

SUMMARY
Autophagy is a catabolic process involving capture of cytoplasmic materials into double-membraned autophagosomes that subsequently fuse with lysosomes for degradation of the materials by lysosomal hydrolases. One of the least understood components of the autophagy machinery is the transmembrane protein ATG9. Here, we report a cryoelectron microscopy structure of the human ATG9A isoform at 2.9-Å resolution. The structure reveals a fold with a homotrimeric domain-swapped architecture, multiple membrane spans, and a network of branched cavities, consistent with ATG9A being a membrane transporter. Mutational analyses support a role for the cavities in the function of ATG9A. In addition, structure-guided molecular simulations predict that ATG9A causes membrane bending, explaining the localization of this protein to small vesicles and highly curved edges of growing autophagosomes.
In Brief
Guardia et al. report a high-resolution cryo-EM structure of human ATG9A, the only transmembrane protein of the core autophagy machinery. The structure shows that ATG9A is a domain-swapped homotrimer with a complex network of internal cavities. Structure-based computational simulations predict that ATG9A has membrane-bending properties.
Graphical Abstract
INTRODUCTION
Macroautophagy (herein referred to as autophagy) is a cytoplasmic degradative process induced in response to a wide range of stimuli (Bento et al., 2016;Gatica et al., 2018). During autophagy, cells enclose cytoplasmic materials into double-membraned autophagosomes that subsequently fuse with lysosomes to form autolysosomes. Cells depend on autophagy for the clearance of abnormal particles, such as damaged organelles, protein aggregates, and intracellular pathogens, as well as for the release of essential nutrients by degradation of normal organelles during starvation (Bento et al., 2016; Gatica et al., 2018). Autophagy is thus essential for the maintenance of cellular homeostasis, particularly under conditions of stress, and its dysfunction is linked to various diseases, such as neurodegenerative disorders and cancer (Levine and Kroemer, 2019). The process of autophagy involves multiple steps, including the formation of a cup-shaped membrane structure termed phagophore in close association with the endoplasmic reticulum (ER), expansion of the phagophore membrane around captured autophagic substrates, closure of the membrane into a mature autophagosome, autophagosome-lysosome fusion, substrate degradation, and lysosome reformation (Bento et al., 2016; Gatica et al., 2018). These steps are mediated by a complex molecular machinery including more than 40 autophagy-related (ATG) proteins; most of which are cytosolic proteins that are recruited to membranes upon activation of autophagy (Bento et al., 2016). An exception is ATG9 (Lang et al., 2000; Noda et al., 2000), a multispanning membrane protein that is delivered to the forming autophagosome by vesicular transport from the trans-Golgi network (TGN) and/or endosomes (Young et al., 2006). This delivery is mediated by small (30–60 nm) vesicles that coalesce at phagophore-assembly sites (PASs) to nucleate other ATG proteins and/or enable the acquisition of lipids for expansion of the nascent autophagosome (Mari et al., 2010; Yamamoto et al., 2012). Despite the unique properties and critical role of ATG9 in autophagy, its exact molecular function remains unknown. A recent study reported a 7.8-Å-resolution structure of ATG9 from the plant Arabidopsis thaliana (Lai et al., 2019). That study concluded that plant ATG9 is a homotrimer, with each protomer having six transmembrane α helices and contributing to the formation of a central pore (Lai et al., 2019). At this resolution, however, the arrangement of the transmembrane helices, the nature of the trimeric interface, and other structural details were difficult to discern. To obtain further insights into the structure of ATG9, we used single-particle cryoelectron microscopy (cryo-EM) to solve the structure of the ubiquitous isoform of human ATG9, named ATG9A, to 2.9-Å resolution. The structure shows that human ATG9A is a homotrimer with a fold thus far not reported in any membrane protein. Unlike the reported structure of Arabidopsis thalianaATG9, our structure shows that each protomer of human ATG9A comprises four transmembrane helices. The homotrimeric interface in ATG9A involves extensive domain swapping in the membrane, and part of the C-terminal domain forms an extended helical platform, presumably for protein-protein interactions. ATG9A has an intriguing internal network of branched cavities throughout the trimer, which is consistent with its hypothetical role as a lipid transport protein. Functional experiments confirm the importance of cavity-lining residues and also reveal a requirement of the folded part of the C-terminal domain for interactions with ATG2A, another critically important protein in the autophagy pathway. Finally, molecular dynamics simulations show that ATG9A has membrane-bending properties that explain its localization to highly curved membranes in the cell.
RESULTS AND DISCUSSION
Homotrimeric Architecture of ATG9A Revealed by High-Resolution Cryo-EM Analysis
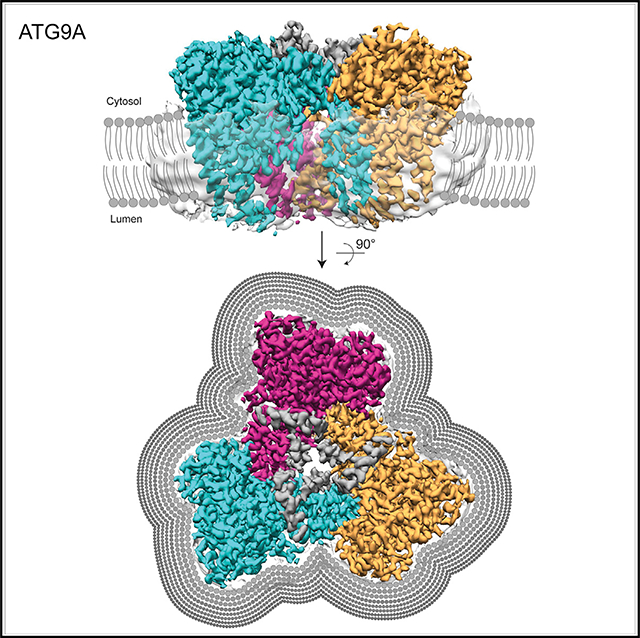
Given its physiological and biomedical importance, we decided to focus on the ubiquitous isoform of human ATG9, named ATG9A (Figure 1A). Extensive extraction and purification trials with fluorescent size-exclusion chromatography (FSEC) (Kawate and Gouaux, 2006; Rana et al., 2018) led to a robust purification scheme in lauryl maltose neopentyl glycol (LMNG). Recombinant human ATG9A was expressed in human HEK293S GnTI− cells (Goehring et al., 2014), followed by solubilization and purification in LMNG (Figure 1B). Cross-linking experiments showed that the purified recombinant protein (Figure 1C) and the endogenous protein (Figure 1D) could form trimers. Indeed, negative-stain EM of the purified preparation revealed a homogeneous sample with clear trimeric architecture (Figure 1E). Cryo-EM images of this preparation were collected using a 300-kV electron microscope, and data processing was performed with RELION3 and cryoSPARC2 (see Method Details; Figures S1–S3; Table S1). Three-dimensional reconstruction confirmed that ATG9A forms a homotrimeric complex with a 3-fold rotational symmetry (Figures 2A and S1). Image classification revealed two main conformations (named states A and B) in which the protomers were rotated by 8° (Figures S1, 2A, and 2B; Video S1). The motion occurring in individual protomers (EMDB: EMD-21877 [state A] and EMD-21878 [state B]) compromised the resolution of cryo-EM 3D reconstruction of the trimers. To further improve the structure of the ATG9A protomer, the signal of each protomer was extracted from cryo-EM images by symmetry expansion and density subtraction for focused refinement of a single protomer (see Method Details; EMDB: EMD-21874 [state A] and EMD-21876 [stateB]). Two models were built, one for each state (PDB: 6WQZ [state A]; PDB: 6WR4 [state B]); because of its better resolution, most of our subsequent structural description focuses on state A. The final model contains residues 36 to 587 (from a total of 839 amino acids) with missing loop residues 96–108 and 536–538. The model also includes two additional helices (αA and αB) in the C-terminal domain that were built as polyalanine helices because of their lack of connection to the rest of the model and because the side chains could not be built ab initio(Figures 2B–2D).
Figure 1. Trimeric Structure of Human ATG9A.

(A) Sequence alignment of the ATG9A core structure. Alignment is numbered according to human ATG9A, comprising amino acids 36–587 (from 839 total). Selected orthologs range in size from 839 to 997 amino acids; depicted are H. sapiens, D. rerio (36–583),C. elegans (102–663), D. melanogaster (87–644), S. cerevisiae (296–843), and A. thaliana (64–620) orthologs. Red boxes highlight 100% sequence identity. Residues highlighted in pink make notable structural contributions. Alignment was generated using the PRALINE webserver, and the figure was adapted from ESPript 3.0 output.
(B) Size-exclusion chromatogram and SDS-PAGE of LMNG-solubilized human ATG9A used for cryo-EM structure determination.
(C) Membranes containing ATG9A-mVenus fusion protein were cross-linked with DSS, run on SDS-PAGE, and imaged with in-gel fluorescence.
(D) Intact HeLa cells were cross-linked with different concentrations of DSS and analyzed by SDS-PAGE and immunoblotting with antibody to endogenous ATG9A. In (C) and (D), the positions of molecular mass markers are indicated on the left, and the positions of ATG9A monomer (mATG9A, Mr ~94 kDa), dimer (dATG9A, Mr ~188 kDa), and trimer (tATG9A, Mr ~282 kDa) are indicated on the right.
(E) Representative negative stain EM image of ATG9A particles, revealing trimeric architecture.
Figure 2. Cryo-EM Structure of ATG9A.

(A) Cryo-EM density map of ATG9A in both conformations (states A and B) is shown in side and top views and colored according to the protomer (cyan, magenta, and orange). Dark gray represents density without assigned sequence, and translucent surface shows disordered detergent molecules surrounding the transmembrane surface of the trimer (state A).
(B) The major conformational changes between the two states involve 8° tilting of each protomer. The central pore and cytosolic face of the trimer is occluded by part of the C-terminal domain that was visualized at low resolution (translucent light gray surface).
(C) Topology of the ATG9A protomer showing numbered α helices as rounded rectangles and β strands as filled arrows. In (A)–(C), the edges of the membrane are shown as gray lines.
(D) Structure of an isolated protomer (cyan) and two α helices from another protomer (magenta). Numbers correspond to α helices. The layers that define the surfaces of the wedge-shape monomer are colored in increasingly darker shades of cyan; the same colors are used in (C) for consistency. Polyalanine helices are colored gray.
ATG9A Protomers Have Four Transmembrane Helices and Assemble by Extensive Domain Swapping
The overall structure of ATG9A consists of a homotrimer in which each protomer resembles a triangular wedge (Figures 2A and 2B). The model for each protomer comprises 22 α helices and 4 β strands, starting with the N-terminal segment facing the cytosol followed by the entire transmembrane domain, and the cytosolic C-terminal domain up to residue 587 (Figures 1A,2C, and 2D). Most of the extramembranous part of the protein lies on the cytosolic side, with relatively short segments connecting the transmembrane helices on the luminal side (Figures 2A–2C). Unlike the published structure of plant ATG9 (Lai et al., 2019), our structure of human ATG9A shows four transmembrane helices (α2, α6, α14, and α15), and two helices (α11 and α19) that are partially embedded in the membrane but do not cross it completely (Figures 2C and 2D). Of these, α11 penetrates deeply into the membrane, and its C-terminal half runs nearly parallel to the transbilayer normal (Figures 2C and 2D). On the other hand, α6 is an unusually long helix that extends from the bilayer well into the cytosol, thus forming a structural anchor that contacts different parts within the protomer (Figures 2C and 2D).
Within the trimer, there is extensive domain-swapping in the transmembrane domain, where a two-helix unit consisting of α14 and α15 from one monomer crosses over and stacks in a parallel fashion with α2 and α6 from a neighboring monomer (Figures 2B–2D and 3A). This stacking forms a planar surface that projects out radially from the center of the trimer and creates one surface of the triangular wedge (Figures 2D and 3A). The two partially membrane-embedded helices (α11 and α19) form a severely kinked unit that binds this planar surface toward the luminal side of the bilayer, predominantly through the C-terminal half of α11, and, in doing so, makes the second, narrower surface of the triangular wedge (Figures 2D and 3A). The kinks in α11 and α19 are formed by highly conserved proline residues, Pro-302 in α11 and Pro-483 in α19 (Figure 1A). The transmembrane helices are linked by predominantly helical connectors and extended loops (Figures 2B–2D and 3A). The third surface of the wedge is formed partly by these helical connectors arranged in two distinct layers (Figures 2B–2D and 3A). One layer is formed by the N-terminal helix α1 on one side and α9 on the other. Although α9 is positioned within the bilayer, it is surrounded by the protein and makes extensive contacts with α11 and α19 through predominantly hydrophobic interactions (Figure 3E). α1 contacts α2 and α6 through similar hydrophobic interactions (Figure 3F). Two polar contacts, one between Asn-250 and Asn-297, and the other one a tripartite contact between Thr-244, Ser-482, and Thr-486, likely strengthening this interface in the hydrophobic environment and helping sequester the partial charges of these polar residues (Figure 3E). All of these residues are either highly conserved or retain similar side-chain chemistry in ATG9A orthologs (Figure 1A). A second layer of helices comprising α7, α8, and α20 on the cytosolic side bridges α2 and α11. α7 makes direct contact with α1, whereas α20 makes direct contact with α11, with α8 exhibiting a bent shape and acting as a helical bridge between α7 and α20. This bent shape exposes a backbone carbonyl oxygen at Arg-211, which is capped by the carboxylate from Asp-44, a residue that is highly conserved across all ATG9A orthologs (Figure 3F). Toward the center of the trimer, there is a small four-helix bundle formed between α13, α16, α17, and α18, which connects a two-helix insertion α14–α15 into the neighboring monomer to form a domain-swapped trimer in the membrane (Figures 2C, 2D, and 3D). After α15, the rest of the C-terminal domain forms an α-helical platform connected by extended loops, which may form sites of engagement for other proteins (Figure 2D).
Figure 3. ATG9A Protomers Share an Extensive Hydrophobic Interface of Trimerization.

(A) Overall trimeric structure of ATG9A, with the protomers colored cyan, orange, and magenta. The majority of the magenta protomer was hidden for clarity. Polyalanine helices A and B are colored gray. The edges of the membrane are shown as gray lines.
(B and C) Interaction between protomers in the cytosolic segment (B) and at the membrane-cytosol interface (C).
(D) Interaction between transmembrane helices of adjacent protomers. Interacting residues are shown in stick representation.
(E) The interface between the different layers of the wedge is formed by helices α9, α11, and α19. The inset shows a close view of the interacting residues with hydrogen bonds as dashed lines.
(F) The second layer is composed of helices α7, α8, and α20. The inset shows a close-up of the interacting region and residues between helices α1 and α8.
Structure and Interactions at the Homotrimeric Interface
The homotrimeric interface comprises two regions—one embedded in the membrane and the other one in the cytosol. The transmembrane interface arises from the domain-swapped interaction between α14–α15 from one monomer and α2–α6 from the neighboring monomer (Figures 2C and 2D). Embedded in the membrane, this interaction is primarily formed by α2 and α14 running in parallel and forming a series of hydrophobic interactions at the interface (Figure 3D). This interface is rich in phenylalanine residues, raising the possibility that conformational changes can be accommodated by rotations of the phenylalanine aromatic ring. The α2–α14 interface is stapled together by α11 running in a perpendicular direction (Figure 3B). On either side of the transmembrane trimeric interface, α11 contacts α6 from one monomer and α15 from another monomer, thus acting as a stapling anchor at that interface (Figure 3B). Together, each transmembrane interface buries 1,239 Å2 of surface area. Vertically above the transmembrane interface, the cytosolic part of the trimeric interface is comparatively small. One prominent interaction is formed by α7 packing against the α17–α18 loop (Figure 3C). The α13–α14 loop packs against the α17–α18 loop with the sidechain of Phe-368, a highly conserved residue. This engages the α17–α18 loop with the α7 of the neighboring monomer, primarily through the engagement between His-457—another highly conserved residue—and Glu-182, which is either an aspartate or a glutamate in most ATG9A orthologs (Figure 1A). The other part of the cytosolic interface at this layer involves the α15–α16 loop packing against the α13 helix of the neighboring monomer through presumably hydrophobic and π-stacking interactions (Figure 3D). This interaction causes a kink in α13, engaging its C terminus with α12 within the same monomer. We tested the predictions from the structure by mutating pairs of residues at different positions in the trimeric interface to cysteines and testing formation of disulfide cross-linked species in nonreducing SDS-PAGE. At the cytosolic interface the N57C,P371C double mutant and at the transmembrane interface, the F68C,N379C double mutant showed disulfide cross-linked trimers, consistent with the structure (Figure S4A and S4B).
ATG9A Has a Network of Internal Cavities and Pores
Intriguingly, our structure shows that the ATG9A trimer has a branched network of internal pores and cavities (Figures 4A–4D); it consists of a central pore that starts at the luminal side of the bilayer and continues across the membrane along the transbilayer normal toward the cytosolic side (Figures 4A–4D). On the cytosolic side, the central pore is occluded by the C-terminal domain (Figures 4B–4D). The central pore branches out into two additional pores within each protomer (Figure 4B). One of them (hereafter referred to as the perpendicular branch) continues toward the cytosolic side, off the central axis of the trimer. The other branch (the lateral branch) moves in a lateral direction along the membrane and ends at the putative membrane-aqueous border on the cytoplasmic side (Figures 4B–4D). ATG9A thus comprises a tree-like network of pores with a single opening on the luminal side and several openings on the cytoplasmic side (Figure 4A). One of the most striking differences between the two conformational states of ATG9A (i.e., states A and B) is the degree of dilation of the central pore (Figures 4E and 4F). The central pore is hydrophilic in nature, being lined with polar residues that could provide favorable contact points for solutes, including the polar headgroups of phospholipids, a possible substrate for ATG9A (Figures 4C and 4D). Both the perpendicular branch and the lateral branch are unoccluded in both conformations of ATG9A (Figures 4G and 4H). Access to the perpendicular branch from the cytoplasmic side may be regulated by conformational changes of the large extramembranous C terminus (Figure 4D). On the other hand, the lateral branch is open to the cytosol but has a constriction immediately before it opens up on the cytoplasmic side (Figure 4D). Similar to the central pore, the lateral branch is also lined with hydrophilic residues (Figures 4D, S4E, and S4F). The central pore, together with the lateral branch, offers a hydrophilic passage connecting both sides of the membrane. One proposed mechanism of transbilayer movement of phospholipids involves translocation of the headgroups across the membrane through a hydrophilic passage created by the protein, which lowers the energy barrier of moving a polar group through the hydrophobic interior of the membrane. In this mechanism, the hydrophobic tails of the lipids are dragged along without necessarily going through a cavity. If ATG9A is involved in movement of lipids across the bilayer, the lateral branch may serve such a function. At the junction of the central pore and the perpendicular branch, we see densities in the cryo-EM reconstructions that fit well with two detergent (LMNG) molecules (Figures S4C–S4F), although we believe that they are not bound in a way a transported lipid would be.
Figure 4. The ATG9A Trimer Shows a Complex Network of Pores and Cavities.

(A and B) Branched network of internal pores in the ATG9A trimer shown in top (A) and side (B) view. Protomers of ATG9A are colored cyan, magenta, and orange. Channels are shown as a solid surface with blue color representing a big radius to permeate molecules larger than water and green color representing channels that could only permeate water. The edges of the membrane are shown as gray lines. Three lateral branches and three perpendicular branches are shown in (A), and only one lateral and one perpendicular branch are shown in (B). N265 and R422 targeted for mutagenesis (see Figure 5) are shown in a ball-stick representation.
(C and D) Electrostatic potential surface of the central pore, lateral branch, and perpendicular branch (two faces, C and D, with 180° rotation are shown). The electrostatic potential scale is shown below the molecules, from negative values (−2 kBT/e) in red, to positive values (+2 kBT/e) in blue. Green, yellow, and pink arrows are used to show the positions of the central pore, lateral branch, and perpendicular branch, respectively, and also the orientations corresponding to the radius calculations in (E), (G), and (H).
(E) Alignment of two conformations of ATG9A. State A is colored cyan and grey for helices αA and αB, and state B is colored deep blue. The blue arrow indicates the shift orientation of the protomer.
(F–H) Radius comparison of central pore (F), lateral branch (G), and perpendicular branch (H) in the two studied conformations.
Structure-Guided Mutagenesis Identifies Critical Cavity-Lining Residues and Determinants for Interaction with ATG2A
The availability of a high-resolution structure allowed us to perform structure-function analyses to assess what parts of ATG9A are required for its function in autophagy. To that end, we made a number of truncations and single amino-acid substitutions in human ATG9A and tested for the ability of the resulting mutants to rescue autophagy in ATG9A-knockout (KO) HeLa cells. ATG9A-KO cells were previously shown to exhibit large cytoplasmic puncta corresponding to aberrant autophagosomes or protein aggregates decorated with the autophagy effector protein LC3B (Runwal et al., 2019). We found that KO of ATG9A in HeLa cells indeed resulted in the appearance of bright LC3B-positive puncta with a diameter that was ∼40% larger than that of LC3B-positive puncta in wild-type (WT) HeLa cells (Figures 5A, arrows, and 5G). Expression of ATG9A-GFP in those cells restored the distribution of LC3B to smaller cytoplasmic puncta (Figures 5B, outlined cells, and 5G). Deletion of distal C-terminal sequences missing in our model and presumably disordered (constructs 1–723) did not affect the ability of ATG9A-GFP to rescue LC3B distribution (Figures 5C and 5G), but complete deletion of the C-terminal domain after the last transmembrane domain, including helices α21, α22, αA, and αB, (constructs 1–522) abrogated that rescue (Figures 5D and 5G), indicating that the C-terminal platform domain of ATG9A is essential for the function of the protein.
Figure 5. Functional Importance of Domains and Amino Acids in ATG9A.

(A) Confocal microscopy of WT and ATG9A-KO HeLa cells immunostained for endogenous LC3B. Notice that ATG9A KO results in the appearance of large, bright LC3B puncta in the cytoplasm (arrows).
(B–F) ATG9A-KO HeLa cells were transfected with plasmids encoding various GFP-tagged ATG9A constructs (WT in B, 1–723 in C, 1–522 in D, R422W in E, or N265W in F) and immunostained for endogenous LC3B. Transfected cells are indicated by the dashed outlines. Scale bar: 10 μm. Notice that ATG9A WT and 1–723 rescue the normal distribution of LC3B but 1–522, R422W, and N265W do not.
(G) Quantification of LC3 puncta size from experiments (A)–(F), using the “analyze particles” plugin in ImageJ (Schneider et al., 2012). Bars represent the means ± SEM from three independent experiments (20–40 cells per condition). Significance: ns, not significant, p > 0.05, ∗∗∗∗p ≤ 0.0001, one-way ANOVA, followed by multiple comparisons using the Dunnett test and WT cells as a control group.
(H) Lysates of HEK293T cells co-transfected with plasmids encoding Spot-Tag-Cerulean-ATG2A and either GFP, ATG9A-GFP, ATG9A-(1–723)-GFP, or ATG9A-(1–522)-GFP were immunoprecipitated with an antibody to the Spot epitope, followed by immunoblotting with an antibody to GFP. Input was adjusted for Spot-Tag-Cerulean-ATG2A expression levels. The positions of molecular mass markers are indicated in the center lane, and the products of the transfected constructs are indicated on the right.
(I) Quantification of ATG9A-GFP levels in immunoprecipitates relative to ATG9A levels in the input and ATG2A amounts immunoprecipitated from experiments, such as that in (H). Bars represent the means ± SEM from three independent experiments. Significance: ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗∗p ≤ 0.0001, one-way ANOVA, followed by multiple comparisons using the Tukey test.
ATG9A is known to interact with another autophagy protein, ATG2A (Gómez-Sánchez et al., 2018; Kotani et al., 2018; Tang et al., 2019), which was recently shown to function as a tunnel for the delivery of phospholipids from the ER to the growing phagophore (Maeda et al., 2019; Osawa et al., 2019; Valverde et al., 2019). We confirmed this interaction by demonstrating specific co-immunoprecipitation of ATG9A-GFP with Spot-Tag-Cerulean-ATG2A expressed by transfection in HEK293T cells (Figures 5H and 5I). We observed that the truncated 1–723 construct showed a partial reduction and the truncated 1–522 construct an almost complete loss of co-immunoprecipitation with ATG2A (Figures 5H and 5I). These experiments indicated that the C-terminal platform domain of ATG9A is required for interaction with ATG2A. The correlation between the functional rescue and co-immunoprecipitation experiments further suggested that the interaction of ATG2A with the cytoplasmic platform domain of ATG9A is essential for the function of ATG9A, possibly by enabling the transfer of lipids from ATG2A to ATG9A for integration into the phagophore membrane.
We also used the functional rescue assay to assess the importance of the lateral branch for ATG9A activity. To that end, we substituted a tryptophan residue for Arg-422 (the R422W construct), which lies at the junction of the lateral branch and the central pore, or Asn-265 (the N265W construct), which is at a place where the lateral branch opens to the membrane-cytosolic interface. We observed that both R422W (Figures 5E and 5G) and N265W constructs (Figures 5F and 5G) failed to rescue the size of LC3B puncta in ATG9A-KO cells. These findings suggest that occlusion of the lateral branch by placement of a bulky tryptophan residue impairs ATG9A activity, possibly by blocking a transport function of that channel. Another interesting observation from the functional rescue experiments was that expression of the inactive ATG9A-GFP constructs (1–522, R422W, and N265W) increased the size of LC3B puncta not only in ATG9A-KO cells (Figure 5G) but also in WT HeLa cells (Figure S5), most likely through a dominant-negative effect. Further experiments will be needed to determine how these ATG9A mutations affect the eventual degradation of LC3B and autophagy cargos in autolysosomes (i.e., autophagy flux).
Molecular Dynamics Simulations Reveal That ATG9A Promotes Membrane Curvature
The presence of ATG9A in small vesicles (Kakuta et al., 2017; Sekito et al., 2009;Yamamoto et al., 2012) and at the membrane edge of expanding phagophores (Gómez-Sánchez et al., 2018; Young et al., 2006) prompted us to hypothesize that ATG9A may have an intrinsic affinity for highly curved membranes. To evaluate that hypothesis, we carried out a structure-based molecular dynamics simulation of a single ATG9A homotrimer embedded in a model phospholipid bilayer (Figure 6A), initially focusing on “state B.” Although the membrane was initially flat, by the end of the calculated trajectory it had relaxed to a deformed state resembling a shallow cone, with ATG9A at its apex (Figure 6B). This perturbation originates at the protein-lipid interface, which is both extensive and consistently tilted relative to the membrane perpendicular. Accordingly, lipid alkyl chains near the protein are also markedly tilted, to appropriately coat the hydrophobic surface of the protein (Figures 6C, 6D, and 6F), resulting in a noticeable deflection of the membrane mid-plane, by about 8° (Figures 6E). The membrane perturbation caused by ATG9A propagates radially away from its surface for ∼100 Å; as a result, the protein is elevated by ∼14 Å (Figure 6B), i.e., about one-third of the membrane width. In view of that result, we asked what membrane morphology might result from the combined effect of multiple copies of the protein. Guided by the 3-fold symmetry of ATG9A, we postulated a system whereby a central ATG9A is surrounded by three concentric hexagonal rings, each populated by 6, 12, and 18 copies of the protein (Figure 7A). In all cases, any two adjacent proteins were initially set to be 160 Å apart (i.e., not in direct contact), reasoning that, at that distance, the only possible interaction between them would be mediated by the membrane itself. This simulation revealed a radical transformation of the membrane morphology, much larger in magnitude than for a single ATG9A. Specifically, we observed the spontaneous emergence of a dome-like structure (Figure 7B; Video S2). At the apex of that dome, the elevation of the membrane relative to the base is about 280 Å (Figure 7C), and the resulting membrane curvature is approximately 1/550 Å (Figure 7D), i.e., comparable to that of ATG9A vesicles. It is worth noting that in that simulation, all ATG9A molecules can tumble and diffuse freely; indeed, the rate of displacement is similar for all molecules and comparable to that of isolated ATG9A (Figures 7E and 7F). However, direct protein-protein contacts were observed rarely; nevertheless, the cluster remained loosely assembled throughout the simulations (Figure 7F), seemingly stabilized by a collective effect on the membrane (Anselmi et al., 2018). Taken together, these data explain why ATG9A proteins localize to regions in which the membrane is strongly curved because this curvature matches their own propensity to bend the bilayer. Indeed, our data indicate that clusters of ATG9A might contribute to sustain highly curved morphologies, as has been noted for other integral membrane proteins known to influence organellar morphology (Davies et al., 2012). These data also show that the interplay between ATG9A and the morphology of the surrounding membrane stems from the specific features of the protein structure revealed by our cryo-EM data and not from other assumptions in our computational analysis (e.g., lipid composition of the bilayer, protein density, etc.). Indeed, identical control simulations of the K+channel KcsA, which is not known to foster and/or favor a specific membrane morphology, showed no evidence of significant bilayer bending or disruption when the protein was examined individually (Figures S6A–S6E). Consistent with that result, when KcsA was simulated in a hypothetical cluster analogous to that evaluated for ATG9A, the protein copies moved randomly and tended to disperse and failed to have any kind of collective effect on membrane morphology (Figures S6F–S6J). It is also worth noting that simulations of the ATG9A homotrimer in the alternate conformer revealed by our cryo-EM data, i.e., “state A,” produced results that were highly similar to those obtained with state B, despite the significant degree of structural flexibility reflected by those two conformations (Figure S7). This consistency underscores the notion that it is the overall architecture of the ATG9A trimer and the amino-acid makeup of its surface that explain its preference for curved membranes. Further experimental corroboration of these results will be an important future direction to establish the role of ATG9A in defining the morphology of the membranes in which it resides.
Figure 6. Molecular Dynamics Simulation of Isolated ATG9A in a POPC Bilayer.

(A) View of the periodic unit cell used to simulate a single copy of ATG9A (gray) in a 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (POPC lipid) bilayer (colors). The configuration shown is that at the end of the 1-μs simulation. Protein and membrane are represented with a coarse-grained forcefield. The system amounts to ~264,000 particles. Solvent is omitted for clarity.
(B) Quantification of the perturbation of the membrane induced by ATG9A. Shown is a map of the mean value of the z coordinate of the center of the bilayer across the x-y plane, calculated from the trajectory data, and relative to the edge of the simulation system, where the membrane is flat. The white space in the center corresponds to the area occupied by ATG9A.
(C) The protein is shown alongside a 3D density map (yellow) for the alkyl chains of the bilayer, calculated from the trajectory data. The map reflects only lipid molecules in the first solvation shell, i.e., 10 Å from the protein surface. The tilt of the protein-lipid interface, which is roughly constant across the perimeter of the protein, is indicated.
(D) Same as (C), viewed from the luminal side.
(E) Same as (A), but for the lipid-alkyl chains within 100 Å from the protein surface. The downward deflection of the membrane plane is indicated.
(F) Mean second-rank-order parameter of the C–C bonds along the lipid alkyl chains, for either the outer or inner layers of the lipid bilayer, across the x-y plane. The second-rank-order parameter of a given C–C bond is defined as 1/2 (3 cos2 < θ > −1), where < θ > denotes the average value of the angle between the bond and the bilayer normal. It is, therefore, a measure of tilt. The data in this figure derive from a simulation of state B; see Figure S7 for results from a simulation of state A.
Figure 7. Molecular Dynamics Simulation of a Hypothetical ATG9A Cluster in a POPC Bilayer.

(A) Configuration of the system at the beginning of the simulation; 36 ATG9A proteins (gray) are arranged around a central trimer in three concentric hexagons (inset). Note the proteins are not in contact. The system amounts to ~11,220,000 particles. Solvent is again omitted for clarity.
(B) Same as (A), at the end of the calculated trajectory of 1 μs. See also Video S2.
(C) Quantification of the membrane perturbation induced by the ATG9A cluster. Note change in scale relative to Figure 6B. White spaces across the map correspond to the area occupied by ATG9A copies.
(D) Cross-section of the dome-like structure shown in (B) (blue curve). The data reflect an average of 36 cross-sections that meet at the apex of the dome and differ by rotations around the z axis in 10° increments. The approximate elevation of the apex is indicated. The cross-section of the dome is compared with a sphere whose curvature fits the simulation data (red curve).
(E) Time-evolution of the x,y, and z dimensions of the simulation system during the calculated trajectory. As the dome emerges, the z dimension increases and the x,y dimension decreases accordingly. The system ceases to change significantly after 0.8 μs. The plot shows time-averages for consecutive 200-ns trajectory fragments and the corresponding SD values.
(F) Free lateral diffusion of the 37 copies of ATG9A during the calculated trajectory of 1 μs. Each trace (colors) represents the center-of-mass of one ATG9A protein. For comparison, a trace is also shown for isolated ATG9A, in the same scale, from the simulation system shown in Figure 6A.
Concluding Remarks
Our structural analysis of human ATG9A thus reveals a fold not reported thus far in any membrane protein, presents a topology different from that proposed from previous studies, elucidates the details of the homotrimeric interface, shows the existence of pores and cavities within the trimer running both parallel and perpendicular to the membrane, and provides a mechanistic explanation for the localization of ATG9A to highly curved membranes.
The structure shows a homotrimeric architecture of wedge-shaped protomers with a network of branched pores throughout the protein. Our functional experiments using mutants demonstrate the importance of the branches and also show that the C-terminal platform domain is crucial for the function and interaction with ATG2A. Previous studies suggested that ATG9A is part of a machinery that forms a conduit for phospholipid transport during autophagosome formation and maturation (Gómez-Sánchez et al., 2018). We think that the cavities identified in our structure are plausible pathways for the polar headgroups of phospholipids across the membrane. The presence of those cavities also offers an intriguing possibility that they help maintain osmotic pressure across the membrane in the relatively small ATG9A vesicles. Finally, another critical aspect of ATG9A function is targeting ATG9 vesicles to the PAS through engagement with other essential protein partners in the autophagy pathway; we think that this targeting may be achieved through the C-terminal platform domain that serves as a site for protein-protein interactions, as shown here for ATG2A.
Finally, our molecular dynamics simulations show that the ATG9A homotrimer deforms the lipid bilayer locally and that the presence of multiple copies of the ATG9A homotrimer amplifies the distortion caused by each, creating a dome shape in the bilayer. This finding is consistent with experiments showing that ATG9 resides on 30–60-nm-diameter vesicles (Kakuta et al., 2017; Yamamoto et al., 2012), membrane tubules (Mari et al., 2010), and the edge of growing autophagosomes (Yamamoto et al., 2012); all of which involve localization of ATG9A to highly curved membranes. We speculate that the uniqueness of ATG9A as the only essential transmembrane protein in autophagy arises out of its regulated protein-lipid and protein-protein interactions in the early stages of the process. The availability of atomic-level insight into the structure of ATG9A will help guide future experiments into the molecular mechanisms in which this protein is involved.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for reagents may be directed to the Lead Contact Anirban Banerjee (anirban.banerjee@nih.gov).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
The relevant structural datasets generated during this study are available at EMDB [ATG9A trimer, state A: EMD-21874; ATG9A monomer, state A: EMD-21877; ATG9A trimer, state B: EMD-21876; ATG9A monomer, state B: EMD-21878] and PDB [ATG9A trimer, state A: 6WQZ; ATG9A trimer, state B: 6WR4].
EXPERIMENTAL MODEL AND SUBJECT DETAILS
HeLa and HEK293T cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (FBS), 100 IU/mL penicillin and 100 μg/mL streptomycin, and 2 mM L-glutamine in a 37°C incubator (5% CO2). The ATG9A-KO HeLa cell line has been described previously (Mattera et al., 2017). Cells used for imaging (Figures 5A–5F and S5) and immunoprecipitation assays (Figure 5G) were transfected using Lipofectamine 2000 following the manufacturer’s recommended conditions. Cells used for in vitro crosslinking experiments (Figures S4B) were transfected using polyethyleneimine (PEI).
Suspension HEK293S GnTI− cells and Sf9 cells were grown and maintained as previously described (Rana et al., 2018). ATG9A-mVenus P0 baculovirus was generated using PEI-mediated transfection of 50 mL Sf9 cells according to established protocols (Rana et al., 2018). The P0 baculovirus was amplified to generate P1 virus by infecting Sf9 cells at a density of 3.0×106 per mL with 1/200th the volume, and harvesting the media 3 days post-transfection. All baculovirus stocks were stored in the dark at 4°C with 1% FBS to prevent proteolysis.
ATG9A-mVenus protein expression was initiated by transducing 1 L of HEK293S GnTI− cells with 100 mL of P1 virus. Approximately 15 h later, sodium butyrate was added to a final concentration of 10 mM. Protein expression was allowed to continue for another 48 h before harvesting the cells by centrifugation. Pellets were washed with 1X TBS and frozen at −80°C for further processing.
METHOD DETAILS
Plasmids
ATG9A-mVenus construct used for large-scale viral transduction and subsequent protein expression and purification (Figures 1B and 1D) was generated by ligating the human ATG9A cDNA using EcoRI and XhoI restriction sites into a modified pEG-BacMam vector with mVenus and 10xHis as a C-terminal fusion tag (Rana et al., 2018), and a PreScission cut site to remove both poly-histidine and fluorescent tags. Mutant cysteines (Figure S6) were introduced into the indicated ATG9A positions, using the ATG9A-mVenus template plasmid. Plasmid encoding human ATG9A-GFP (Figure 5) was described previously (Mattera et al., 2017). All the mutants and truncations were generated by site-directed mutagenesis and verified by DNA sequencing. Human ATG2A cDNA was subcloned into a derivative of pEG BacMam for CMV promoter-controlled expression. Spot-Tag fused to mCerulean was cloned into the plasmid using overlap extension PCR. ATG2A was then amplified by PCR and fused to the 3′ end of mCerulean using Gibson assembly; fidelity of the complete Spot-Tag-mCerulean-ATG2A coding region was verified by DNA sequencing.
Protein Purification
HEK293S GnTI− cell pellets from ∼3 L suspension culture were suspended in 200 mL lysis buffer (50 mM Tris-HCl, pH 8.0, 500 mM NaCl, 5 mM β-ME, 2 mM EDTA, 1 mM DTT, 10 mM MgCl2) followed by protease inhibitor supplementation (1 μg/mL pepstatin, 2.5 μg/mL leupeptin, 50 μg/mL soy trypsin inhibitor, 10 KIU/mL aprotinin, 1 mM AEBSF, 1 mM PMSF, 3 mM benzamidine), and 10 μg/mL DNase. All subsequent preparative steps were performed at 4°C or on ice. The cell suspension was sonicated in ice water at 80% amplitude for a total 3 min on-time by cycling 5 s on/20 s off with additional 0.25 mM PMSF added every 12 cycles. Sonicated lysate was centrifuged at 9000 × g for 10 min; supernatant was collected, and pellets resuspended up to 100 mL in lysis buffer. Cell suspension was again sonicated but for a total 30 s on-time. Lysate was similarly spun again to remove debris and supernatant harvested. Both membrane supernatants were then pooled and ultracentrifuged at 181,000 × g for 1.5 h. Membranes were then resuspended to 50 mL in extraction buffer (100 mM Tris-HCl, pH 8.0, 400 mM NaCl, 4 mM TCEP, 10 mM β-ME, 20% glycerol), homogenized using a 55 mL Potter-Elvehjem, and frozen at −80°C.
For detergent extraction, a 25 mL aliquot of ATG9A-mVenus membranes was thawed and supplemented with 100 μL protease inhibitor cocktail III, EDTA-free. Membrane slurry was stirred to homogeneity and another 100 μL protease inhibitor cocktail and 1 mM PMSF added. Membrane suspension was then diluted 1:1 by addition of 3.2% LMNG in water and stirred for 2 h. Insoluble debris was removed by centrifugation at 38,000 × g for 30 min. Clarified ATG9A-mVenus extract was applied to 2 mL Talon cobalt affinity resin (Takara) and rotated for 2 h. Resin was collected by gravity flow in a disposable column then washed with twelve bed volumes Talon buffer (1 × TBS, 2 mM TCEP, 5 mM β-ME, 25 mM imidazole, 10% glycerol, 0.14 mM LMNG). ATG9A-mVenus was then eluted with 4 bed volumes elution buffer (Talon buffer + 175 mM imidazole), DTT supplemented up to 2 mM from a 1 M stock, and 1 mM EDTA added. ATG9A-mVenus eluate was concentrated to ∼220 μL using a 100 kDa MWCO concentrator, transferred to a 1.5 mL tube, 20 μL PreScission protease (1 mg/mL) added to cleave the mVenus-His tag fusion, and rotated overnight. ATG9A was finally subjected to size exclusion chromatography using a Superdex 200 Increase 10/300 column (GE Healthcare) equilibrated in SEC buffer (1 × TBS, 2 mM DTT, 1 mM EDTA, 5% glycerol, 0.09 mM LMNG). Peak fractions containing 0.9–1.0 mg/mL ATG9A were used for cryo-EM grid preparations without further concentration (Figure 1B).
Cryo-EM Sample Preparation and Data Collection
The peak fraction from size exclusion chromatography of the ATG9A sample (Figure 1B) was collected for cryo-EM grid preparation. 3 μL of sample was loaded onto a glow-discharged holey carbon grid (Quantifoil 300 mesh Cu R1.2/1.3). The grid was blotted for 5 s at 15°C and 100% relative humidity using a FEI Vitrobot Mark IV plunger before being plunge-frozen in liquid nitrogen-cooled liquid ethane. Cryo-EM data were collected using a Titan Krios G3 electron microscope (Thermo-Fisher) operated at 300kV. Micrographs were acquired at the nominal magnification of 130,000x (calibrated pixel size of 1.06 Å on the sample level) using a Gatan K2 Summit direct electron detection camera equipped with a Gatan Quantum LS imaging energy filter with the slit width set at 20 eV. The dose rate on the camera was set to 8 e‒/pixel/s. The total exposure time of each micrograph was 10 s fractionated into 50 frames with 0.2 s exposure time for each frame. The data collection was automated using the Leginon software package (Suloway et al., 2005). A total of 14,071 micrographs were collected from 3 data collection sessions.
Image Processing
The beam-induced image motion between the frames of each dose-fractionated micrograph was corrected with MotionCor2 (Zheng et al., 2017). Two average images, one with dose weighting and the other one without, were generated from MotionCor2 for each micrograph. The average images without dose weighting were used for defocus determination using CTFFIND4 (Rohou and Grigorieff, 2015) and those with dose weighting were used for particle picking and extraction. Quality of the micrographs was evaluated using the results from CTFFIND4. The micrographs with poor resolution as reported by CTFFIND4 were removed and a total of 10,431 micrographs were selected for further processing. The single particle data analysis was performed following the standard procedures in RELION3 (Zivanov et al., 2018) and cryoSPARC2 (Punjani et al., 2017) with few modifications as summarized in Figures S1 and S2. A subset of 600 micrographs was used for initial template-free particle picking using Gautomatch (https://www2.mrc-lmb.cam.ac.uk/research/locally-developed-software/zhang-software/#gauto). The particles were processed using cryoSPARC2 for 2D classification and ab-initio reconstruction to generate starting models. The best starting model was then used as the template to pick particles from all micrographs using Gautomatch. A total of 6,926,538 particles were picked and extracted in 96×96 pixels with 2x binning (pixel size 2.12 Å). The particles were processed using RELION3 for 2D and 3D classifications. Some of the 2D class averages show strong features of a 3-fold symmetry (Figure S1B), therefore the C3 symmetry was used in the following 3D classifications and 3D refinements until the symmetry expansion. The best particles were selected iteratively by selecting the 2D class averages and 3D reconstructions that had interpretable structural features. A total number of 2,943,366 particles were selected using the above procedures and aligned to the center using a single-class 3D classification. After re-centering and removing overlapping particles, 2,938,157 particles were re-extracted in 192×192 pixels without binning (pixel size 1.06 Å). Another batch of 2D classification on these particles resulted in 2,692,183 selected particles. These particles were then 3D-classified into 4 classes using smaller angular sampling rates (3.7° for first 25 iterations followed by 1.8° for another 25 iterations). Comparison among these 4 reconstructions revealed that two of them, which are with the best resolution and quality, are in different conformations (named state A and state B). The particles in these two classes were separated for further processing carried out using identical procedures. Briefly, the particles were subjected to 3D autorefinement and then particle polishing using the first 30 frames. The polished “shiny” particles were sent to a final 2D classification to clean up few bad particles before the 3D autorefinement using RELION3 or the non-uniform refinement using cryoSPARC2. The cryoSPARC2 non-uniform refinement with the C3 symmetry achieved 2.8 Å and 2.9 Å resolutions for state A and B, respectively.
Comparison of the 3D reconstructions of the two conformations suggested that the complex is “breathing” with the 3 subunits tilting toward or from the center of the complex. Despite two discrete conformations being separated by 3D classification, particles within each class might be heterogeneous because of the continuous motion of each subunit in the complex. Therefore, we used a combination of symmetry expansion and density subtraction to refine the 3D reconstruction of an individual subunit of the complex in order to improve the resolution. Briefly, each polished particle was expanded into 3 particles using the program relion_particle_symmetry_expand. These expanded particles were considered “asymmetric” and therefore no symmetry was applied in the afterward procedures. A soft mask that included only two subunits and the detergent micelle was carefully constructed following the instructions (Bai et al., 2015) for density subtraction. Another soft mask that included the remaining subunit was also constructed for the following 3D classification and refinement. During RELION3 3D autorefinement, local search was forced by setting the parameter “initial angular sampling” to 0.9°. After the 3D autorefinement, a 3D classification without image alignment (skip align) was carried out to separate the particles into 4 classes. The particles in each class were then subjected to 3D autorefinement with local search. The best class was further processed using the local refinement (with the non-uniform refinement option turned on) in cryoSPARC2. The final resolution of the single-subunit reconstruction using cryoSPARC2 is 2.9 Å for both conformations. Symmetry expansion substantially improved the quality of the reconstruction of the subunit as demonstrated in Figure S2C.
Model Building
Models of ATG9A in different conformations were manually built from scratch (independently by TL and XT) using Coot (Emsley et al., 2010). Models were built based on the monomer maps due to the better side chain density and connectivity. Transmembrane helices α11 and α14 were selected as starting points separately of model building to further ensure the accuracy of the models. The density of the last 2 helices in the C-terminal part, αA and αB, was very poor, so polyalanines were fitted into the map without assigning residues only in state A. Three monomer models were fitted into the trimer map and merged together. ATG9A trimer models were further checked and refined in the trimer maps. PHENIX (Liebschner et al., 2019) was used to do the real space refinement, and models were validated by comprehensive validation in PHENIX. Structural figures were generated using PyMOL and UCSF Chimera (Pettersen et al., 2004). The HOLE program was used to calculate pore radius and generate channel figures (Smart et al., 1996).
In Vitro ATG9A Crosslinking
Each reaction step was performed at 4°C unless otherwise noted. Membranes from the purification of ATG9A-mVenus were diluted 4-fold in reaction buffer (20 mM HEPES pH 8, 140 mM NaCl, 1 mM TCEP·HCl, 1 mM DTT) and pelleted at 100,000 g for 10 min. Membranes were then resuspended in reaction buffer and pelleted once more. Pellets were homogenized in reaction buffer with a Dounce homogenizer and aliquoted on ice. DSS was diluted to the listed concentrations (Figure 1C) from DMSO stocks, resulting in a 20% dilution of the aliquoted membranes. DMSO was used in place of DSS for the negative control. Crosslinking reactions were performed on ice for 10 min, and then quenched for 1 h with 1 mM glycine in reaction buffer. Quenched reactions were mixed with SDS loading dye and run on SDS-PAGE. Imaging was performed using a Biorad ChemiDocMP imager by detecting in-gel mVenus fluorescence.
In Vivo Endogenous ATG9A Crosslinking
HeLa cells (2 × 104) were scraped, washed with 500 μL of PBS and pelleted (3 min, 500 g) twice. Cells were then resuspended and incubated under gentle rocking in 1 mL of the listed DSS dilutions in PBS (Figure 1D) for 30 min at room temperature. After crosslinking, cells were pelleted and washed twice with PBS. To quench the excess of DSS, cells were incubated in 1 mL of 50 mM Tris pH 7.5 for 10 min at room temperature before lysing the pellet in 600 μL 2X LDS loading buffer. Lysates were incubated at 37°C for 15 min, sonicated, and analyzed by SDS-PAGE and immunoblotting with antibody to ATG9A.
ATG9A Engineered Disulfide Crosslinking
Plasmids encoding WT ATG9A and variants (Figure S4B) were transfected into HEK293T cells (one 10 cm dish per variant, grown to ∼70% confluence) and transfected with PEI (10 μg plasmid DNA was complexed with 30 μg PEI). Complexes were added dropwise to each HEK293T dish, and incubated at 37°C for 48 h. Cells were harvested, washed with PBS, and the cell pellet snap-frozen in liquid N2, then stored at −80°C. All ATG9A purification steps were performed at 4°C. Cell pellets were thawed on ice, then resuspended in 800 μL extraction buffer (50 mM HEPES, pH 7.4, 150 mM NaCl, 5 mM β-ME, protease inhibitors, 1 μg/mL DNase I). Cells were mechanically lysed by ten passages through a 27-gauge needle, then debris and unbroken cells were pelleted by centrifugation at 600 g. Membrane-containing supernatants were transferred to fresh 1.5 mL tubes and 200 μL 10% LMNG in extraction buffer was added (2% LMNG final concentration) and tubes rotated for 2 h. ATG9A-containing extracts were centrifuged at 21,000 g for 10 min, supernatant applied to 50 μL LMNG-equilibrated TALON resin, and rotated for 2 h. Resins were collected in a mini-column by gravity flow and washed with 600 μL wash buffer (20 mM HEPES, pH 7.4, 150 mM NaCl, 10 mM imidazole, 10% glycerol, 0.2 mM LMNG). ATG9A variants were eluted with 200 μL wash buffer supplemented with 250 mM imidazole. Eluted ATG9A and variants were treated with 10 mM DTT and incubated at 4°C for 1 h. Protein samples were quenched by addition of 20 mM NEM (from 1 M stock in ethanol) then SDS-PAGE loading buffer (without reductant) added. Samples were run on SDS-PAGE and in-gel fluorescence of ATG9A-mVenus fusions imaged using a BioRad ChemiDocMP.
Immunoblotting
The cell lysates were separated by SDS-PAGE and transferred to nitrocellulose membrane. The membrane was blocked with 5% nonfat milk before incubation with primary and secondary antibodies (1 h at room temperature each incubation, followed by 3 washes of the membrane with PBS supplemented with 0.1% Tween-20). Bound antibodies were visualized using Clarity Western ECL substrate.
Immunoprecipitation
Transfected HEK293T cells (Figure 5G) were washed with ice-cold PBS and lysed in lysis buffer (150 mM NaCl, 10 mM Tris-HCl, pH 7.4, 0.5 mM EDTA, 0.5% NP-40) with a protease inhibitor cocktail. Cell lysates were clarified by centrifugation at 20,000 g for 10 min and incubated with BC2 nanotrap agarose beads (Braun et al., 2016) at 4°C for 1 h. After 3 washes with wash buffer (150 mM NaCl, 10 mM Tris-HCl, pH 7.4, 0.5 mM EDTA, 0.2% Triton X-100), the precipitates were eluted with 2X LDS loading buffer. The precipitated samples were analyzed by immunoblotting with the indicated antibodies.
Immunofluorescence Microscopy
HeLa cells were seeded on coverslips in 24-well plates at 40,000 cells per well in normal culture medium. After transfection (Figures 5A–5F and S5), cells were fixed in −20°C methanol for 5 min, and blocked in 0.2% BSA in PBS for 30 min at 37°C. The cells were sequentially incubated with primary antibody to LC3B and secondary antibodies diluted with 0.2% BSA in PBS for 30 min at 37°C. Coverslips were mounted on glass slides using Fluoromount-G with DAPI. Cells were imaged using a Zeiss LSM780 confocal microscope (Carl Zeiss AG, Oberkochen, Germany). The final composite images were created and analyzed using ImageJ (Schneider et al., 2012).
Molecular Dynamics Simulations
All simulations were carried out with GROMACS 4.5.5 (Hess et al., 2008) and the coarse-grained (CG) MARTINI 2.1 forcefield (Marrink et al., 2007) at constant pressure (1 atm) and temperature (298 K), with periodic boundary conditions. The pressure was applied isotropically in the X and Y dimensions, i.e., the membrane plane. Non-bonded interactions were calculated using shifted Lennard-Jones Coulombic potential, both cut off at 12 Å, with a relative dielectric constant of 15. Prior to collecting data for analysis, each system was energy-minimized and equilibrated in a sequence of short simulations using integration timesteps of 1, 5 and 10 fs, and the Berendsen thermostat and barostat. The production trajectories were calculated using an integration timestep of 10 fs, using the Berendsen thermostat and the Parrinello-Rahman barostat. The duration of the trajectories was 1 μs each.
The secondary and tertiary structures of both ATG9A (in either ‘State A’ or ‘State B’) and KcsA were preserved throughout the simulations by applying a set of harmonic distance restraints. These restraints act on the distance between any two carbon-alpha atoms separated by more than 5 Å and less than 9 Å; the force constant of these harmonic distance restraints was 1.2 kcal/mol Å−2, so as to permit some variability relative to the experimental starting structure (Figures S7E–S7F). Protein sidechains could however move freely. As a whole, both proteins could also tumble and diffuse freely in the membrane.
All simulations used a hydrated bilayer of 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (POPC) lipids. Counter-ions were added to neutralize the charge of the proteins. The dimensions of the simulation systems for the isolated ATG9A trimers (in either ‘State A’ or ‘State B’) are approximately 500 × 500 × 120 Å. These systems include ∼7,500 lipids and ∼170,000 solvent molecules, for a total of ∼264,000 CG particles. The analogous KcsA system has approximately the same dimensions but includes ∼7,600 lipids and ∼171,000 solvent molecules. The dimensions of the simulation systems containing 37 copies of ATG9A (only ‘State B’) or KcsA are 1,940 × 1,940 × 340 Å. The ATG9A system includes ∼110,000 lipids and ∼9,7000,000 solvent molecules, for a total of ∼11,220,000 particles. The KcsA system includes ∼121,000 lipids and ∼9,800,000 solvent molecules, for a total ∼11,250,000 particles.
The 2D map shown in Figure 6B (and in Figures S6E and S7D) was calculated from analysis of all simulation snapshots saved for the isolated ATG9A trimer (and the isolated KcsA tetramer). To correct for the lateral diffusion and tumbling of the protein, all averages were calculated after transforming the coordinates in the system in a manner that results in the optimal superposition (least-squares fitting) of the backbone of the trimer onto a reference structure (the cryo-EM structure) that was centered in the simulation box and aligned with the XY plane. Note that this procedure preserved all relative atomic coordinates. After processing all snapshots, the average membrane mid-plane was found to be flat beyond ∼240 Å from the protein center; the value of Z at that distance was therefore considered the zero-level in Figure 6B. A similar but not identical procedure was used to calculate the 2D map in Figure 7C (and Figure S6I). Here, the transformation of coordinates prior to averaging merely re-positioned the center of the 37-ATG9A cluster, so that it coincided with the center of the simulation box (this explains why the area occupied by the ATG9A proteins in Figure 7C is in most cases blurred, while that in Figure 6B is highly defined).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical tests of significance were performed with GraphPad Prism 8.3.0 as indicated in the figure legends (Figure 5). In particular, Figure 5G shows represent the mean ± SEM from 3 independent experiments (20–40 cells per condition). One-way ANOVA, followed by multiple comparisons using the Dunnett test and WT cells as a control group was used to compare the different conditions. n represents the total number of LC3-positive puncta analyzed (WT: 5079; ATG9A KO: 1610; ATG9A-GFP: 1685; ATG9A(R422W)-GFP: 754; ATG9A(N265W)-GFP: 88; ATG9A(1–522)-GFP: 2835; ATG9A(1–723)-GFP: 3189). For Figure 5I, bars represent the mean ± SEM (n = 3). Here, n represents three independent biological replicates of each coimmunoprecipitation experiment. One-way ANOVA, followed by multiple comparisons using the Tukey test was used to compare the different conditions.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| LC3B (rabbit, monoclonal) | Cell Signaling Technology | 3868; RRID: AB_2137707 |
| ATG9A (rabbit, monoclonal) | Abcam | Ab108338; RRID: AB_10863880 |
| GFP, HRP-conjugated (mouse, monoclonal) | Miltenyi Biotec | 130-091-833; RRID: AB_247003 |
| Anti-rabbit, HRP-conjugated | Mab Technologies | NEF812001EA; RRID: AB_2571640 |
| Anti-rabbit, Alexa Fluor 555-conjugated | Thermo Fisher Scientific | A-31572; RRID: AB_162543 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Disuccinimidyl suberate (DSS) | Sigma-Aldrich | S1885 |
| Glycine | Sigma-Aldrich | 50046 |
| 2-mercaptoethanol (β-ME) | Sigma-Aldrich | M6250 |
| N-ethylmaleimide (NEM) | Sigma-Aldrich | E3876 |
| Dithiothreitol (DTT) | Sigma-Aldrich | D0632 |
| Bovine serum albumin (BSA) | Sigma-Aldrich | A7030 |
| Imidazole | Sigma-Aldrich | I0250 |
| Tween-20 | Sigma-Aldrich | P9416 |
| Triton X-100 | Sigma-Aldrich | T9284 |
| Soy trypsin inhibitor | Sigma-Aldrich | T9003 |
| Sodium butyrate | Sigma-Aldrich | B5887 |
| DNase I | Roche | 4716728001 |
| EDTA-free protease inhibitor cocktail | Roche | 11873580001 |
| Tris(2-carboxyethyl)phosphine hydrochloride (TCEP·HCl) | GoldBio | TCEP25 |
| Benzamidine | GoldBio | B-050 |
| NP-40 | Fluka | 74385 |
| Paraformaldehyde | Electron Microscopy Sciences | 15714 |
| Protease inhibitor cocktail III, EDTA-free | EMD Milipore | 539134 |
| Lauryl maltose neopentyl glycol (LMNG) | Anatrace | NG310 |
| TALON metal affinity resin | Takara | 635653 |
| Pepstatin | RPI | P30100 |
| Leupeptin | RPI | L22035 |
| 4-(2-Aminoethyl)-benzenesulfonyl fluoride, HCl (AEBSF) | RPI | A-540 |
| Phenylmethylsulfonyl fluoride (PMSF) | RPI | P20270 |
| Aprotinin | Fisher Scientific | BP250340 |
| Tris-buffered saline (TBS) | KD Medical | RGF-3385 |
| Phosphate-buffered saline (PBS) | Corning | 21-040-CV |
| Polyethyleneimine (PEI) | Polysciences | 24765-1 |
| Deposited Data | ||
| ATG9A Homotrimer State A (map) | This paper | EMDB: EMD-21874 |
| ATG9A Homotrimer State A (model) | This paper | PDB: 6WQZ |
| ATG9A Homotrimer State B (map) | This paper | EMDB: EMD-21876 |
| ATG9A Homotrimer State B (model) | This paper | PDB: 6WR4 |
| ATG9A monomer State A (map) | This paper | EMDB: EMD-21877 |
| ATG9A monomer State B (map) | This paper | EMDB: EMD-21878 |
| Critical Commercial Assays | ||
| Gibson Assembly Cloning Kit | New England Biolabs, Inc. | E5510S |
| QuikChange II XL Site-Directed Mutagenesis Kit | Agilent Technologies | 200522 |
| Experimental Models: Cell Lines | ||
| HeLa | ATCC | CCL-2 |
| ATG9A KO HeLa | Mattera et al., 2017 | N/A |
| HEK293S GnTI− | ATCC | CRL-3022 |
| HEK293T | ATCC | CRL-11268 |
| Sf9 | Expression Systems | 94-001 |
| Recombinant DNA | ||
| pEG BacMam mVenus vector | Rana et al., 2018 | N/A |
| pEG BacMam ATG9A-mVenus | This paper | N/A |
| pEG BacMam ATG9A-mVenus (N57C + P371C) | This paper | N/A |
| pEG BacMam ATG9A-mVenus (F68C + N379C) | This paper | N/A |
| pEG BacMam ATG9A-mVenus (P371C) | This paper | N/A |
| ATG9A-GFP | Mattera et al., 2017 | N/A |
| ATG9A-GFP (1-723) | This paper | N/A |
| ATG9A-GFP (1-522) | This paper | N/A |
| ATG9A-GFP (N265W) | This paper | N/A |
| ATG9A-GFP (R422W) | This paper | N/A |
| pMRXIP-GFP-hATG2A | Addgene | 114462 |
| pEG BacMam Spot-Tag-mCerulean-ATG2A | This paper | N/A |
| Software and Algorithms | ||
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| Leginon | Suloway et al., 2005 | https://emg.nysbc.org/redmine/projects/leginon/wiki/Leginon_Homepage |
| MotionCor2 | Zheng et al., 2017 | https://emcore.ucsf.edu/ucsf-motioncor2 |
| CTFFIND4 | Rohou and Grigorieff, 2015 | https://grigoriefflab.janelia.org/ctf |
| RELION3 | Zivanov et al., 2018 | https://github.com/3dem/relion |
| cryoSPARC2 | Punjani et al., 2017 | https://cryosparc.com/ |
| Gautomatch | Kai Zhang Personal Webpage | https://www2.mrc-lmb.cam.ac.uk/research/locally-developed-software/zhang-software/#gauto |
| PHENIX | Liebschner et al., 2019 | https://www.phenix-online.org/download/ |
| HOLE | Smart et al., 1996 | http://www.holeprogram.org/ |
| PyMOL | Schrödinger | https://pymol.org/2/ |
| UCSF Chimera | Pettersen et al., 2004 | https://www.cgl.ucsf.edu/chimera/ |
| GROMACS 4.5.5 | Hess et al., 2008 | http://www.gromacs.org/ |
| Prism 8.3.0 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Microsoft Excel | Microsoft | https://www.microsoft.com/en-us/microsoft-365/excel |
| Coot | Emsley et al., 2010 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/binaries/ |
| Other | ||
| Dulbecco’s modified Eagle’s medium | Corning | 15-013-CV |
| Hyclone CDM4HEK293 media | GE Healthcare | SH30858 |
| ESF 921 media | Expression Systems | 96-001 |
| Fetal bovine serum (FBS) | Corning | 35-011-CV |
| L-Glutamine | Corning | 25-005-CI |
| Antibiotics (penicillin and streptomycin) | Corning | 30-002-CI |
| Lipofectamine 2000 transfection reagent | Thermo Fisher Scientific | 11668019 |
| BC2 nanotrap agarose beads | Braun et al., 2016 | N/A |
| Fluoromount-G with DAPI | Electron Microscopy Sciences | 17984-24 |
| 300 mesh Cu R1.2/1.3 grids | Quantifoil Micro Tools GmbH | N1-C14nCu30-01 |
Highlights.
The transmembrane autophagy protein ATG9A is a domain-swapped homotrimer
Each ATG9A protomer comprises four transmembrane domains
The ATG9A homotrimer exhibits an internal network of branched cavities
Molecular dynamics simulations show that ATG9A trimers deform membranes
ACKNOWLEDGMENTS
This work used the NIH Multi-Institute Cryo-EM Facility (MICEF) and the computational resources of the NIH HPC Biowulf facility (https://hpc.nih.gov). We thank Huaibin Wang and Haifeng He for technical support on the NIH MICEF Titan Krios electron microscope and Michal Jarnik for help with the preliminary negative-stain EM. This work was supported by the Intramural Research Program of the National Heart, Lung, and Blood Institute (J.D.F.G. and J.J.) and Eunice Kennedy Shriver National Institute of Child Health and Human Development (J.S.B. project ZIA HD001607 and A.B. project ZIA HD008928), National Institutes of Health.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interest.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107837.
REFERENCES
- Anselmi C, Davies KM, and Faraldo-Gómez JD (2018). Mitochondrial ATP synthase dimers spontaneously associate due to a long-range membraneinduced force. J. Gen. Physiol 150, 763–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai XC, Rajendra E, Yang G, Shi Y, and Scheres SHW (2015). Sampling the conformational space of the catalytic subunit of human γ-secretase. eLife 4, e11182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bento CF, Renna M, Ghislat G, Puri C, Ashkenazi A, Vicinanza M, Menzies FM, and Rubinsztein DC (2016). Mammalian Autophagy: How Does It Work? Annu. Rev. Biochem 85, 685–713. [DOI] [PubMed] [Google Scholar]
- Braun MB, Traenkle B, Koch PA, Emele F, Weiss F, Poetz O, Stehle T, and Rothbauer U (2016). Peptides in headlock—a novel high-affinity and versatile peptide-binding nanobody for proteomics and microscopy. Sci. Rep 6, 19211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies KM, Anselmi C, Wittig I, Faraldo-Gómez JD, and Kühlbrandt W (2012). Structure of the yeast F1Fo-ATP synthase dimer and its role in shaping the mitochondrial cristae. Proc. Natl. Acad. Sci. USA 109, 13602–13607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatica D, Lahiri V, and Klionsky DJ (2018). Cargo recognition and degradation by selective autophagy. Nat. Cell Biol 20, 233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goehring A, Lee CH, Wang KH, Michel JC, Claxton DP, Baconguis I, Althoff T, Fischer S, Garcia KC, and Gouaux E (2014). Screening and large-scale expression of membrane proteins in mammalian cells for structural studies. Nat. Protoc 9, 2574–2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Sánchez R, Rose J, Guimarães R, Mari M, Papinski D, Rieter E, Geerts WJ, Hardenberg R, Kraft C, Ungermann C, and Reggiori F (2018). Atg9 establishes Atg2-dependent contact sites between the endoplasmic reticulum and phagophores. J. Cell Biol 217, 2743–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess B, Kutzner C, van der Spoel D, and Lindahl E (2008). GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput 4, 435–447. [DOI] [PubMed] [Google Scholar]
- Kakuta S, Yamaguchi J, Suzuki C, Sasaki M, Kazuno S, and Uchiyama Y (2017). Small GTPase Rab1B is associated with ATG9A vesicles and regulates autophagosome formation. FASEB J. 31, 3757–3773. [DOI] [PubMed] [Google Scholar]
- Kawate T, and Gouaux E (2006). Fluorescence-detection size-exclusion chromatography for precrystallization screening of integral membrane proteins. Structure 14, 673–681. [DOI] [PubMed] [Google Scholar]
- Kotani T, Kirisako H, Koizumi M, Ohsumi Y, and Nakatogawa H (2018). The Atg2-Atg18 complex tethers pre-autophagosomal membranes to the endoplasmic reticulum for autophagosome formation. Proc. Natl. Acad. Sci. USA 115, 10363–10368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai LTF, Yu C, Wong JSK, Lo HS, Benlekbir S, Jiang L, and Lau WCY (2019). Subnanometer resolution cryo-EM structure of Arabidopsis thaliana ATG9. Autophagy 16, 575–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang T, Reiche S, Straub M, Bredschneider M, and Thumm M (2000). Autophagy and the cvt pathway both depend on AUT9. J. Bacteriol 182, 2125–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, and Kroemer G (2019). Biological Functions of Autophagy Genes: A Disease Perspective. Cell 176, 11–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebschner D, Afonine PV, Baker ML, Bunkóczi G, Chen VB, Croll TI, Hintze B, Hung LW, Jain S, McCoy AJ, et al. (2019). Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D Struct. Biol 75, 861–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S, Otomo C, and Otomo T (2019). The autophagic membrane tether ATG2A transfers lipids between membranes. eLife 8, e45777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mari M, Griffith J, Rieter E, Krishnappa L, Klionsky DJ, and Reggiori F (2010). An Atg9-containing compartment that functions in the early steps of autophagosome biogenesis. J. Cell Biol 190, 1005–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrink SJ, Risselada HJ, Yefimov S, Tieleman DP, and de Vries AH (2007). The MARTINI force field: coarse grained model for biomolecular simulations. J. Phys. Chem. B 111, 7812–7824. [DOI] [PubMed] [Google Scholar]
- Mattera R, Park SY, De Pace R, Guardia CM, and Bonifacino JS (2017). AP-4 mediates export of ATG9A from the trans-Golgi network to promote autophagosome formation. Proc. Natl. Acad. Sci. USA 114, E10697–E10706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda T, Kim J, Huang WP, Baba M, Tokunaga C, Ohsumi Y, and Klionsky DJ (2000). Apg9p/Cvt7p is an integral membrane protein required for transport vesicle formation in the Cvt and autophagy pathways. J. Cell Biol 148, 465–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osawa T, Kotani T, Kawaoka T, Hirata E, Suzuki K, Nakatogawa H, Ohsumi Y, and Noda NN (2019). Atg2 mediates direct lipid transfer between membranes for autophagosome formation. Nat. Struct. Mol. Biol 26, 281–288. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Punjani A, Rubinstein JL, Fleet DJ, and Brubaker MA (2017). cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296. [DOI] [PubMed] [Google Scholar]
- Rana MS, Wang X, and Banerjee A (2018). An improved strategy for fluorescent tagging of membrane proteins for overexpression and purification in mammalian cells. Biochemistry 57, 6741–6751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohou A, and Grigorieff N (2015). CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol 192, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runwal G, Stamatakou E, Siddiqi FH, Puri C, Zhu Y, and Rubinsztein DC (2019). LC3-positive structures are prominent in autophagy-deficient cells. Sci. Rep 9, 10147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekito T, Kawamata T, Ichikawa R, Suzuki K, and Ohsumi Y (2009). Atg17 recruits Atg9 to organize the pre-autophagosomal structure. Genes Cells 14, 525–538. [DOI] [PubMed] [Google Scholar]
- Smart OS, Neduvelil JG, Wang X, Wallace BA, and Sansom MSP (1996). HOLE: a program for the analysis of the pore dimensions of ion channel structural models. J. Mol. Graph 14, 354–360, 376. [DOI] [PubMed] [Google Scholar]
- Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, Quispe J, Stagg S, Potter CS, and Carragher B (2005). Automated molecular microscopy: the new Leginon system. J. Struct. Biol 151, 41–60. [DOI] [PubMed] [Google Scholar]
- Tang Z, Takahashi Y, Chen H, Young MM, and Correspondence H-GW (2019). TOM40 targets Atg2 to mitochondria-associated ER membranes for phagophore expansion. Cell Rep. 28, 1744–1757.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valverde DP, Yu S, Boggavarapu V, Kumar N, Lees JA, Walz T, Reinisch KM, and Melia TJ (2019). ATG2 transports lipids to promote autophagosome biogenesis. J. Cell Biol 218, 1787–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, Kakuta S, Watanabe TM, Kitamura A, Sekito T, KondoKakuta C, Ichikawa R, Kinjo M, and Ohsumi Y (2012). Atg9 vesicles are an important membrane source during early steps of autophagosome formation. J. Cell Biol 198, 219–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young ARJ, Chan EYW, Hu XW, Köchl R, Crawshaw SG, High S, Hailey DW, Lippincott-Schwartz J, and Tooze SA (2006). Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J. Cell Sci 119, 3888–3900. [DOI] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, and Agard DA (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJH, Lindahl E, and Scheres SHW (2018). New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The relevant structural datasets generated during this study are available at EMDB [ATG9A trimer, state A: EMD-21874; ATG9A monomer, state A: EMD-21877; ATG9A trimer, state B: EMD-21876; ATG9A monomer, state B: EMD-21878] and PDB [ATG9A trimer, state A: 6WQZ; ATG9A trimer, state B: 6WR4].