

Abstract
Prenatal microcephaly is posited to arise from aberrant mitosis of neural progenitors, which disrupts both neuronal production and survival. Although microcephaly has both a genetic and environmental etiology, the mechanisms by which dysregulation of mitosis causes microcephaly are poorly understood. We previously discovered that prolonged mitosis of mouse neural progenitors, either ex vivo or in vitro, directly alters progeny cell fate, resulting in precocious differentiation and apoptosis. This raises questions as to whether prolonged progenitor mitosis affects cell fate and neurogenesis in vivo, and what are the underlying mechanisms? Towards addressing these knowledge gaps, we developed an in vivo model of mitotic delay. This uses pharmacological inhibition to acutely and reversibly prolong mitosis during cortical development, and fluorescent dyes to label direct progeny. Using this model, we discovered that a causal relationship between mitotic delay of neural progenitors and altered progeny cell fate is evident in vivo. Using transcriptome analyses to investigate the state of delayed cells and their progeny, we uncovered potential molecular mechanisms by which prolonged mitosis induces altered cell fates, including DNA damage and p53 signaling. We then extended our studies to human neural progenitors, demonstrating that lengthened mitosis duration also directly alters neuronal cell fate. This study establishes a valuable new experimental paradigm towards understanding mechanisms whereby lengthened mitosis duration may explain some cases of microcephaly.
Keywords: mitosis, progenitor, cortex, cell fate, neurogenesis
Introduction
The cerebral cortex is a highly ordered and complex structure which is critical for our higher cognitive functions including analytical thought and complex learning and language. In the developing mouse cortex, neurons are generated between embryonic day (E)10.5 and E18.5 (1). In mice, cortical excitatory neurons are primarily generated from radial glial cells (RGCs). RGCs can produce neurons directly, or indirectly via production of intermediate progenitors (IPs), which themselves divide one to two times before generating neurons (2–4). In mice, RGCs can undergo symmetric proliferative divisions generating two new RGCs, symmetric neurogenic generating two new neurons, or asymmetric generating any of the following pairs: RGC/IP; RGC/Neuron; Neuron/IP. RGCs produce cortical excitatory neuron subtypes sequentially with the earliest born neurons populating deep cortical layers VI–V and the late born neurons residing in the upper layers IV–II. Neurogenesis is influenced by many parameters, including cell cycle kinetics of progenitors (5, 6). For example, over the course of development mitosis (M phase) increases (7) and shortened S phase is linked to differentiation (8). Further, manipulation of either G1 or M phase kinetics impacts neurogenesis (9–11).
Genetic or environmental perturbations to embryonic cortical development can result in devastating neurodevelopmental disorders, including microcephaly and autism spectrum disorder (12–14). Individuals diagnosed with microcephaly have reduced head circumference more than 3 standard deviations below the mean. Microcephaly is accompanied by varying degrees of cognitive deficiency. Primary microcephaly is present at birth in contrast to postnatal degenerative microcephaly which may have a distinct etiology. While autosomal recessive primary microcephaly (MCPH) is relatively rare, microcephaly syndromes are highly prevalent, affecting between 1–2% of the population. To date, MCPH has been linked to mutations in 25 loci (15). Remarkably, the vast majority of these genes encode proteins associated with cell division (15). This has led to a long-standing hypothesis in the field that disruptions to mitosis explain microcephaly. Indeed, a link between aberrant mitosis and microcephaly has been borne out in human organoid, cell culture and mouse models. In human organoid models, defective progenitor mitosis is associated with altered neuron and progenitor number and massive apoptosis (16, 17). Further, mouse studies of human microcephaly genes reveal consistent phenotypes of excessive apoptosis as well as imbalanced numbers of more neurons and fewer progenitors (18–24).
Beyond a genetic basis, microcephaly is also caused by viral infection, notably cytomegalovirus (CMV) or Zika virus (ZIKV) infection during pregnancy (25). Several studies have shown that acute ZIKV infection can also impair neural progenitor mitosis and in some cases is associated with aberrant neurogenesis and apoptosis in human and mouse models (26–29). Thus, both genetic and viral causes of microcephaly are linked to progenitor mitosis defects and similar defects in neurogenesis and cell survival. However, despite this clear clinical link, the mechanisms by which mitotic defects underlying this pathology are poorly understood.
We previously uncovered one potential explanation for how progenitor mitotic defects could cause microcephaly, with the discovery that progenitors undergoing a delayed mitosis (specifically M phase) directly produce aberrant cell progeny (11, 30). Using live imaging of brain slices and primary progenitors from a mouse microcephaly model called Magoh+/−, we determined that mitotically delayed mutant progenitors produced significantly fewer neural progenitors and more apoptotic progeny. We extended this correlative data by employing two distinct drugs, STLC and nocodazole, to reversibly and acutely prolong mitosis in wild-type ex vivo organotypic brain slices and in vitro primary progenitors. In both paradigms, progenitors with prolonged mitosis alone directly generated neurons at the expense of progenitors; exceedingly long mitoses led to production of apoptotic progeny. While these data indicate prolonged mitosis is relevant for microcephaly, they raise several important questions. Is a causal link between prolonged mitosis and altered neurogenesis evident in vivo? How do alterations in mitotic length drive rapid changes in differentiation? Do these mechanisms hold true in human progenitors?
To address these questions, in this study we developed and characterized a new in vivo model for prolonged progenitor mitosis during cortical development. With this experimental paradigm, an acute injection of a pharmacological mitotic inhibitor into the embryonic cortex delays mitosis of progenitors transiently, reversibly, and reproducibly. This was coupled with FlashTag, to selectively label the delayed progenitors and their direct progeny. Using this strategy to lineage trace daughter cells of delayed progenitors we discovered that acute mitotic delay of RGCs in vivo results in reduced progenitor number, concomitant increased neuron number, as well as apoptosis of progeny. We then applied this strategy to isolate the delayed progenitors and progeny to assess early transcriptome changes associated with a single mitotic delay in the context of otherwise normal development. Among the observed transcriptome changes, our data highlight p53 signaling as a notable pathway linked to mitotic delay, with well-studied p53 targets prominently upregulated. Finally, using human embryonic stem cell (ES) derived neural progenitors, we demonstrate that mitotic delay is also linked to altered cell fates in human progenitors similar to that we observed in vitro and in vivo in the mouse. Taken together, our study further establishes prolonged mitosis as a driver of apoptosis and neuronal fates in vivo and reinforces that increased mitotic length provides a plausible mechanism to explain some cases of microcephaly. Our study thus describes and characterizes a valuable new model for understanding the etiology of microcephaly.
Materials and Methods:
Mice:
All mice were fed a standard chow. All alleles were maintained on a C57BL/6 background. Bax heterozygotes were bred to produce knockout embryos, which were genotyped at time of experiment. P53 cKO (Emx1-Cre; p53lox/lox) mice were bred as homozygotes, F1 in-breeding was avoided. Embryogenesis was timed, calling embryonic day 0.5 at time of observed vaginal plugging the next morning. All experiments were approved by Duke IACUC.
Mouse Injections:
Intraventricular injections were performed on E14.5 embryos. Pregnant dams were anesthetized with Isoflurane. 0.8uL of DMSO or STLC (80uM, 150uM, 250uM, dissolved in DMSO) with 0.01% Fast Green in dH20 was injected into the right ventricle of each embryo using a kd Scientific Legato 100 microinjector. For some experiments (Figures 1, 2A–H, 3, 5, 6, S1) we co-injected with 0.8uL of 1mM FlashTag (CFSE CellTrace®, Thermo Fisher C34570, stock prepared according to manufacturer’s recommendations then diluted 1/10 in injection solution). For some experiments (Figures 2I–K, 4, S2, S3, S4) we performed a sequential injection in which STLC or DMSO was first injected followed 2 hours later by FlashTag injection into the same brain. Briefly, between surgeries, the dam was placed in her home cage and allowed to recover. For the second injection, sutures were removed, injection was performed, then the surgical opening was again sutured closed. The dam was placed in her home cage and allowed to recover. Both surgeries were performed in the same manner. The sequential injection was optimal to label cells at the time of their delay in prometaphase at the ventricular surface. At various timepoints post-injection embryos were dissected, cortices were dissected, isolated, and prepared for downstream applications described below (FACS, Cell Culture, RNA-seq, fixed analysis).
Figure 1. A model for acute inducible prolonged mitosis in vivo.
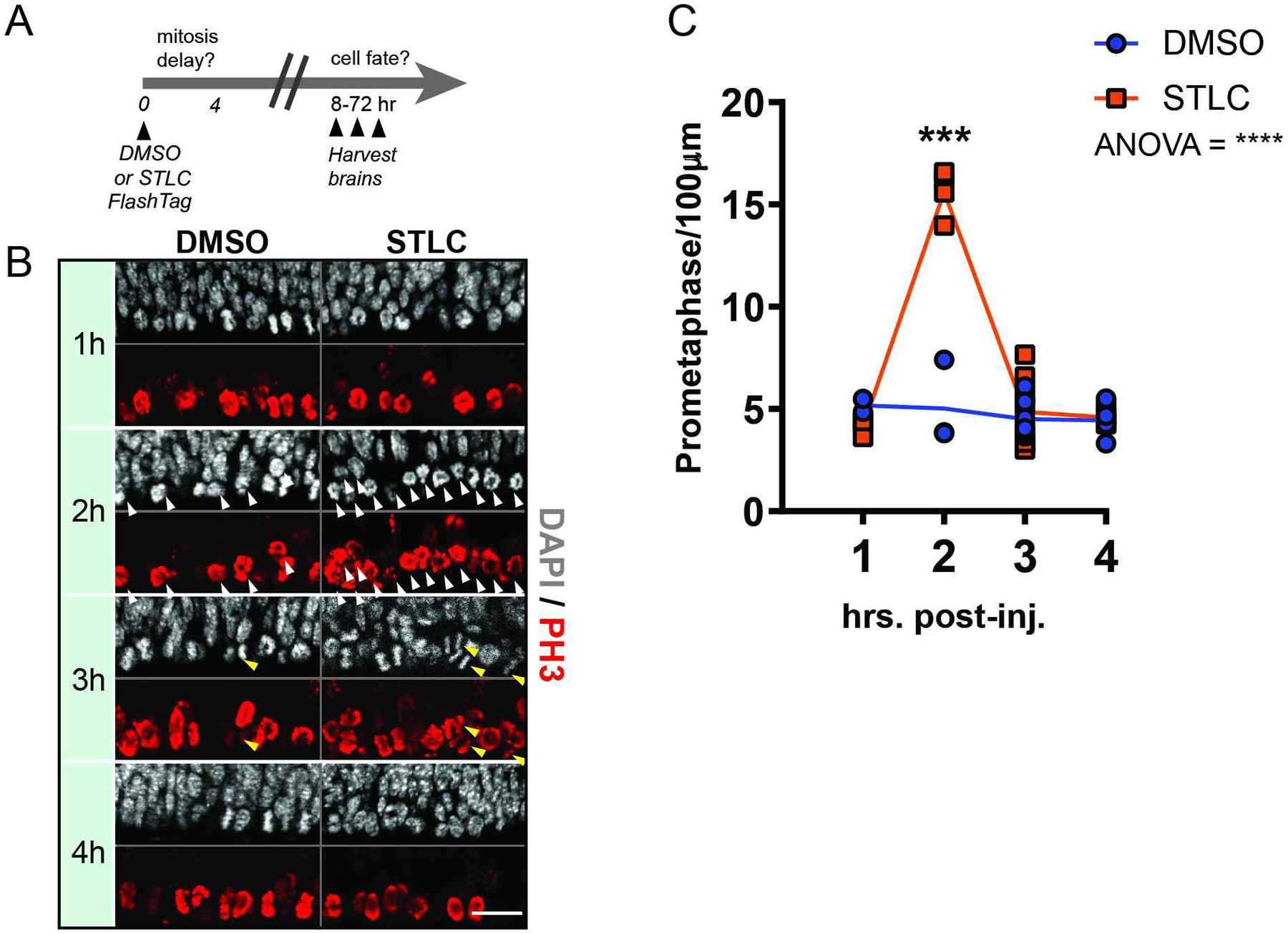
(A) Experimental paradigm with injection of either DMSO or STLC at timepoint 0 and harvesting of brains for fate analysis between 8–73 hours later. (B) Immunofluorescence depicting PH3+ cells (red) and DAPI (white) at the ventricle of an E14.5 cortex, at different time points following injection of DMSO or STLC. Scale bar: 25 μm. White arrowheads: prometaphase cells; yellow arrowheads:- anaphase cells. (C) Quantification of the average number of prometaphase cells per 100 μm, along the ventricular surface of the cortex. N ≥ 3 embryos for each condition, at each timepoint. N=37 total embryos. Statistics: Anova analysis, post-hoc student’s t-test. ***, p=0.0005. ****, p<0.0001. Error bar=s.d.
Figure 2. Effects of prolonged mitosis in vivo on cell fate acquisition.
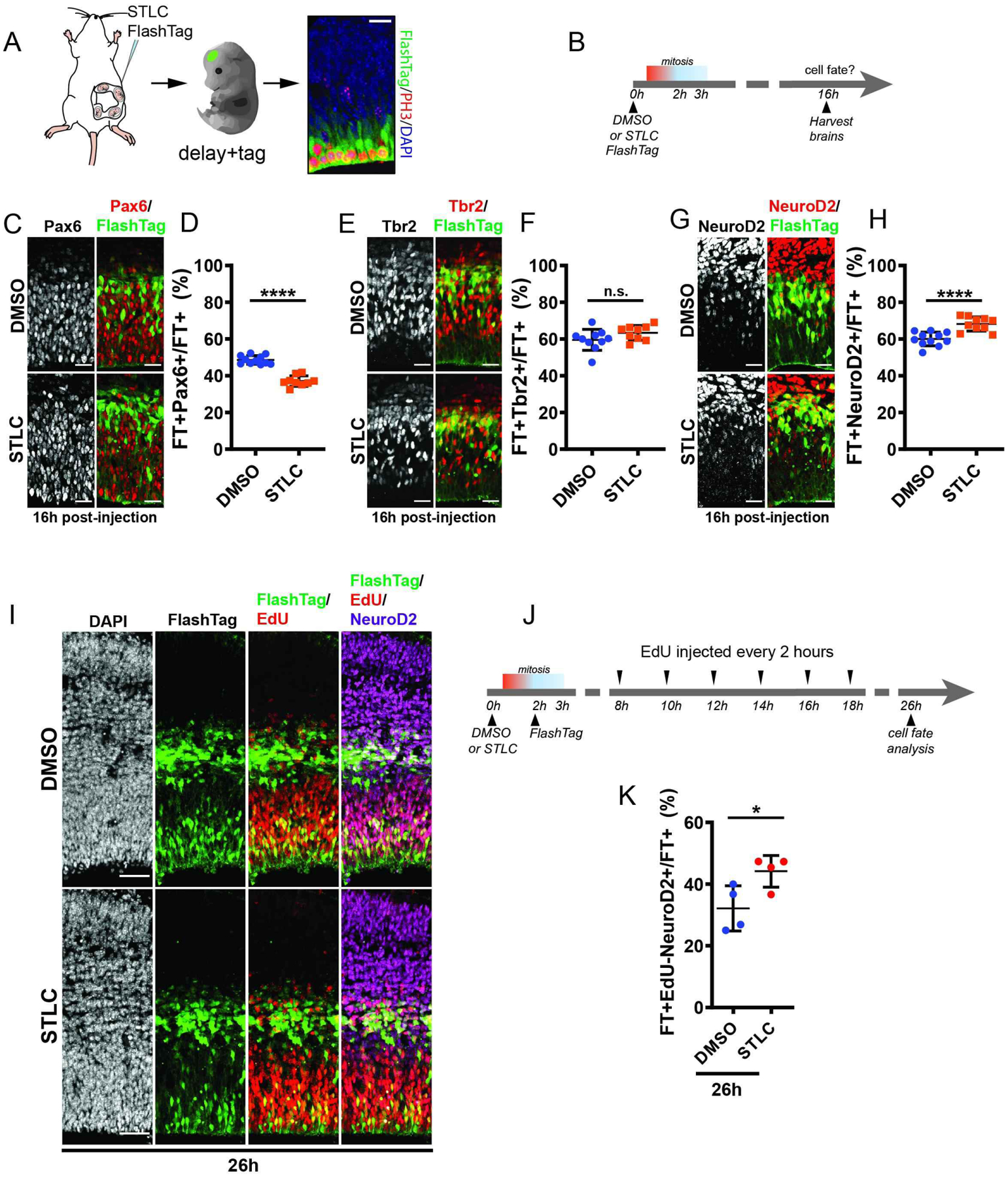
(A) Scheme of injection paradigm used in this study. B) Time course of injection paradigm used to assess cell fate following prolonged mitosis. C, E, G) Images depicting E14.5 brains 16 hrs after injection with either STLC or DMSO and stained for C) Pax6 (white, red) (C) , Tbr2 (white, red) (E) , Neurod2 (white, red) (G) along with FlashTag (green). D, F, H) Quantification of the fraction of FT+ cells which are Pax6 (D), Tbr2 (F), and Neurod2 (G) 16h after injection of DMSO or STLC. Pax6 analysis DMSO: n=11 embryos; STLC: n=9 embryos, n=2 sections each. Tbr2 analysis, DMSO: n=10 embryos, STLC: n=9 embryos, n= 2 or more sections each. NeuroD2 analysis, DMSO: n=11 embryos, STLC: n=10 embryos, n=2 or more sections each. Statistics: student’s t-test. I) Images depicting FT (green), EdU (red), and NeuroD2 (purple) 26h post-injection of DMSO or STLC. J) EdU labeling scheme following injection of DMSO or STLC, analysis at 26h post-injection. K) Quantification of the fraction of FT cells which are EdU-NeuroD2+FT+ 26h after injection of DMSO or STLC. NeuroD2 **: p=0.0037. Pax6 **** p<0.0001. EdU/NeuroD2 *: p=0.04. Scale bars: A: 20 μm. E,C, G, 25 μm; J, 100 μm. Error bar=s.d.
Figure 3. Prolonged mitosis in vivo results in apoptosis in daughter cells.
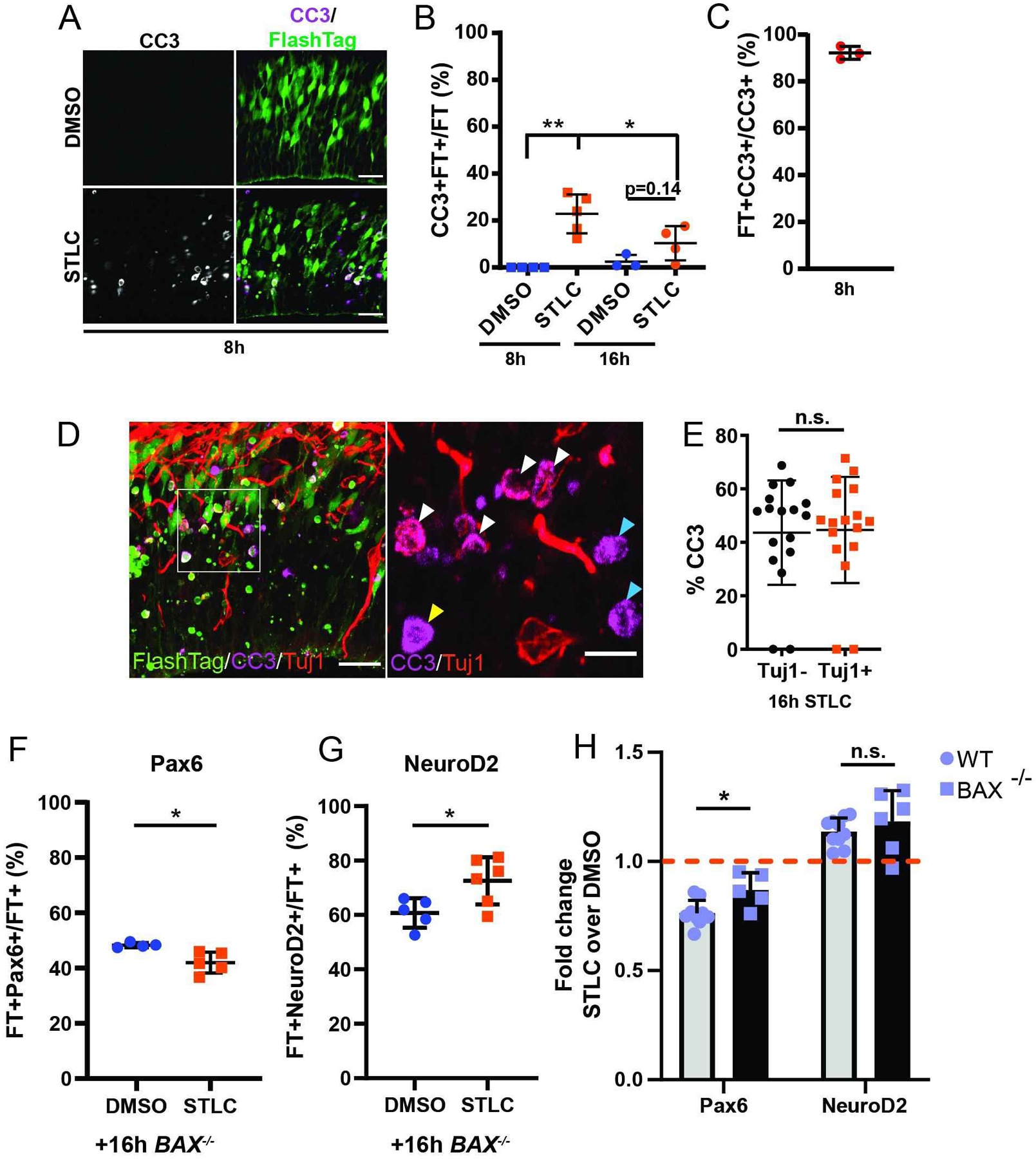
A) Images depicting the VZ/SVZ with FT (green) and CC3 (white) 8h after injection of DMSO or STLC. B) Quantification of the fraction of FT cells which are CC3+FT+ cells in 200 μm wide radial columns. C) Quantification of CC3+ cells that are FT+ at 8h in STLC condition. Dots represent individual embryos, n= 2 or more sections each. D) Images depicting the VZ/SVZ +16h after injection of STLC, depicting FT (green), CC3 (purple), and Tuj1 (red). Inset, right: ROI depicting CC3 and Tuj1. White arrowheads: FT+CC3+Tuj1+; Yellow arrowheads: FT+CC3+Tuj1−; Blue arrowheads: FT+CC3+Tuj1−. E) Quantification of the fraction of CC3+ cells that are either Tuj1+ or Tuj1− 16h after STLC injection, in 200 μm wide radial columns. N>10 sections. F and G) Quantification of the fraction of FT+ cells which are Pax6 (F) or NeuroD2 (G) 16h after injection of DMSO or STLC in E14.5 Bax−/− embryos; in 200 μm wide radial columns. Pax6 analysis, DMSO: n=4 embryos; STLC: n=5 embryos, n=3 sections each. NeuroD2 analysis, DMSO: n=5 embryos, STLC: n=6 embryos, n= 3 sections each. H) Comparison of fold changes in expression of Pax6 or NeuroD2 between WT and Bax−/− embryos at 16h following STLC injection. CC3 8h ***: p=0.001, 8h v 16h: p=0.05. BAX Pax6 *: p=0.015, NeuroD2 *: p=0.03. Pax6 BAX v WT *: p=0.01. Students t-test. Scale bars: A, D, 25 μm; D, 10 μm, Error bar=s.d.
Figure 5. Upregulated P53 pathway and DNA damage following prolonged mitosis.
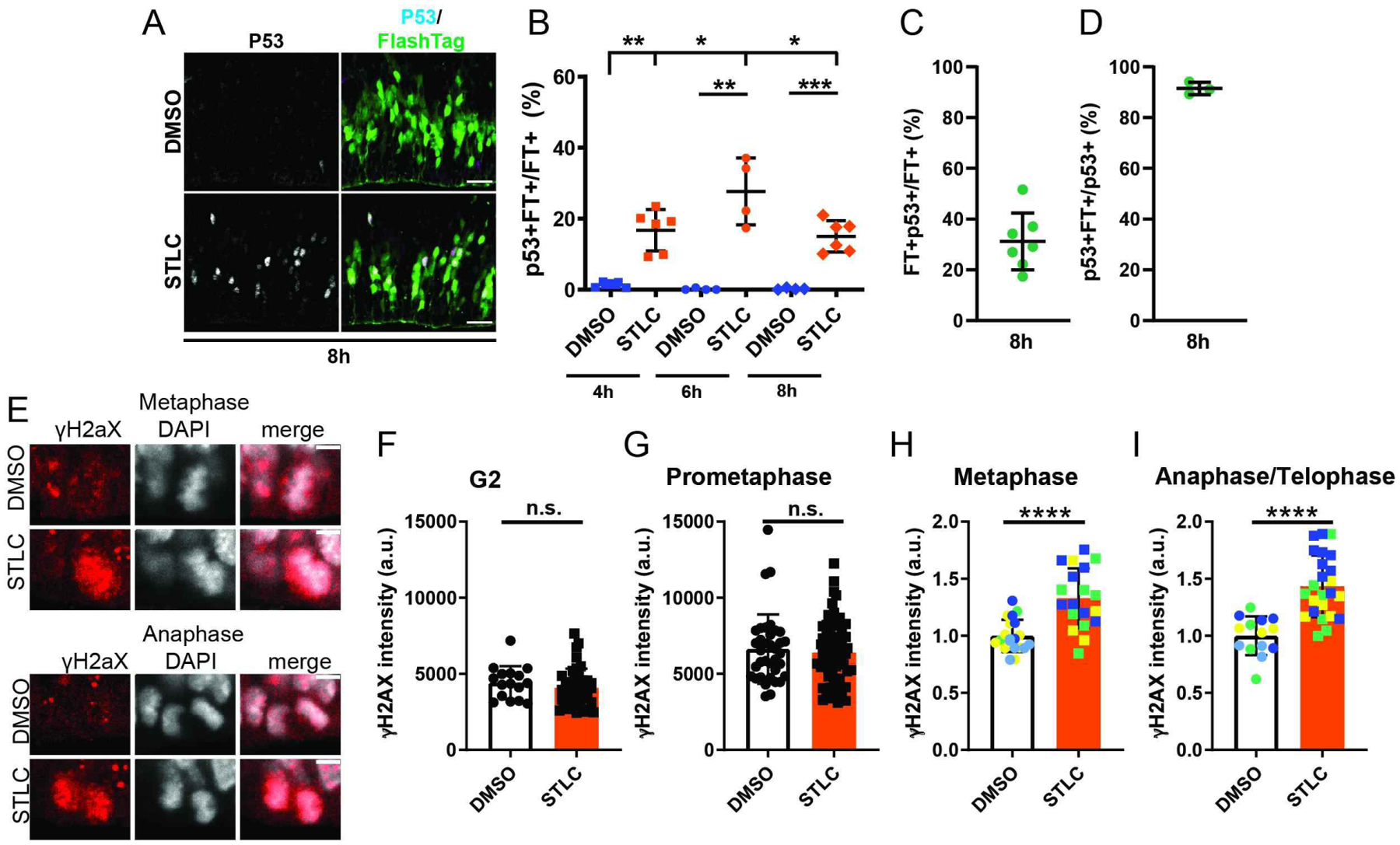
A) Images of the VZ/SVZ depicting FT (green) and p53 (white) 8h after injection of DMSO or STLC. b) Quantification of the fraction of FT+ cells that are CC3+FT+ cells 4h, 6h, and 8 h after STLC injection via analysis of 200micron cortical columns. Dots represent individual embryos, average of 2 or more sections per embryo. 6h**: p=0.004, 8h ****: p<0.0001, 10h ***: p=0.0003, 6h v 8h *:p=0.02, 8h v 10h: p=0.04. C) Quantification of the fraction of FT+ cells that are p53+ at 8h in STLC condition. C) Percent of p53+ cells labeled by FlashTag 8h after STLC injection. N=3 embryos, average of 3 sections per embryo. E) Images of H2AX (red) and DAPI (white) in mitotic cells at the ventricle 3h after injection of DMSO or STLC. F) Quantification of H2AX intensity of mitotic cells at the ventricle 3h after DMSO or STLC injection. Dots represent individual cells across two or more embryos. Note for STLC, primarily FT+ cells were quantified in order to enrich for mitotic cells. Statistics: student’s t-test. metaphase ****: p<0.0001. anaphase/telophase ****: p<0.0001. Scale bar: A, 25 μm, E, 5 μm, Error bar= s.d.
Figure 6. Prolonged mitosis in human neural progenitors directly alters cell fate.
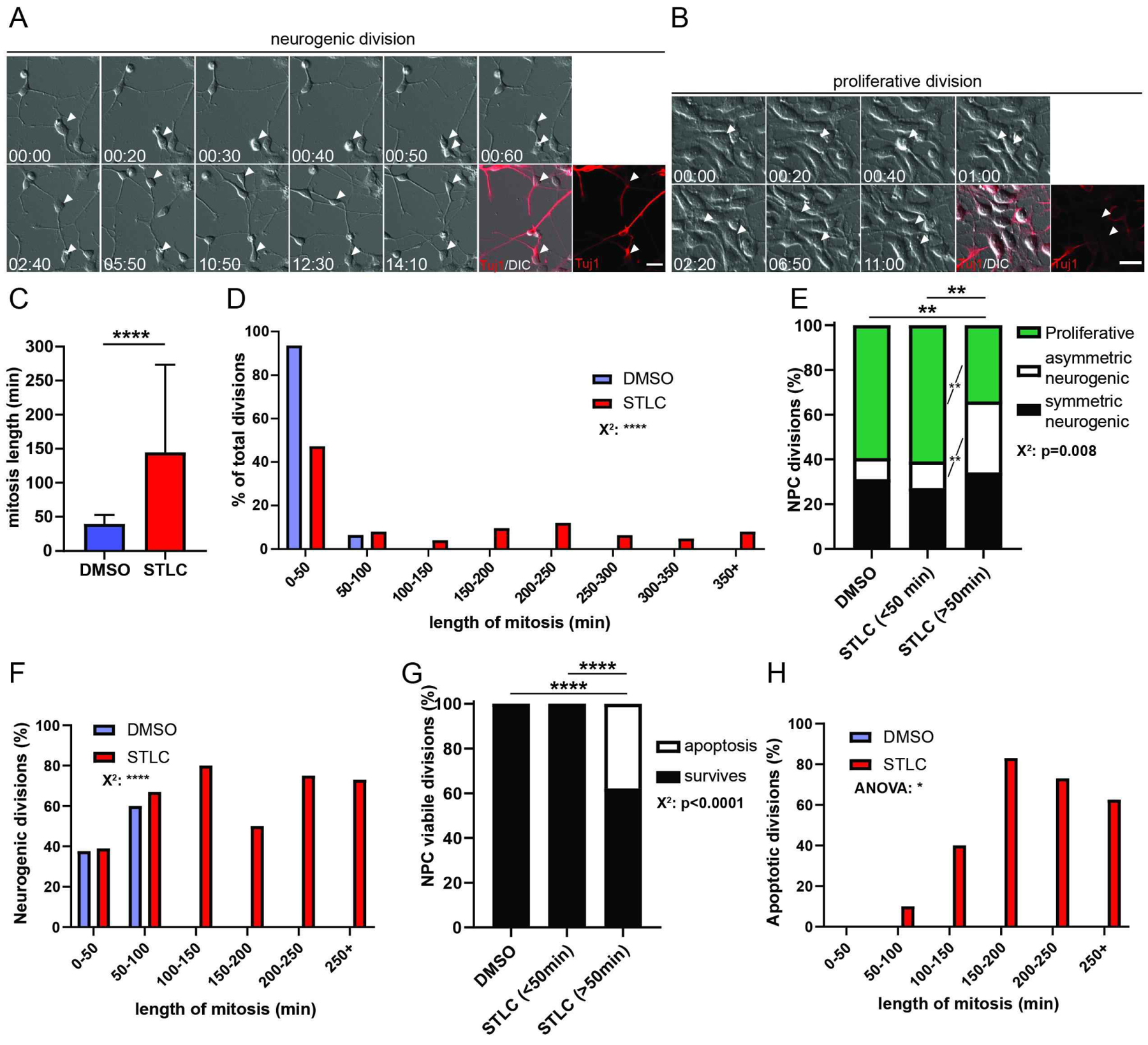
A, B) Panels of live imaging timeseries depicting neurogenic (A) and proliferative divisions (B) with immunostaining for Tuj1 (red) and DIC. Arrowheads indicate 1 cell over time which divides to become 2 cells. C) Quantification of average mitosis duration for NPCs treated with either DMSO (blue) or STLC (red). Student’s t-test. ****: p<0.0001. D) Histogram of NPC mitosis duration, using 50min bins. Chi-square, ****: p<0.0001. E) Quantification of fraction of NPC divisions which are proliferative (Tuj1−; green), asymmetric neurogenic (1 Tuj1−, 1Tuj1+, white), or symmetric neurogenic (both Tuj1+, black), under different conditions and graphed by mitosis duration. DMSO: n=74 daughter pairs, STLC <50 mins: n=58 daughter pairs, STLC >50 mins: n= 41 cells. Chi-square with Bonferroni correction followed by post-hoc analysis. DMSO v STLC delayed **: p=0.006, STLC normal v delayed **: p=0.009, post hoc proliferative fates **: p=0.001, asymmetric neurogenic **: p=0.003. F) Histogram of neurogenic divisions (% of total at each mitotic duration) in 50 min bins. Chi-square ****: p<0.0001. G) Quantification of the fraction of NPC divisions generating viable (black) or apoptotic (white) progeny. DMSO: n=74 daughter pairs, STLC <50 mins: n=58 daughter pairs, STLC >50 mins: n= 66 cells. Statistics: Chi-square and post-hoc chi-square with Bonferroni correction. DMSO v Delayed STLC ****: p<0.0001, STLC normal v delayed STLC ****: p<0.0001. H) Histogram of proportion of apoptosis in progeny. ANOVA *: p=0.02, Error bar=s.d., Scale Bar= A, B: 20 μm.
Figure 4. Transcriptome analysis following prolonged mitosis.
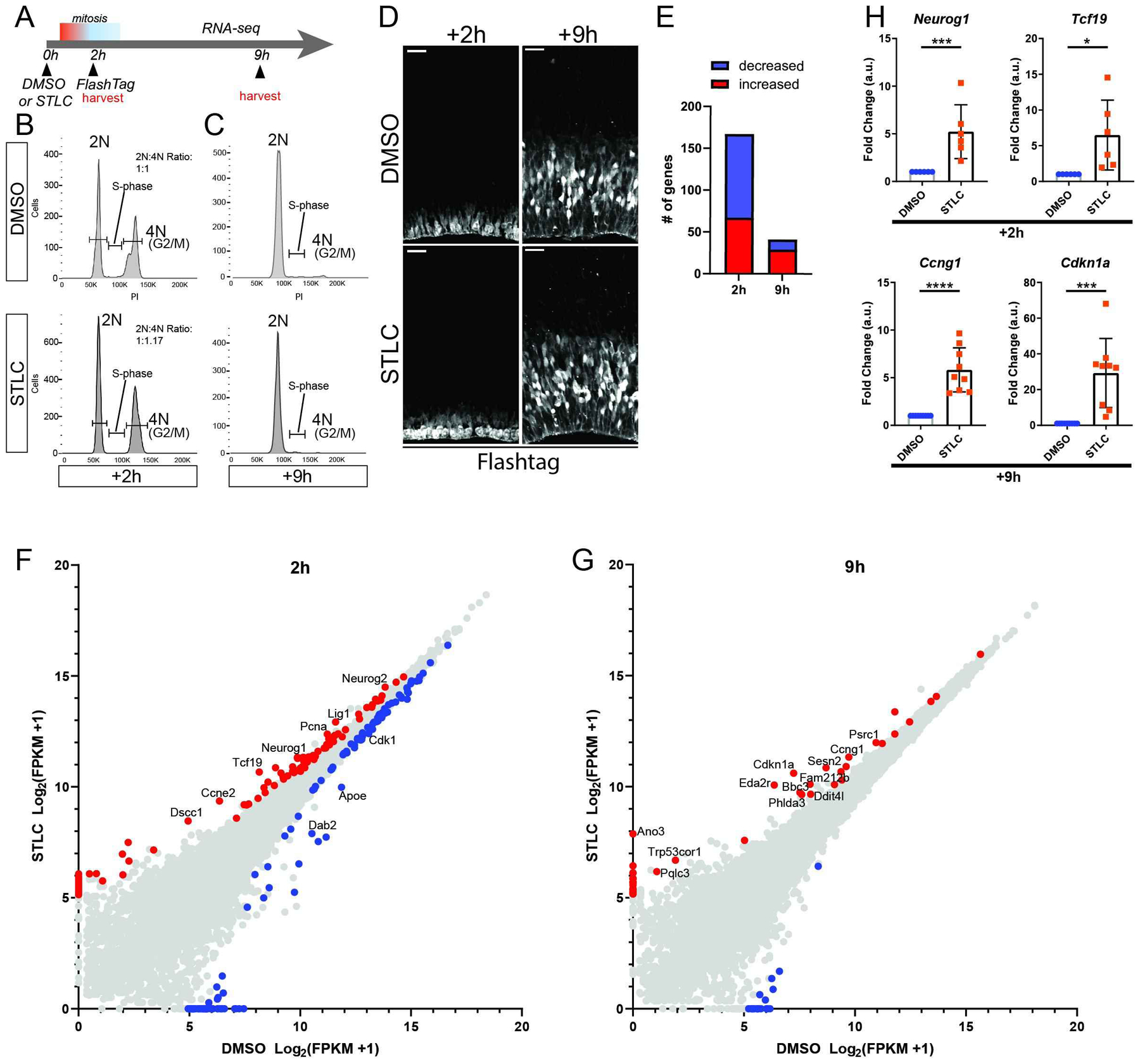
A) Schematic of RNA-seq analysis. B) Images depicting cortices stained for FT (white) 2 and 9 hrs after injection with either DMSO and STLC conditions, corresponding to RNA-seq timepoints. C, D) Cell cycle plots of FACS sorted populations by condition and timepoint. Y axis: cell count, X axis: Propidium Iodide. E) Graph of total significant transcript changes at both timepoints, >2-fold expression difference STLC v DMSO, p<0.05. F, G) Scatterplots of gene expression changes at +2h (F) and +9h (G) timepoints. Red: p<0.05, enriched in STLC. Blue: p<0.05, enriched in DMSO. Transcripts of notable change or associated with significant pathways have been annotated. H) qPCR for select RNA-seq candidates at +2h and +9h timepoints. Neurog1 ***: p=0.004, Tcf19 ***: p=0.02, Ccng1 ***: p<0.0001, Cdkn1a ***: p=0.0005. Dots equal independent biological replicates. At +2h, samples were identical to those use for FACs, whereas at +9h, 3 brains were from FACS and remaining were independent replicates. Error bar=s.d.
Fluorescence activated cell sorting of FlashTag+ cells and cell cycle analysis:
Embryos were dissected into ice cold HBSS (Life Technologies, Without Ca or Mg). Cortices were isolated and meninges removed in ice cold HBSS. As necessary, tails were reserved from embryos for genotyping and placed in lysis buffer. Cortices were placed one or two hemispheres at a time into 250 μL of cold 0.25% Trypsin/EDTA (Life Technologies) with RNase-Free DNase I (New England Biolabs, 1:1000). Cortices were incubated in trypsin for 10 minutes at 37°C. Trypsin was inactivated with addition of 500 μL of ice cold HBSS containing propidium iodide, and tissue was passed through a glass pipette (VWR) 6 times, then passed through a 30 micron filter. Dissociated cells were sorted at 6°C with gates for FSC, SSC, PI and CFSE, dead cells and doublets discarded, and live cells collected in either HBSS (without Ca or Mg) with 10% FBS, HBSS (no FBS), RLT buffer (Qiagen) or Tri-Reagent (Sigma). For Cell Cycle analysis, cells were sorted at 4°C into ice cold HBSS with 10% FBS. Cells were spun at 200g at 4°C, supernatant removed, and resuspended in 300uL PBS (no Ca no Mg). 650 μL of 100% EtOH was added dropwise to fix cells. Cells were fixed for 1 hour, then spun down at 200g and washed 3x with 5% BSA (fraction V, Thermo Fisher) in 1x PBS (no Ca no Mg). Cells were incubated with RNAse A 1:1000 (Sigma) for 30 minutes at room temp, and Propidium Iodide added at 1:1000. Cells were incubated for a further 30 min at room temp then stored at 4°C until sorted. Cells were sorted and analyzed using a FACSCantoII™ Flow Cytometer (BD Biosciences) capturing FSC, SSC, CFSE, and PI. Output files were analyzed in FlowJo Version X. Cell cycle graphs were created in FlowJo. Briefly, output files were gated for single cells comparing FSC v SSC and PI intensity plotted in a histogram to determine proportion of cells 2N, S/G2, and 4N. For +2h, we isolated the top 1.5–2.3% of CFSE+ cells for control and the top 14–20% of CFSE+ cells for STLC. For +9h, we isolated the top 25% of CFSE+ cells for control and for STLC.
qPCR of FACS sorted cells:
Cells were sorted into RLT buffer and lysates frozen at −80°C. Samples were thawed for 15 minutes, mixed, and RNA was purified using Qiagen RNEasy Mini Plus columns into 15 μL elution buffer (Qiagen). cDNA was generated using iScript (BioRad). qPCR was performed using template specific primers (Table 1) with iScript SYBR GREEN (Biorad) in 15 μL reactions in an Applied Biosystems StepOnePlus or in 2uL reactions performed in Applied Biosystems QuantStudio6 Flex. For 2ul reactions, qpcr plate was loaded from a source plate using a manual template on a LabCyte Echo 550 Acoustic Liquid Handler.
Immunofluorescence:
Embryonic brains were fixed overnight in 4% PFA at 4°C, then suspended in 30% sucrose at 4°C for at least 4h. Brains were submerged in Neg-50™ (thermo scientific) and frozen on dry ice, then placed at −80. 20 micron sections were cryosectioned and slides were stored at −80°C. Sections were washed with PBS and incubated in 0.25% Triton for 15 minutes. If antigen retrieval was performed, sections were then submerged in sodium citrate buffer, heated to 100°C for 30 minutes, taken off heat, and allowed to equilibrate to room temperature for ~30 minutes. Then sections were blocked in 1x PBS with 10% NGS for 1h. Primary antibodies, listed in Table S3, were incubated at room temperature for 2h. Sections were washed 3x then secondary antibodies, along with DAPI, were incubated for 20 min. Sections were washed and mounted in mowiol. Immunofluorescence images were acquired by epifluorescence on a Zeiss AxioObserver with Apotome, or on a Zeiss 780 Inverted Confocal microscope and images were analyzed in Fiji software. Fiji quantitative data was transferred into Excel spreadsheets and statistical analyses were performed with GraphPad Prism.
Quantifications:
In 150, 200, or 250 micron cortical columns of the dorsal cortex the number of cells expressing FT and markers for RGCs (Pax6/Sox2), IPs (Tbr2) or neurons (NeuroD2) were calculated using Fiji Cell Counter. For layer marker analysis, in 250 micron cortical columns the abundance and colocalization of FT and various pyramidal neuron markers was quantified. For prometaphase and mitosis quantifications, PH3 positive cells with condensed DNA were counted in blinded sections and the number of mitotic cells (PH3+ cells with condensed DNA) and the number of prometaphase cells (PH3+, DAPI without a metaphase plate, not in anaphase or telophase) determined by (#cells/microns)*100). For γH2AX analysis, immunofluorescence was performed using antigen retrieval, and images were captured on a Zeiss 780 inverted confocal microscope. Z-sections were acquired every 2 microns. All processing and analysis was performed in ImageJ (FIJI). 2 z-sections of each image were combined in a maximum intensity projection for analysis. Phase of mitosis was called for each cell using DAPI. For analysis of metaphase and prometaphase cells, an oval of 25–26 micron perimeter was drawn around each cell. For analysis of anaphase cells an ellipse of 28–29 micron perimeter was drawn around each cell. Mean, minimum, and maximum intensity of pixels were measured. For γH2AX, analysis was completed on sections prepared 3h after injection and for pATR, analysis completed 4h after injection. For STLC condition, all FlashTag+ cells at the ventricle were measured. For DMSO, FlashTag+ and FlashTag− cells were measured.
EdU time course:
E14.5 embryos were injected as described (mouse injections), first with DMSO or STLC, and 2h later, injected with FlashTag. 6h after injection, EdU was administered by IP injection at 10mg/kg every 2 hours until 18h to cumulatively label dividing cells in this period. 24h later, cortices were dissected, fixed in 4% PFA, and sectioned (as above). Prior to antibody staining, slides containing sections were washed twice in 1x PBS for 8 min each, permeabilized with 0.5% Triton in 1x PBS for 2 min, washed three times in 1x PBS 2 min, and immunofluorescence staining for EdU was carried out using Life Technologies Click-It Plus Kit (#C10749) following manufacturer’s instructions.
Transcriptome analysis:
E14.5 embryos were injected with FlashTag and DMSO or STLC using the sequential labeling approach described above. Following treatment and labeling of RGCs, at +2h and +9h following delay induction cortices were dissected and RGCs dissociated as described above. FlashTag+ cells were sorted into RLT lysis buffer and stored at −80°C. RNA was purified from sorted RGCs using RNeasy Plus Micro Kit (Qiagen) and stored at −80°C. cDNA libraries were prepared using Nugen Universal Plus mRNA-seq and sequenced using an Illumina Hiseq 4000. RNA-seq data was processed as previously (30). In short we used the TrimGalore toolkit which employs Cutadapt to trim low-quality bases and Illumina sequencing adapters from the 3’ end of the reads. Only reads that were 20nt or longer after trimming were kept for further analysis. Reads were mapped to the GRCm38v73 version of the mouse genome and transcriptome using the STAR RNA-seq alignment tool. Reads were kept for subsequent analysis if they mapped to a single genomic location. Gene counts were compiled using the HTSeq tool. Only genes that had at least 10 reads in any given library were used in subsequent analysis. Normalization and differential expression were carried out using the DESeq2 Bioconductor package with the R statistical programming environment. Within individual analysis, genes were further filtered if more than 4 samples had zero reads. The false discovery rate was calculated to control for multiple hypothesis testing. Gene set enrichment analysis (Standard GSEA) was performed using normalized counts for the entire gene set averaged from biological triplicates per condition to identify gene ontology terms and pathways associated with altered gene expression for each of the comparisons performed. Gene identification was matched to Ensembl mouse CHIP file IDs.
Primary cultures:
live imaging of RGCs and quantifying cell fate was performed as previously described, with slight modifications. Briefly, before dissection, primary culture media was assembled and MatTek glass coverslip bottom culture plates were coated with 50ug/mL Poly-L-Lysine for at least 30 minutes at 37°C. Plates were washed 3x with sterile water. Primary culture media recipe: 500μl N2 Supplement (Thermo Fisher), 1mL B27 Supplement (without Vitamin A, Thermo Fisher), 500μl of 100mM N-acetyl-Cysteine (prepared in sterile water), and 50uL of 10ug/mL mouse bFGF (R&D Systems 3139-FB025, prepared using manufacturer’s instructions) was added to cold 48.95mL high-glucose DMEM (with Sodium Pyruvate, Glutamine, Thermo Fisher 11995–065). Media was kept at 4°C for up to one week. Only enough media needed for each day was heated at 37°C. E13.5 cortices were dissected in ice-cold HBSS, and the meninges removed, before placing cortices into ice-cold 0.25% trypsin-EDTA (Gibco). Cortices were digested for 10 minutes at 37°C, then trypsin was inactivated by adding 1x trypsin inhibitor (Glycine max soybean, Sigma) diluted in primary cell culture media. Cells were spun down at 200g for 5 minutes, and supernatant removed. Cells were washed once with primary culture media, supernatant was removed, and cells were diluted in primary cell culture media. Cells were counted with a cell counter, and 250,000 cells were plated onto MatTek plates with glass coverslip bottoms in 1mL primary culture media. The culture plate was placed in a 37°C incubator with 5% CO2, and cells allowed to settle and attach to plate bottom for 1 hour before proceeding.
In vitro live imaging:
A Zeiss Axiovision inverted A1 microscope fitted with a Pecon incubation chamber was warmed to 37°C and atmosphere conditioned to 5% CO2, via sensors and integrated Zen software. For drug application, each drug was diluted to appropriate concentration in 500uL warmed primary culture media (STLC 1uM, AZ20 1uM, VE821 1uM). 500uL of primary culture media was removed from cultures, and media with DMSO, STLC, individual ATR inhibitors or drug cocktail was added to each well, respectively. The culture plate was placed into the microscope under a Pecon plate cover attached to a regulated, humidified CO2 line providing 5% CO2. Typically, 80–120 x,y,z imaging positions were chosen in Zen software (~20 per well), and DIC images acquired with 10 minute intervals between frames. Z focus was periodically adjusted as needed. Cells were incubated for 3h, then STLC (STLC wash off) removed from all conditions by removal of media, 1x wash with primary culture media, and application of new media. New media containing either DMSO or ATR inhibitors was added, and the plate was taken back to the microscope to resume imaging. After an additional 3h ATR inhibitors were removed (ATRi wash off) by replacing all the cell culture media. The culture plate was taken back to microscope to resume imaging, z-focus was adjusted after 30 minutes, and imaging proceeded for 16 hours.
In vitro fixation and fate analysis:
following live imaging capture, acquisition was paused (to maintain x,y,z positions), the culture plate was removed, media was removed, and cells were fixed in 4% PFA for 20 minutes at 4°C. Cells were washed 3x in 1x PBS for 2 min each. Cells were permeabilized in 0.1% Triton for 10 minutes. Cells were washed 2x in 1x PBS 2 min each, then blocked in 3% BSA in 1x PBS for 30 minutes. Cells were then incubated with primary antibodies diluted in 3% BSA/1x PBS for 1–2 hours at room temperature. Cells were washed 3x with 1x PBS 5 minutes each. Cells were incubated in secondary antibody with DAPI for 20 minutes at room temperature, then washed 3x in 1x PBS 5 minutes each, finally adding 500 μL PBS to each well prior to image acquisition. The plate was then placed back into the incubation chamber on the microscope, allowed to equilibrate to chamber temperature for 30 minutes, and z-plane was adjusted for each x,y position. Images were acquired in Zen software. Fixed imaging was then correlated to the last frame of live imaging to call fates for individual daughter cell pairs. The total number of frames a cell was in mitosis was multiplied by 10 to achieve mitosis duration. This was correlated to cell fates of daughters and data compiled in Microsoft Excel. Data was analyzed in Excel and graphed in Excel or GraphPad Prism.
Differentiation and live imaging of hNPCs:
hNPCs were differentiated from ESC or iPSCs as previously described (31). Briefly, ESCs or iPSCs were cultured until 100% confluent on Matrigel-coated 6-well plates in mTesR media. Cells were differentiated by replacing mTesR media with hNPC differentiation media (1:1 N2/B27 media with dual SMAD inhibitors Dorsomorphin and SB431542). 2.5mL of media was replaced daily for 10 days. Cells were then passaged with Dispase and plated onto 60mm dishes coated with Polyornithine (Sigma) and Laminin (Sigma ) in 1mL of N2/B27 with Dorsomorphin, SB431542, and ROCK inhibitor (Y-27632, StemCell Technologies. The next day, media was replaced with N2/B27 media. Cells were maintained in this medium until formation of neural rosettes was observed, then media was supplemented with bFGF for up to 4 days. bFGF was removed around day 18–20, and cells were cultured an additional 2 days. Cells were passaged with accutase and plated in N2/B27 media onto 60mm plates coated with poly-ornithine and laminin. Cells were passaged with accutase at 1:4 ratio, maintaining cultures around 50–80% confluence. For live imaging, cultures between differentiation days 24–36 were plated onto poly-ornithine/laminin coated MatTek 24-well glass-bottom plates.
Results
A paradigm for in vivo delay of progenitor mitosis
Increased mitotic length of embryonic neural progenitors is associated with microcephaly, yet the underlying mechanisms by which mitosis duration impairs neurogenesis are poorly understood. Further, whether there prolonged progenitor mitosis alters cell fate in vivo is unknown. To address this gap, we developed an in vivo experimental paradigm to acutely lengthen mitosis duration of progenitors in the developing mouse cortex (Figure 1A).
To acutely prolong progenitor mitosis, we injected the small molecule STLC (s-trityl-l-cysteine) directly into the ventricle of the developing cortex at E14.5. STLC specifically and potently inhibits activity of the kinesin Eg5, which is critical for bipolar spindle formation (32). Importantly, STLC application reversibly arrests cells in prometaphase and has been widely used to induce prometaphase delay in immortalized cell lines without affecting other cell cycle stages. Furthermore, our previous studies showed that acute STLC treatment of embryonic brain slices or primary progenitor cultures was sufficient to prolong mitosis without inducing non-specific cell death or aneuploidy (11).
In an asynchronous dividing population, we predicted that injection of STLC would result in a buildup of mitotic cells at the ventricle in a dosage-dependent fashion. To test this, we first optimized STLC drug dosage to induce a short mitotic delay in vivo. For this, we injected either DMSO or STLC into the ventricle of E14.5 embryos using volumes of 0.8 μL at a starting concentration of 80 μM (Figure 1). We then assessed the kinetics of mitosis by quantifying mitotic cells at 1, 2, 3, and 4 hours (h) following injection of 0.8u μL 80 μM STLC (Figure 1B). To quantify prometaphase ventricular cells we stained sections for phosphohistone H3 (PH3) expression and DAPI. We specifically measured progenitors dividing at the ventricle (presumed RGCs) which were PH3+ and showed condensed DNA, characteristic of prometaphase (11) (Figure 1B). Cells were quantified over a 100 μm wide radial column (Figure 1C). In STLC treated brains, the number of prometaphase RGCs at the ventricle significantly increased 2h after injection, then largely receded by 3h and 4h, when the fraction of ventricular progenitors in mitosis had returned to control levels. In addition to measuring prometaphase we extended our analysis to all mitotic stages by quantifying cells for PH3 expression and presence of condensed DNA (Figure S1C). This revealed a peak of mitosis at 2h which was sustained at 3h, relative to control, Importantly, at 3h, this likely demarcates PH3+ cells which are transiting anaphase (Figure 1A, yellow arrowheads). Thus, the number of ventricular RGCs in mitosis increased at 2h and 3h after injection and resolved to normal rates at 4 h.
To reinforce if this was an appropriate concentration at which to use STLC, we tested higher dosages and measured mitosis at 4h. Notably the number of prometaphase cells was significantly higher with increasingly higher STLC concentration, indicating a correlated dosage-response to the Eg5 inhibitor in vivo (Figure S1A,B). Mitotically-delayed cells were evident both apically at the ventricle and basally in the higher concentrations. While these higher concentrations provide a more potent delay of mitosis, they may be less physiologically relevant than the delay induced with 80 μM. Thus, for remaining studies we opted to use 80 μM. These data establish a reproducible paradigm to acutely and reversibly extend the length of mitosis, and specifically prometaphase, in RGCs of the in vivo developing mouse cortex.
Acute mitotic delay of RGCs alters progenitor and neuronal fate of progeny in vivo
Having established an assay to acutely and reversibly delay RGCs in mitosis, we next aimed to examine the effects of this acute cell cycle alteration. To label mitotic RGCs and their progeny we employed FlashTag, also termed CFSE (carboxyflourescin succinimidyl ester). Previous studies have demonstrated the utility of FlashTag in the developing cortex for labeling RGCs adjacent to the ventricle, and in their progeny (33, 34). Importantly, because RGCs at the ventricle are largely in G2/M, this allows us to label mitotic RGCs. Indeed, we were able to recapitulate utility of this labeling paradigm (Figure 2A).
With this dual mitotic delay and labeling paradigm, we then sought to assess the direct effects of prolonged mitosis on RGCs in vivo (Figure 2B). Our prior ex vivo and in vitro studies indicate that prolonged progenitor mitosis directly alters cell fate (11). Thus, we hypothesized that mitotic delay in vivo would alter the neuronal and progenitor fates of progeny in the cortex. In the E14.5 cortex, RGCs cycle about 19 h on average, and divide either symmetrically or asymmetrically to produce RGCs, IPs, and excitatory neurons (8, 35). We unilaterally injected STLC or DMSO (control) along with FlashTag into the cortices of E14.5 embryos. We then quantified the fate of FlashTag+ (FT) daughter cells 16 h after injection, reasoning this would allow us to examine the immediate progeny of delayed and labeled progenitors. Colocalization of FT with markers for RGCs (Pax6) and IPs (Tbr2) was quantified. At 16 h post-injection, in control FT+ progeny, about 50% of cells were Pax6+ (Figures 2C,D). Compared to DMSO, in STLC treated brains, there was a significant 24.6% decrease in FT+Pax6+ RGCs (p=0.01). In contrast, there was no significant change in newborn Tbr2+ IP number between control and STLC conditions (p=0.24)(Figures 2E,F). These data indicate that mitotic delay in vivo preferentially reduces the number of RGCs, without having a striking impact upon IP number.
Given the significant reduction of RGC number following a prolonged progenitor mitosis we next asked if there was a concomitant increase in newborn daughter neurons. To monitor newborn neurons, we quantified the expression of NeuroD2, a neurogenic transcription factor (36). NeuroD2 is expressed in the VZ, SVZ, and CP soon after neurons are born. In control brains about 60% of FT+ newborn progeny were NeuroD2 positive (Figures 2G,H). The proportion of daughter cells expressing NeuroD2 showed a slight, but significant 1.14-fold increase following prolonged prometaphase (p<0.0001). This increase was especially evident in rostral regions of the developing cortex (Figure S1D). Low expression of NeuroD2 can also label Tbr2+ IPs. However, we observed no change in Tbr2+ number following prolonged prometaphase, and thus posit that increased number of NeuroD2+ cells reflect more newborn neurons.
We aimed to further probe this possibility and discriminate between NeuroD2+ proliferative versus differentiated daughter cells. To assess the number of newborn neurons following prolonged mitosis, we labeled proliferative daughters using cumulative EdU injection every 2 hours, starting 8 hours after injection of either STLC or DMSO and FT (Figures 2I, J). With this approach, any FT+ daughters exiting the cell cycle would be EdU−, while proliferative progeny would be EdU+. We quantified the proportion of NeuroD2+EdU-FT+ cells that exited the cell cycle 26 h post-injection (Figure 2K). Compared to control, in STLC treated brains, there was a significant increase in cell cycle exit of NeuroD2+ cells (compare 32% to 44%, respectively, p=0.035). Thus, prolonged mitosis of RGCs alters neurogenesis in vivo, resulting in fewer progenitors and more neurons. Notably these phenotypes of skewed proliferative to neurogenic fates are commonly seen in microcephaly disease models (18–24).
Given the pro-neurogenic outcome linked to increased mitosis duration, we next asked whether acute mitotic delay would result in a longer-term impact upon cortical fates, by quantifying laminar markers 72 h after injection. In order to discriminate direct and indirect progeny derived from delayed progenitors, we performed cumulative EdU labeling following either STLC or DMSO injection (37) (Figure S2A). We quantified SATB2 and RORβ as these primarily mark neurons of layers II/III and IV, respectively, which are generated at or after E14.5, when mitotic delay was induced (38). Although we noted slight trends toward more upper layer neurons, these were not quite significant (Figures S2B,C). This suggests that an acute lengthening of mitosis in RGCs in this paradigm is not sufficient to dramatically alter lamination patterns of cortical neurons.
Acute mitotic delay of RGCs independently increases apoptosis of newborn progeny in vivo
Another phenotype commonly linked to microcephaly is apoptosis. Hence, we asked whether cell death of daughter cells was evident following prolonged mitosis of RGCs. Analysis of brains 8 hours after STLC injection revealed apoptotic-like puncta evident in FT+ cells, but not in controls (Figure 3A). To monitor apoptosis, we performed immunostaining for cleaved-Caspase 3 (CC3) at 8h and 16h after injection of DMSO or STLC (Figure 3A). 8h post-injection, CC3 expression was significantly increased in 22% of FlashTag+ daughter cells in STLC compared to control (p=0.0021) (Figure 3B). CC3 expression was highly specific to FT+ cells (92.2%+/−2.7%)(Figure 3C). This indicates that STLC injection does not cause a non-cell autonomous increase in apoptosis in the tissue, similar to that seen in our prior studies (11). By 16 h after prolonged mitosis, CC3+FT+cells were inconsistently evident in STLC brains, and was not significantly different than control. However, in STLC-treated brains, the average number of apoptotic progeny was significantly different between 8 and 16 h. These data indicate that apoptosis of newborn progeny is high 8 h after injection. Based upon the kinetics of mitotic delay (Figures 1, S1) this indicates that apoptosis is evident in progeny which are estimated to be on average about 5 hours old.
One possible explanation for reduced progenitor number and increased neuron number 16 h after injection is selective cell death of progenitors. To assess this, we quantified the fraction of CC3 cells which expressed a neuronal marker. Given that apoptotic CC3+ cells lose nuclear structure we used a cytoplasmic neuronal marker, Tuj1. 16 h after injection, in the small number of apoptotic cells present, we quantified similar fractions of Tuj1 positive and Tuj1 negative FT+ progeny (Figures 3D, E). This suggests that apoptosis affects progenitors and neurons equivalently at a time point when cell fate is altered. We could not quantify the fates of cells that may have died earlier given variable onset of fate marker expression. Nevertheless, these results are in line with the finding that prolonged mitosis directly promotes neuron generation, reduces progenitor production, and causes excess apoptosis in progeny.
How does apoptosis affect the overall composition of progeny following prolonged progenitor mitosis? We further probed the distinct fates of apoptosis and differentiation by measuring the impact of prolonged mitosis in a Bax−/− model. These models are valuable for eliminating apoptosis, especially that which occurs during normal development (39). Thus, we used this model to ask if it could eliminate apoptosis associated with prolonged mitosis. We repeated either DMSO or STLC injections and collected brains for immunofluorescence analysis 16 h later. There was a significantly reduced fraction of Pax6+FT+ cells in Bax−/− animals compared to DMSO-treated Bax−/− mice (Figure 3F). Concomitantly, there was an increased proportion of NeuroD2+ daughter cells from progenitors stalled in prometaphase (Figure 3G). While the relative fold changes in Neurod2+ cells were similar in WT and Bax−/−, the Pax6+ reduction following STLC was less striking in the Bax mutant compared to control (Figure 3H). We noted variable apoptosis in the Bax−/− brains 16h after STLC treatment, similar to that seen in WT brains. Thus, it is possible that Bax does not completely eliminate apoptosis, perhaps due to redundancy with other apoptotic regulators. Nonetheless, the quantification of equivalent apoptosis in progenitors and neurons suggests that the increase in neurons is not entirely explained by selective progenitor apoptosis.
Prolonged prometaphase alters the transcriptome of mitotic progenitors and their progeny
Having established that RGC mitotic delay alters the differentiation state and survival of progeny, we sought to understand the cascade of molecular events which accompany cell fate changes. Specifically, we asked what are the downstream alterations in the transcriptome following an acute prolonged progenitor prometaphase? We thus took advantage of our model system for the ability to assess gene expression changes as a result of a single delay event. Towards this, we injected E14.5 brains with STLC or DMSO, followed by injection of FT two hours later (Figures 4A, S4, see methods). This paradigm was used in order to label populations of RGCs at their peak of prometaphase delay (Figure 1) and label their direct progeny (Figure 4B). Importantly, this paradigm recapitulated the results observed previously with co-injection of FlashTag and STLC (compare Figure 2 to S4B). Further, this sequential injection paradigm resulted in higher fold cell fate alterations (Figure S4B).
We isolated cortices either immediately after FT injection to enrich for mitotic RGCs (+2h) or several hours later to enrich for newborn progeny (+9h). We then performed FACS, collecting 20,000 cells/condition or 40,000 cells/condition for +2 and +9h respectively (Figures 4B, C). Cells were sorted based upon FT signal to enrich relatively equivalent cell populations using a 5% threshold for control and 1% for STLC (Figures S3A, B). Using propidium iodide staining we assessed DNA content as a proxy for cell cycle stage (Figures 4B, C). Of those cells derived from either STLC or DMSO brains at +2h about 50% had a 4N DNA content (indicative of G2/M) and about 50% had a 2N DNA content (indicative of G0/G1/S). Given the timing of mitotic delay, an important caveat of this approach is that the control would be expected to have later G1 stages versus earlier stages in STLC. We then carried out transcriptome analysis in triplicate at each timepoint. With this we obtained >5×106 reads/sample. We noted that between time points there was a significant increase in neurogenic transcripts in WT brains, correlating with increased differentiation of newborn daughter cells. This reflects the utility of our assay in labeling progenitors and newborn progeny (Figure S3C, Table S1).
At +2h, we detected differential expression of a small number of transcripts including 221 transcripts (q<0.1); or 167 transcripts with a stricter statistical cutoff (q<0.05) (Figures 4E, F, Table S1). Of these, the majority of transcripts were downregulated (e.g. 100 in q<0.05). Gene set enrichment analysis (GSEA) (cutoff of FDR<0.1, fold change +/−2) revealed several differentially expressed pathways (Table S2). This included several categories related to cell cycle including DNA replication, G2/M transition and G1. We also noted enrichment for DNA damage pathways, including several genes assigned to “S phase enrichment”.
At +9h, we detected less differential expression which included changes in 63 transcripts (q<0.1) or 41 transcripts (q<0.05) (Figures 4E,G). Similar to +2h, the majority of transcripts were upregulated (e.g. 29 in q<0.05) (Figure 4G). Gene set enrichment analysis (GSEA) (cutoff of FDR<0.1, fold change +/−2) revealed several differentially expressed pathways including those linked to cell cycle and translation (Table S2). Strikingly, at +9h, the P53 pathway was significantly enriched in the upregulated transcript list. This overlaps with multiple studies which have discovered upregulation of P53 targets, including irradiation-induced microcephaly of the embryonic brain (40, 41).
To validate these findings qPCR was performed on transcripts from the +2h and +9h timepoints. For the +2h timepoint we assessed levels of Neurog1 and Tcf19, two transcripts linked to differentiation and showing >2 fold change from RNA-seq. Both were significantly upregulated in STLC-treated brains relative to control, consistent with the transcriptomic findings (Figure 4H). For the +9h timepoint, we measured levels of Cdkn1a (P21) and Ccng1, which are known P53 targets. Both transcripts showed potent increases in STLC versus control, also consistent with results from RNA-seq at this timepoint (Figure 4H).
In sum, transcriptome analysis of STLC-delayed RGCs and their progeny point to differential gene expression changes in just a small fraction of transcripts expressed in the E14.5 cortex, indicating that overall gene expression is largely unaffected. This provides a valuable and manageable list of transcripts which may be relevant for cell fate specification in response to mitotic delay.
P53 is a major signaling pathway altered following RGC mitotic delay in vivo
The P53 pathway was strongly upregulated in newborn progeny following mitotic delay. We thus analyzed expression of stable nuclear P53 in brains following either DMSO or STLC injection, at +4, +6, and +8h following injection of either DMSO or STLC (Figures 5A, B). At all timepoints, P53 levels were significantly elevated in FT+ positive progeny, showing peak elevation 6h after injection (Figure 5D). P53 expression 8h after injection was evident in 38% of FT+ daughter cells, when daughters are ~5 h old (Figure 5C). Importantly, P53 expression was also highly specific to FT+ cells 8h after injection (91.5%+/−2.4%, n=3) (Figure 5D). We thus asked if p53 loss would impact cell fate changes associated with prolonged mitosis. We generated Emx1-Cre; p53lox/lox mice and repeated experiments to prolong mitosis by acute injection of STLC or DMSO followed by FT 2 hours later and harvesting brains at +16h. In the absence of p53 we noted no significant difference in Pax6 or Neurod2+ cells between DMSO and STLC treated p53 mutant brains (Figure S4C). These experiments suggest that with in our in vivo paradigm, p53 signaling is activated and may influence daughter cells born from mitotically delayed progenitors.
Given the increase in P53 signaling and apoptosis following prolonged mitosis, we sought to understand upstream mechanisms triggering this cascade of events. One of the pathways associated with increased p53 signaling is DNA damage response, which was also upregulated in our transcriptome analysis. Our previous studies in vitro showed that DNA damage accumulates following mitotic delay but it was unclear at which stage this occurred (11). We thus assessed DNA damage in progenitors during prolonged prometaphase in vivo. For this we immunostained tissue sections with ɣH2AX, a marker of double strand DNA breaks (Figure 5E). FT+ and - RGCs were quantified 2 h and 3h after STLC or DMSO injection, in order to assess ɣH2AX levels in cells transiting mitosis. There was no difference in ɣH2AX signal in either G2 or prometaphase cells, indicating that DNA damage does not accumulate at the time of delay (Figures 5F,G). In contrast, quantification of ɣH2AX+ cells in metaphase and anaphase/telophase stages showed significant increases following STLC injection relative to the control (Figures 5H,I). These experiments demonstrate that prometaphase delay in vivo results in increased DNA damage during metaphase and continuing into telophase.
Two pathways that are activated by DNA damage are ATM and ATR. We examined activation of the ATR pathway, using an antibody against phosphorylated ATRS345. 4 h after STLC injection we quantified higher pATR levels, suggesting that newborn progeny have activation of the ATR pathway (Figure S5A,B). We thus asked whether ATR activation explains altered cell fate following prolonged prometaphase. To inhibit ATR signaling, we used two well characterized small molecules AZ20 and VE821(42, 43). We first established conditions in which the drugs effectively inhibited ATR, as monitored by western blot analysis. AZ20 or VE821 inhibition of ATR decreased pChk1 phosphorylation in mouse NIH3T3 cells (Figure S5C,D), and in primary cultures derived from E14.5 cortices (Figure S5E,F). To assess the impact of ATR signaling on cortical fates, we employed in vitro live imaging of progenitors, an approach we have used to live image single RGCs and their daughter cells (11, 44). Primary cells were live imaged with co-incubation of STLC or DMSO and select ATR inhibitors AZ20 and VE821. After 3 h, STLC was washed out, and cells progressed through mitosis in the presence of ATR inhibitors for 3 h, after which time, cells were allowed to divide in normal media. We examined fates of daughter cells at E13.5 from either delayed (STLC treated, >50 min in mitosis) or not delayed (DMSO treated) progenitors cultured with AZ20 (1μM, control) or VE821 (160nM) (Figures S5G, H). After 18h we stained for cell fate markers Sox2, NeuroD2, and Tuj1, and apoptosis. By quantifying individual fate events, we noted no difference in either neurogenic divisions or viability regardless of whether STLC-delayed progenitors had ATR activity or not. This suggests that ATR activation does not explain apoptosis and cell fate changes following progenitor prolonged mitosis.
Prolonged mitosis alters fate of human neural progenitors
Our findings, together with previous studies, demonstrate that prolonged mitosis alters cell fate of mouse neuronal progeny in vivo, ex vivo and in vitro. Finally, to further probe the relevance of prolonged mitosis for altered cell fate we asked whether human neural progenitor cells (NPCs) show the same sensitivity to mitosis duration. We used human embryonic stem cells (ESCs) to generate cortical NPCs. These NPCs were plated onto poly-ornithine/laminin coated 24-well plates in media containing either DMSO (control) or STLC to induce prolonged mitosis. Progenitors were then imaged live, over a 24-hour period, and DMSO or STLC was washed off after the first 4 hours (Figures 6A,B). We first quantified NPC mitosis duration, which significantly increased following STLC treatment (Figure 6C). The vast majority of control NPCs proceeded to complete division within 50 minutes (Figure 6D). Indeed, the average mitosis length for cells dividing in the STLC condition was 144 minutes, and encompassed a range of mitosis lengths, from 40 to over 250 minutes.
Fixed cells were then assessed for cell fate changes associated with mitotic delay. We quantified progeny expressing the neuronal marker Tuj1, and cell fates were retrospectively assigned to parental progenitors. Only in the delayed cohort did we observe increased neurogenic divisions and fewer proliferative divisions (Figure 6E). In contrast, non-delayed human NPCs showed no difference in the neurogenic divisions, compared to control (Figure 6E). This reinforces that STLC acts upon cell fate by delaying NPCs in mitosis, and does so with specificity. In the STLC conditions, however, there was an increased proportion of asymmetric neurogenic divisions compared to control or non-delayed divisions. Accordingly, symmetric proliferative divisions occurred at a significantly lower frequency across different mitotic durations (Figure 6F). This suggests that mitotic delay of human NPCs resulted in significantly more neurogenic divisions at the expense of symmetric proliferative divisions.
We also assayed cell death by quantifying cell morphology after 18h. Apoptosis occurred in 38% of delayed cells and was absent in control divisions or in STLC conditions where divisions were shorter than 50 minutes (Figure 6G). Again, we noted that apoptosis was evident across all mitotic durations with a significant increase that correlated with longer mitosis (Figure 6H). These data demonstrate that human NPCs are subject to fate changes following prolonged mitosis in a similar manner to that seen in mice. Thus, cell biological and developmental programs responding to prolonged mitosis are conserved across species and relevant for human progenitors.
Discussion
Mutations in mitotic regulators result in human microcephaly, which has led to the long-standing model that mitotic defects underlie this disease. However, how perturbations to mitosis impact cortical development is unknown. In this study we demonstrate that acute prolonged mitosis of neural progenitors disrupts neurogenesis in vivo. We establish and characterize a new experimental model for transiently and acutely lengthening progenitor mitosis and for monitoring cell fate. This experimental paradigm allowed us to gain new molecular insights linking prolonged mitosis to microcephaly associated phenotypes. Further, we show that prolonged mitosis of human neural progenitors also alters cell fate, further supporting the value of investigating links between mitosis duration and fate of progeny. This experimental paradigm offers a valuable approach for interrogating mechanisms of microcephaly and related disorders. Further our findings bolster the notion that mitotic delay of progenitors is a relevant mechanism to explain some cases of microcephaly.
Over 25 loci are implicated in the etiology of human microcephaly and a majority of these encode mitotic regulators (15). This rich human genetics has continued to support the long-standing model that mitotic defects in progenitors underlie microcephaly (45–48). Indeed, this notion has also been borne out experimentally. Across a variety of models including mice, organoids and cultured cells, studies have shown that mitotic defects manifest in genetic and ZIKV etiology of microcephaly. However very few studies have assessed how mitosis is perturbed. Most studies use only fixed analyses to show abundant mitotic cells, without normalizing this to progenitor cell cycle or number. In this regard, the use of live imaging, and detailed cell fate tracing in vivo, as we have done here, provides invaluable insights into the nature of mitotic defects. Along with prior studies (11), our data strongly argue that lengthened mitosis in mice in vivo and in human cells has detrimental impacts upon neurogenesis, and specifically abundant apoptosis, imbalance of progenitors and neurons. Importantly these same phenotypes are often evident in microcephaly models which co-present with mitotic delay.
In the future, implementation and variations on the experimental model we developed may be valuable to further understand the pathogenesis of microcephaly and to understand the role of mitotic delay in cell fate. There are several notable strengths of our model, as it allows one to acutely and reversibly delay RGCs in mitosis and then to trace cell fates in vivo. We also showed how this paradigm may be applied to different genetic backgrounds in order to interrogate genetic mechanisms by which prolonged mitosis impacts cell fate. Altogether, the neurogenesis defects we observed following acute mitotic delay in vivo were significant. It is important to note that the experiments in the current study used a single injection of STLC at E14.5, thus focusing the potential phenotypic impact to a single division. While technically challenging, it is exciting to consider how multiple injections across different stages could lead to more potent and significant impact upon cortical lamination and brain size, thus further modeling a ZIKV or genetic microcephaly. Further, our studies also indicate that higher dosages may have a more profound impact on mitosis duration. Likewise, it would be interesting to apply this acute delay model to earlier or later stages of development to evaluate roles for delayed progenitor mitosis in the neuroepithelial to RGC transition or astrocyte differentiation, respectively.
Our findings reinforce prior studies from our lab linking prolonged mitosis to cell fate in ex vivo and in vitro studies. The parallels we observe are remarkable given that the current experiments are all performed in vivo. There were some phenotypic differences; in comparison to slice experiments which led to 1.5 fold more neurons and 1.25 fold less Tbr2 cells, the current experiments resulted in 1.2 fold more neurons, with no change in IPs (11). Cell fate was comparable in DMSO and STLC treated p53 mutants, whereas in comparison, in slices and primary cells, p53 did not rescue cell fate following prolonged mitosis (11). We posit that these phenotypic disparities may be attributable to differences in experimental paradigms. Here, we used a conditional p53 model in which p53 is eliminated in RGCs at E9.5 with analysis at E14.5, whereas prior studies used a germline model and assayed cell fate one day earlier at E13.5. Neuronal fates in both studies were also monitored with different markers and different lineage tracers. Thus, experimental and phenotypic differences may explain the observed results. The BAX−/− embryos retained variable cell death following prolonged mitosis, whereas p53 experiments reinforce that apoptosis is a major cause of altered cell populations in this paradigm. However. quantification of equivalent apoptosis in progenitors and neurons suggests that the increase in neurons is not entirely explained by selective apoptosis.
Mechanisms linking mitotic delay to microcephaly
Our results firm up a role for p53 signaling in microcephaly. Indeed, it is striking that across the literature, p53 is reproducibly identified as a unifying downstream pathway in microcephaly models (11, 41, 49–55). Many genes mutated in primary microcephaly and microcephaly syndromes function in key aspects of mitosis including bipolar spindle formation and centrosome assembly (eg. CEP152, CEP135, WDR62, CDK5RAP2, ASPM, SAS4/CENPJ, CENPE, SAS6). Of these, in particular, loss of Sas4 results in a prolonged mitosis and p53 activation, and these mice have severe microcephaly (56). Thus, from these data and this current work, we argue that prolonged mitosis is a plausible underlying cause of cell fate alterations in microcephaly. Indeed, this suggests that functional classification of microcephaly loci into those that lengthen mitosis, and particularly prometaphase, may be especially prone to the mechanisms described here.
A key question going forward is what are the signals that transduce the response to prolonged prometaphase, activating p53, altering cell fates, and potentiating apoptosis? We found that prolonged mitosis induces DNA Damage in progenitors, followed by detectable pATR expression in newborn progeny, and p53 signaling within hours of cell birth, followed by cell death and altered neurogenic fates. Indeed, DNA Damage, in certain contexts, can result in increased P53 activity and at times has been found to trigger cell cycle checkpoint arrest, or cell cycle exit. Here, we discover that increased DNA damage is upregulated in delayed progenitors, most notably at metaphase and anaphase. However, ATR inhibition did not affect outcomes linked to delay; thus alternative pathways may explain why P53 upregulation occurs in newborn progeny.
A plausible path to discover pathways upstream of p53 is by interrogating molecular changes evident before P53 upregulation. Notably, our transcriptome analysis highlighted a number of candidates whose levels were significantly altered immediately following delay (+2h). Amongst these are several transcripts associated with promoting neurogenesis, such as Neurogenin1, which would be of strong interest to interrogate further. Likewise, other omics-based approaches focused on changes at the protein level may give valuable insights as well into possible translational and post-translational changes at play during mitotic delay.
Taken all together our study further reinforces the notion that prolonged mitosis of progenitors has deleterious impact on the developing brain. This has implications for developmental conditions like human microcephaly where prolonged mitosis is an important phenotypic occurrence.
Supplementary Material
Acknowledgement
We thank members of the Silver lab for insightful comments. We also thank Charlie Sheehan and Carly Newman for technical assistance with mouse husbandry.
Funding sources
This work was supported by the following: NIH R01NS083897, NIH R21NS098176, NIH R01NS110388 (DLS); and NIH F31NS101785 (to AMD).
Footnotes
Statement of Ethics
All mouse procedures were approved by the Duke IACUC and followed international guidelines
Disclosure Statement
The authors have no conflicts of interest.
References
- 1.Sousa AMM, Meyer KA, Santpere G, Gulden FO, Sestan N. Evolution of the Human Nervous System Function, Structure, and Development. Cell. 2017;170(2):226–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kowalczyk T, Pontious A, Englund C, Daza RAM, Bedogni F, Hodge R, et al. Intermediate neuronal progenitors (basal progenitors) produce pyramidal-projection neurons for all layers of cerebral cortex. Cerebral cortex (New York, NY : 1991). 2009;19(10):2439–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mihalas AB, Elsen GE, Bedogni F, Daza RAM, Ramos-Laguna KA, Arnold SJ, et al. Intermediate Progenitor Cohorts Differentially Generate Cortical Layers and Require Tbr2 for Timely Acquisition of Neuronal Subtype Identity. CellReports. 2016;16(1):92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vasistha NA, García-Moreno F, Arora S, Cheung AFP, Arnold SJ, Robertson EJ, et al. Cortical and Clonal Contribution of Tbr2 Expressing Progenitors in the Developing Mouse Brain. Cerebral Cortex. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takahashi T, Nowakowski RS, Caviness VS Jr.. The cell cycle of the pseudostratified ventricular epithelium of the embryonic murine cerebral wall. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1995;15(9):6046–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McConnell SK, Kaznowski C. Cell cycle dependence of laminar determination in developing neocortex. Science (New York, NY). 1991;254(5029):282–5. [DOI] [PubMed] [Google Scholar]
- 7.Haydar TF, Ang E, Rakic P. Mitotic spindle rotation and mode of cell division in the developing telencephalon. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(5):2890–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arai Y, Pulvers JN, Haffner C, Schilling B, Nüsslein I, Calegari F, et al. Neural stem and progenitor cells shorten S-phase on commitment to neuron production. Nature Communications. 2011;2:154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lange C, Huttner WB, Calegari F. Cdk4/cyclinD1 overexpression in neural stem cells shortens G1, delays neurogenesis, and promotes the generation and expansion of basal progenitors. Cell stem cell. 2009;5(3):320–31. [DOI] [PubMed] [Google Scholar]
- 10.Pilaz L-J, Patti D, Marcy G, Ollier E, Pfister S, Douglas RJ, et al. Forced G1-phase reduction alters mode of division, neuron number, and laminar phenotype in the cerebral cortex. Proceedings of the National Academy of Sciences. 2009;106(51):21924–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pilaz L-J, McMahon JJ, Miller EE, Lennox AL, Suzuki A, Salmon E, et al. Prolonged Mitosis of Neural Progenitors Alters Cell Fate in the Developing Brain. Neuron. 2016;89(1):83–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Faheem M, Naseer MI, Rasool M, Chaudhary AG, Kumosani TA, Ilyas AM, et al. Molecular genetics of human primary microcephaly: an overview. BMC medical genomics. 2015;8 Suppl 1(Suppl 1):S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gilmore EC, Walsh CA. Genetic causes of microcephaly and lessons for neuronal development. Wiley Interdisciplinary Reviews: Developmental Biology. 2012;2(4):461–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jayaraman D, Bae BI, Walsh CA. The Genetics of Primary Microcephaly. Annu Rev Genomics Hum Genet. 2018;19:177–200. [DOI] [PubMed] [Google Scholar]
- 15.Degrassi F, Damizia M, Lavia P. The Mitotic Apparatus and Kinetochores in Microcephaly and Neurodevelopmental Diseases. Cells. 2019;9(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang W, Yang SL, Yang M, Herrlinger S, Shao Q, Collar JL, et al. Modeling microcephaly with cerebral organoids reveals a WDR62-CEP170-KIF2A pathway promoting cilium disassembly in neural progenitors. Nat Commun. 2019;10(1):2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lancaster MA, Renner M, Martin C-A, Wenzel D, Bicknell LS, Hurles ME, et al. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501(7467):373–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marthiens V, Rujano MA, Pennetier C, Tessier S, Paul-Gilloteaux P, Basto R. Centrosome amplification causes microcephaly. Nature cell biology. 2013;15(7):731–40. [DOI] [PubMed] [Google Scholar]
- 19.Silver DL, Watkins-Chow DE, Schreck KC, Pierfelice TJ, Larson DM, Burnetti AJ, et al. The exon junction complex component Magoh controls brain size by regulating neural stem cell division. Nature Neuroscience. 2010;13(5):551–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Asami M, Pilz GA, Ninkovic J, Godinho L, Schroeder T, Huttner WB, et al. The role of Pax6 in regulating the orientation and mode of cell division of progenitors in the mouse cerebral cortex. 2011;138(23):5067–78. [DOI] [PubMed] [Google Scholar]
- 21.Gruber R, Zhou Z, Sukchev M, Joerss T, Frappart P-O, Wang Z-Q. MCPH1 regulates the neuroprogenitor division mode by coupling the centrosomal cycle with mitotic entry through the Chk1-Cdc25 pathway. Nature cell biology. 2011;13(11):1325–34. [DOI] [PubMed] [Google Scholar]
- 22.Lizarraga SB, Margossian SP, Harris MH, Campagna DR, Han A-P, Blevins S, et al. Cdk5rap2 regulates centrosome function and chromosome segregation in neuronal progenitors. 2010;137(11):1907–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McIntyre RE, Lakshminarasimhan Chavali P, Ismail O, Carragher DM, Sanchez-Andrade G, Forment JV, et al. Disruption of mouse Cenpj, a regulator of centriole biogenesis, phenocopies Seckel syndrome. PLoS genetics. 2012;8(11):e1003022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yingling J, Youn YH, Darling D, Toyo-oka K, Pramparo T, Hirotsune S, et al. Neuroepithelial Stem Cell Proliferation Requires LIS1 for Precise Spindle Orientation and Symmetric Division. Cell. 2008;132(3):474–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lazear HM, Diamond MS. Zika Virus: New Clinical Syndromes and Its Emergence in the Western Hemisphere. Journal of Virology. 2016;90(10):4864–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li H, Saucedo-Cuevas L, Yuan L, Ross D, Johansen A, Sands D, et al. Zika Virus Protease Cleavage of Host Protein Septin-2 Mediates Mitotic Defects in Neural Progenitors. Neuron. 2019;101(6):1089–98 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li R, Sun L, Fang A, Li P, Wu Q, Wang X. Recapitulating cortical development with organoid culture in vitro and modeling abnormal spindle-like (ASPM related primary) microcephaly disease. Protein Cell. 2017;8(11):823–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Souza BS, Sampaio GL, Pereira CS, Campos GS, Sardi SI, Freitas LA, et al. Zika virus infection induces mitosis abnormalities and apoptotic cell death of human neural progenitor cells. Sci Rep. 2016;6:39775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wolf B, Diop F, Ferraris P, Wichit S, Busso C, Misse D, et al. Zika virus causes supernumerary foci with centriolar proteins and impaired spindle positioning. Open Biol. 2017;7(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sheehan CJ, McMahon JJ, Serdar LD, Silver DL. Dosage-dependent requirements of Magoh for cortical interneuron generation and survival. Development. 2020;147(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shi Y, Kirwan P, Livesey FJ. Directed differentiation of human pluripotent stem cells to cerebral cortex neurons and neural networks. Nature protocols. 2012;7(10):1836–46. [DOI] [PubMed] [Google Scholar]
- 32.Skoufias DA, DeBonis S, Saoudi Y, Lebeau L, Crevel I, Cross R, et al. S-trityl-L-cysteine is a reversible, tight binding inhibitor of the human kinesin Eg5 that specifically blocks mitotic progression. The Journal of biological chemistry. 2006;281(26):17559–69. [DOI] [PubMed] [Google Scholar]
- 33.Telley L, Govindan S, Prados J, Stevant I, Nef S, Dermitzakis E, et al. Sequential transcriptional waves direct the differentiation of newborn neurons in the mouse neocortex. Science (New York, NY). 2016:1–8. [DOI] [PubMed] [Google Scholar]
- 34.Govindan S, Oberst P, Jabaudon D. In vivo pulse labeling of isochronic cohorts of cells in the central nervous system using FlashTag. Nat Protoc. 2018;13(10):2297–311. [DOI] [PubMed] [Google Scholar]
- 35.Namba T, Huttner WB. Neural progenitor cells and their role in the development and evolutionary expansion of the neocortex. Wiley Interdisciplinary Reviews: Developmental Biology. 2016:1–16. [DOI] [PubMed] [Google Scholar]
- 36.Olson JM, Asakura A, Snider L, Hawkes R, Strand A, Stoeck J, et al. NeuroD2 is necessary for development and survival of central nervous system neurons. Dev Biol. 2001;234(1):174–87. [DOI] [PubMed] [Google Scholar]
- 37.Vitali I, Fievre S, Telley L, Oberst P, Bariselli S, Frangeul L, et al. Progenitor Hyperpolarization Regulates the Sequential Generation of Neuronal Subtypes in the Developing Neocortex. Cell. 2018;174(5):1264–76 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Greig LC, Woodworth MB, Galazo MJ, Padmanabhan H, Macklis JD. Molecular logic of neocortical projection neuron specification, development and diversity. Nature reviews Neuroscience. 2013;14(11):755–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Southwell DG, Paredes MF, Galvao RP, Jones DL, Froemke RC, Sebe JY, et al. Intrinsically determined cell death of developing cortical interneurons. Nature. 2012;491(7422):109–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Quintens R, Verreet T, Janssen A, Neefs M, Leysen L, Michaux A, et al. Identification of novel radiation-induced p53-dependent transcripts extensively regulated during mouse brain development. Biology Open. 2015;4(3):331–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mao H, McMahon JJ, Tsai Y-H, wang Z, Silver DL. Haploinsufficiency for Core Exon Junction Complex Components Disrupts Embryonic Neurogenesis and Causes p53-Mediated Microcephaly. PLoS genetics. 2016;12(9):e1006282–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Foote KM, Blades K, Cronin A, Fillery S, Guichard SS, Hassall L, et al. Discovery of 4-{4-[(3R)-3-Methylmorpholin-4-yl]-6-[1-(methylsulfonyl)cyclopropyl]pyrimidin-2-y l}−1H-indole (AZ20): a potent and selective inhibitor of ATR protein kinase with monotherapy in vivo antitumor activity. J Med Chem. 2013;56(5):2125–38. [DOI] [PubMed] [Google Scholar]
- 43.Prevo R, Fokas E, Reaper PM, Charlton PA, Pollard JR, McKenna WG, et al. The novel ATR inhibitor VE-821 increases sensitivity of pancreatic cancer cells to radiation and chemotherapy. Cancer Biol Ther. 2012;13(11):1072–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pilaz L-J, Silver DL. Live imaging of mitosis in the developing mouse embryonic cortex. Journal of visualized experiments : JoVE. 2014(88):e51298–e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bond J, Roberts E, Mochida GH, Hampshire DJ, Scott S, Askham JM, et al. ASPM is a major determinant of cerebral cortical size. Nature Genetics. 2002;32(2):316–20. [DOI] [PubMed] [Google Scholar]
- 46.Jackson A, Eastwood H, Bell S, Adu J, Toomes C, Carr I, et al. Identification of microcephalin, a protein implicated in determining the size of the human brain. American journal of human genetics. 2002;71(1):136–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kumar A, Markandaya M, Girimaji S. Primary microcephaly: microcephalin and ASPM determine the size of the human brain. J Biosci. 2002;27(7):629–32. [DOI] [PubMed] [Google Scholar]
- 48.Mochida GH. Genetics and biology of microcephaly and lissencephaly. Seminars in pediatric neurology. 2009;16(3):120–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Little JN, Dwyer ND. p53 deletion rescues lethal microcephaly in a mouse model with neural stem cell abscission defects. Hum Mol Genet. 2019;28(3):434–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shi L, Qalieh A, Lam MM, Keil JM, Kwan KY. Robust elimination of genome-damaged cells safeguards against brain somatic aneuploidy following Knl1 deletion. Nat Commun. 2019;10(1):2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.El Ghouzzi V, Bianchi FT, Molineris I, Mounce BC, Berto GE, Rak M, et al. ZIKA virus elicits P53 activation and genotoxic stress in human neural progenitors similar to mutations involved in severe forms of genetic microcephaly and p53. 2016;7(10):e2440–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Breuss M, Fritz T, Gstrein T, Chan K, Ushakova L. Mutations in the murine homologue of TUBB5 cause microcephaly by perturbing cell cycle progression and inducing p53-associated apoptosis. ??? 2016. [DOI] [PubMed] [Google Scholar]
- 53.Breuss M, Fritz T, Gstrein T, Chan K, Ushakova L, Yu N, et al. Mutations in the murine homologue of TUBB5 cause microcephaly by perturbing cell cycle progression and inducing p53 associated apoptosis. Development. 2016:1–28. [DOI] [PubMed] [Google Scholar]
- 54.cacute MM, nchez-Huertas CSa, eacute BT, mez RioGo, Scheel JF, Pacheco S, et al. CEP63 deficiency promotes p53-dependent microcephaly and reveals a role for the centrosome in meiotic recombination. Nature Communications. 2015;6:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Insolera R, Bazzi H, Shao W, Anderson KV, Shi S-H. Cortical neurogenesis in the absence of centrioles. Nature Neuroscience. 2014;17(11):1528–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bazzi H, Anderson KV. Acentriolar mitosis activates a p53-dependent apoptosis pathway in the mouse embryo. Proceedings of the National Academy of Sciences. 2014;111(15):E1491–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.