 site who were excluded because of extensive violation of the protocol and consequent unreliability of the data, and 2 participants because they discontinued before taking any study medication. It was unclear to which groups the 16 participants were assigned. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was in blocks by study site. Each participant was assigned a unique treatment number, which corresponded to the blinded randomised treatment. |
| Allocation concealment (selection bias) | Low risk | The treatment identity (LEV/PCB) and dosage (LEV1000 mg/day or 3000 mg/day) for each participant were contained in sealed envelopes labelled with the trial identification and the treatment number. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | All study medication was blinded. The PCB and both strengths of LEV were identical in appearance, shape, size, colour, and smell. Unbroken tablets were equal in taste. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Investigators described as blinded. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Although reported as an ITT analysis, before breaking the blinding, 18 randomised participants were excluded, including all 16 participants at 1 site who were excluded because of extensive violation of the protocol and consequent unreliability of the data, and 2 participants because they discontinued before taking any study medication. It was unclear to which groups the 16 participants were assigned or what their outcomes were. Therefore, the reason for missing outcome data could be related to true outcome, with either imbalance in numbers or reasons for missing data across intervention groups. In a study of this size, the proportion of missing outcomes compared with observed event risk would be enough to induce clinically relevant bias in intervention effect estimate. |
| Selective reporting (reporting bias) | Low risk | All outcomes measured (that were also relevant to this review) were published. |
Inoue 2015.
| Study characteristics | ||
| Methods | Randomised, double‐blind, PCB‐controlled, parallel trial Multicentre, across Japan 5 treatment arms: 4 LEV and 1 PCB Randomisation concealment: method not specified. Random list generation: method not specified. Blinding: study described as double blind. Method not specified. Baseline: 12 weeks. Treatment period: 16 weeks (4 weeks' titration, 12 weeks' maintenance). Followed by 4 weeks' downtitration or transition period. |
|
| Participants | All adults Total randomised 352 adults; 351 analysed (1 excluded due to drug dispensing error) 71 adults to LEV 500 mg; 70 adults to LEV 1000 mg; 70 adults to LEV 2000 mg; 70 adults to LEV 3000 mg; 70 adults to PCB 54% men in LEV; 47% men in PCB Age range: 16–64 years Other AEDs: 1–3 ≥ 12 focal seizures during 12‐week baseline, ≥ 2 every 4 weeks History of partial seizure > 2 years, with EEG confirmation within 1 year prior to trial entry Mean duration of epilepsy (years): LEV 500 mg: 16.4 (SD 10.9); LEV 1000 mg: 14.5 (SD 8.9); LEV 2000 mg: 13.8 (SD 9.6); LEV 3000 mg: 15.2 (SD 10.3); PCB: 16.3 (SD 11.9) Median baseline seizure frequency per week: LEV 500 mg: 2.67; LEV 1000 mg: 2.75; LEV 2000 mg: 3.21; LEV 3000 mg: 2.65; PCB: 3.00 |
|
| Interventions | 4 tablets taken in each arm LEV 500 mg/day LEV 1000 mg/day LEV 2000 mg/day LEV 3000 mg/day PCB The dosage for participants assigned to LEV 500 mg/day or 1000 mg/day was not uptitrated; they received 3 PCB tablets plus 1 LEV 250 mg (for LEV 500 mg/day) or 500 mg tablet (for LEV 1000 mg/day) twice daily. The dosage for participants randomised to LEV 2000 mg/day or 3000 mg/day was uptitrated in the following manner: all participants received LEV 1000 mg/day (500 mg tablet twice daily) and had their dosage increased by 1000 mg/day every 2 weeks until the target dose was reached |
|
| Outcomes | ≥ 50% reduction in seizure frequency Treatment withdrawal Adverse effects |
|
| Notes | 1 participant excluded due to drug dispensing error. The denominators given for the 50% or greater reduction in seizure frequency outcomes were 68 for LEV 500 mg, 68 for LEV 1000 mg, 68 for LEV 2000 mg, 69 for LEV 3000 mg, and 69 for PCB. These were fewer participants than were randomised to each of these groups (see above) and it was unclear what the outcomes were for each of the unreported participants. This resulted in the study being graded as high risk for incomplete outcome data biases. The missing participants contribute to our best‐case and worst‐case analyses. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method not specified. |
| Allocation concealment (selection bias) | Unclear risk | Method not specified. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Method not specified. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Method not specified. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 1 randomised participant excluded due to drug dispensing error. It was unclear to which group they were assigned or their outcome. The denominators given for the 50% or greater reduction in seizure frequency outcomes were 68 for LEV 500 mg, 68 for LEV 1000 mg, 68 for LEV 2000 mg, 69 for LEV 3000 mg, and 69 for PCB. These were fewer participants than were randomised to each of these groups (see above) and it was unclear what the outcomes were for each of the unreported participants. |
| Selective reporting (reporting bias) | Low risk | All outcomes measured (that were also relevant to this review) were published. |
Levisohn 2009.
| Study characteristics | ||
| Methods | Randomised, double‐blind, PCB‐controlled trial Multicentre (28) across the US, Canada, and South Africa 2 treatment arms: 1 LEV and 1 PCB Randomisation concealment: method not stated. Random list generation: no explicit statement of sequence‐generation method, but participants were randomised either to LEV or PCB in a 2:1 ratio. Randomisation was stratified for age (4–7 years, 8–12 years, 13–16 years) and number of concomitant AEDs (1 or 2). Blinding: descried as double‐blind without further specification aside from stating that neurocognitive testing was carried out by the same experienced, blinded neuropsychologist. Baseline: 4 weeks historical, 1 week prospective. Treatment period: 12 weeks (4 weeks' titration, 8 weeks' maintenance) |
|
| Participants | All children Total randomised 98 children 64 children to LEV 60 mg/kg/day; 34 children to PCB 61% boys in LEV; 50% boys in PCB Age range: 4.1–16.7 years Other AEDs: 1 or 2 ≥ 1 focal seizure during 4 weeks before screening Diagnosis of focal epilepsy made ≥ 6 months before screening Mean duration of epilepsy (years): not given Median baseline seizure frequency per week: LEV: 0.9 (IQR 0.4–1.9); PCB: 1.4 (IQR 0.4–5.2) |
|
| Interventions | LEV 60 mg/kg/day PCB add‐on Uptitration dosages: 20 mg/kg/day orally twice daily as tablets or 10% solution, uptitrated in increments of 20 mg/kg/day every 2 weeks |
|
| Outcomes | ≥ 50% reduction in seizure frequency Treatment withdrawal Adverse effects Cognitive effects Behavioural and emotional functioning |
|
| Notes | Cognitive assessment using Leiter‐R AM, WRAML‐2, and Leiter‐R ERS. Behavioural and emotional functioning assessed using CBCL and CHQ‐PF50. Cognitive, behavioural, and emotional function results shown only for the per‐protocol population: 46 in LEV, 27 in PCB. A few participants had generalised‐onset (1 in LEV, 1 in PCB) or unclassified seizures (1 in PCB), or both, in addition to partial‐onset seizures. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method not stated. |
| Allocation concealment (selection bias) | Unclear risk | Method not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Method not stated. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Method not stated. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 99 participants randomised, with 1 (1%) randomised participant not analysed. A clear reason was given that this was because they had not taken any study medication although their outcome was not clarified. Overall, this was unlikely to have introduced bias. All other participants included in the ITT were analysed in the groups they were initially assigned. |
| Selective reporting (reporting bias) | Low risk | All outcomes measured (that were also relevant to this review) were published. |
Peltola 2009.
| Study characteristics | ||
| Methods | Randomised, double‐blind, PCB‐controlled trial Multicentre: 7 centres in Finland, India, Mexico, Russia, South Africa, and Ukraine 2 treatment arms: 1 LEV XR and 1 PCB Randomisation concealment: interactive voice response system. Random list generation: randomised 1:1 using interactive voice response system. Blinding: identical tablets and packages, all study personnel and participants were described as being blinded to treatment assignment. Baseline: 8 weeks. Treatment period: 12 weeks (no uptitration took place) |
|
| Participants | All adults Total randomised: 158 adults 79 adults to LEV XR 1000 mg; 79 adults to PCB 66% men in LEV XR; 59% men in PCB Age range 12–70 years Other AEDs 1–3 ≥ 8 focal seizures during 8‐week baseline within which ≥ 2 focal seizures per 4 weeks Diagnosis of uncontrolled focal epilepsy made ≥ 6 months before screening Mean duration of epilepsy (years): LEV XR 13.11 (SD 10.87) (range 0.8–42.6); PCB: 16.43 (SD 11.93) (range 0.7–53.5) Mean baseline seizure frequency per week: LEV XR: 40.7 (SD 66.0); PCB: 30.6 (SD 52.5) |
|
| Interventions | LEV XR 1000 mg PCB |
|
| Outcomes | ≥ 50% reduction in seizure frequency Treatment withdrawal Adverse effects |
|
| Notes | 2 participants randomised to LEV XR did not receive any medication; therefore, they were excluded from the safety population, leaving 77 participants treated with LEV XR and 79 treated with PCB in the safety analysis dataset. Baseline level for determining reduction in seizure frequency was derived from 74 participants in LEV XR group and 78 in PCB group. A few participants had other seizure types in addition to partial‐onset seizures. "Study personnel" were taken to mean investigators. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants randomised 1:1 (using an interactive voice response system) to receive either the targeted dose of 1000 mg/day LEV XR (as 2 × 500 mg oral tablets) or 2 matching PCB tablets once‐daily (each evening). |
| Allocation concealment (selection bias) | Low risk | Voice response system used. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Both LEV XR 500 mg and PCB tablets were identical in shape, size, taste, and colour, and all study personnel and participants were blinded to treatment assignment. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | All study personnel and participants were blinded to treatment assignment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants randomised into the ITT were analysed in the groups they were initially assigned. |
| Selective reporting (reporting bias) | Low risk | All outcomes measured (that were also relevant to this review) were published. |
Shorvon 2000.
| Study characteristics | ||
| Methods | Randomised, double‐blind, PCB‐controlled, crossover trial Multicentre across Europe 3 treatment arms: 2 LEV and 1 PCB Randomisation concealment: allocated sequentially sealed, numbered packages containing either LEV or PCB. Random list generation: random permuted blocks (size 6) Blinding: identical tablets and packages. Investigators and staff were described as blinded to treatment assignment. If treatment code was broken, the participant had to be removed from the trial. Baseline: 8–12 weeks. Treatment period: 16 weeks (4 weeks' titration, 12 weeks' maintenance) |
|
| Participants | All adults Total randomised 324 adults; all with drug‐resistant focal epilepsy but few also had generalised‐onset or unclassified seizures, or both. 106 adults to LEV 1000 mg; 106 adults to LEV 2000 mg; 112 adults to PCB 49% men Age range: 14–69 years Other AEDs: 1 or 2 ≥ 4 focal seizures per 4 weeks during 8‐ or 12‐week baseline ≥ 2‐year history of uncontrolled focal epilepsy Mean duration of epilepsy (years): LEV 1000 mg: 23.8 (SD 12.3); LEV 2000 mg: 23.6 (SD 13.3); PCB: 23.2 (SD 11.0); overall: 23.6 (SD 12.2) Mean baseline seizure frequency per week: 2.62; range: 0.3–102.7 |
|
| Interventions | LEV 1000 mg LEV 2000 mg PCB Uptitration dosages: LEV was titrated upwards in twice‐daily increments of 500 mg at 2‐week intervals until participants were stabilised on their assigned dosages (1000 mg/day or 2000 mg/day). The 1000‐mg LEV group received PCB for 2 weeks before initiation of active drug |
|
| Outcomes | ≥ 50% reduction in seizure frequency Treatment withdrawal Adverse effects QoL and cognitive effects |
|
| Notes | A few participants had generalised‐onset or unclassified seizures, or both, in addition to partial‐onset seizures. QoL assessed using the ESI‐55 for 92 participants each in LEV 1000 mg and LEV 2000 mg groups, and 89 participants in PCB group. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random permuted blocks (size 6). |
| Allocation concealment (selection bias) | Low risk | Allocated sequentially sealed, numbered packages containing either LEV or PCB. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Identical tablets and packages. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Investigators and staff were described as blinded to treatment assignment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants randomised into the ITT were analysed in the groups they were initially assigned to according to the published data, although in the unpublished protocols 2 participants were said to have been excluded from 50% responder analysis: 1 in LEV 2000 mg; 1 in PCB. Overall, this was unlikely to have introduced bias as missing outcome data balanced in numbers across intervention groups. |
| Selective reporting (reporting bias) | Low risk | All outcomes measured (that were also relevant to this review) were published. |
Tsai 2006.
| Study characteristics | ||
| Methods | Randomised, double‐blind, PCB‐controlled trial Multicentre in Taiwan 2 treatment arms: 1 LEV and 1 PCB Randomisation concealment: allocated sequentially sealed, numbered packages containing either LEV or PCB. Random list generation: random permuted blocks (size 4). Blinding: identical tablets and packages. Investigators were described as blinded to treatment assignment. Baseline: 8 weeks. Treatment period: 14 weeks (2 weeks' titration, 12 weeks' maintenance) |
|
| Participants | All adults Total randomised 94 adults 47 adults to LEV 2000 mg; 47 adults to PCB 36% men in LEV; 53% men in PCB Age range: 16–60 years Other AEDs: 1–3 ≥ 4 focal seizures during 8‐week baseline Diagnosis of uncontrolled focal epilepsy made ≥ 6 months before study Mean duration of epilepsy (years): LEV: 18.6 (SD 8.5); PCB: 18.7 (SD 10.7) Mean baseline seizure frequency per week: LEV: 4.0 (SD 14.1); PCB: 4.3 (SD 7.0) |
|
| Interventions | LEV 2000 mg/day PCB Uptitration dosages: initial LEV dose was 500 mg twice daily, which was increased to 1000 mg twice daily after 2 weeks. |
|
| Outcomes | ≥ 50% reduction in seizure frequency Treatment withdrawal Adverse effects |
|
| Notes | 1 participant (LEV group) excluded from 50% responder analysis. A minority of participants also had generalised or unclassified, or both, seizures in addition to partial‐onset seizures. 14 participants required dose reduction (11 in LEV; 3 in PCB). |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The random treatment‐allocation sequence was generated in Belgium by the Clinical Drug Supply Unit of the sponsor on 22 May 2000, by using a randomisation program (permuted blocks of size 4) generated by a statistician of the sponsor. |
| Allocation concealment (selection bias) | Low risk | Treatments assigned sequentially to participants at every site by the investigators. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Study drug containers were labelled in Belgium by the sponsor using an identical method for both treatments. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Investigators were described as blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 1 participant (LEV group) excluded from 50% responder analysis because they had missing data for weekly frequency of partial seizures. This reason for missing outcome data was unlikely to be related to true outcome. Remaining randomised participants were analysed in the group they were assigned. |
| Selective reporting (reporting bias) | Low risk | All outcomes measured (that were also relevant to this review) were published. |
Wu 2009.
| Study characteristics | ||
| Methods | Randomised, double‐blind, PCB‐controlled trial Multicentre, 6 centres in China 2 treatment arms: 1 LEV and 1 PCB Randomisation concealment: method not stated. Study medications were supplied and packaged by UCB S.A. Pharma. Method of sequence generation: not stated. Blinding: "matched placebo" was used. No further specification. Baseline: 8 weeks. Treatment period: 16 weeks (4 weeks' titration, 12 weeks' maintenance) |
|
| Participants | All adults Total randomised 206 adults 103 adults to LEV; 103 adults to PCB 50% men in LEV; 54% men in PCB Age range: 16–70 years Other AEDs: 1 or 2 ≥ 8 focal seizures during 8‐week baseline Diagnosis of focal epilepsy made ≥ 6 months before screening Mean duration of epilepsy (years): LEV: 16.5 (SD 12.7); PCB: 17.3 (SD 12.1) Median baseline seizure frequency per week: LEV 1.81 (IQR 1.13–3.38); PCB 1.75 (IQR 1.13–4.00) |
|
| Interventions | LEV 3000 mg PCB Uptitration dosages: started with 500 mg (1 tablet) twice daily and was uptitrated in twice‐daily increments of 500 mg (1 tablet) at 2‐week intervals; the dose was increased to 2000 mg/day after 2 weeks and to 3000 mg/day after an additional 2 weeks. |
|
| Outcomes | ≥ 50% reduction in seizure frequency Treatment withdrawal Adverse effects |
|
| Notes | 4 participants excluded from 50% responder analysis: 1 in LEV 3000 mg (lost to follow‐up) and 3 in PCB (2 lost to follow‐up and 1 adverse effect). A few participants (1 in LEV, 2 in PCB) had primary generalised‐onset seizures in addition to partial‐onset seizures. 1 participant (1.0%) in the LEV group and 2 (1.9%) in the PCB group temporarily discontinued the study drug, while 8 (7.8%) in the LEV and 2 (1.9%) in the PCB groups reduced the dosage because of adverse events. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method not given. |
| Allocation concealment (selection bias) | Unclear risk | Method not given. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Matched placebo" was used. No further specification. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Method not given. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 4 participants excluded from 50% responder analysis: 1 in LEV 3000 mg (lost to follow‐up) and 3 in PCB (2 lost to follow‐up and 1 adverse effect). The reasons for missing outcome data may possibly be related to true outcome (e.g. lost to follow‐up due to improvement), but these reasons are largely equally distributed between the intervention groups. |
| Selective reporting (reporting bias) | Low risk | All outcomes measured (that were also relevant to this review) were published. |
Xiao 2009.
| Study characteristics | ||
| Methods | Randomised, double‐blind, PCB‐controlled trial Single centre in China 2 treatment arms: 1 LEV and 1 PCB Randomisation concealment: numbered containers containing either LEV or PCB. Random list generation: randomisation codes were generated by the study sponsor. Each participant who qualified to receive double‐blind treatment was assigned a randomisation number and given LEV or PCB accordingly. Blinding: identical tablets and packages. Investigators were described as blinded to treatment assignment. Baseline: 8 weeks. Treatment period: 16 weeks (4 weeks' titration, 12 weeks' maintenance) |
|
| Participants | All adults Total randomised 56 adults; all with drug‐resistant focal epilepsy 28 adults to LEV 3000 mg; 28 adults to PCB 42.9% men in LEV; 42.9% men in PCB Age range: 16–70 years Other AEDs: 1 or 2 ≥ 4 focal seizures per month over preceding 2 months ≥ 10 weeks' background AED treatment Mean duration of epilepsy (years): LEV: 14.1 (SD 9.4) (range 2–40); PCB: 16.1 (SD 12.5) (range 2–48) Mean baseline seizure frequency per week: LEV 4.9 (range 1–23.6); PCB 5.6 (range 1–50) |
|
| Interventions | LEV 3000 mg/day PCB Uptitration dosages: received LEV 1000 mg/day (administered twice daily) and increased to 2000 mg/day after 2 weeks, and to 3000 mg/day after another 2 weeks |
|
| Outcomes | ≥ 50% reduction in seizure frequency Treatment withdrawal Adverse effects |
|
| Notes | 2 LEV‐treated participants decreased dose to 2000 mg (owing to adverse effects). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation codes generated by study sponsor. |
| Allocation concealment (selection bias) | Low risk | Numbered containers containing either LEV or PCB. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Identical tablets and packages. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Investigators described as blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants randomised were analysed in the group they were assigned. |
| Selective reporting (reporting bias) | Low risk | All outcomes measured (that were also relevant to this review) were published. |
Yagi 2010.
| Study characteristics | ||
| Methods | Randomised, double‐blind, PCB‐controlled, parallel trial Multicentre in Japan 3 treatment arms: 2 LEV and 1 PCB Randomisation concealment: method not stated. Random list generation: method not stated. Blinding: method not stated. Baseline: 12 weeks. Treatment period: 16 weeks (4 weeks' titration, 12 weeks' maintenance) |
|
| Participants | All adults Total randomised 216 adults 73 adults to LEV 1000 mg/day; 71 adults to LEV 3000 mg/day; 72 adults to PCB 51.2% men Age range: 16–65 years Other AEDs: 1–3 Mean duration of epilepsy (years): LEV 1000 mg/day: 23.05 (SD 10.80); LEV 3000 mg/day: 20.79 (SD 10.80); PCB: 22.54 (SD 11.57) Median baseline seizure frequency per week: LEV 1000 mg/day: 3.66 (IQR 1.75–6.71); LEV 3000 mg/day 3.21 (IQR 1.93–6.54); PCB: 2.61 (IQR 1.93–5.38) |
|
| Interventions | LEV 1000 mg/day LEV 3000 mg/day PCB |
|
| Outcomes | ≥ 50% reduction in seizure frequency Treatment withdrawal Adverse effects |
|
| Notes | The numbers randomised to ITT were 73 LEV 1000 mg/day; 71 LEV 3000 mg; and 72 PCB, but in the analysis the denominators were 64 LEV 1000 mg/day; 63 LEV 3000 mg/day; and 65 PCB. It was reported that 2 participants were not analysed due to misallocation from PCB and 1 from LEV 1000 mg/day due to no study medication taken. The reasons for exclusion from analysis and outcomes of the remaining missing participants were not clear. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method not stated. |
| Allocation concealment (selection bias) | Unclear risk | Method not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Method not stated. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Method not stated. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Denominators changing in the analysis section with large numbers of missing participant data between those randomised and those actually analysed. |
| Selective reporting (reporting bias) | Unclear risk | Method not stated. |
Zheng 2009.
| Study characteristics | ||
| Methods | Randomised, double‐blind, PCB‐controlled trial Single centre in China 2 treatment arms: 1 LEV and 1 PCB Randomisation concealment: method not stated Random list generation: method not stated Blinding: method not stated Baseline: 8 weeks. Treatment period: 16 weeks (4 weeks' titration, 12 weeks' maintenance) |
|
| Participants | All adults Total randomised 30 adults, 3 dropouts, 27 completed the trial; all with drug‐resistant focal epilepsy 18 adults to LEV; 9 adults to PCB 61.1% men in LEV; could not determine % men in PCB Age range: could not determine Other AEDs: 1–4 ≥ 8 focal seizures during 8‐week baseline ≥ 12 weeks' background AED treatment Mean duration of epilepsy: not stated Mean baseline seizure frequency per year: unknown |
|
| Interventions | LEV 3000 mg/day PCB |
|
| Outcomes | ≥ 50% reduction in seizure frequency Treatment withdrawal Adverse effects |
|
| Notes | Translated data 3 participants quit the trial before measurement (1 for adverse effects and 2 because they thought treatment was invalid). It is unclear which treatment groups the participants withdrew from. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method not stated. |
| Allocation concealment (selection bias) | Unclear risk | Method not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Method not stated. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Method not stated. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 3 participants quit the trial before measurement (1 for adverse effects and 2 because they thought treatment was invalid). It was unclear to which groups they were randomised. These reasons for missing outcome data were likely related to true outcome, and in a small study such as this, such incomplete outcome data would be likely to introduce bias. |
| Selective reporting (reporting bias) | Low risk | All outcomes measured (that were also relevant to this review) were published. |
Zhou 2008.
| Study characteristics | ||
| Methods | Randomised, double‐blind, PCB‐controlled trial Single centre in China 2 treatment arms: 1 LEV and 1 PCB Randomisation concealment: participants received an exclusive random number consecutively on entry into the study, and received treatment on the basis of this random number. Random list generation: random numbers table. Blinding: described as double‐blind with no further specification. Medications were supplied and packaged by UCB S.A. Pharma. Baseline: 8 weeks. Treatment period: 16 weeks (4 weeks' titration, 12 weeks' maintenance) |
|
| Participants | All adults. Total randomised 28 adults 14 adults to LEV 3000 mg; 14 adults to PCB 54% men in LEV; 55% men in PCB Age range: 16–70 years Other AEDs: 1–2 ≥ 8 seizures during 8‐week baseline with 2 per 4 weeks Mean duration of epilepsy (years): LEV: 8.7 (SD 6.4); PCB 16.5 SD 7.2) Mean baseline seizure frequency per week: LEV: 6.55 (SD 10.79); PCB: 6.15 (SD 11.20) |
|
| Interventions | LEV 3000 mg/day PCB add‐on Uptitration dosages: 500 mg twice daily in the first 2 weeks, 1000 mg twice daily in the third and fourth weeks) |
|
| Outcomes | ≥ 50% reduction in seizure frequency Treatment withdrawal Cognitive function QoL |
|
| Notes | Cognitive function assessment using a battery of neuropsychological tests: Wisconsin Card Sorting Test, Verbal Fluency, Trail Making Test, Digit Symbol, Stroop Color–Word Interference Task, Logic Memory, Delayed Logic Memory, Visual Memory, Delayed Visual Memory, Calculation. QoL assessment using QOLIE‐31 Dropouts (1 in LEV, 3 in PCB) excluded from the study author's analysis. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random numbers table used. |
| Allocation concealment (selection bias) | Low risk | Participants received an exclusive random number consecutively on entry into the study, and received treatment on the basis of that random number. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Described as double‐blind with no further specification. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not mentioned. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 4 participants (1 from LEV group and 3 from PCB group) withdrew from the trial due to non‐compliance. They were not analysed and their outcomes were not stated. The reasons for missing outcome data here were likely to be related to true outcome, with imbalance in numbers of missing data across intervention groups. |
| Selective reporting (reporting bias) | Low risk | All outcomes measured (that were also relevant to this review) were published. |
AED: antiepileptic drug; CBCL: Achenbach Child Behavior Checklist; CHQ‐PF50: Child Health Questionnaire‐Parent Form 50; EEG: electroencephalography; ESI‐55: Epilepsy Surgery Inventory scale; IQR: interquartile range; ITT: intention to treat; Leiter‐R AM: Leiter International Performance Scale‐Revised Attention and Memory; Leiter‐R ERS: Leiter International Performance Scale‐Revised, Examiner’s Rating Scale; LEV: levetiracetam; PCB: placebo; QoL: quality of life; QOLIE: Quality Of Life In Epilepsy inventory; SD: standard deviation; WRAML‐2: Wide Range Assessment of Memory and Learning‐2; XR: extended release.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Boon 2002 | Cross‐over trial where separate data pertaining to the first treatment period only were not available. Therefore, could not analyse the first treatment period as if it were a parallel trial (see Unit of analysis issues). |
Characteristics of studies awaiting classification [ordered by study ID]
NCT01392768.
| Methods | Double‐blind, phase III, multicentre, randomised, PCB‐controlled, parallel trial 2 treatment arms: 1 LEV; 1 PCB Randomisation concealment: described as concealed, method not given. Random list generation: described as randomised, method not given. Blinding: participant, care provider, investigator, outcomes assessor stated as blinded. Method not given. Baseline: not stated. Treatment period: 24 weeks (titration and maintenance period not stated) |
| Participants | 126 participants Numbers randomised to each arm: unknown Sex: unknown Eligible age range: 4–65 years Other AEDs: 1–3 ≥ 4 focal seizures per month over preceding 2 months ≥ 2‐year history of seizures Mean duration of epilepsy: unknown Mean baseline seizure frequency per week: unknown |
| Interventions | LEV PCB |
| Outcomes | ≥ 50% reduction in seizure frequency Treatment withdrawal Adverse effects |
| Notes | Results not yet posted. |
AED: antiepileptic drug; LEV: levetiracetam; PCB: placebo.
Differences between protocol and review
The original version of the review was published in 2001 (Chaisewikul 2001), and the review was last updated in 2012 (Mbizvo 2012). Therefore changes have been made to the format and content of the methods and the review from the protocol and from the last update to this version of the review, in line with current MECIR standards (MECIR 2012) and the Cochrane Style Manual (community.cochrane.org/style-manual).
Previous versions of the review considered the inverse of the primary outcome, that is, the proportion of people who did not achieve 50% or greater reduction in focal seizure frequency, termed 'non‐responders.' This analysis was not deemed to be reflective of clinical practice for the 2019 update so the analysis of 'non‐responders' was not performed in the current review. The dose–response analysis was also updated and simplified for this update. Additionally, this update investigated for the presence of publication bias using funnel plots.
The term 'partial' has been replaced by 'focal', in accordance with the most recent classification of epilepsies of the International League Against Epilepsy (Scheffer 2017).
Contributions of authors
GKM, BC, SJN, and PD were involved in all stages of conducting and writing of this review, assessing trials for inclusion, extracting data, assessing trials for bias, and evaluating the overall certainty of evidence. These steps were each conducted independently before collaboration with any disagreements resolved by discussion with AGM. JH oversaw data analysis.
Sources of support
Internal sources
No sources of support supplied
External sources
-
National Institute of Health Research (NIHR), UK
This review update was funded by the National Institute for Health Research (NIHR) (Clinically effective treatments for central nervous system disorders in the NHS, with a focus on epilepsy and Movement Disorders (SRPG project 16/114/26)). The views expressed are those of the review authors and not necessarily those of the NIHR or the Department of Health and Social Care.
Declarations of interest
GKM was a speaker at two UCB‐sponsored meetings in 2019 as a PhD student and received honoraria to attend these. The work presented was for educational purposes, was not sponsored by UCB, and remains unrelated to both the current review and to UCB. GKM declares no conflicts of interest.
BC: none known.
SJN: none known.
PD: a consortium of pharmaceutical companies (GSK, EISAI, UCB Pharma) funded the National Audit of Seizure Management in Hospitals (NASH) through grants paid to the University of Liverpool.
JLH was a non‐executive director of USS, so might be associated indirectly with companies which make or supply antiepileptic drugs. JLH provides medico‐legal reports on life expectancy which sometimes refer to epilepsy.
AGM: a consortium of pharmaceutical companies (GSK, EISAI, UCB Pharma) funded the National Audit of Seizure Management in Hospitals (NASH) through grants paid to the University of Liverpool. Professor Tony Marson is part funded by the Applied Research Collaboration North West Coast (ARC NWC).
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Ben‐Menachem 2000 {published and unpublished data}
- Ben-Menachem E, Falter U. Efficacy and tolerability of levetiracetam 3000 mg/d in patients with refractory partial seizures: a multicenter, double-blind, responder-selected study evaluating monotherapy. European Levetiracetam Study Group. Epilepsia 2000;41(10):1276-83. [DOI: 10.1111/j.1528-1157.2000.tb04605.x] [PMID: ] [DOI] [PubMed] [Google Scholar]
- UCB. Evaluation of the efficacy and tolerability of UCB L059 (1500 mg b.i.d., 500 mg tablets) monotherapy in epileptic patients with complex partial onset seizures, having experienced improved seizure control under add-on treatment: a 60-week (maximum), double-blind, multicenter, responder-selected trial (phase III study, placebo comparator). UCB S.A. Pharma Sector.
Betts 2000 {published and unpublished data}
- Betts T, Waegemans T, Crawford P. A multicentre, double-blind, randomized, parallel group study to evaluate the tolerability and efficacy of two oral doses of levetiracetam, 2000 mg daily and 4000 mg daily, without titration in patients with refractory epilepsy. Seizure 2000;9(2):80-7. [DOI: 10.1053/seiz.2000.0380] [PMID: ] [DOI] [PubMed] [Google Scholar]
- UCB. A 24-week multicentered, double-blind, parallel group, add-on study to compare the efficacy and the tolerability of 2 oral daily doses of UCB L059 (2000 mg and 4000 mg tablets) with placebo in patients with refractory epilepsy, followed by a 24-week open-label active treatment. UCB S.A. Pharma Sector.
Cereghino 2000 {published and unpublished data}
- Cereghino JJ, Biton V, Abou-Khalil B, Dreifuss F, Gauer LJ, Leppik I, and the United States Levetiracetam Study Group. Levetiracetam for partial seizures: results of a double-blind, randomized clinical trial. Neurology 2000;55(2):236-42. [DOI: 10.1212/wnl.55.2.236] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Cramer JA, Arrigo C, Van Hammee G, Gauer LJ, Cereghino JJ. Effect of levetiracetam on epilepsy-related quality of life: N132 study group. Epilepsia 2000;41(7):868-74. [DOI: 10.1111/j.1528-1157.2000.tb00255.x] [PMID: ] [DOI] [PubMed] [Google Scholar]
- UCB. Evaluation of the efficacy and tolerability of UCB L059 (500 and 1500 mg bid, tablets) add-on treatment in epileptic patients with partial onset seizures: a 38-week, double-blind, placebo-controlled, parallel-group multicenter trial. UCB S.A. Pharma Sector.
Glauser 2006 {published data only}
- Glauser TA, Ayala R, Elterman RD, Mitchell WG, Van Orman CB, Gauer LJ, et al. Double-blind placebo-controlled trial of adjunctive levetiracetam in pediatric partial seizures. Neurology 2006;66(11):1654-60. [DOI: 10.1212/01.wnl.0000217916.00225.3a] [PMID: ] [DOI] [PubMed] [Google Scholar]
Inoue 2015 {published data only}
- Inoue Y, Yagi K, Ikeda A, Sasagawa M, Ishida S, Suzuki A, et al, Japan Levetiracetam N01221 Study Group. Efficacy and tolerability of levetiracetam as adjunctive therapy in Japanese patients with uncontrolled partial-onset seizures. Psychiatry and Clinical Neurosciences 2015;69(10):640-8. [DOI: 10.1111/pcn.12300] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Yamada M, Yoshida K, Suzuki A. Adjunctive therapy of levetiracetam in adult Japanese patients with uncontrolled partial-onset seizures: pooled data from two double-blind, placebo-controlled, randomized trials. Therapeutic Research 2015;36(8):787-97. [Google Scholar]
Levisohn 2009 {published data only (unpublished sought but not used)}
- Levisohn PM, Mintz M, Hunter SJ, Yang H, Jones J. Neurocognitive effects of adjunctive levetiracetam in children with partial-onset seizures: a randomized, double-blind, placebo-controlled, noninferiority trial. Epilepsia 2009;50(11):2377-89. [DOI: 10.1111/j.1528-1167.2009.02197.x] [PMID: ] [DOI] [PubMed] [Google Scholar]
- la Loge C, Hunter SJ, Schiemann J, Yang H. Assessment of behavioral and emotional functioning using standardized instruments in children and adolescents with partial-onset seizures treated with adjunctive levetiracetam in a randomized, placebo-controlled trial. Epilepsy & Behavior 2010;18(3):291-8. [DOI: 10.1016/j.yebeh.2010.04.017] [PMID: ] [DOI] [PubMed] [Google Scholar]
Peltola 2009 {published data only}
- Peltola J, Coetzee C, Jimenez F, Litovchenko T, Ramaratnam S, Zaslavaskiy L, et al. Once-daily extended-release levetiracetam as adjunctive treatment of partial-onset seizures in patients with epilepsy: a double-blind, randomized, placebo-controlled trial. Epilepsia 2009;50(3):406-14. [DOI: 10.1111/j.1528-1167.2008.01817.x] [PMID: ] [DOI] [PubMed] [Google Scholar]
Shorvon 2000 {published and unpublished data}
- Shorvon SD, Lowenthal A, Janz D, Bielen E, Loiseau P, for the European Levetiracetam Study Group. Multicenter double-blind, randomized, placebo-controlled trial of levetiracetam as add-on therapy in patients with refractory partial seizures. Epilepsia 2000;41(9):1179-86. [DOI: 10.1111/j.1528-1157.2000.tb00323.x] [PMID: ] [DOI] [PubMed] [Google Scholar]
- UCB. Evaluation of the efficacy and tolerability of UCB L059 (500 and 1000 mg b.i.d., tablets) add-on treatment in refractory epileptic patients with partial onset seizures: a 32-week double-blind placebo-controlled crossover multicenter trial. UCB S.A. Pharma Sector.
Tsai 2006 {published data only}
- Tsai JJ, Yen DJ, Hsih MS, Chen SS, Hiersemenzel R, Edrich P, et al. Efficacy and safety of levetiracetam (up to 2000 mg/day) in Taiwanese patients with refractory partial seizures: a multicenter, randomized, double-blind, placebo-controlled study. Epilepsia 2006;47(1):72-81. [DOI: 10.1111/j.1528-1167.2006.00372.x] [PMID: ] [DOI] [PubMed] [Google Scholar]
Wu 2009 {published data only (unpublished sought but not used)}
- Wu XY, Hong Z, Wu X, Wu LW, Wang XF, Zhou D, et al. Multicenter double-blind, randomized, placebo-controlled trial of levetiracetam as add-on therapy in Chinese patients with refractory partial-onset seizures. Epilepsia 2009;50(3):398-405. [DOI: 10.1111/j.1528-1167.2008.01729.x] [PMID: ] [DOI] [PubMed] [Google Scholar]
Xiao 2009 {published data only}
- Xiao Z, Li JM, Wang XF, Xiao F, Xi ZQ, Lv Y, et al. Efficacy and safety of levetiracetam (3,000 mg/day) as an adjunctive therapy in Chinese patients with refractory partial seizures. European Neurology 2009;61(4):233-9. [DOI: 10.1159/000197109] [PMID: ] [DOI] [PubMed] [Google Scholar]
Yagi 2010 {published data only}
- Yagi K, Kameyama S, Kaneko S, Murasaki M, Yamauchi T. Multicenter, double-blind, randomized, placebo-controlled study of levetiracetam as add-on therapy in Japanese patients with uncontrolled partial seizures. Journal of the Japan Epilepsy Society 2010;28(1):3-16. [Google Scholar]
- Yamada M, Yoshida K, Suzuki A. Adjunctive therapy of levetiracetam in adult Japanese patients with uncontrolled partial-onset seizures: pooled data from two double-blind, placebo-controlled, randomized trials. Therapeutic Research 2015;36(8):787-97. [Google Scholar]
Zheng 2009 {published data only (unpublished sought but not used)}
- Zheng XZ, Wu SJ, Xia M, Li Q. Study on the therapeutic effect of levetiracetam as an additive therapy for refractory partial epilepsy and the relativity between levetiracetam and multidrug resistance gene: a randomised double-blind placebo-controlled trial. Chinese Journal of Contemporary Neurology and Neurosurgery 2009;9(2):173-7. [DOI: 10.3969/j.issn.1672-6731.2009.02.016] [DOI] [Google Scholar]
Zhou 2008 {published data only (unpublished sought but not used)}
- Zhou B, Zhang Q, Tian L, Xiao J, Stefan H, Zhou D. Effects of levetiracetam as an add-on therapy on cognitive function and quality of life in patients with refractory partial seizures. Epilepsy & Behavior 2008;12(2):305-10. [DOI: 10.1016/j.yebeh.2007.10.003] [PMID: ] [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Boon 2002 {published data only}
- Boon P, Chauvel P, Pohlmann-Eden B, Otoul C, Wroe S. Dose–response effect of levetiracetam 1000 and 2000mg/day in partial epilepsy. Epilepsy Research 2002;48(1):77-89. [DOI: 10.1016/S0920-1211(01)00323-0] [PMID: ] [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
NCT01392768 {unpublished data only}
- NCT01392768. Efficacy and safety of levetiracetam in partial seizures control, with or without secondary generalization (Mozart). clinicaltrials.gov/ct2/show/NCT01392768 (first received 13 July 2011).
Additional references
Asconapé 2001
- Asconapé JJ, Gerardot JM, Da Costa G. Behavioral changes associated with levetiracetam use in patients with epilepsy. Epilepsia 2001;42 (Suppl 7):299. Abstract no: J.08. [Google Scholar]
Berg 2010
- Berg AT, Berkovic SF, Brodie MJ, Buchhalter J, Cross JH, Emde Boas W, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005-2009. Epilepsia 2010;51(4):676-85. [DOI] [PubMed] [Google Scholar]
BNF 2019a
- Joint Formulary Committee. British National Formulary. London: BMJ Group and Pharmaceutical Press, 2019. [Google Scholar]
BNF 2019b
- Joint Formulary Committee. British National Formulary for Children. London: BMJ Group and Pharmaceutical Press, 2019. [Google Scholar]
Brodie 2010
Burneo 2002
- Burneo JG, Montori VM, Faught E. Magnitude of the placebo effect in randomized trials of antiepileptic agents. Epilepsy & Behavior 2002;3(6):532-4. [DOI] [PubMed] [Google Scholar]
Chen 2017
- Chen B, Choi H, Hirsch LJ, Katz A, Legge A, Buchsbaum R, et al. Psychiatric and behavioral side effects of antiepileptic drugs in adults with epilepsy. Epilepsy & Behavior 2017;76:24-31. [DOI] [PubMed] [Google Scholar]
Chen 2019
- Chen D, Bian H, Zhang L. A meta-analysis of levetiracetam for randomized placebo-controlled trials in patients with refractory epilepsy. Neuropsychiatric Disease and Treatment 2019;15:905-17. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Cochrane 1998
- Cochrane HC, Marson AG, Baker GA, Chadwick DW. Neuropsychological outcomes in randomized controlled trials of antiepileptic drugs: a systematic review of methodology and reporting standards. Epilepsia 1998;39(10):1088-97. [DOI] [PubMed] [Google Scholar]
Cock 2011
- Cock HR. Established Status Epilepticus Treatment Trial (ESETT). Epilepsia 2011;52(Suppl 8):50-2. [DOI] [PubMed] [Google Scholar]
Coppola 2017
- Coppola G, Iapadre G, Operto FF, Verrotti A. New developments in the management of partial-onset epilepsy: role of brivaracetam. Drug design, development and therapy 2017;11:643-57. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Crepeau 2010
- Crepeau AZ, Treiman DM. Levetiracetam: a comprehensive review. Expert Review of Neurotherapeutics 2010;10(2):159-71. [DOI] [PubMed] [Google Scholar]
Fisher 2015
- Fisher RS. Redefining epilepsy. Current Opinion in Neurology 2015;28(2):130-5. [DOI] [PubMed] [Google Scholar]
Fisher 2017a
- Fisher RS, Cross JH, French JA, Higurashi N, Hirsch E, Jansen FE, et al. Operational classification of seizure types by the International League Against Epilepsy: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017;58(4):522. [DOI] [PubMed] [Google Scholar]
Fisher 2017b
- Fisher RS, Cross JH, D'Souza C, French JA, Haut SR, Higurashi N, et al. Instruction manual for the ILAE 2017 operational classification of seizure types. Epilepsia 2017;58(4):531. [DOI] [PubMed] [Google Scholar]
Gambardella 2008
- Gambardella A, Labate A, Colosimo E, Ambrosio R, Quattrone A. Monotherapy for partial epilepsy: focus on levetiracetam. Neuropsychiatric Disease and Treatment 2008;4(1):33-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Gibbs 2006
- Gibbs JE, Walker MC, Cock HR. Levetiracetam: antiepileptic properties and protective effects on mitochondrial dysfunction in experimental status epilepticus. Epilepsia 2006;47(3):469-78. [PMID: ] [DOI] [PubMed] [Google Scholar]
Gillard 2006
Guekht 2010
- Guekht AB, Korczyn AD, Bondareva IB, Gusev EI. Placebo responses in randomized trials of antiepileptic drugs. Epilepsy & Behavior 2010;17(1):64-9. [DOI] [PubMed] [Google Scholar]
Halma 2014
- Halma E, Louw AJ, Klinkenberg S, Aldenkamp AP, IJff DM, Majoie M. Behavioral side-effects of levetiracetam in children with epilepsy: a systematic review. Seizure 2014;23(9):685-91. [DOI] [PubMed] [Google Scholar]
Hanaya 2016
- Hanaya R, Arita K. The new antiepileptic drugs: their neuropharmacology and clinical indications. Neurologia Medico-chirurgica 2016;56(5):205-20. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2005
- Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions 4.2.4 (updated March 2005). Chichester, UK: John Wiley & Sons, Ltd, 2005. [Google Scholar]
Higgins 2011
- Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.handbook.cochrane.org.
Kerr 2011
- Kerr C, Nixon A, Angalakuditi M. The impact of epilepsy on children and adult patients' lives: development of a conceptual model from qualitative literature. Seizure 2011;20(10):764-74. [DOI] [PubMed] [Google Scholar]
Kwan 2010
- Kwan P, Arzimanoglou A, Berg AT, Brodie MJ, Allen Hauser W, Mathern G, et al. Definition of drug resistant epilepsy: consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia 2010;51(6):1069-77. [DOI] [PubMed] [Google Scholar]
Lefebvre 2011
- Lefebvre C, Manheimer E, Glanville J. Chapter 6: Searching for studies. In: Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.handbook.cochrane.org.
Lukyanetz 2002
Lynch 2004
- Lynch BA, Lambeng N, Nocka K, Kensel-Hammes P, Bajjalieh SM, Matagne A, et al. The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proceedings of the National Academy of Sciences of the United States of America 2004;101(26):9861-6. [DOI] [PMC free article] [PubMed]
Lyseng‐Williamson 2011
- Lyseng-Williamson KA. Levetiracetam: a review of its use in epilepsy. Drugs 2011;71(4):489-514. [DOI] [PubMed] [Google Scholar]
Maguire 2011
- Maguire M, Marson AG, Ramaratnam S. Epilepsy (partial). BMJ Clinical Evidence 2011;2011:1214. [PMID: ] [PMC free article] [PubMed]
Manni 2000
- Manni R, Tartara A. Evaluation of sleepiness in epilepsy. Clinical Neurophysiology 2000;111(Suppl 2):S111-14. [PMID: ] [DOI] [PubMed] [Google Scholar]
MECIR 2012
- Higgins JP, Lasserson T, Chandler J, Tovey D, Thomas J, Flemyng E, et al. Methodological Expectations of Cochrane Intervention Reviews. London (UK): Cochrane, 2012. [Google Scholar]
NICE 2012
- National Institute for Health and Clinical Excellence. The epilepsies: the diagnosis and management of the epilepsies in adults and children in primary and secondary care, January 2012. www.guidance.nice.org.uk/CG137 (accessed 3 August 2012).
Niespodziany 2001
Pellock 2001
Penovich 2004
- Penovich PE. Much ado about something or nothing: behavioral problems with levetiracetam use in epilepsy patients. Epilepsy Currents / American Epilepsy Society 2004;4(4):145-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Rigo 2002
- Rigo JM, Hans G, Nguyen L, Rocher V, Belachew S, Malgrange B, et al. The anti-epileptic drug levetiracetam reverses the inhibition by negative allosteric modulators of neuronal GABA- and glycine-gated currents. British Journal of Pharmacology 2002;136(5):659-72. [DOI] [PMC free article] [PubMed]
SANAD‐II
- SANAD-II. A pragmatic randomised controlled trial comparing the effectiveness and cost effectiveness of levetiracetam and zonisamide versus standard treatments for epilepsy: a comparison of Standard And New Antiepileptic Drugs (SANAD-II). www.sanad2.org.uk/ (accessed 13 August 2015). [DOI] [PMC free article] [PubMed]
Scheffer 2017
- Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, et al. ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017;58(4):512. [DOI] [PMC free article] [PubMed] [Google Scholar]
Schünemann 2009
- Schünemann H, Brożek J, Oxman A. GRADE handbook for grading quality of evidence and strength of recommendation. Version 3.2 (updated March 2009). The GRADE Working Group, 2009. Available from www.cc-ims.net/gradepro (accessed 3 August 2012).
Shorvon 1996
- Shorvon SD. The epidemiology and treatment of chronic and refractory epilepsy. Epilepsia 1996;37(Suppl 2):S1-3. [DOI] [PubMed] [Google Scholar]
Snoeck 2007
- Snoeck E, Stockis A. Dose–response population analysis of levetiracetam add-on treatment in refractory epileptic patients with partial onset seizures. Epilepsy Research 2007;73(3):284-91. [DOI] [PubMed] [Google Scholar]
Stata [Computer program]
- Stata. Version 14. College Station, TX, USA: StataCorp, 2015. Available at www.stata.com.
Tomson 2015
- Tomson T, Marson A, Boon P, Canevini MP, Covanis A, Gaily E, et al. Valproate in the treatment of epilepsy in girls and women of childbearing potential. Epilepsia 2015;56(7):1006-19. [DOI] [PubMed] [Google Scholar]
Trinka 2011
- Trinka E. What is the evidence to use new intravenous AEDs in status epilepticus? Epilepsia 2011;52(Suppl 8):35-8. [DOI] [PubMed] [Google Scholar]
Turner 2000
- Turner RM, Omar RZ, Yang M, Goldstein H, Thompson SG. A multilevel model framework for meta-analysis of clinical trials with binary outcomes. Statistics in Medicine 2000;19(24):3417-32. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Chaisewikul 2001
- Chaisewikul R, Privitera MD, Hutton JL, Marson AG. Levetiracetam add-on for drug-resistant localization related (partial) epilepsy. Cochrane Database of Systematic Reviews 2001, Issue 1. Art. No: CD001901. [DOI: 10.1002/14651858.CD001901] [DOI] [PubMed] [Google Scholar]
Mbizvo 2012
- Mbizvo GK, Dixon P, Hutton J, Marson A. Levetiracetam add-on for drug-resistant focal epilepsy: an updated Cochrane Review. Cochrane Database of Systematic Reviews 2012, Issue 10. Art. No: CD001901. [DOI: 10.1002/14651858.CD001901.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]