

Abstract
Human hepatocellular carcinoma (HCC) is a common malignant tumor of the digestive tract that is prevalent worldwide. Improving diagnosis methods for HCC helps to improve patient survival rate. The present study aimed to identify novel HCC biomarkers for the diagnosis of HCC through analyzing gene changes on peripheral blood mononuclear cells (PBMCs) and verifying these in additional samples. The gene expression profiles GSE49515 (including 10 specimens from normal patients and 10 specimens from patients with HCC) and GSE58208 (including 5 specimens from normal patients and 10 specimens from patients with HCC) were downloaded from the online Gene Expression Omnibus database (GEO). Differentially expressed genes (DEGs) in PBMCs between healthy controls and patients with HCC were identified using R software. A total of 935 DEGs, including 686 upregulated DEGs and 249 downregulated DEGs, were identified in the present study. In order to identify any internal associations, these DEGs were used to construct weighted gene co-expression networks (WGCNA). Gene Ontology (GO) enrichment analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis of genes in each module were conducted using the online database DAVID. Furthermore, hub genes with high module membership were identified in a co-expression network and receiver operating characteristic curves were used to verify the diagnostic values of these eight hub genes. Furthermore, the expression and diagnosis value of the eight hub genes were also verified in additional samples. The results of the present study suggested that secreted protein acidic and cysteine rich(SPARC), transmembrane protein 40 (TMEM40), solute carrier family 25 member 44, formyl peptide receptor 2 (FPR2), complement C8 β chain, N-myristoyltransferase 1, protein kinase C δ(PRKCD) and protein phosphatase, Mg2+/Mn2+ dependent 1M(PPM1M) were hub genes. SPARC, TMEM40, FPR2, PRKCD and PPM1M had prominent diagnostic value according to the results from the GEO data and the additional samples. The present study demonstrated that these hub genes may help to improve the diagnosis of HCC.
Keywords: hepatocellular carcinoma, peripheral blood mononuclear cells, Gene Expression Omnibus, bioinformatics analysis, weighted gene co-expression network analysis
Introduction
Hepatocellular carcinoma (HCC) is a highly lethal tumor, which is the third most frequent cause of cancer-associated mortality with an increasing incidence rate worldwide (1). Accurate and timely diagnoses of HCC are often challenging. Diagnosis of HCC relies on a number of different diagnostic methods, including ultrasound, computed tomography, biopsy and blood tests (2). Liver biopsy is regarded as the ‘gold standard’ for the diagnosis of HCC; however, liver biopsies are invasive procedures where the patient often experiences complications, such as bacteremia or death (risk of death, ~0.01%) (3). Similarly, there are certain disadvantages to ultrasound and computed tomography, which limit their effectiveness in the diagnosis. Currently, the serum α-fetoprotein (AFP) level is regarded as a biomarker for HCC and is used widely in the clinical setting. However, the specificity and sensitivity of AFP against HCC remains unsatisfactory. As presented in the studies performed by Lok et al (4) and Marrero et al (5), the sensitivity of AFP in the diagnosis of HCC when using a threshold of 20 ng/ml was 61%, while when using a threshold of 200 ng/ml the sensitivity decreased to 22%. Therefore, novel HCC biomarkers are urgently required.
Peripheral blood mononuclear cells (PBMCs) are immune cells that include lymphocytes and monocytes, and play a key role in the host immune system to fight against various abnormal conditions such as infection and carcinoma (6). Various studies have demonstrated that the gene expression level of PBMCs was significantly changed in patients with tumors and detection of changes in PBMCs may help to improve the diagnosis (7,8). Ciarloni et al (9) demonstrated the potential for developing a noninvasive and accurate inspection to identify patients at an average risk of colorectal cancer based on gene expression analysis of PBMCs segregated from patient blood samples. Similarly, Baine et al (10,11) demonstrated that eight genes, including ARG1, were differently expressed in the PBMCs of patients with pancreatic cancer when compared with healthy controls. These genes could potentially be developed as novel biomarkers for the early diagnosis of pancreatic cancer. Furthermore, Mishra et al (12) revealed that microRNA (miR)-195-5p and miR-495 were downregulated in the PBMCs of patients with breast cancer when compared with healthy controls, and suggested that these two miRs could be used for the early detection of breast cancer.
In the present study, eight hub genes from two gene expression profiles were identified and it was revealed that these hub genes have good diagnostic value for the diagnosis of HCC. The diagnosis value of five hub genes was also prominent in the additional samples used in the present study, including secreted protein acidic and cysteine rich (SPARC), transmembrane protein 40 (TMEM40), formyl peptide receptor 2 (FPR2), protein kinase C δ(PRKCD) and protein phosphatase, Mg2+/Mn2+ dependent 1M(PPM1M). These hub genes may have the potential to be novel biomarkers for HCC.
Materials and methods
Data collection
The gene expression data files of GSE49515 and GSE58208 were downloaded from the Gene Expression Omnibus database (GEO) database (http://www.ncbi.nlm.nih.gov/geo/). The platform used to analyze these two data files was GPL570 (Affymetrix Human Genome U133 Plus 2.0 Array). The dataset profile of GSE49515 contained 10 PBMC specimens from normal patients and 10 PBMC specimens from patients with HCC. The dataset profile of GSE58208 contained 5 PBMC specimens from normal patients and 10 PBMC specimens from patients with HCC.
Data pretreatment and identification of DEGs
The present study first integrated all samples of GSE49515 and GSE58208 to significantly improve the number of samples 5 normal samples vs. 20 tumor samples) and to avoid generating less relable results, followed by batch normalization in the R computing environment using the sva package (13). The differential anaysis (|Log2FC| > 0.5, adjusted P<0.05) was then performed by comparing tumor tissues with normal tissues in the R computing environment [version 3.5.2; The R Foundation for Statistical Computing (14)] using the limma package (version 2.13; Bioconductor). DEGs were visualized in a heatmap.
Weighted gene co-expression networks (WGCNA) and module analysis
WGCNA is a systems biology method for identifying patterns that have relevance among genes in microarray samples. In the WGCNA network, modules of highly correlated genes can be investigated; as such, clusters using a module eigengene or an intra-modular hub gene can be summarized; modules relationship with one another and to external sample traits can be identified, and module membership measures can be calculated (15). In order to identify the interactions between the DEGs in the present study, the WGCNA was performed to identify the co-expression modules with a threshold of power, 16. A hierarchical clustering dendrogram of the topological overlap matrix was then constructed using the average distance with a minimum size threshold of 30 to classify the similar gene expression profiles into different gene modules. The modules with an average distance<0.2 were merged. Following co-expression, modules were identified in the WGCNA network and heatmaps and bar charts were used to describe module eigengenes.
Functional enrichment analysis of modules
The database for annotation, visualization and integrated discovery (DAVID; version 6.8; http://david-d.ncifcrf.gov/) was used in the study to perform functional annotation for genes in four modules, including GO [biological processes (BP), molecular function (MF) and cellular components (CC)] and KEGG pathway analysis. Analysis results were extracted when P<0.05. Only the top five GO terms were visualized using R software if there were more than five terms, as with the results of the KEGG pathway analysis.
Co-expression network construction and identification of hub genes
First, the co-expression data of all genes in each module with a threshold weight >0.05 were exported into a textfile. Secondly, the top 10 genes with highest module membership in each module were selected to construct the co-expression network. Co-expression network visualization was performed using Cytoscape (version 3.5.1; National Institute of General Medical Sciences). Genes with the highest module membership (MM) and degree scores in the co-expression network were regarded as hub genes.
Diagnostic value of hub genes
The sensitivity and specificity of the hub genes identified in the present study were evaluated through the receiver operating characteristic (ROC) curve. The ROC curve was created using SPSS 20.0 (IBM, Corps.). When the genes had an area under the curve (AUC) >0.7, they were considered to have good diagnosis value.
Patient blood samples
The clinical blood samples from normal patients (female=9, male=33; mean age: 54±9) or patients with HCC (female=6, male=22; mean age: 56±14) in the present study were obtained from Renmin Hospital of Wuhan University (Wuhan, China), between April 2019 and June 2019. All patients who had samples taken had provided informed consent. The present study was approved by the Ethics Committee of the Renmin Hospital of Wuhan University and was performed in accordance with the Declaration of Helsinki.
PBMC isolation
Patient blood samples were collected in anticoagulant tubes containing EDTA. PBMCs were isolated by density gradient centrifugation using Ficoll-Paque Plus (Invitrogen; Thermo Fisher Scientific, Inc.) and cultured in RPMI 1640 medium (Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.) at 37˚C with 5% CO2.
Reverse transcription-quantitative PCR
Total RNA was extracted from tissues and cells using TRIzol® reagent (Yeasan). The PrimeScript RT Reagent kit (Yeasan) was used to perform the cDNA synthesis. The following primers were used: TMEM40: Forward, 5'-CAGAGCAACCGGAAAACATCG-3'; reverse, 5'-TCATCCTTCAAAACGTCAGGC-3'; SPARC: Forward, 5'-TGAGGTATCTGTGGGAGCTAATC-3'; reverse, 5'-CCTTGCCGTGTTTGCAGTG-3'; solute carrier family 25 member 44 (SLC25A44): Forward, 5'-ATGGAGGACAAACGCAACATC-3'; reverse, 5'-ACACTGACACGGATCATCATTG-3'; FPR2: Forward, 5'-AGTCTGCTGGCTACACTGTTC-3'; reverse, 5'-TGGTAATGTGGCCGTGAAAGA-3' (reverse); complement C8 β chain(C8B): Forward, 5'-ATTCCTTTGGGTCAAATGCAGT-3'; reverse, 5'-GGACCAACTAGACAGCTCACA-3'; N-myristoyltransferase 1 (NMT1): Forward, 5'-GGTCAGGGACCTGCCAAAAC-3'; reverse, 5'-CATGGGTGTTCACCACTTCG-3'; PRKCD: Forward, 5'-GTGCAGAAGAAGCCGACCAT-3'; reverse, 5'-CCCGCATTAGCACAATCTGGA-3'; PPM1M: Forward, 5'-CTTGGTGCGGAGAGATGAGAT-3'; reverse, 5'-GCTCAGGATAGACAAAGGCCAG-3' and β-actin: Forward, 5'-CATGTACGTTGCTATCCAGGC-3'; reverse, 5'-CTCCTTAATGTCACGCACGAT-3'.
The reaction was performed under the following conditions: 95˚C 30 sec for the first initial cycle; 95˚C for 30 sec and 40 subsequent cycles at 60˚C for 30 sec each. β-actin was used as the loading control. The relative level of gene expression was calculated using the 2-ΔΔCt method (16).
Statistical analysis
GraphPad Prism (version 6.0; GraphPad Software, Inc.) was used in the present study to perform the statistical analysis. Data was presented as mean ± standard deviation. Independent-sample t-tests were used to analyze the data. P<0.05 was considered to indicate a statistically significant difference.
Results
Identification of DEGs
The present study first integrated all samples of GSE49515 and GSE58208 by sva package to form a novel gene profile, including the gene expression profile of PBMCs from 15 normal controls and 20 patients with HCC. A total of 935 DEGs (including 686 upregulated DEGs and 249 downregulated DEGs) were identified under the threshold of |logFC| >0.5 and adjusted P<0.05. These 935 DEGs were presented in a heatmap (Fig. 1) and then were used for subsequent analysis.
Figure 1.

Heatmap presenting the up- and downregulated genes in peripheral blood mononuclear cells from patients with hepatocellular carcinoma. Red represents the upregulated genes and green represents the downregulated genes. GEO, Gene Expression Omnibus.
Construction of weighted gene co-expression modules
In order to identify any associations between the 935 DEGs, WGCNA, a systems biology method, was used to screen potential biomarkers and therapeutic targets via gene co-expression network construction. DEGs involved in similar pathways or with the same biological function tended to have the same expression cluster. In the present study, β =16 was set to ensure high scale independence (~0.9) and low mean connectivity (~0.0) (Fig. 2). The dissimilarity of the modules was set as 0.2 and a total of four co-expressed modules were identified with a module size cut-off ≤30. The grey colored clusters represent the non-clustering genes in WGCNA (Fig. 3A). Furthermore, the module eigengene diagrams for blue, green, brown and yellow modules all exhibited a higher average expression profile in HCC samples (Fig. 3B-E).
Figure 2.

Construction of the weighted gene co-expression network of DEGs. (A) Analysis of the scale-free fit index for various soft-thresholding powers (β). (B) Analysis of the mean connectivity for various soft-thresholding powers. (C) Histogram of connectivity distribution when β=16. (D) Checking the scale-free topology when β=16. DEG, differentially expressed genes.
Figure 3.

Eigengene analysis. (A) Dendrogram of all differentially expressed genes clustered based on a dissimilarity measure (1-topological overlap matrix). Heatmap and bar graph presenting the eigengene in each module. (B) Blue module. (C) Green module. (D) Brown module. (E) Yellow module. N, peripheral blood mononuclear cells from normal patients; H, peripheral blood mononuclear cells from patients with hepatocellular carcinoma.
GO analysis for the modules
There were 165 genes in the blue module and the top five GO terms that the genes were enriched in were: Extracellular exosome (GO category, CC), focal adhesion (GO category, CC), cell surface (GO category, CC), cytoskeleton (GO category, CC) and platelet degranulation (GO category, biological BP; Fig. 4A). In the green module, there were 73 genes and the top five GO terms that they were enriched in were: Protein binding (GO category, MF); extracellular exosome (GO category, CC); signal transduction (GO category, BP); inflammatory response (GO category, BP) and immune response (GO category, BP) (Fig. 4B). The brown module had 503 genes, which were enriched in: Protein binding (GO category, MF); nucleus (GO category, CC); cytosol (GO category, CC); nucleoplasm (GO category, CC) and transcription (GO category, BP) (Fig. 4C). The yellow module had 111 genes and the top five GO terms the genes enriched in were: Extracellular exosome (GO category, CC); membrane (GO category, CC); intracellular signal transduction (GO category, BP); cytoplasmic vesicle (GO category, CC) and electron carrier activity (GO category, MF; Fig. 4D).
Figure 4.

Circle plot presenting the top five GO term that each of the module genes are enriched in. (A) Blue module. (B) Green module. (C) Brown module. (D) Yellow module. Z-score represents the overall change in the expression of genes that are enriched in the GO term. Red represents increasing gene expression, while white represents decreasing gene expression. GO, Gene Ontology.
KEGG analysis for the modules
A KEGG analysis was performed using DAVID. The results revealed that the genes in the blue module were enriched in platelet activation, focal adhesion, regulation of actin cytoskeleton, hypertrophic cardiomyopathy and dilated cardiomyopathy pathway, while the genes in the green module were enriched in the Toll-like receptor signaling pathway, measles, hepatitis B, influenza A and the estrogen signaling pathway (Fig. 5A and B). The genes in the brown modules were enriched in herpes simplex infection, measles, influenza A, the cell cycle and the rheumatoid arthritis-associated pathway (Fig. 5C). Furthermore, the genes in the yellow module were enriched in the calcium signaling pathway, the neurotrophin signaling pathway, osteoclast differentiation and the long-term potentiation assocated pathway (Fig. 5D).
Figure 5.

KEGG analysis for genes in the four modules. (A) Blue module. (B) Green module. (C) Brown module. (D) Yellow module. The top five KEGG pathways are presented in the bubble chart. The size of bubble represents the enrichment score, while colors indicate enrichment significance (P-value). KEGG, Kyoto Encyclopedia of Genes and Genomes.
Identification of hub genes and their diagnosis value based on the gene expression profiles
Genes in the blue, green, brown and yellow modules with module membership scores ranked in the top 10 were used to construct co-expression networks and were visualized in Cytoscape. It was demonstrated that SPARC and TMEM40 were hub genes in the blue module, SLC25A44 and FPR2 were hub genes in the green module, C8B and NMT1 were hub genes in the brown module, and PRKCD and PPM1M were hub genes in the yellow module (Fig. 6). ROC curves were created and it was revealed that all hub genes had diagnostic value: SPARC (AUC, 0.917; 95% CI, 0.811-1.000), TMEM40 (AUC, 0.887; 95% CI, 0.770-1.000), SLC25A44 (AUC, 0.943; 95% CI, 0.874-1.000), FPR2 (AUC, 0.903; 95% CI 0.796-1.000), C8B (AUC, 0.810; 95% CI, 0.654-0.966), NMT1 (AUC, 0.920; 95% CI 0.832-1.000), PRKCD (AUC, 0.923; 95% CI, 0.829-1.000) and PPM1M (AUC, 0.963; 95% CI, 0.901-1.000; Fig. 7).
Figure 6.

Identification of hub genes in the four modules. Top 10 genes in each module with the highest module membership were used to construct the co-expressed network. The co-expressed network was visualized using Cytoscape. Genes with higher module memberships in module and degree score in the co-expressed network were identified as hub genes. (A) Blue module. (B) Green module. (C) Brown module. (D) Yellow module. Rhombus in the protein-protein interaction network represents the hub gene.
Figure 7.

Receiver operator characteristic curve of 8 hub genes (SPARC, TMEM40, SLC25A44, FPR2, C8B, NMT1, PRKCD and PPM1M). The curves were calculated according to the two gene expression profiles and the corresponding clinical state. AUC, area under the curve. SPARC, secreted protein acidic and cysteine rich; TMEM40, transmembrane protein 40; SLC25A44, solute carrier family 25 member 44; C8B, complement C8 β chain; NMT1, N-myristoyltransferase 1; PRKCD, protein kinase C δ; PPM1M, protein phosphatase, Mg2+/Mn2+ dependent 1M.
Expression and diagnostic value in patient blood samples
In order toverify the results obtained from the prediction analyses, reverse transcription-quantitative PCR was performed on the additional blood samples. The results revealed that the expression levels of TMEM40, SPARC, FPR2, PRKCD and PPM1M were significantly increased in the PBMCs from patients with HCC compared with the normal patients (P<0.05), while the expression levels of SLC25A44, C8B and NMT1 were not significantly different (Fig. 8). ROC curves were also calculated and the results revealed that TMEM40, SPARC, FPR2, PRKCD and PPM1M all had good diagnostic value using a cut-off AUC value of 0.7. The results of the AUC were as follows: TMEM40 (AUC, 0.796; 95% CI, 0.692-0.900), SPARC (AUC, 0.789; 95% CI 0.678-0.900), SLC25A44 (AUC, 0.626; 95% CI, 0.496-0.756), FPR2 (AUC, 0.865; 95% CI, 0.769-0.960), C8B (AUC, 0.635; 95% CI, 0.498-0.773), NMT1 (AUC, 0.626; 95% CI, 0.493-0.759), PRKCD (AUC, 0.827; 95% CI, 0.725-0.929) and PPM1M (AUC, 0.761; 95% CI, 0.645-0.877; Fig. 9).
Figure 8.

PCR was preformed to detect the expression of 8 hub genes (SPARC, TMEM40, SLC25A44, FPR2, C8B, NMT1, PRKCD and PPM1M). Blue dots represent the expression levels of genes in normal patients (n=42), red dots represent the expression levels of genes in patients with HCC (n=28). SPARC, secreted protein acidic and cysteine rich; TMEM40, transmembrane protein 40; SLC25A44, solute carrier family 25 member 44; C8B, complement C8 β chain; NMT1, N-myristoyltransferase 1; PRKCD, protein kinase C δ; PPM1M, protein phosphatase, Mg2+/Mn2+ dependent 1M.
Figure 9.
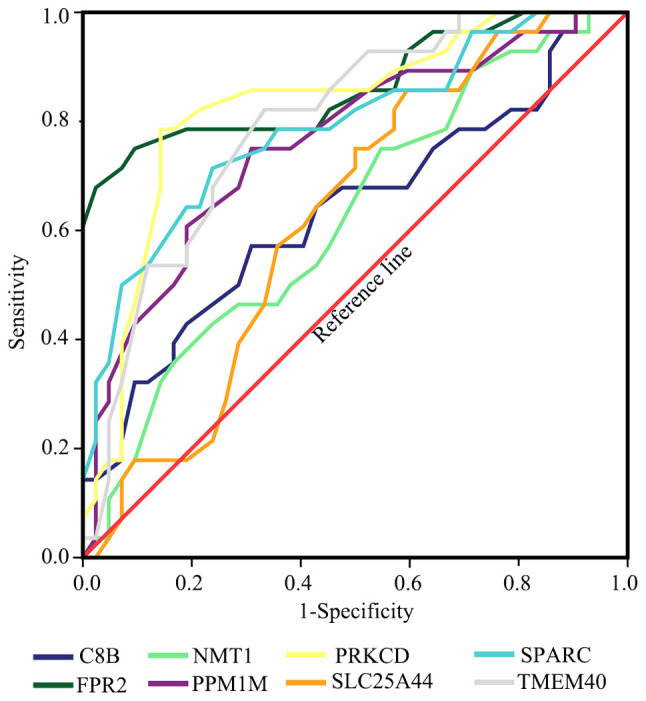
Receiver operator characteristic curve of 8 hub genes (SPARC, TMEM40, SLC25A44, FPR2, C8B, NMT1, PRKCD and PPM1M). The curves were calculated according to the results of the PCR and the corresponding clinical state. SPARC, secreted protein acidic and cysteine rich; TMEM40, transmembrane protein 40; SLC25A44, solute carrier family 25 member 44; FPR2, formyl peptide receptor 2; C8B, complement C8 β chain; NMT1, N-myristoyltransferase 1; PRKCD, protein kinase C δ; PPM1M, protein phosphatase, Mg2+/Mn2+ dependent 1M.
Discussion
HCC is one of the most malignant tumors of the digestive tract worldwide. Surgical excision is currently the main treatment method for patients with HCC. Early effective diagnosis for HCC is critical for the success of surgical excision and improving patient survival rate. However, the commonly used biomarkers, including AFP, have significant diagnostic limitations and novel biomarkers identified from patient blood samples would be valuable in screening for HCC. Furthermore, previous studies have suggested that analyzing the genetic changes in PBMCs may be a valid method for identifying novel biomarkers (7,8).
In the present study, 935 DEGs were identified in the gene expression profiling analyses using the limma algorithms. Furthermore, WGCNA was used to construct a free-scale gene co-expression network to investigate the associations between different sets of DEGs. To clarify the different clusters of WGCNA, functional enrichment analysis, co-expression network and associated experiments were performed. It was revealed that SPARC, TMEM40, SLC25A44, FPR2, C8B, NMT1, PRKCD and PPM1M were hub genes and five of them had good diagnostic value according to the results from the two gene expression profiles and the additional samples, including SPARC, TMEM40, FPR2, PRKCD and PPM1M.
SPARC encodes an extracellular protein that plays key roles in various cancer cell processes, such as cell proliferation, migration, matrix cell adhesion, angiogenesis and tissue remodeling (17). A number of studies have identified the impact of SPARC overexpression in patients with certain types of tumor and indicated that high expression levels of SPARC were associated with poor outcome, such as in pancreatic cancer (18). Furthermore, SPARC can activate the PI3K pathway and promote oral squamous cell carcinoma proliferation and metastasis (19). TMEM40 encodes a 23 kDamulti-pass membrane protein. A previous study revealed that TMEM40 is localized at chromosome 3p25.2 and is believed to play a key role in collagen-induced arthritis (20). The expression levels of TMEM40 were significantly higher in bladder cancer tissues and were associated with clinical grade (21). FPR2 encodes a seven transmembrane G protein-coupled receptor, which was first identified in phagocytic leukocytes and plays an important role in the host defense due to its effect on mediating leukocyte chemotaxis upon activation by bacterial and host-derived agonists (22). Recently, evidence has suggested that FPR2 is associated with certain types of cancer and that high mRNA levels of FPR2 are associated with poor prognosis in these types of cancer (23,24). PRKCD is a member of the protein kinase C family of serine- and threonine-specific protein kinases, which is both a tumor suppressor and a positive regulator of cell cycle progression. In breast cancer, PRKCD phosphorylated and activated the transcription factor STAT3, leading to increased expression levels of interleukin 6 and NANOG, two key mediators of breast cancer stem cells (25). However, in esophageal squamous cell carcinoma, increased PRKCD enhances the stability of p21 and inhibits cell proliferation (26). PPM1M is a phosphoprotein phosphatase that is involved in RNA polymerization. The effect of PPM1M on immunoreactions has been demonstrated to be associated with Herpes Simplex virus-1 infection (27). However, the effect of PPM1M in cancer progression remains unknown.
Overall, the present study identified that the functions of the indicated hub genes primarily include cell proliferation, metastasis, inflammation and immune response. These results suggested that, when compared with healthy individuals, the PBMCs of patients with HCC were more likely to be activated with increased DNA, RNA synthesis, inflammatory factor secretion and enzymatic activity. It was speculated that all these changes in the PBMCs of patients with HCC may be caused by environmental stress triggered by the local development of HCC, or the host's response against the tumor. Identification of these hub genes and the functional changes that they were involved in may contribute to the diagnosis of HCC.
However, there were limitations to the present study; the primary limitation was that these hub genes remain to be verified by experiments. In addition, the diagnostic value evaluation was performed using small sample sizes and therefore, additional samples are required for further research. Furthermore, the molecular mechanism of the change of key genes requires investigation.
Acknowledgements
Not applicable.
Funding
The present study work was supported by Tackling Key Problems in Science and Technology (grant no. 2007AA301B35-2), the Natural Science Foundation of Hubei Province of China (grant no. 2010CDB06807), and the Important Project of Wuhan Administration of Science &Technology (grant no. 2.008E+11).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
ZY, ZZ and YS were responsible for data collection, analysis and interpretation of the results. ZC wrote the manuscript. All authors have read and approved the final version of the article.
Ethics approval and consent to participate
All patients who had samples taken had provided informed consent. The present study was approved by the Ethics Committee of the Renmin Hospital of Wuhan University and was performed in accordance with the Declaration of Helsinki.
Patient consent for publication
Patients provided written consent for the publication of the present study.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Raoul JL, Raimbourg J, Hiret S, Adhoute X, Senellart H. Hepatocellular carcinoma: Increase in incidence or future plague? Bull Cancer. 2018;105:502–507. doi: 10.1016/j.bulcan.2018.02.003. (In French) [DOI] [PubMed] [Google Scholar]
- 2.Gupta M, Gabriel H, Miller FH. Role of imaging in surveillance and diagnosis of hepatocellular carcinoma. Gastroenterol Clin North Am. 2018;47:585–602. doi: 10.1016/j.gtc.2018.04.013. [DOI] [PubMed] [Google Scholar]
- 3.Sparchez Z, Mocan T. Contemporary role of liver biopsy in hepatocellular carcinoma. World J Hepatol. 2018;10:452–461. doi: 10.4254/wjh.v10.i7.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lok AS, Sterling RK, Everhart JE, Wright EC, Hoefs JC, Di Bisceglie AM, Morgan TR, Kim HY, Lee WM, Bonkovsky HL, et al. Des-gamma-carboxy prothrombin and alpha-fetoprotein as biomarkers for the early detection of hepatocellular carcinoma. Gastroenterology. 2010;138:493–502. doi: 10.1053/j.gastro.2009.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marrero JA, Feng Z, Wang Y, Nguyen MH, Befeler AS, Roberts LR, Reddy KR, Harnois D, Llovet JM, Normolle D, et al. Alpha-fetoprotein, des-gamma carboxyprothrombin, and lectin-bound alpha-fetoprotein in early hepatocellular carcinoma. Gastroenterology. 2009;137:110–118. doi: 10.1053/j.gastro.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang JX, Yu C, Li ZP, Xiao J, Zhang H, Chen MY, Sun CY. Insights into significant pathways and gene interaction networks in peripheral blood mononuclear cells for early diagnosis of hepatocellular carcinoma. J Cancer Res Ther. 2016;12:981–989. doi: 10.4103/0973-1482.154081. [DOI] [PubMed] [Google Scholar]
- 7.Li H, Mao Y, Xiong Y, Zhao HH, Shen F, Gao X, Yang P, Liu X, Fu D. A comprehensive proteome analysis of peripheral blood mononuclear cells (PBMCs) to identify candidate biomarkers of pancreatic cancer. Cancer Genomics Proteomics. 2019;16:81–89. doi: 10.21873/cgp.20114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Papageorgiou SG, Kontos CK, Diamantopoulos MA, Bouchla A, Glezou E, Bazani E, Pappa V, Scorilas A. MicroRNA-155-5p overexpression in peripheral blood mononuclear cells of chronic lymphocytic leukemia patients is a novel, independent molecular biomarker of poor prognosis. Dis Markers. 2017;2017(2046545) doi: 10.1155/2017/2046545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ciarloni L, Ehrensberger SH, Imaizumi N, Monnier-Benoit S, Nichita C, Myung SJ, Kim JS, Song SY, Kim TI, van der Weg B, et al. Development and clinical validation of a blood test based on 29-gene expression for early detection of colorectal cancer. Clin Cancer Res. 2016;22:4604–4611. doi: 10.1158/1078-0432.CCR-15-2057. [DOI] [PubMed] [Google Scholar]
- 10.Baine MJ, Chakraborty S, Smith LM, Mallya K, Sasson AR, Brand RE, Batra SK. Transcriptional profiling of peripheral blood mononuclear cells in pancreatic cancer patients identifies novel genes with potential diagnostic utility. PLoS One. 2011;6(e17014) doi: 10.1371/journal.pone.0017014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baine MJ, Menning M, Smith LM, Mallya K, Kaur S, Rachagani S, Chakraborty S, Sasson AR, Brand RE, Batra SK. Differential gene expression analysis of peripheral blood mononuclear cells reveals novel test for early detection of pancreatic cancer. Cancer Biomark. 2011-2012;11:1–14. doi: 10.3233/CBM-2012-0260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mishra S, Srivastava AK, Suman S, Kumar V, Shukla Y. Circulating miRNAs revealed as surrogate molecular signatures for the early detection of breast cancer. Cancer Lett. 2015;369:67–75. doi: 10.1016/j.canlet.2015.07.045. [DOI] [PubMed] [Google Scholar]
- 13.Leek JT. doi: 10.1093/nar/gku864. Svaseq: Removing batch effects and other unwanted noise from sequencing data. Nucleic Acids Res 42: 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. 2013; ISBN 3-900051-07-0. [Google Scholar]
- 15.Zhao W, Langfelder P, Fuller T, Dong J, Li A, Hovarth S. Weighted gene coexpression network analysis: State of the art. J Biopharm Stat. 2010;20:281–300. doi: 10.1080/10543400903572753. [DOI] [PubMed] [Google Scholar]
- 16.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 17.Vaz J, Ansari D, Sasor A, Andersson R. SPARC: A potential prognostic and therapeutic target in pancreatic cancer. Pancreas. 2015;44:1024–1035. doi: 10.1097/MPA.0000000000000409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arqueros C, Salazar J, Arranz MJ, Sebio A, Mora J, Sullivan I, Tobeña M, Martín-Richard M, Barnadas A, Baiget M, Páez D. SPARC gene variants predict clinical outcome in locally advanced and metastatic pancreatic cancer patients. Med Oncol. 2017;34(136) doi: 10.1007/s12032-017-0993-3. [DOI] [PubMed] [Google Scholar]
- 19.Jing Y, Jin Y, Wang Y, Chen S, Zhang X, Song Y, Wang Z, Pu Y, Ni Y, Hu Q. doi: 10.1002/jcp.28205. SPARC promotes the proliferation and metastasis of oral squamous cell carcinoma by PI3K/AKT/PDGFB/PDGFRβ axis. J Cell Physiol 2019 (Epub ahead of print). [DOI] [PubMed] [Google Scholar]
- 20.Zhang Q, Huang D, Zhang Z, Feng Y, Fu M, Wei M, Zhou J, Huang Y, Liu S, Shi R. High expression of TMEM40 contributes to progressive features of tongue squamous cell carcinoma. Oncol Rep. 2019;41:154–164. doi: 10.3892/or.2018.6788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang ZF, Zhang HR, Zhang QY, Lai SY, Feng YZ, Zhou Y, Zheng SR, Shi R, Zhou JY. High expression of TMEM40 is associated with the malignant behavior and tumorigenesis in bladder cancer. J Transl Med. 2018;16(9) doi: 10.1186/s12967-017-1377-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alessi MC, Cenac N, Si-Tahar M, Riteau B. FPR2: A novel promising target for the treatment of influenza. Front Microbiol. 2017;8(1719) doi: 10.3389/fmicb.2017.01719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hou XL, Ji CD, Tang J, Wang YX, Xiang DF, Li HQ, Liu WW, Wang JX, Yan HZ, Wang Y, et al. FPR2 promotes invasion and metastasis of gastric cancer cells and predicts the prognosis of patients. Sci Rep. 2017;7(3153) doi: 10.1038/s41598-017-03368-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xiang Y, Yao X, Chen K, Wang X, Zhou J, Gong W, Yoshimura T, Huang J, Wang R, Wu Y, et al. The G-protein coupled chemoattractant receptor FPR2 promotes malignant phenotype of human colon cancer cells. Am J Cancer Res. 2016;6:2599–2610. [PMC free article] [PubMed] [Google Scholar]
- 25.Lan J, Lu H, Samanta D, Salman S, Lu Y, Semenza GL. Hypoxia-inducible factor 1-dependent expression of adenosine receptor 2B promotes breast cancer stem cell enrichment. Proc Natl Acad Sci U S A. 2018;115:E9640–E9648. doi: 10.1073/pnas.1809695115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wen J, Hu Y, Liu Q, Ling Y, Zhang S, Luo K, Xie X, Fu J, Yang H. miR-424 coordinates multilayered regulation of cell cycle progression to promote esophageal squamous cell carcinoma cell proliferation. EBioMedicine. 2018;37:110–124. doi: 10.1016/j.ebiom.2018.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yue L, Guo S, Zhang Y, Liu L, Wang Q, Wang X, Shen D, Wang L, Sun L, Wang J, et al. The modulation of phosphatase expression impacts the proliferation efficiency of HSV-1 in infected astrocytes. PLoS One. 2013;8(e79648) doi: 10.1371/journal.pone.0079648. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.