

Abstract
Purpose
Pheochromocytomas/paragangliomas (PHEOs/PGLs) are rare in children with only a few SDHB mutation-related cases. Previous studies on children were conducted in small cohorts. This large set of pediatric patients provides robust data in the evaluation of clinical outcomes.
Methods
Sixty-four pediatric PHEO/PGL patients with SDHB germline mutations were included in the present study. The clinical presentation, disease course, and survival rate were evaluated.
Results
Thirty-eight males and 26 females were diagnosed with PHEO/PGL at a median age of 13 years. The majority of patients displayed norepinephrine hypersecretion and 73.44% initially presented with a solitary tumor. Metastases developed in 70% of patients at the median age of 16 years and were mostly diagnosed first 2 years and in years 12–18 post-diagnosis. The presence of metastases at the time of diagnosis had a strong negative impact on survival in males but not in females. The estimated 5-, 10-, and 20-year survival rates were 100%, 97.14%, and 77.71%, respectively.
Conclusion
The present report has highlighted several important aspects in the management of pediatric patients with SDHB mutations associated-PHEO/PGL. Initial diagnostic evaluation of SDHB mutation carriers should be started at age of 5–6 years with initial work-up focusing on abdominal region. Thorough follow-up is crucial first 2 years post-diagnosis and more frequent follow-ups are needed in years 10–20 post-diagnosis due to the increased risk of metastases. Although this age group developed metastasis as early as 5 years from diagnosis, we have shown that the overall 20-year prognosis and survival are good.
Keywords: Pheochromocytoma, Paraganglioma, SDHB mutation, Pediatric oncology
Introduction
Pheochromocytoma (PHEO) and paraganglioma (PGL), catecholamine-secreting neoplasms of neural crest origin, carry the highest degree of heritability in human cancers (Fishbein et al. 2017). Clinical hallmarks of the hereditary diseases include early-onset, bilaterality, multifocality, extra-adrenal locations, and, often, malignancy. In approximately 30–40% of patients, these neuroendocrine tumors were found to be genetically inherited (Gimenez-Roqueplo et al. 2012; Vicha et al. 2013) and 10–39% of apparently sporadic PHEOs/PGLs result from somatic mutations (Fishbein et al. 2017). Up-to-date, more than 20 genes predisposing to PHEO/PGL development have been identified (Fishbein et al. 2017).
Approximately, 10–20% of all PHEOs/PGLs are diagnosed in pediatric population (Barontini et al. 2006; Choat et al. 2014) with germline mutations reported in up to 80% of them with the predominance of VHL (von Hippel-Lindau gene) followed by SDHB, SDHD (succinate dehydrogenase subunit B and D), and NF1 (neurofibromin 1) mutations (Armstrong et al. 2008; Barontini et al. 2006; Bausch et al. 2014; Cascon et al. 2013; Ciftci et al. 2001; De Krijger et al. 2006; King et al. 2011; Neumann et al. 2002). Malignancy occurs in approximately 12% of pediatric/adolescent patients, with 39% incidence rate of the familial type (Barontini et al. 2006). With the rare incidence of 2–8 per million (Timmers et al. 2009), data in the pediatric age group are still insufficient as studies were limited by a few cases (Bissada et al. 2008; Cascon et al. 2013; Ciftci et al. 2001; De Krijger et al. 2006; Ein et al. 1990; Kaufman et al. 1983; Neumann et al. 2002; Pham et al. 2006; Ross 2000; Stackpole et al. 1963). More recent reports have observed a cohort of 26 (23 SDHB- and 3 SDHD-mutated) and 42 (25 SDHB- and 17 SDHD-mutated) young patients with familial PGL syndrome types 1 and 4 with finding of more frequent disease recurrence in patients harboring SDHD germline mutations (Bausch et al. 2014), and significant increase in the metastasis incidence and mortality in patients with SDHB germline mutations compared to other germline mutations or sporadic tumors (Amar et al. 2005; King et al. 2011). Another study on 49 patients with SDHB mutations and 35 sporadic cases demonstrated earlier onset of PHEO/PGL, increased local recurrence, distance metastasis, and lesser median disease-free interval in patients with SDHB mutations (Assadipour et al. 2017). Recently, there have been interesting developments related to risk factors predicting the development of metastatic disease. Previous studies proposed SDHB mutations an independent prognostic factor for survival in patients with metastatic PHEO/PGL (Amar et al. 2007; Assadipour et al. 2017; Turkova et al. 2016), but Hescot et al. (2019) in their study did not confirm these findings in a large cohort of metastatic PHEO/PGL patients. However, SDHB mutation status was demonstrated to be independently associated with metastatic disease by Crona et al. (2019). Moreover, both studies (Hescot et al. 2019; Crona et al. 2019) showed that hypersecreting tumors (e.g. those presenting with elevated norepinephrine, dopamine, or chromogranin A) are associated with metastasis development and survival. Reported metastatic rate in patients with SDHB-related PHEO/PGL is found to be consistently high at 34–71% (Amar et al. 2005; Benn et al. 2006; Jochmanova et al. 2017; Neumann et al. 2004; Timmers et al. 2007) and thus, warrants long-term follow-up.
We believe that further diagnostic, prognostic and therapeutic recommendations in pediatric hereditary PHEO/PGL can be only made with extensive data on large studies that focus on a specific pathogenic genetic mutation. Thus, the present study aimed to provide additional detailed clinical data, presentation, and outcomes on SDHB-mutation-related pediatric PHEOs/PGLs. Our research was a single-center study, the second largest so far on these pediatric tumors, which allowed us to outline some important conclusions for the management of pediatric patients with SDHB mutations.
Methods
Patients
Sixty-six SDHB mutation carriers with histologically documented PHEOs/PGLs who presented with their primary tumor before 20 years of age, which was defined as a pediatric/adolescent tumor, were selected for this study. An additional 59 pediatric/adolescent patients included those who carried non-SDHB germline mutations or those with no known germline mutations associated with PHEO/PGL. The patients included were selected from the larger group of 2177 patients with PHEOs/PGLs who were referred and recorded at the National Institutes of Health (NIH) registry between January 1st, 2000 and April 30th, 2019 (Fig. 1). This study was approved by the Institutional Review Board of the National Institutes of Child Health and Human Development at the NIH and all patients gave written informed assent prior to testing and signed permission from one or both parents were obtained for all the individual participants included in the study.
Fig. 1.
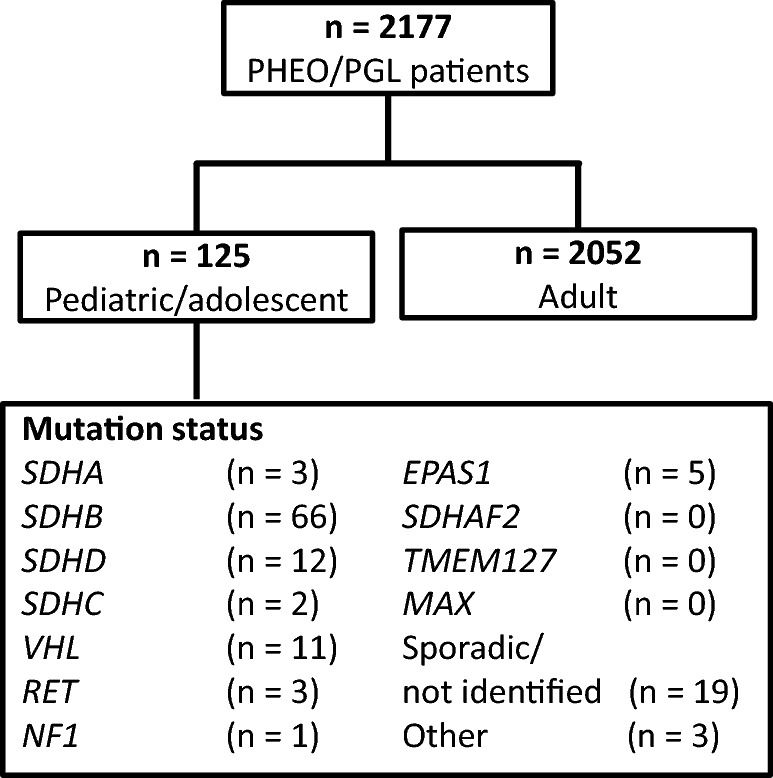
Study population
Genetic analysis
Genetic testing for germline mutations in the SDHB, SDHC, SDHD, and VHL genes was performed at the Department of Human Genetics of the Pittsburgh University Medical Center, Children’s Hospital of Philadelphia, and at Mayo Clinic Laboratories, Minnesota, USA as described elsewhere (Baysal et al. 2002; Maher et al. 2002). Not all currently known susceptibility genes for PHEO/PGL were tested in our patients if a known pathogenic germline mutation associated with PHEO/PGL has been identified. Furthermore, testing for TMEM127, SDHA, SDHAF2, and MAX genes mutations was not performed in all patients (Comino-Mendez et al. 2011; Hensen and Bayley 2011). Patients with typical clinical phenotype were tested for the presence of EPAS1 and RET mutations. In 22 patients, the mutations in the aforementioned genes were not identified (Fig. 1).
Laboratory tests and imaging
Evaluation of patients included biochemical analysis with plasma and/or urine metanephrines and catecholamines followed by full-body anatomical and functional imaging as clinically indicated. For biochemical testing, patients were asked to abstain from acetaminophen for 5 days and caffeinated and decaffeinated products, smoking, and alcohol for 24 h prior to blood extraction and 24-h urine collection.
For plasma catecholamines and metanephrines (MNs) determination, a cannula was inserted into the forearm for intravenous access. Patients rested in the supine position without a pillow in a quiet room for 20–30 min. before and during collection. As soon as blood was collected, it was placed on ice and stored at − 80 °C until testing. Basal plasma levels of catecholamines and MNs were measured by high-performance liquid chromatography (HPLC).
For urinary catecholamine and MN determination, total volume collected over 24 h was used and measured by HPLC or liquid chromatography–tandem mass spectrometry. Hypersecretion of plasma or urinary catecholamines or MNs was defined as any elevation above the upper reference limit.
Anatomical localization of PHEO/PGL was performed by axial images of the neck, chest, abdomen, and pelvis using magnetic resonance imaging (MRI) or computed tomography (CT). One or more of the functional imaging studies such as 18F-FDG PET/CT, 18F-FDA PET/CT, 18F-FDOPA PET/CT, and 123I-MIBG scintigraphy were performed in our patients depending on the indications and possible treatment options. Since the study involved pediatric patients, the researchers made all attempts to minimize radiation exposure. Laboratory and imaging studies performed at different institutions prior to referral to NIH were likewise reviewed if available.
Outcome measurements
Demographic and clinical data, including gender, signs and symptoms, age at initial symptoms and at diagnosis, family history together with plasma and/or urine catecholamine levels, imaging studies, and germline mutation analysis, if previously done, were obtained from medical records. PHEO/PGL tumors were classified according to size, location (adrenal, extra-adrenal, head and neck), and tumor presentation at initial diagnosis (solitary, multiple, metastatic). Metastasis is defined as the presence of tumors in non-chromaffin-containing tissues such as lymph nodes and bones (WHO Classification of Tumours of Endocrine Organs). Patients were followed-up at 6–12-month intervals depending on clinical indications. Treatment modalities such as surgery, chemotherapy, 131I-MIBG and radiation therapy, radiofrequency ablation, and experimental therapy were noted. Records of treatment and follow-up at outside institutions were also obtained. Age and appearance of recurrence and metastatic lesions were recorded as well as the date of death, if applicable. Time to metastases among males and females was calculated according to age at diagnosis in the first 5 and 10 years.
Statistical analysis
The endpoints evaluated in study included: age at initial symptoms, age of initial diagnosis, age at diagnosis of recurrent disease, interval between initial diagnosis and recurrence, age at diagnosis of metastatic disease, interval between initial diagnosis and occurrence of metastases (or last follow-up if no metastatic disease), interval between initial diagnosis and death (or last follow-up if still alive), and the effect of the type of SDHB mutation on patients’ outcome. Categorical variables were summarized as frequency counts and percentages. Continuous variables were summarized as the median and range. For plotting metastases-free survival (i.e., the time from initial diagnosis to development of metastases or last follow-up) of all patients included (both patients who developed and did not develop metastases), Kaplan–Meier plots for censored survival data were calculated along with their associated confidence bands; small vertical marks on the Kaplan–Meier curves showed the follow-up times for patients who did not develop metastases. All P values were two sided.
Metastasis-free survival, time to metastasis to specified organs, time to death following the initial diagnosis of PHEO/PGL and associated 95% confidence intervals (CI) were computed using standard survival methods, including Kaplan–Meier curves and Greenwood’s formula for the associated standard error and confidence interval. Estimates of median values were based on interpolating values from the Kaplan–Meier estimates. Comparisons of percentages in cross-tabulated tables to assess independence or association of factors used Fisher’s exact test for both simple 2 × 2 contingency tables and those with more rows and/or columns. This test assumes that neither the rows nor columns have an inherent ordering, as was the case in our applications. Wilcoxon/rank-sum test was used for numeric value comparisons. All reported p values are two sided.
Results
Patient characteristics
Of the 2177 patients with PHEO/PGL evaluated at NIH between January 2000 and April 2019, 125 (5.74%) patients were diagnosed with PHEO/PGL before the age of 20 years. Among the group of patients with pediatric/adolescent PHEO/PGL, 66 (52.8%) were found to have germline SDHB mutations. From the remaining 59 patients, in 40 patients germline mutations in other PHEO/PGL susceptibility genes were identified (Fig. 1). The pathogenic germline mutations associated with PHEO/PGL could not be found in 19 (15.2%) patients (Fig. 1). Patients with SDHB mutations (N = 66) were selected for further analyses in this study. For two patients, no sufficient clinical data were available, thus, they were excluded from further analyses. The basic demographic and clinical data of patients with SDHB germline mutations are depicted in Table 1.
Table 1.
Characteristics of 66 pediatric PHEO/PGL patients with SDHB mutations
| Total | Male | Female | p value | |
|---|---|---|---|---|
| Patients (N, %) | 64 | 38 (59.38%) | 26 (40.63%) | 0.13 |
| Age of initial symptomsa | 11 (4–19) | 11 (4–19) | 12 (5–18) | 0.20 |
| Age of initial diagnosisa | 13 (6–19) | 12 (6–19) | 13 (8–19) | 0.21 |
| Biochemistry done at the time of diagnosis (N, %) | 40 (62.5%) | 24 (63.15%) | 16 (61.54%) | 0.90 |
| Norepinephrine/normetanephrine (N, %b) | 31 (88.57%) | 18 (85.1%) | 13 (92.26%) | 0.53 |
| Epinephrine/metanephrine (N, %b) | 10 (29.41%) | 5 (23.81%) | 5 (38.46%) | 0.37 |
| Dopamine (N, %b) | 10 (55.56%) | 9 (69.23%) | 1 (20.0%) | 0.08 |
| Chromogranin A (N, %b) | 9 (75.0%) | 6 (75.0%) | 3 (75.0%) | 1.00 |
| Vanillylmandelic acid (N, %b) | 12 (85.71%) | 5 (83.33%) | 7 (87.5%) | 0.83 |
| Normal (N, %b) | 4 (8.70%) | 2 (7.41%) | 2 (10.53%) | 0.71 |
| Primary tumor location | ||||
| Adrenal (N, %) | 10 (15.63%) | 6 (15.79%) | 4 (15.38%) | 0.97 |
| Extra-adrenal sympathetic (N, %) | 50 (78.13%) | 29 (76.32%) | 21 (80.77%) | 0.67 |
| Extra-adrenal parasympathetic (N, %) | 1 (1.56%) | 0 | 1 (3.85%) | 0.22 |
| Adrenal and extra-adrenal sympathetic (N, %) | 2 (3.13%) | 2 (5.26%) | 0 | 0.23 |
| Extra-adrenal sympathetic + parasympathetic (N, %) | 1 (1.56%) | 1 (2.63%) | 0 | 0.41 |
| Primary tumor size [cm, median (range)] | 5.7 (1.5–15) | 5.6 (1.7–14) | 5.7 (1.5–15) | 0.45 |
| Recurrence (N, %) | 13 (20.31%) | 8 (21.05%) | 5 (19.23%) | 0.86 |
| Age at recurrencea | 16 (13–36) | 16 (13–32) | 16 (13–36) | 0.87 |
| Diagnosis to recurrence intervala | 2 (0–26) | 3 (0–17) | 1 (1–26) | 0.36 |
| Metastatic disease (N, %) | 45 (70.31%) | 27 (71.05%) | 18 (69.23%) | 0.88 |
| Synchronous metastases (N, %) | 12 (18.75%) | 6 (15.79%) | 6 (23.08%) | 0.47 |
| Age of metastasis diagnosisa | 16 (8–39) | 16 (10–39) | 17 (8–36) | 0.72 |
| Diagnosis to metastasis intervala | 4 (0–26) | 5 (0–24) | 4 (0–26) | 0.75 |
| Deceased (N, %) | 8 (12.50%) | 6 (15.79%) | 2 (7.69%) | 0.34 |
| Diagnosis to death intervala | 21 (8–31) | 21 (8–28) | 25 (19–31) | 0.74 |
| Other tumors (N, %) | 2 (3.13%) | 0 | 2 (7.69%) | 0.09 |
N number
aYears, median (range)’ bof patients in whom particular biochemical marker was measured
Clinical characteristics
Sixty-four [26 (40.63%) females and 38 (59.38%) males] pediatric patients with SDHB-related PHEO/PGL were included in further analyses. Patients’ characteristics are summarized in Table 1.
The most common presenting symptom was hypertension (76.56%), followed by headache (68.75%), sweating/diaphoresis (51.56%), palpitations (40.63%), nausea and/or vomiting (31.25%), tumor-related pain (26.56%), and flushing (25.0%). The first occurrence of symptoms was reported at the median age of 11 years, ranging from 4 to 19 years. The median age of diagnosis was 13 (6–19) years. At the time of initial diagnosis, in 40 (62.5%) patients, a biochemical testing was performed for at least one of the following tests: plasma or urine catecholamines and/or MNs, urine vanillylmandelic acid, urine and/or plasma dopamine, and chromogranin A. For additional 22 (34.38%) patients, we were not able to obtain the results of initial biochemical testing and in 2 (3.13%) patients, biochemical testing was not done. Available data showed in a higher percentage of patients increased plasma norepinephrine/normetanephrine (88.57%), vanillylmandelic acid (85.71%), dopamine (55.56%), and chromogranin A (22.5%). Four patients (8.70%) had non-secreting tumors at initial diagnosis (Table 1).
Tumor characteristics
Forty-seven patients (73.44%) had a solitary tumor at first presentation; five patients (7.81%) initially presented with multiple tumors and 12 (18.75%) with metastatic disease. The median size of the primary tumor was 5.7 cm, range from 1.5 to 15 cm. Ten (15.62%) patients presented with adrenal PHEO, 50 (78.12%) with extra-adrenal sympathetic tumor, and 1 (1.56%) patient presented with extra-adrenal parasympathetic (head and neck) PGL. Five patients with multiple tumors at initial diagnosis presented as follows: 2 (3.12%) patients had multiple extra-adrenal sympathetic PGLs, in other 2 (3.12%) patients, there was a combination of adrenal PHEO and extra-adrenal sympathetic PGL, and in 1 (1.56%) patient, extra-adrenal sympathetic and parasympathetic tumors were present (Table 1).
Outcomes
Recurrent disease
Recurrence of PHEO/PGL was diagnosed in 13 (20.31%) of patients at a median age of 16 years (range 13–36 years). The median time from the first diagnosis of the disease to the recurrence was 2 (0–26) years (Table 1). In 5 (38.46%) patients, recurrent tumors occurred in adrenal glands; in 11 (84.62%) patients, sympathetic extra-adrenal recurrent tumors were identified. Ten (76.92%) of patients with recurrent disease also developed metastases.
Metastatic disease
Forty-five [70.31%; 27 (71.05%) males and 18 (69.23%) females] patients developed metastatic lesions at a median age of 16 years (range 8–39 years). The median age at initial PHEO/PGL diagnosis in this group was 12 years (range 6–19 years) and the median interval between diagnosis of a primary tumor and of metastatic disease was 4 years, ranging from 0 to 26 years. This interval did not differ significantly between males and females (median 5 vs. 4 years, p = 0.75) (Table 1). In 12 (18.75%, 6 (15.79%) males and 6 (23.08%) females) patients, metastases were present at the time of diagnosis. Most common sites of metastases included the skeleton (88.89%), retroperitoneum (60%), lungs (44.44%), lymph nodes (28.89%), chest/mediastinum (28.89%), and liver (24.44%). Some patients had more than one site of metastases.
We evaluated the primary tumor size to establish an optimal size cut-off associated with the increased risk of metastasis. Patients with primary tumors ≤ 5 cm (N = 25) had a median time to develop metastases of 7 years (range 0–24 years) while those with larger tumors (> 5 cm; N = 37) had a median time of only 2 years (range 0–16 years) (p = 0.014, Fig. 2a). We found no association between the interval of PHEO/PGL-related signs/symptoms and diagnosis to surgical management and the onset of metastasis (p = 0.54 and p = 0.86, respectively).
Fig. 2.

Kaplan–Meier curves. a Proportion free of metastases based on the tumor size. b Kaplan–Meier curves for development of metastases post-diagnosis in subset of pediatric SDHB mutation-positive patients without metastases at diagnosis based on the presence of tumor pain at the time of diagnosis
The analysis of factors such as gender (p = 0.36), mode of inheritance whether paternal or maternal (p = 0.20), age at diagnosis (p = 0.69), biochemical hypersecretion (p = 0.28), presence of symptoms at diagnosis (p = 0.43), did not show any statistical significant association with the metastatic disease development. However, most patients with metastatic disease presented with noradrenergic (17; 37.78%) and mixed noradrenergic and dopaminergic (16; 35.56%) biochemical phenotype, 2 (4.44% displayed dopaminergic and 1 (2.22%) adrenergic phenotype. Eight (17.78%) patients with metastases had non-secreting tumors. Elevated chromogranin A levels were present in 31 (68.89%) patients with metastatic disease. Earlier development of metastatic disease was observed in patients who presented with sweating (p = 0.0073) at initial diagnosis. Sub-analysis of the 52 patients who did not have metastatic lesions at initial presentation showed that patients who experienced tumor pain at diagnosis had metastatic lesions at an earlier interval compared to the rest (p = 0.0088, Fig. 2b).
Kaplan–Meier analysis of metastasis-free survival showed that 50% of pediatric patients with SDHB mutation will develop metastasis in 7 years from diagnosis (Fig. 3a). When looking at the risk of metastasis development using hazard function, the probability of developing metastasis the first year post-diagnosis was 28% (Fig. 3a). After excluding subjects with metastatic disease at the time of diagnosis, this probability drops to roughly 10%. The estimated risk of metastatic disease between years 1 and 5 post-diagnosis is about 2.84% and this risk does not increase except for the years from 12 to 18, where it seems to be 30% higher (Fig. 3b).
Fig. 3.

Kaplan–Meier curves. a Analysis of metastasis-free survival. b Hazard function for the development of metastases
The median age at initial diagnosis in the 19 (29.69%) pediatric/adolescent patients who did not develop metastatic disease was 13 (range 10–18) years and it did not differ significantly from the age of initial diagnosis in patients who developed metastatic disease. Cox regression analysis of the effect of age at diagnosis on the interval from diagnosis to the development of metastases (or last follow-up) showed no impact of the age at diagnosis on this endpoint (p = 0.36). A trend log-rank test showed a modest effect of tumor size on the development of metastases post-diagnosis (p = 0.046), with larger tumors having a progressively worse outcome.
Effect of type of SDHB genetic mutation
In the present study, patients had 31 different SDHB mutations (Table 2). Ten (15.63%) had large deletion, 3 (4.69%) had frameshift mutations, 26 (40.63%) had missense mutation, 20 (31.25%) had nonsense mutation, and 4 patients (6.25%) had splice-site mutation. In 1 (1.56%) patient, exact type of mutation was not known (Table 2). Five mutations (p. Val140Phe, p. Arg90*, exon 4 and 5 deletion, p. Arg46*, and exon 1 deletion), identified in the largest number of probands (29; 18 males, 11 females) were examined independently. There were no differences in the time to death in patients harboring these five mutations (p = 0.36). Exon 1 deletion and p. Val140Phe mutation might be associated with the earlier development of metastatic disease, however, the analyzed group of patients was too small to confirm this association (Fig. 4).
Table 2.
Germline mutations in the SDHB Gene in the pediatric PHEO/PGL patients
| Exon/intron | SDHB mutation (cDNA nucleotide) | SDHB mutation/protein change | Type | Pathogenicitya | Casesb |
|---|---|---|---|---|---|
| 1–8 | Exon 1–8 deletion | p.0 | Large deletion | AF | 2 |
| 1 | Exon 1 deletion | p.? | Large deletion | AF | 4 |
| 1 | c.26 T>A | p.Leu9X | Nonsense | AF | 1 |
| IVS 1 | c.73-9A>G | IVS1–9A>G | Splice site | VUS | 1 |
| 2 | c.79C>A | p.Arg27X | Nonsense | AF | 1 |
| 2 | c.136C>T | p.Arg46X | Nonsense | AF | 6 |
| 2 | c.137G>A | p.Arg46Gln | Missense | AF | 1 |
| 2 | c.183 T>G | p.Tyr61X | Nonsense | AF | 1 |
| 2 | c.194 T>A | p.Leu65His | Missense | VUS | 1 |
| 3 | c.268C>T | p.Arg90X | Nonsense | AF | 7 |
| 3 | c.271A>T | p.Arg91X | Nonsense | AF | 1 |
| 3 | c.274 T>C | p.Ser92Pro | Missense | VUS | 1 |
| 4–5 | Exon 4–5 deletion | p.? | Deletion | likely AF | 3 |
| 4 | c.287G>A | p.Gly96Asp | Missense | likely AF | 2 |
| 4 | c.293G>A | p.Cys98Tyr | Missense | AF | 1 |
| 4 | c.330_331del | p.Leu111SerfsX7 | Frameshift | AF | 1 |
| 4 | c.380 T>G | p.Ile127Ser | Missense | likely AF | 2 |
| 4 | c.418G>T | p.Val140Phe | Missense | AF | 12 |
| 5 | c.445C>T | p.Gln149X | Nonsense | AF | 1 |
| 5 | c.445-447delCAinsGGTATCT | p.Gln149Leufsa159 | Frameshift | VUS | 1 |
| 5 | c.490C>T | p.Gln164X | Nonsense | AF | 1 |
| IVS 5 | c.541-2A> G | IVS5-2A >G | Splice site | AF | 1 |
| 6–8 | Exon 6–8 deletion | p.? | Deletion | VUS | 1 |
| 6 | c.553G>T | p.Glu185X | Nonsense | VUS | 1 |
| 6 | c.575G>A | p.Cys192Tyr | Missense | AF | 1 |
| 6 | c.590C>G | p.Pro197Arg | Missense | AF | 1 |
| 6 | c.600G>T | p.Trp200Cys | Missense | AF | 1 |
| IVS 6 | c.642 + 1G>A | IVS6 + 1G>A | Splice site | AF | 2 |
| 7 | c.689G>A | p.Arg230His | Missense | AF | 1 |
| 7 | c.725G>A | p.Arg242His | Missense | AF | 1 |
| 7 | c.736A>T | p.Ile246Phe | Missense | AF | 1 |
| 7 | c.761insC | p.Pro254fsX255 | Frameshift | VUS | 1 |
AF affects function, PHEO/PGL pheochromocytoma/paraganglioma, VUS variant of unknown significance
aAs reported by LOVD and NCBI ClinVar databases
bMutation types reported are for 63 pediatric SDHB mutation-positive PHEO/PGL patients. For three patients we were not able to obtain exact mutation type
Fig. 4.

Kaplan–Meier analysis of metastasis-free survival in pediatric SDHB mutation-positive patients with 5 most common mutation types
Mortality
Eight (12.5%) patients with SDHB mutations died during the study period. All had metastatic disease and survived for a median of 7.5 (range 4–19) years after the diagnosis of metastatic disease. Remaining 37 (82.22%) patients with metastatic disease have been followed for a median of 10.5 (range 0.9–56) years after the initial diagnosis of disease and 5 (range 0.9–50) years after the diagnosis of metastatic disease. Nineteen (29.69%) patients without metastatic disease have been followed for a median of 4.5 (range 1–31) years after the initial diagnosis of PHEO/PGL. Among all 64 patients, Kaplan–Meier estimates of the 5-, 10-, 20-, 30, and 40-year survival rates were 100% (95% CI, undefined), 97.14% (95% CI 81.40–99.59%), 77.71% (95% CI 49.40–91.38%), 43.71% (95% CI 12.99–71.50%), and 21.86% (95% CI 1.38–58.28%), respectively, as shown in Fig. 5. The median time to death after diagnosis is estimated at 28 years (95% CI: 20–years to infinity). The presence of metastatic disease at diagnosis has a strong negative impact on survival when compared with solitary or multiple primary tumors (p = 0.0043, Fig. 6a). Interestingly, this association between metastatic disease and a shorter disease-specific survival was present only in males (p < 0.0001) but not in females (p = 1, Fig. 6b, c). When we looked at the size of the primary tumor and time to death, we found a statistically significant difference (p = 0.0147); the time to death is getting progressively shorter with the increasing size of the primary tumor (Fig. 7a). When patients were divided according to sex, time to death was significantly shorter with larger tumors in males (p = 0.0129; Fig. 7b) but not in females (p = 0.414; Fig. 7c). No significant differences were found in males and females in the time from diagnosis to death (p = 0.24).
Fig. 5.

Kaplan–Meier analysis of survival rates post-diagnosis
Fig. 6.

Kaplan–Meier analysis of survival rates based on the first tumor presentation
Fig. 7.

Kaplan–Meier analysis of survival rates based on the size of primary tumor
Discussion
PHEO/PGL has an overall incidence rate estimated at 2–8 cases per million per year (Timmers et al. 2009), which classifies the disease as rare, especially in pediatric patients. There has been a lack of large population-based registries and studies that investigate clinical outcomes of pediatric patients with SDHB-related PHEO/PGL. This study evaluated one of the largest pediatric populations with a total of 64 children with a specific mutation and disease, here SDHB. In our patient cohort, PHEO/PGL diagnosis was established at or after the age of 6 years with median of 13 years. This warrants regular screening for the disease in SDHB mutation carriers older than 6 years. Most of the patients (78.13%) presented with extra-adrenal sympathetic tumors and median size of primary tumor was 5.7 cm (Table 1). Our results are concordant with previous studies that describe SDHB-related PHEO/PGL as predominant in adolescent males with a median age of 12 years old. Since the most common localization of the primary tumor was in the abdominal region (in 82.22% of our group of pediatric patients with SDHB mutation), initial diagnostic work-up should be directed towards abdomen. If abdominal imaging is negative, evaluation of pelvic, chest, and head and neck regions needs to follow.
A recent study established that measurements of plasma-free normetanephrine and metanephrine provide a superior test for diagnosis of pheochromocytoma than measurements of the deconjugated metabolites in urine (Darr et al. 2012; Pamporaki et al. 2013). A review of this pediatric group’s biochemical phenotype showed mainly noradrenergic hypersecretion (Table 1). This is consistent with previous knowledge, being that extra-adrenal PGLs rarely secrete epinephrine, which reflects a decreased expression of phenylethanolamine-N-methyltransferase, the enzyme that converts norepinephrine to epinephrine (Timmers et al. 2007). With the regard to its clinical presentation, a high clinical suspicion for PHEO/PGL is considered among adults with headache, sweating, and palpitation on top of persistent hypertension. Similarly, these symptoms were most commonly reported in our cohort of pediatric/adolescent patients with SDHB-related PHEO/PGL.
Sustained hypertension without paroxysm has been a stable sign in 63% of pediatric patients with pheochromocytoma (Armstrong et al. 2008). In this study, hypertension remains to be the most common symptom (76.56%). Sweating and/or tumor pain at diagnosis were remarkably associated with metastatic lesions at an earlier interval compared to the rest of the symptoms (p = 0.0073 and p = 0.0088, respectively). Approximately, 10–20% of cases are diagnosed during childhood at an average age of 11 years, with a high predominance in boys, particularly those under the age of ten (Barontini et al. 2006; Bausch et al. 2014; Beltsevich et al. 2004; Ciftci et al. 2001; Gimenez-Roqueplo et al. 2012; Ross 2000; Spoudeas 2005; Stackpole et al. 1963). Those studies, however, evaluated pediatric patients with no regard to genetic background. In this study, focused on SDHB-mutated pediatric population, we did not find significant differences in disease occurrence or ages at diagnosis between males and females, although the number of male patients was slightly higher compared to females. This seems to be in disagreement with our recent study (Jochmanova et al. 2017) on 344 SDHB mutation carriers (including 143 pediatric and adult PHEO/PGL patients), where we reported an earlier progression to disease as well as tendency to earlier development of metastatic disease in males when compared to females. But when looking at the results, it is clear that these differences appear to be apparent later in life. If gender differences in older patients with SDHB mutations will be confirmed by other studies, we need to clarify why they do occur.
Adjusting for age and gender, having solitary or multiple tumors at initial diagnosis did not influence the time from diagnosis to the development of metastasis (p = 0.79, based on Cox regression with three covariates: age, gender, solitary/multiple). Similarly, we did not find any association between biochemical profile or tumor location and the risk of metastatic disease, although recent studies found noradrenergic and dopaminergic secretory phenotype to be linked to development of metastases (Crona et al. 2019; Hescot et al. 2019). Most of our patients with metastatic disease— 77.78%—presented with either noradrenergic, dopaminergic, or mixed (noradrenergic and dopaminergic) biochemical phenotype. However, the numbers of patients with dopaminergic or adrenergic biochemical profiles in our cohort were too small to get valid information on association of the secretory phenotype and the risk of metastatic disease development.
In a study on adult patients with SDHB-related primary PHEO/PGL, those with tumor sizes ≥ 4.5 cm developed metastases earlier and those with primary tumor sizes ≥ 5.5 cm had worse survival rates than those with smaller tumors (Schovanek et al. 2014). Another study showed primary tumor size of > 3 cm to be associated with lower disease-free interval (Assadipour et al. 2017) and in the recent work of Hescot et al., (Hescot et al. 2019) 76% patients with metastatic disease had primary tumors larger than 5 cm. Our study on pediatric patients showed that a cut-off of ≥ 5 cm in primary tumor size could be a risk factor for metastasis, which was shown within 5 years from diagnosis. This seems to support the same observation among adult SDHB-related PHEOs/PGLs (Schovanek et al. 2014). Moreover, in our cohort, metastases developed in most of the patients with recurrent disease (76.92%). Our findings are similar to the recent work of Mei et al. (2019) where association of metastatic disease with multiple tumors was described. Based on this data, we can conclude that the total tumor burden as well as the size of primary tumor predict the development of metastases.
The risk of development of metastatic disease seems to be the highest during the first 2 years post-diagnosis and then again between years 12 and 18 post-diagnosis. Although the apparent high rate of metastatic disease development during the years 1 and 2 post-diagnosis might be caused by possibility that metastases were present but not detected, these data suggest the need of more frequent follow-ups in pediatric population with PHEO/PGL during first 2 years post-diagnosis and between years 10 and 20 post-diagnosis.
Remarkably, bearing the SDHB mutation is an independent risk factor for metastatic disease with a reported rate of 85.2% if a primary PHEO/PGL was found in the abdomen before age 20 years (King et al. 2011). Recent study of Jochmanova et al. (2017) and Niemeijer et al. (2017) reported the development of metastases in 59.44% and 7.7% of patients with SDHB mutations, respectively. Metastases occurred in 70.31% of our patients. Adrenal primary tumor was present in 6 (13.33%), primary abdominal tumor in other location was found in 31 (68.88%), pelvic primary tumor in 3 (6.66%), and chest primary tumor in 5 (11.11%) out of 45 SDHB mutation-positive pediatric patients with metastases. These findings are important for establishing the best diagnostic algorithm and are again strongly suggestive of the need to focus diagnostic process primarily to the abdominal region. Sites of metastases are also of interest since they can give us a lead on the most effective follow-up schedule in pediatric patients with SDHB mutations. Most common site of metastases were bones, followed by retroperitoneum and lungs. CT/MRI are most common imaging methods used in pediatric PHEO/PGL population, with per-lesion detection rate around 80% (Jha et al. 2018). The detection of PHEO/PGL with DOTA analogs is very useful in determining patients’ eligibility for peptide receptor radionuclide therapy (PRRT) (Kong et al. 2017) and/or cold somatostatin analog therapy. Currently, 68Ga-DOTATATE PET/CT shows the highest per-lesion detection rate (93.5%) of primary and metastatic lesions compared to 18F-FDG PET/CT and CT/MRI scans. However, the use of 68Ga-DOTATATE PET/CT seems to be limited to non-abdominal, especially bone lesions. Abdominal PHEO/PGL detection by 68Ga-DOTATATE PET/CT was suboptimal in pediatric population with SDHx mutations, probably due to reduced expression of SSTR2 in abdominal PHEOs/PGLs (Jha et al. 2018).
There seems to be no significance in relation to the mode of inheritance in this study. Recent studies suggest there might be potential differences in disease presentation in patients with particular SDHB mutations (Jochmanova et al. 2017; Rijken et al. 2016; Solis et al. 2009). Hence, we analyzed the clinical presentation of PHEO/PGL in patients harboring the five most represented mutations in our cohort of pediatric patients: p.Val140Phe, p.Arg46*, p.Arg90*, exon 1 deletion, and exon 4 and 5 deletion. We did not find statistically significant association of any of these mutations with time to death or development of metastatic disease, however, the analyzed group of patients was too small to draw any conclusions.
In previous report on children with SDHB mutation, the 5-, 10-, and 20-year survival rates were 95.8%, 95.8%, and 71.9%, respectively (King et al. 2011). Our study, which includes also 27 pediatric patients reported by King et al., showed 5-, 10- and 20-year survival rates of 100%, 97.14%, and 77.71%, respectively. Overall, there was an estimate of 22.29% (95% CI 8.62–50.60%) patients at risk of death after 20 years. Interestingly, presence of metastases at diagnosis was associated with decreased survival rate in males, but not in females, when compared to solitary or multiple tumor disease presentation (p < 0.0001). However, due to the low numbers of patients (especially females) with solitary or multiple tumors at the time of PHEO/PGL diagnosis, this finding needs to be evaluated on larger group of patients. With these risk factors in mind, this group with SDHB mutations still warrants aggressive follow-up.
Similar to previous studies on pediatric PHEO/PGL, we recognize limitations associated with potential for referral bias and retrospective nature of the study. Since our institution is a tertiary care center, potential referral bias of more complicated patients with extensive and metastatic disease is more likely. Moreover, small number of patients harboring particular SDHB mutations does not allow to perform genotype–phenotype analysis for each mutation. Multi-institutional international long-term prospective studies are needed. Nonetheless, the present data emphasize the need for early screening of pediatric population harboring SDHB mutation.
PHEO/PGL is rare in pediatric population and it is usually associated with germline mutation in one of the PHEO/PGL susceptibility genes, even in patients with negative family history for this tumor. Recent study found germline mutation to be present in 66% of pediatric patients without family history of PHEO/PGL (Babic et al. 2017). Mutations in SDHB are associated with aggressive disease and high risk of metastases (Amar et al. 2007; Brouwers et al. 2006; Gimenez-Roqueplo et al. 2003). Although there are not specific recommendations about genetic testing for pediatric population, based on the present data, we recommend genetic testing in every pediatric patient diagnosed with PHEO/PGL, even if there is no family history of PHEO/PGL. Knowledge of underlying genetic mutation allows to predict outcomes and to plan appropriate therapeutic and follow-up care in PHEO/PGL patients. If germline mutation is identified in patient with PHEO/PGL, it is reasonable to perform genetic testing in first degree relatives to identify yet healthy mutation carriers, who should be screened for the disease and followed-up regularly. Based on present results, we recommend initiating annual biochemical screening for PHEO/PGL in pediatric SDHB mutation carriers at age of 5–6 years. If biochemistry comes back positive, it should be followed by anatomical imaging to localize the tumor. Since only 6.67% of pediatric patients presented with biochemically silent tumors in our study, imaging of patients with negative biochemical results is most likely not warranted. However, any unusual pain or discomfort that could be related to potential tumor warrants further examination, including imaging despite negative biochemical testing. Around 19% of pediatric patients with SDHB mutation-related PHEO/PGL presented with metastatic disease at the initial diagnosis, which warrants whole body studies to be performed at initial imaging evaluation. Long-term follow-up of pediatric patients with SDHB-related PHEO/PGL is essential due to the high risk of malignancy and recurrence. After initial diagnosis, we still recommend that biochemical testing to be done on the annual basis in those patients with normalization of positive biochemical tests after primary tumor removal and anatomical imaging (CT/MRI). If any biochemical tests, preferable the measurement of plasma metanephrines, become positive, imaging is warranted. Currently, there is no consensus whether CT or MRI would be preferable but to limit radiation exposure, MRI is advised in younger patients. Nevertheless, if MRI provides suboptimal results, CT scan needs to be performed. This can be also coupled with the use of functional imaging, currently the use of DOTA analogs (Jha et al. 2018). Based on present study, closer follow-up is warranted between 10 and 20 years post-diagnosis: even in patients with negative annual biochemical screening, we recommend anatomical imaging once in 2 years during this 10-year period. If stable disease is present, reflecting unchanged biochemical test results and at least two anatomical scans, 6–12-month biochemical interval testing is recommended. Any rise of biochemical test results calls for immediate imaging to further detect disease progression. The last part of this algorithm is also recommended to be used in patients in whom either metastatic disease is found at the initial presentation or in whom the normalization of biochemical tests after the removal of a primary tumor does not occur. Currently, there is no consensus or solid data how to perform long-term imaging follow-up in patients with repeatedly normal biochemical tests after the initial operation or in those with unchanged positive biochemical tests and stable disease. It should be also noted, that about 7% of patients who initially present with negative biochemical tests despite the presence of PHEO/PGL need a different follow-up algorithm from those described above. Here, adding the measurement of chromogranin A and periodic imaging screening is necessary and remains to be determined on which time interval basis.
Conclusion
In summary, our study has highlighted several important aspects in the management of pediatric patients with PHEO/PGL associated with SDHB mutations. The SDHB mutations seem to be an independent risk factor for metastatic disease, as demonstrated by previous studies (Assadipour et al. 2017; Crona et al. 2019; King et al. 2011). Although patients with SDHB mutations develop metastases as early as 5 years from diagnosis, we have shown that the 20-year prognosis and survival is good. Metastases in pediatric patients with SDHB mutations develop earliest in the bones followed by lymph nodes, lungs, and liver. A tumor size ≥ 5 cm and multiple/recurrent tumors warrant closer follow-up for earlier detection of metastatic lesions. Although noradrenergic and dopaminergic tumor secretory phenotypes were linked to metastatic disease development in some recent studies, larger cohorts of pediatric patients are needed to confirm those findings. This study supplements previous recommendations on appropriate and timely clinical screening for carriers of SDHB mutations to optimize long-term outcomes.
Author contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by IJ, AMTA, RJSG, CLPM, RW, TP, and KP. The first draft of the manuscript was written by IJ and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the Intramural Research Program of the Eunice Kennedy Shriver NICHD, NIH (Grant Z1AHD008735).
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Informed consent
Informed assent from the patients and signed permission from one or both parents were obtained for all the individual participants included in the study.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Amar L, Bertherat J, Baudin E et al (2005) Genetic testing in pheochromocytoma or functional paraganglioma. J Clin Oncol 23:8812–8818. 10.1200/JCO.2005.03.1484 [DOI] [PubMed] [Google Scholar]
- Amar L, Baudin E, Burnichon N et al (2007) Succinate dehydrogenase B gene mutations predict survival in patients with malignant pheochromocytomas or paragangliomas. J Clin Endocrinol Metab 92:3822–3828. 10.1210/jc.2007-0709 [DOI] [PubMed] [Google Scholar]
- Armstrong R, Sridhar M, Greenhalgh KL et al (2008) Phaeochromocytoma in children. Arch Dis Child 93:899–904. 10.1136/adc.2008.139121 [DOI] [PubMed] [Google Scholar]
- Assadipour Y, Sadowski SM, Alimchandani M et al (2017) SDHB mutation status and tumor size but not tumor grade are important predictors of clinical outcome in pheochromocytoma and abdominal paraganglioma. Surgery 161:230–239. 10.1016/j.surg.2016.05.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babic B, Patel D, Aufforth R et al (2017) Pediatric patients with pheochromocytoma and paraganglioma should have routine preoperative genetic testing for common susceptibility genes in addition to imaging to detect extra-adrenal and metastatic tumors. Surgery 161:220–227. 10.1016/j.surg.2016.05.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barontini M, Levin G, Sanso G (2006) Characteristics of pheochromocytoma in a 4- to 20-year-old population. Ann NY Acad Sci 1073:30–37. 10.1196/annals.1353.003 [DOI] [PubMed] [Google Scholar]
- Bausch B, Wellner U, Bausch D et al (2014) Long-term prognosis of patients with pediatric pheochromocytoma. Endocr Relat Cancer 21:17–25. 10.1530/ERC-13-0415 [DOI] [PubMed] [Google Scholar]
- Baysal BE, Willett-Brozick JE, Lawrence EC et al (2002) Prevalence of SDHB, SDHC, and SDHD germline mutations in clinic patients with head and neck paragangliomas. J Med Genet 39:178–183. 10.1136/jmg.39.3.178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltsevich DG, Kuznetsov NS, Kazaryan AM, Lysenko MA (2004) Pheochromocytoma surgery: epidemiologic peculiarities in children. World J Surg 28:592–596. 10.1007/s00268-004-7134-9 [DOI] [PubMed] [Google Scholar]
- Benn DE, Gimenez-Roqueplo AP, Reilly JR et al (2006) Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. J Clin Endocrinol Metab 91:827–836. 10.1210/jc.2005-1862 [DOI] [PubMed] [Google Scholar]
- Bissada NK, Safwat AS, Seyam RM et al (2008) Pheochromocytoma in children and adolescents: a clinical spectrum. J Pediatr Surg 43:540–543. 10.1016/j.jpedsurg.2007.10.038 [DOI] [PubMed] [Google Scholar]
- Brouwers FM, Eisenhofer G, Tao JJ, Kant JA, Adams KT, Linehan WM, Pacak K (2006) High frequency of SDHB germline mutations in patients with malignant catecholamine-producing paragangliomas: implications for genetic testing. J Clin Endocrinol Metab 91:4505–4509. 10.1210/jc.2006-0423 [DOI] [PubMed] [Google Scholar]
- Cascon A, Inglada-Pérez L, Comino-Méndez I et al (2013) Genetics of pheochromocytoma and paraganglioma in Spanish pediatric patients. Endocr Relat Cancer 20:L1–6. 10.1530/ERC-12-0339 [DOI] [PubMed] [Google Scholar]
- Choat H, Derrevere K, Knight L, Brown W, Mack EH (2014) SDHB-associated paraganglioma in a pediatric patient and literature review on hereditary pheochromocytoma-paraganglioma syndromes. Case Rep Endocrinol 2014:502734. 10.1155/2014/502734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciftci AO, Tanyel FC, Senocak ME, Buyukpamukcu N (2001) Pheochromocytoma in children. J Pediatr Surg 36:447–452. 10.1053/jpsu.2001.21612 [DOI] [PubMed] [Google Scholar]
- Comino-Mendez I, Gracia-Aznárez FJ, Schiavi F et al (2011) Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat Genet 43:663–667. 10.1038/ng.861 [DOI] [PubMed] [Google Scholar]
- Crona J, Lamarca A, Ghosal S, Welin S, Skogseid B, Pacak K (2019) Genotype-phenotype correlations in pheochromocytoma and paraganglioma. Endocr Relat Cancer. 10.1530/ERC-19-0024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darr R, Zöphel K, Eisenhofer G et al (2012) Combined use of 68Ga-DOTATATE and 18F-FDG PET/CT to localize a bronchial carcinoid associated with ectopic ACTH syndrome. J Clin Endocrinol Metab 97:2207–2208. 10.1210/jc.2012-1402 [DOI] [PubMed] [Google Scholar]
- De Krijger RR, Van Nederveen FH, Korpershoek E, De Herder WW, De Muinck Keizer-Schrama SM, Dinjens WN (2006) Frequent genetic changes in childhood pheochromocytomas. Ann NY Acad Sci 1073:166–176. 10.1196/annals.1353.017 [DOI] [PubMed] [Google Scholar]
- Ein SH, Shandling B, Wesson D, Filler R (1990) Recurrent pheochromocytomas in children. J Pediatr Surg 25:1063–1065. 10.1016/0022-3468(90)90219-y [DOI] [PubMed] [Google Scholar]
- Fishbein L, Leshchiner I, Walter V et al (2017) Comprehensive molecular characterization of pheochromocytoma and paraganglioma. Cancer Cell 31:181–193. 10.1016/j.ccell.2017.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimenez-Roqueplo AP, Favier J, Rustin P et al (2003) Mutations in the SDHB gene are associated with extra-adrenal and/or malignant phaeochromocytomas. Cancer Res 63:5615–5621 [PubMed] [Google Scholar]
- Gimenez-Roqueplo AP, Dahia PL, Robledo M (2012) An update on the genetics of paraganglioma, pheochromocytoma, and associated hereditary syndromes. Horm Metab Res 44(5):328–333. 10.1055/s-0031-1301302 [DOI] [PubMed] [Google Scholar]
- Hensen EF, Bayley JP (2011) Recent advances in the genetics of SDH-related paraganglioma and pheochromocytoma. Fam Cancer 10:355–363. 10.1007/s10689-010-9402-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hescot S, Curras-Freixes M, Deutschbein T et al (2019) Prognosis of malignant pheochromocytoma and paraganglioma (MAPP-Prono Study): a European network for the study of adrenal tumors retrospective study. J Clin Endocrinol Metab 104:2367–2374. 10.1210/jc.2018-01968 [DOI] [PubMed] [Google Scholar]
- Jha A, Ling A, Millo C et al (2018) Superiority of (68)Ga-DOTATATE over (18)F-FDG and anatomic imaging in the detection of succinate dehydrogenase mutation (SDHx )-related pheochromocytoma and paraganglioma in the pediatric population. Eur J Nucl Med Mol Imaging 45:787–797. 10.1007/s00259-017-3896-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jochmanova I, Wolf KI, King KS et al (2017) SDHB-related pheochromocytoma and paraganglioma penetrance and genotype-phenotype correlations. J Cancer Res Clin Oncol 143:1421–1435. 10.1007/s00432-017-2397-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman BH, Telander RL, van Heerden JA, Zimmerman D, Sheps SG, Dawson B (1983) Pheochromocytoma in the pediatric age group: current status. J Pediatr Surg 18:879–884. 10.1016/s0022-3468(83)80040-2 [DOI] [PubMed] [Google Scholar]
- King KS, Prodanov T, Kantorovich V et al (2011) Metastatic pheochromocytoma/paraganglioma related to primary tumor development in childhood or adolescence: significant link to SDHB mutations. J Clin Oncol 29:4137–4142. 10.1200/JCO.2011.34.6353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong G, Grozinsky-Glasberg S, Hofman MS et al (2017) Efficacy of peptide receptor radionuclide therapy for functional metastatic paraganglioma and pheochromocytoma. J Clin Endocrinol Metab 102:3278–3287. 10.1210/jc.2017-00816 [DOI] [PubMed] [Google Scholar]
- Lloyd RV, Osamura RY, Klöppel G, Rosai J (eds) (2017) WHO classification of tumours of endocrine organs WHO/IARC classification of Tumours, 4th edn. International Agency for Research on Cancer, Lyon [Google Scholar]
- Maher ER, Eng C (2002) The pressure rises: update on the genetics of phaeochromocytoma. Hum Mol Genet 11(2347–2354):1. 10.1093/hmg/11.20.2347 [DOI] [PubMed] [Google Scholar]
- Mei L, Khurana A, Al-Juhaishi T, Faber A, Celi F, Smith S, Boikos S (2019) Prognostic factors of malignant pheochromocytoma and paraganglioma: a combined SEER and TCGA databases review. Horm Metab Res 51:451–457. 10.1055/a-0851-3275 [DOI] [PubMed] [Google Scholar]
- Neumann HP, Bausch B, McWhinney SR et al (2002) Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med 346:1459–1466. 10.1056/NEJMoa020152 [DOI] [PubMed] [Google Scholar]
- Neumann HP, Pawlu C, Peczkowska M (2004) Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA 292:943–951. 10.1001/jama.292.8.943 [DOI] [PubMed] [Google Scholar]
- Niemeijer ND, Rijken JA, Eijkelenkamp K et al (2017) The phenotype of SDHB germline mutation carriers: a nationwide study. Eur J Endocrinol 177:115–125. 10.1530/EJE-17-0074 [DOI] [PubMed] [Google Scholar]
- Pamporaki C, Därr R, Bursztyn M et al (2013) Plasma-free vs deconjugated metanephrines for diagnosis of phaeochromocytoma. Clin Endocrinol (Oxf) 79:476–483. 10.1111/cen.12191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham TH, Moir C, Thompson GB et al (2006) Pheochromocytoma and paraganglioma in children: a review of medical and surgical management at a tertiary care center. Pediatrics 118:1109–1117. 10.1542/peds.2005-2299 [DOI] [PubMed] [Google Scholar]
- Rijken JA, Niemeijer ND, Corssmit EP, Jonker MA, Leemans CR, Menko FH, Hensen EF (2016) Low penetrance of paraganglioma and pheochromocytoma in an extended kindred with a germline SDHB exon 3 deletion. Clin Genet 89:128–132. 10.1111/cge.12591 [DOI] [PubMed] [Google Scholar]
- Ross JH (2000) Pheochromocytoma. Special considerations in children. Urol Clin N Am 27:393–402. 10.1016/s0094-0143(05)70088-4 [DOI] [PubMed] [Google Scholar]
- Schovanek J, Martucci V, Wesley R et al (2014) The size of the primary tumor and age at initial diagnosis are independent predictors of the metastatic behavior and survival of patients with SDHB-related pheochromocytoma and paraganglioma: a retrospective cohort study. BMC Cancer 14:523. 10.1186/1471-2407-14-523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solis DC, Burnichon N, Timmers HJ et al (2009) Penetrance and clinical consequences of a gross SDHB deletion in a large family. Clin Genet 75:354–363. 10.1111/j.1399-0004.2009.01157.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spoudeas HA (ed) (2005) Paediatric endocrine tumours. A multi-disciplinary consensus statement of best practice from a working group convened under the auspices of the BSPED and UKCCSG (rare tumour working groups). Novo Nordisk Ltd, West Sussex [Google Scholar]
- Stackpole RH, Melicow MM, Uson AC (1963) Pheochromocytoma in children Report of 9 case and review of the first 100 published cases with follow-up studies. J Pediatr 63:314–330. 10.1016/s0022-3476(63)80345-5 [DOI] [PubMed] [Google Scholar]
- Timmers HJ, Kozupa A, Eisenhofer G et al (2007) Clinical presentations, biochemical phenotypes, and genotype-phenotype correlations in patients with succinate dehydrogenase subunit B-associated pheochromocytomas and paragangliomas. J Clin Endocrinol Metab 92:779–786. 10.1210/jc.2006-2315 [DOI] [PubMed] [Google Scholar]
- Timmers HJ, Gimenez-Roqueplo AP, Mannelli M, Pacak K (2009) Clinical aspects of SDHx-related pheochromocytoma and paraganglioma. Endocr Relat Cancer 16:391–400. 10.1677/ERC-08-0284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turkova H, Prodanov T, Maly M et al (2016) Characteristics and outcomes of metastatic SDHB and sporadic pheochromocytoma/paraganglioma: an national institutes of health study. Endocr Pract 22:302–314. 10.4158/EP15725.OR [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicha A, Musil Z, Pacak K (2013) Genetics of pheochromocytoma and paraganglioma syndromes: new advances and future treatment options. Curr Opin Endocrinol Diabetes Obes 20:186–191. 10.1097/MED.0b013e32835fcc45 [DOI] [PMC free article] [PubMed] [Google Scholar]