

Abstract
Multiple sclerosis (MS) is widely acknowledged to be an autoimmune disease affecting the neuronal myelin structure of the CNS. Autoantigens recognized as the target of this autoimmune process are: myelin basal protein, anti-proteolipid protein, antimyelin-associated glycoprotein and antimyelin-based oligodendrocytic basic protein. Ample evidence supports the idea of a dysregulation of immunological tolerance towards self-antigens of neuronal myelin structure triggered by one or more viral or bacterial microbial agents in predisposed HLA gene subjects. Genetic predisposition to MS has been highlighted by numerous studies associating the disease to specific HLA haplotypes. Moreover, a wide range of evidence supports the fact that MS may be consequence of one or more viral or bacterial infections such as measles virus, EBV, HHV6, HZV, Chlamydia pneumoniae, Helicobacter Pylori, and other microbial agents. Microbiota elements also seems to have a role on the determinism of the disease as a pathogenic or protective factor. The autoimmune pathogenetic process could arise when a molecular mimicry between a foreign microbial antigen and an auto-antigen occurs in an HLA gene subject competent for that particular antigen. The antigen-presenting cells in this case would induce the activation of a specific Th clone causing a cross-reaction between a foreign antigen and an autoantigen resulting in an autoimmune response.
Highlights
-
•
A multifactorial ethiopathogenetic model based on immunomediation is a reliable hypothesis for multiple sclerosis.
-
•
Evidence found in the scientific literature makes it possible to reconstruct this etiopathogenetic hypothesis for MS.
-
•
HLA gene predisposition, correlation with infections, molecular mimicry and other immunological data are reported.
1. Introduction
Multiple sclerosis (MS) is considered an autoimmune inflammatory demyelinating disease characterized by a destruction of the neuronal axonal myelin of the central nervous system. Characteristic lesions appear as plaques representing areas of demyelination of the corpus callosum, brainstem, spinal cord, periventricular areas, cerebellum, etc. Myelin damage brings a slowing down/blocking of nerve impulse conduction resulting in a neurological deficit. Multiple sclerosis is thought to occur in genetically predisposed people due to one or more environmental factors. It mainly affects young adults mostly in females, leading to progressive disability [1].
MS is common in northern Europe, continental north America and Australasia, but not in the Far East, the Arabian Peninsula, Africa, continental South America or India [2]. In a recent epidemiological evaluation conducted on data collected between 1990 and 2016, the estimates of prevalence of multiple sclerosis, standardized by age, were highest in North America (164.6 per 100,000), Western Europe (127.0 per 100,000) and Australasia (91.1 per 100,000) and lowest were sub-Saharan East Africa s (3.3 per 100,000), in central sub-Saharan Africa (2.8 per 100,000) and Oceania (2.0 per 100,000) [3].
Multiple sclerosis lesions have been characterized by infiltrates of T-lymphocytes, small numbers of B-lymphocytes, plasma cells and activated microglia or macrophages. Furthermore, in myelinated lesions, the expression and production of numerous related immune molecules such as cytokines, adhesion molecules and histocompatibility antigens have been defined. These patterns suggest that in this disease a T-cell mediated immune response is at the basis of an inflammation of nerve tissue and a subsequent demyelination. Furthermore, a variable degree of axonal destruction that significantly leads to permanent functional deficit has been detected. The extent and severity of the inflammatory reaction is related to the amount of axonal damage. In experimental models, axonal damage may be induced by cytokines produced by activated macrophages. These cells determine neuronal damage through specific excitotoxins acting on glutamate receptors which are distributed on dendrites and neuronal pericarya rather than on the axons themselves. Nitric oxide also contributes to axonal damage, especially in demyelinated fibers [4].
In the following paragraphs, I will describe the scientific reports data that support the immune-mediated pathogenesis of multiple sclerosis. These data indicate the fulcrum of this disease pathogenesis, lies in the dysregulation of the balance between self-tolerance and autoimmunity according to the following multifactorial model: a genetic predisposition factor, linked to the histocompatibility complex gene HLA allows the triggering of the disease by an infectious factor that induce an autoimmune reaction towards the structures of the myelin sheath of the nervous system.

The predisposing HLA molecules support the triggering action through the process of antigen presentation of an infectious agent exerted by the APCs, coinciding with the molecular mimicry between a microbial antigen and the myelin autoantigen. The activated T and B lymphocytes induce the cross-reaction which leads to autoimmunity. Activation of self-reactive CD4+ T cells and their differentiation into a Th1 phenotype are crucial events in the early stages, and these cells probably also play an important role in the evolution of the disease. Damage to the central nervous system is, however, most likely mediated by other components of the immune system, such as antibodies, complement, CD8+ T cells, and factors produced by innate immune cells. Disorders in immunomodulatory networks that include Th2 cells, CD4 + Treg cells, NK cells and others may be partly responsible for relapse-remitting or chronic progressive nature of the disease [5].
2. Genetic predisposition and immunopathogenetic function of HLA
GWAS (genome Wide Association Studies) recently pointed to HLA-DRB1 in the MHC Class II region as the main susceptibility locus for MS. The best documented HLA allele is DRB1*15:01, which is the highest risk factor for MS. HLA-DRB1*15:01 has the strongest effect with an average odds (OR) ratio of 3,08 and an additional risk in homozygous subjects [6]. These results were confirmed by 2 meta-analyses [7,8].
An independent combination for DRB1*08:01 was observed in an Ashkenazi cohort when patients were divided into clinical subgroups, with a weak but significant combination reported only for primary patients with progressive MS [9].
A well-established, though not very strong (OR = 1:7), association was found for DRB1*03:01, a very common allele all over the world. HLA-DRB1*13:03 is a very rare allele worldwide, therefore it is clearly a risk allele only when found at higher frequencies, as in the Israeli population [[9], [10]], in Sardinia [11,12] or in studies with thousands of individuals. As for African-Americans, allele DRB1*15:03 seems to be the allele DR2 predominantly associated in this population with MS (OR = 1:6) [13]. Another risk allele for MS is DRB1*04:05, which has been recognized as a risk variant for Asian types of MS (OR = 2:23), characterized by early onset, mild course and reduced brain lesions [14]. These results were partially reproduced in another Asian cohort [15]. HLA-DRB1*04:05 was also found to be associated with multiple sclerosis in Sardinia, where the incidence of multiple sclerosis is quite high [16], as well as among African Americans [17] and Sicilians [18].
As regards protective alleles for multiple sclerosis, DRB1*14 and DRB1*07 showed protective effects against multiple sclerosis in a meta-analysis of 37 case studies, all in Caucasian populations [8]. Class II allele – consistently associated with MS protection in European populations – is DRB1*14:01. DRB1*14-mediated protection appears to be dominant, reducing the predisposing effect of DRB1*15:01when both alleles are present in the same individual (OR = 0.2) [19].
HLA-DRB1*11 was also found to be protective in the Brazilian [20], Canadian [21] and Greek [22] populations. The protective role of the HLA-A*02:01 has been confirmed in many different populations and remains the most widely studied HLA Class I allele in MS [23].
Susceptibility to MS determined by HLA genes and detected by the statistical association data reported above is expressly due to the immunological function of HLA molecules and for this reason they play a primary role in the molecular mechanism of immunopathogenesis of this autoimmune disease.
Class II HLA molecules expressed on the APCs perform the receptor function towards peptides derived from the processing of exogenous antigens ingested via phagocytosis or endocytosis. APCs expose the processed antigen on the cell surface by means of the HLA receptor and then presents it to the T lymphocytes which in turn recognize the antigen by binding it specifically to its receptor (TCR) [24]. In addition, T-cells require a second signal to activate and initiate the adaptive response. This second signal is performed by membrane molecules such as CD 28 of the T naive cells as well as CD86, CD80 of the APC. This molecular T-cell activation mechanism involving 2 categories of membrane molecules (HLA and CD) has been termed as “two-signal model of T-cell activation” [25] (Fig. 1). The intervention of specific cytokines then determines the evolution of the naive T-cell and activate depolarized responses Th1, Th2, Th17 etc., but when there is no second signal, T reg cells are activated inducing tolerance. The systemic administration of nanoparticles coated with autoimmune peptides relevant to specific diseases, linked to HLA class II molecules has been shown to trigger the generation and expansion of CD1 + type T antigen-specific regulatory cells, leading to the resolution of phenomena autoimmune in different animal models. Regardless of their genetic background, ten MHCII-based nanomedicines show similar biological effects, prevalence of the T cell population or MHC restriction. The nanoparticles coated with autoimmune peptides, bound to HLA class II molecules are a means for avoiding the second signal given by the membrane molecules that would be present if the antigenic peptides were presented to the T cells by APCs [26].
Fig. 1.
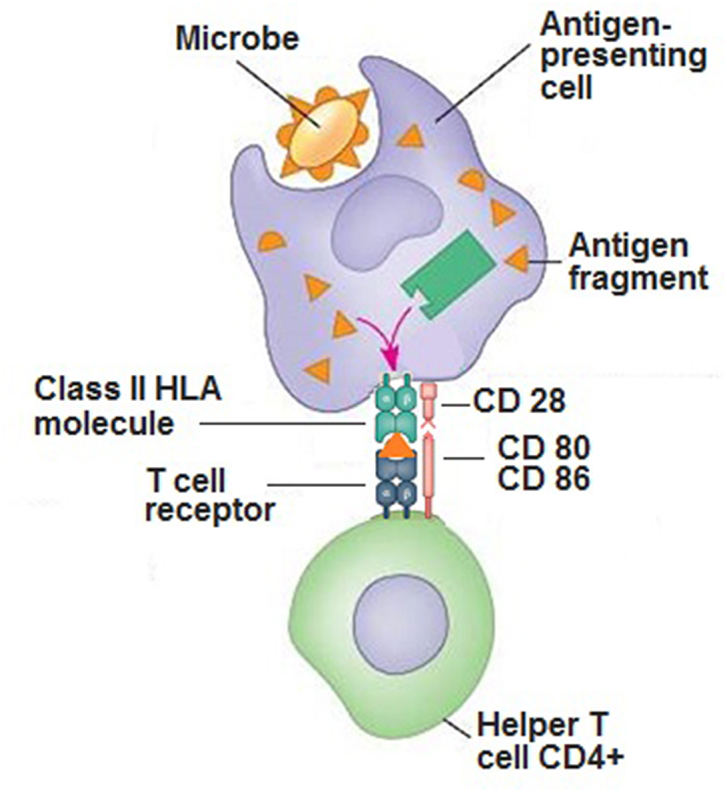
Molecular function of class II HLA in antigen presentation.
It may be noted that in the absence of HLA molecules capable of binding a specific antigen, the antigen presentation function cannot be performed by APCs. Consequently, autoreactive T-cells cannot be activated, which from a molecular point of view, explains why subjects who do not have HLA predisposing alleles for a given disease are protected throughout their existence.
3. Self-reactive T-cells
Most studies on the role of T-cells in MS have focused on peripheral T-cells activated by myelin and limited by the main class II HLA molecules. In animal models of multiple sclerosis such as experimental autoimmune encephalomyelitis (EAE) and a demyelinating disease induced by murine Theiler’s encephalomyelitis virus (TMEV-IDD), pro-inflammatory Th1 cells and Th17 cells have been shown to play a decisive role in the exacerbation phases of the disease, while Th2 cells and regulatory T-cells (Treg) can contribute to remission [27]. Tests for CD8 (+) T cell populations were also submitted. Despite the evidence in animals and human studies supporting the idea that self-reactive T cells are involved in disease induction, myeloid lineage cells, antibodies and complement as well as processes intrinsic to the central nervous system appear to determine the actual stages of tissue damage [28]. Activated T-cells attracted by chemokine release pass through the permeated blood-brain barrier (BBB); also B cells and monocytes/macrophages migrate into the CNS where the myelin autoantigen is presented by APS (macrophages, microglia and or astrocytes). In this way, the auto-reactive T-cells are induced to produce cytokines that activate other inflammatory cells such as macrophages which damage the central nervous system and phagocytize myelin [29]. An overexpression of Th1-dependent cytokines such as IFN-γ and TNF-α in MS plaques was observed. Therefore, Th1 cytokines can sustain a chronic state of inflammation by mediating a continuous recruitment of activated T-cells or directly inducing myelin disruption, as demonstrated for TNF-α. Furthermore, the active expression of TNF-α in peripheral blood mononuclear cells (PBMC) has been shown to be associated with MS activity [30]. Numerous EAE studies have contributed considerably to clarifying the immunopathogenic role of T-cells in demyelinating diseases. In summary, after immunization with CFA, whooping cough toxin and myelin antigens, the dendritic cells are activated in the lymph nodes by toll-like receptor agonists (TLR) and present the myelital antigen to naïve T-cells. Specific T-cells activated for myelin enter the bloodstream and the CNS. The rupture of the BBB allows the recruitment of other inflammatory cells into the CNS. T-cells entering the CNS encounter their related myelin antigens and are reactivated by the local APC. T cells expand and release inflammatory mediators that help recruit other immune cells at the inflammation site. The activation of local microglial cells and infiltrating cells involves the production of proteases, glutamates, reactive oxygen species and other cytotoxic agents that promote myelin destruction. Damage to the myelin sheath of the surrounding axons is followed by axonal damage and neurological impairment [31] (Fig. 2).
Fig. 2.

Migration and effector function of T cells in the central nervous system (CNS) during experimental autoimmune encephalomyelitis (EAE) After immunization with myelin antigens, complete Freund’s adjuvant (CFA) and pertussis toxin, dendritic cells (DC) are activated in the lymph nodes by Toll-like receptor (TLR) agonists within the mycobacterium tuberculosis component of CFA, and present myelin antigen to naive T cells The activated myelin-specific T cells enter the bloodstream and traffic to and enter the CNS Breakdown of the blood-brain barrier (BBB) occurs, allowing recruitment of other inflammatory cells into the CNS. T cells entering the CNS encounter their cognate myelin antigens and become reactivated by local APC. T cells expand and release inflammatory mediators which help recruit other immune cells to the site of inflammation. Activation of local microglial cells and infiltrating cells results in production of proteases, glutamate, reactive oxygen species and other cytotoxic agents which promote myelin breakdown. Damage to the myelin sheath surrounding axons is followed by axonal damage and neurological impairment.
4. Auto-antigens targets of self-reactive T-cells
Several studies indicate myelin antigens as the specific targets of self-reactive T-cells. Myelin basic protein (MBP) is the first and most widely studied myelin protein involved in MS. Other myelinated proteins, such as the proteolipid protein (PLP), myelin-associated glycoprotein (MAG) and myelin-associated basic oligodendrocytic protein (MOG) have been proposed as potential autoantigens in MS. In a 1993 study, Joshi N. et al. found that (1) comparable frequencies of MBP-reactive T-cell lines were obtained from the peripheral blood of patients with MS and their healthy siblings. The pairs of siblings with an identical HLA predisposition but different as regards MS disease presence showed similar frequencies of reactive MBP T cell lines. (2) A broad spectrum of MBP epitopes has been recognized in the T cell lines of all the individuals studied. (3) MBP epitopes were recognized in the context of several class II HLA alleles. At least four DR alleles each served as restrictive elements for the recognition of the P82-101 or MBP terminal carboxic region –two regions considered important in the response of human T-cells to the molecule [32].
As regards PLP, the data shown in the review by Tuohy VK, indicate that: (1) PLP is an important encephalitogen specific to the central nervous system (CNS); (2) the reactivity of CD4 + T-cells to discrete PLP peptide determinants can mediate the development of acute, chronic recurrent and chronic progressive autoimmune encephalomyelitis (EAE); and (3) the responsiveness of T-cells to multiple determinants of PLP occurs in patients with multiple sclerosis (MS) – the main demyelinating disease of the human CNS [33].
In a 1999 study by Weerth S et al., the capacity of myelin-associated glycoprotein (MAG) was demonstrated to initiate a self-aggressive T-cell response in the Lewis rat, supporting the concept that MAG-specific autoimmune responses can play an important role in the pathogenesis of demyelinating diseases in humans. In fact, it has been observed that, when injected into the Lewis rat, specific T-cells reactive to certain MAG epitopes mediated a pathological inflammatory response in the nervous system. In particular, clinical disease was observed in those animals injected with T-cells specific to sequence a. a. 20–34 (MP1.1), which also started an inflammatory response in the peripheral nervous system (PNS). T-cells specific to MP1.1 together with 8–18C5 monoclonal antibodies specific to myelin-associated basic oligodendrocytic protein MOG increased the severity of the disease and induced widespread CNS demyelination [34].
A 2010 study by Kaushansky N et al. showed that MOBP is an important target antigen candidate in MS. Studies indicate that: T-cells autoreactive to MOBP can be detected in patients with MS; T-cells reactive against MOBP can be pathogenic in several mouse strains as well as in “humanized” HLA-DR15-Tg mice; HLA-DQ6-restricted, but not HLA-DR15-restricted, MOBP-reactive T-cells in HLA-DR15-Tg mice cause clinical disease similar to the one associated with perivascular and parenchymal infiltration, demyelination, axonal loss, and optical neuritis. Consequently, MOBP should be considered a primary target antigen in MS, in addition to MBP, PLP and MOG [35].
5. The role of B cells
Many studies have been conducted on the mechanisms by which B cells contribute to the pathogenesis of MS due to their role as antigen-presenting cells (APC), secretion and proinflammatory cytokines and chemokines, as well as their production of autoantibodies against the myelin sheath and neuronal axons [36].
Structures similar to lymph node follicles containing B cells and Follicular dendritic cells (FDC) in CNS (ectopic germinal centers) can be found in mice with recurrent progressive EAE. The formation of these ectopic follicles indicates that B cells migrate to the brain, are activated locally, have antigens and differentiate into memory B cells or plasmablasts within the central nervous system. The presence of these germinal structures of B cells reflect a greater impact on the integrity of cortical structures [37]. Similar structures have also been found in the meninges of patients with progressive secondary multiple sclerosis and their presence is associated with a younger age onset of disease and a severe cortical pathology [38]. The formation of ectopic follicles indicates that B cells migrate to the brain, are activated locally with APC function and differentiate into memory B cells or plasmablasts within the central nervous system [39]. Recent clinical studies with Rituximab, however, have shown that the administration of this monoclonal anti-CD20 B cell antibody in RRMS patients rapidly reduces B cells, as well as myelin lesions and clinical recurrences with an effect lasting 3–12 months [40].
In MS, the balance of cytokines is aimed at inflammation rather than tolerance, with an increase in IL-1, IL-2, TNF and IFNγ levels and a decrease in anti-inflammatory cytokines such as IL10 and TGF-β [41]. Activated B cells can produce a variety of immunomodulating cytokines and growth factors [42]. In the ectopic germinal centers that form in the CNS lesions of patients with multiple sclerosis, both TNF and LT have been detected [43]. B cells from MS patients produce less IL-10 than B cells from healthy individuals, but high levels of IL-10 and TGF-β produced by new generation B cells have been detected following treatment with Rituximab [44].
6. Auto-antibodies
Much effort has been made to determine the specificity of antibodies found in the lesions, serum and cerebrospinal fluid (CSF) of patients with MS and other demyelinating diseases related to a wide variety of myelin sheath and axon antibody targets. Oligoclonal IgG bands are detectable in over 95% of MS patients and increased IgG concentrations in CSF are found in up to 70% of cases [45]. The oligoclonal nature of these antibodies suggests an antigen-induced process and the determination of the specificity of these antibodies could therefore provide insights into the causes of MS. Several microbial antigens and autoantigens have been proposed as targets [46].
Antibodies specific to myelin [47,48] and axonal proteins [49,50] have been eluted from multiple sclerosis lesions. Increased frequencies of anti-MOG antibodies have been reported in the serum and cerebrospinal fluid of MS patients compared to controls [51]. Other studies conducted on serums or CSF of MS patients have detected antibodies against other myelin components including MBP, PLP, MAG and cyclic nucleotide phosphodiesterase (CNP) [52].
A recent study has shown that axonal protein is also targeted for autoantibodies in a subset of MS patients, assuming that auto-antibody response is involved in axonal damage. About a third of MS sera contained antibodies to neurofascin – an adhesion molecule expressed by oligodendrocytes and neurons that localizes on the axon-myelin interface to Ranvier’s nodes. Systemic administration of anti-neurofascin antibodies results in a rapid worsening of EAE due to axonal damage [53].
In the majority of patients with NMO (73%) or an optic-spinal form of MS (58%), IgG serum reactivity against the blood-brain barrier of the CNS tissue of mice was found, with a 91–100% specificity for optic-spinal demyelinating conditions [54]. Aquaporins are a family of water channels responsible for maintaining cellular water balance, and they are also expressed by astrocytes in the CNS located on the BBB. The high specificity and sensitivity of anti-Aqp4 tests have been confirmed in larger-scale studies of patients with NMO and other autoimmune neurological diseases, including pediatric and adult-onset MS [[55], [56], [57]]; the evaluation of anti-Aqp4 has since been incorporated into the diagnostic criteria for NMO [58]. Spinal cord and optic nerve cells have been observed to express Aqp4 at high levels and this explains why anti-aquaporin antibodies play an active role in the progression of NMO [59].
7. Infectious factor
As pointed out by numerous reports, among the environmental etiological factors of MS, a determining role has been attributed to the infectious factor. The idea that an infection could be the cause of MS arises from both epidemiological data and data on immunological changes found in this disease. Some data are based on the observed MS epidemics and on the geographical distribution of the disease linked to the endemic presence of an infectious agent while other data are based on biological observation that reflect an immune response to an indeterminate antigen. From the data reported in these reports, infection is the moment of activation of a self-reactive immune response often due to a phenomenon of antigenic molecular mimicry, resulting in chronic autoimmunity. Furthermore, infections subsequent to the onset of the disease were found to cause evolution and clinical recurrences.
Many infectious microbial agents have been implicated in the triggering or progression of MS, most notably: Epstein-Barr Virus, Human Herpes Virus 6 and Chlamydophila (Chlamydia) pneumoniae [60]. Other important viruses and bacteria considered in this area are: Measles virus, Human Zoster-varicella virus and Helicobacter pylori, Mycobacterium avium para-TBC, Mycoplasma pneumoniae, Clostridium perfringens type B, elements of the human microbiota: Euryarchaeota, Firmicutes and Proteobacteria (Sutterella). (Table 1).
Table 1.
| Virus and bacteria in MS trigger or progression |
|---|
|
|
|
|
|
|
|
|
|
|
8. Post-infection autoimmune activation
Several mechanisms by which infections can cause demyelination have been proposed. For example, in progressive multifocal leukoencephalitis (PML), the JC virus can infect oligodendrocytes and cause their destruction and subsequent demyelination [61]. But the most accredited mechanisms provide for the induction of an autoimmune response are: molecular mimicry, bystander activation, epitope spreading and superantigens. In case of molecular mimicry, the sequence or confomational homologies between infectious microbial antigens and myelin antigens in an HLA-predisposed individual determine the activation of cross-reactive T-cells (pathogen and self-reactive T-cells) with subsequent autoimmune demyelination. In the bystander activation model, APCs activated by microbial antigens result in the activation of self-reactive CD44 + T memory cells through the production of cytokines. Bacterial superantigens can cause self-reactive T-cell activation for an aspecific link between class II HLA and TCR and the onset and/or exacerbation of autoimmune diseases. Epitope spreading occurs when tissue damage caused by a new infection in an already present autoimmune disease or by chronic autoimmune inflammation determines the exposure of a new antigenic epitope and thus its presentation by APC to autoreactive T-cells, thereby promoting an exacerbation of the disease [62] (Fig. 3).
Fig. 3.

Molecular mechanisms of pathogen-induced autoimmunity. (A) Pathogen-activated antigen-presenting cells can display self-antigens from dying cells to autoreactive T lymphocytes in a process known as bystander activation. (B) Activation of the immune system resulting from stimulation of pattern recognition receptors by infectious agents can lead to expression of proinflammatory mediators and triggering of autoreactive lymphocytes. (C) Microbial superantigens cross-link MHC class II molecules with TCRs inducing antigen unspecific activation of autoreactive T cells. (D) Certain pathogen-derived antigens share structural similarities with self-peptides causing activation of autoreactive T cells through molecular mimicry. (E) The process of epitope spreading can enhance autoimmune responses by activating autoreactive! T cells to “new” self-antigens during the progression of the disease. (F) Viral agents can enhance the activation state of autoantigen presenting cells and induce the survival of autoreactive lymphocytes. As an example, persistent infection of microglial cells with Theiler’s murine encephalomyelitis virus (TMEV) was shown to upregulate expression of MHC and co-stimulatory molecules and enhance the ability of these cells to function as effective A PCs [34]. Furthermore, EBV infection could assist in the survival of autoreactive B cells [36]. APC antigen-presenting cell: MHC, major histocompatibility complex: PAMP, pathogen-associated molecular pattern: TCR, T-cell receptor, TLR, Toll-like receptor.
The study of virus-induced demyelinating autoimmunity has produced significant evidence in favor of the possible role of microbial agents as immuno-pathological triggers. The involvement of molecular mimicry has been demonstrated in the induction of EAE in rabbits, by means of hepatitis B virus polymerase peptides homologous with MBP. It has also been demonstrated that MBP-specific T-cell clones are reactive to mimic peptides of a herpesvirus group (Epstein-Barr virus [EBV], herpes simplex virus [HSV] and cytomegalovirus [CMV]), flu virus and papillomavirus.
Epitope spreading implies that auto-antigenic epitopes are continuously discovered during the autoimmune inflammatory reaction. It is interesting to note that epitope spreading both in EAE and in MS was described in a study showing that the initial self-reactivity to a primary autoantigen decreased and disappeared, being replaced by secondary self-reactivity to new antigens during disease progression. Many studies have identified several linear and conformational sequence homologies between myelin structure peptides and antigens of different viral and bacterial species in MS. This leads to the conclusion that the structural similarity between microbic antigenic epitopes and auto-peptides could induce a self-aggressive response by T-cells ([63,64]).
The structural similarity between viral epitopes recognized by T-cells and auto-peptides is the molecular mimicry criterion that induces a self-aggressive response by T-cells. A data-base research was conducted considering the molecular homologies of the structural type (molecular mimicry motif) rather than linear sequence. Subsequently, a panel of 129 peptides that corresponded to the molecular mimicry motif was tested on seven MBP-specific T-cell clones from MS patients. Seven viral peptides and one bacterial peptide efficiently activated three of these clones. Only one peptide could be identified by sequence alignment as a molecular mimic. The observation that a single T-cell receptor is able to recognize fairly distinct but structurally related peptides from multiple atoms has important implications for understanding the pathogenesis of autoimmunity. The peptides – DQ1 or DR2 restricted and recognized by 3 different MBP-Specific T-Cell Clones (MBP 85–99) – are 10 protein peptides belonging to 6 different viruses and one bacterium: Herpes simplex UL15 protein; Adenovirus type 12 ORF; Pseudomonas phosphomannomutase; Human papillomavirus type 7 L2 protein; EBV DNA polymerase; Influenza type A hemagglutinin; Reovirus type 3 sigma 2 protein; EBV DNA polymerase; influenza type A hemagglutinin; Herpes simplex DNA polymerase [65] (Tab le2).
Table 2.
Sequence alignment of viral/bacterial mimicry peptides that stimulate MBP-Specific T cell clones that are DQ1 or DR2 restricted.
| Peptides Recognized by Clone Hy.1B11 (DO1 Restricted) | ||||
| 85 | 90 | 94 | 99 | |
| MBP(85–99) Herpes simplex, UL15 protein Adenovirus type 12, ORFPseudomonas, phosphomannomutaseHuman papillomavirus type 7, L2 protein |
ENPVVHFFKNIVTPR FRQLVHFVRDFAQLL DFEVVTFLKDVLPEF DRLLMLFAKDWSRN IGGRVHFFKDISPI A | |||
| Peptides Recognized by Clone Hy.2E11 (DR2 Restricted) | ||||
| 85 | 90 | 94 | 99 | |
| MBP(85–99) EBV, DNA polymerase Influenza type A, hemagglutinin Reovirus type 3, sigma 2 protein |
ENPVVHFFKNIVTPR TGGVYHFVKKHVHES YRNLVWFIKKNTRYP MARAAFLFKTVGFGG | |||
| Peptides Recognized by Clone Hy.1G11 (DR2 Restricted) | ||||
| 85 | 90 | 94 | 99 | |
| MBP(85–99)EBV, DNA polymeraseInfluenza type A, hemagglutininHerpes simplex, DNA polymerase | ENPVVHFFKNIVTPR TGGVYHFVKKHVHES YRNLVWFIKKNTRYP GGRRLFFVKAHVRES | |||
Only the human papillomavirus peptide has obvious sequence similarity with the MBP(85–99) peptide (residues identical in the 89–95 segment are underlined). All mimicry peptides that stimulate the DQ1-restricted clones have aspartic acid (D) at position 94 (a putative TCR contact); hydrophobic residues were selected at position 95 (a putative MHC contact). In contrast, two of three mimicry peptides for the DR2-restricted clones have a positive charge (lysine) at position 94.
Kai If. Wucherpfennig and Jack L. Strominger. Molecular Mimicry in T Cell-Mediated Autoimmunity. Cell. Vol. 80.695–705. March 10.199.
8.1. Measles virus
Much attention has been paid by researchers to the ability of the measles virus (MV) to induce post-infectious encephalitis and subacute sclerosing panencephalitis. High anti-MV antibody indices have been reported by many researchers in serum and in (CSF) samples of MS patients, suggesting that the immune response to this virus plays a role in the pathogenesis of MS.
In 1965, when measuring the antibody titers of MV in the serum and in the CSF by means of neutralization and complement fixation tests, Adams et al. found higher antibody titers in MS patients compared to control subjects. The most significant data concerned CSF from MS patients, with viral antibody titers in over 75% of the cases tested, while no antibodies were present in the control subjects [66].
In a serological study conducted by Cendrowski in 1976, in 159 of 161 patients with multiple sclerosis (MS), a significant increase in the level of measles antibodies against hemagglutinin (HI) was found in serum and in 92 of these the presence of anti-HI measles antibodies in CSF was significantly more frequent than in the controls [67]. In the same study, a higher titer of measles antibodies in the CSF was found in patients with a malignant disease course (P less than 0.001). It may thus be concluded that persistent infection with proviruses or non-specific stimulation of some lymphocyte clones in individuals with genetic susceptibility results in an excessive synthesis of viral antibodies in MS patients [68]. In another study conducted utilizing hemagglutination inhibition testing to measure the rate of measles antibodies on serums from over 300 patients (133 MS patients and 172 control subjects), the mean measles antibody titers in MS patients were twice as high as in the controls. The authors conclude that these results suggest the persistence of the virus in some form in some patients with multiple sclerosis, but that the final evidence of a possible causal relationship between measles and multiple sclerosis cannot be determined until the MV is isolated from patients with MS [69]. Other studies on measles virus antibodies have confirmed the presence of high antibody titers in MS patients compared to healthy controls [70,71] with a higher titre in the CSF compared to serum [72].
After a healthy person has been infected or vaccinated against measles, normally the titers of measles antibodies tend to decrease over time. A research presented At the Fifth Joint Triennial Congress of the European and American Committees for the treatment and research in multiple sclerosis, the results of research conducted to evaluate the variation in MV antibody levels in MS patients according to age and disease duration were presented. 161 patients with multiple sclerosis aged between 15 and 49 years (age of MS onset between 10 and 39 years) and a control group of 50 healthy volunteers aged between 18 and 57 years were studied as regards on antibody levels of measles in serum and CSF by means of enzyme immunoassays. However, most patients with MS, in addition to having higher measles antibody titers in serum and CSF compared to healthy controls, showed an increase in these titres over time and in relation to the duration of the disease [73].
The numerous investigations on the relationship between MV and multiple sclerosis have produced results that demonstrate the presence of high rates of anti-MV antibodies especially in the CSF, but no information on the virus isolate as such in MS patients is available as concerns the CSF, nervous system tissues. This leads us to conclude that if there is a causal relationship between MV and MS, it is likely that it is not to be due to a persistent infection but rather to an immune-mediated trigger of the disease in which the high antibody titers only play an immuno-pathogenetic role.
8.2. Epstein Barr virus
The hypothesis that the Epstein-Barr virus (EBV) may play a role in the pathogenesis of MS is supported by the observation of the increased risk of MS in subjects with a history of mononucleosis [74,75] or with high EBNA antigen antibody titers in subjects affected by MS [[76], [77], [78], [79]], as well as by the higher prevalence of EBV infection in MS cases compared to controls [[80], [81], [82], [83]].
Ascherio and Munger analyzed data from 13 studies and found that, compared to people with EBV in childhood, those infected in adolescence and adulthood had a 23 times higher chance of getting MS, while those who they were EBV negative 10 times less likely to have MS [84]. In a study published in Neurology, Annette Langer-Gould et al. showed that EBNA-1 seropositivity was independently associated with a greater likelihood of MS in all 3 racial/ethnic groups studied (p < 0.001 for blacks and Caucasians, p = 0.02 for Hispanics) [85].
A “case-control” study of serum samples from 305 individuals with MS and 610 healthy controls from the collection of sera taken from Active-duty US Army, Navy, and Marines personnel and kept in the Department of Defense Serum Repository (DoDSR) was conducted to determine whether the risk of multiple sclerosis (MS) increases following primary infection with the Epstein-Barr virus (EBV). EBV infection time was determined by measuring the antibody titers in serum samples collected before MS onset in cases and controls on several occasions. Ten (3.3%) cases and 32 (5.2%) controls were initially negative for EBV. All 10 EBV negative cases became EBV positive prior to the onset of MS; in contrast, only 35.7% (10) of the 28 controls with seroconverted follow-up samples (exact p-value = 0.0008) did so. The authors conclude that the risk of multiple sclerosis is extremely low among non-EBV infected individuals, but increases sharply in these individuals following EBV infection [86].
A systematic review as well as a case-control meta-analysis and cohort studies on the prevalence of anti-EBV antibodies in MS were conducted. 39 research studies on Medline and Embase from 1960 to 2012 were considered. Mantel-Haenszel (OR) probability ratios for serum-positivity of anti-EBV antibodies were calculated and meta-analyses were conducted. Quality assessment was performed using a modified version of the Newcastle Ottawa scale. It was found that most studies reported an acceptable selection and comparability of cases and controls. The majority of studies found a higher prevalence of anti-EBNA IgG and anti-VCA IgG in cases than controls. The meta-analysis showed a significant OR for serum positivity to anti-EBNA and anti-VCAIgG Ig in MS cases (4.5 [95% confidence interval (CI) 3.3 to 6.6, p, 0.00001] and 4.5 [95% CI 2.8 to 7.2, p, 0.00001] respectively). However, an examination of the funnel diagram suggested a publication distortion as concerns the reporting of anti-EBNA IgG. No significant difference in OR was found for serum positivity to anti-EA IgG (1.4 [95% CI 0.9–2.1, p = 0.09]). These results support previous systematic reviews, although a distortion of publication cannot be ruled out. The methodological conduct of the studies could be improved, in particular as regards the communication and carrying out of laboratory analyses [87].
A study of 145 cases of MS collected in 1970 and 145 controls investigated the association between MS and exposure to uncommon viruses or infection at an older age with 1 or more common viruses. There was a strong positive association with a history of infectious mononucleosis (IM) in older subjects exposed to the Epstein-Barr virus. Significant positive associations were also found for the number of different domiciles before adulthood and for travel abroad. This suggests an increased probability of exposure to uncommon viruses or multiple strains of a common agent [88].
Comparing a cohort of 494 cases of infectious mononucleosis (MI) with a multiple sclerosis (MS) registry, 3 MS cases were found, with a 12-year MI-MS interval and a relative 3.7 risk of MS occurring after MI (p = 0.05) [89].
EBV-specific cellular immune responses were studied using enzyme-linked proliferation and immunospotic assays and humoral immune responses by means of anti-EBV antibody analysis, in a cohort of 164 subjects, including 108 patients at different stages of MS, 35 with other neurological diseases and 21 healthy control subjects. In addition, the cohort was tested against cytomegalovirus (CMV), another neurotropic herpes virus that was not convincingly associated with MS, nor believed to be harmful to the disease. All age data were corrected using linear regression analysis on the total cohorts of EBV- and CMV-infected subjects. In the entire cohort, the rate of EBV and CMV infections was 99% and 51% respectively. The frequency of EBV-specific CD8 + T cells secreting IFN-gamma in MS patients was significantly higher than that of healthy controls. EBV specific CD8 + T cell responses were found to decrease in 12/13 patients with clinically isolated syndrome (CIS) followed prospectively for 1.0 ± 0.2 years. These data show high levels of CD8 + T cell activation against EBV - but not CMV - early in the course of MS, thus supporting the hypothesis that EBV may be associated with the onset of this disease [90].
8.3. Human herpes virus 6
HHV-6A/B has been studied extensively as a potential viral trigger agent of multiple sclerosis. HHV-6A exhibits a particular tropism for the central nervous system (CNS) it infects and where it can remain latent. Numerous clinical studies have shown a correlation between MS and several parameters that evaluate HHV-6A/B infection and the possible biological mechanisms of triggering the disease by the virus have also been studied [91]. A relationship between HHV-6A and MS was initially suggested by the immunohistochemical demonstration of the viral antigen in oligodendrocytes of MS white matter lesions but not in the control brain [92]. HHV-6 DNA was detected at higher frequencies in cerebrospinal fluid and mononucleate cells of peripheral blood in MS patients [93,95]. An analysis of brain biopsies and autopsy brain tissue indicated that HHV-6A/B DNA was more frequently present in demyelinating brain lesions of MS patients than in control brain tissues. Immunohistochemistry analyses confirmed the presence at a higher frequency of viral proteins in the oligodendrocytes and astrocytes of demyelinating plaques in the brains of MS patients [[96], [97], [98]]. Serum levels of IgG and IgM HHV-6A/B in CSF were higher in numerous studies in patients with MS [[93], [94]]. In addition, viral charges and high levels of specific HHV-6A/B IgG were mostly detected in patients whose disease conditions were exacerbated [99,100]. Finally, it appears that HHV-6A is more frequent than HHV-6B in the serological studies of MS patients [101].
Recent studies on the biology of HHV-6A have provided indications of the mechanism by which HHV-6A can intervene in MS pathogenesis. Molecular mimicry has been proposed as the mechanism for activating the autoimmune process. In this regard, cross-reactivity between basic myelin protein and HHV6 in MS has been demonstrated. One study reported that 15%–25% of HHV-6 specific T-cell clones obtained from healthy donors or multiple sclerosis patients were cross-reactive to basic myelin protein (MBP) [102]. Close sequence homology was found between MBP (residues 96–102) and the U24 protein of HHV-6 (residues 4–10) and over 50% of the T-cells of MS patients reacted crosswise with MBP and at a synthetic peptide corresponding to residues 1 to 13 of U24 HHV-6. Higher antibody titers were found in these patients, both for the U24 peptide and for the MBP peptide, compared to those of the healthy controls, suggesting the sensitization of B-cells to the two antigens [103]. This specific cross-reactivity was further confirmed by a more recent Chinese study in which the presence of cytotoxic CD8 + cross-reactive T-cells was more significant [104]. Overall, these studies suggest that HHV-6A infection can activate T-cell responses, which can be simultaneously directed against the myelin sheath.
In particular, it has been suggested that by binding HHV-6 to the CD46 receptor, T-cells activate the autoimmune reaction. CD46 is the cellular receptor for numerous bacteria and viruses including HHV-6A [105] and has also been related to T-cells regulation. In fact, CD46/CD3 costimulation has been shown to strongly promote T-cell proliferation, leading to the conclusion that CD46 acts as a potent costimulatory molecule for human T-cells [106]. Furthermore, CD46 can modulate T-cell responses, depending on which cytoplasmic tail is expressed [107] and can induce CD4 + T-cells towards a type 1 regulatory T-cells (Tr1) phenotype with high IL-10 production [108].
It could therefore be hypothesized that HHV-6A/B can modulate the functions of the CD46 receptor by binding to it. A clinical study indicated that the increase in HHV-6A/B viral load was related to an increase in CD46 expression in MS patients with consequent changes in its functions, i.e. a strong reduction in IL-10 secretion from part of the T-cells [109], increased production of IL-23 by DC and expression of IL-17 by T cells [110,111].
Soluble form levels of CD46 have been found to increase in the serum of patients with autoimmune disorders, including MS [112]. In addition, a physical association was found between the HHV-6 virion and CD46 in the form of HHV-6/CD46 complexes in the serum of MS patients, but not in the control serum [113]. Thus, if HHV-6 is attached to CD46 in its soluble form, several epitopes of the virus that are homologous to myelin proteins [114] and, being more exposed, they are more easily recognized by the cells that activate the autoimmune response. Furthermore, CD46 is highly expressed in the blood brain barrier (BBB) [115] and this can mediate HHV-6 access to the brain.
Finally, CD46 also regulates the complement system by inactivating C3b/C4b deposited on the membrane of autologous cells; therefore, the CD46/HHV6 bond can favor the activation of the complement system in multiple sclerosis, both at the peripheral and intrathecal level, thereby contributing to the pathogenesis of the disease [116,117].
8.4. Varicella zoster virus
Both MS and chickenpox have a higher prevalence in temperate zones and both are rare in countries closest to the equator. Migration studies suggest the hygienic hypothesis that an infectious agent acquired before the age of 14 would play a role in the risk of contracting MS. The Hutterites, who practice home-schooling, have been found to have a lower incidence of chicken pox, shingles and multiple sclerosis. In line with these epidemiological observations, compared to their neighbors, they have reduced varicella-zoster seropositivity. Instead, MS patients report a higher incidence of shingles at an early age compared to a group of non-MS patients [118]. In Mexico, a strong and progressive increase in the incidence of multiple sclerosis has been reported over the past 20 years. A case-control study was conducted in this country using a questionnaire that included demographic, nutritional, infectious and personal antecedents to identify potential risk factors for MS. A previous chickenpox infection in the clinical history of MS patients was found to be the most significant risk factor for this disease [119]. In a Canadian research study, 633 MS patients were interviewed regarding a possible history of varicella zoster virus (VZV) infection. The controls were healthy subjects or patients with other neurological diseases (OND). In the MS group with a positive/probable history of herpes zoster (HZ), the HZ/MS rate was 106/633 (16.8%) with a VZV infection occurring at an early age. In healthy subjects, the rate was 192/3534 (5.4%); and among patients with OND it was 42/616 (6.8%). More than one attack of HZ was also more common in the MS group. This study adds evidence that MS patients have a peculiar relationship with the shingles virus [120]. In a study of 82 patients with relapsing-remitting MS, VZV DNA was investigated by PCR, as well as serum IgG and IgM antibodies against the virus. Viral DNA was found in the mononuclear cells of 13 (87%) of 15 MS patients during acute relapse. All patients who were tested during remission (n = 67) were DNA negative. All healthy control patients (n = 20) and those with other neurological diseases (n = 100) were negative. There were no differences in serum antibodies against VZV. The discovery of VZV DNA in MS patients during the clinical recurrence of the disease suggests its participation in the etiopathogenesis of multiple sclerosis [121]. The presence of VZV DNA, herpes simplex virus 1 and 2; EBV and human herpes-virus-6 (HHV6) in mononuclear cells of patients with MS during the relapse phase (n = 40), during remission (n = 131) and healthy controls (n = 125) were investigated. VZV DNA was found in 95% of MS patients during relapse and in 17% during remission; all controls were negative; in contrast, HHV6 DNA was found in 24% of MS patients during relapse and in 2% during remission; Herpes simplex virus DNA was not found in any subject; and DNA from EBV was found in a similar percentage of subjects from all groups. In conclusion, the conspicuous presence of VZV during relapses of MS may indicate a period of active infection and suggests the involvement of VZV in the pathogenesis of MS [122]. In an Iranian research study, the prevalence of the VZV was studied in patients with RRMS and in a group of healthy subjects for comparison. Plasma and peripheral blood mononuclear cells collected from MS patients (n = 82) and controls (n = 89) were screened as regards the presence of anti-VZV IgG antibodies and VZV DNA by means of ELISA methods and PCR. Of all MS patients, 78 (95.1%) and 21 (25.6%) tested positive anti-VZV and VZV DNA, respectively. Statistical analysis of PCR results showed a significant correlation for the presence of VZV DNA (P < 0.001) among MS patients, but not for the anti-VZV antibody rate. These results support the hypothesis that VZV may contribute to MS in establishing a systemic infection process and inducing an immune response [123]. Evidence supporting the association of VZV with RRSM was considered in a 2010 publication. Regarding laboratory investigations, consistent evidence emerged as regards the presence of DNA from VZV in the leukocytes and CSF of patients with MS limited to the period of clinical recurrence. The above observations and the characteristics of neurotropism and long latency periods of VZV support the idea that VZV is involved in the etiology of MS [124].
8.5. Chlamydia pneumoniae
Chlamydia pneumoniae (Cpn) is a common respiratory pathogen that is now implicated in a number of chronic illnesses such as cardio and cerebro vascular diseases. Several studies have focused attention on Cpn’s ability to infect the CNS. The tropism of C. pneumoniae on neural tissue suggests that it could play a role in several neurological diseases [125]. Some research has identified Chlamydia DNA pneumoniae in the CSF of some people with MS. In a study conducted using bacterial cultures, Cpn was isolated from 64% of CSF samples from MS patients and only 11% of healthy controls. PCR identified the outer Cpn membrane protein gene in the CSF of 97% of multiple sclerosis patients and 18% of controls and ELISA showed that 86% of MS patients had Cpn antibodies in their CSF [126].
DNA and mRNA transcripts of Chlamydophila pneumoniae were studied by PCR and RT-PCR in fresh CSF and peripheral blood mononuclear cell (PBMC) samples cocultured in Hep-2 cell lines and collected from 14 patients with defined MS RR and 19 patients with other inflammatory disorders (OIND) and non-inflammatory (NIND) neurological controls. C. pneumoniae DNA and mRNA positivity was detected in CSF and PBMC of 9 patients with MS RR (64.2%) showing evidence of disease activity, while only 3 controls were positive for chlamydia DNA. These preliminary results suggest that C. pneumoniae may occur in a persistent and metabolically active state both peripherally and intrathecally in MS, but not in OIND and NIND [127].
A meta-analysis of research assessing the presence of Cpn among patients with multiple sclerosis (MS) and other patients with neurological diseases or healthy controls identified 26 studies with 1332 MS patients and 1464 controls. Using random effect methods, MS patients were found to be more likely to have detectable levels of Cpn DNA (OR = 3.216; 95% CI: 1.204, 8.585) in their CSF and immunoglobulins synthesized intrathecally (OR = 3.842; 95% CI: 1.317, 11.212), compared to other patients with neurological diseases. However, there is no evidence of an increase in serum immunoglobulin levels (OR = 1.068; 95% CI: 0.745, 1.530). Similarly, there is no evidence for the association of immunoglobulins against Cpn in CSF (OR = 3.815; 95% CI: 0.715, 20.369). In random-effect meta-regressions, adjusting for the confounding effect of gender differences results in stronger and statistically significant associations of multiple sclerosis with detectable levels of Cpn DNA, immunoglobulins synthesized intrathecally and immunoglobulins in the CSF. The authors conclude that although the presence of Cpn is clearly more likely in MS patients, these results are not sufficient to establish an etiological relationship [128].
After molecular and culture tests of Cpn CSF of MS patients, the association between C. pneumoniae and MS has been intensively studied with controversial results. Seroepidemiological reports showed no strong association between Cpn infection and MS risk, neither when using bacterial isolation techniques in the CSF and in the brain tissue of MS patients nor when DNA was isolated by means of PCR in the CSF or intrathecal synthesis of anti-Cpn. Moreover, Cpn IgG were not detectable in MS patients or, when present, they were not selectively associated with MS. However, an association between PCR positivity for Cpn in CSF and disease activity was found in a subset of MS patients. Intrathecal production of high affinity anti-Cpn IgG especially in the progressive forms of MS and metabolically active Cpn has been identified in CSF. Cpn has been recognized in CSF and brain tissue at the immunohistochemical, molecular and ultrastructural level. Cpn was also able to induce the animal model of MS. This growing set of data suggests that Cpn infection may play a role in the pathogenesis in a subset of MS patients [129].
8.6. Helicobacter pylori
In the past, several studies have suggested that Helicobacter pylori (HP), is a potential trigger of chronic autoimmune diseases [130], in particular of various neurological disorders such as Alzheimer’s, Parkinson’s, seizure disorders, cerebrovascular diseases, mild cognitive impairment, migraine and ophthalmic disorders and MS/NMO [131].
A prospective comparative study evaluated the incidence of active Hp infection from histology and endoscopic abnormalities in 44 patients with relapsing-remitting MS and 20 anemic controls.
The overall prevalence of histologically confirmed active Hp infection in 44 patients with multiple sclerosis was 86.4% against 50% in 20 matched anemic control participants (P = 0.002, odds ratio 6.33, IC 95% 1.85–21.64). Concomitant autoimmune diseases, including hypothyroidism and ulcerative colitis, were present only in patients with MS. In addition, a trend towards an increased presence of endoscopic findings of related HP gastro-esophageal pathologies has been observed in MS patients. In conclusion, HP infection appears to be more frequent in MS patients, indicating HP infection as a possible causal factor for the development of MS [132]. A study in Japanese multiple sclerosis patients investigated the association between HP neutrophil activation protein (HP-NAP) and anti-aquaporin-4-induced neural damage (AQP4) in MS/NMO patients. There are two distinct subtypes of multiple sclerosis (MS) in Asians: opticospinal (OSMS) (similar to NMO optic neuromyelitis) and conventional MS (CMS). The serums from 162 patients with multiple sclerosis, 37 patients with other inflammatory neurological diseases (OIND) and 85 healthy subjects have been studied. Half of the patients with OSMS had the NMO-Immunoglobulin G (IgG)/anti-AQP4 antibody. HP seropositivity rates were significantly higher in patients with MS/NMO positive for anti-AQP4 antibodies (19/27, 70.4%) than in CMS patients negative for anti-AQP4 antibodies (22/83, 26, 5%). Among HP infected subjects, the anti-HP-NAP antibody was significantly more common in patients with AQP4 +/MS and AQP4 -/OSMS than in healthy subjects (36.8%, 34.6% versus 2,8%). Among patients with AQP4 +/MS, a significant positive correlation was found between anti-HP-NAP antibody levels and final scores on the Kurtzke expanded disability status scale [133]. Long et al. determined the status of HP infection in a cohort of 42 patients with MS, 2 NMO, 17 at high risk for NMO, and 27 healthy controls. HP antibodies were found in 90.4% of NMO, in 95.8% of high risk NMO, in 73.8% of MS and in 59.3% of controls. There was no statistically significant difference between MS and the control group (P = 0.726). Interestingly, 93% of patients with aquaporin-4 antibodies were also HIV positive for HP [134]. Another study showed that the role of HP’s Heat Shock Protein antigen (HSP60) is inconsistent in the pathogenesis of MS. On the contrary, antibodies against HP’s antigens different from HSP prove to be factors rupturing tolerance towards MS autoantigens. The prevalence and clinical significance of antibodies (ab) against 14 HP antigens in 139 patients with relapsing-remitting MS (RRMS, n = 102) or secondary progressive (SPMS, n = 37) and 68 healthy controls (CS) were studied. Anti-VacA ab was shown to be more frequent in SPMS than in CS; the anti-p54, anti-p29-UreA and anti-p26 correlated with extended disability scale (EDSS)); ati-p26 and anti-p17 correlated with the number of relapses. On the contrary, the anti-flagellin and anti-p41 in MS were less frequent than in CS. The antibodies against 5 of the 14 antigens were less frequent in CSRMS, including p41, p54-flagellin, p29-UreA, p67-FSH and p120-CagA. The difference in antibody responses between MS and CS and between RRMS and SPMS, being prevalent in SPMS compared to RRMS, suggest an association between anti-Hp and SPMS SM [135].
A meta-analysis was conducted on 9 selected studies on PubMed and EMBASE to determine the prevalence of HP infection in MS patients. These studies included a total of 1553 MS patients and 1253 controls. Overall, the prevalence of HP infection in MS patients was lower than that in the control groups (24.66% versus 31.84%, OR = 0.69, 95% CI: 0.57–0.83, P < 0.0001). A subgroup analysis revealed that HP infection levels among MS patients were lower than control subjects in western countries (11.90% versus 16.08%, OR = 0.63, CI at 95%: 0.43–0.91, P = 0.01), but were not statistically significant in the eastern countries (39.39% against 43.82%, OR = 0.79, 95% CI: 0.55–1.14, P = 0.20). These data show that HP infection and MS are negatively correlated, especially in western countries [136]. Other perspectives have even reported a low prevalence of anti-Hp antibodies in a wide range of MS, concluding that it may even have a protective role for MS [137,138].
In particular, a complete database search, including PubMed/MEDLINE and EMBASE, was performed on the studies available until January 2016. The inclusion criteria were observational studies conducted on adults to evaluate the association between HP infection and multiple sclerosis. The authors demonstrated a significantly lower prevalence of helicobacter pylori infection in patients with multiple sclerosis, concluding that this HP could be a protective factor for the development of multiple sclerosis [139].
The conflicting data at our disposal, coming from research on the relationship between HP and MB, suggest that the matter is rather complex. Probably the genetic predisposition of the subjects affected by MS and the different immune response of the subjects predisposed to the different HP antigens play an important role in this phenomenology and could represent a confounding distortion factor in the results of the different studies. In fact, it turns out that only HLA DRB1 *1602 subjects develop MS triggered by HP [140].
9. Other bacteria implicated
Other bacteria have been found to be involved in various ways in the pathogenesis of multiple sclerosis, some as triggering or aggravating factors, others even with protective or curative effects. Below we report a small roundup of reports on this subject:
-
-
Mycobacterium avium paratuberculosis (MAP): in Sardinian patients with MS, MAP DNA was found and humoral immune response against the MAP2694 protein was observed. Moreover, the intrathecal synthesis of anti-MAP IgG associated to MBP antibodies has been reported [141,142] and self-reactive T-cells appear to be activated in the CNS [[143], [144], [145], [146], [147], [148]].
-
-
Mycoplasma pneumoniae (MycPn): antibodies in the serum and intrathecal synthesis of IgG in the anti-MycPn cerebrospinal fluid have been identified in MS patients [149]. Furthermore, MycPn has been isolated from brain and urine samples of MS patients [150], and MycPn invasions in the brain seem to cause demyelination [151].
-
-
Clostridium perfringens: Immunoreactivity to epsilon toxin in PBMC and CSF was found and the bacterium was isolated in the feces of MS patients [152]. Neurotoxin appear to affect endothelial cells, myelinated fibers and CNS oligodendrocytes [153].
-
-
Among the microbiota: Euryarchaeota, Firmicutes, Proteobacteria (Sutterella) have been demonstrated as aggravating factors of the EAE [154]; an increased incidence of these species has been demonstrated in a spontaneous mouse model of demyelinating disease [155]. Furthermore, these bacterial species are most frequent in the microbiota of patients with MS compared to healthy controls [[155], [156], [157]]. Pro-inflammatory response and reduced IL10 production [154] due to staphylococcal enterotoxin B has been proposed as one of the possible mechanisms of action of this pathogenic effect [158].
9.1. Protective effect
-
-
Mycobacterium bovis BCG has been shown to be a disease suppression factor [159,160]. Reduced cerebral MRI activity and low antibody titer in MS patients compared to controls was shown [161,162] with skin vaccination or direct inoculation in the brain or intestines of mice [159,160,163]. Antigenic competition, diversion of autoreactive T-cells into granulomas has been proposed as a mechanism of action [159].
-
-
Among the microbiota: A lower frequency of Firmicutes (Clostridia), Actinobacteria, Bacteroides has been shown in the microbiota of MS patients compared to healthy controls [164]. Furthermore, reduced susceptibility to the disease following the oral administration of Bacteroides fragilis PSA has been reported. Anti-inflammatory response, expansion of CD39 Treg cells and increased IL10 expression seem to be at the basis of this protective effect [154].
10. Conclusions
The hypothesis of an immune-mediated etiopathogenesis of MS in HLA gene predisposed subjects is supported by the wide range of evidence gathered in this article. The results of these studies confirm the correlation of certain HLA haplotypes (DQ1 and DR2) with MS, suggesting that these genes play an immunological role in disease susceptibility. Similarly, many studies support the idea that certain viruses and bacteria intervene in determining MS (Measles virus, EBV, HHV6, HZV, Chlamydia pneumoniae and Helicobacter Pylori). Finally, studies on molecular mimicry suggest that microbial antigenic epitopes responding to structural requirements for both class II MHC binding and TCR recognition are probably able to activate cross-reactive T-cells capable of generating a self-reactive immune response to myelin epitopes that exhibit linear or conformational sequence homologies with these microbial antigens. This evidence supports the hypothesis that the autoimmune reaction triggering MS can be effectively developed by an immunomediated process set off by a previous infection in HLA-predisposed subjects due to a condition of molecular mimicry between the microbial antigens and auto antigens of the myelin structure.
Declaration of competing interest
The author certify that they have NO affiliations with or involvement in any organization or entity with any financial interest (such as honoraria; educational grants; participation in speakers’ bureaus; membership, employment, consultancies, stock ownership, or other equity interest; and expert testimony or patent-licensing arrangements), or non-financial interest (such as personal or professional relationships, affiliations, knowledge or beliefs) in the subject matter or materials discussed in this manuscript.
Acknowledgments
I wish to thank Prof. Angelo Micozzi for his formulation of the chronic disease model in an immunomediated nature.
I also wish to thank prof. Marco Mancini for the research he conducted on this topic.
References
- 1.Compston A., Coles A. Multiple sclerosis. Lancet. 2008;372:1502–1517. doi: 10.1016/S0140-6736(08)61620-7. [PubMed] [DOI] [PubMed] [Google Scholar]
- 2.Alastair Compston Ian McDonald John Noseworthy Hans Lassmann David Miller Kenneth Smith Hartmut Wekerle Christian Confavreux. McAlpine’s Multiple Sclerosis. Imprint: Churchill Livingstone.
- 3.Wallin M.T., Culpepper W.J., Nichols E., Bhutta Z.A., Gebrehiwot T.T., Hay S.I., Khalil I.A., Krohn K.J., Liang X., Naghavi M., Mokdad A.H., Nixon M.R., Reiner R.C., Sartorius B., Smith M., Topor-Madry R., Werdecker A., Vos T., Feigin V.L., Murray C.J.L. Global, regional, and national burden of multiple sclerosis 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019 Mar;18(3):269–285. doi: 10.1016/S1474-4422(18)30443-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lassmann H.1. Neuropathology in multiple sclerosis: new concepts. Mult. Scler. 1998 Jun;4(3):93–98. doi: 10.1177/135245859800400301. [DOI] [PubMed] [Google Scholar]
- 5.M1 Sospedra, Martin R. Immunology of multiple sclerosis. Annu. Rev. Immunol. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- 6.Schmidt H., Williamson D., Ashley-Koch A. HLA-DR15 haplotype and multiple sclerosis: a HuGE review. Am. J. Epidemiol. 2007;165:1097–1109. doi: 10.1093/aje/kwk118. [DOI] [PubMed] [Google Scholar]
- 7.Patsopoulos N.A., Barcellos L.F., Hintzen R.Q., Schaefer C., van Duijn C.M. Fine-mapping the genetic association of the major histocompatibility complex in multiple sclerosis: HLA and non-HLA effects. PLoS Genet. 2013;9 doi: 10.1371/journal.pgen.1003926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Q., Lin C.Y., Dong Q., Wang J., Wang W. Relationship between HLA-DRB1 polymorphism and susceptibility or resistance to multiple sclerosis in Caucasians: a meta-analysis of non-family-based studies. Autoimmun. Rev. 2011;10:474–481. doi: 10.1016/j.autrev.2011.03.003. [DOI] [PubMed] [Google Scholar]
- 9.Kwon O.J., Karni A., Israel S., Brautbar C., Amar A. HLA class II susceptibility to multiple sclerosis among Ashkenazi and non-Ashkenazi Jews. Arch. Neurol. 1999;56:555–560. doi: 10.1001/archneur.56.5.555. [DOI] [PubMed] [Google Scholar]
- 10.Karni A., Kohn Y., Safirman C., Abramsky O., Barcellos L. Evidence for the genetic role of human leukocyte antigens in low frequency DRB1*1501 multiple sclerosis patients in Israel. Mult. Scler. 1999;5:410–415. doi: 10.1177/135245859900500i607. [DOI] [PubMed] [Google Scholar]
- 11.Cocco E., Sardu C., Pieroni E., Valentini M., Murru R. HLA-DRB1-DQB1 haplotypes confer susceptibility and resistance to multiple sclerosis in Sardinia. PloS One. 2012;7 doi: 10.1371/journal.pone.0033972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cocco E., Murru R., Costa G., Kumar A., Pieroni E. Interaction between HLA-DRB1-DQB1 haplotypes in Sardinian multiple sclerosis population. PloS One. 2013;8 doi: 10.1371/journal.pone.0059790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oksenberg J.R., Barcellos L.F., Cree B.A., Baranzini S.E., Bugawan T.L. Mapping multiple sclerosis susceptibility to the HLA-DR locus in African Americans. Am. J. Hum. Genet. 2004;74:160–167. doi: 10.1086/380997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yoshimura S., Isobe N., Matsushita T., Masaki K., Sato S. Genetic and infectious profiles influence cerebrospinal fluid IgG abnormality in Japanese multiple sclerosis patients. PloS One. 2014;9 doi: 10.1371/journal.pone.0095367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matsuoka T., Matsushita T., Osoegawa M., Kawano Y., Minohara M. Association of the HLA-DRB1 alleles with characteristic MRI features of Asian multiple sclerosis. Mult. Scler. 2008;14:1181–1190. doi: 10.1177/1352458508097818. [DOI] [PubMed] [Google Scholar]
- 16.Granieri E., Casetta I., Govoni V., Tola M.R., Marchi D. The increasing incidence and prevalence of MS in a Sardinian province. Neurology. 2000;55:842–848. doi: 10.1212/wnl.55.6.842. [DOI] [PubMed] [Google Scholar]
- 17.Isobe N., Gourraud P.A., Harbo H.F., Caillier S.J., Santaniello A. Genetic risk variants in African Americans with multiple sclerosis. Neurology. 2013;81:219–227. doi: 10.1212/WNL.0b013e31829bfe2f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brassat D., Salemi G., Barcellos L.F., McNeill G., Proia P. The HLA locus and multiple sclerosis in Sicily. Neurology. 2005;64:361–363. doi: 10.1212/01.WNL.0000149765.71212.0A. [DOI] [PubMed] [Google Scholar]
- 19.Barcellos L.F., Sawcer S., Ramsay P.P., Baranzini S.E., Thomson G. Heterogeneity at the HLA-DRB1 locus and risk for multiple sclerosis. Hum. Mol. Genet. 2006;15:2813–2824. doi: 10.1093/hmg/ddl223. [DOI] [PubMed] [Google Scholar]
- 20.Kaimen-Maciel D.R., Reiche E.M., Borelli S.D., Morimoto H.K., Melo F.C. HLA-DRB1* allele-associated genetic susceptibility and protection against multiple sclerosis in Brazilian patients. Mol. Med. Rep. 2009;2:993–998. doi: 10.3892/mmr_00000204. [DOI] [PubMed] [Google Scholar]
- 21.Ramagopalan S.V., Morris A.P., Dyment D.A., Herrera B.M., DeLuca G.C. The inheritance of resistance alleles in multiple sclerosis. PLoS Genet. 2007;3:1607–1613. doi: 10.1371/journal.pgen.0030150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anagnostouli M.C., Manouseli A., Artemiadis A.K., Katsavos S., Fillipopoulou C. HLA-DRB1* allele frequencies in pediatric, adolescent and adult-onset multiple sclerosis patients, in a hellenic sample. Evidence for new and established associations. J Mult Scler. 2014;1 [Google Scholar]
- 23.Brynedal B., Duvefelt K., Jonasdottir G., Roos I.M., Akesson E. HLA-A confers an HLA-DRB1 independent influence on the risk of multiple sclerosis. PloS One. 2007;2:e664. doi: 10.1371/journal.pone.0000664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paola Cruz-Tapias, John Castiblanco, and Juan-Manuel Anaya. Major histocompatibility complex: antigen processing and presentation. Autoimmunity: from Bench to Bedside, (Chapter 10). El Rosario University Press. [PubMed]
- 25.Bernard Alain, Lamy Laurence, Alberti Isabelle. The two-signal model of t-cell activation after 30 years. January 15, 2002;73(1):S31–S35. doi: 10.1097/00007890-200201151-00011. Copyright © 2002 by Lippincott Williams & Wilkins, Inc. Supplement. [DOI] [PubMed] [Google Scholar]
- 26.Clemente-Casares1 X., Blanco J., Ambalavanan P., Yamanouchi J., Singha S., Fandos C., Tsai S., Wang J., Garabatos N., Izquierdo C., Agrawal S., Michael B., Keough V., Yong W., James E., Moore A., Yang Y., Stratmann T., Serra P., Santamaria P. Expanding antigen-specific regulatory networks to treat autoimmunity. Nature. 2016 Feb 25;530(7591):434–440. doi: 10.1038/nature16962. Epub 2016 Feb 17. [DOI] [PubMed] [Google Scholar]
- 27.Sato Fumitaka, Omura Seiichi, Martinez Nicholas E., Tsunoda Ikuo. Chapter 3 - Animal Models of Multiple Sclerosis. second ed. Academic Press; 2018. Neuroinflammation; pp. 37–72. [Google Scholar]
- 28.McFarland H.F.1, Martin R. Multiple sclerosis: a complicated picture of autoimmunity. Nat. Immunol. 2007 Sep;8(9):913–919. doi: 10.1038/ni1507. [DOI] [PubMed] [Google Scholar]
- 29.Barnett M.H., Henderson A.P.D. J W Prineas The macrophage in MS: just a scavenger after all? Pathology and pathogenesis of the acute MS lesion. Mult. Scler. 2006 Apr;12(2):121–132. doi: 10.1191/135248506ms1304rr. [DOI] [PubMed] [Google Scholar]
- 30.Lock Christopher, Jorge Oksenberg, Steinman Lawrence. The role of TNFa and lymphotoxin in demyelinating disease. Ann. Rheum. Dis. 1999;58(Suppl I):I121–I128. doi: 10.1136/ard.58.2008.i121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fletcher J.M., Lalor S.J., Sweeney C.M., Tubridy N., Mills K.H.G. T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin. Exp. Immunol. 2010 Oct;162(1):1–11. doi: 10.1111/j.1365-2249.2010.04143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Joshi N., Usuku K., Hauser S.L. The T-cell response to myelin basic protein in familial multiple sclerosis: diversity of fine specificity restricting elements, and T-cell receptor usage. Ann. Neurol. 1993;34:385–393. doi: 10.1002/ana.410340313. [DOI] [PubMed] [Google Scholar]
- 33.Tuohy V.K. Peptide determinants of myelin proteolipid protein (PLP) in autoimmune demyelinating disease: a review. Neurochem. Res. 1994 Aug;19(8):935–944. doi: 10.1007/BF00968703. [DOI] [PubMed] [Google Scholar]
- 34.Weerth S., Berger T., Lassmann H., Linington C. Encephalitogenic and neuritogenic T cell responses to the myelin-associated glycoprotein (MAG) in the Lewis rat. J. Neuroimmunol. 1999;95:157–164. doi: 10.1016/s0165-5728(99)00004-1. [DOI] [PubMed] [Google Scholar]
- 35.N1 Kaushansky, Eisenstein M., Zilkha-Falb R., Ben-Nun A. The myelin-associated oligodendrocytic basic protein (MOBP) as a relevant primary target autoantigen in multiple sclerosis. Autoimmun. Rev. 2010 Feb;9(4):233–236. doi: 10.1016/j.autrev.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 36.Katherine A., McLaughlin, Wucherpfennig Kai W. B cells and autoantibodies in the pathogenesis of multiple sclerosis and related inflammatory demyelinating diseases. Adv. Immunol. 2008;98:121–149. doi: 10.1016/S0065-2776(08)00404-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Magliozzi Roberta, Howell Owain, Vora Abhilash, Serafini Barbara, Nicholas Richard. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain. April 2007;130(Issue 4):1089–1104. doi: 10.1093/brain/awm038. [DOI] [PubMed] [Google Scholar]
- 38.B1 Serafini, Rosicarelli B., Magliozzi R., Stigliano E., Aloisi F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. 2004 Apr;14(2):164–174. doi: 10.1111/j.1750-3639.2004.tb00049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lyons1 Jeri-Anne, San1 Manuel, Happ2 Mary Pat, Anne H., Cross B cells are critical to induction of experimental allergic encephalomyelitis by protein but not by a short encephalitogenic peptide. Eur. J. Immunol. 1999;29:3432–3439. doi: 10.1002/(SICI)1521-4141(199911)29:11<3432::AID-IMMU3432>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 40.Hauser S.L., Waubant E., Arnold D.L., Vollmer T., Antel J., Fox R.J., Bar-Or A., Panzara M., Sarkar N., Agarwal S., Langer-Gould A., Smith C.H., HERMES Trial Group B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N. Engl. J. Med. 2008 Feb 14;358(7):676–688. doi: 10.1056/NEJMoa0706383. [DOI] [PubMed] [Google Scholar]
- 41.Cannella B., Raine C.S. The adhesion molecule and cytokine profile of multiple sclerosis lesions. Ann. Neurol. 1995 Apr;37(4):424–435. doi: 10.1002/ana.410370404. [DOI] [PubMed] [Google Scholar]
- 42.Pistoia V. Production of cytokines by human B cells in health and disease. Immunol. Today. 1997 Jul;18(7):343–350. doi: 10.1016/s0167-5699(97)01080-3. [DOI] [PubMed] [Google Scholar]
- 43.Selmaj K., Raine C.S., Cannella B., Brosnan C.F. Identification of lymphotoxin and tumor necrosis factor in multiple sclerosis lesions. J. Clin. Invest. 1991 Mar;87(3):949–954. doi: 10.1172/JCI115102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Duddy M.E., Alter A., Bar-Or A. Distinct profiles of human B cell effector cytokines: a role in immune regulation? J. Immunol. 2004 Mar 15;172(6):3422–3427. doi: 10.4049/jimmunol.172.6.3422. [DOI] [PubMed] [Google Scholar]
- 45.Link H., Huang Y.M. Oligoclonal bands in multiple sclerosis cerebrospinal fluid: an update on methodology and clinical usefulness. J. Neuroimmunol. 2006 Nov;180(1–2):17–28. doi: 10.1016/j.jneuroim.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 46.Correale J. De los Milagros Bassani Molinas M Oligoclonal bands and antibody responses in multiple sclerosis. J. Neurol. 2002 Apr;249(4):375–389. doi: 10.1007/s004150200026. [DOI] [PubMed] [Google Scholar]
- 47.Genain C.P., Cannella B., Hauser S.L., Raine C.S. Identification of autoantibodies associated with myelin damage in multiple sclerosis. Nat. Med. 1999 Feb;5(2):170–175. doi: 10.1038/5532. [DOI] [PubMed] [Google Scholar]
- 48.O’Connor K.C., Appel H., Bregoli L., Call M.E., Catz I., Chan J.A., Moore N.H., Warren K.G., Wong S.J., Hafler D.A., Wucherpfennig K.W. Antibodies from inflamed central nervous system tissue recognize myelin oligodendrocyte glycoprotein. J. Immunol. 2005 Aug 1;175(3):1974–1982. doi: 10.4049/jimmunol.175.3.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Warren K.G. Catz I Autoantibodies to myelin basic protein within multiple sclerosis central nervous system tissue. J. Neurol. Sci. 1993 Apr;115(2):169–176. doi: 10.1016/0022-510x(93)90221-j. [DOI] [PubMed] [Google Scholar]
- 50.Mathey E.K., Derfuss T., Storch M.K., Williams K.R., Hales K., Woolley D.R., Al-Hayani A., Davies S.N., Rasband M.N., Olsson T., Moldenhauer A., Velhin S., Hohlfeld R., Meinl E., Linington C. Neurofascin as a novel target for autoantibody-mediated axonal injury. J. Exp. Med. 2007 Oct 1;204(10):2363–2372. doi: 10.1084/jem.20071053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kennel De March A., De Bouwerie M., Kolopp-Sarda M.N., Faure G.C., Béné M.C., Bernard C.C. Anti-myelin oligodendrocyte glycoprotein B-cell responses in multiple sclerosis. J. Neuroimmunol. 2003 Feb;135(1–2):117–125. doi: 10.1016/s0165-5728(02)00434-4. [DOI] [PubMed] [Google Scholar]
- 52.Hafler D.A., Slavik J.M., Anderson D.E., O’Connor K.C., De Jager P., Baecher-Allan C. Multiple sclerosis. Immunol. Rev. 2005 Apr;204:208–231. doi: 10.1111/j.0105-2896.2005.00240.x. [DOI] [PubMed] [Google Scholar]
- 53.Mathey E., Breithaupt C., Schubart A.S., Linington C. Commentary: sorting the wheat from the chaff: identifying demyelinating components of the myelin oligodendrocyte glycoprotein (MOG)-specific autoantibody repertoire. Eur. J. Immunol. 2004 Aug;34(8):2065–2071. doi: 10.1002/eji.200425291. [DOI] [PubMed] [Google Scholar]
- 54.Lennon V.A., Kryzer T.J., Pittock S.J., Verkman A.S., Hinson S.R. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J. Exp. Med. 2005 Aug 15;202(4):473–477. doi: 10.1084/jem.20050304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Banwell B., Tenembaum S., Lennon V.A., Ursell E., Kennedy J., Bar-Or A., Weinshenker B.G., Lucchinetti C.F., Pittock S.J. Neuromyelitis optica-IgG in childhood inflammatory demyelinating CNS disorders. Neurology. 2008 Jan 29;70(5):344–352. doi: 10.1212/01.wnl.0000284600.80782.d5. [DOI] [PubMed] [Google Scholar]
- 56.Paul F., Jarius S., Aktas O., Bluthner M., Bauer O., Appelhans H., Franciotta D., Bergamaschi R., Littleton E., Palace J., Seelig H.P., Hohlfeld R., Vincent A., Zipp F. Antibody to aquaporin 4 in the diagnosis of neuromyelitis optica. PLoS Med. 2007 Apr;4(4):e133. doi: 10.1371/journal.pmed.0040133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Takahashi T., Fujihara K., Nakashima I., Misu T., Miyazawa I., Nakamura M., Watanabe S., Shiga Y., Kanaoka C., Fujimori J., Sato S., Itoyama Y. Anti-aquaporin-4 antibody is involved in the pathogenesis of NMO: a study on antibody titre. Brain. 2007 May;130(Pt 5):1235–1243. doi: 10.1093/brain/awm062. [DOI] [PubMed] [Google Scholar]
- 58.Wingerchuk D.M., Lennon V.A., Pittock S.J., Lucchinetti C.F., Weinshenker B.G. Revised diagnostic criteria for neuromyelitis optica. Neurology. 2006 May 23;66(10):1485–1489. doi: 10.1212/01.wnl.0000216139.44259.74. [DOI] [PubMed] [Google Scholar]
- 59.Nielsen S., Nagelhus E.A., Amiry-Moghaddam M., Bourque C., Agre P., Ottersen O.P. Specialized membrane domains for water transport in glial cells: high-resolution immunogold cytochemistry of aquaporin-4 in rat brain. J. Neurosci. 1997 Jan 1;17(1):171–180. doi: 10.1523/JNEUROSCI.17-01-00171.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pawate Siddharama, Sriram Subramaniam. The role of infections in the pathogenesis and course of multiple sclerosis. Ann. Indian Acad. Neurol. 2010 Apr-Jun;13(2):80–86. doi: 10.4103/0972-2327.64622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weber T. Progressive multifocal leukoencephalopathy. Neurol. Clin. 2008;26:833–854. doi: 10.1016/j.ncl.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 62.Steelman A. Infection as an environmental trigger of multiple sclerosis disease exacerbation. Front. Immunol. 2015 Oct 19;6:520. doi: 10.3389/fimmu.2015.00520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Oskari Virtanen Jussi, Jacobson Steve. Viruses and multiple sclerosis. CNS Neurol. Disord. - Drug Targets. 2012 August;11(5):528–544. doi: 10.2174/187152712801661220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kakalacheva Kristina, Münz Christian, Jan D., Lünemann Viral triggers of multiple sclerosis. Biochim. Biophys. Acta. 2011;1812:132–140. doi: 10.1016/j.bbadis.2010.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wucherpfennig Kai W., Strominger Jack L. Molecular mimicry in T cell-mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell. March 10, 1995;80:695–705. doi: 10.1016/0092-8674(95)90348-8. Copyright © 1995 by Cell Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Adams Jm, Imagawa Dt. Measles antibodies in multiple sclerosis. Proc Soc Exp Biol Med. 1962 Dec;111:562–566. doi: 10.3181/00379727-111-27855. [DOI] [PubMed] [Google Scholar]
- 67.Cendrowski W., Polna I., Nowicka K. Measles virus infection and multiple sclerosis: serological studies. J. Neurol. 1976 Oct 4;213(4):369–376. doi: 10.1007/BF00316278. [DOI] [PubMed] [Google Scholar]
- 68.Cendrowski W., Polna I., Niedzielska K. Serum measles antibodies in multiple sclerosis. J. Neurol. Neurosurg. Psychiatr. 1973;36:57–60. doi: 10.1136/jnnp.36.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Adams J.M., Brooks M.B., Fisher E.D., Tyler C.S. Measles antibodies in patients with multiple sclerosis and with other neurological and nonneurological diseases. Neurology. 1970 Oct;20(10):1039–1042. doi: 10.1212/wnl.20.10.1039. [DOI] [PubMed] [Google Scholar]
- 70.Reunanen M., Arstila P., Hakkarainen H., Nikoskelainen J., Salmi A., Panelius M. A longitudinal study on antibodies to measles and rubella viruses in patients with multiple sclerosis. A preliminary report. Acta Neurol. Scand. 1976 Oct;54(4):366–370. doi: 10.1111/j.1600-0404.1976.tb04366.x. [DOI] [PubMed] [Google Scholar]
- 71.Paul Brown, Francoise Cathala1, D. Carleton Gajdusek, Clarence J. Gibbs, Jr.Measles antibodies in the cerebrospinal fluid of patients with multiple sclerosis. PSEBM (Proc. Soc. Exp. Biol. Med.), Volume: 137 issue: 3, page(s): 956-961. [DOI] [PubMed]
- 72.Erling Norrby M.D., Hans Link M.D., Jan-Ervin Olsson M.D. Measles virus antibodies in multiple sclerosis comparison of antibody titers in cerebrospinal fluid and serum. Arch. Neurol. 1974;30(4) doi: 10.1001/archneur.1974.00490340013002. 285-2. [DOI] [PubMed] [Google Scholar]
- 73.Ahlgren C1., Odén A., Bergström T., Lycke J. Serum and CSF measles antibody levels increase over time in patients with multiple sclerosis or clinically isolated syndrome. J. Neuroimmunol. 2012 Jun 15;247(1–2) doi: 10.1016/j.jneuroim.2012.03.014. 70-4. doi: 10.1016/j.jneuroim.2012.03.014. Epub 2012 Apr 11. [DOI] [PubMed] [Google Scholar]
- 74.Thacker E.L., Mirzaei F., Ascherio A. Infectious mononucleosis and risk for multiple sclerosis: a meta-analysis. Ann. Neurol. 2006;59:499–503. doi: 10.1002/ana.20820. [DOI] [PubMed] [Google Scholar]
- 75.Nielsen T.R., Rostgaard K., Nielsen N.M. Multiple sclerosis after infectious mononucleosis. Arch. Neurol. 2007;64:72–75. doi: 10.1001/archneur.64.1.72. [DOI] [PubMed] [Google Scholar]
- 76.Ascherio A., Munger K.L., Lennette E.T. Epstein-barr virus antibodies and risk of multiple sclerosis: a prospective study. J. Am. Med. Assoc. 2001;286:3083–3088. doi: 10.1001/jama.286.24.3083. [DOI] [PubMed] [Google Scholar]
- 77.Sundstrom P., Juto P., Wadell G. An altered immune response to Epstein-Barr virus in multiple sclerosis: a prospective study. Neurology. 2004;62:2277–2282. doi: 10.1212/01.wnl.0000130496.51156.d7. [DOI] [PubMed] [Google Scholar]
- 78.Levin L.I., Munger K.L., Rubertone M.V. Temporal relationship between elevation of Epstein Barr virus antibody titers and initial onset of neurological symptoms in multiple sclerosis. J. Am. Med. Assoc. 2005;293:2496–2500. doi: 10.1001/jama.293.20.2496. [DOI] [PubMed] [Google Scholar]
- 79.DeLorenze G.N., Munger K.L., Lennette E. Epstein-Barr virus and multiple sclerosis: evidence of association from a prospective study with long-term follow-up. Arch. Neurol. 2006;63:839–844. doi: 10.1001/archneur.63.6.noc50328. [DOI] [PubMed] [Google Scholar]
- 80.Ascherio A., Munch M. Epstein-Barr virus and multiple sclerosis. Epidemiology. 2000;11:220–224. doi: 10.1097/00001648-200003000-00023. [DOI] [PubMed] [Google Scholar]
- 81.Alotaibi S., Kennedy J., Tellier R. Epstein-Barr virus in pediatric multiple sclerosis. J. Am. Med. Assoc. 2004;291:1875–1879. doi: 10.1001/jama.291.15.1875. [DOI] [PubMed] [Google Scholar]
- 82.Pohl D., Krone B., Rostasy K. High seroprevalence of Epstein-Barr virus in children with multiple sclerosis. Neurology. 2006;67:2063–2065. doi: 10.1212/01.wnl.0000247665.94088.8d. [DOI] [PubMed] [Google Scholar]
- 83.Ascherio A., Munger K.L. Environmental risk factors for multiple sclerosis. Part I: the role of infection. Ann. Neurol. 2007;61:288–299. doi: 10.1002/ana.21117. [DOI] [PubMed] [Google Scholar]
- 84.Ascherio, Munger K.L., Lennette E.T., Spiegelman D., Hernán M.A., Olek M.J., Hankinson S.E., Hunter D.J. Epstein–Barr virus antibodies and risk of multiple sclerosis: a prospective study. J. Am. Med. Assoc. : J. Am. Med. Assoc. 2001;286:3083–3088. doi: 10.1001/jama.286.24.3083. [DOI] [PubMed] [Google Scholar]
- 85.Langer-Gould Annette, Wu Jun, Lucas Robyn, Smith Jessica, Gonzales Edlin, Amezcua Lilyana, Haraszti Samantha, Chen Lie Hong, Hong Quach, James Judith A., Barcellos Lisa F., Xiang Anny H. Epstein-Barr virus, cytomegalovirus, and multiple sclerosis susceptibility a multiethnic study. Neurology September. 2017;26 doi: 10.1212/WNL.0000000000004412. 89 (13) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Levin Lynn I., PhD M.P.H.1, Kassandra L., Munger ScD2, Eilis J., O’Reilly, ScD, Kerstin I., Falk PhD4, Alberto Ascherio. Primary infection with the epstein-barr virus and risk of multiple sclerosis. Ann. Neurol. 2010 June;67(6):824–830. doi: 10.1002/ana.21978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Almohmeed1 Yahya H., Avenell1 Alison, Aucott2 Lorna, Vickers Mark A. Systematic review and meta-analysis of the sero-epidemiological association between Epstein barr virusand multiple sclerosis. PLOS one April. 2013;8(Issue 4) doi: 10.1371/journal.pone.0061110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Operskalski E.A., Visscher B.R., Malmgren R.M., Detels R. A case–control study of multiple sclerosis. Neurology. 1989;39:825–829. doi: 10.1212/wnl.39.6.825. [DOI] [PubMed] [Google Scholar]
- 89.Lindberg C., Andersen O., Vahlne A., Dalton M., Runmarker B. Epidemiological investigation of the association between infectious mononucleosis and multiple sclerosis. Neuroepidemiology. 1991;10:62–65. doi: 10.1159/000110248. [DOI] [PubMed] [Google Scholar]
- 90.Jilek S., Schluep M., Meylan P., Vingerhoets F., Guignard L., Monney A. Strong EBV-specific CD8+ T-cell response in patients with early multiple sclerosis. Brain. 2008;131:1712–1721. doi: 10.1093/brain/awn108. [DOI] [PubMed] [Google Scholar]
- 91.Broccolo Francesco, Fusetti Lisa, Ceccherini-Nelli Luca. Possible role of human herpesvirus 6 as a trigger of autoimmune disease. The ScientificWorld Journal. 2013:7. doi: 10.1155/2013/867389. Article ID 867389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Challoner P.B., Smith K.T., Parker J.D. Plaque-associated expression of human herpes virus 6 in multiple sclerosis. Proc. Natl. Acad. Sci. U.S.A. 1995;92:7440–77444. doi: 10.1073/pnas.92.16.7440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Soldan S.S., Berti R., Salem N. Association of human herpes virus 6 (HHV-6) with multiple sclerosis: increased IgM response toHHV-6 early antigen and detection of serumHHV-6 DNA. Nat. Med. 1997;3(12):1394–1397. doi: 10.1038/nm1297-1394. [DOI] [PubMed] [Google Scholar]
- 94.Friedman J.E., Lyons M.J., Cu G. The association of the human herpesvirus-6 and MS. Mult. Scler. 1999;5(5):355–362. doi: 10.1177/135245859900500509. [DOI] [PubMed] [Google Scholar]
- 95.Chapenko S., Millers A., Nora Z., Logina I., Kukaine R., Murovska M. Correlation between HHV-6 reactivation and multiple sclerosis disease activity. J. Med. Virol. 2003;69(1):111–117. doi: 10.1002/jmv.10258. [DOI] [PubMed] [Google Scholar]
- 96.Alvarez-Lafuente R., De LasHeras V., Bartolom'e M., Picazo J.J., Arroyo R. Relapsing-remitting multiple sclerosis and human herpesvirus 6 active infection. Arch. Neurol. 2004;61(10):1523–1527. doi: 10.1001/archneur.61.10.1523. [DOI] [PubMed] [Google Scholar]
- 97.Simpson S., Taylor B., Dwyer D.E. Anti-HHV-6 IgG titer significantly predicts subsequent relapse risk in multiple sclerosis. Mult. Scler. 2012;18:799–806. doi: 10.1177/1352458511428081. [DOI] [PubMed] [Google Scholar]
- 98.Fillet A.-M., Lozeron P., Agut H. HHV-6 and multiple sclerosis. Nat. Med. 1998;4(5):537–538. doi: 10.1038/nm0598-537a. [DOI] [PubMed] [Google Scholar]
- 99.Sanders V.J., Felisan S., Waddell A., Tourtellotte W.W. Detection of Herpesviridae in postmortem multiple sclerosis brain tissue and controls by polymerase chain reaction. J. Neurovirol. 1996;2(4):249–258. doi: 10.3109/13550289609146888. [DOI] [PubMed] [Google Scholar]
- 100.Goodman A.D., Mock D.J., Powers J.M., Baker J.V., Blumberg B.M. Human herpesvirus 6 genome and antigen in acute multiple sclerosis lesions. JID (J. Infect. Dis.) 2003;187(9):1365–1376. doi: 10.1086/368172. [DOI] [PubMed] [Google Scholar]
- 101.Akhyani N., Berti R., Brennan M.B. Tissue distribution and variant characterization of human herpesvirus (HHV)- 6: increased prevalence of HHV-6A in patients with multiple sclerosis. JID (J. Infect. Dis.) 2000;182(5):1321–1325. doi: 10.1086/315893. [DOI] [PubMed] [Google Scholar]
- 102.Cirone M., Cuomo L., Zompetta C. Human herpesvirus6 andmultiple sclerosis: a study of T cell cross-reactivity to viral and myelin basic protein antigens. J. Med. Virol. 2002;68(2):268–272. doi: 10.1002/jmv.10190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tejada-Simon M.V., Zang Y.C.Q., Hong J., Rivera V.M., Zhang J.Z. Cross-reactivity with myelin basic protein and human herpesvirus-6 in multiple sclerosis. Ann. Neurol. 2003;53(2):189–197. doi: 10.1002/ana.10425. [DOI] [PubMed] [Google Scholar]
- 104.Santoro F., Kennedy P.E., Locatelli G., Malnati M.S., Berger E.A., Lusso P. CD46 is a cellular receptor for human herpesvirus 6. Cell. 1999;99(7):817–827. doi: 10.1016/s0092-8674(00)81678-5. [DOI] [PubMed] [Google Scholar]
- 105.Cheng W., Ma Y., Gong F. Cross-reactivity of autoreactive. T cells with MBP and viral antigens in patients with MS. Front. Biosci. 2012;17(5):1648–1658. doi: 10.2741/4010. [DOI] [PubMed] [Google Scholar]
- 106.Astier M.-C., Trescol-Bi'emont, Azocar O., Lamouille B., Rabourdin-Combe C. Cutting edge: CD46, a new costimulatory. molecule for T cells, that induces p120(CBL) and LAT. phosphorylation. J. Immunol. 2000;164(12):6091–6095. doi: 10.4049/jimmunol.164.12.6091. [DOI] [PubMed] [Google Scholar]
- 107.Viglietta V., Baecher-Allan C., Weiner H.L., Hafler D.A. Loss of functional suppression by CD4+ CD25+ regulatory T cells in patients withmultiple sclerosis. J. Exp. Med. 2004;199(7):971–979. doi: 10.1084/jem.20031579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Marie J.C., Astier A.L., Rivailler P., Rabourdin-Combe C., Wild T.F., Horvat B. Linking innate and acquired immunity: divergent role of CD46 cytoplasmic domains in T cell-induced inflammation. Nat. Immunol. 2002;3(7):659–666. doi: 10.1038/ni810. [DOI] [PubMed] [Google Scholar]
- 109.Kemper C., Chan A.C., Green J.M., Brett K.A., Murphy K.M., Atkinson J.P. Activation of human CD4+ cells with CD3 and CD46 induces a T-regulatory cell 1 phenotype. Nature. 2003;421(6921):388–392. doi: 10.1038/nature01315. [DOI] [PubMed] [Google Scholar]
- 110.Astier L., Meiffren G., Freeman S., Hafler D.A. Alterations in CD46-mediated Tr1 regulatory T cells in patients withmultiple sclerosis. J. Clin. Invest. 2006;116(12):3252–3257. doi: 10.1172/JCI29251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Vaknin-Dembinsky, Murugaiyan G., Hafler D.A., Astier A.L., Weiner H.L. Increased IL-23 secretion and altered chemokine production by dendritic cells upon CD46 activation in patients with multiple sclerosis. J. Neuroimmunol. 2008;195(1–2):140–145. doi: 10.1016/j.jneuroim.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kawano M., Seya T., Koni I., Mabuchi H. Elevated serum levels of soluble membrane cofactor protein (CD46, MCP) in patients with systemic lupus erythematosus (SLE) Clin. Exp. Immunol. 1999;116(3):542–546. doi: 10.1046/j.1365-2249.1999.00917.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Soldan S.S., Fogdell-Hahn A., Brennan M.B. Elevated serum and cerebrospinal fluid levels of soluble human herpesvirus type 6 cellular receptor, membrane cofactor protein, in patients withmultiple sclerosis. Ann. Neurol. 2001;50(4):486–493. doi: 10.1002/ana.1135. [DOI] [PubMed] [Google Scholar]
- 114.Fogdell-Hahn S.S., Soldan S. Shue. Co-purification of soluble membrane cofactor protein (CD46) and human herpesvirus 6 variant A genome in serum from multiple sclerosis patients. Virus Res. 2005;110(1–2):57–63. doi: 10.1016/j.virusres.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 115.Alvarez-Lafuente R., Garc'ıa-Montojo M., De las Heras V., Bartolom'e M., Arroyo R. Clinical parameters and HHV-6 active replication in relapsing-remitting multiple sclerosis patients. J. Clin. Virol. 2006;37:S24–S26. doi: 10.1016/S1386-6532(06)70007-5. [DOI] [PubMed] [Google Scholar]
- 116.Liszewski M.K., Atkinson J.P. Complement regulator CD46: genetic variants and disease associations. Hum. Genom. 2015;9:7. doi: 10.1186/s40246-015-0029-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Charvet Benjamin, Reynaud1 Josephine M., Gourru-Lesimple1 Geraldine, Perron Hervé, Marche Patrice N., Horvat Branka. Induction of proinflammatory multiple sclerosis-associated retrovirus envelope protein by human herpesvirus-6A and CD46 receptor engagement. Front. Immunol. 2018 Dec 6;9:2803. doi: 10.3389/fimmu.2018.02803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ross R.T. The varicella-zoster virus and multiple sclerosis. J. Clin. Epidemiol. 1998 Jul;51(7):533–535. doi: 10.1016/s0895-4356(98)00030-4. [DOI] [PubMed] [Google Scholar]
- 119.Tarrats R., Ordoñez G., Rios C., Sotelo J. Varicella, ephemeral breastfeeding and eczema as risk factors for multiple sclerosis in Mexicans. Acta Neurol. Scand. 2002;105:88–94. doi: 10.1034/j.1600-0404.2002.1o077.x. [DOI] [PubMed] [Google Scholar]
- 120.Ross R.T., Cheang M., Landry G., Klassen L., Doerksen K. Herpes zoster and multiple sclerosis. The Canadian Journal of Neurological Sciences Le journal canadien des sciences neurologiques. 1999;26:29–32. [PubMed] [Google Scholar]
- 121.Ordoñez G., Pineda B., Garcia-Navarrete R., Sotelo J. Brief presence of varicella–zoster viral DNA in mononuclear cells during relapses of multiple sclerosis. Arch. Neurol. 2004;61:529–532. doi: 10.1001/archneur.61.4.529. [DOI] [PubMed] [Google Scholar]
- 122.Sotelo J., Ordoñez G., Pineda B. Varicella–zoster virus at relapses of multiple sclerosis. J. Neurol. 2007;254:493–500. doi: 10.1007/s00415-006-0402-x. [DOI] [PubMed] [Google Scholar]
- 123.Najafi Saeideh, Ghane Masood, Yousefzadeh-Chabok Shahrokh, Amiri Mehdi. The high prevalence of the varicella zoster virus in patients with relapsing-remitting multiple sclerosis: a case-control study in the north of Iran. Jundishapur J. Microbiol. 2016;9(3) doi: 10.5812/jjm.34158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Julio Sotelo 1. Corona Teresa. Varicella zoster virus and relapsing remitting multiple sclerosis. Multiple Sclerosis International. 2011:5. doi: 10.1155/2011/214763. |Article ID 214763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yucesan C., Sriram S. Chlamydia pneumoniae infection of the central nervous system. Curr. Opin. Neurol. 2001;14:355–359. doi: 10.1097/00019052-200106000-00015. [PubMed] [DOI] [PubMed] [Google Scholar]
- 126.Sriram S., Stratton C.W., Yao S., Tharp A., Ding L., Bannan J.D. Chlamydia pneumoniae infection of the central nervous system in multiple sclerosis. Ann. Neurol. 1999;46:6–14. ([PubMed]) [PubMed] [Google Scholar]
- 127.Fainardi Enrico, Silva Seraceni, Granieri Enrico, Contini Carlo. 2008. Chlamydophila Pneumoniae DNA and mRNA Transcript Levels in Peripheral Blood Mononuclear Cells and Cerebrospinal Fluid of Patients with Multiple Sclerosis Neuroscience Research. [DOI] [PubMed] [Google Scholar]
- 128.Bagos P.G., Nikolopoulos G., Ioannidis A. Chlamydia pneumoniae infection and the risk of multiple sclerosis: a meta-analysis. Mult. Scler. 2006;12:397–411. doi: 10.1191/1352458506ms1291oa. [DOI] [PubMed] [Google Scholar]
- 129.E1 Fainardi, Castellazzi M., Seraceni S., Granieri E., Contini C. Under the microscope: focus on Chlamydia pneumoniae infection and multiple sclerosis. Curr. Neurovascular Res. 2008 Feb;5(1):60–70. doi: 10.2174/156720208783565609. [DOI] [PubMed] [Google Scholar]
- 130.Smyk D.S. Helicobacter pylori and autoimmune disease: cause or bystander. World J. Gastroenterol. 2014 Jan 21;20(3):613–629. doi: 10.3748/wjg.v20.i3.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Deretzi G., Kountouras J., Polyzos S.A., Zavos C., Giartza-Taxidou E., Gavalas E., Tsiptsios I. Gastrointestinal immune system and brain dialogue implicated in neuroinflammatory and neurodegenerative diseases. Curr. Mol. Med. 2011;11:696–707. doi: 10.2174/156652411797536660. [DOI] [PubMed] [Google Scholar]
- 132.Gavalas E.1, Kountouras J.1, Boziki M.2, Zavos C.1, Polyzos S.A.1, Vlachaki E.1, Venizelos I.1, Tsiptsios D.3, Deretzi G.3. Relationship between Helicobacter pylori infection and multiple sclerosis. Ann. Gastroenterol. 2015;28:353–356. [PMC free article] [PubMed] [Google Scholar]
- 133.Li W. Association of anti-Helicobacter pylori neutrophil-activating protein antibody response with anti-aquaporin-4 autoimmunity in Japanese patients with multiple sclerosis and neuromyelitis optica. Mult. Scler. 2009 Dec;15(12):1411–1421. doi: 10.1177/1352458509348961. [DOI] [PubMed] [Google Scholar]
- 134.Long Y. Helicobacter pylori infection in neuromyelitis optica and multiple sclerosis. Neuroimmunomodulation. 2013;20:107–112. doi: 10.1159/000345838. [DOI] [PubMed] [Google Scholar]
- 135.Efthymiou Georgios, Dardiotis Efthymios, Liaskos Christos, Marou Emmanouela, Tsimourtou Vana, Rigopoulou Eirini I., Scheper Thomas, Daponte Alexandros, Meyer Wolfgang, Sakkas Lazaros I., Hadjigeorgiou Georgios, Dimitrios P. Bogdanos Immune responses against Helicobacter pylori-specific antigens differentiate relapsing remitting from secondary progressive multiple sclerosis. Sci. Rep. 2017 Aug 11;7(1) doi: 10.1038/s41598-017-07801-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yao G.1, Wang P.2, Luo X.D.3, Yu T.M.4, Harris R.A.5, Zhang X.M.5. Meta-analysis of association between Helicobacter pylori infection and multiple sclerosis. Neurosci. Lett. 2016 May 4;620:1–7. doi: 10.1016/j.neulet.2016.03.037. [DOI] [PubMed] [Google Scholar]
- 137.Cook K.W. Helicobacter pylori infection reduces disease severity in an experimental model of multiple sclerosis. Front. Microbiol. 2015;6:52. doi: 10.3389/fmicb.2015.00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Pedrini M.J. Helicobacter pylori infection as a protective factor against multiple sclerosis risk in females. J. Neurol. Neurosurg. Psychiatr. 2015 Jun;86(6):603–607. doi: 10.1136/jnnp-2014-309495. [DOI] [PubMed] [Google Scholar]
- 139.Jaruvongvanich V.1, Sanguankeo A.2, Jaruvongvanich S.3, Upala S.4. Association between Helicobacter pylori infection and multiple sclerosis: a systematic review and meta-analysis. Mult Scler Relat Disord. 2016 May;7:92–97. doi: 10.1016/j.msard.2016.03.013. [DOI] [PubMed] [Google Scholar]
- 140.Yoshimura S.1, Isobe N., Matsushita T., Yonekawa T., Masaki K., Sato S., Kawano Y., Kira J., South Japan J. Distinct genetic and infectious profiles in Japanese neuromyelitis optica patients according to anti-aquaporin 4 antibody status. Neurol Neurosurg Psychiatry. 2013 Jan;84(1):29–34. doi: 10.1136/jnnp-2012-302925. [DOI] [PubMed] [Google Scholar]
- 141.Mameli G., Cocco E., Frau J., Marrosu M.G., Sechi L.A. Epstein Barr Virus and Mycobacterium avium subsp. paratuberculosis peptides are recognized in sera and cerebrospinal fluid of MS patients. Sci. Rep. 2016;6:22401. doi: 10.1038/srep22401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Mameli G., Cossu D., Cocco E., Masala S., Frau J., Marrosu M.G. bEpstein-Barr virus and Mycobacterium avium subsp. paratuberculosis peptides are cross recognized by anti-myelin basic protein antibodies in multiple sclerosis patients. J. Neuroimmunol. 2014;270:51–55. doi: 10.1016/j.jneuroim.2014.02.013. [DOI] [PubMed] [Google Scholar]
- 143.Cossu D., Cocco E., Paccagnini D., Masala S., Ahmed N., Frau J. Association of Mycobacterium avium subsp. paratuberculosis with multiple sclerosis in Sardinian patients. PloS One. 2011;6 doi: 10.1371/journal.pone.0018482. 10.1371/journal.pone. 0018482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Cossu D., Masala S., Cocco E., Paccagnini D., Frau J., Marrosu M.G. Are Mycobacterium avium subsp. paratuberculosis and Epstein-Barr virus triggers of multiple sclerosis in Sardinia? Mult. Scler. 2012;18:1181–1184. doi: 10.1177/1352458511433430. [DOI] [PubMed] [Google Scholar]
- 145.Cossu D., Masala S., Sechi L.A. A Sardinian map for multiple sclerosis. Future Microbiol. 2013;8:223–232. doi: 10.2217/fmb.12.135. [DOI] [PubMed] [Google Scholar]
- 146.Cossu D., Mameli G., Galleri G., Cocco E., Masala S., Frau J. Human interferon regulatory factor 5 homologous epitopes of Epstein-Barr virus and Mycobacterium avium subsp. paratuberculosis induce a specific humoral and cellular immune response in multiple sclerosis patients. Mult. Scler. 2015;21:984–995. doi: 10.1177/1352458514557304. [DOI] [PubMed] [Google Scholar]
- 147.Cossu D., Yokoyama K., Hattori N. Conflicting role of Mycobacterium species in multiple sclerosis. Front. Neurol. 2017;8:216. doi: 10.3389/fneur.2017.00216. 10.3389/fneur.2017.00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Cossu D., Yokoyama K., Tomizawa Y., Momotani E., Hattori N. Altered humoral immunity to mycobacterial antigens in Japanese patients affected by inflammatory demyelinating diseases of the central nervous system. v Sci. Rep. 2017;7:3179. doi: 10.1038/s41598-017-03370-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Bahar M., Ashtari F., Aghaei M., Akbari M., Salari M., Ghalamkari S. Mycoplasma pneumonia seroposivity in Iranian patients with relapsingremitting multiple sclerosis: a randomized case-control study. J. Pakistan Med. Assoc. 2012;62:S6–S8. [PubMed] [Google Scholar]
- 150.Harbo H.F., Gold R., Tintoré M. Sex and gender issues in multiple sclerosis. Ther. Adv. Neurol. Disord. 2013;6:237–248. doi: 10.1177/1756285613488434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lindsey J., Patel S. PCR for bacterial 16S ribosomal DNA in multiple sclerosis cerebrospinal fluid. Mult. Scler. 2008;14:147–152. doi: 10.1177/1352458507082149. [DOI] [PubMed] [Google Scholar]
- 152.Dorca-Arévalo J., Soler-Jover A., Gibert M., Popoff M.R., Martín-Satué M., Blasi J. Binding of epsilon-toxin from Clostridium perfringens in the nervous system. Vet. Microbiol. 2008;131:14–25. doi: 10.1016/j.vetmic.2008.02.015. [DOI] [PubMed] [Google Scholar]
- 153.Sakai R.E., Feller D.J., Galetta K.M., Galetta S.L., Balcer L.J. Vision in multiple sclerosis: the story, structure-function correlations, and models for neuroprotection. J. Neuro Ophthalmol. 2011;31:362–373. doi: 10.1097/WNO.0b013e318238937f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Carabotti M., Scirocco A., Maselli M.A., Severi C. The gut-brain axis: interactions between enteric microbiota, central and enteric nervous systems. Ann. Gastroenterol. 2015;28:203–209. [PMC free article] [PubMed] [Google Scholar]
- 155.Shahi S.K., Freedman S.N., Mangalam A.K. Gut microbiome in multiple sclerosis: the players involved and the roles they play. Gut Microb. 2017;8:607–615. doi: 10.1080/19490976.2017.1349041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Colpitts S.L., Kasper L.H. Influence of the gut microbiome on autoimmunity in the central nervous system. J. Immunol. 2017;198:596–604. doi: 10.4049/jimmunol.1601438. [DOI] [PubMed] [Google Scholar]
- 157.Tremlett H., Waubant E. The multiple sclerosis microbiome? Ann. Transl. Med. 2017;5:53. doi: 10.21037/atm.2017.01.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Brocke S., Gaur A., Piercy C., Gautam A., Gijbels K., Fathman C.G. Induction of relapsing paralysis in experimental autoimmune encephalomyelitis by bacterial superantigen. Nature. 1993;365:642–644. doi: 10.1038/365642a0. [DOI] [PubMed] [Google Scholar]
- 159.Sewell D.L., Reinke E.K., Co D.O., Hogan L.H., Fritz R.B., Sandor M. Infection with Mycobacterium bovis BCG diverts traffic of myelin oligodendroglial glycoprotein autoantigen-specific T cells away from the central nervous system and ameliorates experimental autoimmune encephalomyelitis. Clin. Diagn. Lab. Immunol. 2003;10:564–572. doi: 10.1128/CDLI.10.4.564-572.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Lee J., Reinke E.K., Zozulya A.L., Sandor M., Fabry Z. Mycobacterium bovis bacille Calmette-Guérin infection in the CNS suppresses experimental autoimmune encephalomyelitis and Th17 responses in an IFNgamma-independent manner. J. Immunol. 2008;181:6201–6212. doi: 10.4049/jimmunol.181.9.6201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Cossu D., Mameli G., Masala S., Cocco E., Frau J., Marrosu M.G. Evaluation of the humoral response against mycobacterial peptides, homologous to MOG35 55, in multiple sclerosis patients. J. Neurol. Sci. 2014;347:78–81. doi: 10.1016/j.jns.2014.09.023. [DOI] [PubMed] [Google Scholar]
- 162.Ristori G., Romano S., Cannoni S., Visconti A., Tinelli E., Mendozzi L. Effects of Bacille Calmette-Guerin after the first demyelinating event in the CNS. Neurology. 2014;82:41–48. doi: 10.1212/01.wnl.0000438216.93319.ab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Moliva J.I., Turner J., Torrelles J.B. Immune responses to Bacillus Calmette-Guérin vaccination: why do they fail to protect against Mycobacterium tuberculosis? Front. Immunol. 2017;8:407. doi: 10.3389/fimmu.2017.00407. 10.3389/fimmu.2017. 00407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Logsdon A.F., Erickson M.A., Rhea E.M., Salameh T.S., Banks W.A. Gut reactions: how the blood-brain barrier connects the microbiome and the brain. Exp. Biol. Med. 2018;243:159–165. doi: 10.1177/1535370217743766. [DOI] [PMC free article] [PubMed] [Google Scholar]