

Abstract
Deep vein thrombosis (DVT) is a type of venous thromboembolism and a clinically complex vascular disease. Oxidative stress serves a key role in the pathogenesis of numerous cardiovascular diseases, particularly in endothelial dysfunction-associated syndromes. Nuclear factor erythroid-2-like 2(Nrf2) transcription factor is the primary regulator of antioxidant responses. The levels of reactive oxygen species (ROS) are regulated by Nrf2 and its suppressor protein Kelch-like ECH-associated protein 1 (Keap1). However, to the best of our knowledge, genetic abnormalites in the Nrf2/Keap1 pathway in DVT syndrome have not been thoroughly investigated. The aim of the present study was to investigate the association between the Nrf2/Keap1 pathway and antioxidant responses in DVT. Mutations and expression levels of genes involved in the Nrf2/Keap1 pathway were measured in 27 patients with DVT via DNA sequencing analysis and reverse transcription-quantitative PCR, respectively. The Polymorphism Phenotyping v2 program was used to identify the pathogenic mutations. Total antioxidant activity levels were determined by measuring the effect of serum samples from 27 patients with DVT on oxidation of the 2,2'-azino-bis (3-ethylbenz-thiazoline-6-sulfonic acid) system. A total of 23 mutations, including seven novel mutations, were detected in the Nrf2/Keap1 pathway in 24 (89%) of the 27 patients with DVT. Keap1 mRNA expression levels were significantly higher compared with Nrf2 expression levels in patients with DVT (P=0.02). Analysis of molecular characteristics and gene expression levels demonstrated that Nrf2/Keap1-associated mutations and total antioxidant levels can be used as precursor markers in the diagnosis of DVT.
Keywords: deep vein thrombosis, oxidative stress, antioxidant levels, nuclear factor erythroid 2-related factor 2/kelch-like ECH-associated protein 1 signaling pathway, mutation, gene expression
Introduction
Endothelial cells regulate vascular homeostasis and form a biologically active blood-tissue barrier, thus serving key roles in regulation of vascular tone, control of inflammatory responses and vascular permeability, and the balance between the coagulation and fibrinolysis systems (1,2). Alterations in the functional and morphological architecture of the endothelium impair normal physiological processes. This is known as endothelial dysfunction and results in disruptions in the barrier function, increased prothrombotic/procoagulant activity and impaired vasomotion due to increased or decreased synthesis of vasoactive substances (3-5). Disturbed vascular homeostasis results in vasoconstriction, leukocyte adhesion, platelet activation, predisposition to thrombosis, coagulation disorders, pro-oxidative changes and vascular inflammation (4). Deep vein thrombosis (DVT) is a clinical condition characterized by coagulation in the veins of the upper and often lower extremities. The pathogenesis of DVT involves endothelial dysfunction (endothelial damage-dysfunction), hypercoagulability and stasis (slow blood flow). These mechanisms may lead to venous obstruction and thromboembolism, resulting in serious consequences ranging from post-thrombotic syndrome to pulmonary thromboembolism (6,7). Oxidative stress is a key factor that contributes to the pathogenesis of numerous cardiovascular diseases, including atherosclerosis, hypertension, heart failure, ischemia/reperfusion injury and thrombosis (8-11). Oxidative stress is important for reactive oxygen species (ROS) production, inflammatory responses, cell growth, proliferation, modulation of extracellular matrix production, maintenance of endothelial-vascular smooth muscle cells, apoptosis and angiogenesis control, particularly in the vascular system (2,12,13).
The nuclear factor erythroid-2-like 2 (Nrf2)/Kelch-like ECH-associated protein 1 (Keap1) signaling pathway protects numerous cell types, tissues, organs and systems from the pathogenic effects of oxidative stress. Therefore, Nrf2 is referred to as a ‘multi-organ protector’ (14). The Nrf2/Keap1 signaling pathway is involved in antioxidant defense mechanisms in cardiovascular diseases, such as atherosclerosis and hypertension (15,16). This pathway controls the cellular response that prevents injury to redox-sensitive cellular components, where Nrf2 has been identified as the main regulator of oxidant/antioxidant balance (16). Nrf2 is a transcription factor that regulates transcription of numerous genes involved in tumorigenesis, inflammation, radiation, detoxification of xenobiotics and inhibition of ROS (17). Keap1 is a suppressor of Nrf2 when cells are not under stress (15,17-19). Upon exposure to oxidative stress, cells release Nrf2, which activates cytoprotective genes and forms heterodimers with Maf family proteins, mediated via transcription factors (20). A nuclear localization sequence mediates binding of the Nrf2 heterodimer to the antioxidant response element (ARE) sequence following translocation to the nucleus. Thus, the transcriptional activation of cytoprotective genes, such as NADPH quinone dehydrogenase 1 (NQO-1), superoxide dismutase 1, heme hxygenase 1 (HO-1) and glutathione S-transferase A2 (GSTA2), begins (21-23). Previous studies have demonstrated that activation of the Nrf2/Keap1 signaling pathway may serve a key role in cardioprotection, and disordered activation of the pathway may increasse susceptibility to endothelial dysfunction (24-28). Further investigations are required to better understand the interaction between the cardiovascular system and the Nrf2/Keap1 signaling pathway, which may be an initiator of pathogenesis in cardiovascular diseases.
To the best of our knowledge, there is currently little research regarding the association between the Nrf2/Keap1 signaling pathway (which acts as a cellular defence against oxidative stress) and DVT, a cardiovascular disease that involves pathogenic endothelial damage. The present study aimed to investigate pathogenic mutations in the Nrf2/Keap1 signaling pathway and to determine whether there is an imbalance in Nrf2 and Keap1 levels in patients with DVT in order to identify the underlying mechanism of pathogenesis in DVT. The objectives of the study were to identify pathogenic mutations and expression levels of genes involved in the Nrf2/Keap1 signaling pathway in patients with DVT; to measure total antioxidant values in blood samples from patients with DVT; and to characterize the association between expression levels of Nrf2 and Keap1 pathogenic mutations, levels of antioxidants and DVT.
Materials and methods
Study population
The study population are of comprised 27 patients (20 men and seven women), aged 20-74 years, who were admitted to Faculty of Medicine, Nigde Omer Halisdemir University (Nigde, Turkey) with the diagnosis of primary (spontaneous) DVT. In addition, a healthy control group consisting of 10 people (six men and four women), aged 20-74 years was included in the present study. The present study was performed in accordance with the Code of Ethics of the World Medical Association (Declaration of Helsinki) for experiments involving humans. The study protocol was approved by Erciyes University School of Medicine Ethics Committee (approval no. 2018/533; Kayseri, Turkey). All patients agreed to participate in the study, which was conducted between March and April 2019. Written informed consent was provided by all patients. Demographic, clinical and genetic data regarding the patient group are summarized in Table I.
Table I.
Demographic, clinical and genetic data of patients with DVT.
| Characteristic | Patient data |
|---|---|
| Sex | |
| Male/Female | 20/7 |
| Mean age, years | 42 (range, 24-70) |
| Site of thrombosis | |
| Lower extremity | 25 |
| Upper extremity | 1 |
| Pulmonary embolism | 1 |
| Oral anticoagulant use | |
| Warfarin | 24 |
| Rivaroxaban | 3 |
| Frequency of Kelch-like ECH-associated protein 1 mutations | 24 (88%) |
| Frequency of nuclear factor erythroid-2-like 2 mutations | 14 (52%) |
| Frequency of FV G/A | 7 (26%) |
| Frequency of FV A/A | 6 (22%) |
| Frequency of PT G/A | 4 (15%) |
| Frequency of PT A/A | 0 |
| Hypertension | 6 (22%) |
| White blood cells, x109/l | 8.22 (5.80-14.40) |
| Red blood cells, x109/l | 5.22 (4.15-6.98) |
| Partial thromboplastin time | 40.27±9.05 |
| Thromboplastin time | 22.66±6.66 |
| International normalized ratio | 1.98±0.57 |
| Hemoglobin, g/dl | 15.01±1.58 |
| Hematocrit, % | 44.68 ±4.52 |
| Mean corpuscular volume, fl | 86.38±8.51 |
| Mean corpuscular hemoglobin, pg | 29.05±3.26 |
| Platelet count, x109/l | 239.8 (139.0-355.0) |
| Red blood cell distribution width, % | 13.86±1.11 |
| Procalcitonin, % | 0.22±0.06 |
| Platelet distribution width, % | 16.01±0.43 |
| Mean platelet volume, fl | 9.11±1.15 |
FV, factor V; PT, prothrombin.
Color Doppler ultrasonography, venography, magnetic resonance imaging and computed tomography methods can be used to diagnose DVT. In the present study, the diagnosis of DVT was based on physical examination and venous color Doppler ultrasonography. The age range of the patients with primary DVT was 24-70 years. Subjects were excluded if they had previously undergone surgical interventions, received or ongoing treatment for cancer within the previous 6 months, had given birth within the previous 6 weeks, were receiving hormonal therapy or using oral contraceptives, had a history of acute myocardial infarction, or were immobile.
The age range of the healthy control group is similar to that of the patient group. The healthy control group was selected from the individuals who came to the hospital for checkup purposes and the group is composed of individuals who do not have any disease in the coagulation system or acute and/or chronic disease, including hypertension, diabetes, obesity and endocrine diseases.
Sample collection
Blood samples (5 ml) were collected from subjects in sterile EDTA tubes for DNA and RNA isolation.
Mutation screening
DNA was isolated from whole blood using the GeneJET Genomic DNA Purification kit (Thermo Fisher Scientific, Inc.) according to the manufacturer's instructions. DNA was then stored at -20̊C until use. DNA concentration was quantified using a Genova Nano 3 spectrophotometer (Jenway; Bibby Scientific Ltd.). The coding regions of Nrf2 and Keap1 genes were amplified via PCR. A total of ~200 ng DNA was used as template for PCR amplification. Primer sequences are listed in Table II. The total volume of the reaction mix was 25 µl, comprising 16.75 µl sterile water, 2.5 µl reaction buffer [200 mM (NH4)2SO4], 1 µl dNTP mix (A, C, G, T; 200 mM), 1.25 µl MgCl2 (25 mM), 1 µl forward and reverse primers (10 pmol), 0.5 µl Taq polymerase (500 units; Fermentas; Thermo Fisher Scientific, Inc.) and 1 µl template DNA (>500 ng/µl for each sample). Amplification reactions were performed using a Veriti VR 96-Well Thermal Cycler (Applied Biosystems; Thermo Fisher Scientific, Inc.). Thermocycling conditions were as follows: Initial denaturation at 94̊C for 5 min, followed by 40 cycles of denaturation for 45 sec at 94̊C, primer annealing for 90 sec at 58̊C, extension for 90 sec at 72̊C, and final extension for 10 min at 72̊C. Following amplification, 10 µl PCR products were separated by electrophoresis on a 2% agarose gel containing GelRed in 1X TAE buffer for 40 min at 90 V. A GeneRuler 100 bp DNA Ladder (Thermo Fisher Scientific, Inc.) was used as a marker. Subsequently, gels were visualized using a Gel Logic 100 system (Kodak). PCR products were then sequenced using a 3500 XL DNA Sequencer (Applied Biosystems; Thermo Fisher Scientific, Inc.). Data were analyzed using data collection GeneMapper™ ID-X Software (v1.6; Applied Biosystems; Thermo Fisher Scientific, Inc.) and Chromas (v2.6.5; Technelysium Pty Ltd.) to determine sequence changes compared with reference Nrf2 and Keap1 sequences obtained from the Ensembl genome browser (https://www.ensembl.org/index.html) (29).
Table II.
Primer sequences and amplicon lengths for Nrf2 and Keap1.
| Gene/exon | Forward primer (5'à3') | Reverse primer (5'à3') | Amplicon, bp |
|---|---|---|---|
| Keap1/2-2 | ATACCAAGCAGGCCTTTGG | ATGGAGATGGAGGCCGTGT | 265 |
| Keap1/2-3 | ATGGAGCGCCTCATTGAA | AGCCCCACTTCCCCGCT | 263 |
| Keap1/3-1 | CCGTCCCACTGTCGCCCTC | TGCAGGATCTCGCACTTC | 260 |
| Keap1/3-2 | AGCTGCAGAAGTGCGAG | GTTGTTCCTGCCGCCCACG | 296 |
| Keap1/4 | CATTTTTCTTACGCCCTTGC | GTTTCACCCCAGGATGGTAG | 290 |
| Nrf2/1 | CGTGTAGCCGATTACCGAGT | CAGACCTTCCCGCAACTTC | 454 |
| Nrf2/2 | CCACCATCAACAGTGGCATA | CCTGCCATAACTTTCCCAAG | 400 |
| Nrf2/2-2 | CCTGAGTTCAGTACCCAACCTAT | TGAGCTTCAGTTGCTCATCG | 281 |
| Nrf2/3 | GCTTACTCATCCCCTGTTGG | TAGCACCCTCCAATCCTTCC | 355 |
| Keap1 (mRNA) | GTGTCCATTGAGGGTATCCACC | GCTCAGCGAAGTTGGCGAT | 211 |
| Nrf2 (mRNA) | TCAGCGACGGAAAGAGTATGA | CCACTGGTTTCTGACTGGATT | 174 |
| RPLP0 (mRNA) | CAGATTGGCTACCCAACTGTT | GGAAGGTGTAATCCGTCTCCC | 97 |
| FV G1691A | TGCCCAGTGCTTAACAAGACCA | CTTGAAGGAAATGCCCCATTA | 110 |
| PT G20210A | CCGCTGGTATCAAATGGGG | CCAGTAGTATTACTGGCTCTTCCTG | 106 |
| FV G1691A Hybridization probes | 5'-LC-Red705-TGTCCTTGAAGTAACCTTTCAGAAATTCTG-3'-PHO | 5'-GGCGAGGAATACAGGTAT-3'-Flu | N/A |
| PT G20210A Hybridization probes | 5'-LC-Red640-TCCCAGTGCTATTCATGGGC-3'-PHO | 5'-CTCAGCGAGCCTCAATG-3'-Flu | N/A |
Nrf2, nuclear factor erythroid-2-like 2; Keap1, Kelch-like ECH-associated protein 1; N/A, Not Applicable.
Genotyping of factor V (FV) 1691 G-A and prothrombin (PT/Factor II) 20210 G-A mutations was performed via Real-Time Polymerase Chain Reaction (RT-PCR), with fluorescence melting curve detection analysis via a Light Cycler system (Roche Diagnostics GmbH). The primers and probes used in the present study were summarized in Table II. Melting point analysis was performed using LightMix® in-vitro diagnostics Factor V (Leiden; cat.no. 40-0594-64) and Factor II G20210A mutation (cat. no. 40-0593-64) detection kits according to the manufacturer's protocols (TIB Molbiol; Syntheselabor GmbH).
In silico analysis of Keap1/Nrf2 pathway mutations
Polymorphism Phenotyping v2 (PolyPhen-2; http://genetics.bwh.harvard.edu/pph2) can predict the potential impact of amino acid substitutions on the stability and function of human proteins using structural and comparative evolutionary considerations. The prediction is based on sequence, phylogenetic and structural features that characterize the substitution. PolyPhen-2 performs functional annotation of single-nucleotide polymorphisms (SNPs), maps coding SNPs to gene transcripts, extracts protein sequence annotations and structural attributes, and builds conservation profiles. The program estimates the probability of the missense mutation being damaging and provides both a qualitative prediction (probably damaging, possibly damaging, benign or unknown) and a quantitative score (30). It includes a high-quality multiple protein sequence alignment pipeline and a prediction method that uses machine-learning classification. In addition, to determine the possible pathogenicity of the identified mutations, the scores were provided by the Catalog of Somatic Mutations in Cancer (COSMIC) databases (https://cancer.sanger.ac.uk/cosmic) (31).
Evolutionary conservation of the detected mutant amino acids was estimated among different species (Homo sapiens, Rattus norvegicus, Mus musculus, Pan troglodytes, Felis domesticus, Canis lupus familiaris, Bos taurus, Sus scrofa and Xenopus tropicalis) using the Homologous Gene-Multiple Seguence Alignment Viewer 1.12.0 tool in the National Center for Biotechnology Information (NCBI) database (https://www.ncbi.nlm.nih.gov/genbank/) .
Except for novel mutations, the registration number of the ones detected before was obtained from Human Gene Mutation Database 2020.1 (HGMD; http://www.hgmd.cf.ac.uk/ac/index.php) (32).
Nrf2/Keap1 pathway gene expression analysis
RNA isolation was isolated from whole blood using PureZOLTM solution, according to the manufacturer's instructions (Bio-Rad Laboratories, Inc.). The Genova Nano 3 nanodrop UV spectrophotometer was used to determine the purity and concentration of RNA samples, which were stored at -80̊C until use. Protein contamination was evaluated via A260/A280 ratio; samples with a ratio of 1.8-2.2 were used for subsequent experimentation. RNA samples were stored in a final volume of 500 ng RNA for use in cDNA synthesis.
Reverse transcription (RT)-qPCR was used to detect the expression levels of Nrf2 and Keap1 target genes. Primer sequences are (Table II) were designed using the Primer3 Input (v0.4.0) program (http://primer3.ut.ee/) (33). cDNA synthesis was performed using a First Strand Transducer c-DNA synthesis kit (Roche Diagnostics GmbH) according to the manufacturer's instructions. The RPLP0 gene was used as the reference gene. mRNA expression levels were detected using a SYBR Green I Master mix (Bio-Rad Laboratories, Inc.) in a Rotor-GeneQ thermocycler (Qiagen, GmbH), thermocycling conditions were as follows: Initial denaturation at 95̊C for 5 min, followed by 40 cycles of denaturation for 15 sec at 95̊C, primer annealing for 60 sec at 60˚C, extension for 30 sec at 72˚C, and 1 cycle of melting curve for 30 sec at 55-95˚C, according to the manufacturer's instructions. Each experiment was repeated three times. mRNA expression levels were calculated according to Pfaffl's method (34). Relative quantification of gene expression levels was performed using the comparative quantification cycle (Cq) method, in which the amounts of the target genes are expressed as 2exp-ΔCq (35).
Analysis of total antioxidant capacity (TAC)
The water-soluble analogue of vitamin E (Trolox; 6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid), 2,2'-azino-bis (3-ethylbenz-thiazoline-6-sulfonic acid) (ABTS), potassium persulfate and potassium phosphate were purchased from Sigma-Aldrich (Merck KGaA). ABTS, potassium persulfate and potassium phosphate and ultra-pure grade were commercially available. Peripheral venous blood from all patients was collected for the measurement of TAC levels. All blood samples were collected in vacutainer tubes without anticoagulant and centrifuged at 1,800 x g for 10 min at room temperature. The serum samples obtained were stored at -20˚C until use.
TAC is based on the bleaching of the dark blue-green-colored ABTS radical when it is reduced in the presence of antioxidant molecules. The bleaching rate of the reaction is determined using a spectrophotometer and is associated with the TAC of the sample. The reaction rate is calibrated using Trolox, which is widely used as the antioxidant standard agent for TAC assays, and results are expressed in µmol Trolox equivalent/l (µmol TE/l).
ABTS (7 mmol/l) was dissolved in PBS, then potassium persulfate (2.45 mmol/l) was added and the solution was mixed at room temperature. The ABTS.+ stock solution was kept in the dark at room temperature for 12-16 h before use. The stock solution of ABTS.+ was diluted with PBS (pH 7.4) to obtain an absorbance of 0.70 at 734 nm, measured using a UV-2700 UV-Vis spectrophotometer (Shimadzu Corporation). The diluted ABTS.+ was then mixed with the aforementioned serum samples and left to stand for 10 min at room temperature. The change in absorbance was monitored at 734 nm using the UV-Vis spectrophotometer. Trolox was used as a control. Results are expressed as µmol TE/L.
Statistical analysis
Data are presented as the mean ± SD. The analysis of TAC and gene expression were repeated for three times. Statistical analysis of gene expression levels was performed by one-way ANOVA followed by Dunnett's test and Student's t-test using SPSS Statistics software (v22.0; IBM Corp.). P<0.05 was considered to indicate a statistically significant difference.
Results
Demographic and clinical analysis of patients with DVT
The age distribution of the patient population was 24-70 years, with a mean age of 42 years. The patient population comprised 20 men and seven women (74 and 26% respectively). Thrombosis was detected in the lower extremity veins of 25 patients, the upper extremity veins of one patient and the pulmonary artery of one patient. A total of eight patients had a history of hypertension. Warfarin was used to treat 24 patients and Rivaroxaban was used to treat three patients. Demographic, clinical and genetic data of the patient group are summarized in Table I.
Results of mutation screening
Mutation analysis was performed using Sanger DNA sequencing of DNA isolated from blood samples of patients with DVT. The mutation analysis identified 23 changes in the Nrf2 and Keap1 genes (11 missense mutations, five synonymous nucleotide changes, two splice site mutations and five non-coding region nucleotide changes). Of these changes, 16 had previously been recorded in the HGMD and seven were novel changes. The detected mutations exhibited heterozygous genotypes. Changes were detected in 24 (89%) patients with DVT (Fig. 1). The mutations detected in the present study (those with accesion number) had not been associated with DVT disease previously.
Figure 1.

Sequencing electropherograms of (A) Keap1 and (B) Nrf2 gene mutations and variants. Arrows indicate localizations of the mutation and variant. Keap1, Kelch-like ECH-associated protein 1; Nrf2, nuclear factor erythroid-2-like 2.
The changes detected in the Keap1 gene were located on the functional intervening region (IVR) and broad-complex, tramtrack and bric-a-brac (BTB) domains of the protein encoded by the Keap1 gene. A total of 10 missense mutations, five synonymous nucleotide substitutions and one splice site mutation was detected in the Keap1 gene. Of these, three were novel nucleotide substitutions. Nucleotide substitutions in the Nrf2/Keap1 pathway are presented in Fig. 1. A schematic representation of domain architecture of the identified proteins and mutations is presented in Fig. 2.
Figure 2.
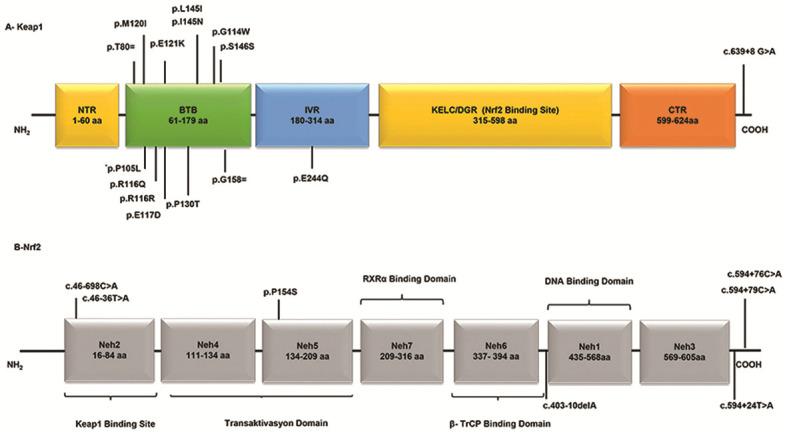
Schematic representation of domain architecture of the Keap1 and Nrf2 proteins and mutations detected in patients with deep vein thrombosis. (A) Human Keap1 is a polypeptide comprising 624 amino acids. There are two different mutation points (c.313/314 C>T) that encode the amino acid at 105 codon. (B) Human Nrf2 is a polypeptide comprising 605 amino acids, which contains seven Neh domains.;Keap1, Kelch-like ECH-associated protein 1; Nrf2, nuclear factor erythroid-2-like 2; Neh, Nrf2-ECH homology; NTR, N-terminal region; BTB, broad-complex, tramtrack and bric-a-brac; IVR, intervening region; KELC/DGR, Kelch/double glycine repeat; CTR, C-terminal region; RXRα, retinoid X receptor α; β-TrCP, β-transducin repeats-containing protein.
It was determined that 12 changes (p.R116Q, p.E121K, p.P105L, p.S146S, p.I145N, p.L145I, p.P130T, p.P105L, p.E244Q, p.E117D, p.M120I and p.G114W) found in Keap1 may be pathogenic, as PolyPhen-2 analysis resulted in scores close to 1. These mutations are listed in the COSMIC database as somatic mutations. To the best of our knowledge, in the present study, p.I145N, p.P105L and p.M120I (missense) changes were identified for the first time. An intronic substitution, c.639 +8 G>A, has previously been identified as a splice site mutation in on the NCBI database (https://www.ncbi.nlm.nih.gov/snp/rs1466903180#clinical_significance). n the present study, seven changes [one missense, one splice site region, one intronic region and four 3' untranslated regions (3' UTRs)] were detected in the Nfr2 gene. Of these detected changes, three had previously been recorded in the HGMD. The present study identified four novel mutations in the 3'UTR (Fig. 2).
The mutations detected in patients with DVT, as well as genotype frequencies of FV 1691 G-A and PT 20210 G-A mutations, are characterized in Table III. Changes in Nrf2 were detected in 14 (52%) patients. The subsititution p.P154S detected in the Nrf2 gene was located on the Nrf2-ECH homology (Neh) 5 transactivation domain. The subsititution c.46-698 C>A is registered in the COSMIC database (reference no. COSM6971525); it was located on the splice site region and detected in 10 (42%) patients. When the Nrf2 genes of different species of organisms were compared, seven functional Neh domains with high evolutionary conservation were detected and named Neh1-7. The c.46-698 C>A and c.46-36 T>A variants detected in the Nrf2 gene were located on the ETGE-Neh2 domain, which is a Keap1 binding site. The Neh2 domain is a large regulatory domain that contains seven lysine residue sequences responsible for two binding sites (ETGE and DLG motifs) that are associated with the regulation of Nrf2 stability and ubiquitin conjugation (18). The missense mutation p.E244Q was detected on the Keap1 IVR domain. This domain contains a conserved nuclear export signal sequence, which is important for localization of Keap1 in the cytoplasm (17,18). The splice site mutations c.639+8 G>A and c. 46-698 C>A were detected in the Keap1 and Nrf2 genes, respectively. As the c.46-698 C>A mutation detected in the Nrf2 gene is located on the first base of the completely conserved splice site across the species in the evolutionary process, this mutation may cause the absence of Nrf2 expression levels. A total of 15 patients carried both Keap1 and Nrf2 substitutions.
Table III.
Mutations of the Nrf2/Keap1 pathway in patients with DVT.
| Case | Age | Sex | Genotype and location | Gene name | Type of mutation | Amino acid change | Clinical significance | Accession no. | Thrombosis | FV G1691A | FII G20210A | Onset age |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 23 | M | c.438 C>T | Keap1- BTB-Domain | Synonymous | p.S146S | 0,83- Pathogenic | Cosm5609243 | Upper Extremity | G/G | G/G | 20 |
| c.460 C>T | Nrf2-Neh5 | Missense | p.P154S | Benign | rs763327009 | |||||||
| 2 | 34 | F | c.438 C>T | Keap1- BTB-Domain | Synonymous | p.S146S | 0,83- Pathogenic | Cosm5609243 | Lower Extremity | G/G | G/A | 29 |
| 3 | 44 | M | c.639+8 G>A | Keap1-Splice site | Intronic variant | - | NA | rs1466903180 | Lower Extremity | A/A | G/G | 37 |
| c.46-698C>A | Nrf2 | Splice acceptor variant | - | Cosm6971525 | ||||||||
| 4 | 23 | M | c.474 T>C | Keap1- BTB- Domain | Synonymous | p.G158G | Neutral | Cosm4131169 | Lower Extremity | G/G | G/G | 19 |
| c.594+76C>Aa | Nrf2-3'UTR | Intronic variant | - | Novel | ||||||||
| 5 | 27 | M | c.474 T>C | Keap1- BTB-Domain | Synonymous | p.G158G | Neutral | Cosm4131169 | Lower Extremity | G/A | G/G | 27 |
| c.46-698C>A | Nrf2 | Splice acceptor variant | - | Cosm6971525 | ||||||||
| 6 | 42 | M | c.348G>A | Keap1- BTB-Domain | Synonymous | p.R116R | Neutral | rs759047759 | Lower Extremity | A/A | G/G | 42 |
| c.347 G>A | Missense | p.R116Q | 0,85-Pathogenic | Cosm3796565 | ||||||||
| c.46-698C>A | Nrf2 | Splice acceptor variant | - | NA | Cosm6971525 | |||||||
| 7 | 55 | M | - | - | - | - | - | - | Lower Extremity | A/A | G/G | 47 |
| 8 | 44 | M | c.313/314 C>Ta | Keap1- BTB Domain | Missense | p.P105L | 0,99- Pathogenic | Novel | Lower Extremity | G/A | G/A | 38 |
| c.348G>A | Missense | p.R116Q | 0,85-Pathogenic | Cosm3796565 | ||||||||
| c. 347G>A | Synonymous | p.R116R | Neutral | rs759047759 | ||||||||
| c.388 C>T | Missense | p.P130T | 1,00- Pathogenic | Cosm6970908 | ||||||||
| c.340 G>T | Missense | p.G114W | 0,94-Pathogenic | Cosm1725916 | ||||||||
| c.361 G>A | Missense | p.E121K | 0,68- Pathogenic | Cosm69679929 | ||||||||
| c.240 A>G | Missense | p.T80T | Neutral | rs1215286054 | ||||||||
| c.46-698C>A | Splice acceptor variant | Cosm6971525 | ||||||||||
| 9 | 56 | F | c.388 C>T | Keap1- BTB-Domain | Missense | p.P130T | 1,00- Pathogenic | Cosm6970908 | Lower Extremity | G/G | G/G | 52 |
| c.347 G>A | p.R116Q | 0,85-Pathogenic | Cosm3796565 | |||||||||
| c.348G>A | Synonymous | p.R116R | Neutral | rs759047759 | ||||||||
| 10 | 20 | M | c.438 C>T | Keap1- BTB-Domain | Synonymous | p.S146S | 0,83- Pathogenic | Cosm5609243 | Lower Extremity | A/A | G/G | 20 |
| c.730 G>C | Keap1- IVR-Domain | Missense | p.E244Q | 0,92- Pathogenic | Cosm6986830 | |||||||
| c.351 G>T | Keap1- BTB-Domain | Missense | p.E117D | 0,97- Pathogenic | Cosm6917923 | |||||||
| c.347 G>A | Synonymous | p.R116R | Neutral | rs759047759 | ||||||||
| c.348G>A | Missense | p.R116Q | 0,85-Pathogenic | Cosm3796565 | ||||||||
| c.361 G>A | Missense | p.E121K | 0,68- Pathogenic | Cosm69679929 | ||||||||
| c.594+24T>Aa | Nrf2-3'UTR | Intronic variant | - | - | Novel | |||||||
| 11 | 37 | F | c.438 C>T | Keap1- BTB-Domain | Synonymous | p.S146S | 0,83- Pathogenic | Cosm5609243 | Lower Extremity | G/G | G/G | 28 |
| c.388 C>T | Missense | p.P130T | 1,00- Pathogenic | Cosm6970908 | ||||||||
| c.348G>A | Synonymous | p.R116R | Neutral | rs759047759 | ||||||||
| 12 | 54 | M | c.474 T>C | Keap1- BTB Domain- | Synonymous | p.G158G | Neutral | Cosm4131169 | Pulmonary Artery | G/G | G/G | 54 |
| c.348G>A | Synonymous | p.R116R | Neutral | rs759047759 | ||||||||
| c.347 G>A | Missense | p.R116Q | 0,85-Pathogenic | Cosm3796565 | ||||||||
| c.46-698C>A | Nrf2 | Splice acceptor variant | Cosm6971525 | |||||||||
| 13 | 57 | M | c.348G>A | Keap1- BTB-Domain | Synonymous | p.R116R | Neutral | rs759047759 | Lower Extremity | G/G | G/G | 47 |
| c.347 G>A | Missense | p.R116Q | 0,85-Pathogenic | Cosm3796565 | ||||||||
| c.46-698C>A | Nrf2 | Splice acceptor variant | - | - | Cosm6971525 | |||||||
| c.594+76C>A | Nrf2-3'UTR | Intronic variant | - | - | Novel | |||||||
| 14 | 22 | M | c.348G>A | Keap1- BTB-Domain - | Synonymous | p.R116R | Neutral | rs759047759 | Lower Extremity | A/A | G/G | 19 |
| c.347 G>A | Missense | p.R116Q | 0,85-Pathogenic | Cosm3796565 | ||||||||
| c.438 C>T | Synonymous | p.S146S | 0,83- Pathogenic | Cosm5609243 | ||||||||
| c.46-698C>A | Nrf2 | Splice acceptor variant | Cosm6971525 | |||||||||
| 15 | 27 | M | - | - | - | - | - | - | Lower Extremity | G/G | G/G | 22 |
| 16 | 60 | M | c.347 G>A | Keap1- BTB-Domain | Missense | p.R116Q | 0,85-Pathogenic | Cosm3796565 | Lower Extremity | G/G | G/G | 54 |
| c.438 C>T | Synonymous | 0,83- Pathogenic | Cosm5609243 | |||||||||
| c.46-698C>A | Nrf2 | Splice acceptor variant | - | - | Cosm6971525 | |||||||
| 17 | 28 | M | c.348G>A | Keap1- BTB-Domain | Synonymous | p.R116R | Neutral | rs759047759 | Lower Extremity | G/G | G/A | 23 |
| c.347 G>A | Missense | p.R116Q | 0,85-Pathogenic | Cosm3796565 | ||||||||
| c.438 C>T | Synonymous | p.S146S | 0,83- Pathogenic | Cosm5609243 | ||||||||
| c.434 T>Aa | Missense | p.I145N | 0,99-- Pathogenic | Novel | ||||||||
| 18 | 48 | F | c.347 G>A | Keap1- BTB-Domain - | Missense | p.R116Q | 0,85-Pathogenic | Cosm3796565 | Lower Extremity | A/A | G/G | 43 |
| c.438 C>T | Synonymous | p.S146S | 0,83- Pathogenic | Cosm5609243 | ||||||||
| c.434 T>A | Missense | p.I145N | 0,99-Pathogenic | Novel | ||||||||
| c.435 C>A | Missense | p.I145L | 0,90- Pathogenic | Cosm6492287 | ||||||||
| c.388 C>T | Missense | p.P130T | 1,00- Pathogenic | Cosm6970908 | ||||||||
| c.46-36T>A | Nrf2 | Intronic variant | rs751650669 | |||||||||
| 19 | 41 | M | c.360 G>Ta | Keap1- BTB-Domain | Missense | p.M120I | 0,81- Pathogenic | Novel | Lower Extremity | G/G | G/G | 33 |
| c.474 T>C | Synonymous | p.G158G | Neutral | Cosm4131169 | ||||||||
| c.46-698C>A | Nrf2 | Splice acceptor variant | - | Cosm6971525 | ||||||||
| 20 | 74 | F | c.348G>A | Keap1- BTBDomain | Synonymous | p.R116R | Neutral | rs759047759 | Lower Extremity | 62 | ||
| c.347 G>A | Missense | p.R116Q | 0,85-Pathogenic | Cosm3796565 | ||||||||
| c.474 T>C | Synonymous | p.G158G | Neutral | Cosm4131169 | ||||||||
| 21 | 44 | M | c.347 G>A | Keap1- BTB-Domain | Missense | p.R116Q | 0,85-Pathogenic | Cosm3796565 | Lower Extremity | G/A | G/G | 37 |
| c.360 G>T | p.M120I | 0,81- Pathogenic | Novel | |||||||||
| c.438 C>T | Synonymous | p.S146S | 0,83- Pathogenic | Cosm5609243 | ||||||||
| 22 | 69 | F | c.360 G>T | Keap1- BTB- Domain | Missense | p.M120I | 0,81- Pathogenic | Novel | Lower Extremity | G/G | G/G | 64 |
| c.438C>T | Synonymous | p.S146S | 0,83- Pathogenic | Cosm5609243 | ||||||||
| c.474 T>C | Synonymous | p.G158G | Neutral | Cosm4131169 | ||||||||
| c.594+79C>Aa | Nrf2-3'UTR | Intronic variant | - | Novel | ||||||||
| 23 | 23 | M | c.360 G>T | Keap1- BTB | Missense | p.M120I | 0,81- Pathogenic | Novel | Lower Extremity | G/G | G/G | 17 |
| c.474 T>C | Synonymous | p.G158G | Neutral | Cosm4131169 | ||||||||
| c.40310delAa | Nrf2-3'UTR | Intronic variant | - | - | Novel | |||||||
| 24 | 64 | M | c.438 C>T | Keap1- BTB-Domain | Synonymous | p.S146S | 0,83- Pathogenic | Cosm5609243 | Lower Extremity | G/G | G/A | 57 |
| c.46-698C>A | Nrf2 | Splice acceptor variant | - | - | Cosm6971525 | |||||||
| 25 | 44 | F | c.438 C>T | Keap1- BTB-Domain | Synonymous | p.S146S | 0,83- Pathogenic | Cosm5609243 | Lower Extremity | G/A | G/G | 36 |
| 26 | 60 | M | - | - | - | - | - | - | Lower Extremity | G/G | G/G | 53 |
| 27 | 23 | M | c.360 G>T | Keap1- BTB-Domain | Missense | p.M120I | 0,81- Pathogenic | Novel | Lower Extremity | G/A | G/G | 21 |
aindicates novel variant. Nrf2, nuclear factor erythroid-2-like 2; Keap1, Kelch-like ECH-associated protein 1; M, male; Neh, Nrf2-ECH homology; BTB, broad-complex, tramtrack and bric-a-brac; F, female; UTR, untranslated region; IVR, intervening region.
Results of in silico analysis of Keap1/Nrf2 pathway mutations
The comparison of amino acid sequences affected by the mutations detected in the present study demonstrated that the missense mutations p.P105L, p.G114W, p.R116Q, p.G114W, p.E117D, p.M120I, p.P130T, p.I145N and p.E244Q of Keap1 exhibited amino acid substitutions at a key point that is conserved throughout the evolutionary process across a number of species, including H. sapiens and X. tropicalis (Fig. 3). The missense mutation p.P154S detected in Nrf2 was also located at this point conserved. According to the results of PolyPhen-2 analysis, the majority of missense mutations detected in Keap1 and Nrf2 may be pathogenic as their pathogenicity scores were close to 1. These missense mutations are detailed in Table III.
Figure 3.

Evolutionary conservation analysis of mutated amino acids in (Aa-c) Keap1 and (B) Nrf2. Red boxes and highlighted residues indicate conserved amino acid resiudes. Keap1, Kelch-like ECH-associated protein 1; Nrf2, nuclear factor erythroid-2-like 2.
Results of Nrf2/Keap1 pathway gene expression level analysis
For gene expression analysis, RT-qPCR was used to determine the expression levels of Nrf2 and Keap1 and the reference gene RPLP0 in 27 patients with DVT and 10 healthy controls. The relative mRNA expression levels of Keap1 were significantly higher than that of Nrf2 in patients with DVT (P=0.02; Fig. 4A). Furthermore, Keap1 mRNA expression levels were higher in the patient group compared with those in the control group (P<0.05; Fig. 4B).
Figure 4.
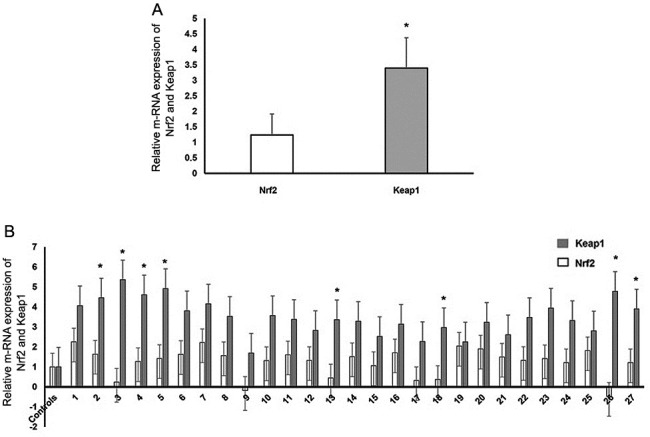
(A) Normalized relative gene expression levels of Nrf2 and Keap1 in patients with deep vein thrombosis. *P<0.05 vs. Nrf2. (B) Nrf2 and Keap1 gene expression levels in patients with deep vein thrombosis normalized to expression levels in healthy subjects. *P<0.05 vs. Controls. Nrf2, nuclear factor erythroid-2-like 2; Keap1, Kelch-like ECH-associated protein 1.
TAC level analysis
TAC levels differed between patients with DVT, with a range of 2.99-1.09 mmol TE/l (Fig. 5). Certain patients (Cases 13, 8, 16, 24 and 12) with splice site mutations demonstrated high TAC values (2.94, 2.66, 2.66, 2.61 and 2.59 mmol TE/l, respectively). However, patients with no mutational change (Cases 15 and 26) exhibited lower TAC levels than the other patients.
Figure 5.

TAC levels in patients with DVT. TAC, total antioxidant capacity; DVT, deep vein thrombosis.
Discussion
Cardiovascular diseases are among the leading causes of global mortality, according to the World Health Organization: Cardiovascular diseases were reported to be responsible for 31% (17.9 million) of global deaths in 2018, and cardiovascular disease-associated mortality is estimated to reach 22.2 million in 2030 (28,36).Venous thromboembolism, including DVT, is the third leading cause of death worldwide associated with cardiovascular disease after coronary artery disease and stroke in 2018(37). In the cardiovascular system, ROS production, vascular tone control, inflammatory responses, cell growth and proliferation, modulation of extracellular matrix production, apoptosis and angiogenesis are important for maintaining the function of endothelial and vascular smooth muscle cells (1,12). Numerous studies have demonstrated that endothelial dysfunction is the first step in the pathogenesis of several cardiovascular diseases (3,28,38). Cellular damage caused by oxidative stress serves a key role in most pathophysiological conditions that occur due to endothelial dysfunction (1,24-26,38).
Numerous antioxidant pathways, including the Nrf2/Keap1 signaling pathway, are involved in cellular redox homeostasis (4,27). Nrf2/Keap1 regulates the expression levels of antioxidants and phase II detoxification enzymes (39-41). Nrf2 is highly expressed in tissues where detoxification reactions take place and is a key regulator of the cellular defense mechanism in several organs, including the brain, lung, bladder, kidney, liver and ovary, as well as macrophages and erythrocytes (4,42). The Nrf2/Keap1 signaling pathway maintains the healthy endothelial phenotype under normal physiological conditions. The endothelium is a type of tissue located at the border between blood and tissues, and is primarily involved in the regulation of vascular tone, thromboresistivity, inflammation of the vascular wall and cellular adhesion (43). Increased ROS production in the endothelium activates Nrf2, which causes increased expression levels of intracellular HO-1, glutathione peroxidase (GPx), GSH, glutamate-cysteine ligase modifier subunit, sulfiredoxin-1, NQO1, protease-activated receptor 4 and oxidative stress-induced growth inhibitor 1 genes in arterial endothelial cells (25).
Studies using animal models have concluded that insufficient levels of Nrf2 in various types of tissue concurrently are associated with susceptibility to a number of diseases, including cardiovascular disease (10,26,42,43). Studies investigating the role of the Nrf2/Keap1 signaling pathway in atherosclerosis, hypertension, myocardial infarction and ischemia have been conducted; however, to the best of our knowledge, there are currently no studies regarding Nrf2/Keap1 signaling in thromboembolism, which is the third leading cause of cardiovascular disease-associated mortality, after coronary artery disease and stroke (25,26,44). In the present study, Sanger DNA sequencing analysis was used to detect mutations/SNPs in the Nrf2/Keap1 signaling pathway genes in 27 patients with DVT. Of 23 mutations detected in the Nrf2/Keap1 signaling pathway in 24 (89%) patients, 11 were missense mutations, five were synonymous nucleotide changes, two were splice site mutations and five were non-coding region nucleotide changes. The present study identified seven novel mutations; 16 mutations had been previously reported. Gene expression level analysis was performed using RT-qPCR. The Keap1 BTB domain is the binding site for the Cullin 3 (Cul3)-E3-dependent ubiquitin ligase complex, which suppresses Nrf2 by promoting Nrf2 ubiquitination and subsequent proteosomal degradation (45,46). Keap1 is known to dimerize via its BTB domain; deletion of the BTB domain from Keap1 prevents binding with Cul3, demonstrating that the BTB domain is required for Keap1-Cul3 binding (46). In the present study, the missense mutations detected in the Keap1 BTB domain may lead to continuous suppression of gene expression levels of Nrf2. Mutations in this domain are associated with ovarian and pancreatic cancer, as well as malignant melanoma (46-48). To the best of our knowledge, however, the association between DVT and mutations in this domain has not yet been investigated. Further studies using larger populations of patients with DVT are required to clarify the association between emerging relapses and Keap1 BTB domain mutations.
In the present study, c.639+8 G>A and c.46-698 C>A splice site mutations were detected in Keap1 and Nrf2 genes, respectively. c.46-698 C>A may cause the absence of Nrf2 expression levels as this mutation is located in the first base of the splice site, which is conserved among species in the evolutionary process. Nrf2 overexpression has been reported to increase the expression levels of ARE-dependent antioxidant enzymes, such as NQO1, GST, HO-1 and GPx, which serve key roles in cellular defense mechanisms against ROS (15,27). Studies using human and animal models have demonstrated that lack of Nrf2 expression levels may be a risk factor in the pathogenesis of numerous disorders, including cardiovascular diseases (10,12,15,47). Nrf2 expression levels in patients with DVT were demonstrated to be lower compared with those of Keap1 in the present study. This may be due to splice site mutations in the Neh2 domain, which is the Keap1 binding site located on Nrf2. This may also be caused by missense mutations on the BTB domain, which allow the formation of the Nrf2 inhibitor complex by binding to the homodimerizable RING box protein 1-bound Cul3 protein on Keap1 (46,48).
In the present study, two patients (Cases 9 and 26) exhibited low Nrf2 expression levels. Case 9 exhibited two missense changes and one synonymous change (p.P130T, p.R116Q and p.R116R, respectively), whereas no changes were detected in Case 26. Furthermore, changes were detected in seven of the eight patients with a history of hypertension. To the best of our knowledge, previous studies have not yet identified the association between blood pressure and the expression of genes involved in the Nrf2/Keap1 signaling pathway; however, Nrf2 may serve a key role in regulating blood pressure (10,12,49). Missense mutations were detected in the Nrf2/Keap1 genes in Cases 27 and 12, who were diagnosed with thrombosis in the upper extremity veins and pulmonary embolism, respectively. A Nrf2 splice site mutation was detected in Case 12. The Neh3, Neh4 and Neh5 domains of Nrf2 are transactivation sites of the Nrf2 molecule. These sites are associated with histone acetyltransferases and bind to the coactivator cAMP-response element binding protein to facilitate Nrf2 transcription (24,50). In the present study, the p.P154S missense mutation was detected on the Neh5 domain of the Nrf2 gene and was classified as benign following functional pathogenicity analysis. Animal models using Nrf2-/- mammals have demonstrated susceptibility to oxidative damage, as Nrf2 serves a key role in the adaptive response of the cell to oxidative stress. A previous study using Nrf2-/- mice demonstrated increased tissue damage and tumorigenesis, severe inflammation and high levels of DNA, lipid and protein oxidation products in experimental animals exposed to chemical or xenobiotic-induced oxidative damage (51). Studies have demonstrated that Nrf2-/- mice are more susceptible not only to cancer but also to asthma, emphysema, pulmonary fibrosis and acute lung injury (46,52,53). In addition, it has been demonstrated that decreased expression levels of Nrf2 are associated with risk of acute lung injury, pulmonary edema and inflammation (20).
Nrf2 expression levels may be controlled by KLF2, which is involved with a different pathway. Krüppel-like factor 2 (KLF2) is a key transcription factor in laminar flow-mediated cytoprotective responses in endothelial cells. Studies have demonstrated that KLF2 transcription is stimulated in endothelium under atheroprotective laminar flow via the MAPK/ERK kinase 5-extracellular signal-regulated protein kinase 5 (ERK5)-MEF2 signaling pathway. ERK5 activation induced by flow may exert cytoprotective effects via Nrf2-ARE-dependent expression of antioxidant genes independently of KLF2(54).
Human Keap1 contains 27 cysteine residues, which convert the protein into a redox sensor for endogenous and environmental oxidative signals and electrophilic reactions (55-57). Under normal physiological conditions, critical cysteine residues (Cys 151, 273 and 288) are present on Keap1; however, in the presence of increased levels of ROS or electrophiles they are oxidized, resulting in the separation of the Keap1 tertiary structure from Nrf2 (19,22,25). The cysteine residues 273, 288 and 297 on the Keap1 IVR domain are important for Nrf2 activation and inhibition (17,19). These cysteine residues serve a key role in altering the conformation of Keap1, which leads to the transcriptional activation of Nrf2 and its targets. In the present study, p.E244Q missense mutation located near these cysteine residues on the IVR domain was identified, with a pathogenic effect score of 0.95. Case 10, the youngest patient in the present study, carried this mutation with the highest number of changes in the Nrf2/Keap1 signaling pathway.
Nrf2 and Keap1 are highly conserved in the evolutionary process. However, cysteine substitutes the amino acid tyrosine located at codon 212 of the Keap1 IVR domain in the sperm whale (P. microcephalus) and in the blind mole rat, which can live >20 years in hypoxic conditions (>3% O2) and whose resistance to cancer has been previously demonstrated (57). This codon change may contribute to the redox sensor property of the Keap1 protein in electrophilic reactions. The cysteine present in this location functions via binding and degrading of Nrf2(58). The presence of this variant in long-lived organisms may affect cellular Nrf2 levels, thus contributing to the emergence of the specific adaptive phenotype by altering gene expression levels of the antioxidant pathway.
DVT arises from complex interactions between genetic and environmental factors. Expression levels of certain genes regulate cellular environmental phenomena, such as redox homeostasis. The Nrf2/Keap1 signaling pathway controls the expression levels of antioxidant enzymes. However, to the best of our knowledge, previous studies have not yet identified an association between Nrf2/Keap1 signaling, antioxidant levels and DVT.
In the present study, antioxidant values of patients with DVT were measured as an indicator of total antioxidant levels. Antioxidant levels were notably higher in patients with splice site mutations, compared with patients that exhibited no mutational change. Patients with splice site mutations exhibited a notable antioxidant defense mechanism, which may be an evolutionary response to avoid the harmful effects of increased levels of free radicals that are generated following oxidative stress. In addition, decreased Nrf2 expression levels were associated with low antioxidant values in the present study. DVT may be associated with oxidative imbalance as the Nrf2 region regulates levels of antioxidant enzymes. Determination of Nrf2/Keap1 and antioxidant levels may present a precursor of DVT that can be used for clinical diagnosis.
The results of the present study indicated that Keap1 missense mutations may be involved in the development of DVT. The amino acid residues affected by these mutations are conserved across species. Furthermore, these mutations may alter the expression levels of genes involved in antioxidant processes, and may also exhibit pathogenic properties, as demonstrated by the functional pathogenic effect analysis.
In summary, 23 changes (11 missense mutations, five synonymous nucleotide changes, two splice site mutations and five non-coding region nucleotide changes) were detected, seven of which were identified for the first time in the present study. However, the present study used a limited sample group. Future investigations using larger samples size are required to clarify the epidemiology of genetic mutations responsible for DVT. The present study identified the frequency and molecular characteristics of mutations in the Nrf2/Keap1 signaling pathway in patients with DVT. In conclusion, as the Nrf2/Keap1 complex serves a key role in preventing oxidative stress-induced injuries, pharmacological agents that stimulate Nrf2 activity may provide a novel approach for the treatment of oxidative stress-associated diseases.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed in the present study are included in the published article.
Authors' contributions
DFAB designed the study. DFAB and SI drafted the manuscript. DFAB, TE, SI and YE performed the experiments. DFAB, SI and TK analyzed the data. All authors participated in the writing and revision of the manuscript. All authors read and approved the final version of the manuscript.
Ethics approval and consent to participate
The present study was approved by the Ethics Committee of Erciyes University (Kayseri, Turkey). All participants provided written informed consent.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Chistiakov DA, Orekhov AN, Bobryshev YV. Endothelial Barrier and Its Abnormalities in Cardiovascular Disease. Front Physiol. 2015;6(365) doi: 10.3389/fphys.2015.00365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Förstermann U. Oxidative stress in vascular disease: Causes, defense mechanisms and potential therapies. Nat Clin Pract Cardiovasc Med. 2008;5:338–349. doi: 10.1038/ncpcardio1211. [DOI] [PubMed] [Google Scholar]
- 3.Loscalzo J. Oxidative stress in endothelial cell dysfunction and thrombosis. Pathophysiol Haemost Thromb. 2002;32:359–360. doi: 10.1159/000073600. [DOI] [PubMed] [Google Scholar]
- 4.Chen B, Lu Y, Chen Y, Cheng J. The role of Nrf2 in oxidative stress-induced endothelial injuries. J Endocrinol. 2015;225:R83–R99. doi: 10.1530/JOE-14-0662. [DOI] [PubMed] [Google Scholar]
- 5.Konukoglu D, Uzun H. Endothelial dysfunction and hypertension. Adv Exp Med Biol. 2017;956:511–540. doi: 10.1007/5584_2016_90. [DOI] [PubMed] [Google Scholar]
- 6.Bengisun U. Basic treatment principles of deep vein thrombosis and pulmonary embolism. TOTBID Derg. 2019;18:505–513. [Google Scholar]
- 7.Kruger PC, Eikelboom JW, Douketis JD, Hankey GJ. Deep vein thrombosis: Update on diagnosis and management. Med J Aust. 2019;210:516–524. doi: 10.5694/mja2.50201. [DOI] [PubMed] [Google Scholar]
- 8.Harrison D, Griendling KK, Landmesser U, Hornig B, Drexler H. Role of oxidative stress in atherosclerosis. Am J Cardiol. 2003;91 (3A):7A–11A. doi: 10.1016/s0002-9149(02)03144-2. [DOI] [PubMed] [Google Scholar]
- 9.Harrison DG, Gongora MC, Guzik TJ, Widder J. Oxidative stress and hypertension. J Am Soc Hypertens. 2007;1:30–44. doi: 10.1016/j.jash.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 10.Howden R. Nrf2 and cardiovascular defense. Oxid Med Cell Longev. 2013;2013(104308) doi: 10.1155/2013/104308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park JG, Oh GT. The role of peroxidases in the pathogenesis of atherosclerosis. BMB Rep. 2011;44:497–505. doi: 10.5483/bmbrep.2011.44.8.497. [DOI] [PubMed] [Google Scholar]
- 12.da Costa RM, Rodrigues D, Pereira CA, Silva JF, Alves JV, Lobato NS, Tostes RC. Nrf2 as a Potential Mediator of Cardiovascular Risk in Metabolic Diseases. Front Pharmacol. 2019;10(382) doi: 10.3389/fphar.2019.00382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Galley JC, Straub AC. Redox control of vascular function. Arterioscler Thromb Vasc Biol. 2017;37:e178–e184. doi: 10.1161/ATVBAHA.117.309945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee JM, Li J, Johnson DA, Stein TD, Kraft AD, Calkins MJ, Jakel RJ, Johnson JA. Nrf2, a multi-organ protector? FASEB J. 2005;19:1061–1066. doi: 10.1096/fj.04-2591hyp. [DOI] [PubMed] [Google Scholar]
- 15.Smith RE, Tran K, Smith CC, McDonald M, Shejwalkar P, Hara K. The Role of the Nrf2/ARE antioxidant system in preventing cardiovascular diseases. Diseases. 2016;4(34) doi: 10.3390/diseases4040034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim J, Cha YN, Surh YJ. A protective role of nuclear factor-erythroid 2-related factor-2 (Nrf2) in inflammatory disorders. Mutat Res. 2010;690:12–23. doi: 10.1016/j.mrfmmm.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 17.Kaspar JW, Niture SK, Jaiswal AK. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic Biol Med. 2009;47:1304–1309. doi: 10.1016/j.freeradbiomed.2009.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kansanen E, Kuosmanen SM, Leinonen H, Levonen AL. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013;1:45–49. doi: 10.1016/j.redox.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baird L, Dinkova-Kostova AT. The cytoprotective role of the Keap1-Nrf2 pathway. Arch Toxicol. 2011;85:241–272. doi: 10.1007/s00204-011-0674-5. [DOI] [PubMed] [Google Scholar]
- 20.Hua CC, Chang LC, Tseng JC, Chu CM, Liu YC, Shieh WB. Functional haplotypes in the promoter region of transcription factor Nrf2 in chronic obstructive pulmonary disease. Dis Markers. 2010;28:185–193. doi: 10.3233/DMA-2010-0700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 22.Robledinos-Antón N, Fernández-Ginés R, Manda G, Cuadrado A. Activators and Inhibitors of NRF2: A Review of Their Potential for Clinical Development. Oxid Med Cell Longev. 2019;2019(9372182) doi: 10.1155/2019/9372182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tebay LE, Robertson H, Durant ST, Vitale SR, Penning TM, Dinkova-Kostova AT, Hayes JD. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic Biol Med. 2015;88 (Pt B):108–146. doi: 10.1016/j.freeradbiomed.2015.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tu W, Wang H, Li S, Liu Q, Sha H. The anti-inflammatory and anti-oxidant mechanisms of the Keap1/Nrf2/ARE signaling pathway in chronic diseases. Aging Dis. 2019;10:637–651. doi: 10.14336/AD.2018.0513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Satta S, Mahmoud AM, Wilkinson FL, Yvonne Alexander M, White SJ. The role of Nrf2 in cardiovascular function and disease. Oxid Med Cell Longev. 2017;2017(9237263) doi: 10.1155/2017/9237263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sarutipaiboon I, Settasatian N, Komanasin N, Kukongwiriyapan U, Sawanyawisuth K, Intharaphet P, Senthong V, Settasatian C. Association of Genetic Variations in NRF2, NQO1, HMOX1, and MT with severity of coronary artery disease and related risk factors. Cardiovasc Toxicol. 2020;20:176–189. doi: 10.1007/s12012-019-09544-7. [DOI] [PubMed] [Google Scholar]
- 27.Korytina GF, Akhmadishina LZ, Aznabaeva YG, Kochetova OV, Zagidullin NS, Kzhyshkowska JG, Zagidullin SZ, Viktorova TV. Associations of the NRF2/KEAP1 pathway and antioxidant defense gene polymorphisms with chronic obstructive pulmonary disease. Gene. 2019;692:102–112. doi: 10.1016/j.gene.2018.12.061. [DOI] [PubMed] [Google Scholar]
- 28.Barančík M, Grešová L, Barteková M, Dovinová I. Nrf2 as a key player of redox regulation in cardiovascular diseases. Physiol Res. 2016;65 (Suppl 1):S1–S10. doi: 10.33549/physiolres.933403. [DOI] [PubMed] [Google Scholar]
- 29.Cunningham F, Achuthan P, Akanni W, Allen J, Amode MR, Armean IM, Bennett R, Bhai J, Billis K, Boddu S, et al. Ensembl 2019. Nucleic Acids Res. 2019;47 (D1):D745–D751. doi: 10.1093/nar/gky1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. 2013;7:7–20. doi: 10.1002/0471142905.hg0720s76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, Boutselakis H, Cole CG, Creatore C, Dawson E, et al. COSMIC: The catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019;47 (D1):D941–D947. doi: 10.1093/nar/gky1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, Hussain M, Phillips AD, Cooper DN. The Human Gene Mutation Database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet. 2017;136:665–677. doi: 10.1007/s00439-017-1779-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG. Primer3--new capabilities and interfaces. Nucleic Acids Res. 2012;40(e115) doi: 10.1093/nar/gks596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29(e45) doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 36. World Health Organization (WHO): Cardiovascular diseases (CVDs). WHO, Geneva, 2017. https://www.who.int/en/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds). Accessed May 17, 2017. [Google Scholar]
- 37.Raskob GE, Angchaisuksiri P, Blanco AN, Buller H, Gallus A, Hunt BJ, Hylek EM, Kakkar A, Konstantinides SV, McCumber M, et al. ISTH Steering Committee for World Thrombosis Day: Thrombosis: A major contributor to global disease burden. Thromb Res. 2014;134:931–938. doi: 10.1160/TH14-08-0671. [DOI] [PubMed] [Google Scholar]
- 38.Heitzer T, Schlinzig T, Krohn K, Meinertz T, Münzel T. Endothelial dysfunction, oxidative stress, and risk of cardiovascular events in patients with coronary artery disease. Circulation. 2001;104:2673–2678. doi: 10.1161/hc4601.099485. [DOI] [PubMed] [Google Scholar]
- 39.Suzuki T, Yamamoto M. Molecular basis of the Keap1-Nrf2 system. Free Radic Biol Med. 2015;88 (Pt B):93–100. doi: 10.1016/j.freeradbiomed.2015.06.006. [DOI] [PubMed] [Google Scholar]
- 40.Barrera-Rodríguez R. Importance of the Keap1-Nrf2 pathway in NSCLC: Is it a possible biomarker? Biomed Rep. 2018;9:375–382. doi: 10.3892/br.2018.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li C, Cheng L, Wu H, He P, Zhang Y, Yang Y, Chen J, Chen M. Activation of the KEAP1-NRF2-ARE signaling pathway reduces oxidative stress in Hep2 cells. Mol Med Rep. 2018;18:2541–2550. doi: 10.3892/mmr.2018.9288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Surh YJ, Kundu JK, Na HK. Nrf2 as a master redox switch in turning on the cellular signaling involved in the induction of cytoprotective genes by some chemopreventive phytochemicals. Planta Med. 2008;74:1526–1539. doi: 10.1055/s-0028-1088302. [DOI] [PubMed] [Google Scholar]
- 43.Vanhoutte PM, Shimokawa H, Feletou M, Tang EH. Endothelial dysfunction and vascular disease - a 30th anniversary update. Acta Physiol (Oxf) 2017;219:22–96. doi: 10.1111/apha.12646. [DOI] [PubMed] [Google Scholar]
- 44.Rangasamy T, Guo J, Mitzner WA, Roman J, Singh A, Fryer AD, Yamamoto M, Kensler TW, Tuder RM, Georas SN, et al. Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J Exp Med. 2005;202:47–59. doi: 10.1084/jem.20050538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kobayashi E, Suzuki T, Yamamoto M. Roles nrf2 plays in myeloid cells and related disorders. Oxid Med Cell Longev. 2013;2013(529219) doi: 10.1155/2013/529219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pandey P, Singh AK, Singh M, Tewari M, Shukla HS, Gambhir IS. The see-saw of Keap1-Nrf2 pathway in cancer. Crit Rev Oncol Hematol. 2017;116:89–98. doi: 10.1016/j.critrevonc.2017.02.006. [DOI] [PubMed] [Google Scholar]
- 47.Konstantinopoulos PA, Spentzos D, Fountzilas E, Francoeur N, Sanisetty S, Grammatikos AP, Hecht JL, Cannistra SA. Keap1 mutations and Nrf2 pathway activation in epithelial ovarian cancer. Cancer Res. 2011;71:5081–5089. doi: 10.1158/0008-5472.CAN-10-4668. [DOI] [PubMed] [Google Scholar]
- 48.Qin JJ, Cheng XD, Zhang J, Zhang WD. Dual roles and therapeutic potential of Keap1-Nrf2 pathway in pancreatic cancer: A systematic review. Cell Commun Signal. 2019;17(121) doi: 10.1186/s12964-019-0435-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Silva-Palacios A, Königsberg M, Zazueta C. Nrf2 signaling and redox homeostasis in the aging heart: A potential target to prevent cardiovascular diseases? Ageing Res Rev. 2016;26:81–95. doi: 10.1016/j.arr.2015.12.005. [DOI] [PubMed] [Google Scholar]
- 50.Nioi P, Nguyen T, Sherratt PJ, Pickett CB. The carboxy-terminal Neh3 domain of Nrf2 is required for transcriptional activation. Mol Cell Biol. 2005;25:10895–10906. doi: 10.1128/MCB.25.24.10895-10906.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ramos-Gomez M, Kwak MK, Dolan PM, Itoh K, Yamamoto M, Talalay P, Kensler TW. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc Natl Acad Sci USA. 2001;98:3410–3415. doi: 10.1073/pnas.051618798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aoki Y, Sato H, Nishimura N, Takahashi S, Itoh K, Yamamoto M. Accelerated DNA adduct formation in the lung of the Nrf2 knockout mouse exposed to diesel exhaust. Toxicol Appl Pharmacol. 2001;173:154–160. doi: 10.1006/taap.2001.9176. [DOI] [PubMed] [Google Scholar]
- 53.Enomoto A, Itoh K, Nagayoshi E, Haruta J, Kimura T, O'Connor T, Harada T, Yamamoto M. High sensitivity of Nrf2 knockout mice to acetaminophen hepatotoxicity associated with decreased expression of ARE-regulated drug metabolizing enzymes and antioxidant genes. Toxicol Sci. 2001;59:169–177. doi: 10.1093/toxsci/59.1.169. [DOI] [PubMed] [Google Scholar]
- 54.Kim M, Kim S, Lim JH, Lee C, Choi HC, Woo CH. Laminar flow activation of ERK5 protein in vascular endothelium leads to atheroprotective effect via NF-E2-related factor 2 (Nrf2) activation. J Biol Chem. 2012;287:40722–40731. doi: 10.1074/jbc.M112.381509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sihvola V, Levonen AL. Keap1 as the redox sensor of the antioxidant response. Arch Biochem Biophys. 2017;617:94–100. doi: 10.1016/j.abb.2016.10.010. [DOI] [PubMed] [Google Scholar]
- 56.Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y, Yamamoto M, Talalay P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci USA. 2002;99:11908–11913. doi: 10.1073/pnas.172398899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schmidt H, Hangmann J, Shams I, Avivi A, Hankeln T. Molecular evolution of antioxidant and hypoxia response in long-lived, cancer-resistant blind mole rats: The Nrf2-Keap1 pathway. Gene. 2016;577:293–298. doi: 10.1016/j.gene.2015.11.038. [DOI] [PubMed] [Google Scholar]
- 58.Taguchi K, Motohashi H, Yamamoto M. Molecular mechanisms of the Keap1–Nrf2 pathway in stress response and cancer evolution. Genes Cells. 2011;16:123–140. doi: 10.1111/j.1365-2443.2010.01473.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed in the present study are included in the published article.