

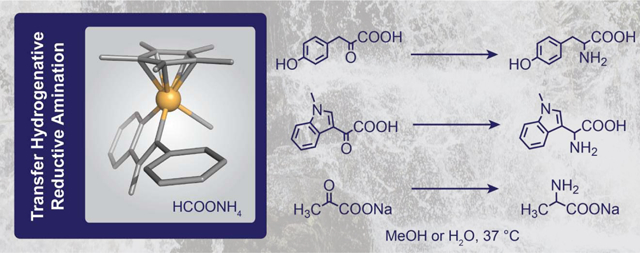
Abstract
The conversion of aldehydes and ketones to 1° amines could be promoted by half-sandwich iridium complexes using ammonium formate as both the nitrogen and hydride source. To optimize this method for green chemical synthesis, we tested various carbonyl substrates in common polar solvents at physiological temperature (37 °C) and ambient pressure. We found that in methanol, excellent selectivity for the amine over alcohol/amide products could be achieved for a broad assortment of carbonyl-containing compounds. In aqueous media, selective reduction of carbonyls to 1° amines was achieved in the absence of acids. Unfortunately, at Ir catalyst concentrations of <1 mM in water, reductive amination efficiency dropped significantly, which suggest that this catalytic methodology might be not suitable for aqueous applications where very low catalyst concentration is required (e.g., inside living cells).
Keywords: Reductive Amination, Green Chemistry, Transfer Hydrogenation, Iridium, Half-Sandwich Complexes
Graphical Astract
Introduction
Primary amines are ubiquitous in nature (e.g., in amino acids, nucleic acids, and alkaloids) and are key components in numerous synthetic products such as dyes, pharmaceuticals, and agrochemicals.1–4 Because of their central importance to both life and industry, the synthesis of 1° amines is a major focus in modern organic chemistry. Conventional methods for the synthesis of 1° amines include reduction of nitrile/amide/nitro groups,5 alkylation of ammonia,6 and hydroamination of alkenes.7 One of the most attractive methods to prepare 1° amines is reductive amination (RA) of aldehydes and ketones because it can be performed under mild reaction conditions and tolerates a broad range of functional groups (Scheme 1).8 The RA process involves condensation between carbonyl compounds with ammonia to generate imine intermediates that are subsequently reduced using a hydride donor. Unfortunately, a common competing side reaction is the reduction of the starting carbonyl compound to the corresponding alcohol.
Scheme 1.

Reductive amination (RA) of carbonyl compounds to give 1° amines
The selection of suitable hydride reagents and reaction conditions are important factors in determining the efficiency and selectivity of RA reactions. The most common reductants used for this application are sodium cyanoborohydride (NaBH3CN),9 sodium triacetoxyborohydride (NaBH(OAc)3),10 organosilanes,11,12 and other similar reagents (Scheme 2).13–15 Such reagents, however, can give poor chemoselectivity and generate potentially non-innocent byproducts (e.g., cyanide and boron-containing species are toxic to cells in biological applications). RA of aldehydes/ketones could also be achieved using metal catalysts in the presence of alkyl amines and hydrogen gas to give 2° amines (hydrogenation)16,17 or ammonium formate to give 1° amines (Leuckart-Wallach reaction).18 A disadvantage of the former is the use of flammable gas and a disadvantage of the latter is the requirement for high temperatures (>120 °C).
Scheme 2.

Methods for conversion of carbonyls to amines
One of the earliest examples of using organometallic catalysts for transfer hydrogenative reductive amination was reported by Ogo/Watanabe and co-workers in 2001, who used pentamethylcyclopentadienyl (Cp*) iridium catalysts and ammonium formate in water to convert aldehydes and ketones to 1° amines.19 Ogo and Fukuzumi also discovered that Cp*Ir complexes were capable of furnishing α-amino acids from α-keto acids using excess ammonium formate (20 equiv. relative to substrate) in aqueous solutions (pH = 5.0–6.5) at 80 °C.20 In 2002, Kitamura and co-workers showed that [Cp*RhCl2]2 in combination with HCOONH4 was highly effective for the chemoselective synthesis of 1° amines from ketones in methanol.21 In 2003, Kadyrov and co-worker achieved the asymmetric RA of aromatic ketones by taking advantage of ruthenium catalysts in the presence of chiral ligands and ammonium formate in ammonia/methanol solutions.22 Lastly, Xiao and co-workers developed cyclometallated Cp*Ir complexes that efficiently provided 2° or 3° amines from ketones, 1° or 2° amines, and azeotropic mixtures of formic acid/triethylamine.23 The synthesis of 1° amines was also achieved.24 Although the examples above demonstrate the remarkable progress made in catalytic amine synthesis in recent years, some of the methods required high temperatures (≥ 80 °C) and have poor chemoselectivity in water. RA in aqueous solutions is particularly challenging because water is unfavorable for the formation of imines, which are key reaction intermediates (Scheme 1).25
Because our research group is interested in biocompatible chemistry, we were intrigued by the prospect of applying transfer hydrogenative RA in biological systems.26–28 A seminal report by McFarland and Francis in 2005 showed that lysozymes could be alkylated via reductive amination between lysine side chains and carbonyl containing substrates using [Cp*Ir(bipyridine)OH2]2+ catalysts in the presence of sodium formate.29 This example was noteworthy because the reactions were carried out under physiologically relevant conditions (pH 7.4 aqueous buffer, 37 °C) and were as efficient as classical protein conjugation methods such as lysine acylation or reductive amination using sodium cyanoborohydride. Given our previous success in demonstrating that [Cp*Ir(N-phenyl-2-pyridinecarboxamidate)Cl] (Ir1) is capable of promoting intracellular transfer hydrogenation,30 we wanted to assess whether such complexes could also mediate transfer hydrogenative RA to afford 1° amines under mild reaction conditions. Recently, Kuwata, Watanabe, and co-workers reported a study showing that Ir1 and related complexes could be used with very low catalyst loading (e.g., substrate/catalyst ratio of up to 20,000) to achieve efficient transfer hydrogenative RA.31 Unfortunately, these reactions were performed at temperatures ≥ 60 °C in methanol, which are not suitable conditions for biological environments. In the present work, we report on the scope and limitations of transfer hydrogenative reductive amination using organoiridium complexes in both methanol and water at 37 °C. Our studies showed that iridium-catalyzed RA reactions could proceed with excellent selectivity for a variety of carbonyl substrates in polar organic solvents; however, catalyst loading below 1.0 mM is not sufficient to achieve appreciable reaction rates for practical synthesis in aqueous media.
Results and Discussion
Catalyst Screening.
Pentamethylcyclopentadienyl iridium complexes are attractive catalysts because they are air and water stable, can be easily prepared, and show excellent transfer hydrogenation activity.19,26,32 To determine whether such catalysts could be employed for synthetically challenging applications, such as in reactions involving thermally sensitive substrates or inside living environments, we evaluated a series of Ir complexes in open air and mild temperatures. Our studies focused primarily on Cp*Ir N-heterocyclic-carboxamidate complexes (Ir1-Ir9, Chart 1) because of their demonstrated ability to catalyze RA at ultra low catalyst loading.31 For comparison, we also screened reactions using [Cp*Ir(N-tosylethylenediamine)Cl] (Ir10),27 [Cp*Ir(bipyridine)Cl]Cl (Ir11),33 and [Cp*Ir(PPh3)Cl2)] (Ir12)34 as examples of other common half-sandwich complexes. All of these Ir complexes were synthesized according to reported procedures.
Chart 1.
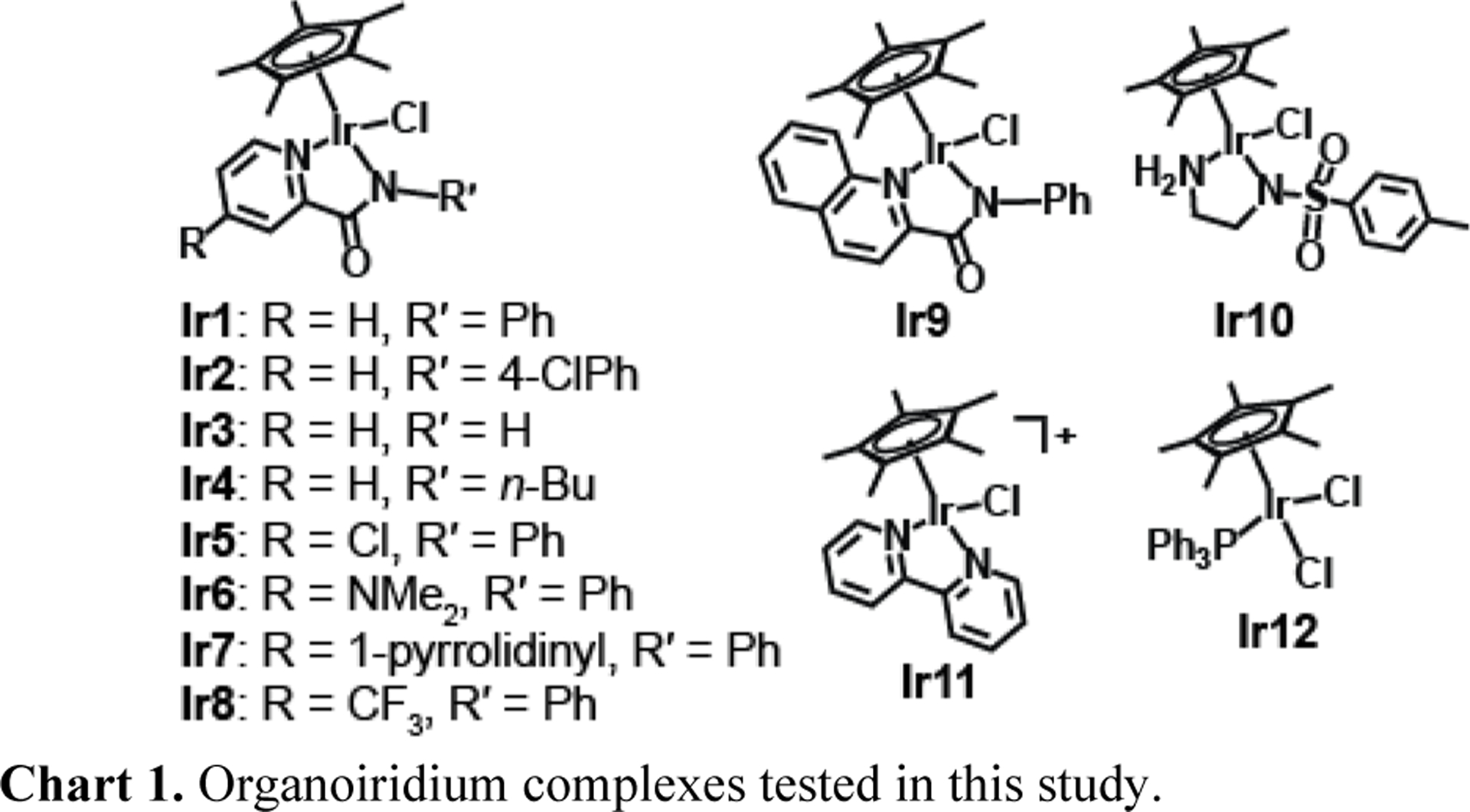
Organoiridium complexes tested in this study.
Our initial studies were conducted in methanol because it provided the best solubility for the catalysts and reagents (Table 1). At 25 °C, reaction of benzaldehyde (1a) with 2 equiv. of HCOONH4 using Ir1 gave only about 40% conversion (Entry 1). The product selectivity was poor, showing 28% benzylamine (2a, product of reductive amination) and 12% benzyl alcohol (3a, product of direct reduction). Only trace amounts of N-benzylformamide (4a) was detected, which is presumably obtained from the condensation of benzylamine with formate. When the reaction temperature was increased to 37 °C (Entries 2–10), complexes Ir1-Ir9 all gave excellent yield and selectivity for benzylamine (> 90%) over benzyl alcohol (< 10%). The complex containing an electron-donating dimethylamino group (Ir6, Entry 7) gave slightly more benzyl alcohol than the one containing an electron-withdrawing trifluoromethyl group (Ir8, Entry 9). This result is consistent with our previous study,26,27 which found that electron rich Ir6 was faster at reducing carbonyls than electron poor Ir8. Because reductive amination is a two-step process (Scheme 1), the faster transfer hydrogenation catalyst Ir6 is actually less ideal for RA because it could reduce carbonyl groups to alcohols before they undergo imine condensation. We found that when the amount of HCOONH4 was increased to 10 equiv., relative to substrate, the formation of 3a by Ir1 in various alcohol solvents was suppressed but significant quantities of 4a were obtained (Entries 14–16). When the solvent was changed to either THF (Entry 17) or acetonitrile (Entry 18), formation of 4a was significantly reduced but various amounts of 3a were observed (25% and 4%, respectively). These results indicate that the chemoselectivity of the Ir catalysts is strongly influenced by their reaction conditions.
Table 1.
Comparison of catalyst efficiency and chemoselectivitya
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Cat. | Solvent | Temp. (°C) | Conv. (%)b | 2a (%)b | 3a (%)b | 4a (%)b |
| 1 | Ir1 | MeOH | 25 | 40 | 28 | 12 | <1 |
| 2 | Ir1 | MeOH | 37 | >99 | 93 | 7 | <1 |
| 3 | Ir2 | MeOH | 37 | >99 | 95 | 5 | <1 |
| 4 | Ir3 | MeOH | 37 | 96 | 92 | 4 | <1 |
| 5 | Ir4 | MeOH | 37 | >99 | 98 | 2 | <1 |
| 6 | Ir5 | MeOH | 37 | >99 | 95 | 5 | <1 |
| 7 | Ir6 | MeOH | 37 | >99 | 90 | 10 | <1 |
| 8 | Ir7 | MeOH | 37 | >99 | 93 | 7 | <1 |
| 9 | Ir8 | MeOH | 37 | >99 | 96 | 4 | <1 |
| 10 | Ir9 | MeOH | 37 | >99 | 96 | 4 | <1 |
| 11 | Ir10 | MeOH | 37 | 80 | 12 | 67 | <1 |
| 12 | Ir11 | MeOH | 37 | 71 | 11 | 59 | <1 |
| 13 | Ir12 | MeOH | 37 | 50 | 21 | 29 | <1 |
| 14c | Ir1 | MeOH | 37 | >99 | 81 | <1 | 18 |
| 15c | Ir1 | EtOH | 37 | >99 | 52 | <1 | 48 |
| 16c | Ir1 | iPrOH | 37 | >99 | 62 | <1 | 38 |
| 17c | Ir1 | THF | 37 | 35 | 10 | 25 | <1 |
| 18c | Ir1 | MeCN | 37 | >99 | 96 | 4 | <1 |
| 19 | none | MeOH | 37 | <1 | <1 | <1 | <1 |
Reagents and conditions: 1a (100 μmol), Ir1 (1.0 μmol), HCOONH4 (200 μmol), solvent (0.5 mL), 15 h.
Determined by 1H NMR spectroscopy using 1,3,5-trimethoxybenzene as an internal standard.
10 equiv. of HCOONH4 was used.
Interestingly, complexes Ir10-Ir12 showed moderate activity (< 80% conversion) and gave more alcohol than amine products (Table 1, Entries 11–13), which demonstrated that Cp*Ir pyridinecarboxamidate complexes are more efficient RA catalysts than other typical Ir half-sandwich complexes.
Benzaldehyde Substrate Scope.
Using an excess of HCOONH4 to maximize the yield of 2/4, we examined the benzaldehyde substrate scope using Ir1 (Table 2). The reaction of benzaldehyde with HCOONH4 (10 equiv.) in methanol at 37 °C for 15 h led to complete consumption of starting material. High chemoselectivity of 2 over 3 was achieved in reactions with benzaldehyde (1a, Entry 1), p-methoxybenzaldehyde (1b, Entry 2), and p-chlorobenzaldehyde (1f, Entry 6). Compounds containing bromo (1c-1e, Entries 3–5), nitro (1h-1j, Entries 8–10), and nitrile (1k, Entry 11) groups gave minor amounts of alcohol in addition to the major amine product. Notably, 4-fluorobenzaldehyde (1g, Entry 7) was converted to significantly larger amounts of alcohol (26%) in comparison to other aldehydes (<10%). The amine/alcohol product ratios for ortho-, meta-, and para-substituted isomers (1c–1e and 1h-1j) varied but with no obvious trend. In most reactions, formamide by-products (4) were detected in different amounts. Because formamide can be hydrolyzed to the free amine during the workup step, its formation is not a major concern from a practical standpoint. Overall, our results suggest that RA of benzaldehyde and its functionalized analogues (except for those bearing fluoro substituents) by Ir catalysts is highly efficient in methanol.
Table 2.
Reductive amination of benzaldehydesa
 | |||||
|---|---|---|---|---|---|
| Entry | Aldehyde | 2 (%)b | 3 (%)b | 4 (%)b | |
| 1 | 1a | R = H | 81 (72c) | <1 | 18 |
| 2 | 1b | R = p-OMe | 79 (92c) | <1 | 21 |
| 3 | 1c | R = p-Br | 87 (73c) | 2 | 11 |
| 4 | 1d | R = m-Br | 72 (84c) | 7 | 21 |
| 5 | 1e | R = o-Br | 97 (89c) | 3 | <1 |
| 6 | 1f | R = p-Cl | 98 (90c) | <1 | <1 |
| 7 | 1g | R = p-F | 48 | 26 | 26 |
| 8 | 1h | R = p-NO2 | 87(81c) | 4 | 9 |
| 9 | 1i | R = m-NO2 | 86 | 4 | 10 |
| 10 | 1j | R = o-NO2 | 71 | 8 | 21 |
| 11 | 1k | R = p-CN | 63 | 9 | 28 |
Reagents and conditions: aldehyde (100 μmol), Ir1 (1.0 μmol), HCOONH4 (1.0 mmol), CH3OH (0.5 mL), 37 °C, 15 h.
Determined by 1H NMR with 1,3,5-trimethoxybenzene as internal standard, each experiment was performed in triplicate and the average yield was calculated.
Isolated yield. Some isolated yields of 2 were higher than their NMR yields because compound 4 was hydrolyzed to 2 during the workup step.
Aromatic Ketone Substrate Scope.
Kuwata, Watanabe, and co-workers demonstrated that high yields of amines could be obtained from reactions of ketones and HCOONH 4promoted by Cp*Ir pyridinecarboxamidate complexes at 60–80 °C.31 Using our standard reaction conditions, we screened a variety of aromatic ketones as substrates (Table 3). We observed that most reactions produced exclusively RA products (i.e., amine 6 and formamide 8), with almost no trace of alcohol 7. Only 4-acetylbenzonitrile 5h gave a minor amount of 7h in addition to 6h (Entry 8). The high selectivity for amines over alcohol products is attributed to the reduced electrophilicity and increased steric bulk of the C=O moieties in ketones compared to those in aldehydes. Furthermore, we have demonstrated previously that reduction of ketones to alcohols by Ir catalysts/formate does not occur readily.26
Table 3.
Reductive amination of aromatic ketonesa
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Ketone | Conv. (%)b | 6 (%)b | 7 (%)b | 8 (%)b | |
| 1 | 5a | R = Ph R’ = Me |
>99 | 99 (96c) | <1 | <1 |
| 2 | 5b | R = p-OMePh R’ = Me |
>99 | >99 (93c) | <1 | <1 |
| 3 | 5c | R = p-OHPh R’ = Me |
>99 | >99 | <1 | <1 |
| 4 | 5d | R = p-BrPh R’ = Me |
>99 | 87 (82c) | <1 | 13 |
| 5 | 5c | R = p-FPh R’ = Me |
81 | 79 | <1 | 2 |
| 6 | 5f | R = p-NO2Ph R’ = Me |
>99 | 92 (87c) | <1 | 8 |
| 7 | 5g | R = m-NO2Ph R’ = Me |
>99 | >99 | <1 | <1 |
| 8 | 5h | R = p-CNPh R’ = Me |
>99 | 96 | 4 | <1 |
| 9 | 5i | R = Ph R’ = CF3 |
>99 | 98 | <1 | <1 |
| 10 | 5j |  |
>99 | 99 | <1 | <1 |
| 11 | 5k |  |
86 | 81 | <1 | <1 |
| 12 | 5l |  |
20 | 20 | <1 | <1 |
| 13 | 5m |  |
64 (>99d) | 63 (70c,d) | <1 | <1 |
| 14 | 5n |  |
<1 | 0 | 0 | 0 |
Reagents and conditions: ketone (100 μmol), Ir1 (1.0 μmol), HCOONH4 (1.0 mmol), CH3OH (0.5 mL), 37 °C, 15 h.
Determined by 1H NMR with 1,3,5-trimethoxybenzene as an internal standard. Each experiment was performed in triplicate and the average yield was calculated.
Isolated yield.
Reaction was run for 24 h.
In contrast to the acetophenone substrates that gave high conversion (Table 3, Entries 1–9), other aromatic ketones were not as easily converted to amines. Although 2-acetylfuran 5j (Entry 10) gave quantitative yield of amine, 2-acetylthiophene 5k (Entry 11) and 3-acetylindole 5l (Entry 12) exhibited only 86 and 20% conversion, respectively. We hypothesized that the reduced yields of the latter substrates might be due to their metal coordinating abilities, which may poison the iridium catalyst. The sterically congested ketone 9-fluorenone 5m was successfully converted to the corresponding amine in 70% yield after 24 h (Entry 13). Although it has a similar structure to that of 9-fluorenone, xanthone 5n did not undergo reductive amination (Entry 14). Benzophenone 5o produced an unexpected product 6o (Scheme 3). This species most likely formed from imine condensation between 5o and an in situ generated amine product. Because 6o has low solubility in methanol, it readily precipitated out of solution and was isolated in 97% yield.
Scheme 3.
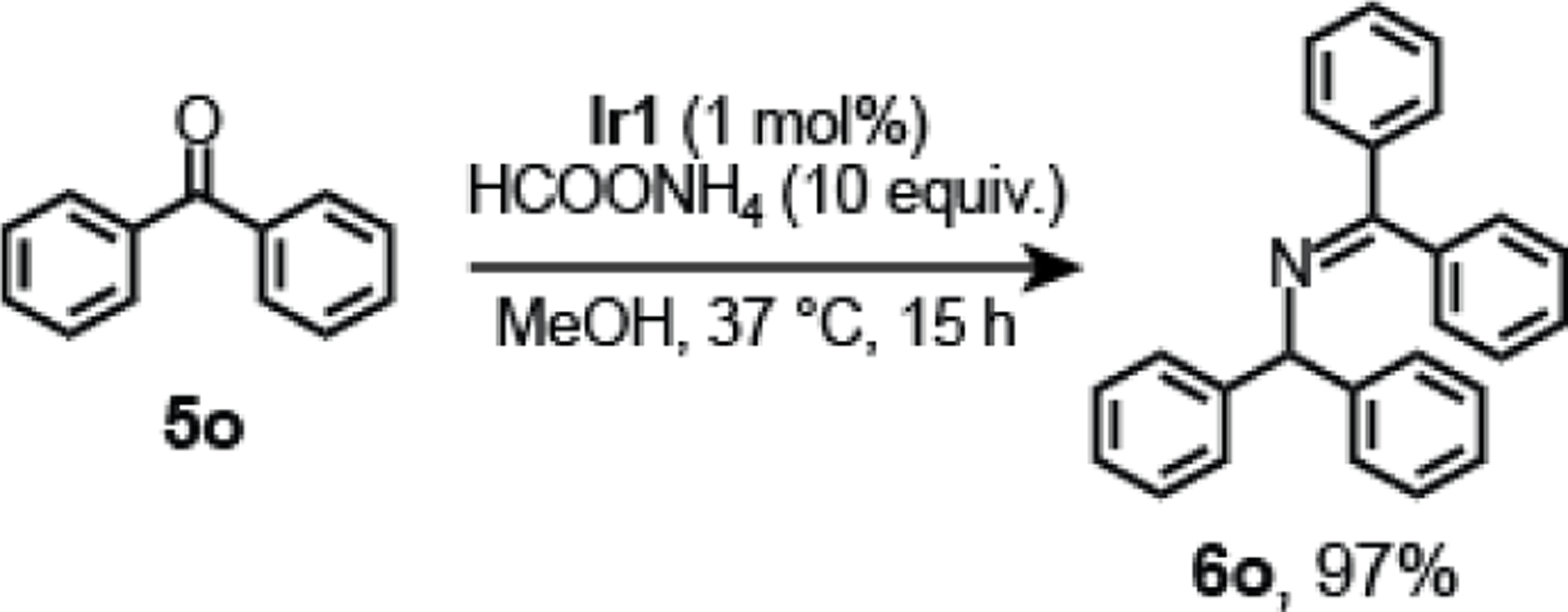
Reductive amination of benzophenone 5o.
Ketoacid Substrate Scope.
To explore further the versatility of transfer hydrogenative RA, we targeted a variety of ketoacid substrates. Because the RA products of α-ketoacids are α-amino acids, this method could provide easy access to both natural and unnatural polypeptide building blocks. In addition, α-amino acids are important precursors for various pharmaceuticals, such as ampicillin,35 amoxicillin,36 resorcinomycin,37 and pheganomycin.37
In the presence of Ir1 and HCOONH4 (10 equiv.) at 37 °C for 6 h, a wide range of ketoacids were converted to the desired amino acids (Scheme 4 and Table S1). The natural α-amino acids glycine (10a), alanine (10b), and tyrosine (10d) were obtained in >90% yield, whereas phenylalanine (10c) and DOPA (10f) were obtained in 62 and 82% yield, respectively. The lower yields of the latter two compounds were attributed to product lost from washing with methanol in the workup step. The unnatural α-amino acids, such as tert-leucine (10e), phenylglycine (10g), 4-methylphenylglycine (10h), 4-methoxyphenylglycine (10i), and N-methyl-3-indoleglycine (10j) were produced in ≥ 95% yield. Several other γ-ketoacids (10k, 10l) and δ-ketoacids (10m, 10n) were also converted into the corresponding amino acids with high efficiency (94–97% yield). Our results were consistent with those reported by Kuwata, Watanabe, and co-workers, who showed that structurally diverse amino acids could be obtained from transfer hydrogenation RA of ketoacids by Ir catalysts and HCOONH4.31 However, since the present studies were performed at 37 °C rather than under reflux, our reaction conditions are more compatible with applications involving thermally sensitive molecules or biological environments.
Scheme 4.

Reductive amination of ketoacids. Reagents and conditions: ketoacid (100 μmol), Ir1 (1.0 μmol), HCOONH4 (1.0 mmol), CH3OH (0.5 mL), 37 °C, 6 h. Reactions were performed in triplicate and the average yields are shown above. Products were quantified by 1H NMR spectroscopy using 1,3,5-trimethoxybenzene as an internal standard. Isolated yields are shown with an asterisk (*).
Reactions in Aqueous Media and Possible Biological Relevance.
Although the results above showed that polar organic solvents (e.g., alcohol or acetonitrile) promoted selective transfer hydrogenative RA, the use of water is preferred in terms of green chemistry38 and biological compatibility.28 However, examples of aqueous RA are still relatively rare,20,32,39,40 particularly those that employ reaction temperatures below 80 °C.41,42 To explore the feasibility of carrying out aqueous RA under physiological conditions, we conducted reactions using Ir1 with formate/formic acid and pyruvic acid (9b) in water at 37 °C. Consistent with other reports,20,32 we found that product selectivity was highly dependent on acid concentration. For example, reaction of Ir1 with pyruvic acid and HCOONH4 produced a mixture containing 58% of alanine (10b) and 42% of lactic acid (11b) (Fig. S2). In contrast, when sodium pyruvate was used instead of pyruvic acid, about 95% of the α-amino acid product was obtained (Fig. S3). Importantly, in pH 7.4 aqueous solutions, such as phosphate buffered saline (PBS) or cell growth media RPMI-1640, > 90% yield of alanine was achieved (Table S2). These results were surprising given that other researchers reported transfer hydrogenative RA was most efficient at ~pH 5.20,32 However, we have not yet conducted a detailed pH dependence study so we cannot comment on the optimal pH range of Ir1 at this time.
Another important point to consider in terms of possible biological relevance is whether the iridium catalysts could operate at appreciable rates in dilute solutions. For example, it is generally recommended that use of metal catalysts inside living cells be restricted to concentrations as low as possible to minimize any potential cytotoxicity issues.28 In these experiments, we kept the stoichiometry of Ir1, HCOONH4, and sodium pyruvate constant in all reactions but only varied their concentrations (Table S3). We observed ≥ 80% reaction conversion after 15 h at 37 °C when the concentration of Ir1 was 2.5 and 1.0 mM. Unfortunately, at 0.5, 0.2, and 0.1 mM catalyst loading, the conversion dropped to 41, 12, and <1%, respectively (Fig. 1). These results were not entirely surprising given we had found in prior work that the overall rate law for transfer hydrogenation was first-order in the Ir1 complex.27 Additionally, because the IC50 (half maximal inhibitory concentration) of Ir1 in NIH-3T3 mouse fibroblast cells is about 0.1 mM,30 our data suggest that keeping Ir1 below its toxic threshold might not be sufficient to enable fast catalysis inside cells. However, due to the complexity of cellular environments, such as the possibility of catalyst aggregation within specific organelles,43 further biological studies are needed to assess the capability of Ir1 to promote intracellular RA reactions.
Figure 1.
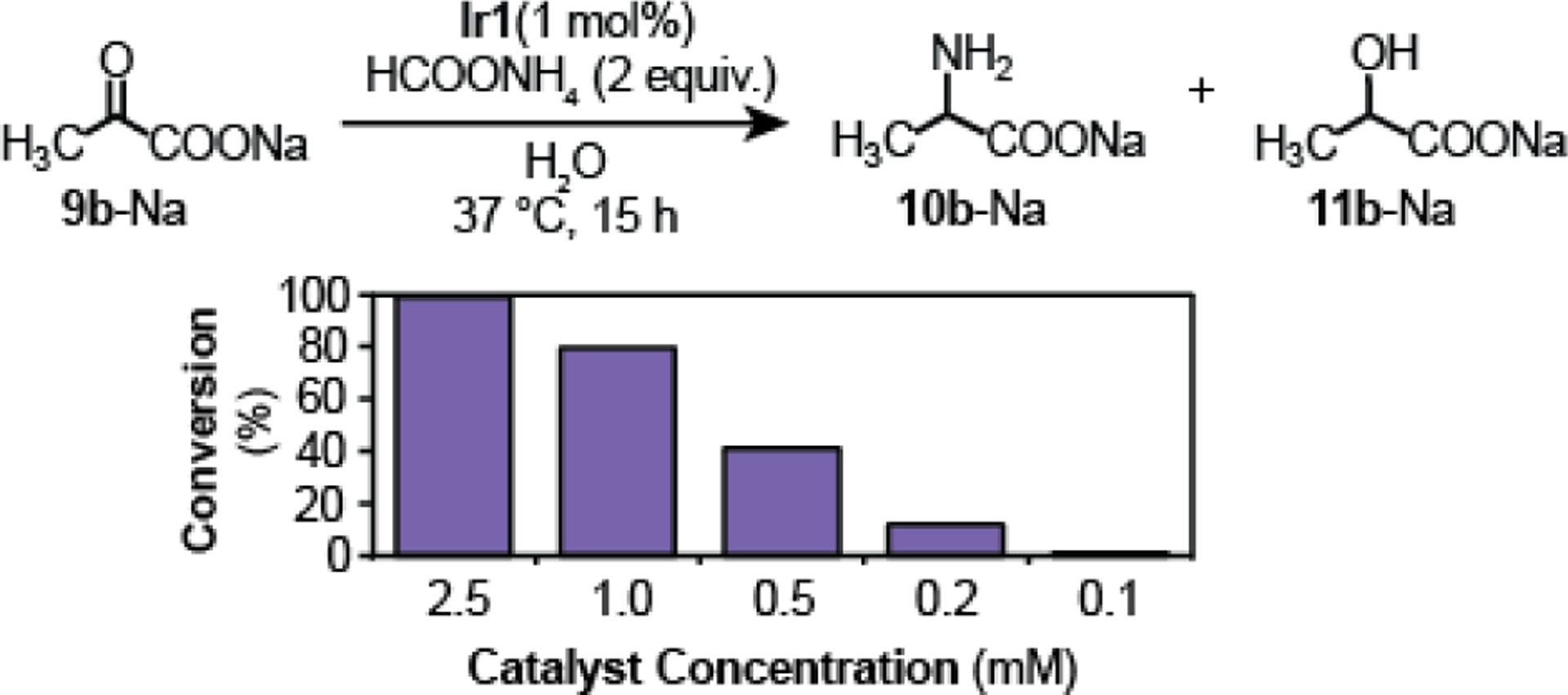
Comparison of reaction efficiency as a function of catalyst concentration. Reaction conditions: Ir1, HCOONH4, and sodium pyruvate in 0.5 mL of H2O at 37 °C for 15 h. See Table S3 for more details.
Conclusion
In summary, we have shown that [Cp*Ir(N-phenyl-2-pyridinecarboxamidate)Cl] and related complexes are excellent catalysts for converting aldehydes and ketones to 1° amines using HCOONH4 as both the hydride and nitrogen donor. This work has expanded upon other studies by demonstrating that aqueous transfer hydrogenative RA could be carried out at mild temperatures (37 °C) and in many cases, with excellent selectivity of amine over alcohol products. A broad range of substrates was tolerated, including ketoacids and sterically hindered carbonyls. Product selectivity in water was highly pH dependent, in which neutral and pH 7.4 buffered solutions gave appreciable yields of the desired amine product. A limitation of these Ir complexes is that they are not efficient catalysts at concentrations below 1 mM in water, which may be problematic for applications requiring low catalyst loading. For intracellular reactions, this problems could perhaps be mitigated by taking advantage of polymeric vesicles44 or other delivery systems to increase catalyst and substrate accumulation inside cells.
Supplementary Material
Highlights.
Half-sandwich Ir complexes show excellent chemoselectivity under optimized reaction conditions.
Reactions could be performed at 37 °C under air.
A variety of natural and unnatural amino acids could be synthesized.
Acknowledgements
We are extremely grateful to the Welch Foundation (Grant No. E-1894) and National Institute of General Medical Science of the NIH (Grant No. R01GM129276) for financial support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https:///
References
- (1).Henkel T; Brunne RM; Müller H; Reichel F Angew. Chem. Int. Ed 1999, 38, 643–647. [DOI] [PubMed] [Google Scholar]
- (2).Berger M; Albrecht B; Berces A; Ettmayer P; Neruda W; Woisetschläger MJ Med. Chem 2001, 44, 3031–3038. [DOI] [PubMed] [Google Scholar]
- (3).Salvatore RN; Yoon CH; Jung KW Tetrahedron 2001, 57, 7785–7811. [Google Scholar]
- (4).Breuer M; Ditrich K; Habicher T; Hauer B; Keßeler M; Stürmer R; Zelinski T Angew. Chem. Int. Ed 2004, 43, 788–824. [DOI] [PubMed] [Google Scholar]
- (5).Satoh T; Suzuki S; Suzuki Y; Miyaji Y; Imai Z Tetrahedron Lett 1969, 10, 4555–4558. [Google Scholar]
- (6).Fujita K.-i.; Furukawa S; Morishima N; Shimizu M; Yamaguchi R ChemCatChem 2018, 10, 1993–1997. [Google Scholar]
- (7).Müller TE; Beller M Chem. Rev 1998, 98, 675–704. [DOI] [PubMed] [Google Scholar]
- (8).Roughley SD; Jordan AM J. Med. Chem 2011, 54, 3451–3479. [DOI] [PubMed] [Google Scholar]
- (9).Barney CL; Huber EV; McCarthy JR Tetrahedron Lett 1990, 31, 5547–5550. [Google Scholar]
- (10).Abdel-Magid AF; Carson KG; Harris BD; Maryanoff CA; Shah RD J. Org. Chem 1996, 61, 3849–3862. [DOI] [PubMed] [Google Scholar]
- (11).Apodaca R; Xiao W Org. Lett 2001, 3, 1745–1748. [DOI] [PubMed] [Google Scholar]
- (12).Patel JP; Li A-H; Dong H; Korlipara VL; Mulvihill MJ Tetrahedron Lett 2009, 50, 5975–5977. [Google Scholar]
- (13).Ranu BC; Majee A; Sarkar AJ Org. Chem 1998, 63, 370–373. [Google Scholar]
- (14).Kim S; Oh CH; Ko JS; Ahn KH; Kim YJ J. Org. Chem 1985, 50, 1927–1932. [Google Scholar]
- (15).Grenga PN; Sumbler BL; Beland F; Priefer R Tetrahedron Lett 2009, 50, 6658–6660. [Google Scholar]
- (16).Werkmeister S; Junge K; Beller M Green Chem 2012, 14, 2371–2374. [Google Scholar]
- (17).Bhor MD; Bhanushali MJ; Nandurkar NS; Bhanage BM Tetrahedron Lett 2008, 49, 965–969. [Google Scholar]
- (18).Alexander ER; Wildman RB J. Am. Chem. Soc 1948, 70, 1187–1189. [DOI] [PubMed] [Google Scholar]
- (19).Ogo S; Makihara N; Kaneko Y; Watanabe Y Organometallics 2001, 20, 4903–4910. [Google Scholar]
- (20).Ogo S; Uehara K; Abura T; Fukuzumi SJ Am. Chem. Soc 2004, 126, 3020–3021. [DOI] [PubMed] [Google Scholar]
- (21).Kitamura M; Lee D; Hayashi S; Tanaka S; Yoshimura MJ Org. Chem 2002, 67, 8685–8687. [DOI] [PubMed] [Google Scholar]
- (22).Kadyrov R; Riermeier TH Angew. Chem. Int. Ed 2003, 42, 5472–5474. [DOI] [PubMed] [Google Scholar]
- (23).Wang C; Pettman A; Bacsa J; Xiao J Angew. Chem. Int. Ed 2010, 49, 7548–7552. [DOI] [PubMed] [Google Scholar]
- (24).Talwar D; Salguero NP; Robertson CM; Xiao J Chem. Eur. J 2014, 20, 245–252. [DOI] [PubMed] [Google Scholar]
- (25).Wei Y; Wu X; Wang C; Xiao J Catal. Today 2015, 247, 104–116. [Google Scholar]
- (26).Ngo AH; Ibañez M; Do LH ACS Catal 2016, 6, 2637–2641. [Google Scholar]
- (27).Ngo AH; Do LH Inorg. Chem. Front 2020, 7, 583–591. [Google Scholar]
- (28).Ngo AH; Bose S; Do LH Chem. Eur. J 2018, 24, 10584–10594. [DOI] [PubMed] [Google Scholar]
- (29).McFarland JM; Francis MB J. Am. Chem. Soc 2005, 127, 13490–13491. [DOI] [PubMed] [Google Scholar]
- (30).Bose S; Ngo AH; Do LH J. Am. Chem. Soc 2017, 139, 8792–8795. [DOI] [PubMed] [Google Scholar]
- (31).Tanaka K; Miki T; Murata K; Yamaguchi A; Kayaki Y; Kuwata S; Ikariya T; Watanabe MJ Org. Chem 2019, 84, 10962–10977. [DOI] [PubMed] [Google Scholar]
- (32).Lei Q; Wei Y; Talwar D; Wang C; Xue D; Xiao J Chem. Eur. J 2013, 19, 4021–4029. [DOI] [PubMed] [Google Scholar]
- (33).Youinou M-T; Ziessel RJ Organomet. Chem 1989, 363, 197–208. [Google Scholar]
- (34).Kang JW; Moseley K; Maitlis PM J. Am. Chem. Soc 1969, 91, 5970–5977. [Google Scholar]
- (35).Rafailidis PI; Ioannidou EN; Falagas ME Drugs 2007, 67, 1829–1849. [DOI] [PubMed] [Google Scholar]
- (36).Todd PA; Benfield P Drugs 1990, 39, 264–307. [DOI] [PubMed] [Google Scholar]
- (37).Ogasawara Y; Ooya K; Fujimori M; Noike M; Dairi TJ Antibiot 2016, 69, 119–120. [DOI] [PubMed] [Google Scholar]
- (38).Simon M-O; Li C-J Chem. Soc. Rev 2012, 41, 1415–1427. [DOI] [PubMed] [Google Scholar]
- (39).Wei Y; Wang C; Jiang X; Xue D; Li J; Xiao J Chem. Commun 2013, 49, 5408–5410. [DOI] [PubMed] [Google Scholar]
- (40).Wang S; Huang H; Bruneau C; Fischmeister C Catal. Sci. Technol 2019, 9, 4077–4082. [Google Scholar]
- (41).Gülcemal D; Gülcemal S; Robertson CM; Xiao J Organometallics 2015, 34, 4394–4400. [Google Scholar]
- (42).Özbozkurt İK; Gülcemal D; Günnaz S; Gökçe AG; Çetinkaya B; Gülcemal S ChemCatChem 2018, 10, 3593–3604. [Google Scholar]
- (43).Conesa JJ; Carrasco AC; Rodríguez-Fanjul V; Yang Y; Carrascosa JL; Cloetens P; Pereiro E; Pizarro AM Angew. Chem. Int. Ed 2019, 59, 1270–1278. [DOI] [PubMed] [Google Scholar]
- (44).Soussan E; Cassel S; Blanzat M; Rico-Lattes I Angew. Chem. Int. Ed 2009, 48, 274–288. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.