

Abstract
Lysosomes figure prominently in theories of aging as the proteolytic system most responsible for eliminating growing burdens of damaged proteins and organelles in aging neurons and other long lived cells. Newer evidence shows that diverse experimental measures known to extend lifespan in invertebrate aging models share the property of boosting lysosomal clearance of substrates through the autophagy pathway. Maintaining an optimal level of lysosome acidification is particularly crucial for these anti-aging effects. The exceptional dependence of neurons on fully functional lysosomes is reflected by the phenotypes seen in congenital lysosomal storage disorders, which commonly present as severe neurodevelopmental or neurodegenerative conditions even though lysosomal deficits are systemic. Similar connections are now being appreciated between risk for late age-onset neurodegenerative disorders and primary lysosomal deficits. In diseases such as Alzheimer’s and Parkinson’s, as in aging alone, primary lysosome dysfunction due to acidification impairment is emerging as a frequent theme, supported by the growing list of familial neurodegenerative disorders that involve primary vATPase dysfunction. The additional cellular roles played by intraluminal pH in sensing nutrient and stress and modulating cellular signaling have further expanded the possible ways that lysosomal pH dysregulation in aging and disease can disrupt neuronal function. Here, we consider the impact of cellular aging on lysosomes and how these changes may create the tipping point for disease emergence in major late-age onset neurodegenerative disorders.
Introduction
Aging lysosomes – a critical precondition for late-age onset neurodegenerative diseases
Lysosomes failure is implicated in the etiology of an increasing number of neurodegenerative diseases across the entire age spectrum(Nixon, 2013; Nixon et al., 2008). For decades, the unusually close relationship between lysosomal dysfunction and neurodegeneration has been appreciated for the family of more than 50 inherited Lysosomal Storage Disorders (LSD) arising prenatally or in childhood. Although the disease impact is systemic for most LSDs, the brain and especially neurons are disproportionately affected and most exhibit neurological disease as the most prominent or exclusive clinical feature(Wolfe et al., 2013) In addition, research has now uncovered pathogenic lysosomal dysfunction in major late age onset neurodegenerative disorders, such as AD, PD, FTD, and possibly ALS, which may be primary in some familial forms of the disease. Underscoring this concept is evidence for homozygous mutations of a single gene that cause a congenital LSD but may cause a late onset neurological disorder when in heterozygous form. For example, glucocerebrosidase (GBA) mutations cause the congenital disorder Gaucher’s disease but, in the elderly, cause Parkinson’s disease. Progranulin (PRGN) mutations, depending on gene dosage, cause either ceroid neuronal lipofuscinosis (NCL) in childhood or Frontotemporal Dementia in adults(Ward et al., 2017). Although early familial Alzheimer’s disease (FAD) arises most frequently in the 4th and 5th decades of life, certain mutations of presenilin 1 (PSEN1), which cause primary defects in lysosomal acidification (Lee et.al. 2010), can induce very early onset AD in young adults. In one report, an early onset form of PSEN1 AD exhibited a neuropathological signature of Kuf’s Disease, a form of NCL that usually arises in adolescence or early adulthood (Larner, 2013).
In most late onset neurodegenerative diseases, the causative misfolded mutant protein is produced throughout life yet only accumulates in the adult or aged brain as clinical disease emerges. This delayed appearance and the further evidence that most of these pathogenic proteins are substrates of lysosomes suggests that a significant, if not dominant, determinant of disease onset in some of these disorders is an aging-related decline in lysosomal system efficiency possibly compounding a disease-related compromise of lysosomes. Why the brain is preferentially vulnerable to systemic insults affecting the lysosomal system reflects in part that neurons are post-mitotic and have less active lysosomal exocytic activity than non-neural cells. Removal of waste by dilution during cell division or by exocytosis are therefore not major options for clearance and neurons must rely more on lysosomes to maintain cellular quality control.
Although research on lysosomes in disease states is now advancing at a rapid pace, efforts to understand the crucial declines in lysosomal efficiency due to brain aging have lagged. Here we will review what is known about the impact of aging on lysosomes in and outside of the brain. Although the evidence is still fragmentary, it establishes lysosomal functional declines as key determinants of senescence and of the vulnerability to neurodegenerative disease. A further intriguing theme emerging from research on lifespan and brain disorders, also addressed here, is the extent to which the various insults during aging converge to dysregulate the process of lysosome acidification. This deficit exerts a pervasive influence on both hydrolytic and signaling functions of lysosomes, synergizing with additional disease-related lysosomal deficits.
Autophagy and lysosome function are key determinants of senescence and longevity
Aging of cells and organisms involves a progressive loss of physiological integrity through multiple pathways(Cuervo and Dice, 2000d). Particularly prominent is the deterioration of proteostasis, a cellular network that governs protein life from synthesis to degradation (Douglas and Dillin, 2010). As the burden of misfolded and damaged constituents grows with age, cells face declining degradative capacity and other counterproductive alterations of intracellular proteolytic systems, Effects on the lysosomal system are arguably the most substantial, as further detailed below, while declines in ubiquitin-proteasome activity develop in aging but are generally less marked (DeMartino and Slaughter, 1999; Shibatani and Ward, 1996). Calpain proteinases are actually hyper-activated in many aging tissues and this change is considered an established marker of aging (Glaser et al., 1994; Saito et al., 1993). Because calpains only carry out limited proteolysis (Carafoli and Molinari, 1998; Glaser et al., 1994), increased generation of protein fragments in aged cells can further overburden lysosomes and the UPS. In addition, over-activated calpains may inhibit autophagy(Menzies et al., 2015; Yousefi et al., 2006). Among effects on the cell’s clearance and quality control systems, the greatest impact of aging is on the lysosome’s role in autophagy. Conversely, lysosomes impairment may well be the most influential determinant of senescence onset and lifespan.
Autophagy is a major lysosomal degradative process responsible for clearance and recycling of cellular constituents and is the only pathway available for intracellular turnover of organelles (Figure 1). While constitutively active in most cells, it can be strongly induced by various forms of stress and nutritional deficiency. In autophagy, cellular substrates may be sequestered and delivered to lysosomes via a variety of complementary mechanisms (Figure 1). The lysosome is the only organelle common to all of these autophagy routes, which include macroautophagy, chaperone-mediated autophagy, and microautophagy. Although it has become common to refer to an “autophagic-lysosomal pathway”, it is important to note, especially for the discussion here, that autophagy requires substrate digestion by lysosomes and that autophagy rates depend as much on the efficiency of lysosomal as on autophagy induction and substrate sequestration rates. Importantly for considerations of lysosomes in aging, growing evidence points to declining lysosomal function as the most influential change in the autophagy pathway that lowers the organism’s longevity (although mitophagy declines imply an importance of sequestration failure as well). Indeed, among the most established markers of neuronal aging are lipofuscin granules containing incompletely digested lysosomal substrates. The role of autophagy is especially critical for aging neurons and other post-mitotic cells, which are unable to use mitosis to dilute waste build-up and have low lysosomal exocytosis capability that allows non-neural cells to jettison waste in bulk. Neurons are further challenged by their asymmetry: distal axons and synapses that actively sequester substrates are often at long distances from lysosomes that are located in cell bodies.
Figure 1. Major routes of substrate delivery to lysosomes .
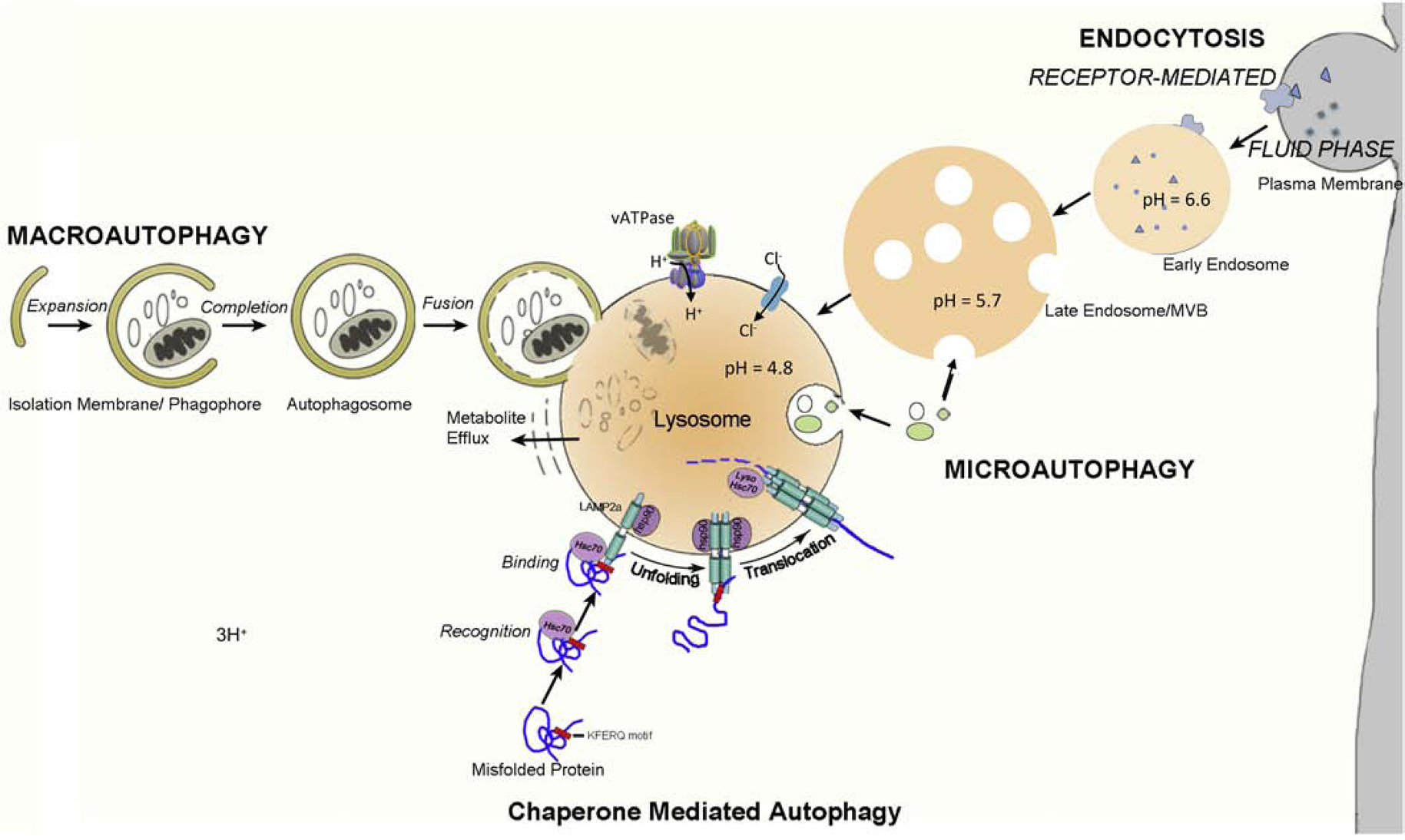
(a) Macroautophagy is characterized by the sequestration of organelles or cytoplasmic constituents targeted for degradation into double-membrane vesicles called autophagosomes. Fully formed autophagosomes fuse with hydrolase-filled lysosomes releasing hydrolases into the lumen of the created autolysosome. Introduction of a proton pump (v-ATPase) induces full acidification of the autolysosomal lumen necessary to activate acid hydrolases. The metabolites resulting from digestion are transported into the cytoplasm and used for synthesis of new macromolecules or for energy. (b) During chaperone-mediated autophagy, proteins carrying the pentapeptide KFERQ-like sequence are recognized by the Hsc70 chaper-one, which then associates with the integral lysosome membrane protein LAMP-2A, triggering its oligomerization. This event leads to the translocation of the bound protein into the lysosome interior through a process that requires Hsc70. (c) Microautophagy involves “bulk” or chaperone-mediated internalization and degradation of cytoplasmic substrates within late endo-some/MVB or lysosomal compartments by a process of membrane invagination followed by membrane scission to release the cargo into the lysosomal lumen for degradation. (d) Heterophagy involves the lysosomal degradation of plasma membrane components and exogenous substrates after they are internalized by receptor-mediated or bulk endocytosis. Selected proteins are sorted to different cellular destinations or recycled to the plasma membrane. Proteins targeted for degradation are trafficked to late endosomes/ MVB, which mature to lysosomes to effect complete degradation.
The dysfunction of autophagy in aging-related neurodegenerative disorders has been extensively reviewed (Boland et al., 2018; Menzies et al., 2017; Nixon, 2016; Nixon, 2013; Scrivo et al., 2018). Although there are remaining questions about the extent to which macroautophagy is induced or impeded at the initial stages of induction, cargo recognition or sequestration, there is a general consensus that the clearance of autophagy substrates through the lysosome is progressively corrupted in Alzheimer’s Disease, and likely in Frontotemporal Dementia and Parkinson’s Disease (Boland et al., 2018; Bordi et al., 2016; Menzies et al., 2017; Nixon, 2013). The activity of CMA, in particular, has been extensively studied and shown to be markedly impaired in multiple neurodegenerative proteinopathies as well as in aging. The decline of CMA in aging involving mainly the declining levels of LAMP2a, the key chaperone for substrate delivery to lysosomes(Cuervo and Dice, 2000b, c), amply reflects the cumulative impact of aging-related processes on this vital function of lysosomes, including the involvement of increased oxidative stress, lipid alterations, and structural damage to the substrates. Experimental disruptions of CMA induce a phenotype of advanced aging in tissues while supra-physiological levels of LAMP2a that enhance CMA efficiency delay the onset of aging phenotypic changes(Kaushik and Cuervo, 2018).
In addition to inefficient proteostasis (Vilchez et al., 2014), contributions to cellular aging include shortening of telomeres (Wright et al., 1996), accumulation of extra-chromosomal DNA (Sinclair and Guarente, 1997) and oxygen free radicals (Harman, 1972), as well as dysregulation of the cell cycle (Afshari and Barret, 1996), signaling by insulin/mTOR, and secretion of proteins (Ivanov et al., 2013). The failure of these aging-associated pathways, in some cases, can be related back to antecedent declines in autophagy. For example, mitochondrial turnover is entirely dependent on autophagy (mitophagy). In aging cells, mitochondria become enlarged and are less efficiently sequestered by mitophagy (Terman et al., 2003) and more vulnerable to acute damage, which can then trigger mitochondrial membrane permeabilization and apoptosis or necrosis. Cell senescence, the state of irreversible cell cycle arrest, is accompanied by progressive nuclear changes including remodeling of chromatin and loss of total histones, which are, in part, lysosome-dependent processes. Autophagy of chromatin released from nuclei may drive senescence by making cell cycle re-entry less likely (Ivanov et al., 2013).
Lysosome acidification – the Achilles heel of aging cells?
The intimate relationship between lysosome activity and cellular aging is reinforced by the evidence that a dozen or so experimental manipulations known to extend lifespan in a wide range of invertebrate organisms share the property of increasing the efficiency of autophagy and, in particular, lysosomes (Chin et al., 2014; Vilchez et al., 2014) (Figure 2). While longevity pathways may involve different upstream triggers, all life-extension mechanisms converge on the autophagy pathway, including, for example, reduced insulin/IGF-1 signaling, mTOR signaling, and mitochondrial respiration, as well as dietary restriction and germline removal. The heightened autophagy activity seen in long-lived animals is required for their longevity(Nakamura and Yoshimori, 2018) while autophagy impairment or lysosomal deficits(Ivy et al., 1984) induce senescence-related changes (Kang et al., 2011; Markaki and Tavernarakis, 2013; Rubinsztein et al., 2011).
Figure 2. Disparate strategies that enhance autophagy or preserve full acidification of lysosomes are able to extend lifespan in lower species.
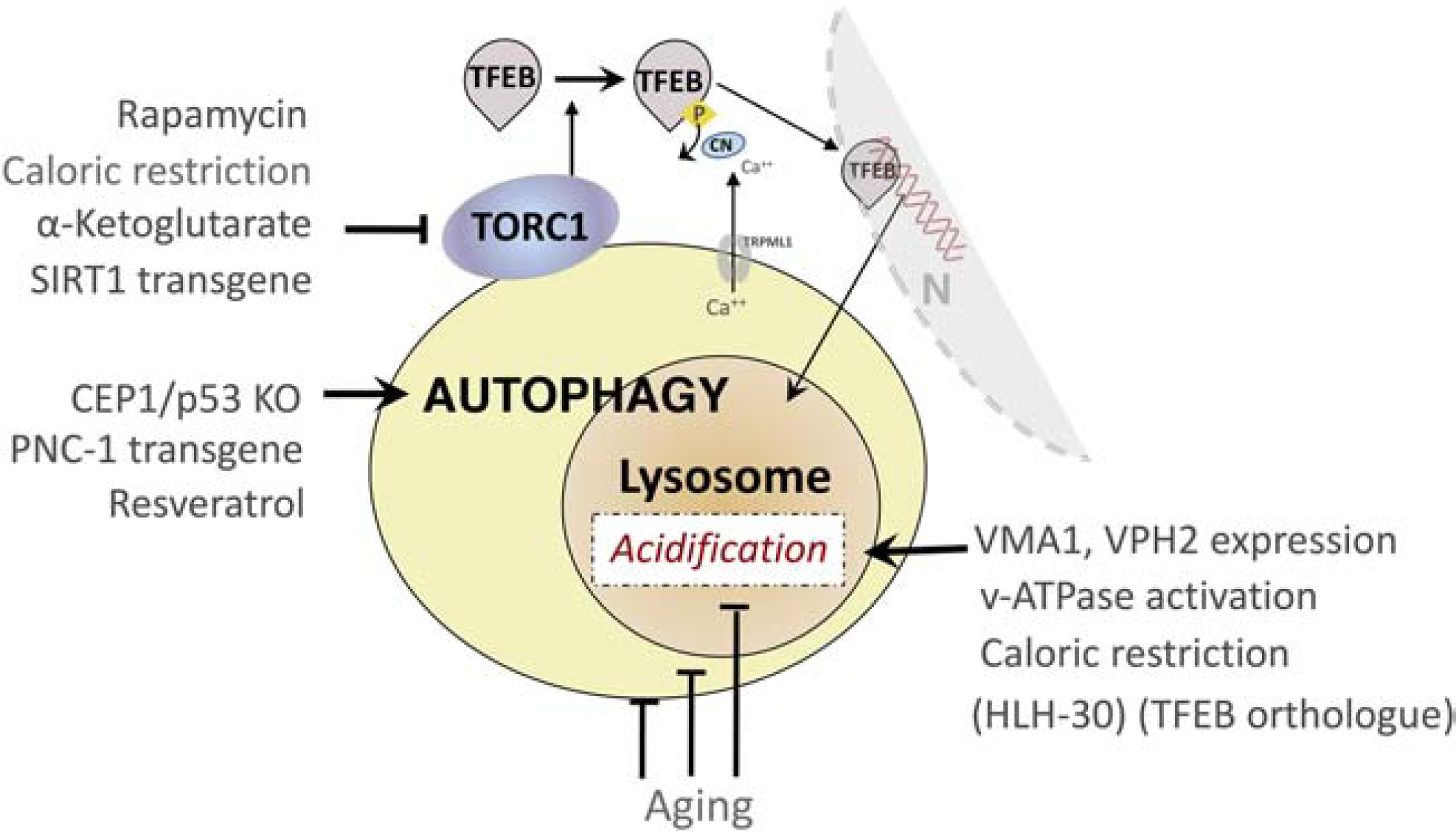
Extensive studies in yeast, C. elegans, flies have identified distinct metabolic pathways modulated by drugs or genetic manipulations that extend lifespan and also share the property of facilitating substrate clearance through autophagy. Many of these pathways are linked to those independently implicated in the evolution of cellular aging, as discussed in this review. Notably, recent evidence from investigations on the vATPase complex establish the special importance of maintaining optimal lysosomal acidification as a key factor mediating the extension of lifespan through autophagy modulation.
The activity of mTOR, a molecular rheostat that balances autophagy and protein synthetic activities, is particularly critical in mediating lifespan extension and is influenced by many different signaling pathways in response to environmental and disease factors (Kenyon, 2010; Zoncu et al., 2011b) . Arguably of greatest importance is the reciprocal signaling between lysosomes and mTOR that binds to the vATPase complex of lysosomes in its inhibited state. mTOR is regulated in part by lysosomal signals that release mTOR enabling it to phosphorylate transcription factors. Among the most important of these factors are TFEB and TFE3, which modulate the expression of genes encoding most lysosomal components and additional components of the autophagosome machinery (Lapierre et al., 2013; Vilchez et al., 2014). TFEB is negatively regulated by mTOR under conditions of changing nutrient supply. mTOR on the lysosome surface senses amino acid supply through several mechanisms, including inside-out signaling through the v-ATPase(Zoncu et al., 2011a) and the lysosomal arginine transporter SLC38A9(Jung et al., 2015; Rebsamen et al., 2015; Wang et al., 2015). Starvation or other stress states inhibit TOR allowing TFEB to be dephosphorylated and translocated into the nucleus where it initiates the transcription of target genes encoding most lysosomal components and additional components of the autophagosome machinery(Lapierre et al., 2013; Vilchez et al., 2014) The activity of v-ATPase via its association with other regulator molecules is vital to the sensing functions of lysosomes: inhibiting-ATPase prevents mTORC1 activation by amino acids(Zoncu et al., 2011a)
Although longevity research has mainly focused on autophagy “flux” (i.e. the complete process of substrate sequestration and digestion), recent studies in yeast have identified lysosomes and specifically the lysosomal proton pump, vATPase as the critical determinants of longevity (Hughes and Gottschling, 2012; Ruckenstuhl et al., 2014; Stephan et al., 2013). When aging yeast cells lose lysosomal acidity, limited lifespan results partly from the inhibition of mitochondrial function(Carmona-Gutierrez et al., 2016). The transport of lipids and possibly nutrients between these organelles involves a protein-tethering complex (vCLAMP) that forms a physical connection between lysosomes and mitochondria(Elbaz-Alon et al., 2014; Honscher et al., 2014). Cells lacking subunits of the v-ATPase or treated with v-ATPase inhibitors have a very short lifespan and exhibit a range of mitochondrial impairments(Dimmer et al., 2002; Hughes and Gottschling, 2012; Merz and Westermann, 2009; Schleit et al., 2013). In the C. elegans model, overexpression of HLH-30, a homolog of TFEB, is sufficient to extend lifespan similarly to pro-longevity signaling by inhibiting insulin/IGF-1 signaling, mitochondrial respiration, TOR signaling, protein translation or germline removal, and is required for longevity(Hughes and Gottschling, 2012; Ruckenstuhl et al., 2014). Acidification of the vacuole, the metazoan equivalent of the lysosome, also seems to be critical in mediating lifespan-extending effects of caloric restriction(Hughes and Gottschling, 2012; Molin and Demir, 2014) and methionine restriction(Ruckenstuhl et al., 2014). Exercise-induced autophagy requires lysosomal Ca2+ release through the lysosomal calcium channel mucolipin 1 (TRPML1) and the subsequent activation of calcineurin, which in turn promotes nuclear translocation of TFEB(Medina et al., 2015). Interestingly, lysosomal exocytosis is modulated by Ca2+ and TFEB(Medina et al., 2011), both of which have regulatory functions during aging. The transcription of most subunits of the v-ATPase complex is also regulated by TFEB(Sardiello et al., 2009) making it likely that the positive effects of TFEB induction in these models involves upregulated v-ATPase function and greater lysosomal acidification. Lysosomal function is also intricately connected to regulation of the PKA pathway(Bond and Forgac, 2008; Dechant et al., 2010; Hlavata et al., 2008) which influences lifespan. PKA pathway stimulation facilitates the ER to lysosome delivery of the ClC7 chloride channel (Lee et al 2020, in press) and its effector. OSTM1(Majumdar et al., 2011). The import of chloride ions promotes lysosomal acidification by reducing the electrogenic gradient caused by vATPase-mediated proton import.
Animal models in which the vATPase complex has been modulated highlight the potential impact of chronic aging related impairments of lysosomal acidification on the aging brain, revealing that v-ATPase defects are sufficient to induce neuropathological phenotypes similar to those observed in AD and PD. A loss-of-function mutation of a v-ATPase subunit in Drosophila induces a phenotype exhibiting failed protein degradation and aging-dependent neurodegeneration(Williamson and Hiesinger, 2010). Loss of the V0a1 subunit, in particular, increases the susceptibility of neurons to abeta- and tau-induced toxicity(Williamson and Hiesinger, 2010), but only in the context of aging or toxic stress. These observations are reminiscent of the delayed synergy among AD-related pathogenic proteins and the striking reduction in v-ATPase function caused by Presenilin-1 mutations (Lee et al., 2010; Nixon, 2017). Conditional deletion of the ATP6AP2 gene, which encodes for a critical v-ATPase-regulating protein, reduces v-ATPase activity(Korvatska et al., 2013) leading to autophagic vacuole accumulation, neurodegeneration and cognitive impairment in both fly and mouse models(Dubos et al., 2015). These results suggest that delayed effects of a partial loss-of-acidification function do not necessarily impede the autophagic-lysosomal system immediately but instead render the system more vulnerable to failure over time, similar to the pattern in aging-related neurodegenerative diseases, such as AD.
Oxidative stress – an independent aging factor corrupting lysosome acidification and hydrolase function
The free radical theory of aging(Hekimi et al., 2011) proposes that reactive oxygen species (ROS), when excessively produced in cells under stress conditions, are detrimental to cell components and homeostasis(Scherz-Shouval and Elazar, 2007). Many of the covalent modifications of proteins common in aging such as oxidation, glycation, phosphorylation, deamidation, carbonyl modification, and misfolding (Gafni, 1997), reduce the proteolytic susceptibility of the proteins delivered to lysosomes (Sukharev et al., 1997) . Besides effects on the proteolytic substrates, oxidative stress associated with cell aging may also adversely impact the lysosome directly. Age-dependent declines in CMA(Cuervo and Dice, 2000a) may be promoted by increased oxidative stress and attempts to eliminate oxidatively damaged proteins (Massey et al., 2006). Cellular iron deficits arise due to lysosomal dysfunction(Diab and Kane, 2013; Kurz et al., 2011) and can significantly impact the functions of mitochondria(Tai et al., 2017), which are enriched in iron requiring enzymes(Stehling and Lill, 2013). Another manifestation of this impact, seen especially in postmitotic cells, is accumulation of the age pigment(Jolly et al., 1993) lipofuscin within lysosomal-related compartments (Terman and Brunk, 2004).
Lipofuscin is an autofluorescent polymeric pigment composed of aldehyde cross linked protein fragments, oxidized lipids, carbohydrates and trace amount of metals, especially iron (Brunk and Terman, 2002; Seehafer and Pearce, 2006), which is stored at high levels in lysosomes. Lipofuscin is formed by the iron-catalyzed peroxidation of decomposed lipids principally from vesicular organelles such as autophagocytosed mitochondria and its accumulation, which is linearly correlated with age in various organisms (Strehler et al., 1959), reflects the impaired intralysosomal degradation of autophagic substrates under conditions of growing oxidative stress within aging cells. Loss of lysosomal enzyme activity, as seen experimentally using inhibitors of protease or acidification, increases the opportunity for lipid peroxidation and accelerates lipofuscin generation as well as other cardinal manifestations of brain aging (Bednarski et al., 1997) (Terman and Brunk, 1998; Terman et al., 2008). (Ivy et al., 1984). Although initially thought to have negligible influence on lysosomal function, further studies demonstrated lipofuscin-dependent decreases in activities of lysosomal cysteine proteases (Amano et al., 1995; Seehafer and Pearce, 2006). Components of lipofuscin may also inhibit v-ATPase, promoting its own accumulation and lysosomal failure(Bergmann et al., 2004). In a model of chronic oxidative stress, lysosomal acidification and autophagic flux were decreased in trabecular meshwork cells(Porter et al., 2013). Similarly, hydrogen peroxide inhibits synaptic vesicle v-ATPase activity and causes impaired uptake of glutamate into synaptic vesicles in isolated bovine brain synaptosomes (Wang and Floor, 1998). The v-ATPase itself is a target of oxidative stress in aging(Barone, 2016; Butterfield et al., 2014b), including carbonylation of the V1B2 subunit in aged rat brain tissue (Di Domenico et al., 2010), increased nitration of the V1E1 subunit in early AD (Butterfield and Sultana, 2007), and increased oxidative modification of the v-ATPase in AD and DS brain(Butterfield et al., 2014a). Nitrative stress in neuronal cells has been shown to reduce the activity of the lysosomal v-ATPase (Colacurcio, unpublished data). Oxidative/nitrative modifications impair the F-type mitochondrial ATPase, a homologue of the v- ATPase(Fujisawa et al., 2009; Haynes et al., 2010).
Lysosomal hydrolase changes in aging and the influence of pH regulation
Scattered reports of aging-related changes in the activities of cathepsins are generally consistent with an overall decline in lysosomal hydrolysis in aging in various tissues(Stoka et al., 2016). This conclusion accords with the accumulation of lipofuscin, most evident in neurons, and the enlargement of the lysosomal compartment (Kurz et al., 2000) which is also possibly indicative of slowed substrate clearance. Published data on individual enzyme activities in lysosomes mainly reflect measurements in tissues rather than lysosomes and are difficult to interpret given the brain’s cellular heterogeneity, changes in neural cell composition in brain during aging and disease, and varied properties of lysosomes even those within a single cell that are due to dynamic changes in intralumenal pH during the lysosome’s “life cycle”. Compounding these measurement challenges are the limited methods available to assess lysosomal activity in vivo. Hydrolase activities measured in vitro are a poor reflection of the total activities within lysosomes in living cells. Age-related changes of lysosomal pH, levels of endogenous lysosomal cysteine protease inhibitors (e.g., cystatins B, C, etc) (Kay et al., 2014), membrane stability (Nakamura et al., 1989), or changes in myriad other possible conditions within the intralumenal environment of the lysosome, may well account for the overall decreased lysosomal activity suspected to develop in aging cells; however, the inter-relationships among these factors and individual hydrolases that establish net in situ activity are difficult to discern when tissues or cells are lysed for protease assays.
The foregoing caveats notwithstanding, the in vitro activities of a few lysosomal hydrolases (eg.cathepsin D and certain lysosomal cysteine proteases) have been reported to be higher in aging rat liver (Keppler et al., 2000) and brain (Nakanishi et al., 1994b; Porta et al., 1995) and in vitro β-galactosidase activity may be elevated in aging human fibroblasts. In brain, certain cell types may possibly account for observed higher activities is whole brain tissue. For example, microglia, comprising up to 20% of the brain’s glial cell population and representing a prominent source of cathepsins, are activated during aging (von Bernhardi et al., 2015). Upregulated expression of cathepsins during brain aging, including cathepsins B, X, and S, has been suggested as a possible mechanism underlying the priming of microglia (Nakanishi, 2003; Nakanishi and Wu, 2009);(Wendt et al., 2007). By contrast, the activity, but not the protein levels, of cathepsin L was reported to be strikingly decreased in all brain regions of aged rats(Nakanishi et al., 1994a) suggesting generation of a catalytically inactive form of cathepsin L due to a higher lysosomal pH(Stoka et al., 2016). Inactivation of cathepsin L in this study was followed by induction of cathepsin D activity and aberrant tau proteolysis (Bednarski and Lynch, 1996). Age-related translocation of neuronal cathepsin D from lysosomes to cytosolic granules seen in about a third of rat cerebral cortex neurons was considered important in leading to age- related in tau proteolysis and cell death (Jung et al., 1999).
The regulation of pH within the lysosomal system plays an outsized role in influencing lysosomal acid hydrolase functions. Newly synthesized lysosomal cathepsins reaching endolysosomal compartments are activated by cleavage of their inhibitory propeptide domain under mild acidic conditions by autocatalysis or by another protease (Fox et al., 1992; Guay et al., 2000). Most cathepsins are inactivated and relatively unstable structurally in the neutral pH of the cytosol and extracellular milieu(Almeida et al., 2001; Turk et al., 1995; Turk et al., 1993; Turk et al., 1994); however, cathepsin S is stable even at neutral pH (Kirschke et al., 1989) and some cathepsins briefly remain somewhat active, which could account for their extra-lysosomal activity in metastasis and other disease states (Jordans et al., 2009; Turk and Turk, 2009). The pH optima of different cathepsins range from 3.5 to 6,0 and prolonged exposure to pH ranges outside these ranges can denature the enzyme and promote its degradation by a second cathepsin (Turk et al., 1999). What seems clear, therefore, is that aging-related changes in pH are likely to dramatically change the balance of proteolytic activities within aging lysosomes and have disruptive effects on the proteolysis of certain substrates.
Aging effects on lysosomal lipids and membrane stability
Lysosomal membrane permeabilization (LMP) is a possible outcome of cumulative insults of lysosomal aging that may herald the degeneration or death of a compromised neuron or other long-lived cell(Gomez-Sintes et al., 2016). LMP is defined as the selective destabilization of the lysosomal membrane allowing translocation of lysosomal contents to the cytoplasm. Some of the lysosomal constituents released, most notably cathepsins, act as the main initiators and executors of lysosome-dependent cell death. LMP is a well-reviewed topic (Boya and Kroemer, 2008; Groth-Pedersen and Jaattela, 2013; Johansson et al., 2010; Repnik et al., 2014; Stoka et al., 2007) and the detailed mechanisms of LMP interface with mechanisms of cell death beyond the scope of this review. The major relevant concept here is that aging of cells provides the key pre-conditions for LMP through routes described earlier, such as damage from free radicals, membrane incorporation of damaged proteins and oxidized lipids, and ionic shifts that alter osmotic equilibrium, which contribute to the increased volume of the lysosomal compartment seen in aging and senescence(Kurz et al., 2000) and is associated with enhanced fragility and LMP (Ono et al., 2003). The demonstration of lipofuscin in cytoplasm suggests the possibility of LMP(Reeg and Grune, 2015). Increasing lysosomal pH using lysosomotropic agents (e.g., certain antibiotics) induces LMP and cell death in several cell types(Boya et al., 2003a; Boya et al., 2003b)
Beyond the foregoing factors, lipids are key to protecting against membrane permeabilization and changes in lipid composition of membranes during aging likely reduce this protection. Liver lysosomes from 22 month old mice, compared to 3 month mice, contain increased levels of cholesterol, ceramide, glucosylceramide, and, notably, lysophosphatidylcholine that shows a comparatively greater increase than the other lipids(Rodriguez-Navarro et al., 2012). The latter change may be relevant to a reported ability of LysoPC to increase lysosomal permeability to potassium ions and protons, an effect that enhances the osmotic sensitivity of these organelles(Hu et al., 2007) and could destabilize older lysosomes and disrupt pH control. Higher levels of ceramide in lysosomes are also associated with membrane destabilization(Blom et al., 2015).The chaperone Hsp70 contributes to lysosomal stabilization by binding to the endolysosomal anionic phospholipid BMP (Kirkegaard et al., 2010), a co-factor that enhances the activity of acid sphingomyelinase, which mediates sphingomyelin catabolism. Consistent with this observation, acid sphingomyelinase deficiency leads to LMP and cytosolic release of cathepsins(Gabande-Rodriguez et al., 2014).
Mucolipin 1 (TRPML1), a major lysosomal calcium channel, is inhibited by high sphingomyelin levels, but is potentiated by sphingomyelinases(Shen et al., 2012). Moreover, abnormally elevated lysosomal pH, which is a prominent feature in Alzheimer’s Disease and other vATPase deficient conditions(Colacurcio and Nixon, 2016) and is suspected to develop during aging, induces release of calcium into the cytosol via TRPML1 channels which, in turn, activates calpains. Calpain cleavage products increase during aging, reflecting the greater activation of calpains, due in part to aging-related declines in its endogenous inhibitor, calpastatin(Nixon et al., 1994). In many circumstances, calpain activation lies upstream of cathepsin release during lysosomal associated cell death. In light induced retinal degeneration photoreceptor cell death, calpain inhibition rescued lysosomal permeabilization and calpain-mediated cleavage of the lysosomal membrane protein LAMP 2A, which was essential for this permeabilization(Villalpando Rodriguez and Torriglia, 2013). Finally, in addition to the role of Hsp70.1 in regulating BMP as described above, oxidative stress-induced carbonylation of Hsp70.1 together with calpain-mediated cleavage, leads to lysosomal destabilization and rupture resulting in neuronal death due to release of cathepsins(Yamashima, 2016).
Conclusion:
Lysosomes in the aging brain – gateway to late-onset neurodegeneration
A remarkable convergence of genetic and molecular- biological data underscores the central importance of the lysosome and its interacting compartments in the pathogenesis of late age-onset neurodegenerative diseases. Given the unique properties of neurons, the heavy reliance on efficient lysosomal clearance mechanisms is not surprising nor is the vulnerability to lysosomal impairments of a postmitotic neuron expected to survive over the lifetime of the organism. Lysosomal vulnerability is amplified by aging, which further impedes efficient substrate delivery and clearance during late life when increasing damage to proteins and membranes requires even greater capacity for protein quality control. In increasing numbers of late age onset neurodegenerative diseases (eg. AD, PD, FTD), milder degrees of primary lysosomal dysfunction related to the disease may remain subclinical until it is compounded by effects of cellular aging. Understanding the critical role evidently played by the lysosomal system in cellular aging and organismal lifespan is, therefore, an important frontier of biology holding many clues to neurodegenerative disease development and therapeutic strategies for new therapies.
Figure 3. Components and activities of the lysosome.
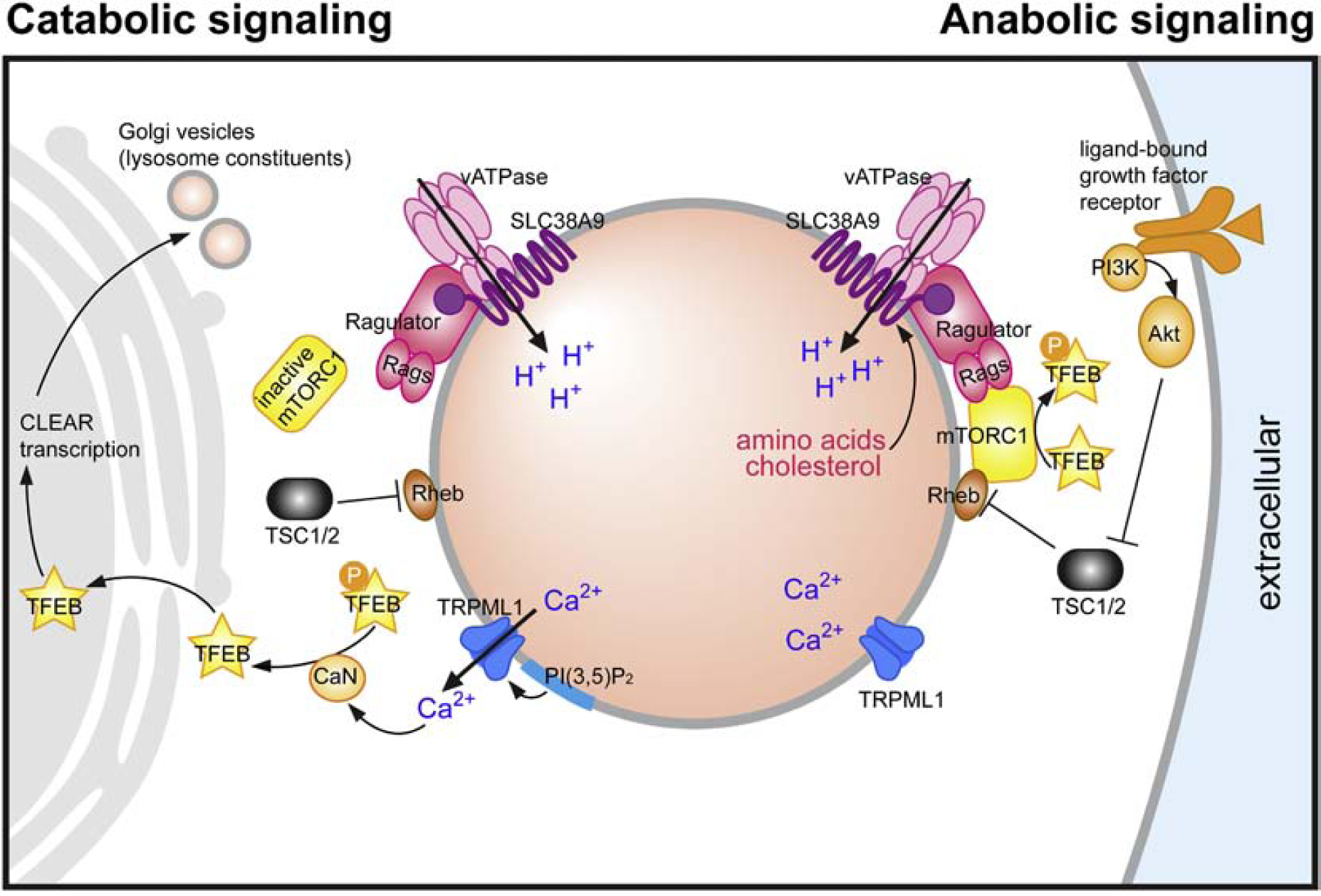
Lysosomes are composed of over two hundred identified proteins and likely hundreds associated with lysosomes but also localized to additional organelles. The diagram here illustrates only a subset relevant to the discussion in this review. In the presence of abundant cellular nutrients, mTORC1 is recruited to the lysosomal surface by the vATPase-SLC38A9-Ragulator-Rag GTPase complex, which senses amino acids and cholesterol levels within lysosomes. These levels are responsive to rates of substrate hydrolysis, which in turn is influenced by intraluminal pH. Both TRPML1-mediated Ca2+ release and the ATP-sensitive TPC-mediated Na+-release are inhibited under nutrient-replete conditions. Conversely, catabolic signaling is favored by nutrient depletion or certain forms of cell stress. Under these conditions, mTORC1 is released from the vATPase SLC38A9-Ragulator-Rag GTPase complex and the inactivated mTORC1 is no longer able to phosphorylate TFEB. PI (3,5)P2-mediated activation of TRPML1 channel triggers lysosomal Ca2+ efflux, activating CaN, which in turn dephosphorylates TFEB and stimulates its nuclear translocation. Nuclear TFEB activates CLEAR gene transcription for lysosome biogenesis. Abnormally elevated lysosomal pH similarly effects Ca2+ release and, when persistent, can over-activate calpains leading to cytotoxicity. Perinuclear redistribution of lysosomes, regulated in part by pH, facilitates the delivery of nascent lysosomal components from the Golgi, thereby promoting lysosomal degradative function. Under low level of ATP, TPC-mediated Na+ release affects the lysosomal membrane potential in a manner that helps maintaining vATPase proton pumping activity and acidification during starvation. (Figure adapted from Lie et al, 2019 with permission)
ACKNOWLEDGEMENTS
Research from the author’s laboratories is supported by the U.S. National Institutes of Health, National Institute on Aging, including current grants NIA P01 AG017617 and R01AG062376 to R.A.N. Contributions of Chitra Hindnavis to manuscript preparation and Dr. Pearl Lie to figure preparation are gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Afshari C, and Barret J (1996). Molecular genetics of in vitro cellular senescence In Cellular Aging and Cell Death, Holbrrok N, Martin G, and Lockshin R, eds. (New York: Wiley-Liss, Inc.), pp. 109–121. [Google Scholar]
- Almeida PC, Nantes IL, Chagas JR, Rizzi CC, Faljoni-Alario A, Carmona E, Juliano L, Nader HB, and Tersariol IL (2001). Cathepsin B activity regulation. Heparin-like glycosaminogylcans protect human cathepsin B from alkaline pH-induced inactivation. The Journal of biological chemistry 276, 944–951. [DOI] [PubMed] [Google Scholar]
- Amano T, Nakanishi H, Kondo T, Tanaka T, Oka M, and Yamamoto K (1995). Age-related changes in cellular localization and enzymatic activities of cathepsins B, L and D in the rat trigeminal ganglion neuron. Mechanisms of ageing and development 83, 133–141. [DOI] [PubMed] [Google Scholar]
- Barone E (2016). Editorial: Oxidative Stress and Alzheimer Disease: Where Do We Stand? Current Alzheimer research 13, 108–111. [DOI] [PubMed] [Google Scholar]
- Bednarski E, and Lynch G (1996). Cytosolic proteolysis of tau by cathepsin D in hippocampus following suppression of cathepsins B and L. Journal of neurochemistry 67, 1846–1855. [DOI] [PubMed] [Google Scholar]
- Bednarski E, Ribak CE, and Lynch G (1997). Suppression of cathepsins B and L causes a proliferation of lysosomes and the formation of meganeurites in hippocampus. JNeurosci 17, 4006–4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann M, Schutt F, Holz FG, and Kopitz J (2004). Inhibition of the ATP-driven proton pump in RPE lysosomes by the major lipofuscin fluorophore A2-E may contribute to the pathogenesis of age-related macular degeneration. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 18, 562–564. [DOI] [PubMed] [Google Scholar]
- Blom T, Li S, Dichlberger A, Back N, Kim YA, Loizides-Mangold U, Riezman H, Bittman R, and Ikonen E (2015). LAPTM4B facilitates late endosomal ceramide export to control cell death pathways. Nature chemical biology 11, 799–806. [DOI] [PubMed] [Google Scholar]
- Boland B, Yu WH, Corti O, Mollereau B, Henriques A, Bezard E, Pastores GM, Rubinsztein DC, Nixon RA, Duchen MR, et al. (2018). Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nature reviews Drug discovery 17, 660–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond S, and Forgac M (2008). The Ras/cAMP/protein kinase A pathway regulates glucose-dependent assembly of the vacuolar (H+)-ATPase in yeast. The Journal of biological chemistry 283, 36513–36521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordi M, Berg MJ, Mohan PS, Peterhoff CM, Alldred MJ, Che S, Ginsberg SD, and Nixon RA (2016). Autophagy flux in CA1 neurons of Alzheimer hippocampus: Increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy 12, 2467–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boya P, Andreau K, Poncet D, Zamzami N, Perfettini JL, Metivier D, Ojcius DM, Jaattela M, and Kroemer G (2003a). Lysosomal membrane permeabilization induces cell death in a mitochondrion-dependent fashion. The Journal of experimental medicine 197, 1323–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boya P, Gonzalez-Polo RA, Poncet D, Andreau K, Vieira HL, Roumier T, Perfettini JL, and Kroemer G (2003b). Mitochondrial membrane permeabilization is a critical step of lysosome-initiated apoptosis induced by hydroxychloroquine. Oncogene 22, 3927–3936. [DOI] [PubMed] [Google Scholar]
- Boya P, and Kroemer G (2008). Lysosomal membrane permeabilization in cell death. Oncogene 27, 6434–6451. [DOI] [PubMed] [Google Scholar]
- Brunk UT, and Terman A (2002). Lipofuscin: mechanisms of age-related accumulation and influence on cell function. Free radical biology & medicine 33, 611–619. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Di Domenico F, Swomley AM, Head E, and Perluigi M (2014a). Redox proteomics analysis to decipher the neurobiology of Alzheimer-like neurodegeneration: overlaps in Down’s syndrome and Alzheimer’s disease brain. The Biochemical journal 463, 177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield DA, Gu L, Di Domenico F, and Robinson RA (2014b). Mass spectrometry and redox proteomics: applications in disease. Mass spectrometry reviews 33, 277–301. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, and Sultana R (2007). Redox proteomics identification of oxidatively modified brain proteins in Alzheimer’s disease and mild cognitive impairment: insights into the progression of this dementing disorder. Journal of Alzheimer’s disease : JAD 12, 61–72. [DOI] [PubMed] [Google Scholar]
- Carafoli E, and Molinari M (1998). Calpain: a protease in search of a function? Biochemical and biophysical research communications 247, 193–203. [DOI] [PubMed] [Google Scholar]
- Carmona-Gutierrez D, Hughes AL, Madeo F, and Ruckenstuhl C (2016). The crucial impact of lysosomes in aging and longevity. Ageing research reviews 32, 2–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin RM, Fu X, Pai MY, Vergnes L, Hwang H, Deng G, Diep S, Lomenick B, Meli VS, Monsalve GC, et al. (2014). The metabolite alpha-ketoglutarate extends lifespan by inhibiting ATP synthase and TOR. Nature 510, 397–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colacurcio DJ, and Nixon RA (2016). Disorders of lysosomal acidification-The emerging role of v-ATPase in aging and neurodegenerative disease. Ageing research reviews 32, 75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo AM, and Dice JF (2000a). Age-related decline in chaperone-mediated autophagy. The Journal of biological chemistry 275, 31505–31513. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, and Dice JF (2000b). Regulation of lamp2a levels in the lysosomal membrane. Traffic 1, 570–583. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, and Dice JF (2000c). Unique properties of lamp2a compared to other lamp2 isoforms. Journal of cell science 113 Pt 24, 4441–4450. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, and Dice JF (2000d). When lysosomes get old. Experimental gerontology 35, 119–131. [DOI] [PubMed] [Google Scholar]
- Dechant R, Binda M, Lee SS, Pelet S, Winderickx J, and Peter M (2010). Cytosolic pH is a second messenger for glucose and regulates the PKA pathway through V-ATPase. The EMBO journal 29, 2515–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMartino GN, and Slaughter CA (1999). The proteasome, a novel protease regulated by multiple mechanisms. The Journal of biological chemistry 274, 22123–22126. [DOI] [PubMed] [Google Scholar]
- Di Domenico F, Perluigi M, Butterfield DA, Cornelius C, and Calabrese V (2010). Oxidative damage in rat brain during aging: interplay between energy and metabolic key target proteins. Neurochemical research 35, 2184–2192. [DOI] [PubMed] [Google Scholar]
- Diab HI, and Kane PM (2013). Loss of vacuolar H+-ATPase (V-ATPase) activity in yeast generates an iron deprivation signal that is moderated by induction of the peroxiredoxin TSA2. The Journal of biological chemistry 288, 11366–11377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimmer KS, Fritz S, Fuchs F, Messerschmitt M, Weinbach N, Neupert W, and Westermann B (2002). Genetic basis of mitochondrial function and morphology in Saccharomyces cerevisiae. Molecular biology of the cell 13, 847–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas PM, and Dillin A (2010). Protein homeostasis and aging in neurodegeneration. The Journal of cell biology 190, 719–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubos A, Castells-Nobau A, Meziane H, Oortveld MA, Houbaert X, Iacono G, Martin C, Mittelhaeuser C, Lalanne V, Kramer JM, et al. (2015). Conditional depletion of intellectual disability and Parkinsonism candidate gene ATP6AP2 in fly and mouse induces cognitive impairment and neurodegeneration. Human molecular genetics 24, 6736–6755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbaz-Alon Y, Rosenfeld-Gur E, Shinder V, Futerman AH, Geiger T, and Schuldiner M (2014). A dynamic interface between vacuoles and mitochondria in yeast. Developmental cell 30, 95–102. [DOI] [PubMed] [Google Scholar]
- Fox T, de Miguel E, Mort JS, and Storer AC (1992). Potent slow-binding inhibition of cathepsin B by its propeptide. Biochemistry 31, 12571–12576. [DOI] [PubMed] [Google Scholar]
- Fujisawa Y, Kato K, and Giulivi C (2009). Nitration of tyrosine residues 368 and 345 in the beta-subunit elicits FoF1-ATPase activity loss. The Biochemical journal 423, 219–231. [DOI] [PubMed] [Google Scholar]
- Gabande-Rodriguez E, Boya P, Labrador V, Dotti CG, and Ledesma MD (2014). High sphingomyelin levels induce lysosomal damage and autophagy dysfunction in Niemann Pick disease type A. Cell death and differentiation 21, 864–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gafni A (1997). Structural modifications of proteins during aging. Journal of the American Geriatrics Society 45, 871–880. [DOI] [PubMed] [Google Scholar]
- Glaser T, Schwarz-Benmeir N, Barnoy S, Barak S, Eshhar Z, and Kosower NS (1994). Calpain (Ca(2+)-dependent thiol protease) in erythrocytes of young and old individuals. Proceedings of the National Academy of Sciences of the United States of America 91, 7879–7883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Sintes R, Ledesma MD, and Boya P (2016). Lysosomal cell death mechanisms in aging. Ageing research reviews 32, 150–168. [DOI] [PubMed] [Google Scholar]
- Groth-Pedersen L, and Jaattela M (2013). Combating apoptosis and multidrug resistant cancers by targeting lysosomes. Cancer letters 332, 265–274. [DOI] [PubMed] [Google Scholar]
- Guay J, Falgueyret JP, Ducret A, Percival MD, and Mancini JA (2000). Potency and selectivity of inhibition of cathepsin K, L and S by their respective propeptides. European journal of biochemistry 267, 6311–6318. [DOI] [PubMed] [Google Scholar]
- Harman D (1972). The biologic clock: the mitochondria? Journal of the American Geriatrics Society 20, 145–147. [DOI] [PubMed] [Google Scholar]
- Haynes V, Traaseth NJ, Elfering S, Fujisawa Y, and Giulivi C (2010). Nitration of specific tyrosines in FoF1 ATP synthase and activity loss in aging. American journal of physiology Endocrinology and metabolism 298, E978–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hekimi S, Lapointe J, and Wen Y (2011). Taking a “good” look at free radicals in the aging process. Trends in cell biology 21, 569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hlavata L, Nachin L, Jezek P, and Nystrom T (2008). Elevated Ras/protein kinase A activity in Saccharomyces cerevisiae reduces proliferation rate and lifespan by two different reactive oxygen species-dependent routes. Aging Cell 7, 148–157. [DOI] [PubMed] [Google Scholar]
- Honscher C, Mari M, Auffarth K, Bohnert M, Griffith J, Geerts W, van der Laan M, Cabrera M, Reggiori F, and Ungermann C (2014). Cellular metabolism regulates contact sites between vacuoles and mitochondria. Developmental cell 30, 86–94. [DOI] [PubMed] [Google Scholar]
- Hu JS, Li YB, Wang JW, Sun L, and Zhang GJ (2007). Mechanism of lysophosphatidylchol-ineinduced lysosome destabilization. The Journal of membrane biology 215, 27–35. [DOI] [PubMed] [Google Scholar]
- Hughes AL, and Gottschling DE (2012). An early age increase in vacuolar pH limits mitochondrial function and lifespan in yeast. Nature 492, 261–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov A, Pawlikowski J, Manoharan I, van Tuyn J, Nelson DM, Rai TS, Shah PP, Hewitt G, Korolchuk VI, Passos JF, et al. (2013). Lysosome-mediated processing of chromatin in senescence. The Journal of cell biology 202, 129–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivy GO, Schottler F, Wenzel J, Baudry M, and Lynch G (1984). Inhibitors of lysosomal enzymes: accumulation of lipofuscin-like dense bodies in the brain. Science 226, 985–987. [DOI] [PubMed] [Google Scholar]
- Johansson AC, Appelqvist H, Nilsson C, Kagedal K, Roberg K, and Ollinger K (2010). Regulation of apoptosis-associated lysosomal membrane permeabilization. Apoptosis : an international journal on programmed cell death 15, 527–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolly RD, Dalefield RR, and Palmer DN (1993). Ceroid, lipofuscin and the ceroid-lipofuscinoses (Batten disease). Journal of inherited metabolic disease 16, 280–283. [DOI] [PubMed] [Google Scholar]
- Jordans S, Jenko-Kokalj S, Kuhl NM, Tedelind S, Sendt W, Bromme D, Turk D, and Brix K (2009). Monitoring compartment-specific substrate cleavage by cathepsins B, K, L, and S at physiological pH and redox conditions. BMC biochemistry 10, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung H, Lee EY, and Lee SI (1999). Age-related changes in ultrastructural features of cathepsin B- and D-containing neurons in rat cerebral cortex. Brain research 844, 43–54. [DOI] [PubMed] [Google Scholar]
- Jung J, Genau HM, and Behrends C (2015). Amino Acid-Dependent mTORC1 Regulation by the Lysosomal Membrane Protein SLC38A9. Molecular and cellular biology 35, 2479–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HT, Lee KB, Kim SY, Choi HR, and Park SC (2011). Autophagy impairment induces premature senescence in primary human fibroblasts. PloS one 6, e23367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik S, and Cuervo AM (2018). The coming of age of chaperone-mediated autophagy. Nature reviews Molecular cell biology 19, 365–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay P, Yang YC, Hiscott P, Gray D, Maminishkis A, and Paraoan L (2014). Age-related changes of cystatin C expression and polarized secretion by retinal pigment epithelium: potential age-related macular degeneration links. Investigative ophthalmology & visual science 55, 926–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon CJ (2010). The genetics of ageing. Nature 464, 504–512. [DOI] [PubMed] [Google Scholar]
- Keppler D, Walter R, Perez C, and Sierra F (2000). Increased expression of mature cathepsin B in aging rat liver. Cell and tissue research 302, 181–188. [DOI] [PubMed] [Google Scholar]
- Kirschke H, Wiederanders B, Bromme D, and Rinne A (1989). Cathepsin S from bovine spleen. Purification, distribution, intracellular localization and action on proteins. The Biochemical journal 264, 467–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korvatska O, Strand NS, Berndt JD, Strovas T, Chen DH, Leverenz JB, Kiianitsa K, Mata IF, Karakoc E, Greenup JL, et al. (2013). Altered splicing of ATP6AP2 causes X-linked parkinsonism with spasticity (XPDS). Human molecular genetics 22, 3259–3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurz DJ, Decary S, Hong Y, and Erusalimsky JD (2000). Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. Journal of cell science 113 ( Pt 20), 3613–3622. [DOI] [PubMed] [Google Scholar]
- Kurz T, Eaton JW, and Brunk UT (2011). The role of lysosomes in iron metabolism and recycling. The international journal of biochemistry & cell biology 43, 1686–1697. [DOI] [PubMed] [Google Scholar]
- Lapierre LR, De Magalhaes Filho CD, McQuary PR, Chu CC, Visvikis O, Chang JT, Gelino S, Ong B, Davis AE, Irazoqui JE, et al. (2013). The TFEB orthologue HLH-30 regulates autophagy and modulates longevity in Caenorhabditis elegans. Nature communications 4, 2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larner AJ (2013). Presenilin-1 mutations in Alzheimer’s disease: an update on genotype-phenotype relationships. Journal of Alzheimer’s disease : JAD 37, 653–659. [DOI] [PubMed] [Google Scholar]
- Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, Wolfe DM, Martinez-Vicente M, Massey AC, Sovak G, et al. (2010). Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 141, 1146–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar A, Capetillo-Zarate E, Cruz D, Gouras GK, and Maxfield FR (2011). Degradation of Alzheimer’s amyloid fibrils by microglia requires delivery of ClC-7 to lysosomes. Molecular biology of the cell 22, 1664–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markaki M, and Tavernarakis N (2013). Metabolic control by target of rapamycin and autophagy during ageing - a mini-review. Gerontology 59, 340–348. [DOI] [PubMed] [Google Scholar]
- Massey AC, Kaushik S, Sovak G, Kiffin R, and Cuervo AM (2006). Consequences of the selective blockage of chaperone-mediated autophagy. Proceedings of the National Academy of Sciences 103, 5805–5810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina DL, Di Paola S, Peluso I, Armani A, De Stefani D, Venditti R, Montefusco S, Scotto-Rosato A, Prezioso C, Forrester A, et al. (2015). Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nature cell biology 17, 288–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina DL, Fraldi A, Bouche V, Annunziata F, Mansueto G, Spampanato C, Puri C, Pignata A, Martina JA, Sardiello M, et al. (2011). Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Developmental cell 21, 421–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzies FM, Fleming A, Caricasole A, Bento CF, Andrews SP, Ashkenazi A, Fullgrabe J, Jackson A, Jimenez Sanchez M, Karabiyik C, et al. (2017). Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 93, 1015–1034. [DOI] [PubMed] [Google Scholar]
- Menzies FM, Garcia-Arencibia M, Imarisio S, O’Sullivan NC, Ricketts T, Kent BA, Rao MV, Lam W, Green-Thompson ZW, Nixon RA, et al. (2015). Calpain inhibition mediates autophagy-dependent protection against polyglutamine toxicity. Cell death and differentiation 22, 433–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merz S, and Westermann B (2009). Genome-wide deletion mutant analysis reveals genes required for respiratory growth, mitochondrial genome maintenance and mitochondrial protein synthesis in Saccharomyces cerevisiae. Genome biology 10, R95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molin M, and Demir AB (2014). Linking Peroxiredoxin and Vacuolar-ATPase Functions in Calorie Restriction-Mediated Life Span Extension. International journal of cell biology 2014, 913071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura S, and Yoshimori T (2018). Autophagy and Longevity. Molecules and cells 41, 65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Takeda M, Suzuki H, Morita H, Tada K, Hariguchi S, and Nishimura T (1989). Lysosome instability in aged rat brain. Neuroscience letters 97, 215–220. [DOI] [PubMed] [Google Scholar]
- Nakanishi H (2003). Microglial functions and proteases. Molecular neurobiology 27, 163–176. [DOI] [PubMed] [Google Scholar]
- Nakanishi H, Tominaga K, Amano T, Hirotsu I, Inoue T, and Yamamoto K (1994a). Age-related changes in activities and localizations of cathepsins D, E, B, and L in the rat brain tissues. Experimental neurology 126, 119–128. [DOI] [PubMed] [Google Scholar]
- Nakanishi H, Tominaga K, Amano T, Hirotsu I, Inoue T, and Yamamoto K (1994b). Age-Related Changes in Activities and Localizations of Cathepsins D, E, B, and L in the Rat Brain Tissues. Experimental Neurology 126, 119–128. [DOI] [PubMed] [Google Scholar]
- Nakanishi H, and Wu Z (2009). Microglia-aging: roles of microglial lysosome- and mitochondria-derived reactive oxygen species in brain aging. Behavioural brain research 201, 1–7. [DOI] [PubMed] [Google Scholar]
- Nixon R (2016). The Lysosome in age-related neurodegenerative diseases In Lysosomes: Biology, Diseases, and Therapeutics, Frederick J.M.W.a.S.L. Maxfield R, ed. (John Wiley & Sons, Inc; ), pp. 137–179. [Google Scholar]
- Nixon RA (2013). The role of autophagy in neurodegenerative disease. Nature medicine 19, 983–997. [DOI] [PubMed] [Google Scholar]
- Nixon RA (2017). Amyloid precursor protein and endosomal-lysosomal dysfunction in Alzheimer’s disease: inseparable partners in a multifactorial disease. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 31, 2729–2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA, Saito KI, Grynspan F, Griffin WR, Katayama S, Honda T, Mohan PS, Shea TB, and Beermann M (1994). Calcium-activated neutral proteinase (calpain) system in aging and Alzheimer’s disease. Annals of the New York Academy of Sciences 747, 77–91. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Yang DS, and Lee JH (2008). Neurodegenerative lysosomal disorders: a continuum from development to late age. Autophagy 4, 590–599. [DOI] [PubMed] [Google Scholar]
- Porta E, Llesuy S, Monserrat AJ, Benavides S, and Travacio M (1995). Changes in cathepsin B and lipofuscin during development and aging in rat brain and heart. Gerontology 41 Suppl 2, 81–93. [DOI] [PubMed] [Google Scholar]
- Porter K, Nallathambi J, Lin Y, and Liton PB (2013). Lysosomal basification and decreased autophagic flux in oxidatively stressed trabecular meshwork cells: implications for glaucoma pathogenesis. Autophagy 9, 581–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebsamen M, Pochini L, Stasyk T, de Araujo ME, Galluccio M, Kandasamy RK, Snijder B, Fauster A, Rudashevskaya EL, Bruckner M, et al. (2015). SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls mTORC1. Nature 519, 477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeg S, and Grune T (2015). Protein Oxidation in Aging: Does It Play a Role in Aging Progression? Antioxidants & redox signaling 23, 239–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repnik U, Hafner Cesen M, and Turk B (2014). Lysosomal membrane permeabilization in cell death: concepts and challenges. Mitochondrion 19 Pt A, 49–57. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Navarro JA, Kaushik S, Koga H, Dall’Armi C, Shui G, Wenk MR, Di Paolo G, and Cuervo AM (2012). Inhibitory effect of dietary lipids on chaperone-mediated autophagy. Proceedings of the National Academy of Sciences of the United States of America 109, E705–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein DC, Marino G, and Kroemer G (2011). Autophagy and aging. Cell 146, 682–695. [DOI] [PubMed] [Google Scholar]
- Ruckenstuhl C, Netzberger C, Entfellner I, Carmona-Gutierrez D, Kickenweiz T, Stekovic S, Gleixner C, Schmid C, Klug L, Sorgo AG, et al. (2014). Lifespan extension by methionine restriction requires autophagy-dependent vacuolar acidification. PLoS genetics 10, e1004347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, Elce JS, Hamos JE, and Nixon RA (1993). Widespread activation of calcium-activated neutral proteinase (calpain) in the brain in Alzheimer disease: a potential molecular basis for neuronal degeneration. Proc Natl Acad Sci U S A 90, 2628–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, et al. (2009). A gene network regulating lysosomal biogenesis and function. Science 325, 473–477. [DOI] [PubMed] [Google Scholar]
- Scherz-Shouval R, and Elazar Z (2007). ROS, mitochondria and the regulation of autophagy. Trends in cell biology 17, 422–427. [DOI] [PubMed] [Google Scholar]
- Schleit J, Johnson SC, Bennett CF, Simko M, Trongtham N, Castanza A, Hsieh EJ, Moller RM, Wasko BM, Delaney JR, et al. (2013). Molecular mechanisms underlying genotype-dependent responses to dietary restriction. Aging Cell 12, 1050–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scrivo A, Bourdenx M, Pampliega O, and Cuervo AM (2018). Selective autophagy as a potential therapeutic target for neurodegenerative disorders. The Lancet Neurology 17, 802–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seehafer SS, and Pearce DA (2006). You say lipofuscin, we say ceroid: defining autofluorescent storage material. Neurobiology of aging 27, 576–588. [DOI] [PubMed] [Google Scholar]
- Shen D, Wang X, Li X, Zhang X, Yao Z, Dibble S, Dong XP, Yu T, Lieberman AP, Showalter HD, et al. (2012). Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nature communications 3, 731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibatani T, and Ward WF (1996). Effect of age and food restriction on alkaline protease activity in rat liver. The journals of gerontology Series A, Biological sciences and medical sciences 51, B175–178. [DOI] [PubMed] [Google Scholar]
- Sinclair DA, and Guarente L (1997). Extrachromosomal rDNA circles--a cause of aging in yeast. Cell 91, 1033–1042. [DOI] [PubMed] [Google Scholar]
- Stehling O, and Lill R (2013). The role of mitochondria in cellular iron-sulfur protein biogenesis: mechanisms, connected processes, and diseases. Cold Spring Harbor perspectives in biology 5, a011312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan J, Franke J, and Ehrenhofer-Murray AE (2013). Chemical genetic screen in fission yeast reveals roles for vacuolar acidification, mitochondrial fission, and cellular GMP levels in lifespan extension. Aging Cell 12, 574–583. [DOI] [PubMed] [Google Scholar]
- Stoka V, Turk V, and Turk B (2007). Lysosomal cysteine cathepsins: signaling pathways in apoptosis. Biological chemistry 388, 555–560. [DOI] [PubMed] [Google Scholar]
- Stoka V, Turk V, and Turk B (2016). Lysosomal cathepsins and their regulation in aging and neurodegeneration. Ageing research reviews 32, 22–37. [DOI] [PubMed] [Google Scholar]
- Strehler BL, Mark DD, and Mildvan AS (1959). GEE MV: Rate and magnitude of age pigment accumulation in the human myocardium. Journal of gerontology 14, 430–439. [DOI] [PubMed] [Google Scholar]
- Sukharev SA, Pleshakova OV, Moshnikova AB, Sadovnikov VB, and Gaziev AI (1997). Age- and radiation-dependent changes in carbonyl content, susceptibility to proteolysis, and antigenicity of soluble rat liver proteins. Comparative biochemistry and physiology Part B, Biochemistry & molecular biology 116, 333–338. [DOI] [PubMed] [Google Scholar]
- Tai H, Wang Z, Gong H, Han X, Zhou J, Wang X, Wei X, Ding Y, Huang N, Qin J, et al. (2017). Autophagy impairment with lysosomal and mitochondrial dysfunction is an important characteristic of oxidative stress-induced senescence. Autophagy 13, 99–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terman A, and Brunk UT (1998). Ceroid/lipofuscin formation in cultured human fibroblasts: the role of oxidative stress and lysosomal proteolysis. Mechanisms of ageing and development 104, 277–291. [DOI] [PubMed] [Google Scholar]
- Terman A, and Brunk UT (2004). Lipofuscin. The international journal of biochemistry & cell biology 36, 1400–1404. [DOI] [PubMed] [Google Scholar]
- Terman A, Dalen H, Eaton JW, Neuzil J, and Brunk UT (2003). Mitochondrial recycling and aging of cardiac myocytes: the role of autophagocytosis. Experimental gerontology 38, 863–876. [DOI] [PubMed] [Google Scholar]
- Terman A, Kurz T, Gustafsson B, and Brunk UT (2008). The involvement of lysosomes in myocardial aging and disease. Current cardiology reviews 4, 107–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turk B, Bieth JG, Bjork I, Dolenc I, Turk D, Cimerman N, Kos J, Colic A, Stoka V, and Turk V (1995). Regulation of the activity of lysosomal cysteine proteinases by pH-induced inactivation and/or endogenous protein inhibitors, cystatins. Biological chemistry Hoppe-Seyler 376, 225–230. [DOI] [PubMed] [Google Scholar]
- Turk B, Dolenc I, Lenarcic B, Krizaj I, Turk V, Bieth JG, and Bjork I (1999). Acidic pH as a physiological regulator of human cathepsin L activity. European journal of biochemistry 259, 926–932. [DOI] [PubMed] [Google Scholar]
- Turk B, Dolenc I, Turk V, and Bieth JG (1993). Kinetics of the pH-induced inactivation of human cathepsin L. Biochemistry 32, 375–380. [DOI] [PubMed] [Google Scholar]
- Turk B, Dolenc I, Zerovnik E, Turk D, Gubensek F, and Turk V (1994). Human cathepsin B is a metastable enzyme stabilized by specific ionic interactions associated with the active site. Biochemistry 33, 14800–14806. [DOI] [PubMed] [Google Scholar]
- Turk B, and Turk V (2009). Lysosomes as “suicide bags” in cell death: myth or reality? The Journal of biological chemistry 284, 21783–21787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilchez D, Simic MS, and Dillin A (2014). Proteostasis and aging of stem cells. Trends in cell biology 24, 161–170. [DOI] [PubMed] [Google Scholar]
- Villalpando Rodriguez GE, and Torriglia A (2013). Calpain 1 induce lysosomal permeabilization by cleavage of lysosomal associated membrane protein 2. Biochimica et biophysica acta 1833, 2244–2253. [DOI] [PubMed] [Google Scholar]
- von Bernhardi R, Eugenin-von Bernhardi L, and Eugenin J (2015). Microglial cell dysregulation in brain aging and neurodegeneration. Frontiers in aging neuroscience 7, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Tsun ZY, Wolfson RL, Shen K, Wyant GA, Plovanich ME, Yuan ED, Jones TD, Chantranupong L, Comb W, et al. (2015). Metabolism. Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science 347, 188–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, and Floor E (1998). Hydrogen peroxide inhibits the vacuolar H+-ATPase in brain synaptic vesicles at micromolar concentrations. Journal of neurochemistry 70, 646–652. [DOI] [PubMed] [Google Scholar]
- Ward ME, Chen R, Huang HY, Ludwig C, Telpoukhovskaia M, Taubes A, Boudin H, Minami SS, Reichert M, Albrecht P, et al. (2017). Individuals with progranulin haploinsufficiency exhibit features of neuronal ceroid lipofuscinosis. Science translational medicine 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendt W, Zhu XR, Lubbert H, and Stichel CC (2007). Differential expression of cathepsin X in aging and pathological central nervous system of mice. Experimental neurology 204, 525–540. [DOI] [PubMed] [Google Scholar]
- Williamson WR, and Hiesinger PR (2010). On the role of v-ATPase V0a1-dependent degradation in Alzheimer disease. Communicative & integrative biology 3, 604–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe DM, Lee JH, Kumar A, Lee S, Orenstein SJ, and Nixon RA (2013). Autophagy failure in Alzheimer’s disease and the role of defective lysosomal acidification. The European journal of neuroscience 37, 1949–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright WE, Brasiskyte D, Piatyszek MA, and Shay JW (1996). Experimental elongation of telomeres extends the lifespan of immortal x normal cell hybrids. The EMBO journal 15, 1734–1741. [PMC free article] [PubMed] [Google Scholar]
- Yamashima T (2016). Can ‘calpain-cathepsin hypothesis’ explain Alzheimer neuronal death? Ageing research reviews 32, 169–179. [DOI] [PubMed] [Google Scholar]
- Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, Brunner T, and Simon HU (2006). Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nature cell biology 8, 1124–1132. [DOI] [PubMed] [Google Scholar]
- Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, and Sabatini DM (2011a). mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 334, 678–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoncu R, Efeyan A, and Sabatini DM (2011b). mTOR: from growth signal integration to cancer, diabetes and ageing. Nature reviews Molecular cell biology 12, 21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]