

Abstract
Endometrial cancer (EC) is the sixth most common cancer in women worldwide. Early diagnosis is critical in recurrent EC management. The present study aimed to identify biomarkers of EC early recurrence using a workflow that combined text and data mining databases (DisGeNET, Gene Expression Omnibus), a prioritization algorithm to select a set of putative candidates (ToppGene), protein-protein interaction network analyses (Search Tool for the Retrieval of Interacting Genes, cytoHubba), association analysis of selected genes with clinicopathological parameters, and survival analysis (Kaplan-Meier and Cox proportional hazard ratio analyses) using a The Cancer Genome Atlas cohort. A total of 10 genes were identified, among which the targeting protein for Xklp2 (TPX2) was the most promising independent prognostic biomarker in stage I EC. TPX2 expression (mRNA and protein) was higher (P<0.0001 and P<0.001, respectively) in ETS variant transcription factor 5-overexpressing Hec1a and Ishikawa cells, a previously reported cell model of aggressive stage I EC. In EC biopsies, TPX2 mRNA expression levels were higher (P<0.05) in high grade tumors (grade 3) compared with grade 1–2 tumors (P<0.05), in tumors with deep myometrial invasion (>50% compared with <50%; P<0.01), and in intermediate-high recurrence risk tumors compared with low-risk tumors (P<0.05). Further validation studies in larger and independent EC cohorts will contribute to confirm the prognostic value of TPX2.
Keywords: endometrial cancer, bioinformatics, biomarkers, recurrence, TPX2
Introduction
Endometrial cancer (EC) is the sixth most common cancer in women worldwide and the second most common gynecologic neoplasm after cervical cancer. According to IARC/GLOBOCAN data (1), a worldwide increase of 52.7% in incidence and 70.6% in mortality is expected in EC for the year 2040.
According to its histopathological characteristics, EC is classified in two categories: Type I or endometrioid endometrial carcinomas (EEC), and type II or non-endometrioid endometrial carcinomas (NEEC) (2). EEC is the most frequent histological type (~80% of all cases); when diagnosed at an early stage [stage I, according to the classification of the International Federation of Gynecology and Obstetrics (FIGO), 2009] (3) and low histological grade, it is associated with a good prognosis and 5-year survival rates of 90% (4,5). However, 13–25% of patients with EEC suffer recurrence and metastatic disease, which is associated with a poor outcome (6–10).
The lower survival rates of patients with relapsed stage I EEC depend on disease distribution. It has been reported that 5-year survival is reduced to 55% for pelvic recurrences and to 17% for extra-pelvic recurrences (11). Notably, patients diagnosed with advanced disease (stage III/IV) have a high risk of recurrence and are more likely to have extra-pelvic metastases at the time of recurrence (12).
Early diagnosis is critical in recurrent EC management. In this context, despite the benefits of the FIGO 2009 surgical-pathological staging system (3), some limitations have been reported. In particular, in Stage I EC, an inaccurate prognostic assessment may lead to unnecessary follow-up for the majority of patients or to suboptimal treatment of patients who will eventually suffer relapse.
The challenge to improve current EC decision-making and management has involved deepening the understanding of the molecular basis of EC, in order to identify molecular entities with altered expression and/or functions, known as biomarkers. In recent decades, a number of tissue biomarkers have been introduced for EC prognosis (13,14). In addition, several studies have proposed EC molecular prognostic signatures (15–17). In particular, the use of high-throughput sequencing technology coupled with bioinformatics has led to a comprehensive genetic and molecular characterization of EC by The Cancer Genome Atlas (TCGA) research network (18). As part of this characterization, four EC molecular subtypes have been identified with distinctive molecular and clinicopathological features (DNA polymerase ε, microsatellite instability, copy number (CN) Low and CN High) (18). Despite its power as a prognostic tool, TCGA classification involves evaluations with a set of techniques (genomics, transcriptomics, proteomics and somatic CN alterations assessment) that require great professional expertise and sophisticated equipment, and thus elevated costs, making its implementation difficult in clinical services worldwide.
To gain further insight into EC biology and the identification of prognostic biomarkers, the present study was conducted to search for differentially expressed genes (DEGs) in EC tissues. These genes were prioritized after comparison with a reference list of EC-related genes gathered from the DisGeNET database. Subsequently, the prioritized genes were subjected to further hierarchical clustering analysis in a different EC cohort. Then, an enrichment analysis of DEGs with information from pathway and/or Gene Ontology databases was performed. In addition, an evaluation was carried out to assess whether DEG-encoded proteins physically interact within the cell or are part of protein molecular networks. A set of genes were further subjected to association analysis with EC clinicopathological parameters and survival analysis. Finally, an expression analysis of one candidate biomarker was conducted using a cell model of EC aggressiveness and a pilot validation study was performed using samples obtained from patients with EC.
Materials and methods
Bioinformatics approaches
Survey of EC-associated genes
To survey current knowledge on EC-related genes, the DisGeNET gene-disease associations database was used (19,20). DisGeNET version 5.0 (online version), containing >500,000 associations between >17,000 genes and 20,000 diseases, was used for this study. DisGeNET uses a vocabulary for diseases given by the Concept Unique Identifiers from the Unified Medical Language System® (UMLS) Metathesaurus® (version UMLS 2018AA) (21). Specifically, genes associated with the following EC disease terms were collected: ‘Endometrial neoplasms’, ‘Endometrial carcinoma’, ‘Stage endometrial cancer’, ‘Recurrent endometrial cancer’, ‘Serous endometrial intraepithelial carcinoma’, ‘Endometrial Squamous Cell Carcinoma’, ‘Endometrial endometrioid adenocarcinoma’, ‘Endometrial intraepithelial neoplasia’, ‘Endometrial serous adenocarcinoma’, ‘Endometrial adenocarcinoma’, ‘Endometrial stromal sarcoma’, ‘Low grade endometrial stromal sarcoma’, ‘Endometrial stromal tumors’ and ‘Malignant neoplasm of endometrium’. To obtain a complete repertoire of EC-associated genes, a search including all identified terms referring to EC was performed and results were compiled in one list.
Analysis of EC DEGs
Affymetrix microarray data from the GSE17025 dataset [n=103 samples; control (atrophic endometrium), n=12; tumor, n=91] (22), was obtained from the Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) database. The clinicopathological characteristics of the patients included in this cohort are detailed in Table SI. Microarray data were analyzed using GPL570 platform (Affymetrix Human Genome U133 Plus 2.0 Array; Affymetrix; Thermo Fisher Scientific, Inc.). DEGs between non-tumor and tumor samples were identified using GEO2R (https://www.ncbi.nlm.nih.gov/geo/geo2r/), where genes with adjusted P<0.05 and |log[fold-change (FC)]|>0.5 were defined as DEGs. Multiple testing corrections were performed through Benjamini-Hochberg False Discovery Rate (FDR)<0.05. In addition, RNAseq (Illumina GA platform; Illumina, Inc.) and clinical data from TCGA Uterine Corpus Endometrioid Cancer (UCEC; n=333 samples; mean age, 63.2 years; age range, 33–90 years; Table SII) dataset was retrieved from the UCSC Xena portal (https://xena.ucsc.edu) in order to address the prognostic potential of EC candidate genes.
Gene prioritization
Gene prioritization and functional enrichment [Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways] analyses of DEGs were performed using ToppGene Suite (http://toppgene.cchmc.org) (23). ToppGene Suite is a portal that allows functional enrichment, prioritization of candidate genes using functional annotations and/or analysis of interaction networks, leading to identification of disease candidate genes. Gene prioritization analysis was carried out considering the following parameters: GO (molecular function, biological process and cellular component), Human phenotype, Mouse phenotype, Pathway, Pubmed, Interaction, Co-expression, Co-expression Atlas and Disease. In all cases, the standard method used to determine statistical significance (P<0.05) was the Hypergeometric probability distribution with Benjamini-Hochberg FDR correction.
Identification of DEGs among tumors of histological grade 1 and 3
To identify a subset of DEGs among tumors of histological grade 1 and 3 among the 3,976 prioritized genes using TCGA UCEC RNAseq data, the unpaired Student's t-test between subjects assuming equal variances was applied (P<0.01). To define sample and gene clusters, non-supervised hierarchical clustering was performed. For all these analyses the MeV software (http://mev.tm4.org/) was used. Functional enrichment analysis of each gene cluster was performed in ENRICHR (http://amp.pharm.mssm.edu/Enrichr/). The ggplot2 R (https://github.com/tidyverse/ggplot2) package was used to create heatmap plots.
Protein-protein interaction (PPI) network analysis construction
A PPI network was built using the Search Tool for the Retrieval of Interacting Genes (STRING) database (https://string-db.org/). In addition, identification of hub genes was conducted using the cytoHubba application in Cytoscape (24).
In silico analysis of TPX2 post-translational modifications
Prediction of the content of potential glycosylation (N and O-glycosylation), as well as phosphorylation sites in the TPX2 protein sequence (NP_036244.2) was performed. For this purpose, glycosylation sites were predicted using the NetNGlyc 1.0 Server (http://www.cbs.dtu.dk/services/NetNGlyc/) and the NetOGlyc 4.0 Server (http://www.cbs.dtu.dk/services/NetOGlyc/) (N and O-glycosylation sites, respectively), and putative phosphorylation sites were predicted using the NetPhos 3.1 Server (http://www.cbs.dtu.dk/services/NetPhos/).
Experimental approaches
Patient samples
Endometrial tissue samples were obtained from patients with EC who underwent surgery before receiving hormonal and/or chemotherapy treatment at Vall d'Hebron Hospital (Barcelona, Spain) between January 2006 and December 2015 (mean age, 66.5 years; age range, 40–82 years). The Institutional Review Boards from Vall d'Hebron Hospital and from IBYME approved the protocol, and written informed consent was provided by all patients participating in the study (approval no. SAF-20083997; CE001/2013). Samples were classified based on the 2009 FIGO staging system. Sample collection and handling was done as previously described (25). Table SIII shows patient samples clinical information.
Cell culture
Hec1a and Ishikawa EC commercial cell lines, as well as Hec1a and Ishikawa cells stably transfected with the human ETS variant transcription factor 5 (ETV5) sequence (Hec1a-ETV5 and Ishikawa-ETV5, respectively) were kindly provided by Dr Reventos and co-workers (Biomedical Research Group in Gynecology at Vall d'Hebron Research Institute, Barcelona, Spain). ETV5-overexpressing Hec1a and Ishikawa cells were generated by stable transfection with the pEGFP-C2 vector (BD Biosciences) containing the human ETV5 coding sequence, as previously reported (26). Hec1a and Hec1a-ETV5 cells were cultured in McCoy's 5A, while parental and ETV5-transfected Ishikawa cells were cultured in DMEM:F12, all supplemented with 10% fetal bovine serum, and handled as described (26). Cell lines were morphologically and genetically authenticated and tested for mycoplasma in accordance with American Association for Cancer Research guidelines as previously reported (27). The mRNA expression levels of ETV5 were monitored in Hec1a, Ishikawa, Hec1a-ETV5 and Ishikawa-ETV5 cells, by means of reverse transcription-quantitative PCR (RT-qPCR) (Fig. S1).
RNA extraction, cDNA synthesis and qPCR
Procedures were performed as previously reported (28). Briefly, total RNA was extracted from cell lines and tissue samples with TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.), according to standard protocols. cDNA synthesis was performed with 1–2 µg total RNA using the SuperScript™ III reverse transcriptase enzyme at 50°C (Thermo Fisher Scientific, Inc.). Negative controls omitting RNA or reverse transcriptase were included and tested in the PCR procedure. Quantitative evaluation of mRNA levels was performed by qPCR using SYBR-Green® PCR Master Mix (Thermo Fisher Scientific, Inc.) with the CFX96 Touch™ unit (Bio-Rad Laboratories, Inc.). Thermocycling conditions were as follows: Stage 1, 2 min at 50°C; stage 2, 10 min at 95°C; stage 3, 15 sec at 95°C; stage 4, 1 min at 60°C; stages 3–4 were repeated 40 times. All samples were run in triplicate; negative controls (the two aforementioned controls of the RT assay and a PCR control where the DNA template was omitted) were tested in all cases. Transcript expression levels were determined as follows: 2−ΔCq, where: DCq=Cq gene under study-Cq housekeeping gene (GAPDH) (29). PCR primers sequences were as follows: Targeting protein for Xklp2 (TPX2), forward 5′-GCGCTCTGATTGGTGCATTC-3′, reverse 5′-TTCTTCCCACGGCTCACCTA-3′ (PCR fragment size, 131 bp); GAPDH, forward 5′-TGCACCACCAACTGCTTAGC-3′ and reverse 5′-GGCATGGACTGTGGTCATGAG-3′ (PCR fragment size, 88 bp); ETV5, forward 5′-TGCTTCAGCTAACCAAGCCT-3′, reverse 5′-ATGGTCCCAGGGAAATCTCG-3′ (PCR fragment size, 150 bp).
Fluorescence immunocytochemistry
Cell monolayers were fixed with 4% paraformaldehyde for 10 min, treated with 0.1% Triton X-100 for 10 min and blocked with 4% bovine serum albumin (Sigma-Aldrich; Merck KGaA) in PBS for 45 min; all of these procedures were performed at room temperature. Subsequently, cells were incubated for 1 h at 37°C with 2 µg/ml anti-TPX2 antibody (1:50; mouse monoclonal, epitope mapping amino acids 635–675 near C-terminus; cat. no. sc-376812; Santa Cruz Biotechnologies, Inc.), followed by a 1-h incubation at room temperature with a fluorescent secondary antibody (1:500; goat anti-mouse IgG-Cy3 conjugate; cat. no. A10521; Invitrogen; Thermo Fisher Scientific, Inc.). Negative controls were run using the same concentration of purified IgG (cat. no. I5381; Sigma-Aldrich; Merck KGaA) from the same species as the primary antibody. Nuclear cell staining was done with Hoechst 33342 (Sigma-Aldrich; Merck KGaA). Cell preparations were analyzed using a Nikon C1 confocal laser microscope (Nikon Corporation).
Sample preparation, SDS-PAGE and western immunoblotting
Cell lysates were prepared in RIPA buffer [20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate] supplemented with a cocktail of protease inhibitors (2 mM p-aminobenzamidine, 1 mM PMSF, 10 µg/ml aprotinin and 10 µg/ml leupeptin). Protein concentration was determined using the Bradford method (Bio-Rad Laboratories, Inc.). Protein extracts (30 µg) from EC cell lines were separated by SDS-PAGE on 10% gels and were electrotransferred to nitrocellulose membranes (Amersham Hybond ECL; GE Healthcare). Membranes were blocked in PBS containing 5% nonfat milk for 1 h at room temperature incubated overnight at 4°C with 2 µg/ml anti-TPX2 antibody (1:100; cat. no. sc-376812; Santa Cruz Biotechnology, Inc.) diluted in blocking solution, and for 1 h at room temperature with secondary antibody [horseradish peroxidase (HRP)-conjugated anti-mouse IgGs; 1:1,000; cat. no. 32430; Thermo Fisher Scientific, Inc.] in blocking solution. Assays were developed with ECL Western Blotting Detection kit (GE Healthcare) chemiluminescence system. Negative controls run using the same concentration of purified IgG from the same species as the primary antibody were included in all cases.
Statistical analysis
In order to determine whether the data followed a Gaussian distribution, the D'Agostino-Pearson test was used. Comparisons involving two groups of samples were analyzed using unpaired Student's t-test or Mann-Whitney test, as shown in each case. For studies involving more than two experimental groups, the Kruskal Wallis test followed by Dunn's post hoc test was applied.
To define gene expression cut-off values, the Cutoff Finder web tool (http://molpath.charite.de/cutoff/) (30) was used (method ‘survival’), and subsequent analysis between categorical variables was performed using the Fisher's Exact Test. Specifically, samples were classified, according to the expression level of each gene included in the analysis, as ‘low’ or ‘high’ expression categories, according to the cut-off value determined by applying the ‘survival’ method in Cutoff Finder. In addition, Receiver Operating Characteristic (ROC) curves were used to determine diagnostic accuracy of selected genes.
For survival analysis, Kaplan-Meier curves were constructed and the differences between them were analyzed by the log-rank test. Additionally, Cox proportional hazards model was used to identify independent survival predictor variables. Analyses were performed on GraphPad Prism version 5 (GraphPad Software, Inc.) and R (version 3.4.4; http://www.r-project.org/). The workflow used in the present study is shown in Fig. 1.
Figure 1.
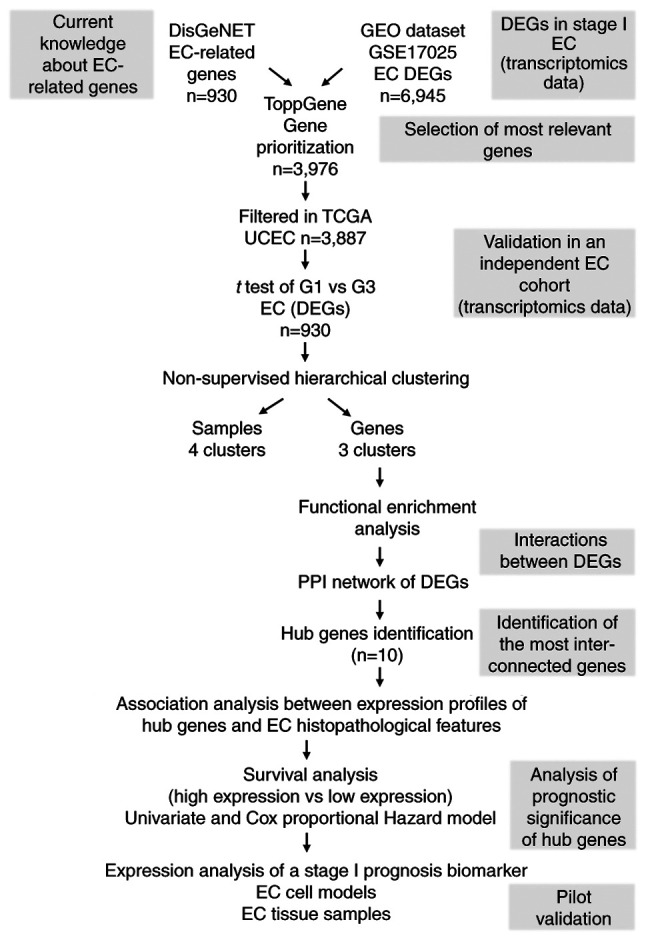
Workflow diagram of text and data mining tools used for the identification of prognostic EC biomarkers. Genes associated with the entire repertoire of EC-related disease terms were gathered from DisGeNET in order to cover a higher spectrum of genes with potential prognostic value in EC. These genes were used as a reference for further selection and prioritization of EC-DEGs retrieved from the GEO microarray dataset GSE17025. The prioritized genes were then subjected to hierarchical clustering analysis using TGCA UCEC RNAseq dataset, which led to the identification of a set of DEGs able to generate sample and gene clusters. Since sample cluster segregation has a lack of prognostic significance, gene clusters were further analyzed. To determine if DEGs belonged to the same pathways, enrichment analysis and PPI network analysis were carried out. A set of ‘hub genes’ were identified, and statistical association analysis with EC clinicopathological parameters and survival analysis was performed. Finally, pilot validation studies were performed using EC cell models and tissue specimens. EC, endometrial cancer; DEGs, differentially expressed genes; GEO, Gene Expression Omnibus; TCGA, The Cancer Genome Atlas; UCEC, Uterine Corpus Endometrioid Cancer; PPI, protein-protein interaction.
Results
Survey of EC-associated genes using the DisGeNET database
A search on DisGeNET was conducted using different EC-related terms to obtain a spectrum of genes associated with EC. The scientific literature shows inconsistencies regarding the terms used when reporting findings, and terms are sometimes not the same to describe a specific phenomenon or variable studied. Consequently, the different source databases that are integrated into DisGeNET annotate the genes associated with EC to different terms present in the controlled vocabularies. While only 10 genes were found to be associated to the disease term ‘endometrial cancer recurrent’, a higher number of genes with potential prognostic significance in EC have been described in the literature, and several of these genes have not been annotated to the term ‘endometrial cancer recurrent’. The spectrum of disease terms related to EC was expanded to retrieve all of the associated genes that could have potential prognostic value in EC. All of these genes were surveyed and compiled in a list. A total of 930 EC-associated genes are listed in Table SIV.
Analysis of EC-DEGs and gene prioritization
Microarray data from the GSE17025 study (GEO repository) was used to identify DEGs between non-tumoral atrophic endometria from postmenopausal women and tumor endometrial samples with the GEO2R tool, resulting in a total of 6,945 DEGs (P<0.05). In order to prioritize these genes according to their relevance for the EC phenotype, the ToppGene tool was used. Specifically, a comparison was done between the list of EC-associated genes retrieved from the DisGeNET database (‘training set’) and the gene list gathered from the GSE17025 dataset (‘test set’). As a result, a list of 3,976 genes associated with the disease was obtained (P<0.05) (data not shown).
Gene expression profiles of candidate genes
To determine the relevance of genes identified in the gene prioritization analysis, their gene expression profiles were evaluated using RNAseq data from TCGA UCEC study. Gene expression profiles of the prioritized genes were analyzed only in tumor samples (n=333). Considering that histological grade has been reported to have a significant impact on recurrence-free survival (RFS) in patients with stage I EC (31), the expression profile of the prioritized genes in tumor tissues was evaluated according to histological grades 1 and 3 (Student's t-test; P<0.05). First, four clusters were identified among tumor samples, clusters 1 (n=41), 2 (n=86), 3 (n=102) and 4 (n=104) (Fig. 2A). A significant association was found between the number of samples in each cluster and tumor histology, stage, grade and molecular subtype (Table SV). Cluster 4 depicted the highest number of serous carcinomas, as well as the highest number of advanced stage (III and IV), grade 3 and CN High subtype carcinomas, although only 13.46 % (14/104) cases showed all four negative prognostic factors. Clusters 1–4 were associated with a significant segregation of endometrial tumors according to their clinicopathological characteristics. However, this segregation was not related to EC patient prognosis, since no significant differences were observed in RFS and overall survival (OS) curves in the cohort studied (RFS, P=0.2465; OS, P=0.6361; Fig. S2). However, the comparison of DEGs between grade 1 and grade 3 tumors revealed the segregation of three distinctive gene clusters (clusters I, II and III; Fig. 2A). Clusters I and II (448 and 293 genes, respectively) contained genes with increased expression in grade 3 tumors compared with grade 1 tumors, whereas cluster III (189 genes) contained genes with a diminished expression in grade 3 tumors compared with grade 1 tumors. Functional enrichment analysis of the genes included in each cluster indicated that genes from clusters I and II were mainly associated with cell cycle regulation, whereas genes from cluster III were related to protein metabolism and regulation of translation (Fig. 2B). Table I lists the most representative functional annotations of each cluster.
Figure 2.

Hierarchical clustering of grade 1 and 3 EC samples. (A) Differential gene expression analysis between grade 1 and 3 EC tumors generated four sample clusters (clusters 1–4) and three gene clusters (clusters I–III). Heatmap representation of the top 100 significant genes of each gene cluster is shown. (B) Functional enrichment analysis of gene clusters showing most significantly represented biological processes for each gene cluster. EC, endometrial cancer.
Table I.
Functional enrichment analysis of gene clusters I–III.
| A, Cluster I | ||
|---|---|---|
| GO term | Adjusted P-value | Genes |
| Cell Cycle_Homo sapiens_R-HSA-1640170 | 9.9122×10−22 | TOP2A; AHCTF1; HSP90AB1; ZWILCH; MCM10; RSF1; SMC4; CDC20; NIPBL; CDC23; EXO1; NUF2; NEK2; KNTC1; YWHAG; RAB2A; HUS1; KIF23; TERF1; MASTL; SMC1A; CKAP5; YWHAZ; CDC25A; PSMA6; TFDP1; PSMA1; NUP50; PSME4; UBE2V2; DNA2; MAPRE1; PSMD12; SEH1L; PSMD11; PSMD14; PKMYT1; CENPA; SKA2; CCNB2; BRIP1; PSMB2; RBBP4; RAD21; E2F3; BUB3; CENPW; RRM2; UBE2C; GINS4; NUP153; TPX2; CENPF; PSMC1; PSMC2; CDK1; TUBGCP3; RAD1; LPIN2; NUP37 |
| Cell Cycle, Mitotic_Homo sapiens_R-HSA-69278 | 2.4984×10−19 | TOP2A; AHCTF1; HSP90AB1; ZWILCH; MCM10; SMC4; CDC20; NIPBL; CDC23; NUF2; NEK2; KNTC1; YWHAG; RAB2A; KIF23; MASTL; SMC1A; CKAP5; CDC25A; PSMA6; TFDP1; PSMA1; NUP50; PSME4; DNA2; MAPRE1; PSMD12; SEH1L; PSMD11; PSMD14; PKMYT1; CENPA; SKA2; CCNB2; PSMB2; RBBP4; RAD21; E2F3; BUB3; RRM2; UBE2C; GINS4; NUP153; TPX2; CENPF; PSMC1; PSMC2; CDK1; TUBGCP3; CENPO; LPIN2; NUP37 |
| M Phase_Homo sapiens_ R-HSA-68886 | 1.7904×10−16 | AHCTF1; PSMD12; PSMD11; SEH1L; PSMD14; ZWILCH; SMC4; CENPA; SKA2; CDC20; CCNB2; NIPBL; CDC23; PSMB2; RAD21; NUF2; KNTC1; BUB3; RAB2A; UBE2C; NUP153; KIF23; MASTL; SMC1A; CKAP5; PSMA6; CENPF; PSMA1; PSMC1; NUP50; PSMC2; PSME4; CDK1; CENPO; MAPRE1; LPIN2; NUP37 |
| Gene Expression_Homo sapiens_R-HSA-74160 | 1.7904×10−16 | ZNF496; TDRKH; GSK3B; SMG1; HNRNPU; ZC3H8; ADAR; EPRS; GLS; MED14; MED13; CASP10; EXO1; XPO5; RIOK1; YWHAG; ZIK1; SUPT16H; TBP; PDPK1; NCBP2; HUS1; BAZ1B; YWHAZ; SRRM1; SAP30; PSMA6; TFDP1; PSMA1; XRN1; TBL1XR1; NUP50; PSME4; TET3; RRAGD; POLR1E; DNA2; FYTTD1; ZNF274; PSMD12; SEH1L; PSMD11; RBM8A; PSMD14; RPN2; DDX21; TARS; BAZ2A; RNPC3; GTF2E1; BRIP1; MED30; PDCD11; PSMB2; RBBP4; PLAGL1; TRA2B; BMS1; ZKSCAN3; EIF4H; TP53BP2; IARS2; RPP14; EXOSC3; SKIL; EIF4B; POLR2K; DCAF13; ZNF420; WWTR1; WTAP; CBX3; UTP3; TAF11; CDC5L; NUP153; LSM4; GATAD2B; LSM3; TPX2; PHF5A; NR6A1; EIF5; POLR3B; PSMC1; PSMC2; CDK1; TAF4B; RAD1; CDK12; DCP1A; TAF2; RAN; FARSB; NUP37; EIF4G1 |
| Separation of Sister | 3.424×10−16 | PSMD12; AHCTF1; PSMD11; SEH1L; PSMD14; ZWILCH; CENPA; |
| Chromatids_Homo sapiens_R-HSA-2467813 | SKA2; CDC20; CDC23; PSMB2; RAD21; NUF2; KNTC1; BUB3; UBE2C; SMC1A; CKAP5; PSMA6; CENPF; PSMA1; PSMC1; PSMC2; PSME4; CENPO; MAPRE1; NUP37 | |
| B, Cluster II | ||
| GO term | Adjusted P-value | Genes |
| Cell Cycle_Homo sapiens_R-HSA-1640170 | 0.00026303 | DIDO1; ANKLE2; YWHAB; CDCA5; CDCA8; FOXM1; PSMA7; LMNA; E2F1; NUP88; PSMF1; PCNT; CSNK2A1; NCAPH2; TUBB; TOP3A; RANGAP1; LEMD2; POLA2; PSMC4; DKC1; MCM3; MCM4; KIF2C; SPC24; LPIN3 |
| Cell Cycle, Mitotic_Homo sapiens_R-HSA-69278 | 0.00061651 | ANKLE2; CSNK2A1; NCAPH2; CDCA5; TUBB; CDCA8; FOXM1; RANGAP1; LEMD2; PSMA7; POLA2; PSMC4; LMNA; MCM3; E2F1; MCM4; NUP88; KIF2C; PSMF1; PCNT; SPC24; LPIN3 |
| Axon guidance_Homo sapiens_R-HSA-422475 | 0.00231835 | BRAP; EPHB6; ROBO3; CSNK2A1; YWHAB; LIMK1; ARAF; FN1; PHB; PSMA7; DNM2; ARPC2; PSMC4; CFL1; GRB2; PSMF1; COL9A2; JAK3; EPHB1; SPTAN1; MYH10; CAMK2G |
| M Phase_Homo sapiens_R-HSA-68886 | 0.00231835 | ANKLE2; CSNK2A1; NCAPH2; CDCA5; CDCA8; RANGAP1; LEMD2; PSMA7; PSMC4; LMNA; NUP88; KIF2C; PSMF1; SPC24; LPIN3 |
| Immune System_Homo sapiens_R-HSA-168256 | 0.00274532 | DCTN5; CUL7; YWHAB; MAPKAP1; ARAF; ICAM5; PHB; UBE2Z; ECSIT; UBE2J2; ELK1; PLD3; PSMA7; ICAM1; ADCY5; RNF216; IRAK1; CFL1; KIF5A; ATP6V0A2; NUP88; PSMF1; JAK3; SPTAN1; CAMK2G; POLR2L; CTSA; BRAP; IL11RA; LIMK1; FN1; DNM2; MAVS; ARPC2; TRIM39; PSMC4; TRAF3; POLR3E; GRB2; UBA1; KIF2C; CRK; RAPGEF3; ATP6V1B1 |
| C, Cluster III | ||
| GO term | Adjusted P-value | Genes |
| Metabolism of proteins_Homo sapiens_R-HSA-392499 | 4.165×10−6 | TSTA3; COX19; ARF1; L3MBTL2; DCTN1; RPL31; POMT1; PARN; PIGV; ZDHHC2; RPS14; POFUT2; EXOSC4; RPL18A; FBXO4; RPL15; EXTL3; RPS10; MPDU1; PIAS4; SEC16B; NFYC; H3F3A; DYNC1LI2; RPS28; CCNE2; EEF1D; EIF3G; PDCL; EXOC3; RPL29; PFDN5 |
| Eukaryotic Translation Elongation_Homo sapiens_R-HSA-156842 | 0.00039761 | RPS14; RPS28; RPL18A; EEF1D; RPL31; RPL15RPL29; RPS10 |
| Selenocysteine synthesis_Homo sapiens_R-HSA-2408557 | 0.00039761 | RPS14; RPS28; RPL18A; RPL31; SECISBP2; RPL15; RPL29; RPS10 |
| Formation of a pool of free 40S subunits_Homo sapiens_R-HSA-72689 | 0.00052927 | RPS14; RPS28; RPL18A; RPL31; EIF3G; RPL15; RPL29; RPS10 |
| Infectious disease_Homo sapiens_R-HSA-5663205 | 0.0005955 | ARF1; RPL31; GTF2F1; RPS14; RPS28; CDK7; PSMB5; PSMC3; RPL18A; RPL15; RPL29; RPS10; VPS28; AP1M1 |
GO, Gene Ontology.
PPI network analysis and identification of hub genes
In order to evaluate the interactions between DEGs obtained as a result of the comparison between grade 1 and 3 EC, a PPI network was constructed using the STRING database. The resulting network contained a total of 930 nodes and 9,392 edges. Subsequently, the network hub genes were identified using the cytoHubba application in Cytoscape. The top 10 genes with the highest degree of connectivity were defined as the hub genes: CCNB2, CDCA8, CDC20, CDK1, CENPF, KIF2C, RRM2, UBE2C, TOP2A and TPX2 (Fig. 3; Table II). Since these genes were also differentially expressed between EC and control samples, to determine their diagnostic potential, a ROC curve analysis was performed using RNAseq data from TCGA UCEC cohort. As a result, all hub genes significantly distinguished patients with EC from control patients (CCNB2, AUC=0.9834; CDCA8, AUC=0.9832; CDC20, AUC=0.9885; CDK1, AUC=0.9486; CENPF, AUC=0.9727; KIF2C, AUC=0.9807; RRM2, AUC=0.9720; UBE2C, AUC=0.9762; TOP2A, AUC=0.9640; TPX2, AUC=0.9775; P<0.0001 for all genes; Fig. S3). Subsequently, the association between the expression levels of hub genes and clinicopathological parameters was evaluated. Firstly, a higher expression of all hub genes was confirmed in grade 3 compared with grade 1–2 tumors. Moreover, CCNB2, CDCA8, CDC20, CENPF, KIF2C, UBE2C, TOP2A and TPX2 expression was significantly associated with tumor histology, with highest mRNA levels in NEEC cases. In addition, CDCA8, CDK1, CENPF, UBE2C and TPX2 transcript levels were higher in stage III–IV than in stage I–II tumors. Finally, CDCA8, CDK1, UBE2C and TPX2 expression levels were higher in tumors with deep myometrial invasion (MI) (Table III).
Figure 3.
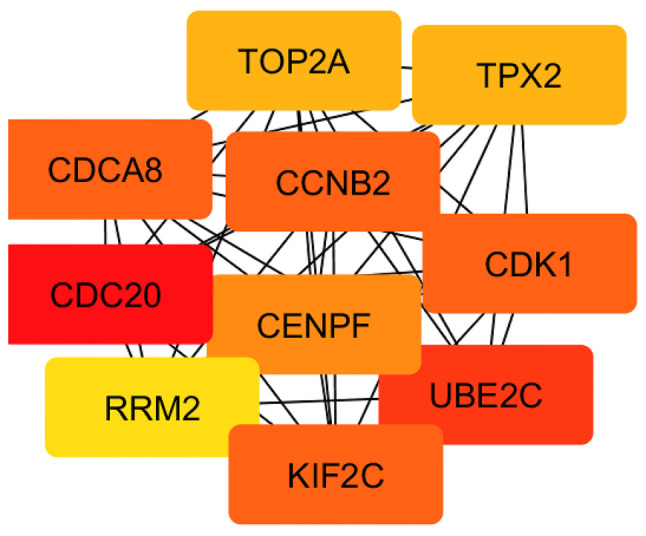
Protein-protein interaction network analysis of grade 1 and 3 differentially expressed genes. Network hub genes identified using the cytoHubba application in Cytoscape. Ten hub genes with the highest degree of connectivity were identified: CCNB2, CDCA8, CDC20, CDK1, CENPF, KIF2C, RRM2, UBE2C, TOP2A and TPX2.
Table II.
Functional enrichment analysis of hub genes.
| A, Biological process (GO) | |||
|---|---|---|---|
| Term/pathway | Description | False discovery rate | |
| GO:1903047 | Mitotic cell cycle process | 2.92×10−13 | |
| GO:0051301 | Cell division | 8.13×10−12 | |
| GO:0000280 | Nuclear division | 1.59×10−7 | |
| GO:0140014 | Mitotic nuclear division | 3.70×10−7 | |
| GO:0051726 | Regulation of cell cycle | 3.82×10−7 | |
| B, Molecular function (GO) | |||
| Term/pathway | Description | False discovery rate | |
| GO:0008022 | Protein C-terminus binding | 0.0122 | |
| GO:0005524 | ATP binding | 0.0122 | |
| GO:0004693 | Cyclin-dependent protein serine/threonine kinase activity | 0.0122 | |
| GO:0042826 | Histone deacetylase binding | 0.0153 | |
| GO:0003682 | Chromatin binding | 0.0153 | |
| C, Cellular component (GO) | |||
| Term/pathway | Description | False discovery rate | |
| GO:0015630 | Microtubule cytoskeleton | 2.43×10−5 | |
| GO:0005819 | Spindle | 2.43×10−5 | |
| GO:0044430 | Cytoskeletal part | 7.34×10−5 | |
| GO:0005815 | Microtubule organizing center | 0.00033 | |
| GO:0005829 | Cytosol | 0.00065 | |
| D, KEGG pathways | |||
| Term/pathway | Description | False discovery rate | |
| hsa04115 | p53 signaling pathway | 9.70×10−5 | |
| hsa04114 | Oocyte meiosis | 0.00023 | |
| hsa04110 | Cell cycle | 0.00023 | |
| hsa04914 | Progesterone-mediated oocyte maturation | 0.0047 | |
| hsa04120 | Ubiquitin mediated proteolysis | 0.0075 | |
GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes.
Table III.
Association analysis between mRNA expression of hub genes and clinicopathological parameters.
| Histology | Stage | MI | Grade | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | NEEC | EEC | P-value | III/IV | I/II | P-value | MI>50% | MI<50% | P-value | Grade 3 | Grade 1/2 | P-value |
| RRM2 | 0.2247 | 0.1653 | 0.2912 | 0.0003a | ||||||||
| High | 12 | 24 | 13 | 23 | 18 | 18 | 31 | 5 | ||||
| Low | 104 | 336 | 110 | 330 | 177 | 263 | 243 | 197 | ||||
| CDC20 | <0.0001a | 0.1928 | 0.1579 | <0.0001a | ||||||||
| High | 33 | 41 | 24 | 50 | 36 | 38 | 66 | 8 | ||||
| Low | 83 | 319 | 99 | 303 | 159 | 243 | 208 | 194 | ||||
| TOP2A | 0.0032b | 0.0636 | 0.0996 | 0.0008a | ||||||||
| High | 13 | 13 | 11 | 15 | 15 | 11 | 23 | 3 | ||||
| Low | 103 | 347 | 112 | 338 | 180 | 270 | 251 | 199 | ||||
| CDCA8 | 0.0001a | 0.0426c | 0.0099b | <0.0001a | ||||||||
| High | 32 | 41 | 26 | 47 | 40 | 33 | 66 | 7 | ||||
| Low | 84 | 319 | 97 | 306 | 155 | 248 | 208 | 195 | ||||
| UBE2C | <0.0001a | 0.0257c | 0.0011b | <0.0001a | ||||||||
| High | 68 | 89 | 51 | 106 | 81 | 76 | 128 | 29 | ||||
| Low | 48 | 271 | 72 | 247 | 114 | 205 | 146 | 173 | ||||
| TPX2 | <0.0001a | 0.0005a | 0.0002a | <0.0001a | ||||||||
| High | 68 | 40 | 42 | 66 | 61 | 47 | 102 | 6 | ||||
| Low | 48 | 320 | 81 | 287 | 134 | 234 | 172 | 196 | ||||
| CDK1 | 0.1159 | 0.0419c | 0.0103c | 0.0022b | ||||||||
| High | 17 | 99 | 19 | 31 | 29 | 21 | 39 | 11 | ||||
| Low | 33 | 327 | 104 | 322 | 166 | 260 | 235 | 191 | ||||
| CCNB2 | 0.0281c | 0.06326 | 0.0733 | <0.0001a | ||||||||
| High | 26 | 49 | 26 | 49 | 38 | 37 | 63 | 12 | ||||
| Low | 90 | 311 | 97 | 304 | 157 | 244 | 211 | 190 | ||||
| KIF2C | 0.0079b | 0.1460 | 0.0843 | <0.0001a | ||||||||
| High | 22 | 34 | 19 | 37 | 29 | 27 | 53 | 3 | ||||
| Low | 94 | 326 | 104 | 316 | 166 | 254 | 221 | 199 | ||||
| CENPF | 0.0001a | 0.0308c | 0.0816 | 0.0004a | ||||||||
| High | 22 | 23 | 18 | 27 | 24 | 21 | 37 | 8 | ||||
| Low | 94 | 337 | 105 | 326 | 171 | 260 | 237 | 194 | ||||
EEC, endometrioid endometrial carcinoma; NEEC, non-endometrioid endometrial carcinoma; MI, myometrial invasion. Statistical analysis was conducted using Fisher's Exact test.
P<0.001
P<0.01
P<0.05.
Next, the relationship between expression of the 10 hub genes and prognosis of patients with EC was assessed in TCGA cohort. A higher expression of CDK1, KIF2C, UBE2C and TPX2 was significantly associated with shorter OS (Fig. 4A), whereas no associations were found for the other hub genes (data not shown). Moreover, CCNB2, CDCA8, CDC20, CDK1, KIF2C, RRM2, UBE2C and TPX2 were significantly associated with RFS (Fig. 4B). No association was found between CENPF and TOP2A genes and RFS (data not shown).
Figure 4.

Kaplan-Meier analysis of selected hub genes. (A) Results of overall survival analysis for CDK1, KIF2C, UBE2C and TPX2 genes. (B) Results of recurrence-free survival analysis for CCNB2, CDCA8, CDC20, CDK1, KIF2C, RRM2, UBE2C and TPX2. P<0.05 was considered to indicate a statistically significant difference.
Cox proportional hazards regression analysis was performed using histology, FIGO stage, grade, MI and expression of the 10 hub genes (data not shown). As a result, histology, grade and MI were the only independent OS predictors (histology: P=0.027, hazard ratio: 1.824; grade: P=0.042, hazard ratio: 2.062; MI: P<0.0001, hazard ratio: 3.729). On the other hand, stage, grade and UB2C were the only RFS predictors (stage: P<0.0001, hazard ratio: 2.571; grade: P=0.0055, hazard ratio: 1.730; UB2C: P=0.005, hazard ratio: 1.955). A similar analysis done for stage I tumors identified UB2C, TPX2, KIF2C and MI as only OS predictors (UB2C: P=0.025, hazard ratio: 0.187; TPX2: P=0.066, hazard ratio: 3.776; KIF2C: P=0.013, hazard ratio: 5.647; MI: P<0.0001, hazard ratio: 5.748). In the case of RFS, TPX2, KIF2C and histology were the only predictors (TPX2: P=0.014, hazard ratio: 3.557; KIF2C: P=0.047, hazard ratio: 2.659; histology: P=0.091, hazard ratio: 0.388).
TPX2 expression analysis in EC cell models and patient samples
Since TPX2 was identified as the best independent RFS predictor in stage I EC, a set of experiments was performed to assess its expression in EC cell models and patient samples.
Firstly, TPX2 mRNA and protein expression analysis was conducted in EC cell models depicting different degrees of aggressiveness. For this purpose, Hec1a and Ishikawa parental cells and ETV5 stable transfectants of both cell lines (Hec1a-ETV5 and Ishikawa-ETV5, respectively) were evaluated. As a result, an increase of 20–25 times in TPX2 transcript expression was detected in cells overexpressing ETV5 compared with parental cells (Fig. 5A). In agreement with these results, an increased expression of TPX2 protein was detected by western blotting (Fig. 5B) and fluorescence immunocytochemistry (Fig. 5C) in Hec1a-ETV5 and Ishikawa-ETV5 when compared with Hec1a and Ishikawa cells, respectively.
Figure 5.

TPX2 expression analysis in EC cell lines and patient tumors. (A) RT-qPCR analysis of TPX2 in parental and ETV5 stable transfectants of Hec1a (left) and Ishikawa (right) EC cell lines (***P<0.001; Student's t-test). (B) Western immunoblotting of TPX2 in total protein extracts of Hec1a and Hec1a-ETV5, Ishikawa and Ishikawa-ETV5 cells. (C) Fluorescent immunocytochemistry of TPX2 in Hec1a and Hec1a-ETV5, Ishikawa and Ishikawa-ETV5 cells. Nuclear staining was done with Hoechst 33342. Magnification, ×600. RT-qPCR analysis of TPX2 mRNA levels in (D) grade 1–2 (n=13) and 3 (n=14) tumors (*P=0.0494; Mann-Whitney U test), E) MI<50% (n=15) and MI>50% (n=11) tumors (**P=0.0051; Mann-Whitney U test), and (F) Low (n=11) and intermediate/high (n=22) risk tumors (*P=0.0374; Mann-Whitney U test). EC, endometrial cancer; RT-qPCR, reverse transcription-quantitative PCR; TPX2, targeting protein for Xklp2; ETV5, ETS variant transcription factor 5; MI, myometrial invasion.
Next, TPX2 mRNA levels were determined in EC tissue biopsies. Firstly, grade 3 tumor samples exhibited higher TPX2 levels than grade 1–2 tumors (Fig. 5D; P<0.05), confirming results from the in silico analysis (Fig. 3; Table II). In addition, a higher expression of TPX2 was detected in samples with deep MI (Fig. 5E; P<0.01). Finally, TPX2 mRNA levels were higher in intermediate-high recurrence risk tumors compared with low risk tumors, classified following guidelines previously established (32) (Fig. 5F; P<0.05).
Discussion
EC risk stratification is currently performed using clinicopathological data (stage, grade, MI and lymphovascular invasion), which are not always representative of the tumor biology (32) and show discrepancies in the final diagnosis (33–35). Therefore, there is a need to identify EC biomarkers that contribute to the prediction of the early evolution of tumors, particularly in stage I tumors that may relapse, with the aim to optimize preoperative disease management and follow-up.
Considering the relevance that bioinformatics has gained in translational medicine, several algorithms have been implemented in recent years for the identification of biomarkers with prognostic value in several types of cancer. As an example, gene expression profile analysis performed using genomic and clinical data stored in public repositories has led to the identification of biomarkers with potential prognostic value in prostate (36) and breast (37) cancer, among others.
DisGeNET has been used in several studies, including for a survey analysis of disease-associated genes (38), prediction of associations between diseases and genes and non-coding sequences (39,40), and associations between diseases (41,42). In the present study, DisGeNET analysis quickly and systematically yielded a total of 930 EC-related genes. This set of genes was used as a ‘training set’ in ToppGene analysis to prioritize the 6,945 EC DEGs retrieved from GEO. Using TCGA UCEC cohort, gene expression profile analysis of the prioritized genes (3,976 genes) identified 930 DEGs in tumors according to histological grade, a clinicopathological parameter significantly associated with EC relapse (31). Gene expression profiles revealed three distinctive gene clusters, with genes upregulated (clusters I and II) or downregulated (cluster III) in grade 3 tumors in comparison to grade 1 tumors. These results are in line with a recent study reporting the identification of genes and microRNAs (miRs) differentially expressed in grade 3 EC and functionally associated with cell cycle regulation (43).
To determine whether the resulting DEGs were associated with a common cellular pathway, two approaches were used. First, an enrichment analysis of DEGs with information from pathway databases and/or the GO was done. A second approach aimed at evaluating whether DEG-encoded proteins may physically interact within the cell or are part of protein molecular networks. These two approaches are complementary; current pathway-curated databases do not capture all the genes in the human genome and consequently several DEGs may not be annotated to any pathway in these databases. The human interactome map has a higher coverage on proteins encoded by human genes; therefore, it can provide additional insights than those based only on pathway databases. Moreover, PPI networks have been used to infer candidate genes for different types of cancer (44–48). These networks are sensitive to disruption of high degree proteins (proteins with numerous interaction partners, also named hub genes) (49,50) since protein degree correlates with its gene essentiality for phenotype survival. PPI network analysis using the STRING database in combination with the cytoHubba application led to the identification of the following hub genes: CCNB2, CDCA8, CDC20, CDK1, CENPF, KIF2C, RRM2, UBE2C, TOP2A and TPX2.
From the ROC curve analysis, it was revealed that all hub genes significantly distinguished EC from control patients, suggesting their diagnostic potential. Further evaluations strengthen the relevance of the identified hub genes in EC, as evidenced by the significant associations found between their expression and tumor grade, stage, histology and MI. Finally, the survival analysis revealed a negative impact of an increased expression of some hub genes on OS and RFS. Among them, TPX2 was found as an independent negative prognostic predictor of RFS and OS in stage I tumors. In line with these observations, increased TPX2 expression (mRNA and protein) was detected in Hec1a and Ishikawa human endometrial adenocarcinoma cell lines overexpressing ETV5. This transcription factor has been found to be overexpressed in EEC tissues, particularly in stage IB tumors (51). Moreover, a higher ETV5 expression has been detected at the invasive front of EC tissues when compared with matched-superficial tumor areas, and has been found to serve a role in MI. In line with these findings, previous studies revealed epithelial to mesenchymal transition molecular changes, as well as a more migratory and invasive phenotype in Hec1a and Ishikawa cells overexpressing ETV5 than parental cell lines (52); since they have been proposed as a model of early stage EC aggressiveness, these cell lines were considered useful for the present study. In line with these findings, TPX2 transcript levels were higher in endometrial tumors with features associated with intermediate-high risk of recurrence. It is worth noting that Ishikawa-ETV5 cells TPX2 Western blot analysis revealed the presence of two protein forms. In this regard, a bioinformatics analysis identified four potential N-glycosylation sites (NetNGlyc 1.0 Server), 75 potential O-glycosylation sites (NetOGlyc 4.0 Server) and ≥50 potential phosphorylation sites (NetPhos 3.1 Server) for TPX2 (data not shown). Regarding the latter, evidence of TPX2 phosphorylation has been reported (53,54).
TPX2 is a key factor for the assembly of the mitotic spindle and of microtubules during apoptosis. It activates Aurora kinase A during mitosis and directs its activity to the mitotic spindle, serving an important role in mitosis. In recent years, it has been shown that both proteins can generate a functional unit with oncogenic properties (55). Increased expression of TPX2 has been associated with the progression of different types of cancer (56). Recent studies have linked TPX2 expression with EC; in particular, two bioinformatics studies have related TPX2 with EC pathogenesis (57,58). In addition, TPX2 was identified as a target of miR-29a-5p, following a mechanism that would regulate EC cell proliferation, invasion and apoptosis (59). In line with these findings, the Aurora kinase gene has been recently identified as one of 13 principal genes involved in the carcinogenesis of poorly differentiated endometrial tumors (60).
Studies based on bioinformatics approaches may have some limitations, mainly due to incomplete knowledge of protein interaction maps in humans, although it has already been demonstrated that even with this incomplete knowledge, it is possible to gain insight on disease-associated modules (61). In addition, they may bias the knowledge on genes associated with diseases, since for many human diseases, the complete catalog of genes and sequence variants is not available, and the information is scattered across different databases and the literature. Moreover, different databases have their own annotation criteria with regards to the use of controlled vocabularies and ontologies. Furthermore, the information that can be found in the literature is often not expressed using controlled vocabularies. This challenge was addressed by using DisGeNET, which is one of the most complete databases publicly available that also includes data text mined from the literature. To overcome the limitation posed by the use of different terms, a broad set of terms to interrogate the database was used. Nevertheless, the information obtained might not be complete due to inherent limitations of the database (update cycle and/or incorrect capture of all the data present in publications). With regard to data mining tools, there is a limited availability of EC transcriptomics datasets, which constitutes an additional limitation, since it makes it difficult to validate candidate genes in independent datasets, which is feasible in other tumor types in which there is larger number of studies. Finally, while PPI networks are a useful tool for the integration of information from text/data mining and pathway databases, the selection of a small set of hub genes in network analysis may have excluded genes/proteins within the network that could be potentially important for the phenotype of interest. However, due to the amount of DEGs and the complexity of the generated PPI, it was necessary to use a computer approach to select a small set of genes of interest. While the selected hub genes depicted a potential diagnostic/prognostic value for EC, further network analysis could be performed to assess the relevance of other modules and sub-networks within the PPI presented as part of this work.
In conclusion, the use of an algorithm that combines a set of bioinformatics tools led to the identification of 10 genes, CCNB2, CDCA8, CDC20, CDK1, CENPF, KIF2C, RRM2, UBE2C, TOP2A and TPX2, associated with EC progression. In particular, to the best of our knowledge, this study was the first to identify TPX2 as an independent prognostic biomarker in stage I EC. Further validation studies in larger and independent EC cohorts will contribute to confirm the prognostic value of TPX2 with the ultimate goal of proposing its use as a complement to current EC management.
Supplementary Material
Acknowledgements
Authors would like to thank Dr Cristian Moiola for his technical assistance.
Funding
Preparation of this manuscript was done with the support of grants from the Agencia Nacional de Promoción Científica y Tecnológica de Argentina (grant no. PICTSU-1072), the Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET; grant no. PIP887), the Instituto Nacional del Cáncer (grant no. INC 2016-2017), Fundación R. Barón (donation) and Fundación Williams (donation) to IBYME (MHVL); [Marie Curie Actions-International Research Staff Exchange Scheme (IRSES); FP7-PEOPLE-2010-IRSES; PROT*BIO*FLUID; grant #269285] funds, provided to JR and MHVL, supported travel expenses.
Availability of data and materials
The datasets generated and/or analyzed during the current study are available in the GEO repository (GSE17025, http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE17025) and UCSC Xena browser (TCGA Endometrioid cancer, UCEC dataset; http://xenabrowser.net/datapages/?cohort=TCGA%20Endometrioid%20Cancer%20(UCEC)&removeHub=https%3A%2F%2Fxena.treehouse.gi.ucsc.edu%3A443).
Authors' contributions
MJB and MHVL were involved in conceptualization of the study. MJB, EL, LIF, RB and MHVL contributed to experimental design. MJB, LM, MCA, EC, AGM, JR, and MHVL collected data from bioinformatics analysis and/or conducted laboratory experiments; MJB, EL, MA, LF, RB and MHVL analyzed data. MJB, EL, LF and MHVL interpreted results. MJB, LIF and MHVL wrote the original draft. MHVL and JR acquired funds. MHVL supervised the entire project. All authors read and approved the final version of the manuscript.
Ethics approval and consent to participate
The Institutional Review Board of Vall d'Hebron Hospital and IBYME approved the protocol, and written informed consent was provided by all patients participating in the study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Globocan 2018, corp-author. International Agency for Research on Cancer 2018. Available from https://www.uicc.org/new-global-cancer-data-globocan-2018. [Google Scholar]
- 2.Bokhman JV. Two pathogenetic types of endometrial carcinoma. Gynecol Oncol. 1983;15:10–17. doi: 10.1016/0090-8258(83)90111-7. [DOI] [PubMed] [Google Scholar]
- 3.Creasman W. Revised FIGO staging for carcinoma of the endometrium. Int J Gynecol Obstet. 2009;105:109. doi: 10.1016/j.ijgo.2009.02.010. [DOI] [PubMed] [Google Scholar]
- 4.Creasman WT, Odicino F, Maisonneuve P, Quinn MA, Beller U, Benedet JL, Heintz AP, Ngan HY, Pecorelli S. Carcinoma of the corpus uteri. FIGO 26th Annual report on the results of treatment in gynecological cancer. Int J Gynecol Obstet. 2006;95(Suppl 1):S105–S143. doi: 10.1016/S0020-7292(06)60031-3. [DOI] [PubMed] [Google Scholar]
- 5.Morice P, Leary A, Creutzberg C, Abu-Rustum N, Darai E. Endometrial cancer. Lancet. 2016;387:1094–1108. doi: 10.1016/S0140-6736(15)00130-0. [DOI] [PubMed] [Google Scholar]
- 6.Siegel RL, Miller KD, Jemal A. Cancer statistics. CA Cancer J Clin. 2018;68:7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 7.Kitchener HC, Trimble EL, Endometrial Cancer Working Group of the Gynecologic Cancer Intergroup Endometrial cancer state of the science meeting. Int J Gynecol Cancer. 2009;19:134–140. doi: 10.1111/IGC.0b013e3181995f90. [DOI] [PubMed] [Google Scholar]
- 8.DeSantis CE, Siegel RL, Sauer AG, Miller KD, Fedewa SA, Alcaraz KI, Jemal A. Cancer statistics for African Americans, 2016: Progress and opportunities in reducing racial disparities. CA Cancer J Clin. 2016;66:290–308. doi: 10.3322/caac.21340. [DOI] [PubMed] [Google Scholar]
- 9.Lheureux S, Wilson M, Mackay HJ. Recent and current Phase II clinical trials in endometrial cancer: Review of the state of art. Expert Opin Investig Drugs. 2014;23:773–792. doi: 10.1517/13543784.2014.907272. [DOI] [PubMed] [Google Scholar]
- 10.Miller KD, Siegel RL, Lin CC, Mariotto AB, Kramer JL, Rowland JH, Stein KD, Alteri R, Jemal A. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin. 2016;66:271–289. doi: 10.3322/caac.21349. [DOI] [PubMed] [Google Scholar]
- 11.Xu Y, Burmeister C, Hanna RK, Munkarah A, Elshaikh MA. Predictors of survival after recurrence in women with early-stage endometrial carcinoma. Int J Gynecol Cancer. 2016;26:1137–1142. doi: 10.1097/IGC.0000000000000733. [DOI] [PubMed] [Google Scholar]
- 12.Connor EV, Rose PG. Management strategies for recurrent endometrial cancer. Expert Rev Anticancer Ther. 2018;18:873–885. doi: 10.1080/14737140.2018.1491311. [DOI] [PubMed] [Google Scholar]
- 13.Jongen V, Briët J, de Jong R, ten Hoor K, Boezen M, van der Zee A, Nijman H, Hollema H. Expression of estrogen receptor-alpha and -beta and progesterone receptor-A and -B in a large cohort of patients with endometrioid endometrial cancer. Gynecol Oncol. 2009;112:537–542. doi: 10.1016/j.ygyno.2008.10.032. [DOI] [PubMed] [Google Scholar]
- 14.Werner HM, Salvesen HB. Current status of molecular biomarkers in endometrial cancer. Curr Oncol Rep. 2014;16:403. doi: 10.1007/s11912-014-0403-3. [DOI] [PubMed] [Google Scholar]
- 15.Catasus L, D'Angelo E, Pons C, Espinosa I, Prat J. Expression profiling of 22 genes involved in the PI3K-AKT pathway identifies two subgroups of high-grade endometrial carcinomas with different molecular alterations. Mod Pathol. 2010;23:694–702. doi: 10.1038/modpathol.2010.44. [DOI] [PubMed] [Google Scholar]
- 16.O'Mara TA, Zhao M, Spurdle AB. Meta-analysis of gene expression studies in endometrial cancer identifies gene expression profiles associated with aggressive disease and patient outcome. Sci Rep. 2016;6:36677. doi: 10.1038/srep36677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun Y, Zou X, He J, Mao Y. Identification of long non-coding RNAs biomarkers associated with progression of endometrial carcinoma and patient outcomes. Oncotarget. 2016;8:52604–52613. doi: 10.18632/oncotarget.17537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cancer Genome Atlas Research Network, corp-author. Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, Robertson AG, Pashtan I, Shen R, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bauer-Mehren A, Rautschka M, Sanz F, Furlong LI. DisGeNET: A Cytoscape plugin to visualize, integrate, search and analyze gene-disease networks. Bioinformatics. 2010;26:2924–2926. doi: 10.1093/bioinformatics/btq538. [DOI] [PubMed] [Google Scholar]
- 20.Piñero J, Queralt-Rosinach N, Bravo À, Deu-Pons J, Bauer-Mehren A, Baron M, Sanz F, Furlong LI. DisGeNET: A discovery platform for the dynamical exploration of human diseases and their genes. Database (Oxford) 2015;2015:bav028. doi: 10.1093/database/bav028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bodenreider O. The Unified Medical Language System (UMLS): Integrating biomedical terminology. Nucleic Acids Res. 2004;32((Database Issue)):D267–D270. doi: 10.1093/nar/gkh061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Day RS, McDade KK, Chandran UR, Lisovich A, Conrads TP, Hood BL, Kolli VS, Kirchner D, Litzi T, Maxwell GL. Identifier mapping performance for integrating transcriptomics and proteomics experimental results. BMC Bioinformatics. 2011;12:213. doi: 10.1186/1471-2105-12-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen J, Bardes EE, Aronow BJ, Jegga AG. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009;37:W305–W311. doi: 10.1093/nar/gkp427. (Web Server issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chin CH, Chen SH, Wu HH, Ho CW, Ko MT, Lin CY. CytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst Biol. 2014;8(Suppl 4):S11. doi: 10.1186/1752-0509-8-S4-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Colas E, Perez C, Cabrera S, Pedrola N, Monge M, Castellvi J, Eyzaguirre F, Gregorio J, Ruiz A, Llaurado M, et al. Molecular markers of endometrial carcinoma detected in uterine aspirates. Int J Cancer. 2011;129:2435–2444. doi: 10.1002/ijc.25901. [DOI] [PubMed] [Google Scholar]
- 26.Monge M, Colas E, Doll A, Gonzalez M, Gil-Moreno A, Planaguma J, Quiles M, Arbos MA, Garcia A, Castellvi J, et al. ERM/ETV5 up-regulation plays a role during myometrial infiltration through matrix metalloproteinase-2 activation in endometrial cancer. Cancer Res. 2007;67:6753–6759. doi: 10.1158/0008-5472.CAN-06-4487. [DOI] [PubMed] [Google Scholar]
- 27.Altadill T, Dowdy TM, Gill K, Reques A, Menon SS, Moiola CP, Lopez-Gil C, Coll E, Matias-Guiu X, Cabrera S, et al. Metabolomic and lipidomic profiling identifies the role of the RNA editing pathway in endometrial carcinogenesis. Sci Rep. 2017;7:8803. doi: 10.1038/s41598-017-09169-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lapyckyj L, Castillo LF, Matos ML, Gabrielli NM, Lüthy IA, Vazquez-Levin MH. Expression analysis of epithelial cadherin and related proteins in IBH-6 and IBH-4 human breast cancer cell lines. J Cell Physiol. 2010;222:596–605. doi: 10.1002/jcp.21974. [DOI] [PubMed] [Google Scholar]
- 29.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 30.Budczies J, Klauschen F, Sinn BV, Győrffy B, Schmitt WD, Darb-Esfahani S, Denkert C. Cutoff Finder: A comprehensive and straight forward Web application enabling rapid biomarker cut off optimization. PLoS One. 2012;7:e51862. doi: 10.1371/journal.pone.0051862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yen MS, Chen TH, Ke YM, Hsu KF, Chen JR, Yu MH, Fu HC, Huang CY, Chiang AJ, Chen CY, et al. Clinicopathologic features and treatment outcomes in patients with stage I, high-risk histology or high-grade endometrial cancer after primary staging surgery: A Taiwanese Gynecologic Oncology group study. J Clin Med. 2018;7:254. doi: 10.3390/jcm7090254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Colombo N, Preti E, Landoni F, Carinelli S, Colombo A, Marini C, Sessa C, ESMO Guidelines Working Group Endometrial cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2011;22(Suppl 6):vi35–vi39. doi: 10.1093/annonc/mdr374. [DOI] [PubMed] [Google Scholar]
- 33.Frumovitz M, Singh D, Meyer L, Smith DH, Wertheim I, Resnik E, Bodurka DC. Predictors of final histology in patients with endometrial cancer. Gynecol Oncol. 2004;95:463–468. doi: 10.1016/j.ygyno.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 34.Neubauer NL, Havrilesky LJ, Calingaert B, Bulusu A, Bernardini MQ, Fleming ND, Bland AE, Secord AA. The role of lymphadenectomy in the management of preoperative grade 1 endometrial carcinoma. Gynecol Oncol. 2009;112:511–516. doi: 10.1016/j.ygyno.2008.11.012. [DOI] [PubMed] [Google Scholar]
- 35.Body N, Lavoué V, De Kerdaniel O, Foucher F, Henno S, Cauchois A, Laviolle B, Leblanc M, Levêque J. Are preoperative histology and MRI useful for classification of endometrial cancer risk? BMC Cancer. 2016;16:498. doi: 10.1186/s12885-016-2554-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rounbehler RJ, Berglund AE, Gerke T, Takhar MM, Awasthi S, Li W, Davicioni E, Erho NG, Ross AE, Schaeffer EM, et al. Tristetraprolin is a prognostic biomarker for poor outcomes among patients with low-grade prostate cancer. Cancer Epidemiol Biomarkers Prev. 2018;27:1376–1383. doi: 10.1158/1055-9965.EPI-18-0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen XH, Zhang WW, Wang J, Sun JY, Li FY, He ZY, Wu SG. 21-gene recurrence score and adjuvant chemotherapy decisions in patients with invasive lobular breast cancer. Biomark Med. 2019;13:83–93. doi: 10.2217/bmm-2018-0396. [DOI] [PubMed] [Google Scholar]
- 38.Abascal MF, Besso MJ, Rosso M, Mencucci MV, Aparicio E, Szapiro G, Furlong LI, Vazquez-Levin MH. CDH1/E-cadherin and solid tumors. An updated gene-disease association analysis using bioinformatics tools. Comput Biol Chem. 2016;60:9–20. doi: 10.1016/j.compbiolchem.2015.10.002. [DOI] [PubMed] [Google Scholar]
- 39.Dalleau K, Marzougui Y, Da Silva S, Ringot P, Ndiaye NC, Coulet A. Learning from biomedical linked data to suggest valid pharmacogenes. J Biomed Semantics. 2017;8:16. doi: 10.1186/s13326-017-0125-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eguchi R, Karim MB, Hu P, Sato T, Ono N, Kanaya S, Altaf-Ul-Amin M. An integrative network-based approach to identify novel disease genes and pathways: A case study in the context of inflammatory bowel disease. BMC Bioinformatics. 2018;19:264. doi: 10.1186/s12859-018-2251-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gomez-Rubio P, Piñero J, Molina-Montes E, Gutiérrez-Sacristán A, Marquez M, Rava M, Michalski CW, Farré A, Molero X, Löhr M, et al. Pancreatic cancer and autoimmune diseases: An association sustained by computational and epidemiological case-control approaches. Int J Cancer. 2019;144:1540–1549. doi: 10.1002/ijc.31866. [DOI] [PubMed] [Google Scholar]
- 42.Santiago JA, Potashkin JA. System-based approaches to decode the molecular links in Parkinson's disease and diabetes. Neurobiol Dis. 2014;72:84–91. doi: 10.1016/j.nbd.2014.03.019. [DOI] [PubMed] [Google Scholar]
- 43.Zang Y, Dong M, Zhang K, Tian W, Wang Y, Xue F. Bioinformatics analysis of key differentially expressed genes in well and poorly differentiated endometrial carcinoma. Mol Med Rep. 2018;18:467–476. doi: 10.3892/mmr.2018.8969. [DOI] [PubMed] [Google Scholar]
- 44.Lin G, Yin G, Yan Y, Lin B. Identification of prognostic biomarkers for malignant melanoma using microarray datasets. Oncol Lett. 2019;18:5243–5254. doi: 10.3892/ol.2019.10914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu J, Yu Z, Sun M, Liu Q, Wei M, Gao H. Identification of cancer/testis antigen 2 gene as a potential hepatocellular carcinoma therapeutic target by hub gene screening with topological analysis. Oncol Lett. 2019;18:4778–4788. doi: 10.3892/ol.2019.10811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shi J, Zhang P, Liu L, Min X, Xiao Y. Weighted gene coexpression network analysis identifies a new biomarker of CENPF for prediction disease prognosis and progression in nonmuscle invasive bladder cancer. Mol Genet Genomic Med. 2019;7:e982. doi: 10.1002/mgg3.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang B, Wu Q, Xu R, Hu X, Sun Y, Wang Q, Ju F, Ren S, Zhang C, Qi F, et al. The promising novel biomarkers and candidate small molecule drugs in lower-grade glioma: Evidence from bioinformatics analysis of high-throughput data. J Cell Biochem. 2019;120:15106–15118. doi: 10.1002/jcb.28773. [DOI] [PubMed] [Google Scholar]
- 48.Zhou M, Zhu Y, Hou R, Mou X, Tan J. Identification of candidate genes for the diagnosis and treatment of cholangiocarcinoma using a bioinformatics approach. Oncol Lett. 2019;18:5459–5467. doi: 10.3892/ol.2019.10904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Callaway DS, Newman ME, Strogatz SH, Watts DJ. Network robustness and fragility: Percolation on random graphs. Phys Rev Lett. 2000;85:5468–5471. doi: 10.1103/PhysRevLett.85.5468. [DOI] [PubMed] [Google Scholar]
- 50.Cohen R, Erez K, ben-Avraham D, Havlin S. Resilience of the internet to random breakdowns. Phys Rev Lett. 2000;85:4626–4628. doi: 10.1103/PhysRevLett.85.4626. [DOI] [PubMed] [Google Scholar]
- 51.Planagumà J, Abal M, Gil-Moreno A, Díaz-Fuertes M, Monge M, García A, Baró T, Xercavins J, Reventós J, Alameda F. Up-regulation of ERM/ETV5 correlates with the degree of myometrial infiltration in endometrioid endometrial carcinoma. J Pathol. 2005;207:422–429. doi: 10.1002/path.1853. [DOI] [PubMed] [Google Scholar]
- 52.Colas E, Muinelo-Romay L, Alonso-Alconada L, Llaurado M, Monge M, Barbazan J, Gonzalez M, Schoumacher M, Pedrola N, Ertekin T, et al. ETV5 cooperates with LPP as a sensor of extracellular signals and promotes EMT in endometrial carcinomas. Oncogene. 2012;31:4778–4788. doi: 10.1038/onc.2011.632. [DOI] [PubMed] [Google Scholar]
- 53.Fu J, Bian M, Xin G, Deng Z, Luo J, Guo X, Chen H, Wang Y, Jiang Q, Zhang C. TPX2 phosphorylation maintains metaphase spindle length by regulating microtubule flux. J Cell Biol. 2015;210:373–383. doi: 10.1083/jcb.201412109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shim SY, Perez de Castro I, Neumayer G, Wang J, Park SK, Sanada K, Nguyen MD. Phosphorylation of targeting protein for Xenopus kinesin-like protein 2 (TPX2) at threonine 72 in spindle assembly. J Biol Chem. 2015;290:9122–9134. doi: 10.1074/jbc.M114.591545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Asteriti IA, Rensen WM, Lindon C, Lavia P, Guarguaglini G. The Aurora-A/TPX2 complex: A novel oncogenic holoenzyme? Biochim Biophys Acta. 2010;1806:230–239. doi: 10.1016/j.bbcan.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 56.Neumayer G, Belzil C, Gruss OJ, Nguyen MD. TPX2: Of spindle assembly, DNA damage response, and cancer. Cell Mol Life Sci. 2014;71:3027–3047. doi: 10.1007/s00018-014-1582-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shen L, Liu M, Liu W, Cui J, Li C. Bioinformatics analysis of RNA sequencing data reveals multiple key genes in uterine corpus endometrial carcinoma. Oncol Lett. 2018;15:205–212. doi: 10.3892/ol.2017.7346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu Y, Hua T, Chi S, Wang H. Identification of key pathways and genes in endometrial cancer using bioinformatics analyses. Oncol Lett. 2019;17:897–906. doi: 10.3892/ol.2018.9667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jiang T, Sui D, You D, Yao S, Zhang L, Wang Y, Zhao J, Zhang Y. MiR-29a-5p inhibits proliferation and invasion and induces apoptosis in endometrial carcinoma via targeting TPX2. Cell Cycle. 2018;17:1268–1278. doi: 10.1080/15384101.2018.1475829. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 60.Zhang K, Li H, Yan Y, Zang Y, Li K, Wang Y, Xue F. Identification of key genes and pathways between type I and type II endometrial cancer using bioinformatics analysis. Oncol Lett. 2019;18:2464–2476. doi: 10.3892/ol.2019.10550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Menche J, Sharma A, Kitsak M, Ghiassian SD, Vidal M, Loscalzo J, Barabási AL. Disease networks. Uncovering disease-disease relationships through the incomplete interactome. Science. 2015;347:1257601. doi: 10.1126/science.1257601. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and/or analyzed during the current study are available in the GEO repository (GSE17025, http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE17025) and UCSC Xena browser (TCGA Endometrioid cancer, UCEC dataset; http://xenabrowser.net/datapages/?cohort=TCGA%20Endometrioid%20Cancer%20(UCEC)&removeHub=https%3A%2F%2Fxena.treehouse.gi.ucsc.edu%3A443).