

SUMMARY
mTORC2 controls glucose and lipid metabolism, but the mechanisms have been unclear. Here, we show that conditionally deleting the essential mTORC2 subunit Rictor in murine brown adipocytes inhibits de novo lipid synthesis, promotes lipid catabolism and thermogenesis, and protects against diet-induced obesity and hepatic steatosis. AKT kinases are the canonical mTORC2 substrates; however, deleting Rictor in brown adipocytes appears to drive lipid catabolism by promoting FoxO1 deacetylation independently of AKT and SGK, and in a pathway distinct from its positive role in anabolic lipid synthesis. This facilitates FoxO1 nuclear retention, enhances lipid uptake and lipolysis, and potentiates UCP1 expression. We provide evidence that SIRT6 is the FoxO1 deacetylase suppressed by mTORC2, and show an endogenous interaction between SIRT6 and mTORC2 in both mouse and human cells. Our findings suggest a new paradigm of mTORC2 function filling an important gap in our understanding of this more mysterious mTOR-complex.
INTRODUCTION
A cell’s ability to balance anabolism and catabolism in response to nutrient availability is essential for metabolic fitness. The mTOR kinase, which senses intracellular nutrients through metabolite-sensing pathways, and systemic nutrients through growth factor signaling, is critical for this metabolic flexibility (Lee et al., 2017, Saxton and Sabatini, 2017). mTOR’s functions are split between two biochemically distinct complexes called mTOR complex 1 (mTORC1) and mTORC2. mTORC1 contains the essential subunit Raptor and is an amino acid and growth factor sensing complex that drives macromolecule biosynthesis and suppresses autophagy through well-defined pathways. mTORC2 uniquely contains the essential subunit Rictor and is implicated in glucose and lipid metabolism, but its regulation and mechanisms of action are more mysterious.
The canonical mTORC2 substrate is AKT, which it phosphorylates in a carboxy-terminal hydrophobic motif (HM) site (Sarbassov et al., 2005). AKT is also phosphorylated by PDK1 in the kinase domain T-loop motif. T-loop phosphorylation is essential for catalytic activity while HM phosphorylation promotes maximum AKT activity (Manning and Toker, 2017). In vitro, knocking down Rictor or acutely treating cells with an mTOR kinase inhibitor reduces T-loop phosphorylation (Sarbassov et al., 2005, Thoreen et al., 2009, Peterson et al., 2009, Feldman et al., 2009). However, HM phosphorylation is not essential for T-loop phosphorylation as genetic models show they can occur independently in vivo (Shiota et al., 2006, Jacinto et al., 2006, Guertin et al., 2006, Lee et al., 2017, Manning and Toker, 2017, Leroux et al., 2018). Moreover, many conditional Rictor knockout models observe profound metabolic phenotypes but often with normal or mild effects on AKT substrate phosphorylation (Hung et al., 2014, Lee et al., 2017, Gaubitz et al., 2016). Thus, the exact function of mTORC2 in AKT signaling is unresolved and whether mTORC2 has AKT-independent functions in metabolism has been unclear.
Brown adipose tissue (BAT) is specialized for non-shivering thermogenesis (NST), which is an adaptive response to cold temperature. NST is stimulated by the sympathetic nervous system and characterized by increased glucose uptake and de novo lipogenesis (DNL) in addition to increased lipolysis, fatty acid (FA) uptake, and FA oxidation (Cannon and Nedergaard, 2004, Scheele and Nielsen, 2017, Sanchez-Gurmaches et al., 2018, Mottillo et al., 2014) NST is also triggered by high fat diet (HFD), termed diet-induced thermogenesis (DIT) to distinguish it from cold induced thermogenesis (CIT). Both modes of thermogenesis are mediated by uncoupling protein 1 (UCP1), which dissipates chemical energy as heat (Rothwell and Stock, 1979, Cannon and Nedergaard, 2004, Feldmann et al., 2009, von Essen et al., 2017). In mice, NST is low in the absence of thermal or dietary stress. Lowering the environmental temperature below the thermoneutral zone stimulates CIT, while DIT is stimulated when thermoneutral housed mice are fed a HFD (Feldmann et al., 2009, Rowland et al., 2016, von Essen et al., 2017, Fischer et al., 2018). While CIT helps maintain euthermia, DIT protects against obesity demonstrated by the fact that Ucp1−/− mice living at thermoneutrality are prone to obesity (Feldmann et al., 2009, von Essen et al., 2017, Kontani et al., 2005).
In mice, interscapular brown adipocytes originate from Myf5-expressing precursors during development (Seale et al., 2008, Sanchez-Gurmaches and Guertin, 2014, Sanchez-Gurmaches et al., 2016). We previously showed that Myf5-cre;Rictor mice have small lipid depleted interscapular brown adipocytes with enlarged mitochondria and altered lipid metabolism (Hung et al., 2014). Unexpectedly, Myf5-cre;Rictor brown adipocytes appear to have normal downstream AKT signaling to many classic substrates. A similar paradox is observed in Adiponectin-cre;Rictor mice, which have impaired insulin-stimulated glucose uptake into white adipose tissue (WAT) and systemic insulin resistance despite seemingly normal AKT signaling (Tang et al., 2016). These findings hint at the existence of undefined mTORC2 mechanisms critical for carbohydrate and lipid metabolism.
Acclimating mice to thermoneutrality lowers their basal metabolism to be more human-like, profoundly influencing many phenotypes (Cannon and Nedergaard, 2011, Gordon et al., 2014, Hylander and Repasky, 2016). Interestingly, Myf5-cre;Rictor mice living at thermoneutrality are protected from diet-induced obesity (Hung et al., 2014), a finding of clinical interest given efforts to develop anti-obesity therapies targeting thermogenesis (Harms and Seale, 2013, Sidossis and Kajimura, 2015, Scheele and Nielsen, 2017). However, Myf5-Cre expresses in many non-brown adipocyte lineages including muscle, several white adipocytes, bones and neurons (Sanchez-Gurmaches et al., 2016) questioning the specificity of this phenotype to brown fat.
Here, we show that inhibiting brown adipocyte mTORC2 cell-autonomously potentiates thermogenesis, which enhances heat production in the cold, and protects against obesity at thermoneutrality. Mechanistically, inhibiting BAT mTORC2 drives lipid catabolism in part by activating the FoxO1 transcription factor, which it does by driving SIRT6-mediated FoxO1 deacetylation independently of AKT and the related mTORC2 substrate SGK. This branch of mTORC2 signaling appears to be independent of mTORC2’s role in promoting de novo lipogenesis. These findings fill an important gap in our understanding of how mTORC2 balances anabolic and catabolic lipid metabolism, and exposes a new aspect of mTORC2 signaling that may be relevant in diabetes and cancer.
RESULTS
mTORC2 suppresses UCP1 expression
Using AKT1-S473 and AKT2-S474 phospho-specific antibodies as reporters, we asked if cold induced thermogenesis (CIT) affects BAT mTORC2 activity. AKT1-S473 phosphorylation is stable between mice living at thermoneutrality (30°C) and mild cold (22°C), but decreases dramatically upon acclimation to severe cold (6°C) [Figure 1A]. Consistent with our previous work, AKT2-S474 phosphorylation and total protein increase at 22°C (Sanchez-Gurmaches and Guertin, 2014). However, while total AKT2 protein remains elevated in severe cold, P-AKT2-S474 markedly decreases [Figure 1A]. The coordinated decrease of P-AKT1-S473 and P-AKT2-S474 in severe cold inversely correlates with UCP1 expression suggesting mTORC2 may suppress thermogenesis.
Figure 1. mTORC2 suppresses UCP1 expression in vitro.

(A) Western blots of the indicated proteins using BAT lysates from wild type 14-week-old male C57/BL6J mice adapted to thermoneutrality (30°C) for 4 weeks, mild cold (22°C), or severe cold (6°C) for 2 weeks. Mice were fed a standard chow diet ad libitum. The (*) indicates isoform-specific phospho-antibodies for AKT1 or AKT2
(B) Experimental strategy for inducing Rictor deletion in mature brown adipocytes. Vehicle or 4-hydroxy-tamoxifen (4-OHT) is administered late during differentiation (Day-6 of differentiation) to achieve fully differentiated cells acutely deleted for Rictor (also see methods).
(C) Western blot showing CL-316,243 (0.1μM, 8h) or Forskolin (1μM, 8h) stimulated UCP1 expression in Rictor-iKO brown adipocytes compared to their isogenic controls. L.E.=long exposure. Arrows indicate the PPARγ1 and 2 isoforms.
(D-H) qRT-PCR analysis showing CL-316,243 (CL, 0.1μM, 8h) or Forskolin (FSK, 1μM, 8h) stimulated gene expression in Rictor-iKO brown adipocytes compared to their isogenic controls (n=3). PBS=phosphate buffered saline control. Data are mean ± SEM. Statistical significance was calculated using two-way ANOVA with the Tukey’s multiple comparisons test; *P < 0.05, **P < 0.01, ***P < 0.001 (D-H: Control vs. Rictor-iKO, PBS treated vs. CL- or FSK- treated)
To acutely examine brown adipocyte mTORC2 function in vitro, we developed a brown adipocyte CreER;rictor model in which 4-hydroxytamoxifen (4-OHT) or vehicle (ethanol) treatment generates Rictor inducible knockout (Rictor-iKO) brown adipocytes and their isogenic control, respectively [Figure 1B]. Rictor-iKO brown adipocytes lack Rictor and AKT-HM phosphorylation [Figure 1C]. To mimic β-adrenergic receptor stimulation, cells were treated with the β3 adrenergic receptor agonist CL316,243, or the adenylyl cyclase activator forskolin. Strikingly, both agonists more potently induce UCP1 mRNA and protein expression in the Rictor-iKO brown adipocytes compared to controls [Figure 1C, 1D]. Expression of the brown fat identity genes pgc1a and cidea is also stimulated by Rictor loss [Figure 1E, 1F], while pparg2 and prdm16 are unaffected [Figure 1G, 1H]. Similar effects are observed in primary adiponectin-cre;rictor brown adipocytes [Figure S1A, S1B], and in Rictor-iKO brite/beige adipocytes [Figure S1C, S1D], the latter additionally having increased fgf21 and pdk4 expression indicative of WAT browning [Figure S1D] (Fisher et al., 2012, Barquissau et al., 2016). These data are consistent with mTORC2 cell-autonomously suppressing thermogenesis.
Conditionally knocking out Rictor in BAT improves cold tolerance
To examine the physiological role of BAT mTORC2, we generated ucp1-cre;rictor conditional knockout mice (hereafter RictorBATKO). RictorBATKO mice living in standard conditions (22ºC; chow diet) have 46% less BAT mass compared to controls [Figure 2A] due to smaller lipid droplets [Figure 2B]. Liver mass is also reduced by 11% while the mass of subcutaneous and visceral white adipose tissue (WAT), skeletal muscle, heart, kidney, spleen, lung and thymus, is normal [Figure 2A]. Western blots confirm loss of Rictor, AKT-HM (S473) phosphorylation, and AKT turn motif (T450) phosphorylation—a growth factor insensitive mTORC2-dependent phosphorylation site affecting AKT mobility (Facchinetti et al., 2008)—only in BAT but not in WAT [Figure 2C, 2D, S2A, S2B]. Despite Rictor-deficient BAT lacking AKT HM phosphorylation, PDK1 still phosphorylates AKT in the T-loop (T308), albeit at slightly lower levels [Figure 2C, S2A]. Consistent with our previously work on adipocytes (Tang et al., 2016, Hung et al., 2014), RictorBATKO mice express 83% lower levels of Chrebpβ [Figure 2E], which encodes a transcriptional activator of de novo lipogenesis (DNL) (Eissing et al., 2013, Herman et al., 2012), and nearly 70–80% lower levels of its target genes Acly (78%), Acc (70%), Fasn (70%), and Elovl6 (70%) [Figure 2E]. This accounts for a dramatic reduction in total ACLY, ACC, and FASN protein [Figure 2C] indicating profound downregulation of the DNL pathway upon mTORC2 loss. Thus, in mice living in standard vivarium conditions, BAT mTORC2 positively regulates BAT size and lipid content.
Figure 2. Increased cold tolerance and UCP1 expression in RictorBATKO mice.

(A) Tissue mass data from RictorBATKO mice and controls under standard conditions (22°C, fed ad libitum with a standard chow diet, 14-week-old males, n=11).
(B) Top: Representative photograph of the interscapular BAT (iBAT) depots from a RictorBATKO mouse and a littermate control living in standard conditions. Bottom: representative H&E staining images. Scale bar, 100μm.
(C) Western blots in triplicate using total BAT lysates from RictorBATKO mice and controls living in standard conditions.
(D) Same as (C) except using subcutaneous adipose tissue (SAT) and visceral adipose tissue (VAT) depots.
(E) -PCR analysis using BAT from RictorBATKO mice and controls living in standard conditions (n=8).
(F) Rectal temperatures during an acute cold challenge (6°C) starting from 22°C (fed ad libitum with a standard chow diet, 10-week-old males, n=9–10).
(G) Infrared thermography of the skin surface temperature directly above the iBAT depot from mice acclimated to 6°C for 7h (acute) or 2 weeks (chronic) (n=4–8).
(H) Representative images from (G). Data are mean ± SEM. Statistical significance was calculated using two-tailed unpaired Student’s t-test (A),(F, right), two-way ANOVA with the Sidak’s multiple comparisons test (E,G), two-way repeated-measures ANOVA with the Sidak’s multiple comparisons test (F, left); *P < 0.05, **P < 0.01, ***P < 0.001 (Control vs. RictorBATKO).
RictorBATKO mice have a normal average body temperature of around 38.2°C in standard living conditions [Figure 2F]. In response to an acute severe cold challenge (6°C for 7 hours), RictorBATKO mice maintain a slightly warmer body temperature [Figure 2F] that correlates with higher BAT heat production determined directly by thermal camera imaging [Figure 2G]. RictorBATKO mice also maintain euthermia for up to two weeks at 6°C, which also correlates with higher BAT heat production [Figure 2G, 2H]. Consistent with the in vitro model [Figure 1C], BAT UCP1 levels are dramatically elevated in RictorBATKO mice [Figure 2C] confirming mTORC2 also suppresses UCP1 expression in vivo. Thus, BAT mTORC2 is dispensable for maintaining euthermia in the cold, and its loss enhances cold induced thermogenesis (CIT).
Prolonged severe cold (6°C) exposure also stimulates subcutaneous WAT browning. The formation of individual brite/beige adipocytes in WAT can be marked in ucp1-cre expressing mice by incorporating the Cre-activated mTmG reporter (Muzumdar et al., 2007)[Figure S2C]. Comparing brite/beige adipocyte formation between ucp1-cre;rictor;mTmG mice and their ucp1-cre;mTmG controls reveals that the Rictor-deficient brite/beige adipocytes are smaller and more multi-locular [Figure S2D]. However, the total number of brite/beige adipocytes (determined by counting mGFP-positive cells) remains the same [Figure S2E]. The ucp1-cre;rictor brite/beige adipocytes also express higher levels of ucp1, pgc1a [Figure S2F], and trending higher UCP1 protein [Figure S2G]. We did not detect a decrease in total Rictor protein in whole SAT lysates from these mice [Figure S2G], but this is perhaps not surprising given the greater heterogeneity of SAT compared to BAT (Roh et al., 2017). Nevertheless, during prolonged severe cold exposure, enhanced WAT browning could also contribute to CIT in ucp1-cre;rictor mice.
RictorBATKO mice are resistant to diet-induced obesity
Next, we acclimated RictorBATKO mice to 30ºC and either kept them on SCD or switched them to high fat diet (HFD) to trigger diet-induced thermogenesis (DIT). Weekly monitoring of body weight reveals no difference between the cohorts on SCD; however, RictorBATKO mice eating HFD gain less weight [Figure 3A]. Living without thermal stress causes whitening of BAT in both the control and RictorBATKO mice eating SCD [Figure 3B]. In contrast, control mice on HFD have abundant multi-locular adipocytes indicating DIT [Figure 3B]; this is strikingly magnified in HFD-fed RictorBATKO mice, which have a BAT size, color, and morphology more akin to cold-stressed mice [Figure 3B, S3A, S3B]. In control mice eating HFD, liver mass increases by 45% over SCD-fed controls [Figure 3C]. Liver mass is unchanged in the SCD-fed RictorBATKO mice compared to controls [Figure 2A] suggesting Rictor-deficient BAT is inert in the absence of thermal or dietary stress. In contrast, RictorBATKO mice on HFD are resistant to hepatomegaly [Figure 3C, 3D] and have less hepatic steatosis as determined by oil red O staining [Figure 3B], and by quantifying total TAGs [Figure 3E]. HFD-fed RictorBATKO mice also have 19% less VAT expansion [Figure 3F], 18% less circulating TAGs [Figure S3C], and improved glucose tolerance [Figure S3D]. There is an interesting trend of less food consumption by RictorBATKO mice, but it is not statistically significant [Figure S3E, S3F]. Thus, conditional BAT Rictor loss may enhance DIT.
Figure 3. RictorBATKO mice are protected from diet-induced obesity at thermoneutrality.

(A) Growth curves of RictorBATKO mice and controls living at 30°C and eating either standard chow diet (SCD) or high fat diet (HFD); n=8–12 male mice per group.
(B) Representative H&E (BAT, VAT) and Oil-Red O (Liver) staining images from mice in (A). Scale bar, 100 μm.
(C) Average liver mass of mice in (A, n=8–12).
(D) Representative photograph of livers from HFD-fed control and RictorBATKO mice in (A).
(E) Corresponding liver TAG quantification for panel (D) (n=7).
(F) Visceral adipose tissue (VAT) mass of mice in (A, n=8–12).
(G) Growth curves of RictorBATiKO (i.e. Ucp1-CreER;Rictor) mice and controls living at 30°C and eating either SCD or HFD. Rictor deletion was induced at 10-weeks of age with Tamoxifen. Mice were then acclimated at 30°C for 1 week before being placed on SCD or HFD; n=13–18 male mice per group.
(H) Representative H&E (BAT, VAT) and Oil-Red O (Liver) staining images from mice in (G). Scale bar, 100 μm.
(I) Blood Free fatty acid (FFA), triglyceride (TAG), and cholesterol, measurements from mice in (G) (n=6–8).
(J) Glucose tolerance test (GTT) of mice in (G) after 10-weeks of HFD. SCD (n=7–9), HFD (n=7–8). Data are mean ± SEM. Statistical significance was calculated using two-way repeated-measures ANOVA with the Tukey’s multiple comparisons test (A,G,J), two-way ANOVA with the Tukey’s multiple comparisons test (C,E,F,I,J); *P < 0.05, **P < 0.01, ***P < 0.001 (HFD-Control vs. HFD-RictorBATKO for A,G,J; SCD vs. HFD, Control vs. RictorBATKO or Control vs. RictorBATiKO for C,E,F,I,J).
We wondered if inducing Rictor deletion in adult mice could achieve the same metabolic benefit. To test this, we generated ucp1-creER;rictor and ucp1-CreER mice, and at 20 weeks of age, briefly administered tamoxifen to generate inducible BAT Rictor knockouts and their respective controls [Figure S3G]. Mice were then moved to 30ºC and kept on SCD, or switched to HFD, for 10-weeks beginning one week after acclimatization. Inducible BAT Rictor knockout mice on HFD gain less weight [Figure 3G], maintain histological features of active BAT [Figure 3H], have less hepatic steatosis [Figure 3H], less VAT expansion [Figure S3H], and less hyperlipidemia (FFAs and TAGs) and hypercholesterolemia [Figure 3I]. Their glucose tolerance is also markedly improved [Figure 3J]. Thus, inducibly inhibiting BAT mTORC2 also protects against diet-induced obesity.
Inhibiting BAT mTORC2 stimulates lipolysis and lipid uptake
Next, we examined the molecular signature of thermoneutral housed RictorBATKO on the different diets. HFD attenuates AKT1-S473 and AKT2-S474 phosphorylation in control BAT, which as expected, are impaired in all RictorBATKO mice [Figure 4A, S4A]; P-AKT-T308 is difficult to detect under these conditions. Deleting Rictor and/or HFD also attenuates SGK1-dependent NDRG1-T346 phosphorylation and NDRG1 total protein [Figure 4A, S4A]. Strikingly, the amount of UCP1 mRNA and protein induced by HFD is dramatically amplified in RictorBATKO mice [Figure 4A, S4A, 4B], indicating that mTORC2 also suppresses UCP1 expression during DIT.
Figure 4. Inhibiting BAT mTORC2 reprograms metabolism to favor lipid uptake and catabolism.

(A) Western blot analysis using BAT lysates mice that were fasted (O/N) then refed for 1 hour. Mice were living at 30°C and eating SCD or HFD for 8 weeks prior to harvesting the tissue (18w males, n=3).
(B) qRT-PCR analysis using BAT from mice in (A) (n=6–8).
(C) Ex vivo lipolysis assay, either basal or after isoproterenol stimulation (ISO), using BAT from RictorBATKO and controls after 8 weeks on HFD at 30°C (n=8).
(D) In vivo 3H-2-deoxy-glucose uptake assay into BAT. Mice were living at 30°C and eating SCD or HFD for 8 to 16-weeks prior to measuring (n=8–10).
(E) In vivo 14bromo-palmitate uptake assay (n=8–10).
(F) In vivo 3H-2-deoxy-glucose uptake assay into skeletal muscle (quadriceps) (n=8–10).
(G) In vivo 14bromo-palmitate uptake assay into skeletal muscle (quadriceps) (n=8–10). Data are mean ± SEM. Statistical significance was calculated using two-way ANOVA with the Tukey’s multiple comparisons test (B-G); *P < 0.05, **P < 0.01, ***P < 0.001 (SCD vs. HFD, Control vs. RictorBATKO, Basal vs. ISO-treated)
Control and RictorBATKO mice eating SCD express low levels of ACLY, ACC and FASN [Figure 4A, S4A] consistent with BAT DNL being low at thermoneutrality (Sanchez-Gurmaches et al., 2018, Trayhurn, 1981). Interestingly, HFD stimulates ACLY, ACC, FASN and UCP1 expression in control BAT [Figure 4A, S4A] indicating that DIT increases DNL similarly to CIT. Moreover, deleting Rictor blocks HFD-induced DNL pathway expression [Figure 4A, S4A], confirming mTORC2 promotes DNL, but also revealing that up-regulated DNL is not required for UCP1 up-regulation in RictorBATKO mice. This led us to hypothesize that BAT mTORC2 loss may potentiate thermogenesis independently of its role in DNL.
Other thermogenesis and lipid catabolism markers were also examined. Pgc1a exhibits the same mRNA signature as ucp1 [Figure 4B]. In contrast, atgl (which encodes the rate limiting regulator of lipolysis) (Zechner et al., 2012) is elevated in HFD-fed RictorBATKO mice [Figure 4B]. The expression of irf4 (which encodes a PGC-1α partner) (Kong et al., 2014), bnip3 (a mitochondria bioenergetics regulator) (Choi et al., 2016), acot4 and ehhadh (FA oxidation regulators) (Schrader et al., 2015), and lpl (a lipase that promotes lipid uptake) (Klingenspor et al., 1996) mirror atgl expression [Figure 4B]. Consistently, ATGL protein level is equivalent between chow-fed control and RictorBATKO mice, but significantly higher in HFD RictorBATKO mice compared to HFD-fed controls [Figure 4A, S4A]. This suggests that mTORC2 suppresses ATGL-mediated lipolysis in the HFD-fed group, which we confirmed in an ex vivo lipolysis assay [Figure 4C]. Thus, in addition to its positive role in anabolic DNL, BAT mTORC2 suppresses lipid catabolism especially during dietary stress.
We also examined lipolytic signaling using the in vitro Rictor-iKO brown adipocyte model. In a time-course experiment following CL316,243 treatment, we observe higher basal ATGL levels in unstimulated Rictor-iKO cells (i.e. at time 0) compared to isogenic controls [Figure S4B]. In contrast, p-HSL and p-Perilipin increase only after 30 minutes of treatment, and to markedly higher levels in the Rictor-iKO cells [Figure S4B]. HSL and Perilipin hyper-phosphorylation precede UCP1 induction, which occurs around 4–8 hours after CL316,243 treatment [Figure S4B]. Cellular cAMP level trend higher in Rictor-iKO cells in the basal state, but are similar to control upon CL316,243 stimulation [Figure S4C]. The increase in lipolytic signaling functionally corresponds to increased lipolysis [Figure S4D]. Thus, inducing brown adipocyte Rictor deletion in vitro enhances basal ATGL expression, but a stimulus is required to potentiate lipolytic signaling and subsequent UCP1 induction. This may partly explain why Rictor loss has little effect in mice that lack thermal or dietary stress.
Finally, we directly measured nutrient uptake into BAT. In the SCD group, there is no difference between control and RictorBATKO mice in the amount of 3H-2-deoxyglucose or 14C-bromopalmitate taken up by BAT [Figure 4D, 4E]. In response to HFD, glucose uptake increases in control BAT by 63% [Figure 4D] while lipid uptake decreases by 37% [Figure 4E] suggesting an increase in de novo lipid synthesis upon dietary stress [Figure 4A, S4A]. In contrast, HFD-diet has minimal effect on BAT substrate utilization in RictorBATKO mice [Figure 4D, 4E] resulting in a nearly 2-fold greater amount of BAT lipid uptake in HFD-fed RictorBATKO mice compare to controls [Figure 4E]. The relative increase in lipid uptake is consistent with higher BAT lpl expression in RictorBATKO mice [Figure 4B]. Interestingly, the HFD-induced increase in BAT glucose uptake in control mice is matched by a decrease in glucose uptake by skeletal muscle, and this too is reversed in RictorBATKO mice [Figure 4F]; muscle FFA utilization is unaffected [Figure 4G]. This may contribute to the improved glucose tolerance of these mice [Figure S3D] and confirms that BAT mTORC2 is influencing system nutrient utilization. We conclude that mTORC2 balances anabolic and catabolic lipid metabolism to maintain metabolic flexibility, and that its absence in BAT removes a brake to lipid catabolism that reprograms systemic nutrient utilization and potentiates thermogenesis.
Inhibiting mTORC2 deacetylates and activates FoxO1 independently of AKT
Next, we investigated the mechanism by which mTORC2 loss reprograms BAT lipid metabolism. We noted that atgl, irf4 and ucp1 expression has been linked to FoxO1, a transcription factor with emerging roles in lipid-related diseases (Chakrabarti and Kandror, 2009, Eguchi et al., 2011, Ortega-Molina et al., 2012, Li et al., 2017). FoxO1 is a classic AKT substrate inhibited by AKT-dependent phosphorylation at T24 and S256 (Manning and Toker, 2017). However, BAT FoxO1 phosphorylation is unaffected in RictorBATKO mice [Figure 5A, 5B]. In fact, many AKT target sites including P-AS160-T642, P-PRAS40-T246, P-GSK3β-S9, and P-TSC2-T1462, are phosphorylated normally when Rictor is deleted [Figure 5A](Hung et al., 2014). Thus, a global AKT signaling defect, and in particular reduced FoxO1 phosphorylation, cannot mechanistically explain the metabolic reprogramming.
Figure 5. mTORC2 loss and β-adrenergic receptor signaling promote FoxO1 deacetylation and nuclear localization.

(A) Western blot analysis using BAT lysates from fasted/refed RictorBATKO and control mice living at 22°C and eating SCD (14w males, n=3).
(B) Western blot analysis using BAT lysates of fasted/refed RictorBATKO and control mice living at 30°C and eating HFD for 8 weeks (18w males, n=3).
(C) Western blot analysis using BAT from RictorBATKO and control mice living at 30°C treated with PBS or CL-316,243 (0.5mg/kg, n=2) for three days. Arrows indicate PPARγ isoforms.
(D) Representative H&E staining images from (C). Scale bar, 100 μm.
(E) Immunoprecipitation assay using an anti-acetyl-lysine antibody (Ace-K) and blotting for total FoxO1 from CL-316,243-stimulated (1μM, 2h) control and Rictor-iKO brown adipocytes. Total cell lysate (TCL) was also probed with the indicated antibodies.
(F) Immunoprecipitation assay as in (E). Cells were pretreated for 1 hour with Torin1 (50nM), MK2206 (0.5μM), or GSK650394 (1μM, O/N) followed by CL-316,243 treatment (1μM, 2h).
(G) Western blot analysis of the cytoplasmic and nuclear fractions of Rictor-iKO brown adipocytes and controls treated with CL-316,243 (1μM) for 1 or 2 hours. Tubulin and Lamin B are used to control for fraction purity.
(H) Immunofluorescence analysis of control and Rictor-iKO brown adipocytes treated with vehicle (PBS) or CL-316,243 (1μM, 1h, n=6). Anti-FoxO1 antibody (green), DAPI (blue), and the corresponding quantification (right panel) is shown. Scale bar, 25 μm
(I) qRT-PCR analysis from Rictor-iKO brown adipocytes and controls, with or without CL-316,243 stimulation and deleted for FoxO1 by CRISPR/Cas9 using 4-independent sgRNAs (n=3).
(J) Corresponding Western blot analysis from (I). Arrows indicate the PPARγ isoforms.
(K) Western blot of lysates from control or Rictor-iKO brown adipocytes in which endogenous FoxO1 was deleted (sgFoxo1) and cells were reconstituted with FoxO1-WT, FoxO1–6KR, or FoxO1–6KQ mutant constructs. Data are mean ± SEM. Statistical significance was calculated using two-way ANOVA with the Tukey’s multiple comparisons test; *P < 0.05, **P < 0.01, ***P < 0.001 (PBS vs. CL-316,243-treated, Control vs. Rictor-iKO, sgControl vs. sgFoxO1). L.E.=long exposure
A less understood FoxO1 regulatory mechanism is reversible lysine (K) acetylation at six sites (K242, K245, K259, K262, K271 and K291), in which deacetylation is thought to promote nuclear retention and activity (Li et al., 2017). Strikingly, BAT FoxO1 is hypo-acetylated in RictorBATKO mice during both CIT [Figure 5A] and DIT [Figure 5B]. FoxO1 deacetylation has not been implicated in thermogenesis; therefore, we administered CL316,243 to wild type mice living at thermoneutrality to test whether FoxO1 deacetylation normally occurs upon BAT stimulation. Indeed, FoxO1 is deacetylated following βAR stimulation [Figure 5C, 5D]. Moreover, while deleting Rictor deacetylates FoxO1 in the basal unstimulated state [Figure 5C], CL316,243 synergizes with Rictor loss to enhance FoxO1 deacetylation and amplify the β-adrenergic response, indicated by higher UCP1 levels [Figure 5C] and reduced lipid storage [Figure 5D]. Thus, FoxO1 is normally deacetylated upon BAT stimulation, and inhibiting mTORC2 appears to establish a pre-thermogenic state by triggering FoxO1 deacetylation in the absence of stimulation.
Mice lacking the major BAT AKT isoform, Akt2 (i.e. Akt2BATKO mice), like RictorBATKO mice, have reduced BAT mass, lipid content and DNL, while Akt1BATKO mice have no obvious phenotype (Sanchez-Gurmaches et al., 2018). However, in contrast to RictorBATKO mice, FoxO1 acetylation is unaffected in Akt2BATKO mice [Figure S5A], and neither Akt2BATKO nor Akt1BATKO mice are resistant to HFD [Figure S5B, S5C, S5D, S5E] suggesting mTORC2 loss may promote FoxO1 deacetylation by an AKT-independent mechanism.
mTORC2-dependent FoxO1 regulation by acetylation, independently of AKT, is recapitulated in vitro. For example, treating control brown adipocytes with CL316,243 triggers FoxO1 deacetylation, indicated by less FoxO1 immuno-purification with a pan-Lys-acetylation antibody, without affecting P-AKT-S473 or P-FoxO1-T24 [Figure 5E]. Moreover, in Rictor-iKO brown adipocytes, FoxO1 is hypo-acetylated without CL316,243 treatment, and this is further reduced by CL316,243 without affecting FoxO1 phosphorylation [Figure 5E]. We further show that treating brown adipocytes with the mTOR kinase inhibitor Torin1 triggers FoxO1 deacetylation to the same extent as CL316,243 [Figure 5F], indicating that inhibiting mTOR catalytic activity is sufficient to stimulate FoxO1 deacetylation. Importantly, neither the pan-AKT inhibitor MK-2206, nor the SGK inhibitor GSK-650394 simulate FoxO1 deacetylation [Figure 5F]. Interestingly, inhibiting AKT protects against CL-316,243-induced deacetylation suggesting that there may be complex interplay between FoxO1 phosphorylation and acetylation. Taken together, these data suggest a non-canonical mechanism by which mTORC2 regulates FoxO1 acetylation.
We also examined FoxO1 localization. Control and Rictor-iKO cells were treated for 0, 1, and 2 hours with CL316,243 then fractionated into cytosolic and nuclear fractions using cytoplasmic tubulin and nuclear lamin B to control for purity [Figure 5G]. In CL316,243-treated controls, cytosolic FoxO1 decreases and nuclear FoxO1 accumulates indicating that β-AR signaling stimulates FoxO1 nuclear localization [Figure 5G]. In Rictor-iKO cells, nuclear FoxO1 levels are elevated (and cytoplasmic levels depleted) without CL316,243, and reach higher nuclear levels upon stimulation [Figure 5G], matching the FoxO1 deacetylation profile. We performed immuno-fluorescence experiments to directly visualize FoxO1 nuclear retention, and consistently, the number of cells with nuclear FoxO1 is similar between CL316,243-treated control cells and unstimulated Rictor-iKO cells (~40%); which is about twice as many compared to the unstimulated controls [Figure 5H]. Moreover, combining Rictor loss and CL316,243 increases the number of cells with nuclear FoxO1 to nearly 60% [Figure 5H]. Thus, inhibiting mTORC2 promotes FoxO1 nuclear retention.
We also asked whether deleting Foxo1 by CRISPR/Cas9 (using 4 independent guides) blocks Atgl and Ucp1 induction upon Rictor loss. Indeed, deleting Foxo1 attenuates atgl and ucp1 mRNA [Figure 5I] and protein expression [Figure 5J] in both control and Rictor-iKO cells. We performed similar experiments with a FoxO1 inhibitor (FoxOi; AS1842856). At high doses (2.5–10μM) FoxO1i blocks Ucp1 induction in both control and Rictor-iKO cells, while at a lower dose (1μM) inhibition is more specific to Rictor-iKO cells [Figure S5F]. We also generated primary adiponectin-Cre;FoxO1 brown adipocytes and confirm in an independent system that ATGL and UCP1 expression, but not PPARγ or ACLY, requires FoxO1 [Figure S5G]. Finally, we reconstituted FoxO1-deleted brown adipocytes with recombinant wild type FoxO1, acetylation deficient FoxO1 (6KR), or an acetylation mimetic FoxO1 (6KQ) [Figure 5K]. Wild type FoxO1 and FoxO1–6KR both rescue ATGL and UCP1 expression in the FoxO1 deleted cells, while FoxO1–6KQ cannot, consistent with acetylation being inhibitory for atgl and ucp1 expression. Moreover, only the wild type and FoxO1–6KR constructs, and not the FoxO1–6KQ construct, support ATGL and UCP1 up-regulation in Rictor-iKO cells in which endogenous Foxo1 is deleted [Figure 5K]. We conclude that FoxO1 deacetylation promotes atgl and ucp1 upregulation upon mTORC2 loss.
SIRT6 interacts with mTORC2 and promotes FoxO1 deacetylation
To identify the FoxO1 deacetylase in mTORC2-deficient brown adipocytes, we first considered class IIa HDACs because two previous studies linked them to FoxO1 (Mihaylova et al., 2011, Masui et al., 2013). The more recent study proposed that mTORC2 controls metabolism through HDAC4/5/7 phosphorylation in glioblastoma cells. Using the same phospho-specific antibodies, and also blotting for total HDAC4/5/7 protein, we do not detect any changes in p-HDAC4/5/7-S246/259/155 or p-HDAC4/5/7-S632/661/486 in control or Rictor-deficient BAT under the same conditions in which FoxO1 is deacetylated and UCP1 expression potentiated [Figure 5E, S6A, S6B, S6C]. While most HDAC4/5/7 is cytoplasmic in brown adipocytes, a sub-fraction is nuclear; however, we see no HDAC redistribution in response to CL316,243 and/or Rictor loss [Figure 5G]. The class II HDAC inhibitor LMK-235 also fails to prevent FoxO1 deacetylation induced by CL316,243 and/or Rictor-deletion [Figure S6D]. Finally, CRISPR-mediated double or triple knockout of class II HDACs (HDAC4/7, HDAC4/5, HDAC4/5/7) does not prevent FoxO1 deacetylation in response to CL316,243 stimulation and/or Rictor loss [Figure S6E]. These data are inconsistent with class IIa HDACs being the FoxO1 deacetylase.
We also considered sirtuins. SIRT1 and SIRT6 are predicted to be nuclear (Houtkooper et al., 2012); however, in brown adipocytes, SIRT1 is predominantly cytoplasmic while SIRT6 is mainly nuclear [Figure 5G]. Using CRISPR/Cas9, we deleted Sirt6, Sirt1, and the mitochondrial sirtuin Sirt3 with three independent guides in CL316,243 stimulated Rictor-iKO brown adipocytes. While the combination of Rictor loss and CL316,243 results in near complete FoxO1 deacetylation [Figure 6A, lane 3], this is completely prevented by deleting Sirt6 [Figure 6A, lane 4–6], and deleting Sirt1 or Sirt3 has no effect [Figure 6A, lane 7–12]. Consistently, deleting Sirt6 attenuates ATGL and UCP1 upregulation in Rictor-iKO cells [Figure 6B]. And deleting SIRT6, but not SIRT1, also prevents CL316,243 stimulated FoxO1 deacetylation in control cells [Figure S6F]. These data implicate SIRT6 as the FoxO1 deacetylase whose activity is enhanced by mTORC2 loss.
Figure 6. SIRT6 interacts with mTORC2 and FoxO1, and is necessary for FoxO1 deacetylation upon β-adrenergic stimulation or Rictor loss.

(A) Immunoprecipitation assay using an anti-acetyl-lysine antibody (Ace-K) and blotting for total FoxO1 from CL-316,243-stimulated (1μM, 2h) Rictor-iKO brown adipocytes in which SIRT6, SIRT1, or SIRT3 were also ablated by CRISPR/Cas9 using 3-independent sgRNAs each.
(B) Western blot analysis of CL-316,243-stimulated (1μM, 8h) Rictor-iKO brown adipocytes expressing 3-independent sgRNAs each targeting SIRT6 or SIRT1.
(C) Endogenous SIRT6 immunoprecipitation (IP) from control and Rictor-iKO brown adipocytes under basal, CL-316,243-stimulated (1μM, 2h), Rictor-deleted, or combined CL-316,243 and Rictor-deleted conditions. Endogenous FoxO1 is also probed in the SIRT6 IP.
(D) Endogenous SIRT6 immunoprecipitation (IP) from control and Rictor-iKO brown adipocytes. Cells were stimulated with CL-316,243-stimulated (1μM, 2h). Endogenous Rictor and mTOR is also probed in the SIRT6 IP.
(E) Endogenous mTOR immunoprecipitation (IP) from control and Rictor-iKO brown adipocytes as in (D). Endogenous Rictor and SIRT6 are also probed in the mTOR IP.
(F) Affinity pull down-MS data from Halo-Tag SIRT6 expressing HEK293 cells identifying mTORC2 subunits (mTOR, Rictor, Sin1) and assembly factors (Tel2, TTI1) as SIRT6 interactors (see also Methods). Spectral counts (SpC) and normalized spectral abundance factors (NSAFs) from two biological replicates are shown for Halo-Tag SIRT6 and Halo-Tag alone control.
(G) Endogenous SIRT6 IP (left) and mTOR IP (right) using HEK293 lysates. IgG is the negative control. L.E.=long exposure, TCL=total cell lysate.
Consistent with SIRT6 being the FoxO1 deacetylase, we detect endogenous SIRT6 in immuno-precipitates (IPs) with endogenous FoxO1 [Figure 6C]. Moreover, the amount of FoxO1 in SIRT6 IPs increases with CL316,243 treatment, or Rictor deletion, and to a greater extent when CL316,243 and Rictor deletion are combined [Figure 6C]. The dynamics of the endogenous SIRT6-FoxO1 interaction precisely mirrors the FoxO1 deacetylation and nuclear retention profiles [Figure 5E, 5G and 5I] supporting SIRT6 as the FoxO1 deacetylase. SIRT6 is also a histone deacetylase and consistent with mTORC2 suppressing SIRT6 activity, its reported histone targets (H3K9 and H3K56) are also hypo-acetylated in Rictor-iKO cells [Figure S6G](Michishita et al., 2009, Yang et al., 2009, Zhong et al., 2010). This suggests mTORC2 might also have epigenetic functions.
We noticed in cell fractionation experiments that some Rictor and mTOR protein co-localize in the nucleus with SIRT6 and FoxO1 [Figure 5G]. This led us to wonder if mTORC2 and SIRT6 physically interact. Indeed, a fraction of endogenous Rictor and mTOR, but not Raptor, Co-IPs with endogenous SIRT6 [Figure 6D]. Notably, deleting Rictor reduces the association between SIRT6 and mTOR, however, some mTOR remains in the SIRT6 immune complexes [Figure 6D]. Reciprocally, endogenous mTOR IPs recover endogenous SIRT6 and Rictor, but not SIRT1, and interestingly, the amount of SIRT6 recovered in mTOR IPs is unaffected by deleting Rictor and destabilized by CL-316,243 [Figure 6E]. Thus, SIRT6 may associate with mTOR independently of Rictor. Finally, using the HaloTag affinity purification system followed by mass spectrometry analysis, mTOR, RICTOR, SIN1 (another mTORC2 subunit), and TTI1 and TEL2 (mTORC2 assembly factors (Kaizuka et al., 2010)) were identified as SIRT6-interactors in HEK293 and HeLa cells [Figure 6F, S6H, and Table S1]. We confirmed the endogenous interaction between human SIRT6 and mTORC2 by co-IP in both cell lines [Figure 6G, S6I]. Thus, mTORC2 appears to interact with SIRT6 in diverse mouse and human cells.
mTORC2 regulates lipid anabolism and catabolism through different pathways
To test the in vivo relevance of FoxO1 downstream of Rictor loss, we generated Ucp1-cre;FoxO1 (FoxO1BATKO) single knockout and Ucp1-cre;Rictor;FoxO1 (FoxO1;RictorBATDKO) double knockout mice. Overall, FoxO1BATKO mice living at 22°C are similar in weight to controls although BAT and WAT mass trend slightly larger [Figure S7A] and this correlates with increased BAT lipid droplet size [Figure 7A]; no differences are observed in other tissues [Figure S7A]. BAT Atgl and irf4 mRNA [Figure 7B] and ATGL protein levels [Figure 7C, S7C] are reduced in FoxO1BATKO mice, consistent with their predicted roles as FoxO1 targets. Expression of pgc1α trends lower [Figure 7B], while Ucp1 mRNA is unchanged [Figure 7B], and UCP1 protein barely decreases [Figure 7C, S7C]. A broader analysis of predicted FoxO1 target genes only further identifies lpl and bnip3 as being significantly reduced under these conditions [Figure S7B]. The DNL pathway (i.e. ACLY, ACC, and FASN expression) is unaffected by FoxO1 loss [Figure 7C, S7C]. Thus, in mild cold adapted mice, deleting BAT FoxO1 mainly affects lipid update and lipolysis regulators, but not the DNL pathway or UCP1.
Figure 7. FoxO1 reprograms catabolic, but not anabolic, lipid metabolism upon mTORC2 loss in vivo.
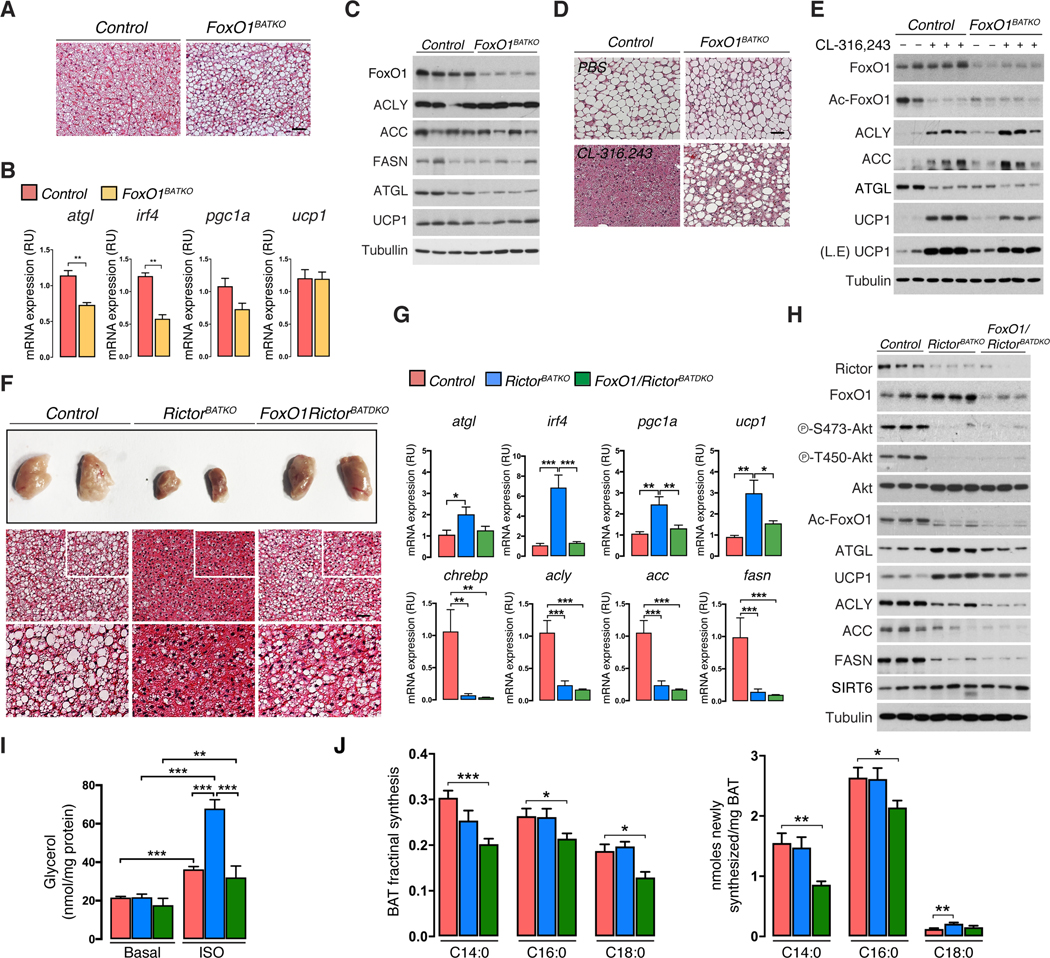
(A) Representative H&E staining images of BAT from FoxO1BATKO and controls under standard condition (22°C, SCD, 8w males). Scale bar, 100 μm.
(B) Quantitative RT-PCR analysis using BAT from fasted/refed FoxO1BATKO and control mice at (22°C, SCD, 8w males, n=5–6).
(C) Western blot analysis corresponding to (B) (8w males, n=4).
(D) Representative H&E staining images of BAT from FoxO1BATKO and control mice living at 30°C and treated with PBS or CL-316,243 (0.5mg/kg) for three days.
(E) Corresponding western blot analysis for panel (D); n=2–3.
(F) Representative photograph and H&E staining images of BAT from control, Rictor BATKO, and FoxO1;RictorBATDKO mice under standard conditions (8w males, n=5–6). White box insert corresponds to the enlarged H&E images below. Scale bar, 100 μm.
(G) qRT-PCR analysis of BAT from (F) (8w males, n=6–8).
(H) Western blot analysis for panel (G), (8w males, n=3).
(I) Ex vivo lipolysis assay of BAT under basal or isoproterenol-stimulated (ISO) conditions (22°C, SCD, 8w males, n=8).
(J) Heavy water (D2O) labeling showing the fraction (left) and quantification (right) of de novo synthesized fatty acid species (C14:0, C16:0, C18:0) in BAT (22°C, SCD, 8w males, n=6–9). Data are mean ± SEM. Statistical significance was calculated using Mann-Whitney test (B), one-way ANOVA with Tukey’s multiple comparisons test (G), two-way ANOVA with Tukey’s multiple comparisons test (I), Multiple t-test (J); *P < 0.05, **P < 0.01, ***P < 0.001 (Control vs. FoxO1BATKO, Control vs. RictorBATKO vs. FoxO1/RictorBATDKO, Basal vs. ISO-treated).
Next, we removed thermal stress by acclimating Ucp1-cre;FoxO1 and control mice to 30°C, then stimulated them with CL316,243 to induced thermogenesis. Without thermal stress, both the control and Ucp1-cre;FoxO1 brown adipocytes are morphologically similar [Figure 7D]. As expected, CL316,243 potently stimulates BAT activity in controls indicated by dramatic lipid depletion [Figure 7D], increased UCP1 levels [Figure 7E], and increased ACLY and ACC expression [Figure 7E]. Deleting FoxO1 partially attenuates CL316,243-induced lipid depletion [Figure 7D], further depletes ATGL levels [Figure 7E], and partially blocks UCP1 induction [Figure 7E], but has no effect on ACLY or ACC induction [Figure 7E]. Thus, BAT FoxO1 is normally required downstream of β-adrenergic signaling to maximally stimulate UCP1 expression, but not to induce DNL.
Finally, we asked if FoxO1 is required for BAT metabolic reprogramming when mTORC2 is inhibited using FoxO1;RictorBATDKO mice. Remarkably, BAT mass and lipid droplet size is mostly restored in FoxO1;RictorBATDKO mice [Figure 7F, S7D], and atgl, irf4, pgc1α, ucp1, and lpl mRNA expression is normalized to near control levels [Figure 7G, S7E]. Deleting Rictor also induces FoxO1-dependent p21 expression, which was unaffected in FoxO1 single knockout mice [Figure S7E]. Western blots confirm Rictor and FoxO1 deletion, and that deleting FoxO1 fully reverts ATGL, and partially reverts UCP1, to control levels in Rictor-deficient BAT [Figure 7H, S7F, S7G]. Ex vivo lipolysis measurements functionally confirm restoration of lipolytic control in the double knockout [Figure 7I] indicating FoxO1 unilaterally drives ATGL expression, and that additional factors likely cooperate with FoxO1 to induce UCP1.
In contrast, Chrebpβ, Acly, Acc, and Fasn levels remain strongly attenuated in both the RictorBATKO and FoxO1;RictorBATDKO mice [Figure 7G] indicating that FoxO1 loss does not rescue the DNL pathway in Rictor-deficient BAT. We confirmed this by Western blotting for ACLY, ACC, and FASN [Figure 7H, S7F, S7G]. To functionally confirm this, we performed in vivo D2O labeling to quantify the de novo synthesized lipids in BAT. No significant difference in D2O labeling is observed between control and RictorBATDKO mice [Figure 7J], which was not unexpected because Rictor-deficient BAT has higher lipid uptake [Figure 4E, S7H] and this technique does not distinguish locally synthesized lipids from those synthesized in other tissues and absorbed from circulation. In vitro, however, acute Rictor loss decreases DNL by 45% [Figure S7I]. If the hypothesis is correct, the doubly deleting FoxO1 should reverse lipid uptake and catabolism thereby revealing the endogenous DNL defect. This is indeed the case; the amount of D2O labeled myristic acid (C14:0), palmitic acid (C16:0) and stearic acid (C18:0) significantly decreases in the FoxO1;RictorBATDKO mice [Figure 7J], consistent with lpl levels returning to normal [Figure S7E], and the DNL pathway remaining down-regulated [Figure 7G, 7H, S7F, S7G]. Thus, we propose a model in which BAT mTORC2 balances anabolic and catabolic lipid metabolism through distinct pathways, and that SIRT6 drives FoxO1 deacetylation and lipid catabolism downstream of mTORC2 loss.
DISCUSSION
Brown adipose tissue is an attractive therapeutic target against obesity and its comorbidities because of its ability to expend energy by non-shivering thermogenesis (Harms and Seale, 2013). Here, we describe a novel mouse model with enhanced BAT activity that protects against over-nutrition and cold temperature, and is driven solely by inhibiting mTORC2 in mature brown adipocytes. We provide evidence of a mechanism in which mTORC2 loss triggers the deacetylation and activation of FoxO1 through SIRT6 in a non-canonical mTORC2 pathway. This appears to lower a transcriptional barrier to adaptive thermogenesis thereby potentiating beta-adrenergic stimulation in response to thermal or dietary stress.
An interesting finding from studying brown and white adipocyte mTORC2 (Hung et al., 2014, Tang et al., 2016), is that while conditionally deleting Rictor profoundly alters metabolism, it appears to do so independently of a major global AKT signaling defect. At least three models that are not-necessarily mutually exclusive could explain this: (1) Following prolonged mTORC2 loss, AKT signaling reprograms to become mTORC2-independent; (2) mTORC2 is more essential for an unidentified subset of AKT functions; or (3) that mTORC2 has AKT independent roles in metabolism. Losing Rictor in adipocytes does not affect mTORC1 activity, nor the ability to stimulate AKT signaling with physiological levels of insulin (Hung et al., 2014, Tang et al., 2016) arguing against compensation by negative feedback or hyperinsulinemia. Lack of an acute and selective mTORC2 inhibitor currently precludes our ability to further test the first model, and while our findings do not rule out the possibility that mTORC2 regulates specific AKT substrates, the data in this study supports the latter hypothesis that mTORC2 can regulate lipid catabolism by AKT (and SGK) independent pathways. The fact that doubly deleting FoxO1 and Rictor normalizes ATGL and partially UCP1 expression, but not DNL, also supports the notion that mTORC2 controls lipid anabolism and catabolism by different mechanisms. The former may be AKT-dependent given that Akt2BATKO mice have a DNL defect (Sanchez-Gurmaches et al., 2018). Defining the acute, essential, and specific roles of mTORC2 in AKT signaling and metabolism remain important goals. Another future goal is to understand how mTORC2 regulates SIRT6. The low Km of SIRT6 for NAD+ (26μM) makes it unlikely that NAD+ availability regulates SIRT6 (Canto et al., 2015, Pan et al., 2011). SIRT6 activity is stimulated by specific lipids (Feldman et al., 2013) whose availability could be regulated by mTORC2. The finding that sub-stoichiometric amounts of SIRT6 co-immunoprecipitate with mTORC2 suggests regulation could be direct, for example by sequestering or phosphorylating SIRT6, although the interaction could also exist though a higher order complex that survives immuno-purification. An interesting speculation is that the interaction between mTORC2 and SIRT6 is mediated by a nuclear mTORC2, which has been predicted (Rosner and Hengstschlager, 2008), but for which no function is assigned. Alternatively, mTORC2 could promote lysine acetyl transferase (KAT) activity towards FoxO1 that counters SIRT6’s deacetylase activity (Pietrocola et al., 2015). Whether SIRT6 histone deacetylase activity (Zhong et al., 2010, Kim et al., 2010), or other unidentified SIRT6 substrates, also contribute to metabolic reprogramming upon mTORC2 loss is another key question.
In sum, our data suggests a novel mechanism by which brown fat mTORC2 controls catabolic lipid metabolism independently of the canonical mTORC2-AKT paradigm to regulate thermogenesis and whole body lipid storage. This mechanism is likely conserved in other tissues and should be explored for its clinical relevance in metabolic diseases and cancer.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and request for resources and reagents should be directed to and will be fulfilled by the Lead Contact, David A. Guertin (david.guertin@umassmed.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All mice used in this study were C57Bl6/J males. Rictorfloxed (Shiota et al., 2006), UCP1-Cre (JAX stock 024670), Adiponectin-Cre (JAX stock 010803), and FoxO1floxed (JAX stock 024756) mice are available from Jackson laboratory. Akt1floxed and Akt2floxed mice are described in (Leavens et al., 2009, Wan et al., 2012). Ucp1-CreER is described in (Rosenwald et al., 2013). Floxed mouse strains were crossed with the different Cre-expressing mice to make tissue specific or inducible tissue specific knockout mice. Cre-negative floxed mice were used as controls. For studies using CreER mice, CreER only mice were included as an additional control.
Unless noted otherwise (e.g. in temperature studies), mice were housed in the UMMS Sherman Center Animal Medicine Facility in a clean room set at 22°C and 45% humidity on a daily 12h light/dark cycle, and kept in ventilated racks fed ad libitum with a standard chow diet, with bedding changed every two weeks.. Mice were sacrificed at 7–30 weeks old depending on the experiment. Please see figure legend for specific age and number of mice used. All animal experiments were approved by the University of Massachusetts Medical School Institutional Animal Care and Use Committee.
Cell culture
Human embryonic kidney (HEK293) and HeLa cell lines were from American Type Culture Collection (ATCC). For Rictor-iKO cell lines, primary brown preadipocytes were isolated from Ubc-CreERT2;Rictor floxed neonates at postnatal day 1 and immortalized with pBabe-SV40 Large T according to a standard protocol (Fasshauer et al., 2001) as described in (Hung et al., 2014). The gender of each neonate was not determined. Cells were maintained in high-glucose (25 mM) DMEM in incubators at 37°C and 5% CO2. Cells stably expressing recombinant proteins were obtained using a retroviral system. For primary, non-immortalized cells (i.e. from Adiponectin-Cre;Rictor and Adiponectin-Cre;FoxO1 mice), the immortalization step was excluded.
METHOD DETAILS
Thermogenic (brown, brite/beige) adipocyte differentiation
For brown adipocyte differentiation, cells were seeded (Day-1) at medium density and allowed to proliferate to confluence in the presence of high-glucose DMEM including 10% FBS, 1% antibiotics, 20 nM insulin and 1 nM T3. After 3 days (Day-4), cells were induced to differentiate by adding induction media (high-glucose DMEM including 10% FBS, 1% antibiotics, 20 nM insulin, 1 nM T3, 0.125 mM indomethacin, 2 μg/mL dexamethasone and 0.5 mM 3-isobutyl-1-methylxanthine (IBMX)) for 2 days. After this, the medium (high-glucose DMEM including 10% FBS and 1% antibiotics) with insulin and T3 was changed every 2 days until Day-11. Rictor deletion was achieved by treating the cells with 4-hydroxytamoxifen (1μM 4-OHT) on Day-6 of differentiation, which by-passes the requirement for Rictor in PPAR-γ induction (Hung et al., 2014). Control cells received an equivalent volume of ethanol, the vehicle used to dissolve 4-OHT. To stimulate β-adrenergic signaling, CL-316,243 (0.1–1 μM), isoproterenol (1–10 μM), or forskolin (1–10 μM), was administered directly to cells in culture that had been given fresh medium (high-glucose DMEM including 10% FBS, 1% antibiotics, 20 nM insulin and 1 nM T3) 24-hours before treatment.
For brite/beige cell analysis, white preadipocytes were from the subcutaneous vascular fraction (SVF) of Ubc-CreERT2;Rictorfloxed mice in 6–8 weeks of age. SVF were isolated by digesting the inguinal WAT in digestion buffer (123 mM NaCl, 5 mM KCl, 1.3 mM CaCl2, 5 mM glucose, 100 mM HEPES, 1% antibiotics and 4% BSA at pH 7.4 containing 1.5 mg/mL of collagenase A). The isolated cells were immortalized by a 3T3 immortalization protocol as previously described (Tang et al., 2016). Cells were maintained in 25 mM glucose, pyruvate-containing DMEM in incubators at 37°C and 5% CO2. For brite/beige adipocyte differentiation, cells were seeded at medium density and allowed to proliferate to confluence in the presence of high-glucose DMEM including 10% FBS, 1% antibiotics (complete medium). Two days after they reached confluency, cells were induced to differentiate by adding induction media (high-glucose DMEM including 10% FBS, 1% antibiotics, 100 nM insulin, 2 μg/mL dexamethasone, 0.5mM 3-isobutyl-1-methylxanthine (IBMX), 1 nM Rosiglitazone and 1 nM T3) for 4 days (Day 1 to 5) and replaced with medium containing 100 nM insulin and T3. Then the complete medium was changed every 2 days until day 9. Rictor deletion was achieved by treating the cells with 4-hydroxytamoxifen (4-OHT, 1 μM), equivalent volume of ethanol for control, on Day-5 of differentiation. To stimulate β-adrenergic signaling, forskolin (10 mM), was administered directly to cells in culture that had been given fresh medium (high-glucose DMEM including 10% FBS, 1% antibiotics, 100 nM insulin and 1 nM T3) 24-hours before treatment.
Fasting/Refeeding
Mice were fasted overnight, then refed by adding food to their cages for 1 hour before dissection.
Tissue Histology
Tissue pieces were fixed in 10% formalin. Embedding, sectioning and Hematoxylin and Eosin (H&E) staining was done by the UMMS Morphological Core facility. Oil red O staining was applied on liver cryo-sections by the UMMS Morphological Core facility.
Whole Mount Confocal Microscopy
Small pieces of adipose tissues were mounted with Fluoromount-G (Southern Biotech) and imaged on a LSM 5 Pascal (Zeiss) point scanner confocal system using a 10x or 40x oil immersion objective. eGFP was excited at 488 nm and detected from 515 to 565 nm.
tdTomato (red fluorescent protein) was excited at 543 nm and detected from 575 to 640 nm.
Cold challenge experiments
For acute cold challenge, 10-week old Ucp1-Cre;RictorFloxed and littermate control mice were transferred early in the morning to pre-chilled cages in a 4°C cold room with free access to pre-chilled food and water. Rectal temperature was measured hourly using a rectal probe (RET-3, ThermoWorks). For chronic cold challenge, the animals were placed in a 6°C thermal chamber (Model RIT330SD Power Scientific) for two weeks with free access to food and water, and maintained on the standard day/night light cycle. BAT and tail temperatures were obtained using an infrared thermal camera (FLIR T420) in lightly anesthetized mice and analyzed with FLIR tools.
Diet challenge experiments
6-week-old Ucp1-Cre;RictorFloxed mice and littermate control mice were transferred to a thermoneutral chamber (30°C) with a standard day/night light cycle and maintained on a standard chow diet (Prolab Isopro RMH3000, LabDiet). At 10-weeks of age, mice were either kept on the chow diet, or switched to a high-fat diet (45% calories from fat; ResearchDiet # D12451). Body weight and food intake were accessed weekly for 16 weeks. For experiments with the Ucp1-CreER;RictorFloxed mice, all mice in both the control and experimental groups were injected (I.P.)with Tamoxifen (100 mg/kg per day) at 20-weeks of age for 6 consecutive days. After tamoxifen treatment, the mice were transferred into a thermoneutral chamber (30°C) for one-week adaptation, then either kept on chow diet or switched to 45% HFD. Total body weight and food intake was monitored weekly. Tamoxifen was dissolved in corn oil/ethanol (9:1 vol/vol) at 2 mg/mL by shaking at 4°C overnight. 6-week-old Ucp1-CreERT2;Akt1Floxed mice, Ucp1-CreERT2;Akt2Floxed mice and respective littermate control mice were injected with tamoxifen (20 mg/mL, at 2 mg/day/mouse 5 times in a period of seven days. At 9-weeks old, mice were transferred to a thermoneutral chamber (30°C) with a standard day/night light cycle and maintained on a standard chow diet (Prolab Isopro RMH3000, LabDiet). At 10-weeks of age, mice were either kept on the chow diet or switched to a high-fat diet (45% calories from fat; ResearchDiet # D12451).
Glucose tolerance test (GTT) and blood metabolite analysis
Overnight fasted animals were subjected to GTT by intraperitoneally injecting glucose at 2 g/kg of body weight, and blood glucose levels were measured by tail bleeding with a commercially available glucose meter. The analysis of blood metabolites was performed by the MMPC at the University of Cincinnati.
Lipolysis assays
Standard ex-vivo lipolysis assays were performed as described in (Schreiber et al., 2017). Briefly, fresh dissected interscapular BAT was put into high-glucose DMEM supplemented with 2% fatty acid (FA)-free BSA. The tissue was minced into small pieces and pre-incubated with DMEM containing 2% FA-free BSA in 96-well plates for 30 minutes. To analyze basal lipolysis, the tissue pieces were transferred into 150 μl of fresh media and incubated for 1 hr in a tissue culture incubator. To analyze agonist-stimulated lipolysis, tissues were pre-incubated in 150μl DMEM containing 2% fatty acid (FA)-free BSA with isoproterenol (ISO, 10 μM) for 30 min then transferred into the same medium for an additional 2 hrs. To determine the amount of basal and stimulated lipolysis, the glycerol content of the medium was analyzed using free glycerol reagent (F6428, Sigma). Protein quantifications of each tissue sample were determined by Bradford assay (Bio-rad, #5000006), and the total amount of lipolysis was calculated as nmol of glycerol per mg/protein. For in vitro lipolysis assay, the same experimental procedure was performed using the cultured brown adipocytes.
Glucose and fatty acid uptake assays
14C-bromopalmitate ([1-14C]-2-bromopalmitic acid) and 3H-deoxyglucose ([1,2–3H(N)]-Deoxy-D-glucose) were used to evaluate NEFA and glucose uptake respectively into BAT, VAT, muscle and liver as previously described in (Menard et al., 2010, Labbe et al., 2016). Both radioactive tracers (Moravek Biochemicals, Inc. Brea, CA, USA) were dissolved in normal saline supplemented with 4% bovine serum albumin (BSA). Following a 6-hr fast, the mice received an intraperitoneal bolus of 10 μCi of each tracer in a total volume of 150 μl. Two hours following the injection, mice were euthanized with an overdose of anesthetic, and tissue samples were collected, weighed and homogenized. Specific fractional uptakes of 14C-bromopalmitate and 3H-deoxyglucose were determined using a scintillation counter (liquid scintillation analyzer Tri-Carb 2900TR, PerkinElmer, Montreal, QC, Canada), as previously described (Ci, 2006; Monge-Roffarello, 2014). Nutrient uptake data is expressed as the percentage of injected dose of [14C] and [3H] per milligram of tissue.
Cellular cAMP measurement assay
Using differentiated brown adipocytes, cellular cAMP level was measured following the instructions of the direct cAMP ELISA Assay kit (Enzo Life Sciences, Inc., Exeter, UK).
Immunofluorescence assay
Cells growing on coverslips in 6-cm dishes were fixed with 4% paraformaldehyde at room temperature for 15 min, followed by blocking with 5% BSA in PBS for 30 min, and then incubation with primary FoxO1 antibody (1:100) at 4°C overnight. After washing with PBS five times, the coverslips were stained with secondary antibodies (AlexaFluor-488-conjugated goat anti-rabbit IgG, Invitrogen, 1:400) at room temperature for 2 hours. Coverslips were also stained with DAPI (Sigma) and mounted on glass slides. Cells were examined with a laser-scanning confocal microscope (Leica).
Subcellular fractionation assay
The Subcellular Protein Fractionation Kit for Cultured Cells (Thermo-Scientific, cat. no. 78840) was used for subcellular fractionations following the manufacturer’s protocol.
Construction of small guide RNAs, lentiviral infection and retroviral infection
Gene-specific custom designed single guide RNAs were cloned into the lentiCRISPRv2 vector (Addgene). The sgRNA sequence was determined by the CRISPR Design Tool (http://chopchop.rc.fas.harvard.edu/) and sequences for sgRNAs targeting the FoxO1, SIRT1, SIRT3, SIRT6, HDAC4, HDAC5 and HDAC7 genes are shown in [Table S2]. To generate lentiviruses, HEK293T cells were transfected with lentiviral vectors expressing the sgRNA of interest in combination with VSV-G envelope plasmid and Delta-Vpr packing plasmid. To generate retroviruses, HEK293T cells were transfected with pBABE-retroviral vectors expressing FoxO1, FoxO1–6KR, and FoxO1–6KQ in combination with the retroviral packaging DNA (pEco), respectively. Culture media was changed 12 hours after transfection and the virus-containing supernatant was collected 48 hours after transfection and passed through a 0.45 μm filter to remove host cells. Brown preadipocytes were infected in medium containing 4 μg/mL of polybrene (Sigma) by centrifugation at 1800 RPM for 30 min. 24 hours after the infection, cells were trypsinized and subjected to antibiotic selection.
Western blot analysis and immunoprecipitation assays
Cells were harvested in cold PBS and lysed in protein lysis buffer (1% Triton X-100, 50 mM Hepes at pH 7.4, 150 mM NaCl, 10mM β-glycerophosphate, 2 mM EDTA, protease/phosphatase inhibitor cocktail). For immunoblot analysis of surgically dissected fat tissue depots, tissues were homogenized and lysed in RIPA buffer (150 mM NaCl, 50 mM Hepes at pH 7.4, 0.1% SDS, 1% Triton X-100, 2 mM EDTA, 0.5% Na-deoxycholate) containing a protease and phosphatase inhibitor cocktail. Protein lysates were mixed with 5X SDS sample buffer and boiled, separated by SDS-PAGE, transferred to polyvinylidene difluoride (PVDF) membrane filters and subjected to immunoblot analysis. For immunoprecipitationfor mTOR complex and SIRT6, cells were lysed in ice-cold mTOR IP buffer (0.3% CHAPS, 40 mM HEPES at pH 7.4, 150 mM NaCl, 5% glycerol, 2 mM EDTA, and protease/phosphatase inhibitor cocktail). 0.5~2mg of cell lysates were incubated with the appropriate primary antibodies (for 12–16 hrs), then with protein G agarose beads (Invitrogen) at 4°C for 2 h. Immuno-complexes were centrifuged, and washed twice with lysis buffer, then eluted from the beads by adding 2X SDS sample buffer and boiling. Immunoblot analysis was subsequently performed using the indicated antibodies.
FoxO1 acetylation assays
To detect endogenous acetylated FoxO1, cells were lysed in protein lysis buffer containing 10 mM Nicotinamide (NAM), 5 μM TSA 0.2% SDS. 0.5~1mg of cell lysates were combined with anti-Acetyl Lysine (CST #9441) antibody and then incubated overnight with gentle rotation. On the next day, 20 μl of protein G agarose beads were added and incubated for an additional 2 hrs. Beads were washed three times using lysis buffer, and the precipitates were eluted in 2X sample buffer and boiled. The acetylated-FoxO1 proteins were detected by immunoblotting using the anti-FoxO1 total protein antibody. The acetylated-FoxO1 protein was also detected in protein lysates using an acetylation-specific FoxO1 (sc-49437) antibody.
Gene expression analysis
Total RNA was isolated from cells or tissues using Qiazol (Qiagen) and an RNeasy kit (Qiagen). Equal amounts of RNA were retro-transcribed to cDNA using a High capacity cDNA reverse transcription kit (#4368813, Applied Biosystems). Quantitative RT-PCR (qRT-PCR) was performed in 10 μL reactions using a StepOnePlus real-time PCR machine from Applied Biosystems using SYBR Green PCR master mix (#4309156, Applied Biosystems) according to manufacturer instructions. Standard and melting curves were run in every plate for every gene to ensure efficiency and specificity of the reaction. TATA-box binding protein (Tbp) gene expression was used as a normalization gene in all conventional RT-PCR experiments. Data analysis was performed on web-based software provided by the manufacturer. Primer sequences are shown in [Table S3]
In vivo measurement of de novo synthesized fatty acid
2H2O method
3-days prior to termination, mice were I.P injected with 0.035 ml/g body weight 0.9% NaCL 2H2O, and drinking water was replaced with 8% 2H2O enriched water. Mice were fasted for 6 hours prior to plasma and tissue collection.
Plasma 2H2O enrichment
The 2H labeling of water from samples or standards was determined via deuterium acetone exchange (McCabe et al., 2006, Yang et al., 1998). 5 μl of sample or standard was reacted with 4 μl of 10N NaOH and 4 μl of a 5% (v/v) solution of acetone in acetonitrile for 24 hours. Acetone was extracted by the addition of 600 μl chloroform and 0.5 g Na2SO4 followed by vigorous mixing. 100 μl of the chloroform was then transferred to a GCMS vial. Acetone was measured using an Agilent DB-35MS column (30 m 3 0.25mm i.d. 3 0.25 mm, Agilent J&W Scientific) installed in an Agilent 7890A gas chromatograph (GC) interfaced with an Agilent 5975C mass spectrometer (MS) with the following temperature program: 60°C initial, increase by 20°C/min to 100°C, increased by 50°C/min to 220°C, and held for 1 min. The split ratio was 40:1 with a helium flow of 1 ml/min. Acetone eluted at approximately 1.5 min. The mass spectrometer was operated in the electron impact mode (70 eV). The mass ions 58 and 59 were integrated and the % M1 (m/z 59) calculated. Known standards were used to generate a standard curve and plasma % enrichment was determined from this. All samples were analyzed in triplicate. Total fatty acids were extracted from tissues and plasma using a bligh and dyer based methanol/chloroform/water extraction with C16 D31 as an internal standard. Briefly, 500 μls MeOH, 500 μls CHCl3, 200 μl H2O and fatty acid isotope internal standards were added to weighed pre-ground tissue. This was vortexed for 10 minutes followed by centrifugation at 10,000 g for 5 minutes. The lower chloroform phase was dried and then derivatized to form fatty acid methyl esters via addition of 500 μls 2% H2SO4 and incubation at 50°C for 2 hours. FAMES were extracted via addition of 100 μl saturated salt solution and 500 μl hexane, and these were analyzed using a Select FAME column (100m x 0.25mm i.d.) installed in an Agilent 7890A GC interfaced with an Agilent 5975C MS using the following temperature program: 80°C initial, increase by 20°C/min to 170°C, increased by 1°C/min to 204°C, then 20°C/min to 250°C and held for 10 min.
Calculations
The % mass isotopomer distributions of each fatty acid was determined and corrected for natural abundance using in-house algorithms adapted from Fernandez et al., (Fernandez et al., 1996). Calculation of the fraction of newly synthesized fatty acids (FNS) was based on the method described by Lee et al., (Lee et al., 2000) where FNS is described by the following equation:
Where ME is the average number of deuterium atoms incorporated per molecule (ME =1 x m1 + 2 x m2 +3 x m3 … ), p is the deuterium enrichment in water and n is the maximum number of hydrogen atoms from water incorporated per molecule. N was determined using the equation:
As described by Lee et al., (Lee et al., 1994) where q is the fraction of hydrogen atoms and p + q = 1. The molar amount of newly synthesized fatty acids was determined by: MNS = FNS x total fatty acid amount (nmoles/mg tissue)
In vitro measurement of de novo synthesized fatty acid
Cells were incubated for 3 days with DMEM in which 0.01% of the total glucose concentration of the medium was comprised of D-[U-14C]-glucose. Chloroform extraction was performed and labeled lipids were measured using a scintillation counter. Each sample was normalized to total protein concentration (Deberardinis et al., 2006)
HaloTag Mammalian Pull-Down Assay
N-terminal HaloTag fusions of human full-length SIRT6 (Q8N6T7) was obtained from Kazusa DNA Research Institute (Kisarazu, Japan) as a pFN21A vector (Promega). HaloTag control vector (Promega G6591) was used for expression of the HaloTag protein alone. HEK293T cells (10 × 106) or HeLa cells (12 × 106) were plated and grown overnight to 70–80% confluency. HaloTag-SIRT6 (Experimental) or HaloTag alone (Control) vectors were then transfected using FuGENE HD Transfection Reagent (Promega) according to manufacturer’s protocol. Cells expressing HaloTag-SIRT6 or HT-Ctrl were harvested and lysed on ice with 50mM Tris-HCl, pH 7.5, 150mM NaCl, 1% Triton X-100, and 0.1% sodium deoxycholate supplemented with Protease Inhibitor cocktail (Promega) and RQ1 RNase-Free DNase (Promega). Lysate was then homogenized with a syringe and centrifuged at 14.000 x g for 5 min. Experimental and control lysates were then incubated with HaloLink Resin (Promega) that had been pre-equilibrated in resin wash buffer (TBS and 0.05% IGEPAL CA-640 (Sigma)) for 15 min at 22oC with rotation to capture HaloTag-SIRT6 and any interacting protein partners. Resin was then washed 5 times with TBS wash buffer and protein interactors were eluted with SDS elution buffer (50mM Tris-HCl, pH 7.5, and 1% SDS).
Mass spectrometry analysis
HaloTag pulldown purified complexes from both HEK293 and HeLa samples were analyzed and processed by MS Bioworks, LLC (Ann Arbor, Michigan). Elution samples were pulsed onto an SDS-PAGE gel and cut into 10 slices. Each gel slice was washed using 25 mM ammonium bicarbonate and acetonitrile, followed by reduction with 10 mM dithiothreitol, and alkylation with 50 mM iodoacetamide. Proteins from each slice treated for 4hr with trypsin (Promega) and quenched with formic acid. Digests were then analyzed by nano LC/MS/MS with a NanoAcquity HPLC (Waters) interfaced with an Orbitrap Velos Pro (Thermo Scientific) tandem mass spectrometer. Resulting data were searched with Mascot (Matrix Science) against the concatenated forward/decoy UniProt Human Database, and Mascot DAT files were visualized and filtered by Scaffold (Proteome Software). Data were filtered using a minimum protein value of 90%, a minimum peptide value of 50% (Protein and Peptide Prophet scores), requiring at least two unique peptides per protein. Spectral counting was performed and normalized spectral abundance factors determined. Data were reported at less than 1% false discovery rate (FDR) at the protein level based on counting the number of forward and decoy matches.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are presented as mean + SEM, unless stated otherwise. Student’s t-test, non-parametric Mann-Whitney test or analysis of variance (ANOVA; one or two ways), as appropriate, were used to determine statistical significance. Statistical analysis was done using GraphPad Prism. The number of mice used per experiment is stated in each figure legend.
Supplementary Material
Table S2. Oligonucleotides used in this study, related to Star Methods
Acknowledgements
We thank Mathieu Laplante for advice with nutrient uptake experiments, Christian Wolfrum for Ucp1-CreER mice, Evan B. Rosen for Adiponectin-Cre and Ucp1-Cre mice, Morris J. Birnbaum for AKT floxed mice, and Jessica L. Feldman for technical advice.
Grants and Support
This work was supported by grants from the NIH (R01DK094004 and R01CA196986) and a Leukemia and Lymphoma Society Career Development Award to D.A.G. S.M.J. is supported by a postdoctoral fellowship from the American Diabetes Association (1–18-PDF-128). J.S.G. was supported by a postdoctoral fellowship from the American Heart Association (15POST25550079). C.M.C. was supported by a postdoctoral fellowship from the American Diabetes Association (1–16-PMF-008). C.M.M. is supported by grant from NIH (R01CA188652).
Footnotes
Conflicts of interests
The authors declare no conflicts of interest.
DATA AND SOFTWARE AVAILABILITY
Original, unprocessed data from this manuscript have been deposited to Mendeley Data: http://
REFERENCES
- BARQUISSAU V, BEUZELIN D, PISANI DF, BERANGER GE, MAIRAL A, MONTAGNER A, ROUSSEL B, TAVERNIER G, MARQUES MA, MORO C, GUILLOU H, AMRI EZ & LANGIN D. 2016. White-to-brite conversion in human adipocytes promotes metabolic reprogramming towards fatty acid anabolic and catabolic pathways. Mol Metab, 5, 352–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CANNON B. & NEDERGAARD J. 2004. Brown adipose tissue: function and physiological significance. Physiol Rev, 84, 277–359. [DOI] [PubMed] [Google Scholar]
- CANNON B. & NEDERGAARD J. 2011. Nonshivering thermogenesis and its adequate measurement in metabolic studies. J Exp Biol, 214, 242–53. [DOI] [PubMed] [Google Scholar]
- CANTO C, MENZIES KJ & AUWERX J. 2015. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab, 22, 31–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAKRABARTI P. & KANDROR KV 2009. FoxO1 controls insulin-dependent adipose triglyceride lipase (ATGL) expression and lipolysis in adipocytes. J Biol Chem, 284, 13296–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHOI JW, JO A, KIM M, PARK HS, CHUNG SS, KANG S. & PARK KS 2016. BNIP3 is essential for mitochondrial bioenergetics during adipocyte remodelling in mice. Diabetologia, 59, 571–81. [DOI] [PubMed] [Google Scholar]
- DEBERARDINIS RJ, LUM JJ & THOMPSON CB 2006. Phosphatidylinositol 3-kinase-dependent modulation of carnitine palmitoyltransferase 1A expression regulates lipid metabolism during hematopoietic cell growth. J Biol Chem, 281, 37372–80. [DOI] [PubMed] [Google Scholar]
- EGUCHI J, WANG X, YU S, KERSHAW EE, CHIU PC, DUSHAY J, ESTALL JL, KLEIN U, MARATOS-FLIER E. & ROSEN ED 2011. Transcriptional control of adipose lipid handling by IRF4. Cell Metab, 13, 249–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EISSING L, SCHERER T, TODTER K, KNIPPSCHILD U, GREVE JW, BUURMAN WA, PINNSCHMIDT HO, RENSEN SS, WOLF AM, BARTELT A, HEEREN J, BUETTNER C. & SCHEJA L. 2013. De novo lipogenesis in human fat and liver is linked to ChREBP-beta and metabolic health. Nat Commun, 4, 1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FACCHINETTI V, OUYANG W, WEI H, SOTO N, LAZORCHAK A, GOULD C, LOWRY C, NEWTON AC, MAO Y, MIAO RQ, SESSA WC, QIN J, ZHANG P, SU B. & JACINTO E. 2008. The mammalian target of rapamycin complex 2 controls folding and stability of Akt and protein kinase C. EMBO J, 27, 1932–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FASSHAUER M, KLEIN J, KRIAUCIUNAS KM, UEKI K, BENITO M. & KAHN CR 2001. Essential role of insulin receptor substrate 1 in differentiation of brown adipocytes. Mol Cell Biol, 21, 319–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FELDMAN JL, BAEZA J. & DENU JM 2013. Activation of the protein deacetylase SIRT6 by long-chain fatty acids and widespread deacylation by mammalian sirtuins. J Biol Chem, 288, 31350–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FELDMAN ME, APSEL B, UOTILA A, LOEWITH R, KNIGHT ZA, RUGGERO D. & SHOKAT KM 2009. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol, 7, e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FELDMANN HM, GOLOZOUBOVA V, CANNON B. & NEDERGAARD J. 2009. UCP1 ablation induces obesity and abolishes diet-induced thermogenesis in mice exempt from thermal stress by living at thermoneutrality. Cell Metab, 9, 203–9. [DOI] [PubMed] [Google Scholar]
- FERNANDEZ CA, DES ROSIERS C, PREVIS SF, DAVID F. & BRUNENGRABER H. 1996. Correction of 13C mass isotopomer distributions for natural stable isotope abundance. J Mass Spectrom, 31, 255–62. [DOI] [PubMed] [Google Scholar]
- FISCHER AW, SCHLEIN C, CANNON B, HEEREN J. & NEDERGAARD J. 2018. Intact innervation is essential for diet-induced recruitment of brown adipose tissue. Am J Physiol Endocrinol Metab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FISHER FM, KLEINER S, DOURIS N, FOX EC, MEPANI RJ, VERDEGUER F, WU J, KHARITONENKOV A, FLIER JS, MARATOS-FLIER E. & SPIEGELMAN BM 2012. FGF21 regulates PGC-1alpha and browning of white adipose tissues in adaptive thermogenesis. Genes Dev, 26, 271–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GAUBITZ C, PROUTEAU M, KUSMIDER B. & LOEWITH R. 2016. TORC2 Structure and Function. Trends Biochem Sci, 41, 532–545. [DOI] [PubMed] [Google Scholar]
- GORDON CJ, AYDIN C, REPASKY EA, KOKOLUS KM, DHEYONGERA G. & JOHNSTONE AF 2014. Behaviorally mediated, warm adaptation: a physiological strategy when mice behaviorally thermoregulate. J Therm Biol, 44, 41–6. [DOI] [PubMed] [Google Scholar]
- GUERTIN DA, STEVENS DM, THOREEN CC, BURDS AA, KALAANY NY, MOFFAT J, BROWN M, FITZGERALD KJ & SABATINI DM 2006. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell, 11, 859–71. [DOI] [PubMed] [Google Scholar]
- HARMS M. & SEALE P. 2013. Brown and beige fat: development, function and therapeutic potential. Nat Med, 19, 1252–63. [DOI] [PubMed] [Google Scholar]
- HERMAN MA, PERONI OD, VILLORIA J, SCHON MR, ABUMRAD NA, BLUHER M, KLEIN S. & KAHN BB 2012. A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature, 484, 333–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOUTKOOPER RH, PIRINEN E. & AUWERX J. 2012. Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol, 13, 225–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUNG CM, CALEJMAN CM, SANCHEZ-GURMACHES J, LI H, CLISH CB, HETTMER S, WAGERS AJ & GUERTIN DA 2014. Rictor/mTORC2 loss in the Myf5 lineage reprograms brown fat metabolism and protects mice against obesity and metabolic disease. Cell Rep, 8, 256–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HYLANDER BL & REPASKY EA 2016. Thermoneutrality, Mice, and Cancer: A Heated Opinion. Trends Cancer, 2, 166–175. [DOI] [PubMed] [Google Scholar]
- JACINTO E, FACCHINETTI V, LIU D, SOTO N, WEI S, JUNG SY, HUANG Q, QIN J. & SU B. 2006. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell, 127, 125–37. [DOI] [PubMed] [Google Scholar]
- KAIZUKA T, HARA T, OSHIRO N, KIKKAWA U, YONEZAWA K, TAKEHANA K, IEMURA S, NATSUME T. & MIZUSHIMA N. 2010. Tti1 and Tel2 are critical factors in mammalian target of rapamycin complex assembly. J Biol Chem, 285, 20109–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KIM HS, XIAO C, WANG RH, LAHUSEN T, XU X, VASSILOPOULOS A, VAZQUEZ-ORTIZ G, JEONG WI, PARK O, KI SH, GAO B. & DENG CX 2010. Hepatic-specific disruption of SIRT6 in mice results in fatty liver formation due to enhanced glycolysis and triglyceride synthesis. Cell Metab, 12, 224–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KLINGENSPOR M, EBBINGHAUS C, HULSHORST G, STOHR S, SPIEGELHALTER F, HAAS K. & HELDMAIER G. 1996. Multiple regulatory steps are involved in the control of lipoprotein lipase activity in brown adipose tissue. J Lipid Res, 37, 1685–95. [PubMed] [Google Scholar]
- KONG X, BANKS A, LIU T, KAZAK L, RAO RR, COHEN P, WANG X, YU S, LO JC, TSENG YH, CYPESS AM, XUE R, KLEINER S, KANG S, SPIEGELMAN BM & ROSEN ED 2014. IRF4 is a key thermogenic transcriptional partner of PGC-1alpha. Cell, 158, 69–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KONTANI Y, WANG Y, KIMURA K, INOKUMA KI, SAITO M, SUZUKI-MIURA T, WANG Z, SATO Y, MORI N. & YAMASHITA H. 2005. UCP1 deficiency increases susceptibility to diet-induced obesity with age. Aging Cell, 4, 147–55. [DOI] [PubMed] [Google Scholar]
- LABBE SM, MOUCHIROUD M, CARON A, SECCO B, FREINKMAN E, LAMOUREUX G, GELINAS Y, LECOMTE R, BOSSE Y, CHIMIN P, FESTUCCIA WT, RICHARD D. & LAPLANTE M. 2016. mTORC1 is Required for Brown Adipose Tissue Recruitment and Metabolic Adaptation to Cold. Sci Rep, 6, 37223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEAVENS KF, EASTON RM, SHULMAN GI, PREVIS SF & BIRNBAUM MJ 2009. Akt2 is required for hepatic lipid accumulation in models of insulin resistance. Cell Metab, 10, 405–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEE PL, JUNG SM & GUERTIN DA 2017. The Complex Roles of Mechanistic Target of Rapamycin in Adipocytes and Beyond. Trends Endocrinol Metab, 28, 319–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEE WN, BASSILIAN S, AJIE HO, SCHOELLER DA, EDMOND J, BERGNER EA & BYERLEY LO 1994. In vivo measurement of fatty acids and cholesterol synthesis using D2O and mass isotopomer analysis. Am J Physiol, 266, E699–708. [DOI] [PubMed] [Google Scholar]
- LEE WN, BASSILIAN S, LIM S. & BOROS LG 2000. Loss of regulation of lipogenesis in the Zucker diabetic (ZDF) rat. Am J Physiol Endocrinol Metab, 279, E425–32. [DOI] [PubMed] [Google Scholar]
- LEROUX AE, SCHULZE JO & BIONDI RM 2018. AGC kinases, mechanisms of regulation and innovative drug development. Semin Cancer Biol, 48, 1–17. [DOI] [PubMed] [Google Scholar]
- LI Y, MA Z, JIANG S, HU W, LI T, DI S, WANG D. & YANG Y. 2017. A global perspective on FOXO1 in lipid metabolism and lipid-related diseases. Prog Lipid Res, 66, 42–49. [DOI] [PubMed] [Google Scholar]
- MANNING BD & TOKER A. 2017. AKT/PKB Signaling: Navigating the Network. Cell, 169, 381–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MASUI K, TANAKA K, AKHAVAN D, BABIC I, GINI B, MATSUTANI T, IWANAMI A, LIU F, VILLA GR, GU Y, CAMPOS C, ZHU S, YANG H, YONG WH, CLOUGHESY TF, MELLINGHOFF IK, CAVENEE WK, SHAW RJ & MISCHEL PS 2013. mTOR complex 2 controls glycolytic metabolism in glioblastoma through FoxO acetylation and upregulation of c-Myc. Cell Metab, 18, 726–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCCABE BJ, BEDERMAN IR, CRONIGER C, MILLWARD C, NORMENT C. & PREVIS SF 2006. Reproducibility of gas chromatography-mass spectrometry measurements of 2H labeling of water: application for measuring body composition in mice. Anal Biochem, 350, 171–6. [DOI] [PubMed] [Google Scholar]
- MENARD SL, CROTEAU E, SARRHINI O, GELINAS R, BRASSARD P, OUELLET R, BENTOURKIA M, VAN LIER JE, DES ROSIERS C, LECOMTE R. & CARPENTIER AC 2010. Abnormal in vivo myocardial energy substrate uptake in diet-induced type 2 diabetic cardiomyopathy in rats. Am J Physiol Endocrinol Metab, 298, E1049–57. [DOI] [PubMed] [Google Scholar]
- MICHISHITA E, MCCORD RA, BOXER LD, BARBER MF, HONG T, GOZANI O. & CHUA KF 2009. Cell cycle-dependent deacetylation of telomeric histone H3 lysine K56 by human SIRT6. Cell Cycle, 8, 2664–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MIHAYLOVA MM, VASQUEZ DS, RAVNSKJAER K, DENECHAUD PD, YU RT, ALVAREZ JG, DOWNES M, EVANS RM, MONTMINY M. & SHAW RJ 2011. Class IIa histone deacetylases are hormone-activated regulators of FOXO and mammalian glucose homeostasis. Cell, 145, 607–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOTTILLO EP, BALASUBRAMANIAN P, LEE YH, WENG C, KERSHAW EE & GRANNEMAN JG 2014. Coupling of lipolysis and de novo lipogenesis in brown, beige, and white adipose tissues during chronic beta3-adrenergic receptor activation. J Lipid Res, 55, 2276–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MUZUMDAR MD, TASIC B, MIYAMICHI K, LI L. & LUO L. 2007. A global double-fluorescent Cre reporter mouse. Genesis, 45, 593–605. [DOI] [PubMed] [Google Scholar]
- ORTEGA-MOLINA A, EFEYAN A, LOPEZ-GUADAMILLAS E, MUNOZ-MARTIN M, GOMEZ-LOPEZ G, CANAMERO M, MULERO F, PASTOR J, MARTINEZ S, ROMANOS E, MAR GONZALEZ-BARROSO M, RIAL E, VALVERDE AM, BISCHOFF JR & SERRANO M. 2012. Pten positively regulates brown adipose function, energy expenditure, and longevity. Cell Metab, 15, 382–94. [DOI] [PubMed] [Google Scholar]
- PAN PW, FELDMAN JL, DEVRIES MK, DONG A, EDWARDS AM & DENU JM 2011. Structure and biochemical functions of SIRT6. J Biol Chem, 286, 14575–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PETERSON TR, LAPLANTE M, THOREEN CC, SANCAK Y, KANG SA, KUEHL WM, GRAY NS & SABATINI DM 2009. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell, 137, 873–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PIETROCOLA F, GALLUZZI L, BRAVO-SAN PEDRO JM, MADEO F. & KROEMER G. 2015. Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab, 21, 805–21. [DOI] [PubMed] [Google Scholar]
- ROH HC, TSAI LT, LYUBETSKAYA A, TENEN D, KUMARI M. & ROSEN ED 2017. Simultaneous Transcriptional and Epigenomic Profiling from Specific Cell Types within Heterogeneous Tissues In Vivo. Cell Rep, 18, 1048–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROSENWALD M, PERDIKARI A, RULICKE T. & WOLFRUM C. 2013. Bi-directional interconversion of brite and white adipocytes. Nat Cell Biol, 15, 659–67. [DOI] [PubMed] [Google Scholar]
- ROSNER M. & HENGSTSCHLAGER M. 2008. Cytoplasmic and nuclear distribution of the protein complexes mTORC1 and mTORC2: rapamycin triggers dephosphorylation and delocalization of the mTORC2 components rictor and sin1. Hum Mol Genet, 17, 2934–48. [DOI] [PubMed] [Google Scholar]
- ROTHWELL NJ & STOCK MJ 1979. A role for brown adipose tissue in diet-induced thermogenesis. Nature, 281, 31–5. [DOI] [PubMed] [Google Scholar]
- ROWLAND LA, MAURYA SK, BAL NC, KOZAK L. & PERIASAMY M. 2016. Sarcolipin and uncoupling protein 1 play distinct roles in diet-induced thermogenesis and do not compensate for one another. Obesity (Silver Spring), 24, 1430–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SANCHEZ-GURMACHES J. & GUERTIN DA 2014. Adipocytes arise from multiple lineages that are heterogeneously and dynamically distributed. Nat Commun, 5, 4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SANCHEZ-GURMACHES J, HUNG CM & GUERTIN DA 2016. Emerging Complexities in Adipocyte Origins and Identity. Trends Cell Biol, 26, 313–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SANCHEZ-GURMACHES J, TANG Y, JESPERSEN NZ, WALLACE M, MARTINEZ CALEJMAN C, GUJJA S, LI H, EDWARDS YJK, WOLFRUM C, METALLO CM, NIELSEN S, SCHEELE C. & GUERTIN DA 2018. Brown Fat AKT2 Is a Cold-Induced Kinase that Stimulates ChREBP-Mediated De Novo Lipogenesis to Optimize Fuel Storage and Thermogenesis. Cell Metab, 27, 195–209 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SARBASSOV DD, GUERTIN DA, ALI SM & SABATINI DM 2005. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science, 307, 1098–101. [DOI] [PubMed] [Google Scholar]
- SAXTON RA & SABATINI DM 2017. mTOR Signaling in Growth, Metabolism, and Disease. Cell, 169, 361–371. [DOI] [PubMed] [Google Scholar]
- SCHEELE C. & NIELSEN S. 2017. Metabolic regulation and the anti-obesity perspectives of human brown fat. Redox Biol, 12, 770–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHRADER M, COSTELLO J, GODINHO LF & ISLINGER M. 2015. Peroxisome-mitochondria interplay and disease. J Inherit Metab Dis, 38, 681–702. [DOI] [PubMed] [Google Scholar]
- SCHREIBER R, DIWOKY C, SCHOISWOHL G, FEILER U, WONGSIRIROJ N, ABDELLATIF M, KOLB D, HOEKS J, KERSHAW EE, SEDEJ S, SCHRAUWEN P, HAEMMERLE G. & ZECHNER R. 2017. Cold-Induced Thermogenesis Depends on ATGL-Mediated Lipolysis in Cardiac Muscle, but Not Brown Adipose Tissue. Cell Metab, 26, 753–763 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SEALE P, BJORK B, YANG W, KAJIMURA S, CHIN S, KUANG S, SCIME A, DEVARAKONDA S, CONROE HM, ERDJUMENT-BROMAGE H, TEMPST P, RUDNICKI MA, BEIER DR & SPIEGELMAN BM 2008. PRDM16 controls a brown fat/skeletal muscle switch. Nature, 454, 961–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHIOTA C, WOO JT, LINDNER J, SHELTON KD & MAGNUSON MA 2006. Multiallelic disruption of the rictor gene in mice reveals that mTOR complex 2 is essential for fetal growth and viability. Dev Cell, 11, 583–9. [DOI] [PubMed] [Google Scholar]
- SIDOSSIS L. & KAJIMURA S. 2015. Brown and beige fat in humans: thermogenic adipocytes that control energy and glucose homeostasis. J Clin Invest, 125, 478–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TANG Y, WALLACE M, SANCHEZ-GURMACHES J, HSIAO WY, LI H, LEE PL, VERNIA S, METALLO CM & GUERTIN DA 2016. Adipose tissue mTORC2 regulates ChREBP-driven de novo lipogenesis and hepatic glucose metabolism. Nat Commun, 7, 11365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THOREEN CC, KANG SA, CHANG JW, LIU Q, ZHANG J, GAO Y, REICHLING LJ, SIM T, SABATINI DM & GRAY NS 2009. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem, 284, 8023–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TRAYHURN P. 1981. Fatty acid synthesis in mouse brown adipose tissue. The influence of environmental temperature on the proportion of whole-body fatty acid synthesis in brown adipose tissue and the liver. Biochim Biophys Acta, 664, 549–60. [DOI] [PubMed] [Google Scholar]
- VON ESSEN G, LINDSUND E, CANNON B. & NEDERGAARD J. 2017. Adaptive facultative diet-induced thermogenesis in wild-type but not in UCP1-ablated mice. Am J Physiol Endocrinol Metab, 313, E515–E527. [DOI] [PubMed] [Google Scholar]
- WAN M, EASTON RM, GLEASON CE, MONKS BR, UEKI K, KAHN CR & BIRNBAUM MJ 2012. Loss of Akt1 in mice increases energy expenditure and protects against diet-induced obesity. Mol Cell Biol, 32, 96–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG B, ZWAANS BM, ECKERSDORFF M. & LOMBARD DB 2009. The sirtuin SIRT6 deacetylates H3 K56Ac in vivo to promote genomic stability. Cell Cycle, 8, 2662–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG D, DIRAISON F, BEYLOT M, BRUNENGRABER DZ, SAMOLS MA, ANDERSON VE & BRUNENGRABER H. 1998. Assay of low deuterium enrichment of water by isotopic exchange with [U-13C3]acetone and gas chromatography-mass spectrometry. Anal Biochem, 258, 315–21. [DOI] [PubMed] [Google Scholar]
- ZECHNER R, ZIMMERMANN R, EICHMANN TO, KOHLWEIN SD, HAEMMERLE G, LASS A. & MADEO F. 2012. FAT SIGNALS--lipases and lipolysis in lipid metabolism and signaling. Cell Metab, 15, 279–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHONG L, D’URSO A, TOIBER D, SEBASTIAN C, HENRY RE, VADYSIRISACK DD, GUIMARAES A, MARINELLI B, WIKSTROM JD, NIR T, CLISH CB, VAITHEESVARAN B, ILIOPOULOS O, KURLAND I, DOR Y, WEISSLEDER R, SHIRIHAI OS, ELLISEN LW, ESPINOSA JM & MOSTOSLAVSKY R. 2010. The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell, 140, 280–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2. Oligonucleotides used in this study, related to Star Methods