

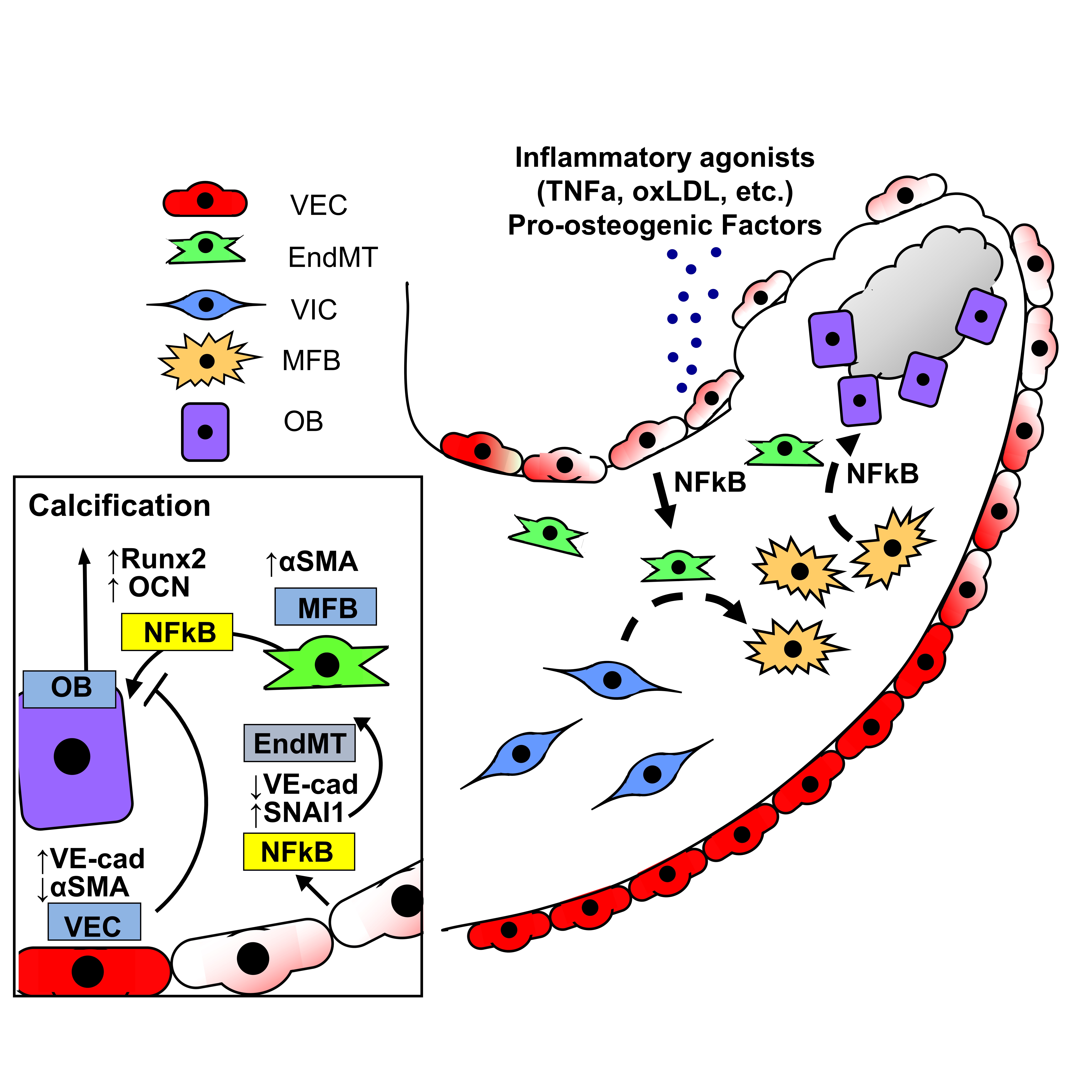
Abstract
Objective:
While often studied independently, little is known how aortic valve endothelial cells (VEC) and valve interstitial cells (VIC) interact collaborate to maintain tissue homeostasis or drive valve calcific pathogenesis. Inflammatory signaling is a recognized initiator of valve calcification, but the cell-type specific downstream mechanisms have not been elucidated. In this study, we test how inflammatory signaling via NFκB activity coordinates unique and shared mechanisms of VEC and VIC differentiation during calcific progression.
Approach and Results:
Activated NFκB was present throughout the CAVD process in both endothelial and interstitial cell populations in an established mouse model of hypercholesterolemia-induced CAVD and in human CAVD. NFκB activity induces EndMT in 3D cultured aortic VEC and subsequent osteogenic calcification of transformed cells. Similarly, 3D cultured VIC calcified via NFκB mediated osteogenic differentiation. NFκB mediated EndMT was directly demonstrated in vivo during CAVD via genetic lineage tracking. Genetic deletion of NFκB in either whole valves or valve endothelium only was sufficient to prevent valve specific molecular and cellular mechanisms of CAVD in vivo despite the persistence of a CAVD inducing environment.
Conclusion:
Our results identify NFκB signaling as an essential molecular regulator for both valve endothelial and interstitial participation in CAVD pathogenesis. Direct demonstration of VEC EndMT transmigration in vivo during CAVD highlights a new cellular population for further investigation in CAVD morbidity. The efficacy of valve-specific NFκB modulation in inhibiting hypercholesterolemic CAVD suggests potential benefits of multi-cell type integrated investigation for biological therapeutic development and evaluation for CAVD.
Keywords: Nuclear factor-kappa B, osteogenesis, heart valve, hypercholesterolemia, endothelial cell
Introduction
Calcific aortic valve disease (CAVD) is a serious and increasingly prevalent pathology, estimated to affect over 500M people worldwide by 20501. CAVD is well recognized as an actively regulated cellular process2 characterized by progressive tissue fibrosis and obstructive mineralization causing aortic stenosis, hypertension and congestive heart failure if not treated. CAVD often has a multi-year progression that affords biological intervention, but clinical trials employing vascular biology-derived biomarkers and therapeutics (e.g. statins) in CAVD have been unsuccessful3–5. There remains a significant need to establish valve specific biological mechanisms regulating tissue homeostasis and calcific degeneration. Much of what is known about CAVD pathogenesis focuses exclusively on the valvular interstitial cell (VIC). The aortic valve is also populated by a unique endothelial (VEC) phenotype, which must collaborate together with VIC to maintain phenotype quiescence within the demanding 3D microenvironment in which they reside6. Very little is known about how each valve cell types engages valvular disease risk factors, nor whether they progress mechanistically along the same molecular/cellular pathway. Such information is critical to develop and evaluate valve specific therapeutic strategies.
Longitudinal PET/CT imaging indicates that inflammation of the valve both precedes calcification7 and progresses proportionally with severity 8. Inflammation has multifaceted origins and signaling pathways, and is also caused by many other conditions that are often shared by patients with CAVD (e.g. renal disease, oxidative stress, hypertension) 9. Recent work from our lab has shown the valve endothelium to be a novel source of oxidative stress driving myofibroblastic activation10, and that inflammatory signaling can drive reactivation of embryonic-like endothelial to mesenchymal transformation (EndMT) as a potential mechanism of early valve dysfunction11. However, it is unknown how specific inflammatory signaling pathways regulate unique and coordinated activities these in two cell populations toward CAVD initiation and/or downstream calcific progression.
Nuclear Factor kappa-light-chain enhancer of activated B cells (NFκB) is a prominent transcription factor family that is involved in many cardiovascular pathologies 12,13. Activated NFκB subunits, including p65/RelA, translocate to the nucleus and directly activate gene transcription controlling cell proliferation, survival, immune, and/or differentiation responses 14. Conversely, NFκB can be sequestered in the cytoplasm via binding to IkBs such as IκBa. The IKK signaling complex is a key convergence site for multiple signaling pathways leading to NFκB activation, with IKK-β participating in the primary pathway of proinflammatory induction.12 Recent evidence from our lab implicates TNFα inflammatory signaling in the induction of endothelial to mesenchymal transformation (EndMT) in aortic valve endothelial cells in vitro11,15. TNFα has also been shown to accelerate VIC calcification16, with NFκB has also been associated with osteoclast and bone remodeling activity17. Taken together, this evidence motivates the hypothesis that NFκB activity coordinates both aortic VEC and VIC molecular and cellular activity throughout the continuum of calcific degeneration, and as such could be a useful valve specific molecular target for disrupting CAVD progression.
In this study, we identify how RelA specific NFκB activity drives VEC and VIC NFκB pathogenic mechanisms in 3D culture. We further employ valve lineage specific Cre mice to test the role of altered p65/RelA NFκB activity in valve cells in normal or established diet-induced hypercholesterolemic CAVD animal model. We additionally employ lineage tracing to map the fate of valve endothelial cells in these conditions. Our findings establish that activation of NFκB is sufficient to induce calcific degenerative processes in both VEC and VIC in vitro and in vivo. Further, genetic inhibition of NFκB activation in valves in vivo mitigates aortic valve specific calcific progression in established CAVD environments. These findings establish that CAVD involves multicellular engagement through diverse cellular events that are controlled at least in part by a shared molecular pathway via p65/RelA.
Methods
Mouse experiments were performed according to the guidelines of the National Institute of Health and the protocols approved by the Institutional Animal Care and Use Committee of Cornell University and Albert Einstein College of Medicine.
Transgenic mouse model
The VEC-specific Nfatc1-enhancer Cre (Nfatc1enCre) mouse line expresses the Cre enzyme in a subset of valve endocardial populations which do not give rise to VIC during embryonic valvulogenesis, thus retaining a VEC phenotype18. The Nfatc1-knock-in Cre (Nfatc1Cre) mouse line expresses Cre in valve endocardium which gives rise to both VEC and VIC populations during embryonic valvulogenesis19.
The Karin lab (University of California, San Diego) kindly provided a mouse with IKKβ-floxed allele20. Cre-mediated excision of IKKβ would thus prevent canonical NFκB p65/RelA activation.
The mice were crossed with LDLR−/− mice from Jackson Labs (#2270). Mice were fed a high fat diet (Harlan Teklad #TD88137, 42% of calories from fat, 0.25% cholesterol) from four to twenty weeks of age, utilized in combination of the homozygous LDLR-knockout background to establish aortic valve disease, as motivated previously21. For fate-tracking, mice were crossed with Rosa26 reporter mice (Jax #003474) expressing floxed-lacZ reporter gene in areas of Cre-activity.
All mice were on a C57BL6J substrain background, backcrossed for genetic homogeneity for > 5 generations. The resulting animals had genomic deletion of the LDL receptor, with or without whole valve or VEC-specific inactivation of the IKKβ gene, and expression of the lacZ reporter in areas of Cre activity.
We utilized these lines to establish the following mouse groups:
wild-type control (LDLR+/+;IKKβ+/+;Nfatc1+/+ with control diet, N=8), CAVD (LDLR−/−, IKKβ+/+;Nfatc1+/+ with high-fat diet, N=17), and CAVD+IKKβvalveKO (LDLR−/−;IKKβf/f;Nfatc1Cre with high-fat diet, N=9), and CAVD+IKKβvecKO(LDLR−/−;IKKβf/f;Nfatc1enCre with high-fat diet, N=15).
Echocardiography
AV function was assessed at five months of age using echocardiography according to established procedures. Mice were anesthetized using inhaled 1.8% isoflurane and cradled in the left lateral recumbent position. Abdominal hair was removed using a chemical depilatory cream and warmed ultrasound transmission gel was applied to the anterior thorax. An ultra-high frequency linear array probe (MS550D 22–55 MHz transducer, Vevo®) was applied in the long-axis position to the chest. The imaging probe was coupled to the Vevo® 2100 system (VisualSonics). Aortic valve leaflet dynamics and ventricular dilation were imaged using the ECG-Gated Kilohertz Visualization (EKV) software (VisualSonics), which captures 5,000 fps in order to create high-resolution videos of cardiac movement. M-mode was used to measure diameter of the aortic root, just above the aortic sinus. Transvalvular velocity and pressure gradients were captured using the pulsed-wave Doppler mode and manual tracing of > 3 velocity time intervals on the resulting plots.
Human Aortic Valves
Non-calcified human aortic valves were obtained from adult (>18 years old) patients undergoing heart transplant for non-valve related diseases at Children’s Hospital in Seattle WA (5 participants total). These valves were free from valvular dysfunction including endothelial activation commonly present in aged “non-diseased” heart transplant patients as previously described10,22. Sclerotic and calcified human valves were obtained from patients undergoing valve replacement surgery at Robert Packer Hospital in Sayre, PA. Patient age range was 65–90 years, with mean age of 76.2 years (21 participants total). Valves were categorized as either sclerotic or calcified based on macroscopic assessment and histological analysis (e.g. Movat Pentachrome, see Supplementary Figures SIA,B). The Institutional Review Board at Seattle Children’s Hospital, Guthrie Institutional Review Board at Robert Packer Hospital, and the Institutional Review Board for Human Participants at Cornell University approved all procedures (IRB#0908–24, “Gene expression and phenotypic changes in stenotic aortic valves”). Written informed consent was obtained from all participants.
Histology
Mouse hearts and human aortic valves were isolated, perfused, fixed, embedded in paraffin, sectioned, and mounted. Slides were deparaffinized through xylene and graded ethanol and rinsed in deionized water. For Alizarin Red S staining, cells were deparaffinized, hydrated, incubated in ARS stain for 2 minutes, rinsed in xylene and xylene-acetone (1 min each), dehydrated in 3 changes of xylene (1 min each), and mounted. Analysis of ARS was done in ImageJ, using thresholding to identify boundaries of the leaflet section. Leaflet section with background subtracted was measured for area and integrated density of ARS stain. Similar deparaffinization and hydration was performed for von Kossa, followed by a 20 minute incubation in 1% aqueous silver nitrate under UV light. Slides were rinsed in water followed by 5% sodium thiosulfate (5 min), rinsed, and counter-stained with 0.1% nuclear fast red (5 min). Dehydration and mounting performed as above. Slides were imaged and thresholded in ImageJ so that only black-brown mineral deposits were selected for analysis. Mineral deposit area was measured and normalized to valve leaflet area. Russell-Movat pentachrome stain was performed according to manufacturer’s instructions, with the following adjustments: 30 seconds in 2% ferric chloride, 20 minutes in 1% alcian blue, and 1 minute in each change of 5% phosphotungstic acid. A color deconvolution algorithm was used to separate individual dyes from the Russell-Movat stained sections into single-channel images. MATLAB© was used to find the integrated density of each single-channel image. Integrated density of each component (GAG = blue, collagen = yellow, elastin = black) was normalized to the area of the valve leaflet section being examined. The resulting output was divided by the average integrated density of each dye in control samples, producing fold change expression of GAG, elastin, and collagen relative to control.
Primary aortic valve cell isolation and culture
Porcine valves are a widely used analog for human adult valve endothelial and interstitial cells. Unlike mice, pigs develop atherosclerosis and valvular lesions without intervention, similar to humans. Large-scale culture of porcine valve cell populations is feasible due to ample tissue size/cell dissociation yields. Valvular endothelial cells (VEC) and valvular interstitial cells (VIC) were harvested from porcine aortic valves (Shirk Meats, Dundee, NY) following collagenase digestion and screened for phenotype and purity using qRT-PCR and immunofluorescence, as demonstrated previously23. Only VEC cultures with consistent CD31 and VE-cadherin expression, cobblestone morphology, and non-detectable αSMA expression were used. αSMA levels were measured via real-time PCR (> 37 cycle threshold), western blot, and immunofluorescence.
All reagents were obtained from Sigma Aldrich, St. Louis, MO, unless otherwise noted. PAVEC were cultured in flasks coated with 50 ug/ml rat-tail collagen I (BD Biosciences, San Jose, CA) at 37C and 5% CO2 in DMEM supplemented with 10% FBS (Invitrogen, Grand Island, NY), 1% penicillin-streptomycin (P/S) (Invitrogen, Grand Island, NY), and 50 U/ml heparin sodium salt. PAVIC were cultured in tissue-culture treated flasks at 37C˚ and 5% CO2 in DMEM supplemented with 10% FBS, 1% P/S. Cells were used at passage 3–5 and 3–6 for PAVEC and PAVIC, respectively.
3D hydrogel culture model preparation
Cellularized type II collagen hydrogels were prepared as previously described24. Briefly, PAVEC were topically seeded at 50,000 cell/cm2 density while PAVIC were encapsulated within the neutralized collagen gel suspension (2 mg/ml) at 1,000,000 cells/ml density. Constructs were allowed to gel for 1 hr at 37C, 5% CO2, and then cultured overnight in their respective basal-medium conditions. For osteogenic differentiation conditions, basal-medium was supplemented with 10mM β-glycerophosphate, 50 ug/ml L-ascorbic acid, and 100nM Dexamethasone, as previously described25. For exogenous TNFα administration, recombinant human TNFα was added to appropriate media condition prior to administration at 30 ng/ml.
Plasmid Transfection
Cell transfections were conducted using plasmid delivery to VEC or VIC using the 100μl Neon Electroporation System (Life Technologies) according to the manufacturer’s instructions. GFP-RelA and empty vector were a gift from Warner Greene (Addgene plasmid #23255). pBabe-GFP-IKBα-mut (super repressor) and empty vector were a gift from William Hahn (Addgene plasmid #15264). Electroporation conditions were 1200V, 20ms, 2 pulses. 10μg of plasmid construct per 1,000,000 cells were used. Cells were trypsinized, electroporated with corresponding plasmid construct, and cultured for 24 hours in 5% serum, antibiotic-free DMEM before being seeded into or onto hydrogels.
Hydrogel Calcification Analysis
To quantify calcified matrix developed in different experimental conditions, an Alizarin Red absorbance assay was implemented. Collagen hydrogels were fixed with paraformaldehyde and then incubated with 40 nmol/L Alizarin Red S (ARS) dye. This dye binds to calcium crystals in cells or matrix fibers, revealing a red color. Unbound solution was washed out overnight under gentle rocking. Bound ARS dye was then released from the gels using 10% acetic acid, followed by neutralization with 10% ammonium hydroxide. The concentration of dye in solution was then quantified using absorbance spectroscopy at 405-nm wavelength. Samples absorbance readings were normalized to basal growth medium (GM) culture condition.
Serum Cholesterol Analysis
Mouse serum cholesterol quantification was performed using the Cholesterol Quantitation Kit as per manufacturer’s instructions (Sigma Aldrich, St. Louis, MO). Murine serum was obtained via right ventricle puncture and blood draw at time of mouse euthanasia, and isolated using BD vacutainer blood collection tubes(BD, Franklin Lakes, NJ).
Quantitative Real-Time Polymerase Chain Reaction
Total RNA was extracted using a RNeasy total RNA purification kit (Qiagen, Valencia, CA) and RNA was reverse transcribed to cDNA using the iScript™ cDNA synthesis kit (Biorad, Hercules, CA). Quantitative real-time PCR was performed on all samples using SYBR Green PCR master mix (Applied Biosystems, Foster City, CA), and a CFX96 or MiniOpticon Real-Time PCR Detection System (Bio-Rad, Hercules, CA). Samples amplifying at > 37 cycles were considered non-detectable.
| Gene | Forward Primer | Reverse Primer | Ascension Number |
|---|---|---|---|
| 18S | TAGAGGGACAAGTGGCGT | AATGGGGTTCAACGGGTT | NR_046261.1 |
| ACTA2 (αSMA) | CAGCCAGGATGTGTGAAGAA | TCACCCCCTGATGTCTAGGA | NM_001164650.1 |
| Osteocalcin | CTCCAGCCACAACATCCTTT | TGGCCTCCAGCACTGTTTAT | NM_001164004.1 |
| PECAM1 | ATCTGCATCTCGTGGGAAGT | GAGCTGAAGTGTCAGCAGGA | NM_213907.1 |
| RUNX2 | GCACTACCCAGCCACCTTTA | TATGGAGTGCTGCTGGTCTG | XM_005666074.3 |
| SNAI1 | GCCCAACTACAGCGAGCTAC | CCAGGAGAGAGTCCCAGATG | XM_021077961.1 |
| CDH5 (VE-Cadherin) | CGTGGTGGAAACACAAGATG | TGTGTACCTGGTCTGGGTGA | NM_001001649.2 |
Immunofluorescence Staining and Analysis
Human aortic valves and whole mouse hearts were fixed in 4% paraformaldehyde overnight at 4°C, and processed for paraffin sectioning. Slides were deparaffinized through xylene and graded ethanol and rinsed in deionized water. Heat mediated antigen retrieval using Citrate buffer, pH 6.0 (Electron Microscopy Sciences, Hatfield, PA), was performed using a commercial pressure cooker for 2 minutes. Tissue sections were subsequently permeabilized with 0.2% Triton-X (VWR International, West Chester, PA).
For in vitro cultures, intact collagen gels were rinsed twice in PBS and fixed in 4% paraformaldehyde for one hour at 25C. Samples were rinsed three times on a rocker with PBS for 15 minutes each, permeabilized with 0.2% Triton-X (VWR International, West Chester, PA) for ten minutes on rocker, and blocked in 10% goat serum for one hour at 25C.
The following antibodies were used at the provided dilution concentrations/ratios:
Human Immunofluorescence (IHC-Paraffin sections)
| Antibody | Manufacturer/Catalog # | Concentration/Dilution Ratio |
|---|---|---|
| mouse anti-human CD31 | Cell Signaling Technology: 3528 | 1:200 |
| rabbit anti-human NFkB p65 | Cell Signaling Technology: 8242 | 1:400 |
Mouse Immunofluorescence (IHC-Paraffin sections)
| Antibody | Manufacturer/Catalog # | Concentration/Dilution Ratio |
|---|---|---|
| mouse anti-human αSMA | Abcam: ab7817 | 10 ug/ml (1:100) |
| rabbit anti-mouse VCAM-1 | Cell Signaling Technology: 32653 | 1:100 |
| rabbit anti-human NFkB p65 | Cell Signaling Technology: 8242 | 1:400 |
| rabbit anti-human ki67 | Abcam: ab15580 | 10 ug/ml (1:100) |
| rabbit anti-mouse CD31 | Abcam: ab28364 | 1:50 |
| rabbit anti-human IKKβ | Abcam: ab124957 | 10 ug/ml (1:100) |
Porcine Immunofluorescence (Whole-mount IF)
| Antibody | Catalog # | Concentration/Dilution Ratio |
|---|---|---|
| mouse anti-human αSMA | Abcam: ab7817 | 10 ug/ml (1:100) |
| rabbit anti-human VE-Cadherin | Cell Signaling Technology: 2500 | 1:200 |
| mouse anti-human Runx2 | Abcam: ab76956 | 10 ug/ml (1:100) |
| mouse-anti human Osteocalcin | Abcam: ab13420 | 10 ug/ml (1:100) |
| rabbit anti-human NFkB p65 | Cell Signaling Technology: 8242 | 1:100 |
Samples were incubated overnight at 4°C, rinsed three times in PBS on rocker for 15 minutes each, and species-specific secondary antibodies raised in goat conjugated to Alexa Fluor® 488 or 568 fluorophores were added at 4 ug/ml (1:500) dilution ratio (Invitrogen, Grand Island, NY).
Samples were incubated for two hours at room temperature, rinsed, then incubated for an additional 30 minutes with Draq5 nuclear stain (Enzo Life Sciences AG, Lausen, Switzerland, 1:1000). Samples were rinsed thoroughly in PBS three times on rocker for 15 minutes each, then imaged using a Zeiss 710 (Thornwood, NY) laser scanning confocal microscope.
TUNEL assay (Invitrogen, Grand Island, NY was performed according to manufacturer’s instructions to label apoptosis, necrosis, and cell nuclei). Cell proliferation was assessed using ki67 immunostaining (1:100 rabbit anti-human (Abcam)). Tissue sections were imaged with a Zeiss 710 laser scanning confocal microscope, analyzed, and quantified using ImageJ. Number of cell nuclei and cells positive for apoptosis/proliferation per section were counted using particle analysis. Percent of cells positive for apoptosis (TUNEL stain) or proliferation (Ki67) were presented.
Nuclear Integrated Density analysis was performed using ImageJ. Slides were imaged and thresholded using IgG isotype controls so that only positive protein-expression regions were selected for analysis. Images were segmented using DAPI stained nuclear positive regions to create an ROI for analysis; MATLAB© was used to find the integrated density for protein expression within the nuclear ROI map. Nuclear Integrated Density was then normalized to cell count within the field of view.
Statistical Analysis
Unless stated otherwise, one-way ANOVA in conjunction with Tukey’s honestly significant difference (HSD) post-hoc test for parametric data sets; non-parametric analysis through Throughout the Study, experimental groups were compared to one another with one-way ANOVA in conjunction with Tukey’s honestly significant difference (HSD) post-hoc test; adjusted P-values less than or equal to 0.05 were considered statistically significant, data are presented as mean ± SEM, unless stated otherwise.
Results
NFκB activity increases in a layer specific manner in proportion to CAVD severity in Human aortic valves
We first quantified changes in NFκB activity in relation to calcific aortic valve pathology and human disease progression. Non-calcified human aortic valve leaflets were thin, exhibited intact and proper tri-layer morphology, and contiguous endothelialization (Suppl. Fig. S1A). Diseased human valves, in contrast, were thickened, exhibited disrupted matrix architecture, and collagen-glycosaminoglycan rich and/or fibrotic-calcified nodule presentation (Suppl. Fig. S1A, S1C). Sclerotic valve leaflets were associated with greater collagen-glycosaminoglycan rich lesions, while calcified valves had greater fibrotic/calcified lesions with greater matrix disruption (Supplementary Figure S1A). Corresponding sections were co-stained for CD31 and NFκB p65 to indicate inflammatory signaling activity in relation to the VEC spatial localization (Figure 1A). In non-calcified valves, nuclear p65 signal was found to be elevated within the fibrosa region, whereas cellular expression within the ventricularis and fibrosa were not statistically different, as assed by normalized nuclear co-localized protein signal integrated density (Figure 1B). In sclerotic valves, cells of the ventricularis region demonstrated lower p65 expression compared to that of the spongiosa or fibrosa. p65 expression was highly colocalized with CD31, consistent with the endothelium undergoing inflammatory dysfunction (Figure 1A). In some of the sclerotic valve sections, we further observed signs of neo-vessel formation to occur within the intimal space (Figure 1A, asterisks), as well as CD31-positive cells independent of neo-vessel formation (Figure 1A, yellow arrows). Assessment of p65 nuclear integrated density indicated the ventricularis of sclerotic valves as having the lowest nuclear colocalized 65 signal as compared to either the spongiosa or fibrosa regions (Figure 1B). Within calcified valves however, this p65 presentation and nuclear localization was more pronounced within the sub-endothelial, interstitial populations (Figure 1A, arrows). Nuclear p65 expression was greater in calcified valves compared to the sclerotic valves regardless of valvular regions (Figure 1B). These results determine that p65 expression and nuclear colocalization in aortic valves increase with calcific disease severity. Furthermore, nuclear active p65 is fibrosa side-specific, but develops significant ventricularis involvement during late stage CAVD.
Figure 1.
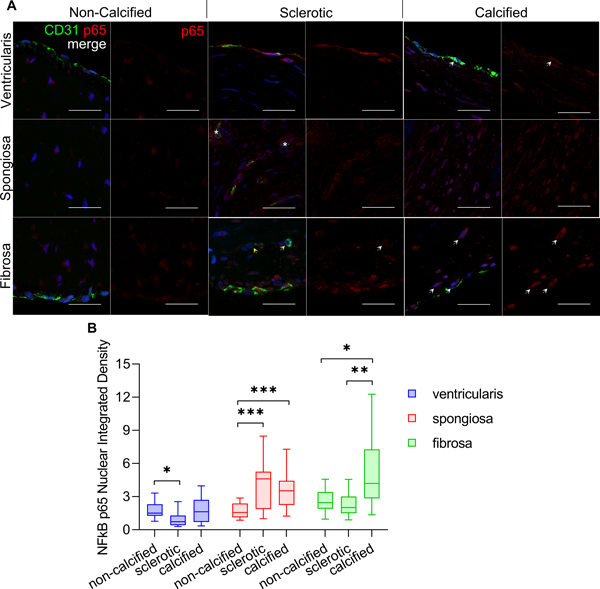
A. Representative images of CD31 (green) and NFkB p65 (red) co-staining of non-calcified, sclerotic, and calcified/stenosed human aortic valves (horizontal axis). Panels indicate expression at ventricularis, spongiosa, and fibrosa regions (vertical axis). White arrows indicate nuclear p65 expression. Asterisks indicate neo-vessel formation in intimal region. Yellow arrows indicate CD31 positive cells in the sub-endothelial region, independent of neo-vessel formation. Scale bar is 100 um. B. Nuclear p65 integrated density, normalized to the sclerotic valve ventricularis expression. Statistical significance indicated comparing different patient groupings for the same valvular-region (e.g. ventricularis of non-calcified vs. sclerotic vs. calcified valves), *: p < 0.05; **: p < 0.005, ***: p < 0.0005).
Valve-specific IKKβ inactivation prevents p65/RelA NFκB signal transduction without perturbing postnatal aortic valve structure or function
To ascertain the role of NFκB signaling contributing to aortic valve homeostasis or CAVD pathogenesis, we first inactivated NFκB by the deletion of IKKβ in VEC and VIC using the valve specific NFATc1 enhancer Cre (Nfatc1Cre) mouse19. We further crossed this mouse with the established hypercholesterolemia-mediated CAVD risk model background (LDLr−/−), which then progressively develops CAVD when fed a Western diet21. We determined that valve specific deletion of IKKβ (Supplementary Fig. SIIA) after embryonic valvulogenesis did not affect heart morphogenesis in age-matched adult mice without a CAVD background (Supplementary Fig. SIIB). Aortic valve peak velocity, peak transvalvular gradient, and mean transvalvular gradient were not affected by IKKβ deletion (IKKvalveKO, thereafter) (Supplementary Fig. SIIC,D,E). Neither aortic root diameter nor valve leaflet thickness significantly differed from their wild-type counterparts (Supplementary Fig. SIIF,G). Cytosolic p65 was expressed primarily in the endothelium with minimal nuclear activation (Figure 2A).
Figure 2.
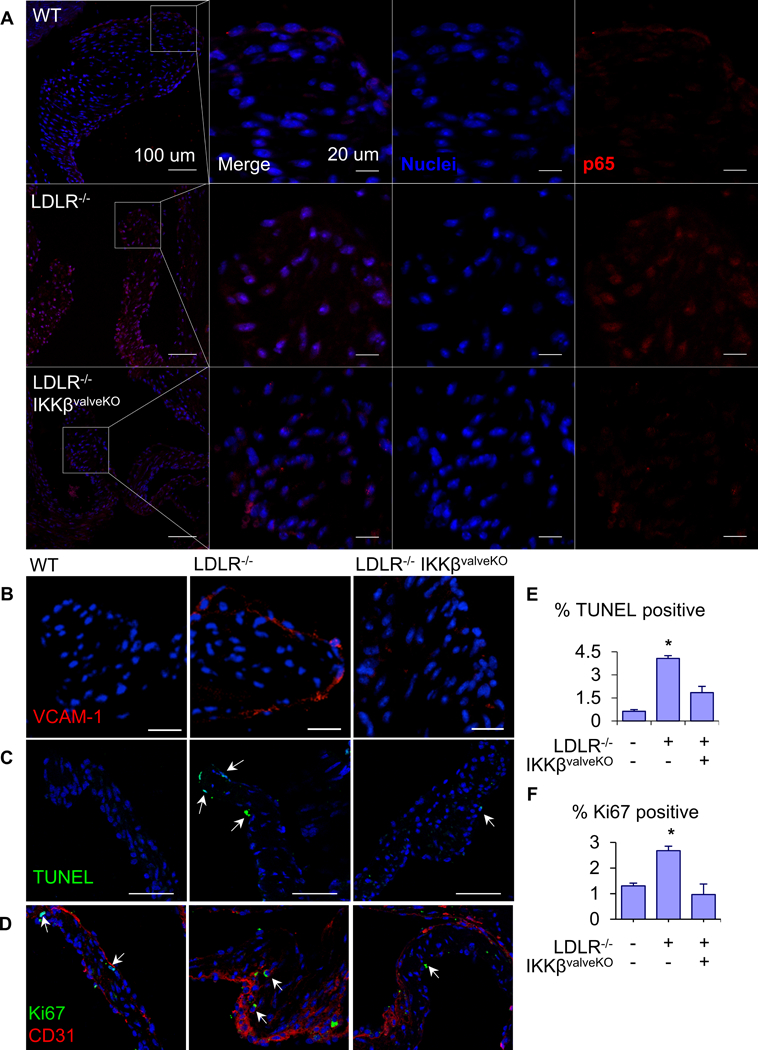
A. Representative images of NFkB p65/RelA (red) protein expression and nuclear localization (arrows) in aortic valve leaflets of 5 month-old WT, AVD, and AVD+IKKβf/f mice. Scale bars are 100μm (left-most column) and 20μm (second column on). B. Representative images of VCAM-1 (red) protein expression from same groups. Scale bar is 100μm. C. TUNEL assay for apoptosis in AoV of same groups, quantification of the percent of cells in the AoV positive for TUNEL staining (n = 5). Scale bar is 100 μm. D. Analysis of proliferation in AoV of same groups via Ki67 staining. Ki 67 (green) and CD31 (red), scale bar is 100μm. Quantification of percent of cells in the AoV positive for nuclear Ki67 (n = 5). Asterisks indicate statistically significant groups (*: p < 0.05).
However, nuclear localization of p65 was significantly increased with mice that develop CAVD (LDLr−/− with western-diet starting at post-natal 4-weeks, thereafter), most prominently in the VEC populations (Figure 2A) consistent with human CAVD. Valvular (VEC and VIC) specific deletion of IKKβ in the same CAVD background significantly reduced levels of p65 expression and nuclear activity across both VEC and VIC in the age-matched CAVD+IKKβvalveKO mice. Inactivation of downstream p65 transcriptional activity was confirmed via immunofluorescence for VCAM-1, a direct transcriptional target of p65 (Figure 2B)12. VCAM-1 was expressed prominently in endothelium of CAVD mice, but not in CAVD+IKKβvaleKO. As the NFκB signaling cascade is an important mechanism of cell survival26, we determined whether its deletion would adversely affect either cell apoptosis (Figure 2C) or proliferation (Figure 2D). As expected, both of these parameters were upregulated in CAVD conditions. However, in CAVD+ IKKβvalveKO, neither cell apoptosis or proliferation were statistically different from their wild-type controls (Figure 2E,F). These results demonstrate that valve specific lack of NFκB signaling is dispensable for murine aortic valve morphogenesis and homeostasis, but NFκB is activated during adult murine CAVD.
Valve-specific inactivation of IKKβ prevents hypercholesterolemia mediated aortic valve pathogenesis
We next determined how valve specific NFκB inactivation modulates hypercholesterolemia induced aortic valve degeneration and hemodynamic stenosis. Echocardiographic analysis revealed increased AoV peak velocity, peak gradient, and mean gradient (Figures 3A,B,C) in CAVD mice in comparison to wildtype controls, but not in CAVD + IKKβvalveKO. Measurement of the aortic root during peak systole and peak diastole (Figure 3D) revealed a loss of aortic distensibility in CAVD that was not prevented by CAVD+ IKKβvalveKO (Figure 3D). CAVD + IKKβvalveKO did however prevent decreased ejection fraction percentage which occurred in CAVD mice compared to wild-type counterparts (Figure 3E). Aortic leaflet thickness measurements indicated that CAVD mice developed significantly thickened leaflets in comparison to either wildtype controls or CAVD + IKKβvalveKO (Figure 3F). Examination of possible sex-differences in CAVD development of hemodynamic dysfunction or its prevention via valve-specific IKKβ deletion did not indicate statistical differences between strain matched males or females (Suppl. Fig. SIII). Histological analysis showed increased calcium and mineral deposition as indicated via Alizarin Red S and von Kossa staining (Figure 4A,B), and collagen content indicated via changes in Russell-Movat pigmentation (Figure 4C) in CAVD mice, but not CAVD+ IKKβvalveKO. We observed a non-significant trend of reduction in glycosaminoglycan (GAG) content with both CAVD and CAVD+ IKKβvalveKO conditions compared to control (Supplementary Fig. SIV). These results support that valve specific NFκB inhibition protects against CAVD within a persistent pro-CAVD environment.
Figure 3.
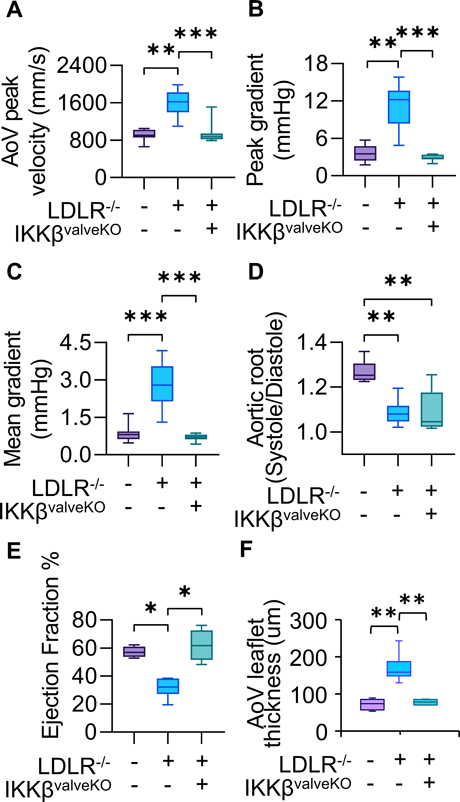
Echocardiographic measurements of AoV function of 5 month old mice, 4 months of high fat diet in all models except WT control. (WT: n=11 LDLR−/−: n=17, LDLR−/−IKKBvalveKO: n=11) A,B,C. Mean AoV peak jet velocity (mm/s), Peak gradient (mmHg), and Mean gradient (mmHg) from Pulsed-wave Doppler imaging, calculated using the average peak from > three cardiac cycles for each mouse. D. Ratio of Aortic Root Systolic:Diastolic ratio calculated from M-mode echocardiographic measurements of aortic root diameter during peak diastole and systole. E. Cardiac Ejection Fraction percentage calculated from parasternal long-axis ventricular volume at peak systole and diastole. Asterisks indicate statistically significance, *: p < 0.05, **: p < 0.005, ***: p < 0.0005).
Figure 4.
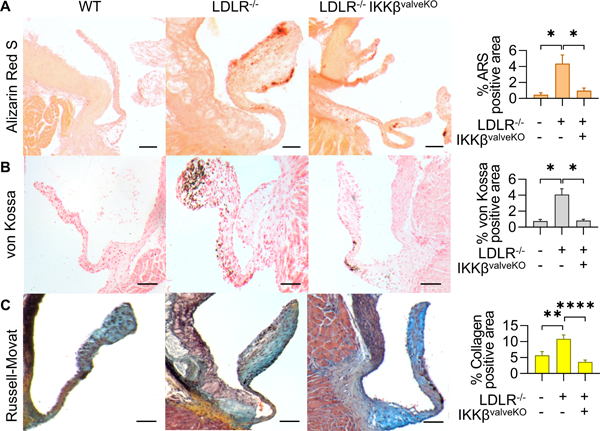
A. Alizarin Red S stain for calcium in AoV. Scale bar is 100μm. B. von Kossa staining for mineral deposition in AoV. Scale bar is 100μm .C. Russell-Movat stain for collagen (yellow), glycosaminoglycans (blue), elastin (black), and mucins (red) in AoV. Quantification indicates percent of the total valve area that was positive for indicated stain, measured from valve hinge-point between annulus and cusp, to the distal tip (n = 5). Asterisks indicate statistically significance (*: p < 0.05, **: p < 0.005, ***: p < 0.0005, ****: p < 0.00005).
p65/RelA activation induces VEC participation in osteogenic differentiation and calcification in vitro
Considering the protective effect of p65 inhibition in VEC and VIC in vivo, we next determined how NFκB signaling modulates VEC phenotype in vitro. Porcine aortic VEC were cultured on 3-D collagen hydrogels under osteogenic differentiation medium (OGM) conditions. Treatment of VEC for 14 days with OGM was unable to induce calcific deposition indicated by Alizarin Red S (ARS) staining (Figure 5A,B). However, co-treatment of OGM with either exogenous TNFα (OGM+TNFα) or forced expression of p65 (OGM+p65) resulted in formation of distinct calcified nodules and significantly increased calcium deposition (Figure 5A,B). Immunofluorescence staining indicated administration of NFkB adduct such as exogenous TNFα is require to induce p65 nuclear localization, where OGM alone is insufficient (Supplementary Fig. SV). Phenotypic characterization was conducted via qRT-PCR and immunofluorescence staining, evaluating changes in expression for myofibroblatic activation marker alpha-smooth muscle actin (αSMA), transcription factor Runx2 required for osteogenic transdifferentiation, and later-stage osteogenic marker osteocalcin (OCN). OGM treatment and p65 overexpression both significantly increased αSMA gene and protein expression, with the combinatorial administration upregulating it even further (Figures 5C,D). Neither OGM or p65 overexpression alone significantly affected Runx2 gene or protein expression (Figures 5E,F). However, the co-administration of +OGM+p65 significantly increased Runx2 gene expression (Figure 5E), with both cytosolic and nuclear protein expression observed (Figure 5E). Similar trends were observed with OCN (Figure 5G,H). Neither OGM nor p65 overexpression resulted in observable protein expression of OCN (Figure 5G). OGM did not significantly alter OCN gene expression, while p65 overexpression resulted in in a significant decrease in OCN gene expression (Figure 5H). However, the co-administration of +OGM+p65 resulted in cytosolic osteocalcin protein expression, and significant increase in OCN gene expression (Figures 5G,H). These results establish that chronic inflammatory mediated NFκB activation is required to induce myofibroblastic and osteogenic differentiation of VEC in vitro.
Figure 5.
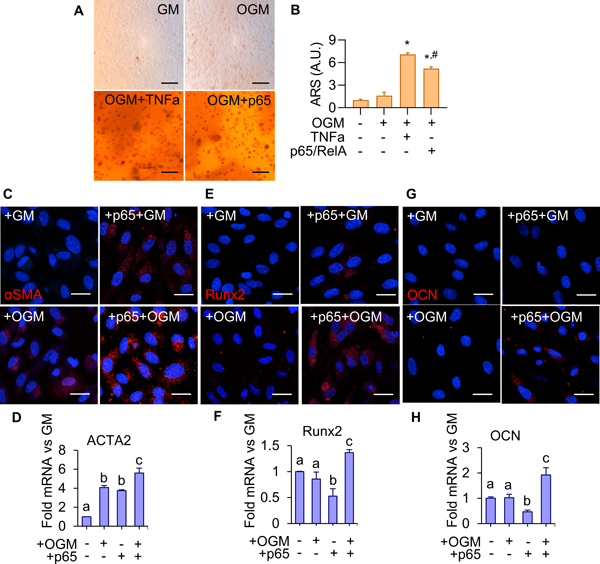
A. Alizarin Red S staining for calcium in 14 day VEC treated with GM, OGM, OGM+TNFα(30 ng/ml), or OGM+p65 over-expression. Scale bar is 100μm. B. Quantification of calcium deposition using ARS dye-extraction assay, normalized to GM + empty vector control (n = 6). (*: significant from groups 1, 2 #: significant from group 3, p < 0.05). C. αSMA protein expression in VEC with or without p65 over-expression with 14-day control or OGM treatment. Scale bar is 20μm. D. ACTA2 mRNA in VEC, same conditions as C (n = 4). E. Runx2 protein expression in VEC, same conditions as C. Scale bar is 20μm. F. Runx2 mRNA in VEC, same conditions as E (n = 4). G. Osteocalcin (OCN) protein expression in VEC, same conditions as C. H. Osteocalcin mRNA in VEC, same conditions as G. Statistical significance between different groups are indicated by unshared letter assignments, p < 0.05. Different letters indicate statistically significant differences between groups (p < 0.05). Error bars indicate SEM.
p65/RelA NFκB signaling is active in pro-osteogenic VIC differentiation, and its inhibition mitigates differentiation and calcification in vitro
We next determined how p65 activity regulates VIC calcification. Porcine VIC were transfected with empty, p65, or IκBα mutant repressor vectors, encapsulated within 3-D collagen hydrogels, and cultured for 14 days with or without OGM treatment. We found that OGM alone induced a significant increase in VIC p65 nuclear expression after 14 days of culture (Supplementary Fig. SVIA,B). Over-expression of p65 exacerbated VIC calcification response to OGM, and inhibition of p65 (+IκBα super repressor) mitigated VIC calcification response as indicated by Alizarin Red S staining and quantification (Figure 6A,B). Alterations to cell phenotype were assessed via qRT-PCR and immunostaining for gene and protein expression. αSMA gene expression was upregulated due to OGM culture, with both control and OGM cells displaying protein expression (Figures 6C,D). OGM cultured VIC displayed significant increases to both Runx2 and OCN gene expression (Figures 6F,H), and observable Runx2 and OCN protein expression compared to controls (Figures 6E,G). Surprisingly, over-expressing p65 resulted in OCN gene expression undergoing an over 100-fold increase compared to control, with observable protein expression (Figures 6G,H) despite the lack of significant changes to αSMA and Runx2 gene or protein expression (Figures 6C-F). The combination of OGM and p65 overexpression (+OGM+p65) exacerbated Runx2 and OCN gene and protein expression, significantly increasing both as compared to OGM or p65 expression alone (Figure 6E-H). αSMA gene expression for +OGM+p65 VIC was not significantly different from control (Figures 6D). Phenotypic assessment for the p65 loss of function (via IκBα overexpression), indicated a reduction of both αSMA and Runx2 expression compared to control (Figures 6E-H). Inhibition of p65 did not mitigate changes in OCN protein expression, despite an over 50-fold increase to gene expression (Figures 6G,H). Inhibition of p65 mitigated increases in αSMA in OGM culture (+IκBα+OGM) (Figure 6C,D). Co-administration (+IκBα+OGM) yielded an intermediary phenotype of VIC in either condition alone (+OGM or + IκBα). Runx2 was elevated and comparable to OGM alone (Figures 6E,F), while gene expression increases for OCN were comparable to IκBα overexpressing VIC (Figures 6H). These results show that NFκB activity exacerbates OGM induced VIC myofibroblastic and osteogenic differentiation in vitro, and its inhibition significantly but not completely reduces this response.
Figure 6.
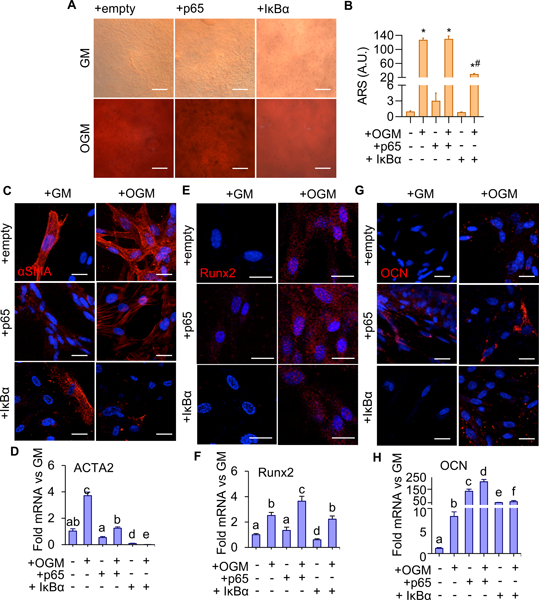
A. Alizarin Red S staining for calcium in 14 day VIC with p65 over-expression (+p65) or inhibition (+IκBα) treated with GM or OGM. Scale bar is 100μm. B. Quantification of calcium deposition using ARS dye-extraction assay, normalized to GM + empty vector control (n = 6). (*: significant from groups 1, 3, 5; #: significant from 2; $: significant from 4, p < 0.05). C. αSMA protein expression in VIC with p65 over-expression (+p65) or inhibition (+IκBα) vectors, with 14-day GM or OGM treatment. Scale bar is 20μm. D. ACTA2 mRNA in VIC, same conditions as in C (n = 4). E. Runx2 protein expression in VEC, same conditions as C. Scale bar is 20μm. F. Runx2 mRNA in VIC, same conditions as E (n = 4). G. Osteocalcin (OCN) protein expression in VIC, same conditions as E. H. OCN mRNA in VIC, same conditions as G (n = 4). Statistical significance between different groups are indicated by unshared letter assignments. Different letters indicate statistically significant differences between groups (p < 0.05). Error bars indicate SEM.
p65 mediates inflammatory EndMT in VEC in vitro and in vivo
The previous data identified reduced susceptibility of VEC to osteogenic media compared to VIC. We therefore next tested whether inflammatory activation of NFκB mediates VEC transformation into a phenotypically distinct plastic state, via endothelial-to-mesenchymal-transition (EndMT). We evauated potential phenotypic transdifferentiation via loss or disruption VE-cadherin concomitant with increased expression of myofibroblastic marker αSMA and increased expression of transcription factor SNAI1, crucial for dissolution of E-cadherin junctions during EndMT. Both exogenous TNFα and p65 over-expression induced EndMT in that period, characterized by disruption of VE-cadherin junctions and reduction in gene expression (CDH5) (Figure 7A,B), concomitant with increased αSMA (Figure 7C,D). SNAI1 gene expression was also significantly increased exogenous TNFα and p65 over-expression (Figure 7E), Canonical p65 inhibition, via the overexpression of IκBα, prevented EndMT induction (Figure 7A-E). These results support that the induction of osteogenic activity in VEC requires a prior transition to a mesenchymal phenotype via EndMT in vitro.
Figure 7.
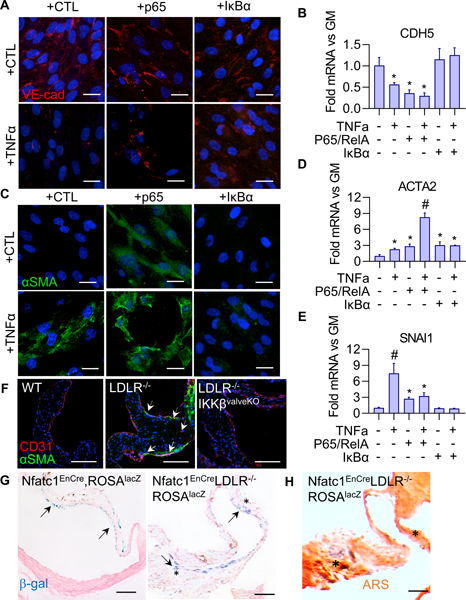
A. VE-cad protein expression in VEC with p65 over-expression or inhibition (+IκBα), with 48 hour GM or GM+TNFα(30 ng/ml) treatment. Scale bar is 20μm. B. CDH5 (VE-cadherin gene) mRNA in VEC, same conditions as A (n = 4). (*: statistically significant from groups 1, 5, 6, p < 0.05). C. αSMA protein expression in VEC, same conditions as A. Scale bar is 20μm. D. ACTA2 (αSMA gene) mRNA in VEC, same conditions as A (n = 4). (*: statistically significant from group 1, p < 0.05), # statistically significant from all other groups, p < 0.05). E. SNAI1 mRNA in VEC, same conditions as A (n = 4). (*: statistically significant from group 1, p < 0.05), # statistically significant from all other groups, p < 0.05). F. CD31 and αSMA expression in 5 month-old mouse AoVs. Arrows indicate cells co-expressing CD31 and αSMA. Scale bar is 100μm. G. β-Galactosidase staining with and Nuclear Fast Red counterstaining in Nfatc1enCreRosa26lacZ reporter mice in either the wild-type or CAVS background. Asterisks indicate β-gal positive cells in interstitial space. Scale bar is 100μm. H. Alizarin Red S staining of calcium deposition. Asterisk indicate β-gal positive cells in interstitial space. Scale bar is 100μm.
EndMT in postnatal AoV in vivo is characterized by co-expression of endothelial (CD31) and mesenchymal (αSMA) phenotypic markers11, but its direct demonstration requires lineage tracking. In the wild-type background, we observed little to no expression of αSMA and a robust, intact CD31-positive endothelial layer (Figure 7F, left). In CAVD mice, αSMA expression was elevated, most prominently within cells co-expressing CD31 and αSMA (Figure 7F, middle, arrows). To further confirm the valve endothelial origins of these EndMT-VEC populations, versus potential circulating progenitor or extravasation of recruited inflammatory populations, the Nfatc1enCre was backcrossed with the LDlr−/−, along with a Rosa26lacZ reporter line. β-galactosidase was restricted to the valve endocardial lining observed in non-CAVD mice (Figure 7G, left), demonstrating that EndMT does not occur during aortic valve homeostasis. However, with CAVD induction via chronic high fat diet in the Ldlr−/− backround, β-galactosidase staining identified substantial disruption of the β-gal+ endothelial lining on the fibrosa surface, with cells that sub-migrated into the tissue interstitium and co-localize with calcified lesions (Figures 7G,H, black arrows and *). Further, CAVD+ IKKβeKO mice had αSMA expression comparable to wildtype controls and no observable cells with co-expression of CD31 and αSMA (Figure 7F, right). These results directly demonstrate the involvement of EndMT in CAVD in vivo, but not during homeostasis, and NFκB inactivation in valves prevents VEC EndMT in vivo.
VEC specific inactivation of IKKβ inhibits hypercholesterolemia mediated aortic valve calcification
To further clarify the role of VEC specific NFκB activity in CAVD degeneration, we performed a VEC-specific deletion of IKKβ in the CAVD background (Nfatc1enCre;LDLr−/−;IKKβfl/fl supplemented with western-diet, henceforth LDLR−/−IKKβvecKO). Echocardiographic analysis of the LDLR−/−IKKβvecKO mice showed significant improvement of valvular hemodynamic function. Aortic valve jet velocity, peak gradient, and mean gradient were improved in the Nfatc1enCre (VEC-specific) versus the CAVD background (Figures 8A,B,C). Aortic root distensibility and ejection fraction percentage for the VEC-specific IKKβ deletion were not statistically different from the CAVD background (Figure 8D,E). As with our whole-valve deletion strategy, we parsed our male and female populations to examine possible sex-specific differences in CAVD induced hemodynamic dysfunction but did not find significant differences between sexes (Suppl. Fig. SVIII). Histological assessment of valvular calcium and mineral deposition via Alizarin Red S and von Kossa staining, respectively, demonstrated that VEC -specific IKKβ deletion was able to achieve a statistically significant reduction of calcium/mineral deposition compared to CAVD background, similarly with our whole-valve specific deletion (Figures 8F,G). Analysis of total serum cholesterol indicated both the whole-valve and VEC-specific IKKβ deletion maintained a hypercholesterolemic state (Suppl. Fig. SVIII). Taken together, these results demonstrate that VEC-specific inhibition ameliorate valvular pathogenesis without affecting affecting associated coronary heart disease.
Figure 8.
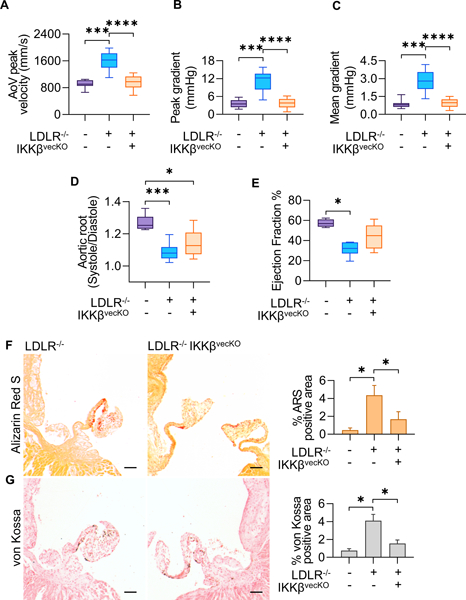
Echocardiographic measurements of AoV function of 5 month old mice, 4 months of high fat diet in all models except WT control. (WT: n=11, LDLR−/−: n=17, LDLR−/−IKKBvecKO: n=15) A,B,C. Mean AoV peak jet velocity (mm/s), Peak gradient (mmHg), and Mean gradient (mmHg) from Pulsed-wave Doppler imaging, calculated using the average peak from > three cardiac cycles for each mouse. D. Ratio of Aortic Root Systolic:Diastolic ratio calculated from M-mode echocardiographic measurements of aortic root diameter during peak diastole and systole. E. Cardiac Ejection Fraction percentage calculated from parasternal long-axis ventricular volume at peak systole and diastole. Asterisks indicate statistically significance (*: p < 0.05, **: p < 0.005, ***: p < 0.0005, ****: p < 0.00005). F. Alizarin Red S staining for calcium in AoV. Scale bar is 100μm. G. von Kossa staining for mineral deposition in AoV. Scale bar is 100μm. Quantification indicates percent of the total valve area that was positive for indicated stain, measured from valve hinge-point between annulus and cusp, to the distal tip (n = 5). Asterisks indicate statistically significance (*: p < 0.05, **: p < 0.005, ***: p < 0.0005, ****: p < 0.00005).
Discussion
The results of this study establish the importance of the p65/RelA NFκB pathway in CAVD pathogenesis and its potential as a target for pharmacological treatment. Inactivation of canonical NFκB pathway via valvular deletion of IKKβ prevented valve endothelial inflammation in vivo in CAVD risk environments. p65 control of pathogenic valvular transitions was conserved in vitro and in vivo, across VEC and VIC, and whether osteogenic medium or hypercholesterolemic diet induced. Inflammation is a hallmark of both early and late stages of CAVD27,28, with important repercussions for both endothelial10,11 and interstitial16 cell participation in disease. The observed mitigation of inflammation (VCAM-1 expression) was not accompanied by lowered serum cholesterol, therefore the downstream improvements in valve function here cannot be attributed to the reduction of general cardiovascular risk factors, in contrast to prior studies that ultimately were not translatable to humans29. Furthermore, specifically targeting VEC IKKβ activity was sufficient in retaining normative valvular function despite a persistent pro-CAVD environment. It should be noted that while IKKβ activity is important to mediate canonical NFkB signal transduction, its pleiotropic activities extend to regulation of processes including nuclear β-catenin and Runx2 signaling30–32. IKKα and IKKβ regulates transcriptional β-catenin signaling, with it being demonstrated that TNF upregulates beta-catenin in the vasculature with atherogenesis33. Nevertheless, the combination of in vitro primary cell culture models coupled with in vivo IKKβ conditional deletion models point to the key role of the IKKβ – p65/RelA relay in VEC and VIC as relevant to CAVD. This demonstrated mechanism between the reduction of inflammatory activation and amelioration of endothelial dysfunction resulting in improved valve function emphasizes the opportunity for early valve endothelial specific diagnostic markers and treatment strategies, but also the utility of in vivo CAVD models for testing valve specific pathogenic mechanisms and their targeting.
Our study demonstrates a role for p65 as a “gatekeeper” for VEC initiation and participation in valvular calcific progression by connecting inflammatory activation to a downstream osteogenic phenotype. Activation of p65 may control a sequential phenotypic shift from quiescent to osteogenic progenitors via EndMT, as supported by studies showing TNFα promotes osteogenic differentiation of mesenchymal stem cells via IKKβ activation34. EndMT has been implicated in coronary arterial pathobiology35,36, and presence of myofibroblast populations are elevated throughout both early and late stages of valvular calcification37. Our results here provide first direct in vivo evidence for NFκB activity driving postnatal EndMT within the aortic valve. This phenomenon could share mechanisms with oncology, as NFκB is known to be essential for EMT in breast cancer38. In postnatal epithelial cells, p65 and Snail1 co-regulate EMT, possibly through p65 nuclear recruitment of Snail139. EndMT of postnatal VEC has been demonstrated in multiple in vitro contexts, including aberrant mechanical stimulation40, consequence of inflammation11, and in response to TGFβ. Similarities between postnatal and developmental EndMT suggests that EndMT may provide a source of interstitial progenitor populations with increased differentiation potential. Recent studies have shown that aortic valves contain a subpopulation of cells displaying phenotypic plasticity, collectively referred to as “mesenchymal progenitors”; VIC displaying differentiation potential into osteogenic, adipogenic, chondrogenic, and myofibroblast lineages41. One source is circulating endothelial progenitor cells, but these account for only 4% of cells with demonstrated phenotypic plasticity in sclerotic valves42. We were unable to identify postnatal VEC undergoing EndMT in healthy valves via genetic lineage tracing, suggesting that VEC EndMT does not participate in postnatal valve homeostasis. In contrast, our findings demonstrate that postnatal VEC in CAVD risk conditions are uniquely re-enabled to undergo EndMT by p65/RelA activation as an initiating component of CAVD.
Amongst valvular interstitial populations, aortic-derived VIC uniquely display an enhanced inflammatory agonist response, expressing higher levels of pro-inflammatory and pro-osteogenic mediators43. We demonstrate that p65 enhances VIC calcific turnover, and inhibition of p65 signaling mitigates that participation. This mechanism may underlie the finding that tissue stiffness induces VIC activation and calcification44, as tissue stiffness is known to activate NFκB in certain cancer cell types mediated by actomyosin contractions45. NFκB could be mechanotransductive signaling bridge between extracellular matrix (ECM) and calcification in VIC, participating in a positive feedback loop via RhoA-p65 interaction, as demonstrated in renal cells46. The mitigating effects of the VEC-specific NFκB inhibition on total valve pathology are possibly due to retained homeostatic function, as we and others have demonstrated the role of VEC in maintaining VIC quiescence and promoting valvular health25,47. VEC actively regulate the structural and mechanical properties of the aortic valve48,49, which leads to downstream changes in both VIC and VEC signaling and disease propagation. Collectively, our findings indicate that diet-induced hypercholesterolemic CAVD initially presents with p65 activity prominently within the VEC which radiates further into the tissue interstitium in later-stage disease. While our study primarily focused on the role of the tissue resident endothelial and interstitial populations, it is also important to note that other cellular constituents may have a role in systemic inflammatory processes underpinning CAVD progression, such as leukocytes and T-cells. Examination of murine and human tissue sections indicate concomitant increase of infiltrating monocyte and macrophage populations with valvular matrix dysregulation/valvular degeneration50. In IL-1Ra deficient mice which develop onset of aortic valve stenosis, it has been indicated that T-cells were thought to be the primary mediators, with pathogenesis being reversed by combined deletion of TNF51. Our results further support the requirement for exogenous cardiovascular inflammation triggering conditions, such as chronic hypercholesterolemia as probed within this body of work and supported in other studies52, to initiate misregulated valvular cell activity mediated via canonical NFκB signaling. From our in vitro results, we further observed that the combination of both p65 activation and osteogenic stimulus is necessary for VEC calcific activity. This suggests that VEC require an exogenous triggering mechanism such as underlying coronary arterial inflammation, to induce an EndMT mediated trans-differentiation to a phenotypically plastic intermediary.
Familial hypercholesterolemia (FH), a common hereditary disease in the Western Hemisphere, is a disorder afflicting lipoprotein metabolism giving rise to chronic systemic inflammation and to increase risk to atherosclerotic development and coronary heart disease53. Less appreciated however is the increased incidence of CAVD in FH patients, with over half of males and 21–41% females with homozygous FH exhibiting aortic regurgitation or advanced valvular dysfunction54,55. Given the relative insufficiency of lipid-lowering or anti-atherogenic therapeutics in addressing the valvular degeneration3,5,56, a closer examination of valvular cell behavior in the context of hypercholesterolemia-induced coronary heart disease is warranted. Furthermore, it is greater appreciated within the field possible sex-differences in valvular calcific progression57,58. However, between our hypercholesterolemia induced CAVD model or corresponding valvular conditional deletion models, we did not denote sex-specific differences in the disease etiology, possibly due to the early timepoint of assessment; such differences may become more apparent at later stages of stenosis/calcific progression. We show here that inhibition of NFκB signaling is sufficient to benefit valve physiologic functionality while underlying hypercholesterolemia and coronary/arterial disease remains. This suggests that NFκB dependent signaling mechanisms downstream of elevated serum cholesterol and subsequent coronary inflammation may be the primary mediator of valvular pathogenesis.
The results of this study present NFκB p65 as a novel therapeutic target for CAVD. Our findings indicate that both VEC and VIC calcific transition and pathogenic activity converge at canonical NFkB mediated signaling, regardless of initiating context. We found that modulating this signaling axis in both VEC and VIC populations, versus VEC alone, to be more effective in preventing CAVD development. We hypothesize this to be due to VIC having their own VEC-independent program of activation and osteogenic transition. The dynamics of these interactions and concomitant activity should be studied further. Supporting this approach of NFκB targeting are recent findings that osteoprotegerin mitigates CAVD calcification59. Osteoprotegerin is a decoy receptor for receptor activator of nuclear factor-kappaB ligand (RANKL)60, which activates the canonical NFκB pathway14. RANKL is necessary for bone formation and is upregulated in calcified aortic valves61, supporting NFκB signaling modulation as the potential mechanism behind the efficacy of osteoprotegerin treatment. While we have demonstrated a role for NFκB in governing valve EndMT and calcification, it has yet to be shown whether pharmacological targeting can mitigate CAVD progression without eliciting adverse systemic effects or toxicity. Furthermore, while our genetic strategy provides some indication NFκB targeting as a potential prophylactic strategy, it must be denoted that Cre-mediated genomic recombination and therefore IKKβ silencing occurs during embryonic valvular development. It remains to be seen whether in the more clinically relevant context of adult disease onset whether these NFκB targeting strategies will be sufficient to ameliorate CAVD progression. In the context of acute cardiovascular injury, pharmacologic agents directly targeting NFκB canonical signaling activity such as pyrrolidine-dithiocarbamamate (PDTC) and BAY 11–7082, have been shown to be effective in treatment of septic shock50 or ischemic reperfusion injury62,63 and therefore may are warranted for evaluation in the context of CAVD. NFκB has been the focus of many studies regarding inflammation in cancer development, resulting in many attempts to modulate its function in vivo64. Cross-talk between oncology and CAVD may lead to future avenues for CAVD treatment via NFκB modulation and may be able to capitalize on the novel NFκB mechanisms of CAVD shown here.
Supplementary Material
{kind=link}
Highlights.
NFκB signaling identified as an essential molecular regulator for both valve endothelial and interstitial participation in CAVD pathogenesis.
Direct demonstration of valve endothelial EndMT transmigration in vivo.
Targeting aortic valve endothelial IKKβ signaling is sufficient to abrogate disease mitigated valve hemodynamic dysfunction.
Acknowledgements
a) Acknowledgements
The authors would like to thank Shirks Meats of Dundee, NY for providing porcine aortic valves. The authors would like to thank Dr. Sanjay Samy, Robert Packer Hospital, Sayre PA, for generous donation of calcified human aortic valves, and Dr. Jonathan Chen, Seattle Children’s Hospital, Seattle, WA for generous donation of non-diseased human aortic valves.
b) Sources of Funding
This work was supported by the National Science Foundation Graduate Research Fellowship (TWG). This work was supported by grants from the National Institutes of Health/National Institute of Heart, Lung and Blood (HL128745 (JTB), HL118672 (JTB), HL143247 (JTB), HL078881 (BZ), HL104441 (BZ), and HL111770 BZ).
Abbreviations:
- ARS
Alizarin Red S
- CAVD
Calcific Aortic Valve Disease
- EndMT
Endothelial to Mesenchymal Transformation
- GM
Basal growth-medium
- GAG
glycosaminoglycan
- OGM
Osteogenic differentiation medium
- OCN
osteocalcin
- PAVEC
Porcine Aortic Valve Endothelial Cells
- PAVIC
Porcine Aortic Valve Interstitial Cells
- VEC
Valve Endothelial Cells
- VIC
Valve Interstitial Cells
Footnotes
C Disclosure
none.
References
- 1.D’Arcy JL, Coffey S, Loudon MA, Kennedy A, Pearson-Stuttard J, Birks J, Frangou E, Farmer AJ, Mant D, Wilson J, Myerson SG, Prendergast BD. Large-scale community echocardiographic screening reveals a major burden of undiagnosed valvular heart disease in older people: The OxVALVE Population Cohort Study. Eur Heart J. 2016;37:3515–3522a. doi: 10.1093/eurheartj/ehw229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mohler ER, Gannon F, Reynolds C, Zimmerman R, Keane MG, Kaplan FS. Bone Formation and Inflammation in Cardiac Valves. Circulation. 2001;103:1522–1528. doi: 10.1161/01.CIR.103.11.1522 [DOI] [PubMed] [Google Scholar]
- 3.Cowell SJ, Newby DE, Prescott RJ, Bloomfield P, Reid J, Northridge DB, Boon NA. A Randomized Trial of Intensive Lipid-Lowering Therapy in Calcific Aortic Stenosis. N Engl J Med. 2005;352:2389–2397. doi: 10.1056/NEJMoa043876 [DOI] [PubMed] [Google Scholar]
- 4.Otto CM. Calcific Aortic Stenosis — Time to Look More Closely at the Valve. N Engl J Med. 2008;359:1395–1398. doi: 10.1056/NEJMe0807001 [DOI] [PubMed] [Google Scholar]
- 5.Rossebø AB, Pedersen TR, Boman K, et al. Intensive Lipid Lowering with Simvastatin and Ezetimibe in Aortic Stenosis. N Engl J Med. 2008;359:1343–1356. doi: 10.1056/NEJMoa0804602 [DOI] [PubMed] [Google Scholar]
- 6.Towler DA. Molecular and cellular aspects of calcific aortic valve disease. Circ Res. 2013;113:198–208. doi: 10.1161/CIRCRESAHA.113.300155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abdelbaky A, Corsini E, Figueroa AL, Subramanian S, Fontanez S, Emami H, Hoffmann U, Narula J, Tawakol A. Early aortic valve inflammation precedes calcification: A longitudinal FDG-PET/CT study. Atherosclerosis. 2015;238:165–172. doi: 10.1016/j.atherosclerosis.2014.11.026 [DOI] [PubMed] [Google Scholar]
- 8.Hjortnaes J, Butcher J, Figueiredo JL, Riccio M, Kohler RH, Kozloff KM, Weissleder R, Aikawa E. Arterial and aortic valve calcification inversely correlates with osteoporotic bone remodelling: A role for inflammation. Eur Heart J. 2010;31:1975–1984. doi: 10.1093/eurheartj/ehq237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li C, Xu S, Gotlieb AI. The progression of calcific aortic valve disease through injury, cell dysfunction, and disruptive biologic and physical force feedback loops. Cardiovasc Pathol. 2013;22:1–8. doi: 10.1016/j.carpath.2012.06.005 [DOI] [PubMed] [Google Scholar]
- 10.Farrar EJ, Huntley GD, Butcher J. Endothelial-derived oxidative stress drives myofibroblastic activation and calcification of the aortic valve. PLoS One. 2015;10:1–19. doi: 10.1371/journal.pone.0123257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mahler GJ, Farrar EJ, Butcher JT. Inflammatory Cytokines Promote Mesenchymal Transformation in Embryonic and Adult Valve Endothelial Cells. Arterioscler Thromb Vasc Biol. 2013;33:121–130. doi: 10.1161/ATVBAHA.112.300504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tak PP, Firestein GS, Tak PP, Firestein GS. NF-kappaB: a key role in inflammatory diseases. J Clin Invest. 2001;107:7–11. doi: 10.1172/JCI11830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van der Heiden K, Cuhlmann S, Luong LA, Zakkar M, Evans PC. Role of nuclear factor κB in cardiovascular health and disease. Clin Sci. 2010;118:593–605. doi: 10.1042/CS20090557 [DOI] [PubMed] [Google Scholar]
- 14.Hoffmann, Alexander; Baltimore D. Circuitry of nuclear factor κB signaling. Immunol Rev. 2006;210:171–186. [DOI] [PubMed] [Google Scholar]
- 15.Farrar EJ, Butcher JT. Heterogeneous Susceptibility of Valve Endothelial Cells to Mesenchymal Transformation in Response to TNFα. Ann Biomed Eng. 2014;42:149–161. doi: 10.1007/s10439-013-0894-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu Z, Seya K, Daitoku K, Motomura S. Tumor Necrosis Factor- alpha Accelerates the Calcification of Human Aortic Valve Interstitial Cells Obtained from Patients with Calcific Aortic Valve Stenosis via the BMP2-Dlx5. … Pharmacol …. 2011:16–23. doi: 10.1124/jpet.110.177915. [DOI] [PubMed] [Google Scholar]
- 17.Crotti TN, Smith MD, Findlay DM, Zreiqat H, Ahern MJ, Weedon H, Hatzinikolous G, Capone M, Holding C, Haynes DR. Factors regulating osteoclast formation in human tissues adjacent to peri-implant bone loss: Expression of receptor activator NFκB, RANK ligand and osteoprotegerin. Biomaterials. 2004;25:565–573. doi: 10.1016/S0142-9612(03)00556-8 [DOI] [PubMed] [Google Scholar]
- 18.Wu B, Wang Y, Lui W, Langworthy M, Tompkins KL, Hatzopoulos AK, Baldwin HS, Zhou B. Nfatc1 coordinates valve endocardial cell lineage development required for heart valve formation. Circ Res. 2011;109:183–192. doi: 10.1161/CIRCRESAHA.111.245035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu B, Zhang Z, Lui W, et al. Endocardial cells form the coronary arteries by angiogenesis through myocardial-endocardial VEGF signaling. Cell. 2012;151:1083–1096. doi: 10.1016/j.cell.2012.10.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Egan LJ, Eckmann L, Greten FR, Chae S, Li Z-W, Myhre GM, Robine S, Karin M, Kagnoff MF. IkappaB-kinasebeta-dependent NF-kappaB activation provides radioprotection to the intestinal epithelium. Proc Natl Acad Sci U S A. 2004;101:2452–2457. doi: 10.1073/pnas.0306734101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Drolet MC, Roussel E, Deshaies Y, Couet J, Arsenault M. A high fat/high carbohydrate diet induces aortic valve disease in C57BL/6J mice. J Am Coll Cardiol. 2006;47:850–855. doi: 10.1016/j.jacc.2005.09.049 [DOI] [PubMed] [Google Scholar]
- 22.Holliday CJ, Ankeny RF, Jo H, Nerem RM. Discovery of shear- and side-specific mRNAs and miRNAs in human aortic valvular endothelial cells. Am J Physiol Circ Physiol. 2011;301:H856–H867. doi: 10.1152/ajpheart.00117.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gould R a Butcher JT. Isolation of valvular endothelial cells. J Vis Exp. 2010:6–10. doi: 10.3791/2158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Butcher JT, Nerem RM. Valvular endothelial cells regulate the phenotype of interstitial cells in co-culture: effects of steady shear stress. Tissue Eng. 2006;12:905–915. doi: 10.1089/ten.2006.12.905 [DOI] [PubMed] [Google Scholar]
- 25.Richards J, El-Hamamsy I, Chen S, Sarang Z, Sarathchandra P, Yacoub MH, Chester AH, Butcher JT. Side-specific endothelial-dependent regulation of aortic valve calcification: Interplay of hemodynamics and nitric oxide signaling. Am J Pathol. 2013;182:1922–1931. doi: 10.1016/j.ajpath.2013.01.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luo J, Kamata H, Karin M. IKK / NF-κB signaling : balancing life and death — a new approach to cancer therapy. J Clin Invest. 2005;115:2625–2632. doi: 10.1172/JCI26322.noreceptors [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coté N, Mahmut A, Bosse Y, Couture C, Pagé S, Trahan S, Boulanger MC, Fournier D, Pibarot P, Mathieu P. Inflammation is associated with the remodeling of calcific aortic valve disease. Inflammation. 2013;36:573–581. doi: 10.1007/s10753-012-9579-6 [DOI] [PubMed] [Google Scholar]
- 28.New SEP, Aikawa E. Molecular imaging insights into early inflammatory stages of arterial and aortic valve calcification. Circ Res. 2011;108:1381–1391. doi: 10.1161/CIRCRESAHA.110.234146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miller JD, Weiss RM, Serrano KM, Brooks RM, Berry CJ, Zimmerman K, Young SG, Heistad DD. Lowering plasma cholesterol levels halts progression of aortic valve disease in mice. Circulation. 2009;119:2693–2701. doi: 10.1161/CIRCULATIONAHA.108.834614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lamberti C, Lin KM, Yamamoto Y, Verma U, Verma IM, Byers S, Gaynor RB. Regulation of β-Catenin Function by the IκB Kinases. J Biol Chem. 2001;276:42276–42286. doi: 10.1074/jbc.M104227200 [DOI] [PubMed] [Google Scholar]
- 31.Al-Huseini I, Ashida N, Kimura T. Deletion of IjB-Kinase β in smooth muscle cells induces vascular calcification through β-Catenin-Runt-related transcription factor 2 signaling. J Am Heart Assoc. 2018;7:1–15. doi: 10.1161/JAHA.117.007405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schröfelbauer B, Polley S, Behar M, Ghosh G, Hoffmann A. NEMO Ensures Signaling Specificity of the Pleiotropic IKKβ by Directing Its Kinase Activity toward IκBα. Mol Cell. 2012;47:111–121. doi: 10.1016/j.molcel.2012.04.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Al-Aly Z, Shao JS, Lai CF, Huang E, Cai J, Behrmann A, Cheng SL, Towler DA. Aortic Msx2-Wnt calcification cascade is regulated by TNF-α-dependent signals in diabetic Ldlr−/− mice. Arterioscler Thromb Vasc Biol. 2007;27:2589–2596. doi: 10.1161/ATVBAHA.107.153668 [DOI] [PubMed] [Google Scholar]
- 34.Hess K, Ushmorov A, Fiedler J, Brenner RE, Wirth T. TNFα promotes osteogenic differentiation of human mesenchymal stem cells by triggering the NF-κB signaling pathway. Bone. 2009;45:367–376. doi: 10.1016/j.bone.2009.04.252 [DOI] [PubMed] [Google Scholar]
- 35.Chen PY, Qin L, Baeyens N, Li G, Afolabi T, Budatha M, Tellides G, Schwartz MA, Simons M. Endothelial-to-mesenchymal transition drives atherosclerosis progression. J Clin Invest. 2015;125:4514–4528. doi: 10.1172/JCI82719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Evrard SM, Lecce L, Michelis KC, et al. Endothelial to mesenchymal transition is common in atherosclerotic lesions and is associated with plaque instability. Nat Commun. 2016;7:11853. doi: 10.1038/ncomms11853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Latif N, Sarathchandra P, Chester AH, Yacoub MH. Expression of smooth muscle cell markers and co-activators in calcified aortic valves. Eur Heart J. 2015;36:1335–1345. doi: 10.1093/eurheartj/eht547 [DOI] [PubMed] [Google Scholar]
- 38.Huber M a, Azoitei N, Baumann B, Grünert S, Sommer A, Pehamberger H, Kraut N, Beug H, Wirth T. NF- κ B is essential for epithelial- mesenchymal transition and metastasis in a model of breast cancer progression. J Clin Invest. 2004;114:569–581. doi: 10.1172/JCI200421358.The [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stanisavljevic J, Porta-de-la-Riva M, Batlle R, de Herreros AG, Baulida J. The p65 subunit of NF-κB and PARP1 assist Snail1 in activating fibronectin transcription. J Cell Sci. 2011;124:4161–4171. doi: 10.1242/jcs.078824 [DOI] [PubMed] [Google Scholar]
- 40.Balachandran K, Alford PW, Wylie-Sears J, Goss JA, Grosberg A, Bischoff J, Aikawa E, Levine RA, Parker KK. Cyclic strain induces dual-mode endothelial-mesenchymal transformation of the cardiac valve. Proc Natl Acad Sci. 2011;108:19943–19948. doi: 10.1073/pnas.1106954108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen JH, Yip CYY, Sone ED, Simmons CA. Identification and characterization of aortic valve mesenchymal progenitor cells with robust osteogenic calcification potential. Am J Pathol. 2009;174:1109–1119. doi: 10.2353/ajpath.2009.080750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Skowasch D, Schrempf S, Wernert N, Steinmetz M, Jabs A, Tuleta I, Welsch U, Preusse CJ, Likungu JA, Welz A, Lüderitz B, Bauriedel G. Cells of primarily extravalvular origin in degenerative aortic valves and bioprostheses. Eur Heart J. 2005;26:2576–2580. doi: 10.1093/eurheartj/ehi458 [DOI] [PubMed] [Google Scholar]
- 43.Yang X, Fullerton DA, Su X, Ao L, Cleveland JC, Meng X. Pro-Osteogenic Phenotype of Human Aortic Valve Interstitial Cells Is Associated With Higher Levels of Toll-Like Receptors 2 and 4 and Enhanced Expression of Bone Morphogenetic Protein 2. J Am Coll Cardiol. 2009;53:491–500. doi: 10.1016/j.jacc.2008.09.052 [DOI] [PubMed] [Google Scholar]
- 44.Yip CYY, Chen JH, Zhao R, Simmons CA. Calcification by valve interstitial cells is regulated by the stiffness of the extracellular matrix. Arterioscler Thromb Vasc Biol. 2009;29:936–942. doi: 10.1161/ATVBAHA.108.182394 [DOI] [PubMed] [Google Scholar]
- 45.Ishihara S, Yasuda M, Harada I, Mizutani T, Kawabata K, Haga H. Substrate stiffness regulates temporary NF-κB activation via actomyosin contractions. Exp Cell Res. 2013;319:2916–2927. doi: 10.1016/j.yexcr.2013.09.018 [DOI] [PubMed] [Google Scholar]
- 46.Wu X, Guo R, Chen P, Wang Q, Cunningham PN. TNF induces caspase-dependent inflammation in renal endothelial cells through a Rho- and myosin light chain kinase-dependent mechanism. Am J Physiol Renal Physiol. 2009;297:F316–F326. doi: 10.1152/ajprenal.00089.2009 [DOI] [PubMed] [Google Scholar]
- 47.El Accaoui RN, Gould ST, Hajj GP, Chu Y, Davis MK, Kraft DC, Lund DD, Brooks RM, Doshi H, Zimmerman K a, Kutschke W, Anseth KS, Heistad DD, Weiss RM. Aortic valve sclerosis in mice deficient in endothelial nitric oxide synthase. Am J Physio Hear Circ Physiol. 2014;306:H1302–13. doi: 10.1152/ajpheart.00392.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aikawa E, Whittaker P, Farber M, Mendelson K, Padera RF, Aikawa M, Schoen FJ. Human semilunar cardiac valve remodeling by activated cells from fetus to adult: Implications for postnatal adaptation, pathology, and tissue engineering. Circulation. 2006;113:1344–1352. doi: 10.1161/CIRCULATIONAHA.105.591768 [DOI] [PubMed] [Google Scholar]
- 49.El-Hamamsy I, Balachandran K, Yacoub MH, Stevens LM, Sarathchandra P, Taylor PM, Yoganathan AP, Chester AH. Endothelium-Dependent Regulation of the Mechanical Properties of Aortic Valve Cusps. J Am Coll Cardiol. 2009;53:1448–1455. doi: 10.1016/j.jacc.2008.11.056 [DOI] [PubMed] [Google Scholar]
- 50.Hulin A, Anstine LJ, Kim AJ, Potter SJ, DeFalco T, Lincoln J, Yutzey KE. Macrophage transitions in heart valve development and myxomatous valve disease. Arterioscler Thromb Vasc Biol. 2018;38:636–644. doi: 10.1161/ATVBAHA.117.310667/-/DC1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Isoda K, Matsuki T, Kondo H, Iwakura Y, Ohsuzu F. Deficiency of interleukin-1 receptor antagonist induces aortic valve disease in BALB/c Mice. Arterioscler Thromb Vasc Biol. 2010;30:708–715. doi: 10.1161/ATVBAHA.109.201749 [DOI] [PubMed] [Google Scholar]
- 52.Nus M, MacGrogan D, Martínez-Poveda B, Benito Y, Casanova JC, Fernández-Avilés F, Bermejo J, De La Pompa JL. Diet-induced aortic valve disease in mice haploinsufficient for the notch pathway effector RBPJK/CSL. Arterioscler Thromb Vasc Biol. 2011;31:1580–1588. doi: 10.1161/ATVBAHA.111.227561 [DOI] [PubMed] [Google Scholar]
- 53.Cuchel M, Bruckert E, Ginsberg HN, et al. Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management. A position paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur Heart J. 2014;35:2146–2157. doi: 10.1093/eurheartj/ehu274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kawaguchi A, Miyatake K, Yutani C, Beppu S, Tsushima M, Yamamura T, Yamamoto A. Characteristic cardiovascular manifestation in homozygous and heterozygous familial hypercholesterolemia. Am Heart J. 1999;137:410–418. doi: 10.1016/S0002-8703(99)70485-0 [DOI] [PubMed] [Google Scholar]
- 55.Rajamannan NM, Edwards WD, Spelsberg TC. Hypercholesterolemic Aortic-Valve Disease. N Engl J Med. 2003;349:717–718. doi: 10.1056/NEJMc031360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Antonini-Canterin F, Hîrşu M, Popescu BA, Leiballi E, Piazza R, Pavan D, Ginghinǎ C, Nicolosi GL. Stage-Related Effect of Statin Treatment on the Progression of Aortic Valve Sclerosis and Stenosis. Am J Cardiol. 2008;102:738–742. doi: 10.1016/j.amjcard.2008.04.056 [DOI] [PubMed] [Google Scholar]
- 57.Porras AM, McCoy CM, Masters KS. Calcific Aortic Valve Disease. Circ Res. 2017;120:604–606. doi: 10.1161/CIRCRESAHA.117.310440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Masjedi S, Lei Y, Patel J, Ferdous Z. Sex-related differences in matrix remodeling and early osteogenic markers in aortic valvular interstitial cells. Heart Vessels. 2017;32:217–228. doi: 10.1007/s00380-016-0909-8 [DOI] [PubMed] [Google Scholar]
- 59.Weiss RM, Lund DD, Chu Y, Brooks RM, Zimmerman KA, Accaoui R El, Davis MK, Hajj GP, Zimmerman MB, Heistad DD. Osteoprotegerin Inhibits Aortic Valve Calcification and Preserves Valve Function in Hypercholesterolemic Mice. PLoS One. 2013;8:1–8. doi: 10.1371/journal.pone.0065201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wright HL, McCarthy HS, Middleton J, Marshall MJ. RANK, RANKL and osteoprotegerin in bone biology and disease. Curr Rev Musculoskelet Med. 2009;2:56–64. doi: 10.1007/s12178-009-9046-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kaden JJ, Bickelhaupt S, Grobholz R, Haase KK, Sarikoç A, Kiliç R, Brueckmann M, Lang S, Zahn I, Vahl C, Hagl S, Dempfle CE, Borggrefe M. Receptor activator of nuclear factor κB ligand and osteoprotegerin regulate aortic valve calcification. J Mol Cell Cardiol. 2004;36:57–66. doi: 10.1016/j.yjmcc.2003.09.015 [DOI] [PubMed] [Google Scholar]
- 62.Liu SF, Ye X, Malik AB. Inhibition of NF- B Activation by Pyrrolidine Dithiocarbamate Prevents In Vivo Expression of Proinflammatory Genes. Circulation. 1999;100:1330–1337. doi: 10.1161/01.CIR.100.12.1330 [DOI] [PubMed] [Google Scholar]
- 63.Kim YS, Kim JS, Kwon JS, Jeong MH, Cho JG, Park JC, Kang JC, Ahn Y. BAY 11–7082, a Nuclear Factor-. 2010:348–353. [DOI] [PubMed] [Google Scholar]
- 64.Lin Yong, Bai Lang, Wenjie Chen SX. The NF-κB activation pathways, emerging molecular targets for cancer prevention and therapy. Expert Opin Ther Pat. 2011;14:45–55. doi: 10.1517/14728220903431069.The [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.