

Abstract
The ornithine transcarbamylase (OTC) gene is on the X chromosome and its product catalyzes the formation of citrulline from ornithine and carbamylphosphate in the urea cycle. About 10–15% of patients, clinically diagnosed with OTC deficiency (OTCD), lack identifiable mutations in the coding region or splice junctions of the OTC gene on routine molecular testing. We collected DNA from such patients via retrospective review and by prospective enrollment. In nine of 38 subjects (24%), we identified a sequence variant in the OTC regulatory regions. Eight subjects had unique sequence variants in the OTC promoter and one subject had a novel sequence variant in the OTC enhancer. All sequence variants affect positions that are highly conserved in mammalian OTC genes. Functional studies revealed reduced reporter gene expression with all sequence variants. Two sequence variants caused decreased binding of the HNF4 transcription factor to its mutated binding site. Bioinformatic analyses combined with functional assays can be used to identify and authenticate pathogenic sequence variants in regulatory regions of the OTC gene, in other urea cycle disorders or other inborn errors of metabolism.
Keywords: ornithine transcarbamylase, ornithine transcarbamylase deficiency, hyperammonemia, gene regulation, promoter mutation, enhancer mutation, urea cycle, hyperammonemia, gene expression
Introduction
The urea cycle functions in the liver where it converts ammonia, a neurotoxic product of protein catabolism, into urea, which is excreted in urine (Brusilow & Horwich, 2001). Ornithine transcarbamylase (OTC; EC 2.1.3.3, MIM# 300461) is a mitochondrial enzyme that catalyzes the second reaction of the urea cycle: the formation of L-citrulline from L-ornithine and carbamylphosphate. The human OTC gene, located on the short arm of the X chromosome (Xp11.4), is 70 kb long and has ten exons which contain a 1062 bp coding sequence (Hata et al., 1986). This gene codes for a protein comprised of 354 amino acids, including a 32 amino acid mitochondrial targeting peptide at its N-terminus (Horwich et al., 1984; Horwich, Kalousek, Fenton, Pollock, & Rosenberg, 1986). Primary OTC deficiency (OTCD; MIM# 311250) is caused by mutations in the OTC gene that lead to either reduced or absent functional OTC enzyme, thus limiting ammonia flux through the urea cycle. This results in the accumulation of blood ammonia, which may manifest as lethargy, vomiting, behavioral and neurological abnormalities, and, in severe cases, coma and death (Breningstall, 1986). In addition to high plasma ammonia, biochemical abnormalities associated with OTCD include elevated plasma glutamine, low or absent citrulline, and increased excretion of orotic acid and orotidine in the urine (Brusilow & Horwich, 2001).
Because the human OTC gene is located on the X-chromosome, severe OTCD primarily affects hemizygous males and accounts for approximately one-half of all urea cycle disorders (Lindgren, de Martinville, Horwich, Rosenberg, & Francke, 1984). Male hemizygotes with mutations that abrogate or severely impair OTC function invariably exhibit hyperammonemia within the first week of life (Ah Mew et al., 2013; McCullough et al., 2000). Males with hypomorphic OTC alleles that retain residual enzyme activity, or female heterozygotes with skewed lyonization (Caldovic, Abdikarim, Narain, Tuchman, & Morizono, 2015; McCullough et al., 2000) typically present symptomatically after the first week of life (Caldovic et al., 2015; Numata et al., 2010). The true prevalence of OTCD is unknown, but it has been estimated to be between 1 in 14,000 and 1 in 76,000 (Balasubramaniam et al., 2010; Brusilow & Maestri, 1996; Dionisi-Vici et al., 2002; Nettesheim et al., 2017; Summar et al., 2013). In 85–90% of patients with the biochemical phenotype of OTCD, a mutation can be identified through commercially available sequencing or deletion/duplication testing. In the remaining 10–15% of affected individuals, a molecular cause of OTCD cannot be ascertained through clinical testing. In a few such instances, mutations were ultimately identified in intronic or regulatory regions (Caldovic et al., 2015; Engel et al., 2008; Luksan, Jirsa, Eberova, Minks, Treslova, Bouckova, Storkanova, Vlaskova, Hrebicek, & Dvorakova, 2010). Mutations in these regions may, in fact, be responsible for a large proportion of OTCD, in patients with no identified variants via current clinical methods. However, sequencing of “deep” intronic or regulatory regions is currently neither routine nor clinically available.
OTC is expressed in the liver and intestine of humans and other mammals (Brusilow & Horwich, 2001). Transcription of the human OTC gene appears to initiate at multiple transcription start sites (TSS) (Luksan et al., 2010), while transcription of the mouse and rat Otc genes initiate 136 bp and 98 bp upstream of the translation initiation codon, respectively (Takiguchi, Murakami, Miura, & Mori, 1987; Veres, Craigen, & Caskey, 1986). In the rat Otc promoter, four regions, A through D, bind transcription factors that regulate expression of the Otc gene (Figure 1; (Murakami, Nishiyori, Takiguchi, & Mori, 1990)). The rat Otc promoter was sufficient to direct expression of transgenes in the liver and intestine of transgenic mice, but expression was higher in the intestine than in the liver (Jones et al., 1990; Murakami, Takiguchi, Inomoto, Yamamura, & Mori, 1989). Expression studies in cultured cells and transgenic animals revealed that an enhancer, located approximately 11 kb upstream of the first exon of the rat Otc gene, is essential for a high level of expression of the Otc gene in the liver (Murakami et al., 1990). In vitro binding studies revealed four sites, designated I-IV, that are important for the function of the rat −11 kb enhancer (Figure 2 (Murakami et al., 1990)).
Figure 1.

Promoter region of the human OTC gene. A. Sequence of the human OTC promoter. Sequence variants identified in this study are shown in red and underlined typeface. Angled arrows indicate transcription start sites in human OTC gene. Sequence variants found in study subjects are: NM_000531.5(OTC_v001):c.−106C>A, NM_000531.5(OTC_v001):c.−116C>T, NM_000531.5(OTC_v001):c.−115C>T, NM_000531.5(OTC_v001):c.−139A>G, NM_000531.5(OTC_v001):c.−142G>A, NM_000531.5(OTC_v001):c.−157T>G, NM_000531.5(OTC_v001):c.−9384G>T. B. LOGO representation of the multiple sequence alignment of OTC promoters from 36 mammals. The height of each letter corresponds to its conservation. Sequences that correspond to experimentally identified transcription factor binding sites in the rat Otc promoter are highlighted in yellow. Known transcription factor binding sites in the rat Otc promoter are shown in cyan and predicted transcription factor binding sites are shown in green.
Figure 2.
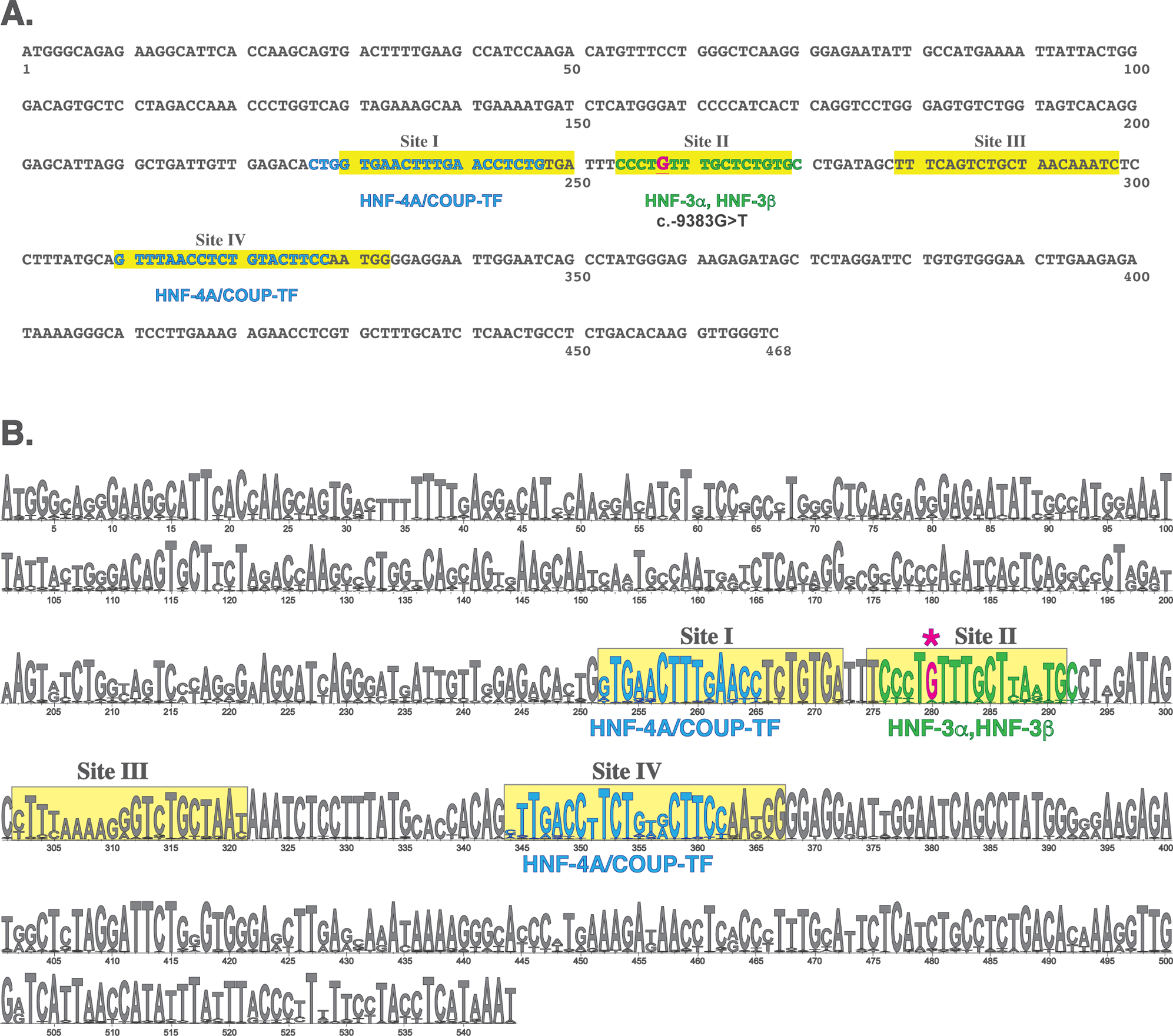
Liver-specific enhancer (LSE) of the human OTC gene. A. Sequence of the human LSE. Sequence variant identified in this study is shown in red and underlined typeface. B. LOGO representation of the multiple sequence alignment of OTC enhancers from 37 mammals. The height of each letter corresponds to its conservation. Sequences that correspond to experimentally identified transcription factor binding sites in the rat Otc enhancer are highlighted in yellow. Known transcription factor binding sites in the rat Otc promoter are shown in cyan and predicted transcription factor binding sites are shown in green.
In this study, we screened conserved upstream regulatory regions of the OTC gene in 38 subjects with clinically diagnosed OTCD, but in whom no deleterious OTC mutation was identified by clinical sequencing of the coding region or splice junctions. Eight of these subjects were found to harbor one of six unique sequence variants in the OTC promoter. One subject had a novel sequence variant in the OTC enhancer. Five sequence variants were within either known or predicted transcription factor binding sites and the remaining two affected base pairs that are highly conserved in vertebrates. Six sequence variants were not found in any of the databases of single nucleotide polymorphisms. Functional testing confirmed that all seven sequence variants described herein cause either reduced expression of reporter gene in cultured cells or reduced binding of the hepatic nuclear factor 4 (HNF4) transcription factor. These results highlight the importance of seeking sequence variants in regulatory regions of genes of patients with OTCD without identifiable mutations as well as the need for better and simpler functional testing of regulatory mutations in disease-causing genes.
Materials and Methods
Study Subjects
This project, which included retrospective and prospective components, was conducted with the approval of the Institutional Review Board of the Children’s National Health System. Inclusion criteria were: suspected urea cycle disorder with reduced or absent OTC enzymatic activity in the liver biopsy and/or urinary orotate above 30 μmoles/mmole of urinary creatinine, but absence of a disease-causing mutation in the exons and intron/exon boundaries of the OTC gene. When available, gender, age of disease onset, enzymatic activity of CPS1 in the liver biopsy, and concentrations of ammonia and citrulline in the plasma were collected for eligible patients. All participants were probands. Participants and their families were also prospectively enrolled into the study after referral by their metabolic physicians. In the retrospective study component, participants were selected from a database of de-identified patients who were screened for mutations in the coding region and intron/exon junctions of the OTC gene at the Children’s National Health System Molecular Genetics Laboratory. Of 1035 patients in the database 106 were eligible for this study; 43 were diagnosed with OTCD based on reduced or absent OTC activity in the liver, and the remaining 63 had a documented history of hyperammonemia and elevated urinary orotate. DNA was available from 24 subjects. Subjects 1–4 were ascertained retrospectively and subjects 5–9 were participants in the prospective study.
Subjects with Sequence Changes in the Regulatory Regions of the OTC Gene.
Subject 1 was a male diagnosed with late-onset OTCD based on OTC activity of 6.5 μmoles min−1 g−1, which was approximately one-tenth of the control value (56.7 μmoles min−1 per gram of liver tissue). The CPS1 activity in the same liver sample was normal (2.9 μmoles min−1 g−1). Values of urinary orotate, plasma ammonia, glutamine, and citrulline were not available.
Subject 2 was a male diagnosed with late onset OTCD based on OTC activity of 10.3 μmoles min−1 g−1, which was approximately one-tenth of the control value (110.6 μmoles min−1 per gram of liver tissue). The CPS1 activity in the same liver biopsy was normal (6.1 μmoles min−1 g−1). His urinary orotate concentration was 77 μmoles/mmole creatinine (normal < 1.2), and plasma ammonia, glutamine and citrulline concentrations were 92 μM (normal < 32), 1064 μM (normal 205 – 756) and 17 μM (normal 12 – 55), respectively.
Subject 3, a male, was included in the study based on the high concentration of urinary orotate (261 μmoles/mmole creatinine), and plasma ammonia, glutamine and citrulline concentrations of 1564 μM, 2715 μM and 37 μM, respectively.
Subject 4 was a male with neonatal onset hyperammonemia and no measurable OTC activity in the liver. The CPS1 activity in the liver biopsy was also absent. His urinary orotate concentration was 1624 μmoles/mmole creatinine, while plasma ammonia and glutamine concentrations were 683 μM, and 4442 μM, respectively.
Subject 5 was an asymptomatic adult male whose brother died from hyperammonemic encephalopathy. Post-mortem examination of the liver revealed decreased OTC activity. At his initial evaluation, the subject’s ammonia was 24 μM, glutamine 731 μM, citrulline 28 μM. Urinary orotate was >50 μmoles/mmole creatinine.
Subject 6 was a male who presented with hyperammonemia after the newborn period and was subsequently hospitalized on multiple occasions for hyperammonemia despite therapy with dietary protein restriction, ammonia scavengers, and arginine. The family history was suggestive of an X-linked recessive hyperammonemia disorder. The proband’s maternal uncle also presented in childhood with a hyperammonemic episode, has not had further episodes, though details of his management were not available.
Subject 7 was a male who presented with neonatal-onset hyperammonemia of 797 μM, glutamine 2082 μM, citrulline 7 μM. Urinary orotate was 434 μmoles/mmole of urinary creatinine. At age 5, on no medical or dietary therapy, this subject’s ammonia was 57 μM, glutamine 1258 μM, citrulline 22 μM and orotic acid 36.4 μmoles/mmole of urinary creatinine.
Subject 8 was a 32-year-old adult male who has had three hospitalizations for altered mental status. Ammonia, only measured on the third hospitalization, was 160 μM, glutamine 1173 μM, citrulline 48 μM. Urine organic acids demonstrated an elevated orotic acid peak which was not quantified. Whole exome sequencing revealed no pathogenic sequence variants. Exome coverage was 100% for OTC and CPS1, 98–100% for the other urea cycle genes, except for NAGS, which was only 73%. However, this patient participated in a clinical trial of N-carbamylglutamate, and did not exhibit the marked enhancement in ureagenesis observed in other NAGS deficient patients (Caldovic et al., 2004; Tuchman et al., 2008).
Subject 9 was a 23-year-old male who presented at age 23 with ammonia > 200 μM, glutamine 1160 μM, Arginine 93 μM (normal 15 – 128), Citrulline 47 μM. Orotic acid was normal at 0.65 μmoles/mmole creatinine, but following allopurinol challenge, peak orotic acid was 51.3 μmoles/mmole creatinine. Standard clinical DNA sequencing of NAGS, CPS1, OTC revealed no mutations. He has had multiple episodes of hyperammonemia. His family history was also notable for a brother who died in childhood due to ‘Reye Syndrome’.
Bioinformatic analysis of the upstream regulatory regions of mammalian OTC genes, identification of transcription factor binding sites, mutation analysis of the conserved regulatory regions of the human OTC gene, DNA-protein pull-down assay, plasmid construction, site-directed mutagenesis, and reporter assays in cultured cells are described in detail in the supplementary material. Nucleotide numbering uses +1 as the A of the ATG translation initiation codon in the reference sequence NM_000531.5. All seven variants have been submitted to the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/) with the following submission numbers: SUB3318191 (c.−106C>A), SUB3336937 (c.−115C>T), SUB3336835 (c.−116C>T), SUB3337149 (c.−139A>G), SUB3337152 (c.−142G>A), SUB3337155 (c.−157T>G), and SUB3337158 (c.−9384G>T).
Results
Regulatory Elements in Mammalian OTC Genes
We used bioinformatic tools to identify regions of conservation within the 5’-flanking sequences of mammalian OTC genes reasoning that sequence variants in the corresponding regions of the human OTC gene are likely to affect its expression. Pair-wise comparisons identified three conserved regions upstream of the first exon of OTC. The first region is approximately 600 bp long and is located immediately upstream of the initiating ATG codon. This area encompasses the OTC promoter and the 5’-untranslated regions of the OTC mRNA. The second region, the human OTC enhancer (Luksan et al., 2010), is approximately 900 bp long and corresponds to the −11 kb enhancer in the rat Otc gene (Murakami et al., 1990; Nishiyori et al., 1994). Because this enhancer is essential for maximal expression of Otc in the liver (Nishiyori et al., 1994), we termed this cis-acting element “liver specific enhancer” (LSE;). Its distance from the OTC promoter varies between 5 and 11 kb in the eight examined mammals. In human OTC gene, the LSE is located approximately 9 kb upstream of the promoter. The third region, which has not been identified before, is located approximately 2 kb upstream of the LSE in eight examined mammals. We termed this sequence “upstream regulatory element” (URE).
Mutation Analysis of the OTC Gene Regulatory Regions in Patients with OTCD
The OTC promoter was amplified and sequenced in all 38 subjects, while the LSE and URE were screened for mutations in 11 participants of the retrospective and 14 participants of the prospective study. LSE and URE could not be analyzed in 13 participants of the retrospective study because PCR of their genomic DNA did not yield sufficient amplification products for sequencing. This was likely due to either deterioration of their genomic DNA during prolonged storage or absence of the LSE and URE from the subjects’ genomic DNA. Sequencing results were compared to a wild-type sequence from an individual without OTCD and to the reference sequence of the human genome. The sequences of URE in 25 subjects were identical to the wild-type sequence and the reference human genome. Six sequence variants were found in the promoters of eight male subjects with OTCD (Figure 1). Variants c.−106C>A, c.−115C>T and c.−116C>T are located in region C, where HNF4 binds and activates transcription of the rat Otc gene (Figure 1) (Kimura et al., 1993; Murakami et al., 1990). Subjects 1, 2 and 5 had a c.−106C>A transversion, which is immediately adjacent to the HNF4 binding site (Figure 1), and therefore it could disrupt the binding of this transcription factor and activation of OTC transcription. Subject 3 had c.−115C>T and subject 8 had the c.−116C>T variant; these two sequence variants affect base pairs that are highly conserved within the HNF4 binding sites (Bolotin, Schnabl, & Sladek) and within mammalian OTC promoters (Figure 1B). Variants c.−139A>G and c.−142G>A, found in subjects 7 and 4 respectively, are located between previously identified regions B and C (Murakami et al., 1990); both sequence variants affect base pairs that are invariant in the alignment of 36 mammalian OTC promoters (Figure 1B) but do not coincide with known or predicted binding sites for transcription factors (Figure 1). Subject 6 has a c.−157T>G sequence variant; his asymptomatic mother is a carrier of the G allele while his asymptomatic brother has the T allele. The c.−157T>G variant affects a highly conserved base pair and is located in the predicted GATA transcription factor binding site (Figure 1).
A novel sequence variant is located 9384 base pairs upstream of the OTC translation initiation site in subject 9. This c.−9384G>T transversion is within a sequence that corresponds to Site II of the rat Otc enhancer that binds the CCAAT/enhancer-binding protein beta (C/EBPβ) transcription factor (Murakami et al., 1990) (Figure 2). The c.−9384G>T variant affects a base pair that is conserved in 33 of 37 mammalian genomes (Figure 2B).
We queried build 147 of dbSNP to determine if the seven novel sequence variants found in our study subjects have been reported previously. The c.−106C>A sequence variant has been observed in one heterozygous female of Northern and Western European descent in the 1000 Genomes Project and annotated as rs7497485506. The other six sequence variants have not been reported in dbSNP147.
Three different measures of evolutionary conservation of the seven affected base pairs was used to evaluate whether they may be important for the OTC gene function: genomic evolutionary rate profiling (GERP) (Cooper et al., 2010; Goode et al., 2010), PhyloP (Siepel et al., 2005), which is a measure evolutionary constraint of each base pair of the human genome, and PhastCons, which estimates the probability that a base pair is within a conserved element (Siepel et al., 2005). The GERP, PhyloP and PhastCons scores were extracted from the UCSC Genome Browser database. The GERP and PhyloP scores of the base pair affected by the c.−106C>A variant are close to zero suggesting little or no evolutionary pressure for conservation of the affected base pair (Table 1). However, the high PhastCons score of base pair affected by the c.−106C>A indicated that it is a part of a conserved element (Table 1) suggesting that replacement of the C at this position could be deleterious to OTC expression. The GERP and PhyloP scores of base pairs affected by variants c.−115C>T, c.−116C>T, c.−139A>G, c.−142G>A, and c.−9384G>T indicate evolutionary constraint against changes, while their PhastCons scores indicate that base pairs affected by these variants are within conserved elements and are likely to be important for regulation of the OTC gene (Table 1). The base pair affected by the c.−157T>G variant has discordant GERP and PhyloP scores (Table 1) while its PhastCons score of zero indicated that affected base pair is not within a conserved element.
Table 1. Conservation scores and predicted effects of sequence variants found in patients with OTCD.
| Subject | Variant | Conservation Score | Predicted Effect | |||
|---|---|---|---|---|---|---|
| GERPa | PhyloP 100Wayb |
PhastCons 100Wayc |
CADD C-score |
Mutation Taster |
||
| 1, 2, 5 | c.−106C>Ae | 0.08 | 0.95 | 0.992 | 13.55 | Polymorphism |
| 3 | c.−115C>T | 5.66 | 3.94 | 1.000 | 6.57 | Disease Causing |
| 8 | c.−116C>T | 5.66 | 3.94 | 1.000 | 14.47 | Disease Causing |
| 7 | c.−139A>G | 5.66 | 3.08 | 1.000 | 14.14 | Disease Causing |
| 4 | c.−142G>A | 5.13 | 4.74 | 1.000 | 20.30 | Disease Causing |
| 6 | c.−157T>G | 4.31 | 0.25 | 0.000 | 19.97 | Disease Causing |
| 9 | c.−9384G>T | 2.57 | 1.55 | 1.000 | 15.33 | nad |
GERP scores range between −12.36 and 6.18.
PhyloP100Way scores range between −20 and 7.532.
PhastCOns100Way scores range between 0 and 1.
Prediction not available
Nucleotide numbering uses +1 as the A of the ATG translation initiation codon in the reference sequence NM_000531.5
Two different mutation effect prediction tools, the Combined Annotation Dependent Depletion (CADD; Webserver v1.3) (Kircher et al., 2014) and MutationTaster2 (Schwarz, Cooper, Schuelke, & Seelow, 2014), were used to evaluate whether sequence variants found in the study subjects could be the cause of OTCD. The C-scores of variants c.−106C>A, c.−116C>T, c.−139A>G, c.−142G>A, c.−157T>G, and c.−9384G>T indicated that the six sequence variants are among 5% most deleterious sequence variants of the human genome (Table 1). The lower C-score of variant c.−115C>T suggests a lower likelihood of deleterious effect on OTC function (Table 1). MutationTaster2 predicted that variants c.−115C>T, c.−116C>T, c.−139A>G, c.−142G>A, and c.−157T>G are disease-causing, while variant c.−106C>A was classified as a polymorphism, most likely because it has been seen in a heterozygous female (Table 1) who could potentially have been an asymptomatic carrier of OTCD.
Functional Testing of OTC Promoter Variants
The predicted effect of each of the six sequence variants on the expression of human OTC was tested in cultured cells using luciferase reporter gene assays. Expression of luciferase reporter gene was controlled by either wild-type or mutant human OTC promoter. Expression plasmid without an OTC promoter insert was used as a negative control. HepG2 and HuH-7 cells were transfected with plasmids harboring either wild-type (phOTC-P) or mutant human OTC promoter and pGL4.74 plasmid containing Renilla luciferase hRluc gene as a transfection efficiency control.
Relative luciferase activity was lower in the HepG2 cells transfected with plasmids containing c.−106C>A, c.−139A>G, c.−142G>A and c.−157T>G mutant promoters than in the HepG2 cells transfected with a plasmid containing wild-type human OTC promoter (Figure 3A). A trend towards lower relative luciferase activity was observed in the HepG2 cells transfected with a plasmid containing c.−116C>T mutant promoter (Figure 3A). Relative luciferase activities in HepG2 cells transfected with plasmids containing either wild-type or c.−116C>T OTC promoter were 1.03±0.03 and 0.94±0.04 (mean±SEM, n=6; p=0.10), respectively. HepG2 cells transfected with plasmids harboring either wild-type or c.−115C>T human OTC promoters had similar relative luciferase activities (Figure 3A). Relative luciferase activity was lower in the HuH-7 cells transfected with any of the plasmids harboring mutated promoters than in the cells that were transfected with a plasmid containing wild-type human OTC promoter (Figure 3B).
Figure 3.

Effects of promoter sequence variants found in study subjects on reporter gene expression in HepG2 (A) and HuH-7 (B) cell lines. All results are averages of either two or three independent experiments, each carried out in triplicate. Results are shown as mean ± SEM. One asterisk (*) indicates P≤0.05, three asterisks (***) indicate P≤0.001, and four asterisks (****) indicate P≤0.0001.
Functional Testing of OTC Enhancer Variant
The effect of c.−9384G>T sequence variant on the expression of human OTC was tested with plasmids P_wtE and P_−9384G>T that harbor the OTC promoter and either wild-type or mutant OTC enhancer. Expression plasmid without promoter and enhancer was used as a negative control and pGL4.74 was a transfection efficiency control. Relative luciferase activity was higher in both HepG2 and HuH-7 cells transfected with plasmid P_wtE than in cells transfected with phOTC-P (Figure 4) indicating that OTC enhancer increases reporter gene expression in both cell types. When HepG2 cells were transfected with plasmid P_−9384G>T, harboring the c.−9384G>T variant, relative luciferase activity was approximately one third lower than in HepG2 cells transfected with plasmid P_wtE (Figure 4A). Relative luciferase activity was approximately six-fold lower in the HuH-7 cells transfected with the P_−9384G>T, which contains the c.−9384G>T variant, than in the HuH-7 cells transfected with the P_wtE (Figure 4B). These results strongly suggest that the c.−9384G>T variant could be the cause of OTCD.
Figure 4.

Effect of the c.−9383G>T sequence variant on expression of the luciferase gene in HepG2 (A) and HuH-7 (B) cell lines. All results are averages of three independent experiments, each carried out in triplicate. Results are shown as mean ± SEM. One asterisk (*) indicates P≤0.05, two asterisks (**) indicate P≤0.01, and four asterisks (****) indicate P≤0.0001.
Pull-Down Assays
Because of their location, variants c.−106C>A, c.−115C>T and c.−116C>T could affect the binding of the HNF4 and chicken ovalbumin upstream promoter transcription factor (COUP-TF) and decrease transcription of the OTC gene. DNA pull-down assays were used to determine if c.−106C>A and c.−115C>T affect binding of HNF4 and COUP-TF to region C of the OTC promoter. Binding of COUP-TF to wild-type region C was not detectable using DNA pull-down assay. Studies in cultured cells have shown that deletion of region C from reporter constructs had a marginal effect on the expression of a reporter gene (Kimura et al., 1993), suggesting that region C may not be important for repression of OTC transcription by COUP-TF. To isolate the effects of c.−106C>A and c.−115C>T variants on region C, wild-type (pWT) and mutated (pSNP1 and pSNP2, respectively) probes were used in DNA pull-down experiments. DNA sequence of similar length located outside OTC promoter and non-biotinylated DNA were used as negative controls. Figure 5 shows results that are representative of three DNA pull-down experiments. The migration of two bands (approximately 44 and 52 kDa), indicative of HNF4 binding to pWT, pSNP1, and pSNP2 (lanes 1, 5 and 7 in Figure 5), were in good agreement with the predicted sizes of two HNF4 isoforms, translated from alternatively spliced transcripts (Bolotin et al.). The bands in lane 5 were approximately 37±7% (mean±SEM; n=3) less intense than bands in lane 1 suggesting that pSNP1, containing c.−106C>A sequence variant, binds about 40% less HNF4 than pWT (Figure 5). The bands in lane 7 were approximately 67±13% (mean±SEM; n=3) less intense than bands in lane 1, suggesting that pSNP2, which contains the c.−115C>T sequence variant, binds less HNF4 than pWT (Figure 5). These data, combined with reduced expression of the reporter gene from the plasmid containing c.−106C>A mutant OTC promoter and absence of the c.−115C>T sequence variant from dbSNP147 suggest that these two variants are likely to cause OTCD by reducing binding of HNF4 to the OTC promoter, leading to reduced transcription of the OTC gene.
Figure 5.

Reduced binding of HNF4 to sites with c.−106C>A and c.−115C>T sequence variants. DNA pull-down assays were used to compare binding of HNF4 to its wild-type and mutated binding sites. Lanes: 1 – biotinylated DNA with wild-type binding site; 2 – unlabeled DNA with wild-type binding site; 3 – non-specific biotinylated DNA; 4 – pull-down without DNA; 5 – biotinylated binding site containing c.−106C>A variant; 6 – non-biotinylated binding site containing c.−106C>A variant; 7 – biotinylated binding site containing c.−115C>T variant; 8 – non-biotinylated binding site containing c.−115C>T variant. Results are representative of three independent experiments.
Discussion
Herein we report six sequence variants in the promoter of the OTC gene and one sequence variant within the LSE. Nine out of 38 screened patients with clinical and biochemical evidence of OTCD, but without identified disease-causing mutations in the coding region or splice junctions of the OTC gene, have potentially pathogenic sequence variants in the OTC promoter or LSE. All nine study subjects were male, three subjects had liver needle biopsy, which was part of diagnostic care, five subjects had elevated urinary orotate, and one had elevated urinary orotate in the allopurinol challenge test.
We compared sequences of the 5’-flanking regions from eight mammalian OTC genes and identified additional conserved element located approximately 2 kb upstream of the LSE (Kimura et al., 1993; Murakami et al., 1990; Nishiyori et al., 1994). This conserved element may be important for regulation of OTC gene expression but additional experiments are needed to investigate this hypothesis.
Mutations in transcription factor binding sites are known to cause disease, including in urea cycle disorders (Heibel et al., 2011). Establishing this kind of causation has required knowledge of either experimentally verified or predicted transcription factor binding sites and functional studies showing decreased binding and/or expression. Alignment of either promoter or enhancer sequences from more than 30 mammals revealed that binding sites for HNF4, COUP-TF, and C/EBP in the promoter and LSE of rat Otc gene (Murakami et al., 1990,Nishiyori, 1990) are conserved in most mammals. Three of the sequence variants, c.−106C>A, c.−115C>T and c.−116C>T, map either within or immediately adjacent to the HNF4/COUP-TF binding sites. Conservation of the base pair affected by the c.−106C>A variant, reduced luciferase activity in both HepG2 and HuH-7 cells transfected with constructs harboring this variant, reduced binding of the HNF4 transcription factor to the probe with the c.−106C>A variant, together with the reduced OTC activity in the livers of study subjects with the c.−106C>A and its absence in healthy males from the 1000 genomes projects, strongly suggest that the presence of the c.−106C>A variant leads to reduced OTC gene expression and OTCD. Variants c.−115C>T and c.−116C>T affect base pairs that are highly conserved in mammalian OTC promoters. The effects of c.−115C>T and c.−116C>T sequence variants on reporter gene expression differed in HepG2 and HuH-7 cells. Luciferase activity was 50% and 20% lower in HuH-7 cells transfected with constructs harboring c.−115C>T and c.−116C>T variants, respectively, while the presence of these two variants did not affect luciferase activity in HepG2 cells. The c.−115C>T sequence variant also decreased HNF4 binding to the mutated binding site in vitro and the study subject with this variant had reduced hepatic OTC activity. These results suggest that c.−115C>T and c.−116C>T variants could cause OTCD in the study subjects.
Sequence variants c.−139A>G, c.−142G>A and c.−157T>G are novel, affect base pairs that are highly conserved in mammalian OTC promoters and located between regions B and C. Variant c.−157T>G maps within the predicted GATA-1 transcription factor binding site. However, a functional role of the GATA-1 transcription factor in the regulation of the OTC gene expression is unlikely because this transcription factor is expressed primarily in hematopoietic cells (Mouthon et al., 1993) and its role in the regulation of hepatic genes remains to be explored. Functional studies in HepG2 and HuH-7 cells revealed deleterious effects of c.−139A>G, c.−142G>A and c.−157T>G variants on reporter gene expression suggesting that these three sequence variants are likely to be the cause of OTCD.
A novel sequence variant c.−9384G>T maps in the LSE, within Site II that has been shown to bind transcription factors in the rat Otc LSE (Murakami et al., 1990). Chromatin immunoprecipitation followed by high throughput sequencing revealed that hepatic nuclear factor 3 beta (HNF3B/FOXA2) and hepatocyte specific transcription factor FOXA1 bind to Site II in HepG2 cells (Gerstein et al., 2012; Wang et al., 2012; Wang et al., 2013). Reporter gene assays in HepG2 and HuH-7 cells revealed that the c.−9384G>T variant abolished enhancer function in both cell types. Therefore, it is likely that the c.−9384G>T variant is the cause of the subject’s OTCD.
Mutation analysis of OTC gene regulatory regions may provide a definitive molecular diagnosis for patients with clinical and biochemical features of OTCD but absence of pathogenic sequence variants in the OTC coding and splice regions. Identification of the causative mutations in such OTCD patients has broad clinical benefits, as it permits diagnosis of at-risk relatives as well as accurate prenatal testing.
We also highlight the development of a model system to ascertain pathogenic OTC regulatory mutations by coupling DNA sequencing of regulatory regions with bioinformatics and functional studies. We anticipate employing this system to rapidly identify and test regulatory sequence variants in patients with a biochemical diagnosis of OTCD but without a molecular diagnosis. Similar methodologies may potentially be applied to identify regulatory mutations in the other urea cycle disorders or hepatic inborn errors of metabolism.
Supplementary Material
Acknowledgments
This work was supported by public health service grants R01DK047870 and R01DK064913 from the National Institute of Diabetes Digestive and Kidney Diseases, National Institutes of Health, Department of Health and Human Services. Dr. Feeney’s work was supported by the Maryland HHMI Undergraduate Research Fellowship. Dr. Ah Mew’s work is generously supported by the Rashid Family Fund. The authors have no conflicts of interest to declare.
Grant Sponsor: R01DK047870 and R01DK064913
References
- Ah Mew N, Krivitzky L, McCarter R, Batshaw M, Tuchman M, & Urea Cycle Disorders Consortium of the Rare Diseases Clinical Research, (2013). Clinical outcomes of neonatal onset proximal versus distal urea cycle disorders do not differ. J Pediatr, 162(2), 324–329 e321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramaniam S, Rudduck C, Bennetts B, Peters G, Wilcken B, & Ellaway C (2010). Contiguous gene deletion syndrome in a female with ornithine transcarbamylase deficiency. Mol Genet Metab, 99(1), 34–41. [DOI] [PubMed] [Google Scholar]
- Bolotin E, Schnabl JK, & Sladek FM (2010, August 4, 2010). HNF4A. Transcription Factor Encyclopedia Retrieved from http://www.cisreg.ca/tfe
- Breningstall GN (1986). Neurologic syndromes in hyperammonemic disorders. Pediatr Neurol, 2(5), 253–262. [DOI] [PubMed] [Google Scholar]
- Brusilow SW, & Horwich AL (2001). Urea Cycle Enzymes In Scriver CR, Beaudet AL, Sly WS, & Valle D (Eds.), The Metabolic & Molecular Bases of Inherited Disease (Vol. 2, pp. 1909–1963): McGraw-Hill. [Google Scholar]
- Brusilow SW, & Maestri NE (1996). Urea cycle disorders: diagnosis, pathophysiology, and therapy. Adv Pediatr, 43, 127–170. [PubMed] [Google Scholar]
- Caldovic L, Abdikarim I, Narain S, Tuchman M, & Morizono H (2015). Genotype-Phenotype Correlations in Ornithine Transcarbamylase Deficiency: A Mutation Update. J Genet Genomics, 42(5), 181–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldovic L, Morizono H, Daikhin Y, Nissim I, McCarter RJ, Yudkoff M, & Tuchman M (2004). Restoration of ureagenesis in N-acetylglutamate synthase deficiency by N-carbamylglutamate. J Pediatr, 145(4), 552–554. [DOI] [PubMed] [Google Scholar]
- Cooper GM, Goode DL, Ng SB, Sidow A, Bamshad MJ, Shendure J, & Nickerson DA (2010). Single-nucleotide evolutionary constraint scores highlight disease-causing mutations. Nat Methods, 7(4), 250–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dionisi-Vici C, Rizzo C, Burlina AB, Caruso U, Sabetta G, Uziel G, & Abeni D (2002). Inborn errors of metabolism in the Italian pediatric population: a national retrospective survey. J Pediatr, 140(3), 321–327. [DOI] [PubMed] [Google Scholar]
- Engel K, Nuoffer JM, Muhlhausen C, Klaus V, Largiader CR, Tsiakas K, Haberle J (2008). Analysis of mRNA transcripts improves the success rate of molecular genetic testing in OTC deficiency. Mol Genet Metab, 94(3), 292–297. [DOI] [PubMed] [Google Scholar]
- Gerstein MB, Kundaje A, Hariharan M, Landt SG, Yan KK, Cheng C, Snyder M (2012). Architecture of the human regulatory network derived from ENCODE data. Nature, 489(7414), 91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goode DL, Cooper GM, Schmutz J, Dickson M, Gonzales E, Tsai M, Sidow A (2010). Evolutionary constraint facilitates interpretation of genetic variation in resequenced human genomes. Genome Res, 20(3), 301–310. doi: 10.1101/gr.102210.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata A, Tsuzuki T, Shimada K, Takiguchi M, Mori M, & Matsuda I (1986). Isolation and characterization of the human ornithine transcarbamylase gene: structure of the 5’-end region. J Biochem, 100(3), 717–725. [DOI] [PubMed] [Google Scholar]
- Heibel SK, Ah Mew N, Caldovic L, Daikhin Y, Yudkoff M, & Tuchman M (2011). N-carbamylglutamate enhancement of ureagenesis leads to discovery of a novel deleterious mutation in a newly defined enhancer of the NAGS gene and to effective therapy. Hum Mutat, 32(10), [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwich AL, Fenton WA, Williams KR, Kalousek F, Kraus JP, Doolittle RF, Rosenberg LE (1984). Structure and expression of a complementary DNA for the nuclear coded precursor of human mitochondrial ornithine transcarbamylase. Science, 224(4653), 1068–1074. [DOI] [PubMed] [Google Scholar]
- Horwich AL, Kalousek F, Fenton WA, Pollock RA, & Rosenberg LE (1986). Targeting of pre-ornithine transcarbamylase to mitochondria: definition of critical regions and residues in the leader peptide. Cell, 44(3), 451–459. [DOI] [PubMed] [Google Scholar]
- Jones SN, Grompe M, Munir MI, Veres G, Craigen WJ, & Caskey CT (1990). Ectopic correction of ornithine transcarbamylase deficiency in sparse fur mice. J Biol Chem, 265(24), 14684–14690. [PubMed] [Google Scholar]
- Kimura A, Nishiyori A, Murakami T, Tsukamoto T, Hata S, Osumi T, Takiguchi M (1993). Chicken ovalbumin upstream promoter-transcription factor (COUP-TF) represses transcription from the promoter of the gene for ornithine transcarbamylase in a manner antagonistic to hepatocyte nuclear factor-4 (HNF-4). J Biol Chem, 268(15), 11125–11133. [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, & Shendure J (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet, 46(3), 310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindgren V, de Martinville B, Horwich AL, Rosenberg LE, & Francke U (1984). Human ornithine transcarbamylase locus mapped to band Xp21.1 near the Duchenne muscular dystrophy locus. Science, 226(4675), 698–700. [DOI] [PubMed] [Google Scholar]
- Luksan O, Jirsa M, Eberova J, Minks J, Treslova H, Bouckova M, Dvorakova L (2010). Disruption of OTC promoter-enhancer interaction in a patient with symptoms of ornithine carbamoyltransferase deficiency. Hum Mutat, 31(4), E1294–1303. [DOI] [PubMed] [Google Scholar]
- McCullough BA, Yudkoff M, Batshaw ML, Wilson JM, Raper SE, & Tuchman M (2000). Genotype spectrum of ornithine transcarbamylase deficiency: correlation with the clinical and biochemical phenotype. Am J Med Genet, 93(4), 313–319. [DOI] [PubMed] [Google Scholar]
- Mouthon MA, Bernard O, Mitjavila MT, Romeo PH, Vainchenker W, & Mathieu-Mahul D (1993). Expression of tal-1 and GATA-binding proteins during human hematopoiesis. Blood, 81(3), 647–655. [PubMed] [Google Scholar]
- Murakami T, Nishiyori A, Takiguchi M, & Mori M (1990). Promoter and 11-kilobase upstream enhancer elements responsible for hepatoma cell-specific expression of the rat ornithine transcarbamylase gene. Mol Cell Biol, 10(3), 1180–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami T, Takiguchi M, Inomoto T, Yamamura K, & Mori M (1989). Tissue- and developmental stage-specific expression of the rat ornithine carbamoyltransferase gene in transgenic mice. Dev Genet, 10(5), 393–401. [DOI] [PubMed] [Google Scholar]
- Nettesheim S, Kolker S, Karall D, Haberle J, Posset R, Hoffmann GF, Swiss Paediatric Surveillance, (2017). Incidence, disease onset and short-term outcome in urea cycle disorders -cross-border surveillance in Germany, Austria and Switzerland. Orphanet J Rare Dis, 12(1), 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyori A, Tashiro H, Kimura A, Akagi K, Yamamura K, Mori M, & Takiguchi M (1994). Determination of tissue specificity of the enhancer by combinatorial operation of tissue-enriched transcription factors. Both HNF-4 and C/EBP beta are required for liver-specific activity of the ornithine transcarbamylase enhancer. J Biol Chem, 269(2), 1323–1331. [PubMed] [Google Scholar]
- Numata S, Koda Y, Ihara K, Sawada T, Okano Y, Matsuura T, Yoshino M (2010). Mutant alleles associated with late-onset ornithine transcarbamylase deficiency in male patients have recurrently arisen and have been retained in some populations. J Hum Genet, 55(1), 18–22. [DOI] [PubMed] [Google Scholar]
- Schwarz JM, Cooper DN, Schuelke M, & Seelow D (2014). MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods, 11(4), 361–362. [DOI] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Haussler D (2005). Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res, 15(8), 1034–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summar ML, Koelker S, Freedenberg D, Le Mons C, Haberle J, Lee HS, Members of the Urea Cycle Disorders Consortium (2013). The incidence of urea cycle disorders. Mol Genet Metab, 110(1–2), 179–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takiguchi M, Murakami T, Miura S, & Mori M (1987). Structure of the rat ornithine carbamoyltransferase gene, a large, X chromosome-linked gene with an atypical promoter. Proc Natl Acad Sci U S A, 84(17), 6136–6140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuchman M, Caldovic L, Daikhin Y, Horyn O, Nissim I, Korson M, Yudkoff M (2008). N-carbamylglutamate markedly enhances ureagenesis in N-acetylglutamate deficiency and propionic acidemia as measured by isotopic incorporation and blood biomarkers. Pediatr Res, 64(2), 213–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veres G, Craigen WJ, & Caskey CT (1986). The 5’ flanking region of the ornithine transcarbamylase gene contains DNA sequences regulating tissue-specific expression. J Biol Chem, 261(17), 7588–7591. [PubMed] [Google Scholar]
- Wang J, Zhuang J, Iyer S, Lin X, Whitfield TW, Greven MC, Weng Z (2012). Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res, 22(9), 1798–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Zhuang J, Iyer S, Lin XY, Greven MC, Kim BH, Weng Z (2013). Factorbook.org: a Wiki-based database for transcription factor-binding data generated by the ENCODE consortium. Nucleic Acids Res, 41(Database issue), D171–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.