

Abstract
Ulinastatin exerts protective effects against lipopolysaccharide (LPS)-induced cardiac dysfunction. Autophagy has been demonstrated to serve an important role in sepsis-induced cardiomyopathy; however, whether ulinastatin has an anti-autophagic effect in sepsis requires further investigation. The present study aimed to determine the protective effects of ulinastatin on cardiac dysfunction and its role in autophagy during sepsis. C57BL/6J mice were randomly divided into a control, LPS and LPS + ulinastatin group, the survival status of the mice was observed every 6 h and the survival rate at each time point was calculated for 7 days. Furthermore, JC-1 dye and ELISAs were used to analyze the mitochondrial membrane potential (MMP) and serum cardiac troponin I (cTnI) levels, respectively. Western blotting and ELISAs were used to measure the levels of tumor necrosis factor (TNF)-α and interleukin (IL)-6. In addition, the cardiac ultrastructure and the number of autophagosomes formed were visualized using transmission electron microscopy, and the pathological changes in the myocardial tissues were analyzed using hematoxylin & eosin staining. Finally, the expression levels of autophagy-related proteins were analyzed using western blotting and immunofluorescence staining. The current study indicated that ulinastatin significantly improved the survival rate of septic mice. It was suggested that ulinastatin may protect against LPS-induced myocardium injury through its anti-inflammatory activity, as decreased cTnI levels, increased MMP and decreased expression levels of TNF-α and IL-6 were all observed following ulinastatin treatment. Furthermore, the number of autophagosomes formed, and the expression levels of microtubule-associated protein light chain 3 and Beclin 1 were significantly decreased following ulinastatin treatment. It was further observed that ulinastatin suppressed LPS-induced autophagosome formation, as indicated by the accumulation of sequestosome 1/p62, and the elimination of lysosome-associated membrane glycoprotein 1. In conclusion, the results of the present study suggested that ulinastatin treatment may improve survival and exert a protective effect over LPS-induced cardiac dysfunction. Furthermore, this protective effect may be associated with its anti-inflammatory and anti-autophagic activity.
Keywords: ulinastatin, autophagy, sepsis, cardiomyopathy
Introduction
Sepsis is a systematic inflammatory response and it is a major cause of mortality in intensive care units (1). Moreover, sepsis is associated with the dysfunction of multiple organs (1). It has been previously documented that 40-50% of patients with sepsis develop cardiac dysfunction (2). Cardiac dysfunction is a major feature of sepsis, with the 50% mortality rate increasing in patients with severe heart failure (3,4). The mortality rate of sepsis is rising despite the increasing numbers of studies developing antimicrobial therapies and circulatory support (5). Therefore, the present study aimed to determine the mechanisms of LPS-induced myocardial injury to facilitate the development of novel treatment strategies.
Ulinastatin is an endogenous inhibitor of proteases that is located in the urine and blood, and inhibits various serine protease (6). Ulinastatin has been previously used to treat sepsis cardiomyopathy, due to its ability to function as a protease inhibitor and its anti-inflammatory effects (7). It was previously demonstrated that ulinastatin enhanced cardiac function, reduced the size of the myocardial infarction and decreased the levels of inflammatory cytokines during hemorrhagic shock and ischemia-reperfusion (IR) injury (8). A previous study has also suggested that the protective effects of ulinastatin are based on its anti-inflammatory, anti-oxidative stress and anti-apoptotic properties (9). Furthermore, ulinastatin was indicated to serve an important role in treating lipopolysaccharide (LPS)-induced myocardial injury (10). However, the specific mechanisms of the role of ulinastatin in sepsis are not fully understood.
Autophagy is a type of programmed cell death and it is an adaptation used to respond to a variety of stress stimuli (11). Previous studies have identified the presence of activated autophagy in a variety of heart diseases, such as IR injury, heart failure and sepsis (12,13). It has been widely demonstrated that autophagy performs two opposing functions in the heart (14); it serves a protective role during nutrient deprivation and cellular stress, whereas the excessive induction of autophagy promotes self-destruction (15). In a model of IR injury, ulinastatin was revealed to protect cardiomyocytes via the activation of the mTOR pathway, which is associated with the process of autophagy (16). A previous study has identified that ulinastatin exerted a protective effect on sepsis through its regulation of autophagy in myocardial IR injury models (16). Therefore, it was hypothesized that ulinastatin may serve a protective role in sepsis-induced myocardial injury by regulating autophagy; however, further research into the role of ulinastatin in sepsis is required. The present study aimed to investigate whether ulinastatin protected against sepsis cardiomyopathy and to determine the possible underlying mechanisms of this interaction.
Materials and methods
Sepsis model
All procedures were approved by The Animal Ethics Committees of The Fourth Military Medical University (Xi'an, China) and were performed in accordance with the guidelines of The China Council of Animal Care. A total of 84 C57BL/6J mice (age, 8-10 weeks; weight, 25-30 g; sex, male) were purchased from The Laboratory Animal Center of The Fourth Military Medical University. Mice were housed at a temperature of 25˚C in ~60% humidity, with a 12 h light/dark cycle. Mice were provided with a standard diet and free access to water. Endotoxemia was induced via an intraperitoneal (i.p.) injection of 18 mg/kg lipopolysaccharide (LPS; cat. no. L2630; Sigma-Aldrich; Merck KGaA) or 10 mg/kg LPS from the Escherichia coli serotype O111:B4(17). Ulinastatin was obtained from Guangdong Tianpu Biochemical Pharmaceutical Co., Ltd. To determine the protective effect of ulinastatin on the survival rate following lethal endotoxemia, 60 mice were divided into two groups: i) A total of 30 mice in the LPS group, where mice were treated with the lethal dose of 18 mg/kg LPS and 0.9% saline; and ii) 30 mice in the LPS + Ulinastatin group, where the mice received 18 mg/kg LPS and i.p. injection of 1x105 U/kg Ulinastatin (i.p.) daily for 4 days, which was determined in a previous study (17).
To investigate the effects of ulinastatin on cardiac function and the levels of autophagy, 24 mice were randomly divided into a control group, a LPS group (mice received 10 mg/kg LPS and 0.9% saline) and a LPS + ulinastatin group [mice received 10 mg/kg LPS and 1x105 U/kg Ulinastatin (i.p.)] (17), with 8 mice in each group. The administered dose of ulinastatin was selected according to a previous study, in which ulinastatin was observed to exhibit a protective effect on sepsis (18). All mice were anesthetized for echocardiography after treated with LPS and/or Ulinastatin for 12 h, following which they were sacrificed for subsequent experiments.
For survival analysis, humane endpoints were established. In the process of observing the survival of mice, once they showed labored breathing, they were euthanized immediately by i.p. injection of 120 mg/kg sodium pentobarbital sodium (20 mg/ml). Conventional anti-shock therapy was given by an intraperitoneal injection of 0.9% saline after 4 days of drug treatment. At the end of the 7 day survival cycle, the rest of the surviving mice in two groups were euthanized using 120 mg/kg sodium pentobarbital sodium (20 mg/ml) through the intraperitoneal route. Following cervical dislocation to ensure death, 600 µl blood samples were collected from the abdominal aorta and the myocardial tissues of mice were collected and frozen at 80˚C for further evaluation. To minimize animal suffering, only qualified personnel were permitted to perform the experiments.
Echocardiography
After anesthesia with an i.p. injection of 60 mg/kg sodium pentobarbital sodium (20 mg/ml), conventional echocardiography of the left ventricle (LV) in each mouse was performed 12 h after an i.p. injection of LPS using a mouse echocardiography system (Vevo 2100 Imaging System; VisualSonics, Inc.) that was equipped with a 30-MHz phased transducer. The following parameters were measured: LV end diastolic pressure (LVEDP), LV developed pressure (LVDP), maximal velocity increase of LV pressure per second (+dP/dtmax) and maximal velocity decrease of LV pressure per second (-dP/dtmax).
ELISAs
Blood samples were centrifuged at 1,500 x g for 15 min at room temperature to collect the serum. Serum cardiac troponin-I (cTnI; cat. no. F00503; Shanghai Westang Bio-Tech Co., Ltd.), cardiac tumor necrosis factor (TNF)-α (cat. no. T7539; Sigma-Aldrich; Merck KGaA) and cardiac interleukin (IL)-6 (cat. no. 555240; BD Biosciences) levels were analyzed using their respective ELISA kits, according to the manufacturer's protocol.
Mitochondrial membrane potential (MMP)
A mitochondria isolation kit (cat. no. C3606; Beyotime Institute of Biotechnology) was used to extract the purified mitochondria from the myocardial tissues, according to the manufacturer's protocol (19). Subsequently, a mitochondrial membrane potential assay kit with JC-1 dye (cat. no. C2006; Beyotime Institute of Biotechnology) and a fluorescence microplate reader (Chromate 4300; Awareness Technology, Inc.) were used to measure MMP, according to the manufacturer's protocol. JC-1 green fluorescence (530 nm) was used to reflect JC-1 monomers and red fluorescence (590 nm) was used to determine the formation of J-aggregates. The ratio of J-aggregates to JC-1 mitochondria were measured using a microplate reader after JC-1 staining. The ratio of monomeric to aggregated JC-1 fluorescence intensity was used to quantify the changes in MMP. Data were presented as the normalized percentage of the average fluorescence intensity and values were normalized to the control group.
Western blotting
Total proteins were extracted using the RIPA protein extraction reagent (Beyotime Institute of Biotechnology) from the myocardial tissues and the protein concentration was determined using the bicinchoninic acid Protein Assay kit (Beyotime Institute of Biotechnology). A total of 20 µg protein/lane was separated via 10-15% SDS-PAGE. The separated proteins were subsequently transferred onto PVDF membranes (EMD Millipore) and blocked with 5% non-fat milk at room temperature for 1 h. The membranes were then incubated with the following primary antibodies overnight at 4˚C: Anti-TNF-α (1:1,000; cat. no. ab6671; Abcam), anti-IL-6 (1:1,000; cat. no. ab9324; Abcam), anti-microtubule-associated protein light chain 3 LC3B (1:1,000; cat. no. ab51520; Abcam), anti-lysosomal-associated membrane protein 1 (LAMP-1; 1:1,000; cat. no. ab24170; Abcam), anti-sequestosome-1 (SQSTM1)/p62 (1:1,000; cat. no. ab56416; Abcam) and anti-GAPDH (1:3,000; cat. no. ab181602; Abcam). Following the primary antibody incubation, the membranes were washed three times with TBS-0.1% Tween 20 and incubated with horseradish peroxidase-conjugated secondary antibody (1:5,000; cat. no. ZB-2301; Beijing Zhongshan Golden Bridge Biotechnology Co., Ltd.) and horseradish peroxidase-conjugated secondary antibody (1:5,000; cat. nos. ab6728; Abcam) for 2 h at room temperature. Protein bands were visualized using an enhanced chemiluminescence kit (EMD Millipore) and expression levels were quantified using a densitometric analysis system (Image Lab software; version 4.0; Bio-Rad Laboratories, Inc.).
Hematoxylin & eosin (H&E) staining
Following the establishment of the LPS model, myocardial tissues were excised, washed with ice-cold PBS, following which 1-mm thick slices were prepared after the hearts were snap frozen at -80˚C for 5 min. The slices were fixed directly in 10% neutral formalin for 24 h at room temperature and were subsequently paraffin-embedded. Sections (3 µm thickness) were then prepared from the paraffin-embedded tissue blocks by dehydration through a graded ethanol series and washing with xylene for H&E staining, for 12 min at room temperature. Morphological changes in the myocardial tissues were evaluated using an Olympus light microscope (magnification x400; Olympus Corporation).
Transmission electron microscopy (TEM)
Myocardial tissues were sliced into 1 mm3 sections and fixed with 2.5% glutaraldehyde overnight at 4˚C. The tissues were immersed in 1% osmium tetroxide for 2 h at room temperature, dehydrated in an ascending ethanol series, followed by dehydration in an 100% acetone for three times at 4˚C and subsequently embedded in epoxy resin. The tissues were then cut into ultrathin sections (60-70 nm) using an ultramicrotome. Sections were stained with 2% uranyl acetate and 2% lead citrate for 30 min at room temperature. Stained sections were visualized using a JEM-1010 TE microscope (magnification x15,000 and x30,000; JEOL, Ltd.) and the amount of autophagosome were quantified using Gatan Digital Micrograph® software (Gatan, Inc.).
Immunofluorescence staining
Myocardial tissues were excised, washed with ice-cold PBS, following which 1-mm thick slices were prepared after the hearts were snap frozen at -80˚C for 5 min. The slices were sequentially fixed with 4% formaldehyde for 30 min at room temperature, permeabilized with 0.2% Triton X-100 (HyClone; GE Healthcare Life Sciences) for 15 min at room temperature and blocked in 5% bovine serum albumin (Beyotime Institute of Biotechnology) for 1 h at room temperature. Sections were then incubated with a rabbit anti-LC3 antibody (1:100; cat. no. ab128025; Abcam) and a rabbit anti-Beclin 1 antibody (1:100; cat. no. ab210498; Abcam) at 4˚C overnight. Following the primary antibody incubation, the sections were incubated with an Alexa Fluor® 488-conjugated goat anti-rabbit IgG secondary antibody (1:10; cat. no. ab150077; Abcam) at 37˚C for 1 h. The cell nucleus was stained with 2 µg/ml DAPI for 30 min at room temperature and 500 ml/l glycerine was used to seal the sections. The slides were observed using a confocal laser scanning microscope (magnification x400) to analyze the expression levels of autophagy-associated proteins.
Statistical analysis
All experiments were independently performed in triplicate. Statistical analysis was performed using SPSS version 19 software (IBM Corp.) and data are presented as the mean ± SEM. Survival rate analysis was analyzed using the Kaplan-Meier method, with a log-rank test used for comparison (20). Statistical differences between groups were determined using a one-way ANOVA, followed by a Student-Newman-Keuls test for multiple comparisons. P<0.05 was considered to indicate a statistically significant difference.
Results
Ulinastatin improves the survival rate of mice with endotoxemia
To determine the protective effect of ulinastatin on the survival rate following lethal endotoxemia, mice were treated with a lethal dose of LPS (18 mg/kg), whereas mice in the LPS + ulinastatin group received 18 mg/kg LPS and ulinastatin (1x105 U/kg) daily for 4 days daily. After 7 days, it was revealed that only 12/30 of mice in the LPS + ulinastatin group survived, thus the survival rate was 40% (Fig. 1). In the LPS group, mice were treated with an equal amount of saline and it was subsequently demonstrated that only 3/30 mice had survived 7 days later, thus demonstrating a survival rate of 10% (Fig. 1). Therefore, these findings suggested that ulinastatin may reduce the mortality rate of lethal endotoxemia.
Figure 1.
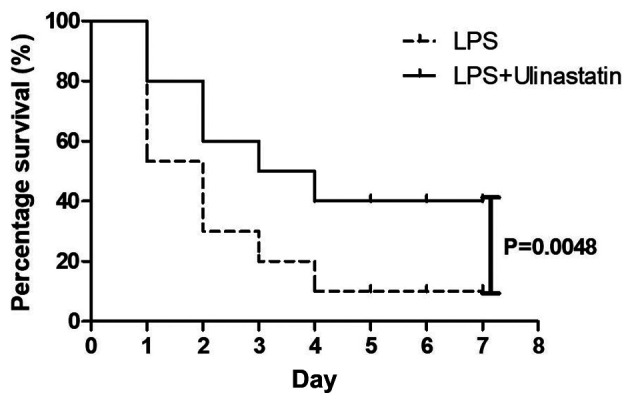
Ulinastatin improves the survival rate of mice with endotoxemia. The survival rate of mice receiving a lethal dose of LPS (18 mg/kg) or LPS (18 mg/kg) + ulinastatin (1x105 U/kg) was analyzed for 7 consecutive days. LPS, lipopolysaccharide.
Effect of ulinastatin on the cardiac function in LPS-stimulated mice
The cardiac function parameters in each experimental group are presented in Table I. Following the establishment of the LPS model mice, it was revealed that the LVEDP was significantly increased compare with that in the control group. The LVDP, +dP/dtmax and -dP/dtmax were all found to be significantly decreased following 12 h of treatment in the LPS group compared with the control group (Table I). Moreover, following ulinastatin treatment, the LVEDP was significantly decreased by 13.3%, whereas the LVDP, +dP/dtmax and -dP/dtmax were increased by 30.5, 15.7 and 44.6%, respectively, compared with the LPS group.
Table I.
Effect of ulinastatin on cardiac function in LPS-induced mice.
| Group | n | Left ventricle end diastolic pressure (mmHg) | Left ventricle developed pressure (mmHg) | Maximal velocity increase of left ventricle pressure per second (mmHg) | Maximal velocity decrease of left ventricle pressure per second (mmHg) |
|---|---|---|---|---|---|
| Control | 8 | 20.7±3.6 | 118.1±11.7 | 8,720.6±135.1 | 6,121.0±212.0 |
| LPS | 8 | 42.1±1.8a | 68.2±7.3a | 6,012.4±312.4a | 3,132.4±254.7a |
| LPS + ulinastatin | 8 | 36.5±1.3a,b | 89.0±5.6a,b | 6,956.2±216.3a,b | 4,528.1±198.5a,b |
aP<0.05 vs. control group;
bP<0.05 vs. LPS group. LPS, lipopolysaccharide.
Effects of ulinastatin on the mice myocardium during endotoxemia
The tissues and serum from individual mice were obtained following 12 h of LPS induction. The ELISA results revealed that cTnI levels were significantly increased in the LPS and LPS + ulinastatin groups compared with the control group (Table II). Of note, the LPS + ulinastatin group had significantly decreased cTnI levels compared with the LPS group (Table II). It was also indicated that the MMP was significantly decreased in the LPS and LPS + ulinastatin groups compared with the control group; however, the LPS + ulinastatin group demonstrated a significantly increased MMP compared with the LPS group (Table II). Previous studies have reported that inflammatory cytokines serve an important role in the progression of sepsis (21). To investigate the protective effects of ulinastatin on the LPS-induced inflammatory response, the levels of TNF-α and IL-6 in the mice myocardial tissue were also investigated. The ELISA results indicated that the levels of TNF-α and IL-6 were significantly increased in the LPS and LPS + ulinastatin groups compared with the control group (Table II). However, following the treatment with ulinastatin, the levels of TNF-α and IL-6 were significantly decreased in the myocardium of mice with endotoxemia compared with the LPS group (Table II).
Table II.
Effect of ulinastatin on the mouse myocardium during endotoxemia.
| Group | n | Mitochondrial membrane potential | Serum cardiac troponin I (ng/ml) | Tumor necrosis factor-α (pg/ml) | Interleukin-6 (pg/ml) |
|---|---|---|---|---|---|
| Control | 8 | 1.00 | 0.136±0.0142 | 39.620±2.140 | 3.820±0.430 |
| LPS | 8 | 0.321±0.067a | 0.919±0.0138a | 268.420±15.920a | 14.560±0.920a |
| LPS + ulinastatin | 8 | 0.673±0.075a,b | 0.585±0.0129a,b | 172.370±8.560a,b | 9.270±0.610a,b |
aP<0.05 vs. control group;
bP<0.05 vs. LPS group. LPS, lipopolysaccharide.
Effects of ulinastatin on the expression levels of inflammatory factors in endotoxemia
The western blotting data revealed that the protein expression levels of TNF-α and IL-6 were significantly increased in the LPS and LPS + ulinastatin groups compared with the control group (Fig. 2A and B). Notably, following treatment with ulinastatin, the expression levels of TNF-α and IL-6 were significantly decreased in the myocardium of mice with endotoxemia in the LPS + ulinastatin group compared with those in the LPS group (Fig. 2A and B). Collectively, these findings suggested that ulinastatin may exert protective effects in the endotoxemia myocardium and may alleviate the inflammatory response through decreasing the expression levels of TNF-α and IL-6.
Figure 2.

Effect of ulinastatin on the expression levels of inflammatory factors in endotoxemia. Western blotting was used to analyze the protein expression levels of (A) TNF-α and (B) IL-6 in myocardial tissues. GAPDH was used as the loading control. Data are presented as the mean ± SEM and statistical differences were determined using a one-way ANOVA and Student-Newman-Keuls post hoc test. #P<0.05 vs. control group; *P<0.05 vs. LPS group. TNF-α, tumor necrosis factor-α; IL-6, interleukin-6; LPS, lipopolysaccharide.
Effects of ulinastatin on the cardiac ultrastructure and pathological changes
TEM was used to observe the cardiac ultrastructure; a normal cardiac structure and mitochondrial distribution were observed in the control group (Fig. 3A), whereas in the LPS group, the myofilaments and mitochondria were disorganized in the myocardial tissue, alongside the presence of autophagy (Fig. 3B). Notably, in the LPS + ulinastatin group, the myocardial tissue exhibited orderly arranged myofilaments, a normal mitochondrial distribution and the presence of small magnitudes of autophagy (Fig. 3C).
Figure 3.

Effect of ulinastatin on attenuating myocardial injuries. LPS-induced endotoxemia mice were treated with saline or ulinastatin, and the left ventricle tissues were collected 12 h later for analysis. Transmission electron microscopy was used to analyze the cardiac ultrastructure in the (A) control, (B) LPS and (C) LPS + ulinastatin groups. Magnification x15,000. Scale bar, 2 µm. White arrow indicates the myofilaments, the red arrow indicates the mitochondria and the yellow arrow indicates the presence of autophagosomes. Hematoxylin & eosin staining was used to determine the pathological changes in the myocardial tissue in the (D) control, (E) LPS and (F) LPS + ulinastatin groups. Magnification x200. LPS, lipopolysaccharide.
H&E staining was subsequently used to assess the pathological changes in the myocardial tissue. In the LPS group, myofibril loss, myocardial cell necrosis and structural abnormalities were all observed, which indicated the occurrence of severe cardiac injury (Fig. 3E). However, in the LPS + ulinastatin group, there was a reduction in the pathological damage observed in the mice hearts compared with the LPS group (Fig. 3F).
Ulinastatin decreases the levels of LPS-induced autophagy
Myocardial tissues from the mice were obtained 12 h after LPS induction. It was subsequently demonstrated that the expression levels of LC3II were significantly increased in the LPS and LPS + ulinastatin group compared with the control group (Fig. 4A). However, the expression levels of LC3II (Fig. 4A), Beclin 1 (Fig. 4B) and LAMP-1 (Fig. 4C) were all significantly decreased following ulinastatin treatment compared with those in the LPS group. In addition, it was revealed that the expression levels of SQSTM1/p62 was significantly decreased in the LPS and LPS + ulinastatin group compared with those in the control group, ulinastatin significantly increased the expression levels of SQSTM1/p62 further compared with those in the LPS group (Fig. 4D). Moreover, TEM was also used to observe the number of autophagosomes formed in the heart of each group. A normal mitochondrial structure and very few autophagosomes were observed in the control group, whereas in the LPS group, myocardial tissues exhibited a significantly increased number of autophagosomes compared with the control group. Notably, ulinastatin treatment was observed to significantly decrease this LPS-induced increase in autophagosome formation (Fig. 4E).
Figure 4.

Effects of ulinastatin on autophagy in mice with endotoxemia. LPS-induced endotoxemia mice were treated with saline or ulinastatin, and the left ventricle tissues were collected 12 h later for analysis. Western blotting was used to analyze the protein expression levels of (A) LC3II, (B) Beclin 1, (C) LAMP-1 and (D) SQSTM1/p62 in the myocardial tissues. GAPDH was used as a loading control. (E) Transmission electron microscopy was used to observe the number of autophagosomes formed in the cardiac tissue. Magnification x30,000. Scale bar, 1 µm. Yellow arrow indicates the autophagosomes, the white arrow indicates the mitochondria and the red arrow indicates the lysosomes. Data are presented as the mean ± SEM. Data were compared using a one-way ANOVA, followed by a Student-Newman-Keuls post hoc test. #P<0.05 vs. control group; *P<0.05 vs. LPS group. LPS, lipopolysaccharide; LC3, microtubule-associated protein light chain 3; LAMP, lysosomal-associated membrane protein 1; SQSTM1, sequestosome-1.
Furthermore, the expression levels of the autophagy associated proteins, LC3 and Beclin 1, were analyzed using confocal microscopy. The expression levels of LC3 and Beclin 1 in the control group were low, whereas under the same conditions, the fluorescent intensities of LC3 and Beclin 1 in the LPS group were significantly increased compared with the control group (Fig. 5). Notably, following the treatment with ulinastatin, the expression levels of LC3 and Beclin 1 were significantly decreased compared with the LPS group (Fig. 5).
Figure 5.

Effect of ulinastatin on autophagy in endotoxemia mice. Endotoxemia mice were treated with vehicle or ulinastatin, and the left ventricle tissues were collected 12 h later for analysis. A confocal microscope was used to analysis the fluorescence distribution of (A) LC3II and (B) Beclin 1 in myocardial tissues. Semi-quantitative analysis of (C) LC3II and (D) Beclin 1 expression levels in myocardial tissues. Magnification x200. Data were analyzed using a one-way ANOVA, followed by a Student-Newman-Keuls post hoc test. #P<0.05 vs. control group; *P<0.05 vs. LPS group. LPS, lipopolysaccharide; LC3, microtubule-associated protein light chain 3.
Discussion
The present study demonstrated that ulinastatin may improve survival rate and exert a protective effect against LPS-induced cardiac dysfunction. It was suggested from the findings of the present study that this protective effect may be associated with the anti-inflammatory activity of ulinastatin and its ability to inhibit autophagy. Therefore, the administration of ulinastatin may exhibit a protective effect in the pathophysiology of sepsis.
A previous study has reported the presence of decreased serum levels of ulinastatin in patients with sepsis, and the levels of ulinastatin were at their lowest during cases of severe sepsis and septic shock (22). Therefore, treatment with ulinastatin may increase the levels of ulinastatin in patients with severe sepsis. The protective effect of ulinastatin in sepsis has been confirmed in a number of previous studies; for example, ulinastatin was observed to serve a protective role in the heart tissue of septic rats, and its mechanism was attributed to its regulatory effect over the stress response, cell signaling transduction, energy metabolism, the immune response and other related genes (23,24). Furthermore, a previous study revealed that ulinastatin contributed to the recovery of cardiac function following reperfusion by reducing mitochondrial dysfunction and maintaining energy production (8). However, the role that ulinastatin serves over the regulation of autophagy during sepsis remains poorly understood. Therefore, in the present study, endotoxemic mice were treated with ulinastatin to investigate its underlying mechanism.
TNF-α and IL-6 are cytokines associated with the inflammatory response (25). A previous study indicated that ulinastatin reduced sepsis-related inflammation by downregulating TNF-α expression levels (26). ELISA and western blotting data from the present study revealed that the serum levels and protein expression levels of TNF-α and IL-6 were significantly increased in endotoxemia mice, and these LPS-induced increases could be significantly decreased following ulinastatin treatment. Therefore, the results of the present study indicated that ulinastatin may protect against LPS-induced myocardial injury by inhibiting the release of TNF-α and IL-6.
Ulinastatin has been proposed to serve as a myocardial protective agent, and it has been reported that ulinastatin improved the myocardial contractility, reduced the myocardial infarct size and decreased the levels of creatine kinase and cTnI in myocardial IR injury (16). cTnI is a highly sensitive and specific marker of myocardial injury (27). Mitochondria are important organelles for cardiomyocytes that not only provide the energy for cells, but can also trigger cell apoptosis (28). Furthermore, decreases in the MMP have been found to be the driving force for mitochondrial dysfunction (29). In the present study, endotoxemic mice were treated with ulinastatin and it was indicated that ulinastatin exhibited a protective effect in the hearts of mice with endotoxemia. This protective effect was demonstrated by decreased cTnI levels, an increased MMP, as well as the presence of an intact, well-ordered cardiac ultrastructure.
Autophagy serves an essential role in cell survival, as well as in cell death (14). Autophagy is a highly regulated intracellular degradation process, by which cells remove cytosolic long-lived proteins and damaged organelles (15). LC3 is the mammalian homologue of yeast Atg8, and as a lipidated form of LC3, LC3II has been widely used as a marker of autophagy by indicating the number of autophagosomes formed (30). Moreover, Beclin 1 has been discovered to serve an important role in autophagosome formation and autolysosomal fusion (31), and LAMP-1 is a lysosome marker that has been associated with the increased accumulation of autophagic vacuoles (32). Furthermore, p62 is a marker of autophagic flux and impaired autophagy is often accompanied by p62 accumulation, which results in large p62/ubiquitinated protein aggregates (33). Previous studies have demonstrated that the LPS-induced autophagic flux and autophagy were at their highest levels 12 h after LPS intraperitoneal injection (34). Therefore, the present study chose to investigate the effect of ulinastatin at this time point. Western blotting and TEM data obtained from the present study revealed that the expression levels of LC3II and the number of autophagosomes formed were significantly decreased following ulinastatin treatment. Furthermore, it was revealed that ulinastatin reduced the LPS-induced autophagosome maturation, which was demonstrated through the decreased protein expression levels of LAMP-1 and the increased expression levels of SQSTM1/p62. Therefore, the present study results suggested that the decreased activation of autophagy may be a critical protective mechanism of ulinastatin in mice with endotoxemia.
In conclusion, the findings of the present study suggested that ulinastatin may serve a cardioprotective role in sepsis, which may be achieved through its ability to suppress inflammation and autophagy. These findings may provide a clinical basis for the use of ulinastatin as a novel therapeutic option for the treatment of sepsis-related cardiac dysfunction. However, there are several limitations to the present study. Whilst the present study was able to observe changes in autophagy, there was a lack of experiments using specific autophagy inhibitors 3-methyladenine or autophagy agonists such as rapamycin. Therefore, future studies should further investigate the effects of ulinastatin on autophagy-related pathways. Additionally, the present study did not investigate the effect of ulinastatin at multiple time points or in in vitro cell models. As the present experimental set-up may have also produced bias in the results, further extensive research is required before solid conclusions can be made.
Acknowledgements
Not applicable.
Funding
The present study was funded by the Foundation of Shanxi Provincial [grant no. 2018SF-095(JK.K)].
Availability of materials and methods
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.
Authors' contributions
PZ and JKK designed the study. PZ, LZ, LFG, QD and QY performed the experiments. Data were collated by LZ and the results of data were discussed by LFG and QY. PZ prepared the figures. PZ and LZ wrote the first draft of the manuscript. All authors read and approved the final version of the manuscript.
Ethics approval and consent to participate
All procedures were approved by The Animal Ethics Committees of The Fourth Military Medical University (Xi'an, China) and were performed in accordance with the guidelines of The China Council of Animal Care.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348:138–150. doi: 10.1056/NEJMra021333. [DOI] [PubMed] [Google Scholar]
- 2.Kumar A, Haery C, Parrillo JE. Myocardial dysfunction in septic shock. Crit Care Clin. 2000;16:251–287. doi: 10.1016/s0749-0704(05)70110-x. [DOI] [PubMed] [Google Scholar]
- 3.Rudiger A, Singer M. Mechanisms of sepsis-induced cardiac dysfunction. Crit Care Med. 2007;35:1599–1608. doi: 10.1097/01.CCM.0000266683.64081.02. [DOI] [PubMed] [Google Scholar]
- 4.You W, Min X, Zhang X, Qian B, Pang S, Ding Z, Li C, Gao X, Di R, Cheng Y, Liu L. Cardiac-Specific expression of heat shock protein 27 attenuated endotoxin-induced cardiac dysfunction and mortality in mice through a PI3K/Akt-dependent mechanism. Shock. 2009;32:108–117. doi: 10.1097/SHK.0b013e318199165d. [DOI] [PubMed] [Google Scholar]
- 5.Fleischmann C, Scherag A, Adhikari NK, Hartog CS, Tsaganos T, Schlattmann P, Angus DC, Reinhart K. Assessment of global incidence and mortality of hospital-treated sepsis. Am J Respir Crit Care Med. 2016;193:259–272. doi: 10.1164/rccm.201504-0781OC. International Forum of Acute Care Trialists. [DOI] [PubMed] [Google Scholar]
- 6.Jönsson-Berling BM, Ohlsson K, Rosengren M. Radioimmunological quantitation of the urinary trypsin inhibitor in normal blood and urine. Biol Chem Hoppe Seyler. 1989;370:1157–1161. doi: 10.1515/bchm3.1989.370.2.1157. [DOI] [PubMed] [Google Scholar]
- 7.Shin IW, Jang IS, Lee SM, Park KE, Ok SH, Sohn JT, Lee HK, Chung YK. Myocardial protective effect by ulinastatin via an anti-inflammatory response after regional ischemia/reperfusion injury in an in vivo rat heart model. Korean J Anesthesiol. 2011;61:499–505. doi: 10.4097/kjae.2011.61.6.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Masuda T, Sato K, Noda C, Ikeda KM, Matsunaga A, Ogura MN, Shimizu K, Nagasawa H, Matsuyama N, Izumi T. Protective effect of urinary trypsin inhibitor on myocardial mitochondria during hemorrhagic shock and reperfusion. Crit Care Med. 2003;31:1987–1992. doi: 10.1097/01.CCM.0000057037.44171.BA. [DOI] [PubMed] [Google Scholar]
- 9.Koga Y, Fujita M, Tsuruta R, Koda Y, Nakahara T, Yagi T, Aoki T, Kobayashi C, Izumi T, Kasaoka S, et al. Urinary trypsin inhibitor suppresses excessive superoxide anion radical generation in blood, oxidative stress, early inflammation, and endothelial injury in forebrain ischemia/reperfusion rats. Neurol Res. 2010;32:925–932. doi: 10.1179/016164110X12645013515133. [DOI] [PubMed] [Google Scholar]
- 10.Yu Z, Rayile A, Zhang X, Li Y, Zhao Q. Ulinastatin protects against lipopolysaccharide-induced cardiac microvascular endothelial cell dysfunction via downregulation of lncRNA MALAT1 and EZH2 in sepsis. Int J Mol Med. 2017;39:1269–1276. doi: 10.3892/ijmm.2017.2920. [DOI] [PubMed] [Google Scholar]
- 11.Mizushima N. The ATG conjugation systems in autophagy. Curr Opin Cell Biol. 2019;31:1–10. doi: 10.1016/j.ceb.2019.12.001. [DOI] [PubMed] [Google Scholar]
- 12.Cursio R, Colosetti P, Gugenheim J. Autophagy and liver ischemia-reperfusion injury. Biomed Res Int. 2015;2015(417590) doi: 10.1155/2015/417590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ho J, Yu J, Wong SH, Zhang L, Liu X, Wong WT, Leung CC, Choi G, Wang MH, Gin T, et al. Autophagy in sepsis: Degradation into exhaustion? Autophagy. 2016;12:1073–1082. doi: 10.1080/15548627.2016.1179410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gurusamy N, Das DK. Is autophagy a double-edged sword for the heart? Acta Physiol Hung. 2009;96:267–276. doi: 10.1556/APhysiol.96.2009.3.2. [DOI] [PubMed] [Google Scholar]
- 15.De Meyer GR, Martinet W. Autophagy in the cardiovascular system. Biochim Biophys Acta. 2009;1793:1485–1495. doi: 10.1016/j.bbamcr.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 16.Xiao J, Zhu X, Ji G, Yang Q, Kang B, Zhao J, Yao F, Wu L, Ni X, Wang Z. Ulinastatin protects cardiomyocytes against ischemiareperfusion injury by regulating autophagy through mTOR activation. Mol Med Rep. 2014;10:1949–1953. doi: 10.3892/mmr.2014.2450. [DOI] [PubMed] [Google Scholar]
- 17.Zhao P, Kuai J, Gao J, Sun L, Wang Y, Yao L. Delta opioid receptor agonist attenuates lipopolysaccharide-induced myocardial injury by regulating autophagy. Biochem Biophys Res Commun. 2017;492:140–146. doi: 10.1016/j.bbrc.2017.06.029. [DOI] [PubMed] [Google Scholar]
- 18.Wang WK, Lu QH, Wang X, Wang B, Wang J, Gong HP, Wang L, Li H, Du YM. Ulinastatin attenuates diabetes-induced cardiac dysfunction by the inhibition of inflammation and apoptosis. Exp Ther Med. 2017;14:2497–2504. doi: 10.3892/etm.2017.4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lan R, Zhang Y, Xiang J, Zhang W, Wang GH, Li WW, Xu LL, Cai DF. Xiao-Xu-Ming decoction preserves mitochondrial integrity and reduces apoptosis after f ocal cerebral ischemiaand reperfusion via the mitochondrial p53 pathway. J Ethnopharmacol. 2014;151:307–316. doi: 10.1016/j.jep.2013.10.042. [DOI] [PubMed] [Google Scholar]
- 20.Qiu SQ, van Rooijen J, Nienhuis HH, van derVegt B, Timmer-Bosscha H, van Leeuwen-Stok E, Walenkamp AME, van Deurzen CHM, de Bock GH, et al. High hepatocyte growth factor expression in primary tumor predicts better overall survival in male breast cancer. Breast Cancer Res. 2020;22(30) doi: 10.1186/s13058-020-01266-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin CM, Chen CR, Wu XQ, Ren JH, Chen SZ, Luo XF, Mei XQ, Shen LY, Guo MX, Ma XD, Yang T. Effects of blood purification on serum levels of inflammatory cytokines and cardiac function in a rat model of sepsis. Blood Purif. 2017;44:40–50. doi: 10.1159/000455060. [DOI] [PubMed] [Google Scholar]
- 22.Lim YP, Bendelja K, Opal SM, Siryaporn E, Hixson DC, Palardy JE. Correlation between mortality and the levels of inter-alpha inhibitors in the plasma of patients with severe sepsis. J Infect Dis. 2003;188:919–926. doi: 10.1086/377642. [DOI] [PubMed] [Google Scholar]
- 23.Han D, Shang W, Wang G, Sun L, Zhang Y, Wen H, Xu L. Ulinastatin and thymosin α1-based immunomodulatory strategy for sepsis: A meta-analysis. Int Immunopharmacol. 2015;29:377–382. doi: 10.1016/j.intimp.2015.10.026. [DOI] [PubMed] [Google Scholar]
- 24.Wang FY, Fang B, Qiang XH, Yu TO, Zhong JR, Cao J, Zhou LX. The efficacy and immunomodulatory effects of ulinastatinand thymosin α1 for sepsis: A systematic review and meta-analysis. Biomed Res Int. 2016;2016(9508493) doi: 10.1155/2016/9508493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gogos CA, Drosou E, Bassaris HP, Skoutelis A. Pro-Versus anti-inflammatory cytokine profile in patients with severe sepsis: A marker for prognosis and future therapeutic options. J Infect Dis. 2000;181:176–180. doi: 10.1086/315214. [DOI] [PubMed] [Google Scholar]
- 26.Chen X, Wang Y, Luo H, Luo Z, Liu L, Xu W, Zhang T, Yang N, Long X, Zhu N, et al. Ulinastatin reduces urinary sepsis-related inflammation by upregulating IL-10 and downregulating TNF-α levels. Mol Med Rep. 2013;8:29–34. doi: 10.3892/mmr.2013.1480. [DOI] [PubMed] [Google Scholar]
- 27.Varvarousis D, Goulas N, Polytarchou K, Psychari SN, Paravolidakis K, Konstantinidou A, Tsoukalas D, Vlad D, Bouki K, Kotsakis A. Biomarkers of myocardial injury and inflammation after permanent pacemaker implantation: The lead fixation type effect. J Atr Fibrillation. 2018;10(1798) doi: 10.4022/jafib.1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shires SE, Gustafsson ÅB. Mitophagy and heart failure. J Mol Med (Berl) 2015;93:253–262. doi: 10.1007/s00109-015-1254-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Joiner ML, Koval OM, Li JD, He B, Allamargot C, Gao Z, Luczak ED, Hall DD, Fink BD, Chen B, et al. CaMKII determines mitochondrial stress responses in heart. Nature. 2012;491:269–273. doi: 10.1038/nature11444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nishida K, Kyoi S, Yamaguchi O, Sadoshima J, Otsu K. The role of autophagy in the heart. Cell Death Differ. 2009;16:31–38. doi: 10.1038/cdd.2008.163. [DOI] [PubMed] [Google Scholar]
- 31.Castillo K, Valenzuela V, Matus S, Nassif M, Oñate M, Fuentealba Y, Encina G, Irrazabal T, Parsons G, Court FA, et al. Measurement of autophagy flux in the nervous system in vivo. Cell Death Dis. 2013;4(e917) doi: 10.1038/cddis.2013.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eskelinen EL. Roles of LAMP-1 and LAMP-2 in lysosome biogenesis and autophagy. Mol Aspects Med. 2006;27:495–502. doi: 10.1016/j.mam.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 33.Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137:1062–1075. doi: 10.1016/j.cell.2009.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao P, Gao JJ, Jiang J, Peng X, Wu W, Zheng L, Yao L. Myocardial cells and mitochondrial autophagy in sepsis mice induced by lipopolysaccharide. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. 2016;32:177–181. (In Chinese) [PubMed] [Google Scholar]