

Abstract
LipoxinA4 (LXA4) is a well-known key mediator of endogenous anti-inflammation and of the resolution of inflammation. Considerable oxidative stress occurs during inflammation due to the generation of reactive oxidative species (ROS). Moreover, high levels of uric acid (UA) contribute to endothelial cell dysfunction, which can promote disease-related morbidity, and NADPH oxidase-derived ROS are crucial regulatory factors in these responses. However, LXA4 also has the potential to reduce oxidative stress. The aim of the present study was to examine whether LXA4 could suppress UA-induced oxidative stress in human umbilical vein endothelial cells (HUVECs) and to investigate its mechanisms of action in vitro. HUVECs were incubated with or without LXA4, followed by the addition of UA. ROS levels were then measured using 2,7-dichlorodihydrofluorescein diacetate and lucigenin-enhanced chemiluminescence was used to evaluate NADPH oxidase activity. p47phox or p22phox small interfering (si)RNA were transfected into HUVECs and protein levels of p47phox were detected using western blot analysis. LXA4 significantly inhibited UA-induced generation of ROS to the same extent as the NADPH oxidase inhibitor, diphenyleneiodonium chloride. Notably, transfection of p47phox siRNA attenuated the generation of ROS and the activation of NADPH oxidase. Cells transfected with p22phox siRNA demonstrated a significant reduction in the expression of p47phox on the membrane. Further experiments demonstrated that LXA4 interfered with the transfer of p47phox from the cytoplasm to the cell membrane. These findings suggested that LXA4 inhibited the release of NADPH oxidase derived ROS in HUVECs stimulated by UA. A potential mechanism of action underlying this effect could be LXA4-mediated suppression of NADPH oxidase activity, leading to inhibition of p47phox translocation from the cytoplasm to the cell membrane.
Keywords: lipoxinA4, uric acid, NADPH, reactive oxidative species, human umbilical vein endothelial cells
Introduction
Uric acid (UA) is the end product of purine metabolism in the human body. The incidence of hyperuricemia is gradually increasing worldwide as the result of various factors, such as high-protein diets, environmental pollution and genetic factors. Hyperuricemia is strongly associated with metabolic syndrome (1), cardiovascular diseases (2) and cerebrovascular diseases (3). Obesity (4) and diabetes (5) can be independent risk factors for these diseases. As an exogenous inflammatory stimulator, UA can cause a systemic inflammatory response that ultimately upregulates pro-inflammatory cytokines through activation of the MAPK signaling pathway and the nuclear factor-κB (NF-κB) signal pathways. These, in turn, contribute to vascular endothelial cell dysfunction and the subsequent development of cardiovascular diseases (6).
Oxidative stress is a state in which reactive oxygen species (ROS) are excessively produced and exceed the clearance capacity of the body. In inflammation (7), toxicity (8), ischemia-reperfusion (9) and other pathological conditions, several ROS are produced and the ability to scavenge ROS is inhibited, leading to an imbalance between oxidation and antioxidation (9,10). While NADPH oxidase-derived ROS are essential for innate immunity and microbial killing, excess production of ROS induces prolonged inflammatory reactions that contribute to cellular dysfunction and inflammatory diseases (7,10-12). ROS can be generated in numerous ways, from a variety of molecules, and participate in pathophysiological processes during disease (7,9,10). A previous study suggested that UA is one of the most important antioxidants in circulation and endothelial cells can benefit from its antioxidant effects (13). However, a growing number of studies have demonstrated that the antioxidant effects of UA are far less than its oxidant effects. Thus, UA-activated oxidative stress can in turn exacerbate the inflammatory response (6,14). UA-induced ROS derived from NADPH oxidase can promote the phosphorylation of MAPK/ERK and PI3-K/AKT and sequentially promote the downstream NF-κB signal pathway, thereby enhancing inflammatory responses (15,16). This pathological mechanism of action was previously identified in endothelial cells (14), adipocytes (17) and smooth muscle cells (18). In addition, Sautin et al (17) previously demonstrated that inflammation induced by high levels of UA was associated with oxidative stress.
NADPH oxidase consists of the cytosolic subunits p47phox, p67phox, p40phox and Rac2, as well as transmembrane subunits p22phox and gp91phox. The fundamental mechanism of NADPH oxidase activation is through the transfer of p47phox to the membrane, which drives other cytoplasmic subunits together and transfers them towards the membrane to complete cell assembly.
Lipoxins are endogenously produced arachidonic acid metabolites, which can act as potent anti-inflammatory agents that suppress the expression of inflammation-related genes and attenuate the activation of inflammatory cells (19). As such, lipoxins are known as the ‘brake signal’ of inflammation. A previous study suggested that the overexpression of lipoxins is fundamental to the control of inflammation in vivo (20). Lipoxin A4 (LXA4), a subtype that best represents the biological activities of lipoxins, restrains ROS generation to reduce the inflammatory response and prevent damage to host cells (21,22). However, to the best of the authors' knowledge, whether LXA4 can inhibit UA-induced oxidative stress in human umbilical vein endothelial cells (HUVECs) has not been previously reported.
Therefore, the aim of the present study was to investigate the impact of LXA4 on the oxidative stress induced by UA in HUVECs, as well as to examine the possible underlying mechanisms in vitro. LXA4 inhibited the release of NADPH oxidase-derived ROS in HUVECs stimulated by UA. A potential mechanism of action underlying this effect could be LXA4-mediated suppression of NADPH oxidase activity, leading to inhibition of p47phox translocation from the cytoplasm to the cell membrane.
Materials and methods
Cell culture and treatment
The HUVEC line was purchased from The American Type Culture Collection and maintained in RPMI-1640 complete medium (Hyclone; GE Healthcare Life Sciences) supplemented with 0.4 µl/ml vascular endothelial growth factor, 100 U/ml penicillin, 100 U/ml streptomycin and 10% fetal bovine serum (Hyclone; GE Healthcare Life Sciences) at 37˚C in a humidified atmosphere of 5% CO2 and 95% atmospheric air. HUVECs were digested with 0.25% trypsin when cells presented their typical morphology (a paving stone shape, clear nucleus, clear membrane and no obvious antennae) and reseeded for further growth. Cells in logarithmic phase were used for experiments. HUVECs were seeded into 6-well plates at a density of 2.5x105 cells/well, then serum-starved for 12 h before further experiments.
HUVECs were incubated for 12 h in the initial experiments with varying concentrations of UA (Sigma-Aldrich; Merck KGaA; 0, 6, 12 or 16 mg/dl) or incubated with 12 mg/dl UA for varying time periods (0, 3, 6, 12, 24 or 48 h). Appropriate concentrations of UA were used for further experiments. Similar to the aforementioned procedure, HUVECs were pre-treated with various concentrations of LXA4 (Cayman Chemical Company; 0, 1, 10 or 100 nM) and time periods (0, 15, 30, 60 and 120 min). A concentration of 100 nM LXA4 was used for further experiments for 1 h. Furthermore, cells were pre-treated for the same time period when pre-treated with diphenyleneiodonium chloride (DPI; 10 µM; Cayman Chemical Company), indomethacin (3 mM; Sigma-Aldrich, Merck KGaA) and rotenone (1 µM; Sigma-Aldrich; Merck KGaA) with LXA4 before the addition of UA under serum-free conditions.
Measurement of intracellular ROS
Total intracellular ROS in HUVECs was determined using the peroxide-sensitive probe 2',7'-dichlorodihydrofluorescein diacetate (DCFH2-DA; Sigma-Aldrich; Merck KGaA) dye as previously described (23). The non-fluorescent form of DCFH2-DA is oxidized by intracellular ROS and forms the highly fluorescent form of DCFH2-DA. The intensity of fluorescence is proportional to the levels of intracellular ROS. HUVECs were cultured and treated according to grouping requirements, and subsequently were washed with PBS and incubated for 30 min with DCFH2-DA at a final concentration of 10 µmol/l. Fluorescence was then measured using a fluorometer (Thermo Fisher Scientific, Inc.) with excitation and emission wavelengths of 480 and 520 nm, respectively. Images were captured using a Nikon fluorescence microscope (magnification, x40; Nikon Corporation).
Measurement of NADPH oxidase activity
The activity of NADPH oxidase was measured using lucigenin-enhanced chemiluminescence. NADPH oxidase was activated to transfer electrons from NADPH to O2, forming O2-. Lucigenin is luminescent when interacting with O2-, with the amount of luminescent electrons indirectly representing the activity of NADPH oxidase, as described previously (24). HUVECs were treated as described, then washed three times with ice-cold PBS (pH 7.4) and centrifuged at 2,000 x g for 5 min at 4˚C. The pellet was resuspended in cell lysis buffer (20 µM K2HPO4; 1 mM EDTA; 1 mM PMSF; 10 µg/ml aprotinin). Cells were lysed and the collected proteins were resuspended to a final concentration of 1 mg/ml. A total of 100 µl of the samples were mixed with 900 µl reaction buffer (50 nM K2HPO4; 1 nM EDTA; 150 µM sucrose; 5 µM lucigenin; 100 µM NADPH). A chemiluminescence analyzer (Thermo Fisher Scientific, Inc.) gathered photon emission every 60 sec for 20 min, with a 5-sec signal integration time.
p47phox small interfering (si)RNA and p22phox siRNA transfection
Transfection of siRNA into HUVECs was performed as previously described (25). The siRNAs (100 nM; Sangon Biotech Co, Ltd.) used in the present study were: p47phox siRNA, 5'-GGACCCAGAACCCAACUAUGCAGGT-3' and 5'-ACCUGCAUAGUUGGGUUCUGGGUCCUC-3'; p22phox siRNA, 5'-GAAGGGCUCCACCAUGGAGTT-3' and 5'-UCCAUGGUGGAGCCCUUCTT-3'; siRNA-scramble, 5'-GCUGCAGTAUGAGGAG-3' and 5'-CGACGC CATUCCGTAGC-3'. Transfections were carried out using Lipofectamine® 2000 transfection reagent (Thermo Fisher Scientific, Inc.), according to the manufacturer's protocol, where subsequent experimentation was performed 48 h after transfection. The mRNA expression of p47phox and p22phox in transfected cells was determined by reverse transcription-quantitative PCR (RT-qPCR).
RT-qPCR
Total RNA was extracted from HUVECs using TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.) according to manufacturer's protocol, cDNA was synthesized from total RNA by Transcriptor First Strand cDNA Synthesis kit (Roche Diagnostics). The temperature protocol was as follows: 10 min at 25˚C, followed by 55˚C for 30 min. The expression of p47phox (forward, 5'-ACGAGAGTGGTTGGTGGTTC-3' and reverse, 5'-TGTAGGCTTTGATGGTGACG-3') and p22phox (forward, 5'-TGCTTGTGGGTAAACCAAGGCCGGTG-3' and reverse, 5'-AACACTGAGGTAAGTGGGGGTGGCTCCTGT-3') were measured using the 2X SYBR-Green Abstart Mix kit (cat. no. B110031; Sangon Biotech Co, Ltd.). GADPH (forward, 5'-GAAGGTGAAGGTCGGAGTC-3' and reverse, 5'-GAAGATGGTGATGGGATTTC-3') as endogenous control. The qPCR reaction system consisted of 10 µl SYBR® Green Master Mix, 0.5 µl upstream and downstream primers, 1 µl cDNA and 8 µl ddH2O. The thermocycling conditions were as follows: Initial denaturation at 95˚C for 3 min, followed by 40 cycles of 95˚C for 30 sec and 58˚C for 1 min. The relative expression of p47phox RNA and p22phox RNA were evaluated using 2-∆∆Cq method (26). GADPH was used as a control for normalization.
Cell membrane and cytoplasm fractionation
HUVECs were cultured and treated as required and subsequently washed three times with ice-cold PBS (pH 7.4). The cells were lysed in RIPA Lysis buffer (Beyotime Institute of Biotechnology) supplemented with protease inhibitor cocktail (Beyotime Institute of Biotechnology). Membrane and cytoplasmic proteins were extracted using a Membrane and Cytosol Protein Extraction kit (Beyotime Institute of Biotechnology) according to the manufacturer's protocol.
Western blot analysis
The concentration of cytoplasmic and membrane proteins extracted from HUVECs was determined using a bicinchoninic acid protein assay kit (Beyotime Institute of Biotechnology) and concentrations were normalized to be electrophoresed. Equivalent masses (5 µg/ml) of protein were electrophoresed on 10% SDS polyacrylamide gels and transferred onto polyvinylidene fluoride membranes (Beyotime Institute of Biotechnology). The transformed membrane was blocked using 5% non-fat milk at 4˚C for 1 h and incubated with primary antibodies against p47phox (cat. no. ab166930; 1:500; Abcam), GAPDH (cat. no. AG019; 1:500; Beyotime Institute of Biotechnology) and Caveolin-1 (cat. no. AF0087; 1:500; Beyotime Institute of Biotechnology) at 4˚C overnight. The hybridized membrane was washed three times with Tris-buffered saline containing 0.05% Tween-20 (TBST) for 10 min and subsequently incubated with horseradish peroxidase-conjugated goat anti-mouse IgG (cat. no. A0216; 1:5,000; Beyotime Institute of Biotechnology) at 4˚C for 2 h. The membrane was washed three times with TBST and the signal was detected using Beyo ECL Plus kit (Beyotime Institute of Biotechnology), densitometric analysis was performed using Image-Pro Plus V software (v7.0; Media cybernetics, Inc.).
Statistical analysis
The SPSS v13.0 statistical software package (SPSS, Inc.) was used for statistical analysis. The results are presented as the mean ± SD. Multigroup comparisons of the means were carried out by ANOVA, followed by post hoc correction with Tukey's test. All experiments were repeated three times. P<0.05 was considered to indicate a statistically significant difference.
Results
UA induces oxidative stress and ROS production by HUVECs, which are reversed by LXA4
UA stimulated HUVECs were treated in a time and concentration-dependent manner to produce ROS. The relative dichlorofluorescein (DCF) fluorescence increased with time and concentration. The maximum signal was reached at 24 h and with 12 mg/dl UA (Fig. 1). In order to determine the effect of LXA4 on ROS generation, HUVECs were incubated with LXA4 for different periods of time and concentrations, then treated with UA. The inhibitory effect of LXA4 was observed after 15 min of pre-incubation and the influence remained significant, with a maximal inhibition reached at 1 h. Pre-incubation with varying concentrations of LXA4 caused a significant reduction in UA-induced ROS generation and a maximum effect was observed for 100 nM (Fig. 1).
Figure 1.

UA-induced oxidative stress induces ROS production and LXA4 affects this response. (A) ROS production was detected in HUVECs following treatment with different concentrations of UA (0, 4, 8, 12 and 16 mg/dl). n=3. (B) ROS production was detected in HUVECs following treatment with UA for varying time periods (0, 3, 6, 12, 24 and 48 h). n=3. (C) ROS were detected in HUVECs incubated with various concentrations of LXA4 (0, 1, 10 and 100 nM) for 1 h following treatment with 12 mg/dl UA for 24 h. The control group was treated with normal saline for 24 h. n=3. (D) ROS were detected in HUVECs incubated with LXA4 for various time periods (0, 15, 30, 60 and 120 min) following treatment with UA. n=3. *P<0.05 vs. 0 mg/dl; &P<0.05 vs. 0 h; #P<0.05 vs. treatment with UA alone; ^P<0.05 vs. 0 min. HUVEC, human umbilical vein endothelial cell; ROS, reactive oxidative species; UA, uric acid; LXA4, lipoxin A4; DCF, dichlorofluorescein.
LXA4 suppresses UA-induced increase in generation of ROS in a NADPH oxidase-dependent manner
The present study adopted various inhibitors which altered the basal superoxide generation when pre-incubated with HUVECs to determine whether the inhibitory effect of LXA4 on ROS generation was NADPH oxidase dependent. HUVECs were incubated with specific inhibitors for cyclooxygenase (indomethacin), mitochondria complex I (rotenone), NADPH oxidase (DPI) and LXA4, results demonstrated that indomethacin and rotenone had a marginal inhibitory effect on ROS generation, although this was not significant. However, DPI abrogated the UA-induced increase in generation of ROS to the same extent as LXA4. This suggested that NADPH oxidase was a major source for ROS production during UA-induced oxidative stress and LXA4 downregulated ROS generation in HUVECs (Fig. 2).
Figure 2.
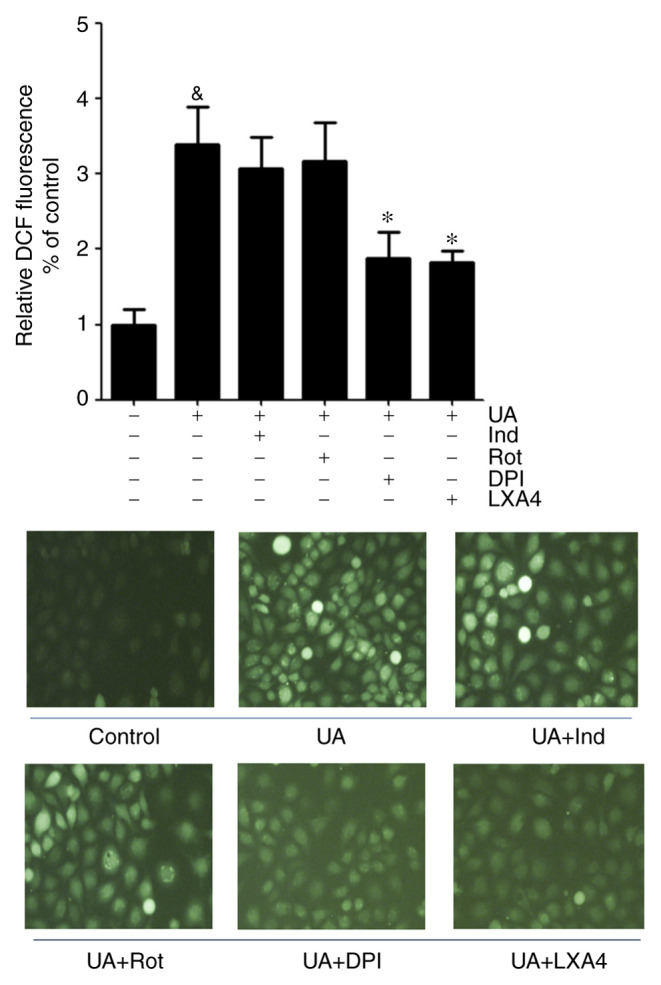
Effect of various inhibitors on the generation of ROS induced by UA in HUVECs. ROS levels were measured in HUVECs incubated with Ind, Rot, DPI or LXA4 following treatment with UA. Data are presented as the mean ± SD. n=3. &P<0.05 vs. control; *P<0.05 vs. UA treatment. Magnification, x40. HUVEC, human umbilical vein endothelial cell; ROS, reactive oxidative species; UA, uric acid; LXA4, lipoxin A4; Ind, indomethacin; Rot, Rotenone; DPI, diphenyleneiodium chloride; DCF, dichlorofluorescein.
LXA4 suppresses UA-induced activation of NADPH oxidase
Having demonstrated that NADPH oxidase was a major source for ROS in the oxidative stress response, NADPH oxidase activity was then measured. Cells were incubated with 100 nM LXA4 for 1 h then with UA (12 mg/dl) for 12 h, and subsequently the NADPH oxidase activity was measured. Compared with the control group, NADPH oxidase activity in HUVECs significantly increased following UA treatment, consistent with the ROS generation. However, when cells were incubated with LXA4 before UA stimulation, a significant reduction in NADPH oxidase activity was observed, compared with that in the UA group (Fig. 3). Thus, LXA4 pre-treatment inhibits NADPH oxidase activity following UA stimulation.
Figure 3.
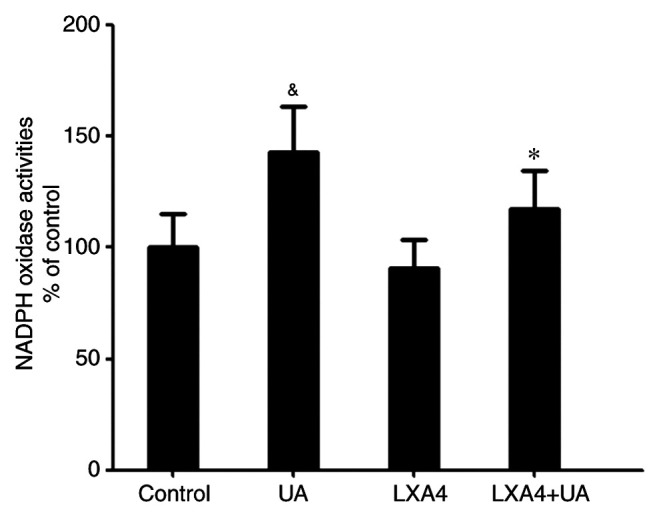
Inhibitory effect of LXA4 on UA-stimulated NADPH oxidase activity in HUVECs. HUVECs were incubated with LXA4, following treatment with UA, then NADPH oxidase activity was detected using lucigenin-enhanced chemiluminescence. Data are presented as the mean ± SD. n=3. &P<0.05 vs. Control; *P<0.05 vs. treatment with only UA. HUVEC, human umbilical vein endothelial cell; UA, uric acid; LXA4, lipoxin A4.
Transfection of p47phox siRNA attenuates UA-induced increase generation of ROS and activation of NADPH oxidase
The mRNA expression levels of p47phox and p22phox were examined by RT-qPCR in transfected HUVECs. The levels of p47phox and p22phox mRNA in HUVECs transfected with p47phox or p22phox siRNA were significantly lower than the blank group and the scrambled group (Fig. 4).
Figure 4.

Expression levels of p47phox mRNA and p22phox mRNA in transfected human umbilical vein endothelial cells. The levels of p47phox mRNA and p22phox mRNA were detected using reverse transcription-quantitative PCR. Data are presented as the mean ± SD. n=3. *P<0.05 vs. scrambled group. siRNA, small interfering RNA.
Activated NADPH oxidase is a membrane-bound enzyme, which depends on the translocation of activated cytoplasmic subunits to the membrane to form the functional complex (26). The cytoplasmic subunit p47phox plays a crucial role in NADPH oxidase activation (27). To ascertain that p47phox is required for the UA-induced oxidative stress response in HUVECs, cells were transfected with p47phox siRNA before exposure to UA (12 mg/dl) for 12 h. Subsequently, ROS generation and NADPH oxidase activity was measured. UA-induced ROS generation and NADPH oxidase activity were both significantly reduced in HUVECs transfected with p47phox siRNA. It was hypothesized that LXA4 suppression of the activation of NADPH oxidase may be interfered with p47phox (Fig. 5).
Figure 5.

Effect of transfection of p47phox siRNA on the UA-induced generation of ROS and activation of NADPH oxidase in HUVECs. (A) ROS were detected following treatment with UA in HUVECs transfected with p47phox siRNA. (B) NADPH oxidase activity was detected following treatment with UA in HUVECs transfected with p47phox siRNA. Data are presented as the mean ± SD. n=3. &P<0.05 vs. control; *P<0.05 vs. UA-treated group. HUVEC, human umbilical vein endothelial cell; ROS, reactive oxidative species; siRNA, small interfering RNA; UA, uric acid; DCF, dichlorofluorescein.
Transfer of p47phox to the cell membrane to activate NADPH oxidase
No apparent change in the total protein expression levels of p47phox was observed in HUVECs in the presence or absence of UA (Fig. 6A). In addition, a previous study demonstrated that p47phox was transferred from the cytoplasm to the membrane and associated with p22phox to activate NADPH oxidase (28). Thus, it was hypothesized an analogous mechanism allowed p47phox to bind to p22phox in HUVECs stimulated with UA. Cells were transfected with p22phox siRNA followed by addition of UA (12 mg/dl) for 12 h. Membrane proteins were extracted and p47phox was detected using western blotting. UA-stimulation demonstrated a marked elevation in the expression of p47phox on the membrane of HUVECs. Cells transfected with p22phox siRNA showed a significant reduction in the expression levels of p47phox on the membrane compared with cells treated with only UA. These findings suggested that UA activated NADPH oxidase by promoting the transfer of p47phox to the membrane, rather than by modulating the expression levels of p47phox (Fig. 6B).
Figure 6.

Effect of transfection of p22phox siRNA on the expression of p47phox in HUVECs. (A) Total expression of p47phox was examined using western blotting assays in HUVECs treated with or without UA. There was no significant difference between the total protein expression of p47phox in HUVECs treated with or without UA. (B) HUVECs were transfected with p22phox siRNA and treated with UA. The levels of membrane bound p47phox were detected using western blotting assays. Data are presented as the mean ± SEM. n=3. &P<0.05 vs. control; *P<0.05 vs. the UA group. HUVEC, human umbilical vein endothelial cell; siRNA, small interfering RNA; UA, uric acid.
LXA4 inhibits NADPH oxidase activity by preventing p47phox translocation
Since UA activated NADPH oxidase by promoting the transfer of p47phox from the cytoplasm to the membrane, the present study aimed to determine whether LXA4 suppressed NADPH oxidase activation by preventing the translocation of p47phox in HUVECs treated with UA. Cells were incubated with 100 nM LXA4 for 1 h, then with UA (12 mg/dl) for 24 h. The cytoplasmic and membrane proteins were extracted and p47phox expression levels were measured using western blotting. The expression levels of p47phox in the cytoplasm significantly decreased in HUVECs stimulated by UA, compared with that of the control cells. However, in cells pre-incubated with LXA4, translocation of p47phox from the cytoplasm was significantly reduced (Fig. 7). Notably, UA treatment increased the levels of p47phox on the membrane and LXA4 interrupted this trend in HUVECs (Fig. 8). These results suggested that LXA4 inhibited the translocation of p47phox from the cytoplasm to the membrane, thereby suppressing NADPH oxidase activity in HUVECs following UA stimulation.
Figure 7.

Effect of LXA4 on the expression of p47phox in the cytoplasm of HUVECs. HUVECs were incubated with LXA4, following treatment with UA. p47phox levels in cytoplasm were measured using western blotting assays. Data are presented as the mean ± SEM. n=3. &P<0.05 vs. control; *P<0.05 vs. the UA group. HUVEC, human umbilical vein endothelial cell; UA, uric acid; LXA4, lipoxin A4.
Figure 8.

Effect of LXA4 on the expression of p47phox on the membrane in HUVECs. HUVECs were incubated with LXA4, following treatment with UA. p47phox levels on the membrane were measured using western blotting assays. Data are presented as the mean ± SEM. n=3. &P<0.05 vs. control; *P<0.05 vs. the UA group. HUVEC, human umbilical vein endothelial cell; UA, uric acid; LXA4, lipoxin A4.
Discussion
In the present study, the impact of LXA4 on oxidative stress induced by UA in HUVECs and the potential mechanism of action underlying the regulation of NADPH oxidase were determined. The primary finding of the present study was that UA-stimulated HUVECs exhibited greater oxidative stress, which contributed to the generation of intracellular ROS, and treatment with LXA4 could protect against this response. It was demonstrated that NADPH oxidase was the dominant source of ROS following treatment with UA in HUVECs. Moreover, LXA4 suppressed the increase in the generation of ROS and this was achieved in an NADPH oxidase-dependent manner. The mechanism of action underlying LXA4-mediated inhibition of NADPH oxidase activation was investigated. It was found that the cytoplasmic subunits of p47phox translocated to the membrane, which resulted in the activation of NADPH oxidase. Furthermore, NADPH oxidase activity was suppressed as result of LXA4 inhibiting the translocation of p47phox. These results provided a mechanistic link between UA and the antioxidant effects of LXA4 in HUVECs.
A previous study demonstrated that UA exerted an antioxidant effect on endothelial cells (13). However, a number of previous studies also suggested that the oxidant properties of UA far exceeded its antioxidant effects in endothelial cells (14), adipocytes (17) and smooth muscle cells (18). Previous studies have demonstrated that high serum levels of UA in vivo for extended periods positively correlated with the incidence of metabolic (1,29), cerebrovascular (3,30) and cardiovascular diseases (2,31), owing to endothelial cell dysfunction and pathological vascular remodeling. A recent study also indicated that prolonged hyperuricemia could reduce peroxide stress and inflammation in body (32). In the present study, UA significantly increased ROS generation in HUVECs, suggesting that UA could promote oxidative stress in these cells. Oxidative stress is involved in several pathophysiological processes (7-10). This is especially the case in inflammatory responses, in which oxidative stress-derived ROS can activate signaling pathways, such as the MAPK/ERK, PI3-K/AKT and NF-κB pathways to upregulate the transcription of genes related to inflammation (10,15,16,33). Thus, the present study aimed to investigate the mechanism of oxidative stress and its prevention, and to identify potential therapeutic targets for inflammatory diseases.
LXA4 is an endogenous anti-inflammatory protein and acts as a ‘brake signal’ for inflammation (19). A variety of inflammation-related genes and inflammatory cytokines, such as tumor necrosis factor-α (TNF-α), interleukin (IL)-β and IL-6 are downregulated by LXA4(34). Li et al (35), reported that LXA4 inhibited the LPS-induced expression of monocyte chemoattractant protein and macrophage colony-stimulating factor in hepatocellular carcinoma cells by switching off the ROS, MAPK and NF-κB pathways and controlling the extent of oxidative stress. Previous studies suggested that LXA4 protected endothelial cells from inflammatory damage, which was associated with the suppression of oxidative stress (22). It is evident from the present study that LXA4 suppressed the generation of ROS induced by UA in HUVECs. ROS have extensive sources in mammalian cells, such as the mitochondrial respiratory chain (36), cyclooxygenases (37) and NADPH oxidase (30,37). The present study examined different inhibitors for mitochondria, cyclooxygenase and NADPH oxidase to explore the main source for ROS in this model. Several inhibitors decreased ROS production, but only to a limited extent. However, both the NADPH oxidase inhibitor DPI and LXA4 significantly decreased ROS production. This suggested that NADPH oxidase was the main source of ROS in this model.
NADPH oxidase is a complex regulator of oxidative stress that is highly expressed in endothelial cells, vascular smooth muscle cells and kidney cells (26). Activated NADPH oxidase provides an electron to O2, creating O2-. Superoxide dismutase catalyzes the conversion of the high concentration OO2-, generating H2O2. H2O2 eventually converts to further ROS (26). In tissues, the excessive production of NADPH oxidase-derived ROS induces tissue inflammation and fibrosis, leading to stress damage (22). NADPH oxidase embedded on the vascular endothelial cell membrane is activated to generate ROS, which triggers multiple pathways downstream of ERK, including generation of NADPH oxidase-derived ROS, transfer of transcriptional activator protein AP-1 to the nucleus and reduction of the levels of the inflammatory cytokine, IL-10(38). Another previous study in BV2 microglia cells suggested that LXA4 acted as an NADPH oxidase inhibitor, which prevented the production of intracellular ROS (39). The present study identified that LXA4 significantly suppressed UA-stimulated NADPH oxidase activity in HUVECs, further strengthening the evidence that LXA4 inhibits ROS in an NADPH oxidase-dependent response.
NADPH oxidase is a multicomponent enzyme system that be activated through the assembly of its cytosolic subunits, p47phox, p67phox, p40phox and Rac2 with the transmembrane subunits p22phox and gp91phox (26). p47phox is considered to be the principal subunit for NADPH oxidase. p47phox transfers to the membrane, and drives other cytoplasmic subunits together and promotes their translocation to the membrane to finish assembly of NADPH oxidase (38,40). Recently, a study have investigated the function of p47phox and the role of NADPH oxidase activation, and it has been identified that ROS generation depends on p47phox regulating the nuclear translocation of FOXO in ischemia cardiac cells. Targeted silencing by p47phox siRNA mitigates this domino effect (41). In p47phox (-/-) murine endothelial cells, NADPH oxidase-derived ROS production was largely suppressed (42). In addition, the p47phox is predominantly involved in the activation of other NADPH oxidase subunits (26). It was hypothesized that LXA4 may suppress the activation of NADPH oxidase through interactions with p47phox. The present study demonstrated that transfection of p47phox siRNA attenuated UA-induced increases in the generation of ROS and the activation of NADPH oxidase. p47phox translocates to the membrane following binding with the membrane subunit p22phox (28). p22phox has a terminal proline-rich tail, which could be used to bind with the cytoplasmic subunits and co-immunoprecipitation assays have demonstrated that p47phox binds with p22phox on the membrane in activated endothelial cells (28,43). In the present study, since UA failed to raise the total protein expression levels of p47phox, it was hypothesized that UA might promote the translocation of p47phox to the membrane from the cytoplasm to activate NADPH oxidase. Moreover, p22phox siRNA was transfected into HUVECs prior to treatment with UA. The expression of p47phox on the membrane significantly decreased in cells transfected with p22phox siRNA. Binding between p22phox and p47phox anchors p47phox at the cell membrane (43). Therefore, it was hypothesized that UA activates NADPH oxidase by promoting the transfer of p47phox to the membrane rather than through the upregulation of p47phox.
Whether LXA4 interfered with the binding of p22phox with the translocated p47phox to suppress UA-induced activation of NADPH oxidase in HUVECs is unclear. Several factors can attenuate NADPH oxidase activity by preventing the assembly of NADPH oxidase, including its p47phox and p22phox subunits (44). Carlo et al (45), demonstrated that the number of complexes of p47phox-p22phox were significantly decreased in activated neutrophils treatment with 15-Epi-lipoxin A4, and suggested that the assembly between p47phox and p22phox might be an effective target for the inhibition of NADPH oxidase activity. In the present study, the expressions of p47phox on the cytoplasm and the membrane were detected, demonstrating that the levels of p47phox significantly decreased in the cytoplasm and significantly increased on the membrane of HUVECs stimulated by UA. In addition, LXA4 could reverse this alteration. It was observed that LXA4 was able to indirectly interfere with the translocation of p47phox from the cytoplasm to the membrane. This mechanism may lead to suppression of NADPH oxidase activation in HUVECs following UA stimulation.
In conclusion, the results of the present study suggested that LXA4 inhibited the UA-induced release of NADPH oxidase-derived ROS in HUVECs. A possible mechanism of action behind this effect involved the suppression NADPH oxidase by preventing the translocation of p47phox from the cytoplasm to the membrane (Fig. 9). Considering the presence of oxidative stress and inflammatory responses downstream in patients with hyperuricemia, the assembly of NADPH oxidase may be considered as a potential therapeutic target to treat hyperuricemia or related diseases and LXA4 may be a therapeutic agent to achieve this effect. However, the present study has limitations in that in vitro experiments were used, which could not fully replicate the internal environment of human tissues. Further research is necessary to follow-up these studies in vivo to investigate the relevance of the observations of the present study for therapeutic intervention.
Figure 9.

Diagram illustrating the process of UA-induced NADPH oxidase activation on the generation of ROS in HUVECs and the impact of LXA4 on the extent of oxidative stress. UA enters the HUVECs and stimulates cytoplasmic subunits. The cytoplasmic subunits migrate to the membrane and bind with the membrane subunits under the guidance of p47phox, which results in the activation of NADPH oxidase. NADPH oxidase then interacts with O2 to produce ROS. LXA4 blocks the transfer of p47phox to the membrane, thus inhibiting the activation of NADPH oxidase and finally reducing the production of ROS. UA, uric acid; LXA4, lipoxin A4; ROS, reactive oxygen species; HUVEC, human umbilical vein endothelial cell.
Acknowledgements
Not applicable.
Funding
The present was supported by The Research Project of Sichuan Provincial Health Planning Commission (grant no. 17PJ067).
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Authors' contributions
YZ was a major contributor in writing the manuscript, performed all experiments and data analysis. HY and AZ performed cell culture and the western blot analyses. XJ designed the present study. ZP, GX and MZ performed data analysis. All authors read and approved the final version of the manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Liu CW, Chen KH, Tseng CK, Chang WC, Wu YW, Hwang JJ. The dose-response effects of uric acid on the prevalence of metabolic syndrome and electrocardiographic left ventricular hypertrophy in healthy individuals. Nutr Metab Cardiovasc Dis. 2019;29:30–38. doi: 10.1016/j.numecd.2018.10.001. [DOI] [PubMed] [Google Scholar]
- 2.Puddu P, Puddu GM, Cravero E, Vizioli L, Muscari A. Relationships among hyperuricemia, endothelial dysfunction and cardiovascular disease: Molecular mechanisms and clinical implications. J Cardiol. 2012;59:235–242. doi: 10.1016/j.jjcc.2012.01.013. [DOI] [PubMed] [Google Scholar]
- 3.Li M, Hou W, Zhang X, Hu L, Tang Z. Hyperuricemia and risk of stroke: A systematic review and meta-analysis of prospective studies. Atherosclerosis. 2014;232:265–270. doi: 10.1016/j.atherosclerosis.2013.11.051. [DOI] [PubMed] [Google Scholar]
- 4.Ogura T, Matsuura K, Matsumoto Y, Mimura Y, Kishida M, Otsuka F, Tobe K. Recent trends of hyperuricemia and obesity in Japanese male adolescents, 1991 through 2002. Metabolism. 2004;53:448–453. doi: 10.1016/j.metabol.2003.11.017. [DOI] [PubMed] [Google Scholar]
- 5.Mortada I. Hyperuricemia, type 2 diabetes mellitus, and hypertension: An emerging association. Curr Hypertens Rep. 2017;19(69) doi: 10.1007/s11906-017-0770-x. [DOI] [PubMed] [Google Scholar]
- 6.Mishima M, Hamada T, Maharani N, Ikeda N, Onohara T, Notsu T, Ninomiya H, Miyazaki S, Mizuta E, Sugihara S, et al. Effects of uric acid on the NO production of HUVECs and its restoration by urate lowering agents. Drug Res (Stuttg) 2016;66:270–274. doi: 10.1055/s-0035-1569405. [DOI] [PubMed] [Google Scholar]
- 7.Lepetsos P, Papavassiliou AG. ROS/oxidative stress signaling in osteoarthritis. Biochim Biophys Acta. 2016;1862:576–591. doi: 10.1016/j.bbadis.2016.01.003. [DOI] [PubMed] [Google Scholar]
- 8.Rasheed NO, Ahmed LA, Abdallah DM, El-Sayeh BM. Nephro-toxic effects of intraperitoneally injected EGCG in diabetic mice: Involvement of oxidative stress, inflammation and apoptosis. Sci Rep. 2017;7(40617) doi: 10.1038/srep40617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Minutoli L, Puzzolo D, Rinaldi M, Irrera N, Marini H, Arcoraci V, Bitto A, Crea G, Pisani A, Squadrito F, et al. ROS-mediated NLRP3 inflammasome activation in brain, heart, kidney, and testis ischemia/reperfusion injury. Oxid Med Cell Longev. 2016;2016(2183026) doi: 10.1155/2016/2183026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kadowaki D, Sakaguchi S, Miyamoto Y, Taguchi K, Muraya N, Narita Y, Sato K, Chuang VT, Maruyama T, Otagiri M, et al. Direct radical scavenging activity of benzbromarone provides beneficial antioxidant properties for hyperuricemia treatment. Biol Pharm Bull. 2015;38:487–492. doi: 10.1248/bpb.b14-00514. [DOI] [PubMed] [Google Scholar]
- 11.Matsuzawa A, Saegusa K, Noguchi T, Sadamitsu C, Nishitoh H, Nagai S, Koyasu S, Matsumoto K, Takeda K, Ichijo H. ROS-dependent activation of the TRAF6-ASK1-p38 pathway is selectively required for TLR4-mediated innate immunity. Nat Immunol. 2005;6:587–592. doi: 10.1038/ni1200. [DOI] [PubMed] [Google Scholar]
- 12.Chen Z, Zhou Q, Zou D, Tian Y, Liu B, Zhang Y, Wu Z. Chloro-benzoquinones cause oxidative DNA damage through iron-mediated ROS production in Escherichia coli. Chemosphere. 2015;135:379–386. doi: 10.1016/j.chemosphere.2015.04.076. [DOI] [PubMed] [Google Scholar]
- 13.Kuzkaya N, Weissmann N, Harrison DG, Dikalov S. Interactions of peroxynitrite with uric acid in the presence of ascorbate and thiols: Implications for uncoupling endothelial nitric oxide synthase. Biochem Pharmacol. 2005;70:343–354. doi: 10.1016/j.bcp.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 14.Xie H, Sun J, Chen Y, Zong M, Li S, Wang Y. EGCG attenuates uric acid-induced inflammatory and oxidative stress responses by medicating the NOTCH pathway. Oxid Med Cell Longev. 2015;2015(214836) doi: 10.1155/2015/214836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Z, Sheng Y, Liu C, Li K, Huang X, Huang J, Xu K. Nox4 has a crucial role in uric acid-induced oxidative stress and apoptosis in renal tubular cells. Mol Med Rep. 2016;13:4343–4348. doi: 10.3892/mmr.2016.5083. [DOI] [PubMed] [Google Scholar]
- 16.Ives A, Nomura J, Martinon F, Roger T, LeRoy D, Miner JN, Simon G, Busso N, So A. Xanthine oxidoreductase regulates macrophage IL1β secretion upon NLRP3 inflammasome activation. Nat Commun. 2015;6(6555) doi: 10.1038/ncomms7555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sautin YY, Nakagawa T, Zharikov S, Johnson RJ. Adverse effects of the classic antioxidant uric acid in adipocytes: NADPH oxidase-mediated oxidative/nitrosative stress. Am J Physiol Cell Physiol. 2007;293:C584–C596. doi: 10.1152/ajpcell.00600.2006. [DOI] [PubMed] [Google Scholar]
- 18.Tang L, Xu Y, Wei Y, He X. Uric acid induces the expression of TNF-α via the ROS-MAPK-NF-κB signaling pathway in rat vascular smooth muscle cells. Mol Med Rep. 2017;16:6928–6933. doi: 10.3892/mmr.2017.7405. [DOI] [PubMed] [Google Scholar]
- 19.Chandrasekharan JA, Sharma-Walia N. Lipoxins: Nature's way to resolve inflammation. J Inflamm Res. 2015;8:181–192. doi: 10.2147/JIR.S90380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weiss GA, Troxler H, Klinke G, Rogler D, Braegger C, Hersberger M. High levels of anti-inflammatory and pro-resolving lipid mediators lipoxins and resolvins and declining docosahexaenoic acid levels in human milk during the first month of lactation. Lipids Health Dis. 2013;12(89) doi: 10.1186/1476-511X-12-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu C, Guan H, Cai C, Li F, Xiao J. Lipoxin A4 suppresses osteoclastogenesis in RAW264.7 cells and prevents ovariectomy-induced bone loss. Exp Cell Res. 2017;352:293–303. doi: 10.1016/j.yexcr.2017.02.018. [DOI] [PubMed] [Google Scholar]
- 22.Nascimento-Silva V, Arruda MA, Barja-Fidalgo C, Fierro IM. Aspirin-triggered lipoxin A4 blocks reactive oxygen species generation in endothelial cells: A novel antioxidative mechanism. Thromb Haemost. 2007;97:88–98. [PubMed] [Google Scholar]
- 23.Peshavariya HM, Taylor CJ, Goh C, Liu GS, Jiang F, Chan EC, Dusting GJ. Annexin peptide Ac2-26 suppresses TNFα-induced inflammatory responses via inhibition of Rac1-dependent NADPH oxidase in human endothelial cells. PLoS One. 2013;8(e60790) doi: 10.1371/journal.pone.0060790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res. 1994;74:1141–1148. doi: 10.1161/01.res.74.6.1141. [DOI] [PubMed] [Google Scholar]
- 25.Maeda M, Hayashi T, Mizuno N, Hattori Y, Kuzuya M. Intermittent high glucose implements stress-induced senescence in human vascular endothelial cells: Role of superoxide production by NADPH oxidase. PLoS One. 2015;10(e0123169) doi: 10.1371/journal.pone.0123169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Δ Δ C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 27.Jaquet V, Scapozza L, Clark RA, Krause KH, Lambeth JD. Small-molecule NOX inhibitors: ROS-generating NADPH oxidases as therapeutic targets. Antioxid Redox Signal. 2009;11:2535–2552. doi: 10.1089/ars.2009.2585. [DOI] [PubMed] [Google Scholar]
- 28.Belambri SA, Rolas L, Raad H, Hurtado-Nedelec M, Dang PM, El-Benna J. NADPH oxidase activation in neutrophils: Role of the phosphorylation of its subunits. Eur J Clin Invest. 2018;48 (Suppl 2)(e12951) doi: 10.1111/eci.12951. [DOI] [PubMed] [Google Scholar]
- 29.Mohandas R, Sautina L, Beem E, Schuler A, Chan WY, Domsic J, McKenna R, Johnson RJ, Segal MS. Uric acid inhibition of dipeptidyl peptidase IV in vitro is dependent on the intracellular formation of triuret. Exp Cell Res. 2014;326:136–142. doi: 10.1016/j.yexcr.2014.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Song C, Zhao X. Uric acid promotes oxidative stress and enhances vascular endothelial cell apoptosis in rats with middle cerebral artery occlusion. Biosci Rep. 2018;38(BSR20170939) doi: 10.1042/BSR20170939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Choi YJ, Yoon Y, Lee KY, Hien TT, Kang KW, Kim KC, Lee J, Lee MY, Lee SM, Kang DH, et al. Uric acid induces endothelial dysfunction by vascular insulin resistance associated with the impairment of nitric oxide synthesis. FASEB J. 2014;28:3197–3204. doi: 10.1096/fj.13-247148. [DOI] [PubMed] [Google Scholar]
- 32.Zhou Y, Zhao M, Pu Z, Xu G, Li X. Relationship between oxidative stress and inflammation in hyperuricemia: Analysis based on asymptomatic young patients with primary hyperuricemia. Medicine (Baltimore) 2018;97(e13108) doi: 10.1097/MD.0000000000013108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xia F, Wang C, Jin Y, Liu Q, Meng Q, Liu K, Sun H. Luteolin protects HUVECs from TNF-α-induced oxidative stress and inflammation via its effects on the Nox4/ROS-NF-κB and MAPK pathways. J Atheroscler Thromb. 2014;21:768–783. doi: 10.5551/jat.23697. [DOI] [PubMed] [Google Scholar]
- 34.Kantarci A, Aytan N, Palaska I, Stephens D, Crabtree L, Benincasa C, Jenkins BG, Carreras I, Dedeoglu A. Combined administration of resolvin E1 and lipoxin A4 resolves inflammation in a murine model of Alzheimer's disease. Exp Neurol. 2018;300:111–120. doi: 10.1016/j.expneurol.2017.11.005. [DOI] [PubMed] [Google Scholar]
- 35.Li Y, Cai L, Wang H, Wu P, Gu W, Chen Y, Hao H, Tang K, Yi P, Liu M, et al. Pleiotropic regulation of macrophage polarization and tumorigenesis by formyl peptide receptor-2. Oncogene. 2011;30:3887–3899. doi: 10.1038/onc.2011.112. [DOI] [PubMed] [Google Scholar]
- 36.Biala AK, Dhingra R, Kirshenbaum LA. Mitochondrial dynamics: Orchestrating the journey to advanced age. J Mol Cell Cardiol. 2015;83:37–43. doi: 10.1016/j.yjmcc.2015.04.015. [DOI] [PubMed] [Google Scholar]
- 37.Uddin MJ, Werfel TA, Crews BC, Gupta MK, Kavanaugh TE, Kingsley PJ, Boyd K, Marnett LJ, Duvall CL. Fluorocoxib A loaded nanoparticles enable targeted visualization of cyclooxygenase-2 in inflammation and cancer. Biomaterials. 2016;92:71–80. doi: 10.1016/j.biomaterials.2016.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lian S, Xia Y, Khoi PN, Ung TT, Yoon HJ, Kim NH, Kim KK, Jung YD. Cadmium induces matrix metalloproteinase-9 expression via ROS-dependent EGFR, NF-кB, and AP-1 pathways in human endothelial cells. Toxicology. 2015;338:104–116. doi: 10.1016/j.tox.2015.10.008. [DOI] [PubMed] [Google Scholar]
- 39.Wang YP, Wu Y, Li LY, Zheng J, Liu RG, Zhou JP, Yuan SY, Shang Y, Yao SL. Aspirin-triggered lipoxin A4 attenuates LPS-induced pro-inflammatory responses by inhibiting activation of NF-κB and MAPKs in BV-2 microglial cells. J Neuroinflammation. 2011;8(95) doi: 10.1186/1742-2094-8-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Park IH, Hwang HM, Jeon BH, Kwon HJ, Hoe KL, Kim YM, Ryoo S. NADPH oxidase activation contributes to native low-density lipoprotein-induced proliferation of human aortic smooth muscle cells. Exp Mol Med. 2015;47(e168) doi: 10.1038/emm.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ter Horst EN, Hahn NE, Geerts D, Musters RJP, Paulus WJ, van Rossum AC, Meischl C, Piek JJ, Niessen HWM, Krijnen PAJ. p47phox-dependent reactive oxygen species stimulate nuclear translocation of the foxO1 transcription factor during netabolic inhibition in cardiomyoblasts. Cell Biochem Biophys. 2018;76:401–410. doi: 10.1007/s12013-018-0847-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen JX, Zeng H, Lawrence ML, Blackwell TS, Meyrick B. Angiopoietin-1-induced angiogenesis is modulated by endothelial NADPH oxidase. Am J Physiol Heart Circ Physiol. 2006;291:H1563–H1572. doi: 10.1152/ajpheart.01081.2005. [DOI] [PubMed] [Google Scholar]
- 43.Chakraborti S, Sarkar J, Chakraborti T. Role of PLD-PKCζ signaling axis in p47phox phosphorylation for activation of NADPH oxidase by angiotensin II in pulmonary artery smooth muscle cells. Cell Biol Int. 2019;43:678–694. doi: 10.1002/cbin.11145. [DOI] [PubMed] [Google Scholar]
- 44.Macías Pérez ME, Hernández Rodríguez M, Cabrera Pérez LC, Fragoso-Vázquez MJ, Correa-Basurto J, Padilla-Martínez II, Méndez Luna D, Mera Jiménez E, Flores Sandoval C, Tamay Cach F, et al. Aromatic regions govern the recognition of NADPH oxidase inhibitors as diapocynin and its analogues. Arch Pharm (Weinheim) 2017;350(1700041) doi: 10.1002/ardp.201700041. [DOI] [PubMed] [Google Scholar]
- 45.Carlo T, Kalwa H, Levy BD. 15-Epi-lipoxin A4 inhibits human neutrophil superoxide anion generation by regulating polyisoprenyl diphosphate phosphatase 1. FASEB J. 2013;27:2733–2741. doi: 10.1096/fj.12-223982. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.