

Abstract
RGD is a prolific example of a tripeptide used in biomaterials for cell adhesion, but the potency of free or surface-bound RGD tripeptide is orders-of-magnitude less than the RGD domain within natural proteins. We designed a set of peptides with varying lengths, composed of fragments of fibronectin protein whose central three residues are RGD, in order to vary their conformational behavior without changing the binding site’s chemical environment. With these peptides, we measure the conformational dynamics and transient structure of the active site. Our studies reveal how flanking residues affect conformational behavior and integrin binding. We find that disorder of the binding site is important to the potency of RGD peptides and that transient hydrogen bonding near the RGD site affects both the energy landscape roughness of the peptides and peptide binding. This phenomenon is independent of longer-range folding interactions and helps explain why short binding sequences, including RGD itself, do not fully replicate the integrin-targeting properties of extracellular matrix proteins. Our studies reinforce that peptide binding is a holistic event and fragments larger than those directly involved in binding should be considered in the design of peptide epitopes for functional biomaterials.
Graphical Abstract

INTRODUCTION
Proteins offer a rich and bountiful array of functions, including binding, signaling, catalysis, and transport. An important target is to create smart biomaterials that are capable of performing diverse functions analogous to proteins themselves.1 One promising route toward this goal is to identify the amino acid sequence in a protein that is directly involved in a biochemical event and covalently tether this short sequence to surfaces.2–5 This approach has shown initial success; however, little is known about the properties and behavior of short peptide sequences when removed from their natural protein environment.
Few peptide motifs garner more attention than those containing the RGD (arginine–glycine–aspartic acid) tripeptide.1–6 This sequence is found in extracellular matrix proteins and is known to bind to integrins: transmembrane proteins important for cell survival and adhesion. RGD is often tethered to hydrogel surfaces to promote cell adhesion, boost biocompatibility, or prevent apoptosis. RGD is also important to cancer therapeutics, where it facilitates drug delivery and inhibits angiogenesis.5 Similar to other peptide sequences taken out of the context of natural proteins, RGD is orders-of-magnitude less effective than its parent proteins, but its simplicity and ease of synthesis have, anyway, led to its use in a vast number of technologies.3,6
Recent improvements in peptide synthesis techniques have enabled high-throughput peptide couplings with dramatic improvements in reaction times and yields and have led to a dramatic improvement in peptide diversity.7,8 As a result, the prospect of producing long RGD-containing peptides that more closely resemble the active domain of their parent protein is now a viable route to scalable integrin-targeting materials with enhanced potencies.
Many RGD peptides work by imposing a particular conformation on their binding site, as demonstrated by molecular dynamics (MD) and nuclear magnetic resonance (NMR) studies of conformation in RGD–integrin complexes.9–11 Very short, integrin-bound RGD peptides have been studied by X-ray crystallography, and their conformations are shown to vary depending on the specific nature of the integrin in question.6 Only a few studies report structural information for complexes between integrins and longer fibronectin-mimetic RGD-containing sequences.12 In solution, MD simulations demonstrate that the RGD site of fibronectin is flexible, changing conformation regularly, and is therefore considered to be intrinsically disordered.13,14 This assessment is consistent with our predictions of disorder in the fibronectin sequence using the PrDOS disorder prediction tool (Figure S6).15,16 The disorder of RGD is difficult to confirm directly; however, disorder has been shown to play an important role in integrin-binding proteins17–20 and other protein-binding peptide systems.21,22
Conformational disorder of the RGD site suggests that the conventional lock-and-key model, in which function is purely dictated by structure, does not adequately describe RGD binding interactions. More recently, two other models, the induced fit and the conformational selection models, have been used to explain the role of conformational distributions in protein binding.23–26 In the induced fit model, the binding conformation of one binding site is induced by the presence of the other, facilitating an interaction.23 In the conformational selection model, some subset of the unbound protein already exists in its binding conformation, and this subset is preferentially selected for binding.24 Both cases appear in nature, and in both cases, the rate of conformational change and the range of conformations adopted by a protein binding site are central to the binding interactions of the protein.25,27–29 Therefore, a detailed understanding of the conformational dynamics of RGD-containing fibronectin fragments is important for designing effective integrin-targeting materials.
Electron paramagnetic resonance (EPR) spectroscopy is used to study dynamic behavior in peptides and proteins. The technique is particularly useful for the study of intrinsically disordered proteins and has been used to analyze conformational behavior in intrinsically disordered human proteins such as the α-synuclein protein implicated in Parkinson’s disease,30 as well as in intrinsically disordered viral31 and algal proteins.32 EPR detects unpaired electron spins, for example, those of radicals or metal ions not normally found in most molecules. In proteins and peptides, spin labels are typically nitroxide radical moieties incorporated by site-directed spin labeling (SDSL) or through the direct synthesis of molecules containing an amino acid with a nitroxide spin label R-group, TOAC (2,2,6,6-tetramethylpiperidine-N-oxide-4-amino-4-carboxylic acid).33,34 Where possible, synthetic introduction of TOAC is preferable to SDSL because, unlike most spin labels, TOAC is directly and rigidly integrated into the backbone of the protein or peptide.33
Continuous-wave EPR (CW-EPR) enables the measurement of the rotational diffusion coefficient (DR) of the spin label according to the stochastic Liouville model.35 This parameter provides information about the local dynamic behavior of the molecule at the site of the probe and has been widely used to study binding and folding behavior in peptides and other molecules.36–45 Both molecular tumbling and conformational changes contribute to DR by causing rotational motion of the TOAC. However, the dominant contribution arises from configurational rearrangements of the peptide, because these occur more frequently. A basic estimate from the Stokes–Einstein relation suggests that molecular tumbling for a 2 kDa molecule (typical for this study) will tumble with a correlation time of ~1.2 ns at 20 °C, whereas the observed rotational correlation times are typically 0.3 ns, indicating that conformational changes are the dominant contributors to rotational diffusion.46 EPR is sensitive to dynamics of analytes at micromolar concentrations, enabling analysis of samples that are prone to aggregation. Since dynamics measurements describe the rate of conformational change, EPR spectra are consequently related to conformational energy landscapes.47–50 In particular, the activation energy of diffusion represents the characteristic energy barrier to conformational change.49
Complementary to CW-EPR is double electron–electron resonance (DEER) spectroscopy, a pulsed EPR technique. DEER requires spin labeling at two chosen sites of a molecule and provides distance distributions between the two spin labels. These distance distributions, in combination with conformational dynamics of single spin label sites, offer a more thorough understanding of the average conformational behavior of a molecule.
MATERIALS AND METHODS
Materials.
Amino acid couplings were performed according to literature using 50 mg of ChemMatrix H-Rink amide resin (0.49 mequiv/g).7 For the TOAC coupling, 3 equiv (0.075 mmol) of 2,2,6,6-tetramethylpiperidine-N-oxyl-4-(9-fluorenylmethyloxycarbonyl-amino)-4-carboxylic acid (Fmoc-TOAC) was coupled overnight (RT) with 2.62 equiv HATU in 2.53 mL of DMF 8% (v/v) DIPEA. Fmoc was deprotected for 20 min in DMF 20% piperidine (RT). The subsequent residue was coupled in flow by a 10 min, 10 mL/min coupling (70 °C) on the peptide synthesizer, and all remaining residues were coupled normally. Peptides were cleaved using a protocol described elsewhere, and precipitated by centrifugation in diethyl ether (−70 °C).51 The identity of each peptide was verified by high-res liquid chromatography/mass spectrometry (LC-MS) analysis on an Agilent 6520 ESI-Q-TOF LC-MS system. Each peptide was then purified by high performance liquid chromatography (HPLC), and the isolated product was tested by LC-MS and lyophilized. LC-MS was performed on a Zorbax 300SB C3 column (2.1 × 150 mm, 5 μm, 0.8 mL/min) using solvents A (water 0.1% (v/v) formic acid) and B (acetonitrile 0.1% (v/v) formic acid) by the following steps: 0–2 min, a 95% A, 5% B wash; 2–11 min, a 5–65% B linear ramp; and 11–12 min, a 65% B wash. Preparative HPLC was performed on an Agilent Zorbax C3 column (21.2 × 250 mm, 7 μm, 5 mL/min) using solvents C (water 0.1% (v/v) trifluoroacetic acid) and D (acetonitrile 0.1% (v/v) trifluoroacetic acid) by the following steps: 0–5 min, a 95% C, 5% D wash; 5–80 min, a 5–45% C linear ramp; 80–85 min, a 45% C wash.
Double Electron–Electron Resonance (DEER).
Solutions of peptides were prepared (water, 30% (v/v) glycerol, 9 mM tris buffer (pH 7.4), 0.07 M NaCl, 1.5% (v/v) H2O2, 0.1 mM sodium tungstate) at 130 μM final concentration 30 min prior to conducting DEER experiments. Hydrogen peroxide and sodium tungstate were included to reoxidize the spin labels from hydroxylamines to nitroxides, and glycerol kept the samples vitreous upon freezing. Samples were centrifuged (3 min at 27,000 × g) to separate any aggregated peptides, and the supernatant was studied. Deuterated glycerol and water were also used for the longest peptide to verify measurement accuracy.52 Samples were loaded into quartz capillaries and frozen in liquid nitrogen.
All DEER measurements were conducted at 17.3 GHz and 60 K using a home-built Ku-band pulse EPR spectrometer.52 Four-pulse DEER experiments utilized for echo detection a π/2−t1−π−t2−π pulse sequence with π/2- and π-pulses having widths of 16 and 32 ns, which was applied at the low-field side of the nitroxide spectrum.53 A 16 ns π-pulse pumped at a 70 MHz lower frequency corresponding to the central maximum of the EPR spectrum. In all DEER measurements, four data records were obtained, advancing each time initial interpulse distances by a quarter period of proton electron spin echo envelope modulation (ESEEM) frequency (26.2 MHz) and summed up to suppress proton modulation. The data collection time was ~50 min per sample. The recorded data were subjected to background removal. The latter points (about half of the record) in the logarithm of DEER data were fit to linear background which was then subtracted out. The resulting linear-scale DEER data for distance reconstruction, V(t), were modified as V′(t) = (V(t) − 1)/V(0) to give the amplitude at zero equal to DEER signal modulation depth and the asymptotic value of zero.54 Based on DEER modulation depth, spin label reoxidation efficiency was 0.8 ± 0.1. All data were processed into distance distributions using either L-curve Tikhonov regularization,55 followed by the maximum entropy regularization method (MEM) refinement56 or denoising/singular value decomposition (SVD) methods57,58 with the outcomes being close enough, so just the Tiknonov distributions are presented in Figure 5.
Figure 5.

Displacement of fluorescently labeled fibronectin, bound to cellular αVβ3, by FMP peptides. Normalized mean fluorescence intensity (MFI) of integrin-bound Alexa647-labeled hFN10 protein is presented as a function of FMP peptide concentration. The longer peptides, relative to FMP9, exhibit a ~5× improvement in their ability to bind to activated integrins. In the longest peptide (FMP27), this improvement is eliminated due to misfolding. The half-maximal inhibitory concentrations (IC50s) observed are FMP9, 1030 ± 208 nM; FMP15, 172 ± 30 nM; FMP21, 218 ± 52 nM; and FMP27, 2740 ± 1070 nM.
Electron Paramagnetic Resonance (EPR).
Measurements were performed using a Bruker EMXplus X-band continuous wave (CW) EPR (9.43 GHz), with a variable temperature unit. Stock peptide solutions were prepared at a concentration of 0.75 mg/mL and were centrifuged through 10,000 MWCO Thermo-Fisher protein concentrator tubes (30 s, 27,000 × g) to remove contaminants and larger aggregates. Peptides were then diluted to a concentration of 150 μM in a aqueous solutions containing either 0% (v/v) or 25% (v/v) DMSO, as well as 1× phosphate buffered saline (PBS, pH 7.4), 1.5% (v/v) hydrogen peroxide (aq) and 0.1 mM sodium tungstate. A total of 5 μL of the supernatant was loaded into a PTFE capillary tube, and sealed using Crytoseal resin. We prepared 2×, 4×, 8×, 16×, and 20× dilutions of these samples in the same media in order to ensure that no concentration effects (such as peptide aggregation) changed dynamics.
EPR spectra for each sample were collected in the range of 275–325 K in increments of 5 K. Spectra were collected over a 150 G range centered at B = 3315 G, with a MW attenuation of 15 dB and a modulation amplitude of 1.5 G. EPR spectra of a background sample containing only water, PBS, hydrogen peroxide and sodium tungstate were subtracted from each peptide spectrum. Finally, samples were frozen to 150 K and frozen spectra were collected. Subsequent LC-MS analysis of the scanned samples confirmed the integrity of each peptide. Using the pepper function in Easyspin, hyperfine (A) and electron g values were collected from spectra of aqueous samples cryogenically frozen to 150 K.59 Virtually no variation was observed as a function of peptide length, so the fitted values of gxx = 2.0081, gyy = 2.0051, gzz = 2.0020, A⊥ = 5.13 G, and A∥ = 37.6 G were assigned to each peptide. Then, NLSL was used to analyze room-temperature data, employing the MOMD model and using an in-house MatLab software package to analyze error.60,61 For each spectrum, 1000 fits were started from a randomized initial guess within a constraint rectangle. In every case, a randomized Gaussian noise (based on background noise from the EPR spectrum) was added to each intensity value in the spectrum to account for experimental error. Error bars were calculated based on the final coordinates of all “good fits”, defined as those with a final χ2 parameter within 50% of the global minimum of χ2. Arrhenius plots were fitted using the linearized diffusion equation
where Q represents the activation energy, D0 is a system-specific constant, R is the universal gas constant, e is Euler’s number, and T is temperature.
Molecular Dynamics Simulations.
MD simulations were performed using Gromacs v5.1.4. Initial peptide inputs were predicted using PEP-FOLD 3, an in silico conformational prediction tool.62,63 The output of these calculations was used as an input for 200 ns simulations of each peptide, using the CHARMM36 force field and explicit water under constant NPT conditions. The temperature was maintained at 310 K. Simulations were performed in dodecahedral unit cells with periodic boundary conditions, and charges were neutralized using sodium/chloride ions, as necessary. An additional 0.154 M sodium chloride concentration was added. The dodecahedral cell was designed to allow a 1.2 nm minimum space between the peptide and the boundary. Simulations were conducted run on a pair of HP Z240 Tower Workstations over the course of 14 days. Trajectories were analyzed using either built-in Gromacs functions or using Visual Molecular Dynamics (VMD). For comparison with DEER results, we corrected for TOAC probes by assuming a rigid, TOAC structure and at each step adding the α-carbon-nitroxide distance vectors to the intercarbon distance vector.
Cell Lines and Cell Culture.
The human erythroleukemia cell line K562 stably expressing recombinant αVβ3 (K562-αVβ3) was described previously.12 Cells were maintained at 37 °C, 5% CO2 in Iscove’s modified Dulbecco’s medium supplemented with 1 mg/mL G418, 10% fetal bovine serum, penicillin, and streptomycin.
Fluorescence Labeling of hFN10.
hFN10 was labeled with N-hydroxysuccinimidyl ester derivative of Fluor 647 (Alexa Fluor 647) from Invitrogen according to the manufacturer instructions. Excess dye was removed using dialysis and buffer exchange into PBS. The final protein concentration and dye/protein molar ratio were measured spectrophotometrically.
Ligand Binding Assays and Flow Cytometry.
K562-αVβ3 cells were harvested and washed three times in HEPES-buffered saline (20 mM HEPES and 150 mM NaCl, pH 7.4) containing 0.1% (w/v) bovine serum albumin (binding buffer, BB). The 0.5 × 106 cells were suspended in 100 μL of BB containing 1 mM each Ca2+ plus Mg2+ or 1 mM Mn2+ and then washed with this solution. For competition studies, K562-αVβ3 were incubated first with serially diluted concentrations of the unlabeled RGD-peptides in Mn2+-containing BB and subsequently with 10 nM of the reporter Alexa647-conjugated hFN10 ligand for an additional 20 min at RT in the dark. Cells were washed with 4 mL of metal ion-containing BB to remove the unbound reporter, centrifuged for 5 min at 525 × g, resuspended, and fixed in 1% paraformaldehyde. They were finally analyzed using a BD-LSRII flow cytometer (BD Biosciences) and processed using FlowJo software. The binding of soluble hFN10 to K562-αVβ3 cells was expressed as mean fluorescence intensity units (MFI), and the mean and standard deviation from three independent experiments were calculated and compared using student’s t-test. The binding profile of each peptide was fit in MatLab using a Hill curve model, in order to determine IC50s and associated uncertainties. Fits are presented in Figure 5, alongside associated data.
RESULTS AND DISCUSSION
We aimed to understand the relationship between dynamic behavior and peptide structure, and to identify what, if any, connection exists between these properties and peptide binding. We used CW-EPR to analyze the conformational behavior of a series of four peptides, shown in Figure 1. These peptides mimic the sequence of human fibronectin (Fn) in the region surrounding an RGD site from the 1FNF fragment reported in the protein data bank.15 We designated these sequences as fibronectin-mimetic peptides (FMPs). Each peptide sequence corresponded to a fragment of the human fibronectin protein centered at residue 1494, the glycine of RGD. We chose four sequences for RGD conformational dynamics and binding analyses: a 9-residue sequence composed of RGD with three flanking residues on either side (FMP9, Figure 1a), a 15-residue sequence with six flanking residues on either side of RGD (FMP15, Figure 1b), a 21-residue sequence with nine residues flanking RGD (FMP21, Figure 1c), and a 27-residue sequence with 12 residues flanking RGD (FMP27, Figure 1d). Although RGD appears in many extracellular matrix proteins, we chose to mimic fibronectin because it is among the most widely studied and biologically relevant. In addition, the interaction between RGD and the αVβ3 integrin targeted by fibronectin is well characterized, and the structure of this integrin is well understood.12
Figure 1.

The structure of RGD-containing fibronectin fragments. Fibronectin fragments studied are highlighted in purple. RGD sites of each peptide are highlighted in yellow. (a) FMP9 includes the RGD site and three flanking residues on either side. (b) FMP15 includes the RGD site and six flanking residues on either side. (c) FMP21 includes the RGD site and nine flanking residues on either side. (d) FMP27 includes the RGD site and 12 flanking residues on either side.
We used CW-EPR spectroscopy and spectral analysis of each peptide to measure conformational dynamics of the FMPs as a function of temperature and peptide length. To accomplish this, we substituted an amino acid spin label (TOAC) for the glycine of the RGD sequence of each FMP peptide. We chose TOAC, rather than another spin label, because its rigid integration into the peptide backbone constrains it, so that only motions of the peptide backbone displace the nitroxide radical. Substituting TOAC into the sequence, like any amino acid substitution, typically affects conformational behavior. However, because TOAC is a relatively small, neutrally charged substituent, the perturbation associated with TOAC substitutions is expected to have a minimal impact on conformational dynamics.49 For simplicity, we refer to the TOAC residue as J. The amino acid sequences, designations, and expected and observed molecular weights of each compound were reported in Table 1. We measured CW-EPR of the TOAC-substituted FMPs at temperatures between −3 and 53 °C and fit each EPR spectrum to find the probe’s rotational diffusion coefficient (DR; see Supporting Information, Figure S7). Lower rotational diffusion rates correspond to slower conformational dynamics at the spin label site.
Table 1.
Fibronectin-Mimetic Peptide (FMP) Designations, Sequences, and Exact Massesa
| Name | Sequence | Exp. MW | Obs. MW |
|---|---|---|---|
| FMP9 | VTGRGDSPA | 857.41 | 857.43 |
| FMP9-J5 | VTGRJDSPA | 998.53 | 998.51 |
| FMP9-J0J5 | JVTGRJDSPA | 1196.66 | 1196.65 |
| FMP15 | VYAVTGRGDSPASSK | 1492.74 | 1492.77 |
| FMP15-J8 | VYAVTGRJDSPASSK | 1633.85 | 1633.83 |
| FMP15-J0J8 | JVYAVTGRJDSPASSK | 1831.99 | 1831.99 |
| FMP21 | TITVYAVTGRGDSPASSKPIS | 2105.09 | 2105.01 |
| FMP21-J11 | TITVYAVTGRJDSPASSKPIS | 2246.20 | 2246.15 |
| FMP21-J0J11 | JTITVYAVTGRJDSPASSKPIS | 2444.34 | 2444.36 |
| FMP27 | VDYTITVYAVTGRGDSPASSKPISINY | 2872.44 | 2872.43 |
| FMP27-J14 | VDYTITVYAVTGRJDSPASSKPISINY | 3013.55 | 3013.52 |
| FMP27-J0J14 | JVDYTITVYAVTGRJDSPASSKPISINY | 3211.69 | 3211.69 |
Each FMP sequence (named in bold) has two associated TOAC peptides, a singly labeled sequence for EPR dynamics measurement, and a twice-labeled sequence used for DEER experiments. The sequences are given with the RGD site in bold, and the position of TOAC spin label residues indicated by a red “J”. Expected molecular weights and observed molecular weights, determined by LC-MS, are presented for each sequence.
Figure 2a shows the Arrhenius plots of each FMP in PBS buffer. The rotational diffusion coefficients in the Arrhenius plots were derived from EPR fitting described in the Supporting Information (Figure S7). Figure 2b presents Arrhenius plots of the same FMPs, except in a solvent mixture of 3:1 PBS/DMSO, where DMSO is a denaturant. The overall slower diffusion of the spin-labeled FMPs in the presence of DMSO results from the greater viscosity of the DMSO/PBS solvent mixture compared to PBS alone. We calculated the activation energy of diffusion (Q) of each peptide as a function of length from the slopes of the Arrhenius plots (Figure 2c) and found Q = 11.3 (FMP9), 19.4 (FMP15), 20.8 (FMP21), and 22.4 kJ/mol (FMP27) in PBS. As expected, the activation energy of diffusion versus peptide length varies continuously when DMSO is present, since no secondary structure is possible. In contrast, a discontinuity is observed in PBS buffer, where the activation energies of diffusion of the shortest FMP (FMP9) is significantly less than that of FMP15, deviating from linear behavior. This discontinuity suggests that a structural change occurs when the number of flanking residues on each side of RGD increases from three to six.
Figure 2.

Dynamic behavior at the RGD site changes discontinuously with length. (a) Arrhenius plots of rotational diffusion of TOAC peptides in PBS buffer, generated from EPR spectral analysis. (b) Arrhenius plots of rotational diffusion determined by EPR in 25% DMSO/75% buffer. The DMSO denaturant prevents secondary structure formation. (c) Activation energy of rotational diffusion in buffer (black data points) and 25% DMSO as a denaturant (red data points).
Few previous EPR studies describe activation energy of diffusion of polymers, peptides, or proteins. One study indicates that polymers activation energies of diffusion exhibit no length-dependence, but in that case, the polymers are much larger than FMPs (33–500 kDa) and are not composed of amino acids. Another report describes the diffusive motion of shorter biopolymers in terms of internal friction effects, which also applies to short peptides such as FMPs, but these results are system-specific and do not indicate the physicality of a linear relationship.64 Therefore, the apparent existence of a linear relationship is unexpected and merits further investigation.
The activation energies of diffusion demonstrated in Figure 2c arise from the characteristic energy barriers associated with conformational changes that displace the nitroxide radical.49 These barriers are influenced partially by hydrogen bonding between the peptide and its hydration shell and partially by the roughness of the conformational free energy landscape.49 The roughness of free-energy landscapes is important for describing protein folding.65,66 The lower the conformational energy barrier, the more smoothly the molecule changes conformation and the lower the conformational dwell-time.49,65 Figure 2c indicates that the longer three peptides are substantially more frustrated than the shortest peptide and are, therefore, more likely to exhibit strong intramolecular interactions in multiple configurations.
To understand the structural origin of dynamic behavior of FMPs, we carried out 200 ns all-atom molecular dynamics (MD) simulations and mapped the probabilities of hydrogen bond formation between residue pairs of the free FMP peptides. Figure 3 shows intramolecular hydrogen bonding within each FMP by indicating the probability of each pair of residues participating in a hydrogen bond. These results show that while transient hydrogen bonding is possible, the RGD site remains disordered, exhibiting several stable conformations, even in the longest peptide. This conclusion is supported by secondary structure timelines (Figure S5) and by Ramachandran plots calculated for the five central residues (GRGDS) of each sequence (Figure S4). Further, the disorder of FMPs is consistent with the EPR results shown in Figure 2, which demonstrate a relatively rapid overall rate of diffusion in all four peptides.
Figure 3.
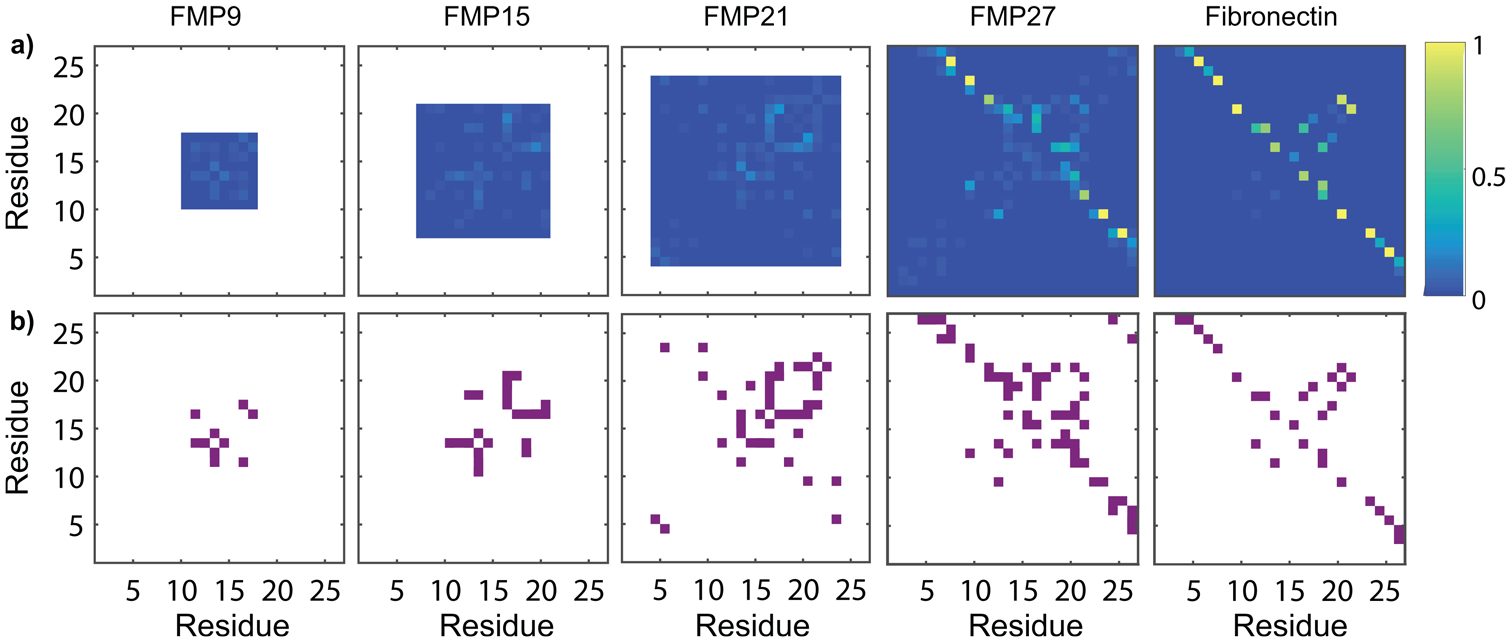
Molecular dynamics simulations illustrate intrachain hydrogen bond formation in FMP peptides and fibronectin protein. (a) Hydrogen bond probability maps of FMP peptides and the corresponding region of fibronectin, determined by MD simulations. Weak hydrogen bonding between residues near to the RGD site (appearing near the y = x line) is observed. Strong hydrogen bonding between flanking chains is observed in FMP27 and fibronectin, but the residues involved in these intramolecular hydrogen bonds differ between FMP27 and fibronectin. (b) Intramolecular hydrogen bonding occurring more than 4% of the time is depicted in purple. A transient hydrogen bonding pattern appears along the y = x line for FMP15, FMP21, and FMP27, which has not fully evolved in FMP9.
Figure 3 indicates that, near the RGD site, the longer three peptides exhibit transient hydrogen bonding that is less pronounced in the shortest peptide. These hydrogen bonds form intermittently between pairs of nearby residues, and do not constitute permanent secondary structure. A hydrogen bonding pattern begins to emerge as RGD is flanked by a greater number of residues. As seen in FMP15, FMP21, and FMP27, the hydrogen bonding probability profile remains similar despite the emergence of more permanent secondary structure in FMP27. This structure is absent in our simulation of a fibronectin protein fragment (labeled as Fibronectin in Figure 3) due to limitations in simulation time given the large number of atoms.
These simulations demonstrate a second change in FMP behavior as a function of length. FMP27 folded permanently due to the formation of strong hydrogen bonds between residues 9–26 and 11–24, whereas the shorter three peptides did not. Although this structure is similar to the hydrogen bonding interactions in the analogous region of fibronectin, there is a slight misalignment between the interactions of FMP27 and the analogous region in fibronectin (which has bonds between residue-pairs 6–26, 8–24, and 10–21). This mismatch preferentially distorts the RGD site into a nonideal conformation for binding. Formation of hydrogen bonds does not appear to affect the activation energy of diffusion of the longest peptide. We attribute this phenomenon to the high stability of these hydrogen bonds, since they are nearly permanent in FMP21. While these interactions modify the overall energy landscape, they appear to have a minimal impact on its roughness.
To support these simulations, double electron–electron resonance (DEER) was used to analyze the distance between the N-terminus and the RGD binding site.53 Table 1 shows the twice-labeled FMPs used for this study. With DEER, we measured interprobe distance distributions (between 1.1 and 10 nm) based on the interference between radical electrons within a molecule, and compared with distributions extracted from our MD simulations. Figure 4 shows the distance distributions between the RGD site. The distance distributions determined by DEER are consistent with MD simulations, showing that the shortest peptide is generally extended, with a uniform distance distribution. As the peptide length increases, the center of the distributions increases. This observation is consistent with the formation of hydrogen bonds in a largely disordered peptide. The general agreement between these MD and DEER distance distributions is high; the average interprobe distance differs by no more than 2 Å in any simulation. Minor deviations between MD and DEER measurements may have resulted from a change in sequence behavior due to the introduction of TOAC, as well as the possibility that MD simulations did not survey the entire conformational space of each peptide.
Figure 4.

Distributions determined by DEER (black) and MD simulations (red) describe the distances between the glycine of RGD and the N-terminus of each FMP peptide. FMP9 shows a narrow distribution centered at 1.9 nm, and FMP15, FMP 21, and FMP27 each show broader distance distributions centered at 2.3, 2.9, and 3.4 nm, respectively.
We measured binding of FMPs to activated αVβ3 integrins on cell surfaces via displacement of fluorophore-labeled hFN10 (a high affinity ligand for the αVβ3 integrin) bound to K562-αVβ3 cells.12 Cell-binding measurements for each peptide are shown in Figure 5, along with the numerical fits used to calculate the half-maximal inhibitory concentration (IC50) for each FMP. Using scrambled analogs of FMPs and RGA (arginine–glycine–alanine) mutations as controls, we demonstrated that specific, rather than nonspecific, binding occurs. All four peptides bind to the activated integrins; however, there are significant differences between the potency of each sequence. FMP15 and FMP21 exhibit the strongest binding, followed by FMP9 and finally FMP27. These results suggest that the dynamic transition associated with hydrogen bonding along the peptide backbone corresponds to increased energy landscape roughness and stronger peptide–integrin binding. We observe weaker binding between integrins and the short FMP that does not form intramolecular hydrogen bonds. We observed the weakest binding between integrins and the FMP that misfolds (FMP27), highlighting the importance of localized secondary structure in a largely disordered peptide.
CONCLUSIONS
Here we presented a strategy for improving the potencies of short peptide signals that were derived from protein active sites. We introduced four peptides that contain the RGD integrin-binding motif. Each peptide contained a different number of residues flanking RGD, with the residue sequences adapted from a fibronectin protein. We showed that binding efficacy of short fibronectin fragments, with RGD as the central three residues, increased with length between nine and 21 residues, but that misfolding occurred in the 27-residue sequence. We used conformational dynamics measurements and molecular dynamics simulations to understand this behavior in the context of free energy landscapes.
The geometric and chemical structure of proteins or peptides has historically been regarded as the key feature that dictates binding efficiencies. In the case of RGD-integrin binding, our results show that dynamic behavior also plays an important role. We performed dynamics experiments on a set of short fibronectin fragments and find that rough conformational energy landscapes are correlated with significant improvements in the potency of RGD peptides. From these results, we suggest energy landscape roughness as a new design parameter for generating more potent bioactive peptides.
We observed two notable features that affect binding of FMP peptides. The first feature is the emergence of transient hydrogen bonding structure. This transient hydrogen bonding increases the energy landscape roughness discontinuously with peptide length and corresponds to an increase in binding affinity of FMPs to integrins in cells. As a control, we observe that in the presence of a denaturant (DMSO), the energy landscape roughness of FMPs varies continuously with peptide length. The second feature corresponds to the formation of permanent intramolecular hydrogen bonding structure within the longest FMP (FMP27), which detrimentally affects binding.
A likely explanation for this phenomenon is that rough energy landscapes correspond to increased dwell-times of peptides in configurations appropriate for binding. A correspondence between these variables is expected because, in a rougher energy landscape, conformational changes requires traversal of a higher energy-barrier, As a result, the dwell-time within any particular conformation will be extended. These longer dwell-times stabilize a favorable conformation of the RGD site, fitting with our understanding of conformation as the primary mechanism by which protein binding occurs. This explanation suggests that intrinsic disorder at the RGD site promotes fibronectin binding, and could reveal why this site is disordered in fibronectin. Notably, permanent folding of FMP27 via hydrogen bonding had little or no bearing on the energy landscape roughness or dynamic behavior at the RGD site. Further studies are needed to confirm that this behavior plays a role in integrin binding by fibronectin proteins.
In summary, we used EPR spectroscopy, quantitative spectral analysis, and molecular dynamics simulations to understand the role of flanking residues in controlling the conformational dynamics and the free energy landscapes of RGD peptides. We observed the emergence of transient secondary structure, which significantly improved RGD binding to integrins. We conclude that energy landscape roughness may contribute to binding of native fibronectin proteins, and should be considered in future analyses of peptide binding. Finally, we suggest that dynamic and transient structure is important for peptide binding, and that binding interactions of peptide-functionalized biomaterials could benefit from consideration of dynamics and incorporation of additional residue, beyond the active binding site.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge the National Biomedical Research Center for Advanced ESR Technology (ACERT) and ACERT personnel for their support with DEER measurements.
Funding
This work was supported in part by the SharedEPR EPR Technology Transfer Experience (ETTE) program. W.R.L. was supported by the National Science Foundation Graduate Research Fellowship Program under Grant No. 1122374. Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number P41GM103521 and National Institute of Diabetes and Digestive and Kidney Diseases and National Heart, Lung and Blood Institute of the National Institutes of Health under the Award Numbers R01DK088327 and R01HL141366.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biomac.0c00506.
Additional data and information describing peptide synthesis and characterization, MD simulations, EPR, and DEER (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.biomac.0c00506
The authors declare no competing financial interest.
Contributor Information
William R. Lindemann, Department of Materials Science and Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, United States.
Alexander J. Mijalis, Department of Chemistry, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, United States
José L. Alonso, Leukocyte Biology and Inflammation Program, Division of Nephrology and Department of Medicine, Massachusetts General Hospital, Charlestown, Massachusetts 02129, United States; Harvard Medical School, Boston, Massachusetts 02115, United States
Peter P. Borbat, Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14850, United States
Jack H. Freed, Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14850, United States.
M. Amin Arnaout, Leukocyte Biology and Inflammation Program, Division of Nephrology and Department of Medicine, Massachusetts General Hospital, Charlestown, Massachusetts 02129, United States; Harvard Medical School, Boston, Massachusetts 02115, United States.
Bradley L. Pentelute, Department of Chemistry, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, United States.
Julia H. Ortony, Department of Materials Science and Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, United States
REFERENCES
- (1).Plow EF; Pierschbacher MD; Ruoslahti E; Marguerie GA; Ginsberg MH The Effect of Arg-Gly-Asp-Containing Peptides on Fibrinogen and von Willebrand Factor Binding to Platelets. Proc. Natl. Acad. Sci. U. S. A 1985, 82 (23), 8057–8061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Plow EF; Haas TA; Zhang L; Loftus J; Smith JW Ligand Binding to Integrins. J. Biol. Chem 2000, 275 (29), 21785–21788. [DOI] [PubMed] [Google Scholar]
- (3).Takada Y; Ye X; Simon S The Integrins. Genome Biol. 2007, 8 (5), 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Desgrosellier JS; Cheresh DA Integrins in Cancer: Biological Implications and Therapeutic Opportunities. Nat. Rev. Cancer 2010, 10 (1), 9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Ley K; Rivera-Nieves J; Sandborn WJ; Shattil S Integrin-Based Therapeutics: Biological Basis, Clinical Use and New Drugs. Nat. Rev. Drug Discovery 2016, 15, 173–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Kapp TG; Rechenmacher F; Neubauer S; Maltsev OV; Cavalcanti-Adam EA; Zarka R; Reuning U; Notni J; Wester HJ; Mas-Moruno C; et al. A Comprehensive Evaluation of the Activity and Selectivity Profile of Ligands for RGD-Binding Integrins. Sci. Rep 2017, 7 (November 2016), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Mijalis AJ; Thomas DA; Simon MD; Adamo A; Beaumont R; Jensen KF; Pentelute BL A Fully Automated Flow-Based Approach for Accelerated Peptide Synthesis. Nat. Chem. Biol 2017, 13 (5), 464–466. [DOI] [PubMed] [Google Scholar]
- (8).Simon MD; Heider PL; Adamo A; Vinogradov AA; Mong SK; Li XL; Berger T; Policarpo RL; Zhang C; Zou Y; et al. Rapid Flow-Based Peptide Synthesis. ChemBioChem 2014, 15 (5), 713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Gilad Y; Noy E; Senderowitz H; Albeck A; Firer MA; Gellerman G Synthesis, Biological Studies and Molecular Dynamics of New Anticancer RGD-Based Peptide Conjugates for Targeted Drug Delivery. Bioorg. Med. Chem 2016, 24 (2), 294–303. [DOI] [PubMed] [Google Scholar]
- (10).McHugh SM; Rogers JR; Solomon SA; Yu H; Lin YS Computational Methods to Design Cyclic Peptides. Curr. Opin. Chem. Biol 2016, 34, 95–102. [DOI] [PubMed] [Google Scholar]
- (11).Yu YP; Wang Q; Liu YC; Xie Y Molecular Basis for the Targeted Binding of RGD-Containing Peptide to Integrin AVβ3. Biomaterials 2014, 35 (5), 1667–1675. [DOI] [PubMed] [Google Scholar]
- (12).Van Agthoven JF; Xiong J-P; Alonso JL; Rui X; Adair BD; Goodman SL; Arnaout MA Structural Basis for Pure Antagonism of Integrin AVβ3 by a High Affinity Form of Fibronectin. Nat. Struct. Mol. Biol 2014, 21 (4), 383–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Krammer A; Craig D; Thomas WE; Schulten K; Vogel V A Structural Model for Force Regulated Integrin Binding to Fibronectin’s RGD-Synergy Site. Matrix Biol. 2002, 21 (2), 139–147. [DOI] [PubMed] [Google Scholar]
- (14).Xie H; Vucetic S; Iakoucheva LM; Oldfield CJ; Dunker AK; Uversky VN; Obradovic Z Functional Anthology of Intrinsic Disorder. I. Biological Processes and Functions of Proteins with Long Disordered Regions. J. Proteome Res 2007, 6 (5), 1882–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Leahy DJ; Aukhil I; Erickson HP 2.0 Å Crystal Structure of a Four-Domain Segment of Human Fibronectin Encompassing the RGD Loop and Synergy Region. Cell 1996, 84 (1), 155–164. [DOI] [PubMed] [Google Scholar]
- (16).Ishida T; Kinoshita K PrDOS: Prediction of Disordered Protein Regions from Amino Acid Sequence. Nucleic Acids Res. 2007, 35 (Suppl. 2), 460–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Monleoń D; Moreno-Murciano MP; Kovacs H; Marcinkiewicz C; Calvete JJ; Celda B Concerted Motions of the Integrin-Binding Loop and the C-Terminal Tail of the Non-RGD Disintegrin Obtustatin. J. Biol. Chem 2003, 278 (46), 45570–45576. [DOI] [PubMed] [Google Scholar]
- (18).Monleón D; Esteve V; Kovacs H; Calvete JJ; Celda B Conformation and Concerted Dynamics of the Integrin-Binding Site and the C-Terminal Region of Echistatin Revealed by Homonuclear NMR. Biochem. J 2005, 387 (1), 57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Daidone I; Aschi M; Patamia M; Bozzi A; Petruzzelli R Structural and Dynamical Properties of KTS-Disintegrins: A Comparison between Obtustatin and Lebestatin. Biopolymers 2013, 99 (1), 47–54. [DOI] [PubMed] [Google Scholar]
- (20).Calvanese L; Falcigno L; Auria GD Essential Dynamics Analysis Captures the Concerted Motion of the Integrin-Binding Site in Jerdostatin, an RTS Disintegrin. Biopolymers 2015, 103 (3), 158–166. [DOI] [PubMed] [Google Scholar]
- (21).Gunasekaran K; Tsai CJ; Kumar S; Zanuy D; Nussinov R Extended Disordered Proteins: Targeting Function with Less Scaffold. Trends Biochem. Sci 2003, 28 (2), 81–85. [DOI] [PubMed] [Google Scholar]
- (22).Hilser VJ; Thompson EB Intrinsic Disorder as a Mechanism to Optimize Allosteric Coupling in Proteins. Proc. Natl. Acad. Sci. U. S. A 2007, 104 (20), 8311–8315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Koshland DE Application of a Theory of Enzyme Specificity to Protein Synthesis. Proc. Natl. Acad. Sci. U. S. A 1958, 44 (2), 98–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Burgen AS Conformational Changes and Drug Action. Fed. Proc 1981, 40 (13), 2723–2728. [PubMed] [Google Scholar]
- (25).Vogt AD; Di Cera E Conformational Selection or Induced Fit? A Critical Appraisal of the Kinetic Mechanism. Biochemistry 2012, 51, 5894–5902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Arai M; Sugase K; Dyson HJ; Wright PE Conformational Propensities of Intrinsically Disordered Proteins Influence the Mechanism of Binding and Folding. Proc. Natl. Acad. Sci. U. S. A 2015, 112 (31), 9614–9619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Schreiber G; Haran G; Zhou H-X Fundamental Aspects of Protein-Protein Association Kinetics. Chem. Rev 2009, 109 (3), 839–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Tompa P Intrinsically Disordered Proteins: A 10-Year Recap. Trends Biochem. Sci 2012, 37 (12), 509–516. [DOI] [PubMed] [Google Scholar]
- (29).Van Agthoven JF; Shams H; Cochran FV; Alonso JL; Kintzing JR; Garakani K; Adair BD; Xiong JP; Mofrad MRK; Cochran JR; et al. Structural Basis of the Differential Binding of Engineered Knottins to Integrins AVβ3 and A5β1. Structure 2019, 27 (9), 1443–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Drescher M; Huber M; Subramaniam V Hunting the Chameleon: Structural Conformations of the Intrinsically Disordered Protein Alpha-Synuclein. ChemBioChem 2012, 13 (6), 761–768. [DOI] [PubMed] [Google Scholar]
- (31).Belle V; Rouger S; Costanzo S; Liquière E; Strancar J; Guigliarelli B; Fournel A; Longhi S Mapping α-Helical Induced Folding within the Intrinsically Disordered C-Terminal Domain of the Measles Virus Nucleoprotein by Site-Directed Spin-Labeling EPR Spectroscopy. Proteins: Struct., Funct., Genet 2008, 73 (4), 973–988. [DOI] [PubMed] [Google Scholar]
- (32).Le Breton N; Martinho M; Mileo E; Etienne E; Gerbaud G; Guigliarelli B; Belle V Exploring Intrinsically Disordered Proteins Using Site-Directed Spin Labeling Electron Paramagnetic Resonance Spectroscopy. Front. Mol. Biosci 2015, 2 (May), 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Schreier S; Bozelli JC; Marín N; Vieira RFF; Nakaie CR The Spin Label Amino Acid TOAC and Its Uses in Studies of Peptides: Chemical, Physicochemical, Spectroscopic, and Conformational Aspects. Biophys. Rev 2012, 4 (1), 45–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Hubbell WL; López CJ; Altenbach C; Yang Z Technological Advances in Site-Directed Spin Labeling of Proteins. Curr. Opin. Struct. Biol 2013, 23, 725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Schneider DJ; Freed JH Calculating Slow Motional Magnetic Resonance Spectra: A User’s Guide. Biol. Magn. Reson 1989, 8, 1–76. [Google Scholar]
- (36).Hubbell WL; Altenbach C Investigation of Structure and Dynamics in Membrane Proteins Using Site-Directed Spin Labeling. Curr. Opin. Struct. Biol 1994, 4, 566–573. [Google Scholar]
- (37).Sato H; Feix JB Peptide-Membrane Interactions and Mechanisms of Membrane Destruction by Amphipathic Alpha-Helical Antimicrobial Peptides. Biochim. Biophys. Acta, Biomembr 2006, 1758, 1245–1256. [DOI] [PubMed] [Google Scholar]
- (38).Nesmelov YE; Thomas DD Protein Structural Dynamics Revealed by Site-Directed Spin Labeling and Multifrequency EPR. Biophys. Rev 2010, 2 (2), 91–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Lappan U; Wiesner B; Scheler U Rotational Dynamics of Spin-Labeled Polyacid Chain Segments in Polyelectrolyte Complexes Studied by CW EPR Spectroscopy. Macromolecules 2015, 48 (11), 3577–3581. [Google Scholar]
- (40).Yu L; Wang W; Ling S; Liu S; Xiao L; Xin Y; Lai C; Xiong Y; Zhang L; Tian C CW-EPR Studies Revealed Different Motional Properties and Oligomeric States of the Integrin B1atransmembrane Domain in Detergent Micelles or Liposomes. Sci. Rep 2015, 5, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Lappan U; Wiesner B; Scheler U Segmental Dynamics of Poly(Acrylic Acid) in Polyelectrolyte Complex Coacervates Studied by Spin-Label EPR Spectroscopy. Macromolecules 2016, 49 (22), 8616–8621. [Google Scholar]
- (42).Skowronska A; Mlotkowska P; Okrasa S; Nielsen S; Skowronski MT Modulatory Effects of Steroid Hormones, Oxytocin, Arachidonic Acid, Forskolin and Cyclic AMP on the Expression of Aquaporin 1 and Aquaporin 5 in the Porcine Uterus during Placentation. J. Physiol. Pharmacol 2016, 67 (2), 311–319. [PubMed] [Google Scholar]
- (43).Oganesyan VS; Chami F; White GF; Thomson AJ A Combined EPR and MD Simulation Study of a Nitroxyl Spin Label with Restricted Internal Mobility Sensitive to Protein Dynamics. J. Magn. Reson 2017, 274, 24–35. [DOI] [PubMed] [Google Scholar]
- (44).López CJ; Oga S; Hubbell WL Mapping Molecular Flexibility of Proteins with Site-Directed Spin Labeling: A Case Study of Myoglobin. Biochemistry 2012, 51 (33), 6568–6583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Tilegenova C; Elberson BW; Cortes DM; Cuello LG CW-EPR Spectroscopy and Site-Directed Spin Labeling to Study the Structural Dynamics of Ion Channels In Potassium Channels; Coetzee WA, Rudy B, Eds.; Springer Science+Business Media, LLC: New York, 2018; pp 279–288. [DOI] [PubMed] [Google Scholar]
- (46).Jeschke G Conformational Dynamics and Distribution of Nitroxide Spin Labels. Prog. Nucl. Magn. Reson. Spectrosc 2013, 72, 42–60. [DOI] [PubMed] [Google Scholar]
- (47).Guzzi R; Babavali M; Bartucci R; Sportelli L; Esmann M; Marsh D Biochimica et Biophysica Acta Spin-Echo EPR of Na, KATPase Unfolding by Urea. Biochim. Biophys. Acta, Biomembr 2011, 1808 (6), 1618–1628. [DOI] [PubMed] [Google Scholar]
- (48).Marsh D; Bartucci R; Guzzi R; Sportelli L; Esmann M Librational Fluctuations in Protein Glasses. Biochim. Biophys. Acta, Proteins Proteomics 2013, 1834, 1591–1595. [DOI] [PubMed] [Google Scholar]
- (49).Lindemann WR; Evans ED; Mijalis AJ; Saouaf OM; Pentelute BL; Ortony JH Quantifying Residue-Specific Conformational Dynamics of a Highly Reactive 29-Mer Peptide. Sci. Rep 2020, 10 (1), 2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Lindemann WR Dynamics Characterization for Designing Functional Soft Materials. Ph.D. Thesis; Massachusetts Institute of Technology, Cambridge, MA, U.S.A., 2020. [Google Scholar]
- (51).Karim CB; Zhang Z; Thomas DD Synthesis of TOAC Spin-Labeled Proteins and Reconstitution in Lipid Membranes. Nat. Protoc 2007, 2 (1), 42–49. [DOI] [PubMed] [Google Scholar]
- (52).Borbat PP; Georgieva ER; Freed JH Improved Sensitivity for Long-Distance Measurements in Biomolecules: Five-Pulse Double Electron-Electron Resonance. J. Phys. Chem. Lett 2013, 4 (1), 170–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Jeschke G DEER Distance Measurements on Proteins. Annu. Rev. Phys. Chem 2012, 63 (1), 419–446. [DOI] [PubMed] [Google Scholar]
- (54).Georgieva ER; Borbat PP; Norman HD; Freed JH Mechanism of Influenza A M2 Transmembrane Domain Assembly in Lipid Membranes. Sci. Rep 2015, 5, 11757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Chiang YW; Borbat PP; Freed JH The Determination of Pair Distance Distributions by Pulsed ESR Using Tikhonov Regularization. J. Magn. Reson 2005, 172 (2), 279–295. [DOI] [PubMed] [Google Scholar]
- (56).Chiang YW; Borbat PP; Freed JH Maximum Entropy: A Complement to Tikhonov Regularization for Determination of Pair Distance Distributions by Pulsed ESR. J. Magn. Reson 2005, 177 (2), 184–196. [DOI] [PubMed] [Google Scholar]
- (57).Srivastava M; Freed JH Singular Value Decomposition Method to Determine Distance Distributions in Pulsed Dipolar Electron Spin Resonance. J. Phys. Chem. Lett 2017, 8 (22), 5648–5655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Srivastava M; Georgieva ER; Freed JH A New Wavelet Denoising Method for Experimental Time-Domain Signals: Pulsed Dipolar Electron Spin Resonance Madhur. J. Phys. Chem. A 2017, 121 (12), 2452–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Stoll S; Schweiger A EasySpin, a Comprehensive Software Package for Spectral Simulation and Analysis in EPR. J. Magn. Reson 2006, 178, 42–55. [DOI] [PubMed] [Google Scholar]
- (60).Budil DE; Lee S; Saxena S; Freed JH Nonlinear-Least-Squares Analysis of Slow-Motion EPR Spectra in One and Two Dimensions Using a Modified Levenberg – Marquardt Algorithm. J. Magn. Reson., Ser. A 1996, 120, 155–189. [Google Scholar]
- (61).Liang ZC; Freed JH An Assessment of the Applicability of Multifrequency ESR to Study the Complex Dynamics of Biomolecules. J. Phys. Chem. B 1999, 103, 6384–6396. [Google Scholar]
- (62).Thévenet P; Shen Y; Maupetit J; Guyon F; Derreumaux P; Tufféry P PEP-FOLD: An Updated de Novo Structure Prediction Server for Both Linear and Disulfide Bonded Cyclic Peptides. Nucleic Acids Res. 2012, 40 (W1), 288–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Lamiable A; Thévenet P; Rey J; Vavrusa M; Derreumaux P; Tufféry P PEP-FOLD3: Faster de Novo Structure Prediction for Linear Peptides in Solution and in Complex. Nucleic Acids Res. 2016, 44 (W1), W449–W454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).de Sancho D; Sirur A; Best RB Molecular Origins of Internal Friction Effects on Protein Folding Rates. Nat. Commun 2014, 79 (2), 211–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Kapon R; Nevo R; Reich Z Protein Energy Landscape Roughness. Biochem. Soc. Trans 2008, 36 (6), 1404–1408. [DOI] [PubMed] [Google Scholar]
- (66).Volk M; Milanesi L; Waltho JP; Hunter CA; Beddard GS The Roughness of the Protein Energy Landscape Results in Anomalous Diffusion of the Polypeptide Backbone. Phys. Chem. Chem. Phys 2015, 17 (2), 762–782. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.