

Abstract
Background
Sickle cell disease is one of the commonest severe monogenic disorders in the world, due to the inheritance of two abnormal haemoglobin (beta globin) genes. Sickle cell disease can cause severe pain, significant end‐organ damage, pulmonary complications, and premature death. Stroke affects around 10% of children with sickle cell anaemia (HbSS). Chronic blood transfusions may reduce the risk of vaso‐occlusion and stroke by diluting the proportion of sickled cells in the circulation.
This is an update of a Cochrane Review first published in 2002, and last updated in 2017.
Objectives
To assess risks and benefits of chronic blood transfusion regimens in people with sickle cell disease for primary and secondary stroke prevention (excluding silent cerebral infarcts).
Search methods
We searched for relevant trials in the Cochrane Library, MEDLINE (from 1946), Embase (from 1974), the Transfusion Evidence Library (from 1980), and ongoing trial databases; all searches current to 8 October 2019.
We searched the Cochrane Cystic Fibrosis and Genetic Disorders Group Haemoglobinopathies Trials Register: 19 September 2019.
Selection criteria
Randomised controlled trials comparing red blood cell transfusions as prophylaxis for stroke in people with sickle cell disease to alternative or standard treatment. There were no restrictions by outcomes examined, language or publication status.
Data collection and analysis
Two authors independently assessed trial eligibility and the risk of bias and extracted data.
Main results
We included five trials (660 participants) published between 1998 and 2016. Four of these trials were terminated early. The vast majority of participants had the haemoglobin (Hb)SS form of sickle cell disease.
Three trials compared regular red cell transfusions to standard care in primary prevention of stroke: two in children with no previous long‐term transfusions; and one in children and adolescents on long‐term transfusion.
Two trials compared the drug hydroxyurea (hydroxycarbamide) and phlebotomy to long‐term transfusions and iron chelation therapy: one in primary prevention (children); and one in secondary prevention (children and adolescents).
The quality of the evidence was very low to moderate across different outcomes according to GRADE methodology. This was due to the trials being at a high risk of bias due to lack of blinding, indirectness and imprecise outcome estimates.
Red cell transfusions versus standard care
Children with no previous long‐term transfusions
Long‐term transfusions probably reduce the incidence of clinical stroke in children with a higher risk of stroke (abnormal transcranial doppler velocities or previous history of silent cerebral infarct), risk ratio 0.12 (95% confidence interval 0.03 to 0.49) (two trials, 326 participants), moderate quality evidence.
Long‐term transfusions may: reduce the incidence of other sickle cell disease‐related complications (acute chest syndrome, risk ratio 0.24 (95% confidence interval 0.12 to 0.48)) (two trials, 326 participants); increase quality of life (difference estimate ‐0.54, 95% confidence interval ‐0.92 to ‐0.17) (one trial, 166 participants); but make little or no difference to IQ scores (least square mean: 1.7, standard error 95% confidence interval ‐1.1 to 4.4) (one trial, 166 participants), low quality evidence.
We are very uncertain whether long‐term transfusions: reduce the risk of transient ischaemic attacks, Peto odds ratio 0.13 (95% confidence interval 0.01 to 2.11) (two trials, 323 participants); have any effect on all‐cause mortality, no deaths reported (two trials, 326 participants); or increase the risk of alloimmunisation, risk ratio 3.16 (95% confidence interval 0.18 to 57.17) (one trial, 121 participants), very low quality evidence.
Children and adolescents with previous long‐term transfusions (one trial, 79 participants)
We are very uncertain whether continuing long‐term transfusions reduces the incidence of: stroke, risk ratio 0.22 (95% confidence interval 0.01 to 4.35); or all‐cause mortality, Peto odds ratio 8.00 (95% confidence interval 0.16 to 404.12), very low quality evidence.
Several review outcomes were only reported in one trial arm (sickle cell disease‐related complications, alloimmunisation, transient ischaemic attacks).
The trial did not report neurological impairment, or quality of life.
Hydroxyurea and phlebotomy versus red cell transfusions and chelation
Neither trial reported on neurological impairment, alloimmunisation, or quality of life.
Primary prevention, children (one trial, 121 participants)
Switching to hydroxyurea and phlebotomy may have little or no effect on liver iron concentrations, mean difference ‐1.80 mg Fe/g dry‐weight liver (95% confidence interval ‐5.16 to 1.56), low quality evidence.
We are very uncertain whether switching to hydroxyurea and phlebotomy has any effect on: risk of stroke (no strokes); all‐cause mortality (no deaths); transient ischaemic attacks, risk ratio 1.02 (95% confidence interval 0.21 to 4.84); or other sickle cell disease‐related complications (acute chest syndrome, risk ratio 2.03 (95% confidence interval 0.39 to 10.69)), very low quality evidence.
Secondary prevention, children and adolescents (one trial, 133 participants)
Switching to hydroxyurea and phlebotomy may: increase the risk of sickle cell disease‐related serious adverse events, risk ratio 3.10 (95% confidence interval 1.42 to 6.75); but have little or no effect on median liver iron concentrations (hydroxyurea, 17.3 mg Fe/g dry‐weight liver (interquartile range 10.0 to 30.6)); transfusion 17.3 mg Fe/g dry‐weight liver (interquartile range 8.8 to 30.7), low quality evidence.
We are very uncertain whether switching to hydroxyurea and phlebotomy: increases the risk of stroke, risk ratio 14.78 (95% confidence interval 0.86 to 253.66); or has any effect on all‐cause mortality, Peto odds ratio 0.98 (95% confidence interval 0.06 to 15.92); or transient ischaemic attacks, risk ratio 0.66 (95% confidence interval 0.25 to 1.74), very low quality evidence.
Authors' conclusions
There is no evidence for managing adults, or children who do not have HbSS sickle cell disease.
In children who are at higher risk of stroke and have not had previous long‐term transfusions, there is moderate quality evidence that long‐term red cell transfusions reduce the risk of stroke, and low quality evidence they also reduce the risk of other sickle cell disease‐related complications.
In primary and secondary prevention of stroke there is low quality evidence that switching to hydroxyurea with phlebotomy has little or no effect on the liver iron concentration.
In secondary prevention of stroke there is low‐quality evidence that switching to hydroxyurea with phlebotomy increases the risk of sickle cell disease‐related events.
All other evidence in this review is of very low quality.
Keywords: Adolescent; Child; Child, Preschool; Humans; Young Adult; Anemia, Sickle Cell; Anemia, Sickle Cell/blood; Anemia, Sickle Cell/complications; Antisickling Agents; Antisickling Agents/adverse effects; Antisickling Agents/therapeutic use; Blood Transfusion; Early Termination of Clinical Trials; Erythrocyte Transfusion; Erythrocyte Transfusion/adverse effects; Hemoglobin, Sickle; Hydroxyurea; Hydroxyurea/adverse effects; Hydroxyurea/therapeutic use; Iron Chelating Agents; Iron Chelating Agents/therapeutic use; Phlebotomy; Phlebotomy/adverse effects; Primary Prevention; Secondary Prevention; Stroke; Stroke/etiology; Stroke/prevention & control
Plain language summary
Long‐term blood transfusions to prevent a stroke in people with sickle cell disease
Review question
We wanted to determine if long‐term blood transfusions given to people with sickle cell disease who are at a higher risk of stroke (primary prevention) or have had a previous stroke (secondary prevention) decreases their risk of a subsequent stroke without causing severe side effects. We compared long‐term blood transfusions to standard treatment or other ways of preventing a stroke. This is an update of a previously published Cochrane Review.
Interventions for silent stroke are addressed in a separate Cochrane Review.
Background
Sickle cell disease is a serious inherited blood disorder where the red blood cells, which carry oxygen around the body, develop abnormally.
Normal red blood cells are flexible and disc‐shaped, but in sickle cell disease they can become rigid, crescent shaped and also stickier. This can lead to blockage of blood vessels, resulting in tissue and organ damage and episodes of severe pain. The abnormal blood cells are more fragile and break apart, which leads to a decreased number of red blood cells, known as anaemia.
Sickled red blood cells can block flow in blood vessels in the brain, leading to strokes.
Strokes occur in up to 10% of children with sickle cell anaemia (HbSS) and can cause limb weakness, slurred speech, seizures, and cognitive impairment.
Two tests have been used in trials to identify children at higher risk of having a first stroke. One (transcranial Doppler ultrasonography) measures the speed of blood flowing through arteries in the brain, and those children with high blood flow are at increased risk of a stroke. The other (magnetic resonance imaging) takes images of the brain to see if there are any small areas of damage (silent strokes), those children with evidence of damage are at increased risk of stroke.
Blood transfusions may help prevent a stroke by reducing the level of anaemia, diluting the sickled red blood cells, and increasing the level of oxygen in the blood.
Blood transfusions can be linked to adverse events, e.g. the development of antibodies to proteins on donor red blood cells (alloimmunisation), accumulation of too much iron in the body from repeated transfusions, increased risk of infection, and extended length of stay in hospital.
Search date
The evidence is current to: 8 October 2019.
Study characteristics
We found five randomised controlled trials which enrolled a total of 660 participants. Three trials compared blood transfusions to no blood transfusions and two trials compared blood transfusion to the drug hydroxyurea. Trials were published between 1998 and 2016 and included children and sometimes adolescents; the majority had one form of sickle cell disease (HbSS).
All trials received government funding.
Key Results
In children who are at a higher risk of having a stroke who have not had previous blood transfusions, a long‐term blood transfusion regime probably reduces clinical strokes, and may also reduce other sickle cell disease‐related complications.
We are very uncertain if stopping blood transfusions in children and adolescents receiving transfusions for a long time (greater than 12 months) increases the risk of stroke.
We are very uncertain whether switching from long‐term transfusions with iron chelation to hydroxyurea with phlebotomy has any effect on stroke, mortality, or sickle cell disease‐related complications in children who have not had a stroke. Hydroxyurea may have little or no effect on iron levels in the liver.
We are very uncertain if switching from long‐term transfusions with iron chelation to hydroxyurea with phlebotomy increases the risk of stroke or mortality in children and adolescents who have had a stroke and were previously receiving regular transfusions. Switching from long‐term transfusions to hydroxyurea may increase some sickle cell disease‐related serious adverse events such as painful crises.
Quality of the evidence
In children at higher risk of stroke who have not had previous long‐term transfusions, there is moderate quality evidence that long‐term red cell transfusions reduce the risk of stroke. The quality of evidence was rated as low to very low for the rest of the outcomes due to trials being at high risk of bias and because there were a small number of trials and a small number of participants included in the trials.
Summary of findings
Summary of findings 1. Long‐term red cell transfusion versus no transfusion in people who are at risk of a primary stroke who have not had previous long‐term red cell transfusions.
| Primary prevention | ||||||
| Patient or population: individuals with sickle cell disease who are at risk of a primary stroke who have not had previous long‐term red cell transfusions Setting: outpatients Intervention: long‐term red cell transfusion Comparison: standard care | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (trials) | Quality of the evidence (GRADE) | Comments | |
| Risk with standard care | Risk with Blood transfusion | |||||
| Clinical stroke follow‐up: mean 24 months | Trial population | RR 0.12 (0.03 to 0.49) | 326 (2 RCTs) | ⊕⊕⊕⊝ Moderate 3 | ||
| 110 per 1000 | 13 per 1000 (3 to 54) |
|||||
| All‐cause mortality | No deaths occurred in either trial arm | ‐ | 326 (2 RCTs) | ⊕⊝⊝⊝ Very low 1 2 3 | ||
| Adverse events associated with transfusion assessed with: alloimmunisation | Moderatea | RR 3.16 (0.18 to 57.17) | 121 (1 RCT) | ⊕⊝⊝⊝ Very low 2 3 4 | ||
| 10 per 1000 | 32 per 1000 (2 to 572) |
|||||
| TIA | Trial population | Peto OR 0.13 (0.01 to 2.11) |
323 (2 RCTs) |
⊕⊝⊝⊝ Very low 3 4 | ||
| 21 per 1000 | 5 per 1000 (0 to 43) |
|||||
| Serious adverse events as a result of sickle cell‐related complications assessed with: ACS | Trial population | RR 0.24 (0.12 to 0.48) | 326 (2 RCTs) | ⊕⊕⊝⊝ Low 2 3 | ||
| 232 per 1,000 | 56 per 1000 (28 to 111) |
|||||
| Moderate | ||||||
| 230 per 1000 | 55 per 1000 (28 to 110) |
|||||
| Measures of neurological impairment assessed with: WASI IQ score | Least square mean 1.7 (SE 95% CI ‐1.1 to 4.4) |
‐ | 166 (1 RCT) |
⊕⊕⊝⊝ Low 2 3 | Author reported data from SIT 2014 | |
| Quality of life assessed with: Child Health Questionnaire Parent Form 50 |
Difference estimate ‐0.54 (‐0.92 to ‐0.17) | ‐ | 196 (1 RCT) |
⊕⊕⊝⊝ Low 2 3 | Author reported data from SIT 2014 | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). Abbreviations: ACS: acute chest syndrome; CI: confidence interval; RCT: randomised controlled trial; RR: risk ratio; OR: odds ratio; TIA: transient ischaemic attack. | ||||||
| GRADE Working Group grades of evidence High quality: we are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: we are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: we have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 We downgraded the quality of evidence by 1 due to imprecision. Rare event. No deaths occurred.
2 We downgraded the quality of the evidence by 1 due to risk of bias. Unblinded trial and cross‐overs, and imbalance between loss to follow‐up between trial arms
3 We downgraded the quality of the evidence by 1 due to indirectness. Only children with HbSS or HbSβº thalassaemia included in trials
4 We downgraded the quality of evidence by 2 due to imprecision. The estimate has very wide CIs
a Based on Chou 2013
Summary of findings 2. Long‐term red cell transfusion versus no transfusion in people who are at risk of a primary stroke who have had previous long‐term red cell transfusions.
| Primary prevention | ||||||
| Patient or population: individuals with sickle cell disease who are at risk of a primary stroke who have had long‐term red cell transfusions to prevent a stroke Setting: outpatients Intervention: long‐term red cell transfusion Comparison: halted transfusion | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (trials) | Quality of the evidence (GRADE) | Comments | |
| Risk with standard care | Risk with blood transfusion | |||||
| Clinical stroke follow‐up: mean 24 months | Trial population | RR 0.22 (0.01 to 4.35) |
79 (1 RCT) | ⊕⊝⊝⊝ Very low 1 2 3 | ||
| 49 per 1000 | 11 per 1000 (0 to 212) |
|||||
| All‐cause mortality | Moderatea | Peto OR 8.00 (0.16 to 404.12) | 79 (1 RCT) | ⊕⊝⊝⊝ Very low 1 2 3 | ||
| 10 per 1000 | 75 per 1000 (2 to 803) |
|||||
| Adverse events associated with transfusion assessed with: alloimmunisation | See comment | 79 (1 RCT) |
‐ | No comparative numbers reported | ||
| TIA | See comment | 79 (1 RCT) |
‐ | No comparative numbers reported | ||
| Serious adverse events as a result of sickle cell‐related complications assessed with: ACS | See comment | 79 (1 RCT) |
‐ | No comparative numbers reported | ||
| Measures of neurological impairment ‐ not reported | Outcome not reported | ‐ | ‐ | ‐ | ||
| Quality of life ‐ not reported | Outcome not reported | ‐ | ‐ | ‐ | ‐ | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). Abbreviations: ACS: acute chest syndrome; CI: confidence interval; RR: risk ratio; OR: odds ratio; TIA: transient ischaemic attack | ||||||
| GRADE Working Group grades of evidence High quality: we are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: we are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: we have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 We did not downgrade the evidence due to risk of bias because the evidence was already very low grade evidence. There was attrition bias. Imbalance between loss to follow‐up between trial arms
2 We downgraded the quality of the evidence by 1 due to indirectness. Only children with HbSS or HbSβº thalassaemia included in trials
3 We downgraded the quality of evidence by 2 due to imprecision. The estimate has very wide CIs
a Assuming a mortality rate of 1%
Summary of findings 3. Long‐term red cell transfusion versus hydroxyurea and phlebotomy in people who are at risk of a primary stroke who have had previous long‐term red cell transfusions.
| Primary prevention | ||||||
| Patient or population: individuals with sickle cell disease who are at risk of a primary stroke who have had long‐term red cell transfusions to prevent a stroke Setting: outpatient Intervention: blood transfusion with iron chelation Comparison: hydroxyurea with phlebotomy | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (trials) | Quality of the evidence (GRADE) | Comments | |
| Risk with hydroxyurea and phlebotomy | Risk with Blood transfusion | |||||
| Clinical stroke | No strokes occurred in either trial arm | ‐ | 121 (1 RCT) |
⊕⊝⊝⊝ Very low 1 2 | ||
| All‐cause mortality | No deaths occurred in either trial arm | ‐ | 121 (1 RCT) |
⊕⊝⊝⊝ Very low 1 2 | ||
| Adverse events associated with transfusion assessed with: Liver iron concentration mg Fe/g dry weight liver |
The mean liver iron concentration was 9.5 mg Fe/g dry weight |
MD 1.8 mg Fe/g dry weight lower (5.16 lower to 1.56 higher) |
‐ | 121 (1 RCT) |
⊕⊕⊝⊝ Low2 3 | Switching to hydroxyurea and phlebotomy may reduce serum ferritin levels compared to continuing to receive red cell transfusions and chelation (MD) ‐1398 μg/L, 95% CI ‐1929 to ‐867; one trial, 121 participants) |
| Incidence of TIA | 49 per 1000 |
50 per 1,000 (10 to 238) |
RR 1.02 (0.21 to 4.84) | 121 (1 RCT) |
⊕⊝⊝⊝ Very low 2 3 4 | |
| Serious adverse events as a result of sickle cell‐related complications assessed with: ACS | Trial population | RR 2.03 (0.39 to 10.69) | 121 (1 RCT) |
⊕⊝⊝⊝ Very low 2 3 4 | ||
| 33 per 1000 |
67 per 1,000 (13 to 350) |
|||||
| Measures of neurological impairment ‐ not reported | Outcome not reported | ‐ | ‐ | ‐ | ‐ | |
| Quality of life ‐ not reported | Outcome not reported | ‐ | ‐ | ‐ | ‐ | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). Abbreviations: ACS: acute chest syndrome; CI: confidence interval; RR: risk ratio; OR: odds ratio; TIA: transient ischaemic attack. | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 We downgraded the quality of the evidence by 2 due to imprecision. Rare event. No deaths or stroke occurred.
2 We downgraded the quality of the evidence by 1 due to indirectness. Only children with HbSS or HbSβº thalassaemia included in trials
3 We downgraded the quality of the evidence by 1 due to risk of bias.Trial was not blinded and stopped early
4 We downgraded the quality of the evidence by 1 due to imprecision. The estimate has very wide CIs
Summary of findings 4. Long‐term red cell transfusion versus hydroxyurea and phlebotomy in people who are at risk of a secondary stroke who have had previous long‐term red cell transfusions.
| Secondary prevention | ||||||
| Patient or population: individuals with sickle cell disease who have had a stroke who have had long‐term red cell transfusions to prevent another stroke Setting: outpatients Intervention: blood transfusion with iron chelation Comparison: hydroxyurea with phlebotomy | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (trials) | Quality of the evidence (GRADE) | Comments | |
| Risk with hydroxyurea and phlebotomy | Risk with Blood transfusion | |||||
| Clinical stroke assessed with: no previous red cell transfusion follow‐up: mean 24 months | Trial population | RR 14.78 (0.86 to 253.66) | 133 (1 RCT) |
⊕⊝⊝⊝ Very low 1 2 3 | ||
| 0 per 1000 |
0 per 1000 (0 to 0) |
|||||
| All‐cause mortality | 15 per 1000 |
15 per 1000 (1 to 198) |
Peto OR 0.98 (0.06 to 15.92) |
133 (1 RCT) |
⊕⊝⊝⊝ Very low 1 2 3 | |
| Transfusion‐related adverse events ‐ assessed with liver iron concentration mg Fe/g dry weight liver |
Hydroxyurea arm: median 17.2 mg IQR 10.0 to 30.6 Transfusion arm: median 17.3 mg IQR 8.8 to 30.7 |
56 (1 RCT) |
⊕⊕⊝⊝ Low 1 2 | P = 0.7920a Switching to hydroxyurea and phlebotomy may reduce serum ferritin levels compared to continuing to receive red cell transfusions and chelation 1994 μg/L, interquartile range (IQR) 998 to 3475, in the hydroxyurea arm and 4064 μg/L, IQR 2330 to 7126, in the transfusion arm; one trial, 133 participants; P < 0.001 a |
||
| Incidence of TIA | Trial population | RR 0.66 (0.25 to 1.74) | 133 (1 RCT) |
⊕⊝⊝⊝ Very low 1 2 3 | ||
| 136 per 1000 |
90 per 1000 (34 to 237) |
|||||
| Serious adverse events as a result of sickle cell‐related complications assessed with: ACS | Trial population |
RR 0.33 (0.04 to 3.08) |
133 (1 RCT) |
⊕⊝⊝⊝ Very low1 2 3 | ||
| 45 per 1000 |
15 per 1000 (2 to 140) |
|||||
| Measures of neurological impairment ‐ not reported | Outcome not reported | ‐ | ‐ | ‐ | ||
| Quality of life ‐ not reported | Outcome not reported | ‐ | ‐ | ‐ | ||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). Abbreviations: ACS: acute chest syndrome; CI: confidence interval; RR: risk ratio; OR: odds ratio; TIA: transient ischaemic attack | ||||||
| GRADE Working Group grades of evidence High quality: we are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: we are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: we have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 We downgraded the quality of the evidence by 1 due to risk of bias. Trial was not blinded and stopped early
2 We downgraded the quality of the evidence by 1 due to indirectness. Only children with HbSS or HbSβº thalassaemia included in trials
3 We downgraded the quality of the evidence by 1 due to imprecision. The estimate has very wide CIs
a Analysis performed by the trial authors
Background
Description of the condition
Sickle cell disease (SCD) is a genetic haemoglobin disorder which can cause severe pain crises and dysfunction of virtually every organ system in the body, ultimately causing premature death. Populations originating from sub‐Saharan Africa, South and Central America, the Caribbean, the Middle East, India and parts of the Mediterranean are predominantly affected. Reductions in infant and child mortality and increasing migration from highly affected countries have made this a worldwide problem (Piel 2012). Over 12,500 people in the UK and 100,000 in the USA suffer from the disease (NICE 2010; Pleasants 2014). A recent study estimated that approximately 305,800 babies were born with SCD in 2010, of which two thirds were born in Africa, and this could increase to approximately 404,200 by 2050 (Piel 2012). In high‐income countries, people with SCD are expected to live into their 40's, 50's and beyond, whereas in low‐income countries including some African nations it is estimated that between 50% to 90% of children born with HbSS die before their fifth birthday (Gravitz 2014; Grosse 2011).
The term 'sickle cell disease' refers to all genotypes that cause the clinical syndrome. There are three main types of SCD. Sickle cell anaemia is the most common form of the disease (up to 70% of cases of SCD in people of African origin) and is due to the inheritance of two beta globin S (S) alleles (haemoglobin (Hb)SS). The second most common genotype (up to 30% of cases in people of African origin) is haemoglobin SC disease (HbSC disease) it is due to the co‐inheritance of the HbS and HbC alleles and tends to be a more moderate form of the disease (Nagel 2003).The third major type of SCD occurs when HbS is inherited with a β‐thalassaemia allele, causing HbS/β‐thalassaemia (Rees 2010). People who have inherited a β‐thalassaemia null mutation along with HbS (HbSβº) have a disease that is clinically indistinguishable from sickle cell anaemia, whereas people with HbSβ⁺ thalassaemia have a milder disorder.
In SCD, under certain conditions in the absence of oxygen, the haemoglobin molecules within the red blood cells can associate as polymers, making the cells rigid and distorted into a variety of shapes, some resembling a sickle. The red blood cells have a shortened life span, resulting in anaemia. They also demonstrate increased adherence to endothelial cells lining the blood vessels, contributing to vaso‐occlusion. In later life, chronic damage to poorly perfused organs becomes apparent (Steinberg 1999). Individual heterogeneity among persons with sickle cell disease make the symptoms highly variable in frequency and severity, but the most common clinical manifestation is the acute sickle pain crisis which occurs when small vessels are blocked, depriving the tissues of oxygen and causing ischaemic damage and pain. Vaso‐occlusion can also occur in some large vessels, such as those in the brain, causing or contributing to stroke.
Stroke, usually ischaemic, occurs in up to 10% of children with sickle cell anaemia (HbSS) (Cohen 1996) and can cause weakness in the limbs, slurring of speech, seizures, coma and cognitive impairment. Recurrent (secondary) strokes occur in a half to two thirds of untreated individuals and are associated with increasing morbidity and mortality (Cohen 1996). 'Silent' cerebral infarctions (SCIs) often go unnoticed but can also cause significant neurological damage and cognitive disability and are present in a further 17% to 27% of children with sickle cell anaemia (Kinney 1999; Kwiatkowski 2009). Transcranial Doppler (TCD) velocities (tests that measure the speed of blood flow through the brain's blood vessels (either the internal carotid artery or the middle cerebral artery) by ultrasound) are used to identify children at high risk of stroke. TCD velocities are classed as normal (less than 170 cm per second); conditional (170 to 199 cm per second); or abnormal (at least 200 cm per second) in (Adams 1998b).
Description of the intervention
The focus in the past has largely been on secondary prevention with long‐term transfusion, as risk factors for first stroke were not well established. However, with the technological breakthrough of the use of TCD cerebral blood flow velocity measurement, screening has become feasible and is currently the standard of care. Abnormally high blood flow in one or more major arteries is associated with vascular narrowing and predicts an increased risk of stroke, allowing preventative treatment (i.e. long‐term red cell transfusion programme) prior to the first stroke (Adams 1998a). The fetal haemoglobin (HbF) stimulating drug hydroxyurea has been substituted successfully for long‐term red cell transfusion for the prevention of secondary stroke in a limited number of cases (Ware 2010). Serial phlebotomy may be highly effective in the reduction of iron overload if transfusions are no longer necessary (Ware 2004).
As well as the direct and indirect costs, long‐term red cell transfusions can have adverse side effects. Iron overload is a problem and requires daily oral iron chelation with deferasirox or deferiprone (or daily subcutaneous or intravenous infusions with desferrioxamine) to avoid the toxic effects of excess iron (Inati 2011). However, compliance with the chelation programmes is often poor, and therefore problems of iron overload are potentially serious. Alloimmunisation occurs when the individual develops antibodies to the foreign red cells (Smith‐Whitley 2012), which is a major problem for future transfusion. Blood products can be contaminated with infective agents such as hepatitis C and HIV, and while this now occurs only rarely in developed countries, the risk is much higher in the developing countries where sickle cell disease is most prevalent. Other problems with transfusions include hyperviscosity of the blood due to over‐transfusion, and haemolytic transfusion reactions, both potentially serious side effects. The regimen is often complex and time‐consuming, requiring monthly transfusions to maintain the HbS at approximately 20% to 30%. In short, blood transfusion is a lengthy and costly process which is not without risks, and these must be balanced against the possible benefits prior to embarking on a long‐term regimen.
How the intervention might work
Red cell transfusions are undertaken in many people with SCD to dilute the circulating sickle cells, thus reducing the risk of vaso‐occlusive episodes and anaemia (Serjeant 1992) and increasing tissue oxygen delivery. Transfusions can be given acutely, in emergency treatment of complications such as acute splenic sequestration, aplastic crisis, and acute chest syndrome (ACS), and are also frequently used in preparation for surgery. In addition, many people with SCD receive chronic transfusion regimens in an attempt to prevent severe vaso‐occlusion and stroke (Smith‐Whitley 2012).
The mechanisms for the reduction in stroke risk from long‐term red cell transfusion are not known (DeBaun 2006). However, a reduction in cells containing high amounts of HbS or an increase in Hb level could have beneficial effects on cerebral blood vessels or interactions between red blood cells and endothelial cells (Adams 1998b). Transfusion does have an immediate haemodynamic effect measured by reduction of middle cerebral artery velocity (Venkatesubramanian 1994).
Hydroxyurea is currently the only approved therapeutic drug for the treatment of sickle cell anaemia (for adults with severe vaso‐occlusive episodes of pain or acute chest syndrome) and its use has become widespread in both children and adults with this condition. In preliminary studies it was substituted successfully for long‐term transfusion in the prevention of secondary strokes, leading to its consideration for use in the phase III SWiTCH trial (SWiTCH 2012).
For the past three decades the standard treatment for iron overload related to long‐term red cell transfusion has been the use of iron chelating agents, including desferrioxamine, deferiprone and deferasirox. Although serial phlebotomy has long been utilized for conditions such as polycythaemia, it has recently been found to be highly effective in the reduction of iron overload from chronic red blood cell transfusion in people who are no longer requiring that treatment (Ware 2004). Pilot information on the combination use of hydroxyurea and phlebotomy led to the development of the SWiTCH trial (SWiTCH 2012).
Why it is important to do this review
In the USA the National Institutes of Health (NIH) guidelines recommend a long‐term red cell transfusion programme for prevention of stroke in people with SCD who have had a prior stroke, or who have an abnormal TCD reading (blood flow velocity equal to or greater than 200 cm per second in the internal carotid artery or the middle cerebral artery). While many studies support the efficacy of this treatment (Adams 1998a; Bernaudin 2011), the optimum regimen and duration of treatment for primary stroke prophylaxis have not been widely agreed upon. This review aims to assess the relative risks and benefits of red cell transfusion regimens for preventing primary strokes in people with SCD, with or without SCIs. Furthermore, older data indicated the need for indefinite continuation of long‐term red cell transfusion and iron removal in the prevention of secondary strokes and its treatment consequences (Wang 1991). The potential substitution of an oral drug (hydroxyurea) for long‐term red cell transfusion and of periodic phlebotomy for oral or subcutaneous iron chelation offered less demanding and potentially less expensive secondary stroke preventative management.
The publication is an update of a Cochrane Review first published in 2002 and most recently updated in 2017 (Hirst 2002; Wang 2013).
Objectives
In this review we aimed to determine whether long‐term red cell transfusion regimens in people with SCD:
reduce occurrence of stroke (primary prevention);
reduce recurrence of stroke (secondary prevention);
reduce mortality;
reduce other complications of SCD including pain crises, ACS and splenic sequestration;
are associated with unacceptable adverse events or costs.
This version of the review does not address the question of whether the risk of silent cerebral infarction is affected by blood transfusion because this is the subject of a separate Cochrane Review (Estcourt 2016).
Methods
Criteria for considering studies for this review
Types of studies
We included randomised controlled trials (RCTs) with no limits on language or publication status.
Types of participants
People with SCD (HbSS, SC, Sβ⁺ and Sβ⁰ proven by electrophoresis, with family studies or DNA tests as appropriate) of all ages and both sexes, whether or not they have a history of prior stroke or transient ischaemic attack (TIA).
Types of interventions
Long‐term red cell transfusion regimens compared to other transfusion regimens, no treatment or the use of hydroxyurea to reduce the incidence of stroke in people with SCD.
Types of outcome measures
We grouped outcome data, where appropriate, into those measured prior to transfusion regimen, one month, one year, five years and more than five years after initiation of transfusion, and one year, five years and more than five years after stopping transfusion.
Primary outcomes
Incidence of clinical diagnosis of any type of stroke (by clinical symptoms and signs, magnetic resonance imaging (MRI) scan, computed tomography imaging (CT) scan or autopsy)
Deaths from any cause in each treatment group
Transfusion‐related complications (e.g. alloimmunisation, infection from blood product, procedural complications, transfusion reactions, reduced immunocompetency, iron overload (measured by serum ferritin, liver iron or quantitative MRI))
Secondary outcomes
Incidence of TIA
Measures of neurological impairment, and measures of neuropsychiatric performance (e.g. full scale intelligence quotient (FSIQ))
Incidence of other sickle cell complications (e.g. pain crises, acute chest syndrome, splenic sequestration)
Quality of life (measured on a validated scale)
Measures of organ damage (e.g. renal function, liver function, and lung function tests)
Haemoglobin level and HbS percentage (mean, pre‐ and post‐transfusion, and at time of event)
Search methods for identification of studies
Electronic searches
We identified relevant trials from the Cochrane Cystic Fibrosis and Genetic Disorders Group’s Haemoglobinopathies Trials Register using the terms: sickle cell OR (haemoglobinopathies AND general) AND stroke AND blood transfusion.
The Haemoglobinopathies Trials Register is compiled from electronic searches of the Cochrane Central Register of Controlled Trials (Clinical Trials) (updated each new issue of the Cochrane Library) and weekly searches of MEDLINE. Unpublished work is identified by searching the abstract books of five major conferences: the European Haematology Association conference; the American Society of Hematology conference; the British Society for Haematology Annual Scientific Meeting; the Caribbean Public Health Agency Annual Scientific Meeting (formerly the Caribbean Health Research Council Meeting); and the National Sickle Cell Disease Program Annual Meeting. For full details of all searching activities for the register, please see the relevant section of the Cochrane Cystic Fibrosis and Genetic Disorders Group's website.
Date of the most recent search of the Group's Haemoglobinopathies Trials Register: 19 September 2019.
We also searched the following databases for RCTs and SRs on 8 October 2019 without language, publication year or publication status restrictions:
the Cochrane Library (CENTRAL) (Issue 10, 2019) (http://www.cochranelibrary.com/);
MEDLINE (OvidSP, Epub Ahead of Print, In‐Process & Other Non‐Indexed Citations, 1946 to 8 October 2019);
Embase (OvidSP, 1974 to 8 October 2019);
CINAHL (EBSCOHost, 1937 to 8 October 2019);
PubMed (for epublications ahead of print, in‐process & other non‐indexed citations only on 8 October 2019) (https://www.ncbi.nlm.nih.gov/pubmed);
Transfusion Evidence Library (1950 to 8 October 2019) (http://www.transfusionevidencelibrary.com/);
LILACS (1982 to 8 October 2019) (LILACS);
IndMed (1986 to 8 October 2019) (IndMed);
KoreaMed (1997 to 8 October 2019) (KoreaMed);
Web of Science (Conference Proceedings Citation Index‐ Science (CPCI‐S) ‐ 1990 to 8 October 2019).
We also searched the following trial databases for ongoing trials on 8 October 2019:
ClinicalTrials.gov (https://clinicaltrials.gov/);
WHO International Clinical Trials Registry Platform (ICTRP) (www.who.int/ictrp/en/);
The full search strategies for each database are available in Appendix 1.
Searching other resources
We augmented database searching with the following.
Handsearching of reference lists
We checked references of all included trials, relevant review articles and current treatment guidelines for further literature. These searches were limited to the ’first generation’ reference lists.
Personal contacts
We contacted authors of relevant trials for unpublished material or further information.
Data collection and analysis
Selection of studies
Two independent review authors (LE, PF) screened all electronically‐derived citations and abstracts of papers identified in the search for relevance. We excluded trials that were clearly irrelevant at this stage based on a review of the abstract. Two independent review authors (LE, PF) formally assessed the full texts of all potentially‐relevant trials for eligibility against the criteria outlined above. We resolved all disagreements by discussion without the need for a third review author. We sought further information from trial authors if the article contained insufficient data to make a decision about eligibility. We used Covidence to screen all abstracts and full‐text articles (Covidence 2015). We recorded the reasons why potentially‐relevant trials failed to meet the eligibility criteria.
Data extraction and management
Since the previous versions of this review, we have updated the data extraction and risk of bias assessment for all included trials (Hirst 2002; Wang 2013). Two review authors (LE, PF) conducted the data extraction according to Cochrane guidelines (Higgins 2011a). We resolved disagreements between the review authors by consensus. The review authors were not blinded to names of authors, institutions, journals, or the outcomes of the trials. We used Covidence (Covidence 2015) to extract data for the two new trials and to assess the risk of bias for all included trials. Two authors (LE, PF) extracted data independently for all the trials. We used the available tables in Review Manager 5 to present extracted data on trial characteristics (Review Manager 5.3).
We extracted the following data.
General information
Review author’s name, date of data extraction, study ID, first author of trial, author’s contact address (if available), citation of paper, objectives of the trial.
Trial details
Trial design, location, setting, sample size, power calculation, treatment allocation, inclusion and exclusion criteria, reasons for exclusion, comparability of groups, length of follow‐up, stratification, stopping rules described, statistical analysis, results, conclusion, and funding.
Characteristics of participants
Age, gender, total number recruited, total number randomised, total number analysed, types of underlying disease, lost to follow‐up numbers, dropouts (percentage in each arm) with reasons, protocol violations, previous treatments, current treatment, prognostic factors, haemoglobin S levels.
Interventions
Experimental and control interventions, method of red cell transfusion (top‐up, partial or full exchange transfusion).
Assessment of bias
Sequence generation, allocation concealment, blinding (participants, personnel, and outcome assessors), incomplete outcome data, selective outcome reporting, other sources of bias.
Outcomes measured
Incidence of clinical diagnosis of any type of stroke (by clinical symptoms and signs, MRI scan, computed tomography imaging (CT) scan or autopsy)
Deaths from any cause in each treatment group
Transfusion‐related complications (e.g. alloimmunisation, infection from blood product, procedural complications, transfusion reactions, reduced immunocompetency, iron overload (measured by serum ferritin, liver iron or quantitative MRI)
Incidence of TIA
Measures of neurological impairment, and measures of neuropsychiatric performance (e.g. full scale intelligence quotient (FSIQ))
Incidence of other sickle cell complications (e.g. pain crises, ACS, splenic sequestration)
Quality of life (measured on a validated scale)
Measures of organ damage (e.g. renal function, liver function, and lung function tests)
Haemoglobin level and HbS percentage (mean, pre‐ and post‐transfusion, and at time of event
We used both full‐text versions and abstracts to retrieve the data. We used one data extraction form per trial, regardless of number of publications relating to that trial. Where these sources did not provide sufficient information, we contacted authors and trial groups for additional details. One review author entered data into Review Manager 5 and a second review author checked these for accuracy Review Manager 5.3) .
Assessment of risk of bias in included studies
We updated the risk of bias assessments from those in the previous versions of this review (Hirst 2002; Wang 2013). Two review authors (LE, PF) assessed all included trials for possible risk of bias (as described in the Cochrane Handbook of Systematic Reviews of Interventions (Higgins 2011b)). The assessment included information about the design, conduct and analysis of the trial. We evaluated each criterion using the Cochrane three‐point scale (low, high, or unclear risk of bias) in the following areas.
Selection bias (random sequence generation and allocation concealment)
Performance bias (blinding of participants and personnel)
Detection bias (blinding of outcome assessment)
Attrition bias (incomplete outcome data)
Reporting bias (selective reporting)
Other bias
If disagreement arose on the assessment of quality of an included trial, we reached a consensus by discussion, without the need for a third review author.
Measures of treatment effect
For continuous outcomes we recorded the mean, standard deviation (SD) and total number of participants in both the treatment and control groups. For those continuous outcomes using the same scale, we performed analyses using the mean difference (MD) with 95% confidence intervals (CIs). There were no continuous outcomes measured using different scales (when we would have used the standardised MD).
For dichotomous outcomes we recorded the number of events and the total number of participants in both the treatment and control groups. We reported the pooled risk ratio (RR) with a 95% CI. Where the number of observed events was small (less than 5% of sample per group), and where trials have balanced treatment groups, we reported the Peto odds ratio (OR) with 95% CI (Deeks 2011).
If data allowed, we undertook quantitative assessments using Review Manager 5 (Review Manager 5.3). If we could not report the available data in any of the formats described above, we performed a narrative report, and if appropriate we presented the data in tables.
Unit of analysis issues
We did not pre‐specify in the protocol how we would deal with unit of analysis issues. We did not expect to encounter unit of analysis issues as cluster randomised trials, cross‐over trials, and multiple observations for the same outcome were not included in this review. Should we have found any trials with these designs we would have treated these in accordance with the advice given in chapter 16 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011c).
Dealing with missing data
We dealt with missing data according to the recommendations in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011c). Where information was missing or unclear, we contacted the primary investigator or where applicable the funding source. In order to allow an intention‐to‐treat (ITT) analysis, irrespective of later exclusion (regardless of cause) or loss to follow‐up, we collected data by allocated treatment groups.
Assessment of heterogeneity
We conducted a meta‐analysis and assessed the statistical heterogeneity if trials were sufficiently homogenous in their design (Deeks 2011). We assessed statistical heterogeneity of treatment effects between trials using a Chi² test with a significance level at P < 0.1 and used the I² statistic to quantify possible heterogeneity (I² value greater than 50% moderate heterogeneity, I² value greater than 75% considerable heterogeneity). If statistical heterogeneity was considerable, we did not report the overall summary statistic. We could not assess potential causes of heterogeneity by sensitivity analyses due to the lack of data (Deeks 2011).
Assessment of reporting biases
We did not perform a formal assessment of potential publication bias (small trial bias) by generating a funnel plot and statistically test using a linear regression test (Sterne 2011) as no meta‐analysis contained 10 or more trials.
Data synthesis
We performed analyses according to Cochrane recommendations (Deeks 2011). We used aggregated data for analysis. For statistical analysis, we entered data into the Review Manager software (Review Manager 5.3). Where meta‐analysis was feasible, we used the fixed‐effect model for pooling the data. We used the Mantel‐Haenszel method for dichotomous outcomes or Peto method as necessary, and the inverse variance method for continuous outcomes. There was no statistical heterogeneity, but if statistical heterogeneity was found to be above 75%, we would identify a reason for clinical heterogeneity and not perform a meta‐analysis but comment instead on the results as a narrative. Even in the absence of statistical heterogeneity, we planned to explore the robustness of any summary measures, particularly with respect to trial methodological quality, but we were unable to perform sensitivity analyses due to inadequate data.
Subgroup analysis and investigation of heterogeneity
We performed subgroup analyses on the following characteristics:
TCD velocities (normal (less than 170 cm/s), conditional (170 to 199 cm/s), abnormal (at least 200 cm/s));
presence or absence of previous SCI on MRI.
We did not perform subgroup analyses on the following characteristics, due to a lack of data:
trials comparing transfusion therapy with no treatment or other treatments separately from those comparing different transfusion regimens;
severity of the disease;
age of the participant (paediatric, adults, older adults (over 60 years)).
If, in future updates, we identify moderate heterogeneity between trials, we will examine subgroups to help explain the reasons for this.
Sensitivity analysis
We planned to use the random‐effects model for sensitivity analyses as part of the exploration of heterogeneity.
Including only those trials with a 'low risk of bias' (e.g. RCTs with methods assessed as low risk for random sequence generation and concealment of treatment allocation)
Including only those trials with less than a 20% dropout rate
We could not do sensitivity analyses due to inadequate data.
Summary of findings table
We used the GRADE approach to create a 'Summary of findings' table, as suggested in chapters 11 and 12 of the Cochrane Handbook for Systematic Reviews of Interventions (Schünemann 2011a; Schünemann 2011b). We used the GRADE approach to rate the quality of the evidence as 'high', 'moderate', 'low', or 'very low' using the five GRADE considerations as follows.
Risk of Bias: serious or very serious
Inconsistency: serious or very serious
Indirectness: serious or very serious
Imprecision: serious or very serious
Publication bias: likely or very likely
We presented summary of findings table on the following outcomes for each intervention comparison.
Incidence of clinical diagnosis of any type of stroke
Deaths due to any cause
Transfusion‐related complications
Incidence of TIA
Measures of neurological impairment,
SCD‐related complications
Quality of life (measured on a validated scale)
We have also rated the quality of evidence with summary of findings table for risk of stroke associated with SCI and TCD velocities above or below the transfusion threshold in primary and secondary prevention of stroke.
Results
Description of studies
See Characteristics of included studies and Characteristics of excluded studies.
Results of the search
See PRISMA flow diagram (Figure 1).
1.
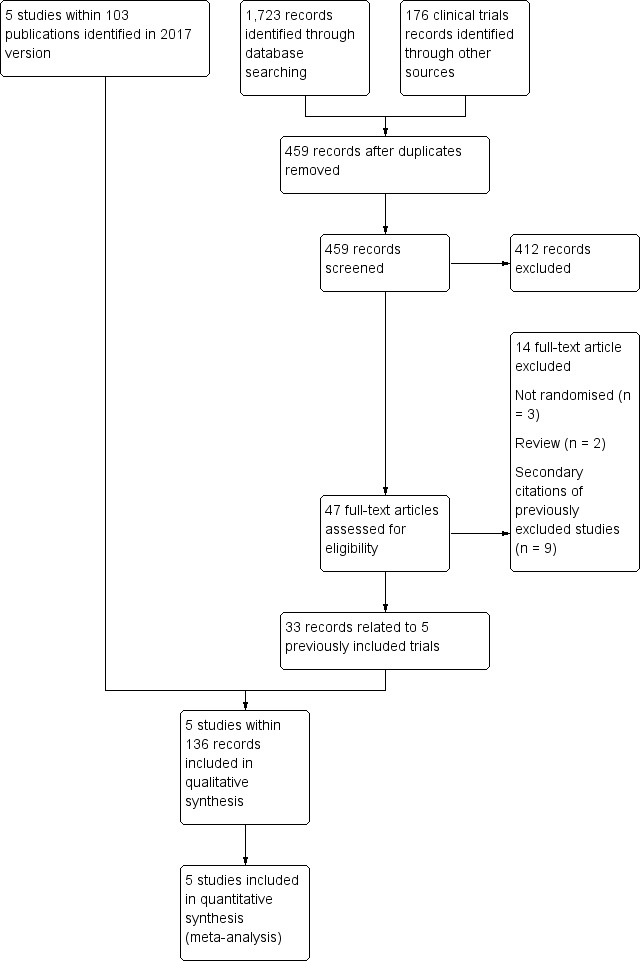
Study flow diagram.
The original review (Hirst 2002) identified two eligible trials (STOP 1998; STOP 2 2005). The first update of the review (Wang 2013) identified one additional completed trial (SWiTCH 2012) and three potentially eligible ongoing trials (SIT 2014; TWiTCH 2016; SCATE 2015).
In the previous version of this review (Estcourt 2017) we assessed 83 full text articles for relevance. We identified two completed trials within 22 publications which were classified as ongoing trials in Wang 2013 (SIT 2014,TWiTCH 2016). We identified 60 additional citations related to previously included trials (STOP 1998; STOP 2 2005, SWiTCH 2012). Therefore there were five included trials (SIT 2014; STOP 1998; STOP 2 2005;SWiTCH 2012; TWiTCH 2016), and no ongoing trials.
The updated searches (conducted 19 September 2019 and 8 October 2019) identified a total of 1899 potentially relevant records (1723 articles and 176 clinical trials). There were 459 records after duplicates were removed. Two review authors (LE, RK) excluded 412 records on the basis of the abstract, and two authors (LE, RK) reviewed 47 full text articles for relevance. We identified no new completed trials. We identified 33 citations related to previously included trials (SIT 2014; STOP 1998; STOP 2 2005, SWiTCH 2012; TWiTCH 2016). We excluded five new trials (Bernaudin 2016; EXTEND 2016; Gwam 2016; SACRED 2018; SPIN 2017). In this update we included the same five trials identified in Estcourt 2017 (SIT 2014; STOP 1998; STOP 2 2005;SWiTCH 2012; TWiTCH 2016). We found no ongoing trials.
Included studies
See Characteristics of included studies for full details of each trial.
Five trials, including 660 participants, met the predefined inclusion criteria (SIT 2014; STOP 1998; STOP 2 2005; SWiTCH 2012;TWiTCH 2016).
Trial design
All five trials were multicentre randomised trials, ranging from 12 centres (STOP 1998; STOP 2 2005) to 29 centres (SIT 2014).
Four trials were terminated early (STOP 1998; STOP 2 2005; SWiTCH 2012; TWiTCH 2016):
STOP 1998 was terminated 16 months early by the trial's data monitoring board when a 92% reduction in incidence of stroke in the transfused group was seen.
STOP 2 2005 was terminated two years early due to safety concerns.
SWiTCH 2012 was stopped early due to futility for the composite primary endpoint.
TWiTCH 2016, a non‐inferiority trial, was stopped after the first scheduled interim analysis because non‐inferiority had been demonstrated.
The SIT trial was the only trial that was not stopped before the planned end of recruitment and follow‐up (SIT 2014).
Trial size
The number of participants enrolled in the five trials ranged from 79 (STOP 2 2005) to 196 (SIT 2014). Power calculations were reported in four trials (SIT 2014; STOP 1998; STOP 2 2005; TWiTCH 2016), three of these studies were stopped early (STOP 1998; STOP 2 2005; TWiTCH 2016). The STOP 2 trial planned to recruit 100 children and the TWiTCH trial planned to recruit 148 children (STOP 2 2005; TWiTCH 2016).
Settings
The trials were published between 1998 (STOP 1998) and 2016 (TWiTCH 2016). All were multicentre trials (12 to 29 recruitment centres). One trial was conducted in the USA (SWiTCH 2012), three trials were conducted in the USA and Canada (STOP 1998; STOP 2 2005; TWiTCH 2016); and one trial was conducted in the USA, Canada, France and the UK (SIT 2014).
Participants
All trials included participants with HbSS disease and HbSβ⁰ thalassaemia. Two trials also included participants with HbS/OArab disease (SWiTCH 2012; TWiTCH 2016). Three trials did not specify the distribution of phenotypes (SIT 2014; STOP 1998; STOP 2 2005). In the SWiTCH trial, 100% of participants in the transfusion arm and 99% in the no transfusion arm had the HbSS phenotype, and in the TWiTCH trial, 97% in the transfusion arm and 100% in hydroxyurea arm had the HbSS phenotype (SWiTCH 2012; TWiTCH 2016).
All participants in the trials were children and adolescents aged from two to 20 years. Two trials included participants over 16 years (STOP 2 2005; SWiTCH 2012). Mean (SD) ages in the trials ranged from the lowest age of eight (three) years in the SIT and STOP trials (SIT 2014;STOP 1998) to a high of 13 (four) years in the SWiTCH trial (SWiTCH 2012). Participants tended to be equally divided between males and females with the highest participation of males (57%) in the SIT trial (SIT 2014) and the lowest (39%) in the TWiTCH trial (TWiTCH 2016).
All trials excluded females who were pregnant or people who had HIV. Other inclusion and exclusion criteria varied depending on the objectives of the trial. In the SWiTCH trial, individuals were included if they had an overt clinical stroke after the age 12 months (SWiTCH 2012), whereas in the other four trials individuals were excluded if they had a clinical history of stroke (SIT 2014; STOP 1998; STOP 2 2005; TWiTCH 2016).
In the SIT trial, children had to have evidence of at least one silent cerebral infarct confirmed on MRI and normal TCD velocities (SIT 2014). In the STOP, STOP 2 and the TWiTCH trials, individuals had to have abnormal TCD velocities prior to any transfusion therapy (greater than or equal to 200 cm/s) (STOP 1998; STOP 2 2005; TWiTCH 2016), and in the STOP 2 trial these abnormal TCD velocities had to have normalised with transfusion therapy (STOP 2 2005). Three trials excluded individuals with a history of seizures (SIT 2014; STOP 1998; STOP 2 2005); two trials excluded individuals with a severe vasculopathy (STOP 2 2005; TWiTCH 2016); and one trial excluded children with a previous TIA (TWiTCH 2016).
Intervention
All trials had a transfusion arm with the aim of keeping HbS to 30% or less with local discretion as to the type of red blood cell transfusion administered (simple, manual exchange or automated exchange). Three trials reported using leucocyte‐depleted red cells and blood matched for C, D, E and Kell antigen (SIT 2014; STOP 1998; STOP 2 2005), the remaining two trials did not report the type of blood component used (SWiTCH 2012; TWiTCH 2016). In the SWiTCH and TWiTCH trials, participants in the transfusion arm also received iron chelation (SWiTCH 2012; TWiTCH 2016), in the SIT and STOP 2 trials, participants received iron chelation if required (SIT 2014; STOP 2 2005) and in the STOP trial, participants did not receive iron chelation (STOP 1998).
Three trials compared long‐term transfusion therapy to standard care with no hydroxyurea (SIT 2014; STOP 1998; STOP 2 2005). In two of these trials participants had not had previous long‐term transfusions (SIT 2014; STOP 1998), and in one trial all participants had previous long‐term transfusions to prevent primary stroke (STOP 2 2005). In the STOP 2 trial, the transfusion halted arm could receive transfusions to treat SCD complications but if hydroxyurea or regular transfusions were initiated it was considered a cross‐over and data were censored at treatment initiation (STOP 2 2005).
Hydroxyurea was the comparator in the SWiTCH and TWiTCH trials, which was initiated at 20 mg/kg/day with escalation to a maximum tolerated dose (MTD) with transfusion overlap for four to nine months until MTD was reached. Once MTD was reached, phlebotomy was commenced with a target of 10 mL/kg of blood removed monthly to reduce iron burden (maximum 500 mL) (SWiTCH 2012; TWiTCH 2016)..
Outcomes
Outcomes varied across trials depending on the objectives.
The primary outcome in SIT was the recurrence of infarct or haemorrhage as determined by neuroimaging. Secondary outcomes were TIAs and changes in cognition (SIT 2014).
In both STOP trials, the primary outcomes were cerebral infarction or intracranial haemorrhage and STOP 2 also included reversion to abnormal velocity on TCD (STOP 1998; STOP 2 2005). In both trials secondary outcomes reported were death, and transfusion‐ and SCD‐related adverse events.
In the SWiTCH trial the primary outcome was a composite primary endpoint of secondary stroke recurrence rate and quantitative liver iron concentration, while non‐stroke neurological events, non‐neurological sickle cell clinical events, quality of life evaluation, and measures of organ function were all secondary outcomes (SWiTCH 2012).
In the TWiTCH trial the primary outcome was TCD time‐averaged mean velocity on the index side, defined as the cerebral hemisphere with the higher mean arterial velocity at baseline assessment (TWiTCH 2016). TCD velocity on the non‐index side, new stroke or non‐stroke neurological events, new brain MRI or MRA lesions, hepatic iron overload, sickle‐related events, neuropsychological status, quality of life, growth, and treatment‐related complications were secondary outcomes.
Funding source
All five trials received government funding.
Ongoing studies
We did not identify any ongoing studies for the update of the review.
Excluded studies
We excluded seven studies or reviews within 17 full text records:
four were not randomised trials (DREPAGREFFE 2017; EXTEND 2016; SACRED 2018; SPIN 2017).
two were reviews (Bernaudin 2016; Gwam 2016)
one trial did not include a transfusion arm (SCATE 2015)
Risk of bias in included studies
Refer to the figures section of the review for visual representations of the assessments of risk of bias across all trials and for each item in the included trials (Figure 2; Figure 3). See the risk of bias section in the Characteristics of included studies section for further information about the bias identified within the individual trials.
2.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
Random sequence generation (selection bias)
We considered four trials to be at low risk of bias, as randomisation was done centrally by the statistical data coordinating centre or was randomly generated or both (SIT 2014; STOP 1998; STOP 2 2005; TWiTCH 2016). We judged the SWiTCH trial to have an unclear risk of bias as the method of randomisation was not adequately reported (SWiTCH 2012).
Allocation concealment (selection bias)
We considered three trials to be at low risk of bias for allocation concealment as assignment was done by a central statistical data centre or allocation was statistically determined or both (SIT 2014; STOP 1998; TWiTCH 2016). We considered two trials to be at unclear risk of bias because no description of allocation concealment was provided (STOP 2 2005; SWiTCH 2012).
Blinding
Blinding of participants and personnel (performance bias)
We considered all five trials to be at high risk of performance bias as it is impractical to mask a blood transfusion intervention so all participants and personnel were unblinded.
Blinding of outcome assessment (detection bias)
We considered all five trials to be at low risk of bias for the outcome assessment of stroke or TIA as these outcomes were adjudicated by experts masked to treatment assignments.
We judged all five trials to be at high risk of bias for all other outcomes except mortality as all trials were unblinded.
Incomplete outcome data
We considered four trials to be at low risk for attrition bias as they all used an ITT analysis and all participants were accounted for in the trials (SIT 2014; STOP 1998; SWiTCH 2012; TWiTCH 2016). We judged the STOP 2 trial to be at high risk for attrition bias as it was not stated if an ITT analysis was used and 17% of participants discontinued or data were censored (STOP 2 2005).
Selective reporting
We considered one trial to be at low risk of reporting bias as a protocol was provided and all planned outcomes were reported (SIT 2014). We rated three trials as unclear risk for reporting bias (STOP 1998; STOP 2 2005; TWiTCH 2016): one trial had no protocol and no prospective trial registration (STOP 1998); one trial did not report any secondary outcomes and it was not clear if all adverse events were reported as some participant data were censored (STOP 2 2005); one trial did not report some secondary outcomes and it was unclear if these outcomes will be reported in future publications (TWiTCH 2016).
We judged one trial to be at high risk for selective reporting bias as several secondary outcomes were not reported (i.e. quality of life, growth and development, organ damage, transfusion‐related, chelation‐related and phlebotomy related complications) (SWiTCH 2012).
Other potential sources of bias
We considered all trials to be at unclear risk for other sources of bias. We judged four trials to have unclear risk due to early termination of these trials (STOP 1998; STOP 2 2005; SWiTCH 2012; TWiTCH 2016). As well, in the TWiTCH trial, children with severe vasculopathy were excluded during screening, so these children might not be suitable candidates for hydroxyurea and longer follow‐up is required to determine if findings are maintained over time (TWiTCH 2016). The SIT trial was considered to have unclear risk because there was a 20% cross‐over rate to either transfusion or hydroxyurea treatment and also because hydroxyurea was started in 17% of participants due to disease severity even though it was part of the exclusion criteria (SIT 2014). In the SWiTCH trial, more participants had moya‐moya in the hydroxyurea arm (11 participants) than the transfusion arm (five participants), it was not known if there was a difference between treatment arms in the number of participants with severe vasculopathy (SWiTCH 2012).
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4
Red cell transfusion versus standard care
Three trials evaluated this comparison (SIT 2014; STOP 1998; STOP 2 2005), all trials assessed the primary prevention of stroke.
Two trials included children who were at higher risk of a primary stroke who had not had previous long‐term red cell transfusions to prevent stroke (SIT 2014; STOP 1998). Children in the SIT trial had evidence of previous silent cerebral infarcts on MRI but normal or conditional TCD velocities (SIT 2014). Children in the STOP trial had abnormal TCD velocities and 35% of these children also had SCIs on MRI (STOP 1998).
One trial included children and adolescents who were at higher risk of a primary stroke (previous history of abnormal TCD velocities) who had previous long‐term red cell transfusions (at least 30 months) to prevent stroke (STOP 2 2005). The majority of participants in the STOP 2 trial had also participated in the earlier STOP trial, participants were included if their TCD velocities had normalised (STOP 1998; STOP 2 2005).
Throughout this review results from the two trials in which participants had not been transfused were not combined with the results from the trial when participants had received previous long‐term transfusions.
Primary outcomes
1. Incidence of clinical stroke (any type)
No previous long‐term red cell transfusion
Long‐term red cell transfusions probably reduce the incidence of clinical stroke in children with a higher risk of stroke (abnormal TCD velocities or previous history of SCIs), compared to children receiving standard care, RR 0.12 (95% CI 0.03 to 0.49) (two trials, 326 participants, moderate quality evidence) (Analysis 1.1; Table 1).
1.1. Analysis.

Comparison 1: Blood transfusion versus standard care, Outcome 1: Clinical stroke
Previous long‐term red cell transfusion
We are very uncertain whether continuing long‐term red cell transfusions reduces the incidence of clinical stroke in children and adolescents whose TCD velocities have normalised, compared to those receiving standard care, RR 0.22 (95% CI 0.01 to 4.35) (one trial, 79 participants, very low quality evidence) (Analysis 1.1;Table 2).
TCD velocity subgroup analysis
Normal TCD velocities ‐ we very uncertain whether red blood cell transfusions reduce the incidence of clinical stroke in children with normal TCD velocities compared to children receiving standard care, RR 0.14 (95% CI 0.02 to 1.12) (one trial, 196 participants, very low quality evidence) (Analysis 1.2). Quality of the evidence was very low due to imprecision of the effect estimate and indirectness (findings only apply to children with HbSS).
1.2. Analysis.

Comparison 1: Blood transfusion versus standard care, Outcome 2: Clinical stroke ‐ velocity
Normalised TCD velocities ‐ we are very uncertain whether continuing long‐term red cell transfusions reduces the incidence of stroke in children and adolescents with normalised TCD velocities compared to children and adolescents receiving standard care, RR 0.22 (95% CI 0.01 to 4.35) (one trial, 79 participants, very low quality evidence) (Analysis 1.2). Quality of the evidence was very low due to imprecision of the effect estimate; risk of bias; and indirectness (findings only apply to children with HbSS).
Abnormal TCD velocities ‐ red cell transfusions may reduce the incidence of clinical stroke in children with abnormal TCD velocities compared to children receiving standard care, RR 0.10 (95% CI 0.01 to 0.73) (one trial, 130 participants, low quality evidence) (Analysis 1.2). Quality of the evidence was low due to risk of bias (imbalance in follow‐up between treatment arms and imbalance in number of participants with alpha thalassaemia trait between treatment arms) and indirectness (findings only apply to children with HbSS).
Since the STOP trial was terminated early due to safety concerns, the groups had different periods of follow‐up, a total of 1229 months in the standard care group, and 1321 in the transfusion group (STOP 1998). The trial authors reported that the rate of stroke per person year of follow‐up was 0.107 in the standard care group, and 0.009 in the transfusion group. Although in the transfusion group the target HbS percentage of 30% was occasionally not met, none of these participants had a stroke.
There was no evidence of subgroup differences (test for subgroup differences: Chi² = 0.20, df = 2 (P = 0.91), I² = 0%).
Presence or absence of SCI on MRI subgroup analysis
Previous lesions (SCIs) on MRI: long‐term red cell transfusions may reduce the incidence of clinical stroke in children with previous SCIs on MRI compared to children receiving standard care, RR 0.11 (95% CI 0.02 to 0.59) (two trials, 243 participants, low quality evidence) (Analysis 1.3). Quality of the evidence was low due to risk of bias (this analysis includes data from a cohort subset who had an MRI in the STOP trial (STOP 1998)) and indirectness (findings only apply to children with HbSS).
1.3. Analysis.

Comparison 1: Blood transfusion versus standard care, Outcome 3: Clinical stroke ‐ SCI
No previous lesions (SCIs) on MRI: we are very uncertain whether red cell transfusions reduce the incidence of stroke in children with no previous SCIs on MRI compared to children receiving standard care, RR 0.27 (95% CI 0.03 to 2.31) (one trial, 79 participants, very low quality evidence) (Analysis 1.3). Quality of the evidence was very low due to: risk of bias (this analysis is derived from data from a cohort subset who had an MRI in the STOP trial (STOP 1998)); imprecision of the effect estimate; and indirectness (findings only apply to children with HbSS).
There was no evidence of subgroup differences (test for subgroup differences: Chi² = 0.41, df = 1 (P = 0.52), I² = 0%).
2. Mortality
No previous long‐term red cell transfusion
No participants died in the SIT and STOP trials (two trials, 326 participants), therefore we have no evidence of any difference in mortality rates between the treatments (very low‐quality evidence) (SIT 2014; STOP 1998; Table 1)
Previous long‐term red cell transfusion
We are very uncertain whether continuing long‐term red cell transfusions reduces mortality in children and adolescents compared to children and adolescents receiving standard care, Peto OR 8.00 (95% CI 0.16 to 404.12) (one trial, 79 participants, very low quality evidence) (Analysis 1.4; Table 2). One participant who was assigned to continue transfusion, died in the STOP 2 trial due to complications of ACS (STOP 2 2005).
1.4. Analysis.

Comparison 1: Blood transfusion versus standard care, Outcome 4: Mortality
We did not perform any subgroup analyses for this outcome because deaths occurred in only one trial (STOP 2 2005).
3. Transfusion related complications
Alloimmunisation
Three trials reported this outcome (SIT 2014; STOP 1998; STOP 2 2005).
No previous long‐term red cell transfusion
From data on transfused participants in the SIT trial, we are very uncertain whether children receiving regular long‐term transfusions have a higher risk for developing alloimmunisations compared to children receiving standard care, RR 3.16 (95% CI 0.18 to 57.17) (one trial, 121 participants, very low quality evidence) (SIT 2014) (Analysis 1.5; Table 1).
1.5. Analysis.

Comparison 1: Blood transfusion versus standard care, Outcome 5: Transfusion‐related adverse events
In the STOP trial, 10 participants in the transfusion arm developed an alloimmunisation, despite a more rigorous matching protocol than usual (STOP 1998). It was not reported if any participants in the standard care developed any alloimmunisations.
Previous long‐term red cell transfusion
In the STOP 2 trial, one participant who was in the continuing transfusion arm developed an alloimmunisation (STOP 2 2005). It was not reported if any participants in the halted transfusion arm developed any alloimmunisations.
Infection from blood component
Two trials reported this outcome (STOP 1998; STOP 2 2005).
No previous long‐term red cell transfusion
In the STOP trial, no participant developed evidence of hepatitis C infection, and all 100 children who were tested were negative for antibodies against HIV and HTLV‐I (STOP 1998).
Previous long‐term red cell transfusion
In the STOP 2 trial, no cases of hepatitis C were identified among the 68 participants who had serologic testing at the end of the trial (STOP 2 2005).
Procedural complications
One trial reported this outcome and in this trial, three participants in the transfusion arm had procedural complications (SIT 2014). A catheter infection developed in one participants, and complications requiring catheter replacement developed in two participants.
Transfusion reactions
Three trials reported this outcome (SIT 2014; STOP 1998; STOP 2 2005).
No previous long‐term red cell transfusion
From data from transfused participants in the SIT trial, we are very uncertain whether children receiving regular long‐term transfusions have a higher risk of developing a transfusion reaction than children not receiving regular transfusions, RR 5.17 (95% CI 0.71 to 37.52) (one trial, 121 participants, very low quality evidence) (SIT 2014) (Analysis 1.5). Quality of the evidence was very low due to imprecision of the effect estimate and indirectness (findings only apply to children with HbSS).
In the STOP trial, 12 participants in the transfusion arm had 16 mild reactions to blood products or transfusion procedures (STOP 1998). This was not reported for the standard treatment arm.
Previous long‐term red cell transfusion
In the STOP 2 trial, seven participants in the continuing transfusion arm had nine reactions to transfusions (STOP 2 2005). One of the reactions was serious and required hospitalisation. This was not reported for the halted transfusion arm.
Reduced immunocompetence
None of the trials reported this outcome.
Iron overload
No previous long‐term red cell transfusion
In the SIT trial, the trial authors reported that there was an increased risk of iron overload (measured by serum ferritin greater than 1500 μg/L) in children receiving long‐term red cell transfusions, incidence rate ratio, 14.42 (95% CI 5.41 to 875.17) (one trial, 121 participants) ‐ this analysis was reported in the SIT trial (SIT 2014) (Table 5).
1. Adverse events per 100 person‐years and incidence rate ratios for transfusion‐related complications.
| Outcomes | Trials | Number of participants with at least one event | Adverse events/100 person‐years |
Incidence rate ratioc (95% CI) |
||
| Transfusion | Standard | Transfusion | Standard | |||
| Transfusion reactions | SIT 2014 | 15 out of 90a | 1 out of 31b | 8.85 | 1.66 | 5.33 (1.67 to 23.52) |
| Ferritin > 1500 μg/L | SIT 2014 | 76 out of 90a | 3 out of 31b | 534.70 | 37.07 | 14.42 (5.41 to 875.17) |
aNine participants who declined transfusion were excluded from the analysis. b31 participants assigned to observation received one or more transfusions. cThe incidence ratio was calculated as the rate of adverse events per 100 person‐years in the transfusion group divided by the rate of adverse events per 100 person‐years in the observation group. The 95% confidence intervals were calculated with the use of the bootstrap method with 10,000 replications.
Abbreviations: CI: confidence interval
In the STOP trial, iron overload developed faster than anticipated in children receiving transfusion, with mean (SD) serum ferritin rising to 1804 (773) μg/L at 12 months and 2509 (974) μg/L at 24 months (Adams 1998b; STOP 1998). This outcome was not reported in the children receiving standard care who had transfusions.
Previous long‐term red cell transfusion
In the STOP 2 trial, mean (SD) baseline serum ferritin levels were similar in both treatment arms (continued transfusion group 3274 (1718) μg/L; halted transfusion group mean 3005 (1504) μg/L) (STOP 2 2005). The mean (SD) serum ferritin levels decreased in the halted transfusion group after 12 months (continued transfusion group 3562 (1536) μg/L; halted transfusion group mean (SD) 1832 (912) μg/L). According to an analysis performed by the trial authors this difference was statistically significant (P = 0.002) (STOP 2 2005).
Secondary outcomes
1. Incidence of transient ischaemic attack (TIA)
No previous long‐term red cell transfusion
Two trials recruiting 323 participants reported this outcome; however, in one trial recruiting 127 participants (STOP 1998), no TIAs occurred in either treatment group so this trial does not contribute to the analysis in this review. In children with SCI we are very uncertain whether long‐term red cell transfusions reduce the incidence of TIAs, compared to children receiving standard care, Peto OR 0.13 (95% CI 0.01 to 2.11) (very low quality evidence) (Analysis 1.6; Table 1).
1.6. Analysis.

Comparison 1: Blood transfusion versus standard care, Outcome 6: TIA
Previous long‐term red cell transfusion
One TIA was reported in one participant in the halted transfusion arm in the STOP 2 trial (STOP 2 2005). It was not reported if there were any TIAs in the group who continued transfusions.
2. Measures of neurological impairment
Two trials reported this outcome, in both trials participants had not had previous long‐term red cell transfusions (SIT 2014; STOP 1998).
In the STOP trial, at the time of discharge, of the 11 children in the standard care group diagnosed with cerebral infarction, two had a major disability, five had mild to moderate disability, two had symptoms but no disability and one was asymptomatic (STOP 1998). Measures of neurological impairment in other participants were not reported in this trial.
The SIT trial reported the difference between trial arms in the Weschler Abbreviated Scale of Intelligence (WASI) in performance, verbal, and full IQ scores (data from baseline to interim and to exit visits were analysed by the trial authors who reported estimated least squares means (LSM) and standard error (SE) based on two‐way repeated measures analysis of variance) (SIT 2014).
The SIT trial authors reported that long‐term red cell transfusion may make little or no difference to WASI performance, verbal or full IQ scores in children with SCIs compared to children receiving standard care (LSM: 1.7, SE 95% CI ‐1.1 to 4.4) (one trial, 166 participants, low quality evidence) (SIT 2014; Table 1).
3. Incidence of other sickle cell complications
Acute chest syndrome
Children with no previous long‐term red cell transfusion
Long‐term red cell transfusions may reduce the incidence of ACS in children compared to children receiving standard care, RR 0.24 (95% CI 0.12 to 0.48) (two trials, 326 participants, low quality evidence) (Analysis 1.7; Table 1).
1.7. Analysis.

Comparison 1: Blood transfusion versus standard care, Outcome 7: Other sickle cell related complications
Children with previous long‐term red cell transfusion
In the STOP 2 trial, the authors state that there were fewer incidences of ACS in participants continuing transfusions (STOP 2 2005). In the halted transfusion group, 18 of 41 participants had at least one episode of ACS. Comparative numbers for the transfusion group were not reported.
Painful crisis
No previous long‐term red cell transfusion
Long‐term red cell transfusions may reduce the incidence of painful crisis in children compared to children receiving standard care, RR 0.62 (95% CI 0.46 to 0.84), two trials, 326 participants (Analysis 1.7). Quality of the evidence was low due to risk of bias and indirectness (findings only apply to children with HbSS).
Previous long‐term red cell transfusion
In the STOP 2 trial, the authors state that there were fewer painful crises in the continuing transfusion arm (STOP 2 2005). Comparative numbers were not reported.
Splenic sequestration
No trials looked at this outcome.
Other sickle cell‐related complications
One trial reported other sickle cell‐related complications (SIT 2014) (Table 6 and Analysis 1.7). In this trial participants had not had previous long‐term red cell transfusions.
2. Adverse events per 100 person years and incidence rate ratios for SCD complications.
| Outcomes | Trials | Number of participants with at least one event | Adverse events/100 person‐years |
Incidence rate ratioa (95% CI) |
||
| Transfusion | Standard | Transfusion | Standard | |||
| Acute chest syndrome | STOP 1998 | 4 out of 63 | 14 out of 67 | 4.8b | 15.3b | ‐ |
| SIT 2014 | 5 out of 99 | 24 out of 97 | 1.81b | 14.35b | 0.41 (0.20 to 0.75) |
|
| Painful crisis | STOP 1998 | 11 out of 63 | 13 out of 67 | 16.2 | 27.6 | ‐ |
| SIT 2014 | 32 out of 99 | 56 out of 97 | 41.58 | 102.21 | 0.13 (0.04 to 0.28) |
|
| Priapism | SIT 2014 | 1 out of 59 | 7 out of 52 | 0.84 | 6.65 | 0.13 (0.03 to 0.55) |
| Symptomatic avascular necrosis of the hip | SIT 2014 | 1 out of 99 | 6 out of 97 | 0.4 | 2.25 | 0.22 (0.05 to 0.85) |
a The incidence ratio was calculated as the rate of adverse events per 100 person‐years in the transfusion group divided by the rate of adverse events per 100 person‐years in the observation group. The 95% confidence intervals were calculated with the use of the bootstrap method with 10,000 replications.
b One child from the standard care group was excluded from these analyses due to a stroke on day 16 of the trial.
Abbreviation: CI: confidence interval
4. Quality of life, inpatient stay, immobility and disability
Quality of life
One trial reported this outcome (SIT 2014). The trial reported quality of life measured on the Child Health Questionnaire Parent Form 50 (CHQ_PF50) completed at baseline and at the occurrence of an overt stroke or at trial exit at 36 months. It is not known what the minimal clinically important differences are for the CHQ_PF50.
The SIT trial authors stated that quality of life may improve slightly in children receiving long‐term red cell transfusions compared to children receiving standard care, (difference estimate ‐0.54, 95% CI ‐0.92 to ‐0.17) (one trial, 196 participants, low quality evidence; analysis performed by the SIT trial authors) (SIT 2014; Table 1).
Inpatient stay
No trials looked at this outcome.
Immobility and disability
The STOP trial noted that after 24 months of transfusion, children receiving transfusion had improved growth (height, weight and body mass index (BMI)) which approached normal, whereas there was no improvement in growth rate in children receiving standard care (STOP 1998; Wang 2005).
5. Measures of organ damage
No trials looked at this outcome.
6. Haemoglobin level and haemoglobin S percentage (mean, pre‐ and post‐transfusion and at time of the event)
Haemoglobin level
One of the trials reported the mean haemoglobin at six months after randomisation (STOP 2 2005). The mean haemoglobin was higher in the group that continued red cell transfusions (Table 7).
3. Mean or median haemoglobin (Hb) levels and HbS percentage.
| Trial | Intervention | Baseline | 6 to 12 months | 12 to 18 months | 18 to 24 months | ||||
| Hb (g/L) | Hb S (%) | Hb (g/L) | Hb S (%) | Hb (g/L) | Hb S (%) | Hb (g/L) | Hb S (%) | ||
| No previous long‐term transfusions | |||||||||
| SIT 2014 | Transfusion | Median 77 IQR (72 to 84) |
Median 85 90% CI (51 to 95) |
‐ | Median 30 90% CI (17 to 43) |
‐ | Median 29 90% CI (16 to 43) |
‐ | Median 30 90% CI (16 to 43) |
| Standard | Median 79 IQR (74 to 89) |
‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | |
| STOP 1998 | Transfusion | Mean (SD) 72 (8) | Mean (SD) 87 (10) | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Standard | Mean (SD) 76 (7) | Mean (SD) 87 (7) | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | |
| Previous long‐term transfusions | |||||||||
| STOP 2 2005 | Transfusion | Mean (SD) 93 (9) |
Mean (SD) 21 (8.6) |
Mean (SD) 94 (9) |
Mean (SD) 25.4 (10.9) |
‐ | ‐ | ‐ | ‐ |
| Standard | Mean (SD) 98 (12) |
Mean (SD) 19 (11) |
Mean (SD) 77 (8) |
Mean (SD) 81.0 (8.6) |
‐ | ‐ | ‐ | ‐ | |
Abbreviations: CI: confidence interval; IQR: interquartile range; SD: standard deviation
None of the trials reported the haemoglobin level pre‐transfusion, post‐transfusion or at the time of the event.
Haemoglobin S level
One of the trials reported the mean haemoglobin S level at six months after randomisation in both trial arms (STOP 2 2005). The mean Hb S levels were lower in the group that continued red cell transfusions (Table 7).
Two trials reported the number of pre‐transfusion HbS level measurements that were above the target HbS level of 30% (STOP 1998; STOP 2 2005). Only a small number of transfusions had a HbS level above the target threshold (Table 8).
4. Pre‐transfusion Haemoglobin S levels.
| Trial | Number of transfusions | Number of HbS levels measured | HbS less than 30% | HbS 30 to 40% | HbS greater than 40% |
| No previous long‐term transfusions | |||||
| STOP 1998 | 1521 | ‐ | ‐ | 101 | 42 |
| Previous long‐term transfusions | |||||
| STOP 2 2005 | 1070 | 988 | 748 (76%) | 192 (19%) | 48 (5%) |
None of the trials reported the haemoglobin S level post transfusion or at the time of the event.
Hydroxyurea and phlebotomy versus transfusions and chelation
Two trials are included in this comparison (254 participants) (TWiTCH 2016; SWiTCH 2012). One trial assessed primary prevention of stroke (TWiTCH 2016) and one trial assessed secondary prevention of stroke (SWiTCH 2012)
Throughout this review results from the two trials assessing primary prevention and secondary prevention will not be combined.
Primary outcomes
1. Incidence of clinical stroke
Primary prevention
No clinical strokes occurred in the TWiTCH trial (TWiTCH 2016); Table 3
In the TWiTCH trial, general stroke adjudications were done for 29 possible new neurological events and no child had a positive adjudication for stroke (TWiTCH 2016).
Secondary prevention
We are very uncertain whether switching to hydroxyurea and phlebotomy increase the risk of stroke compared to red cell transfusions and chelation in children and adolescents with a history of stroke, RR 14.78 (95% CI 0.86 to 253.66) (one trial, 133 participants, very low quality evidence) (Analysis 2.1; Table 4).
2.1. Analysis.

Comparison 2: Hydroxyurea and phlebotomy versus standard treatment (transfusions/chelation), Outcome 1: Clinical stroke ‐ Secondary prevention
Seven participants, all in the hydroxyurea and phlebotomy group, experienced a stroke and all had baseline MRA exams which showed severe vasculopathy, including two with moya‐moya. There were more participants with moya‐moya in the hydroxyurea arm (11 participants) compared to the transfusion arm (five participants), the number of participants with severe vasculopathy in both treatment arms was not reported.
2. Mortality
Primary prevention
No deaths occurred in the TWiTCH trial (TWiTCH 2016); Table 3
Secondary prevention
We are very uncertain whether switching to hydroxyurea and phlebotomy reduces mortality compared to red cell transfusions and chelation in children and adolescents with a history of stroke, Peto OR 0.98 (95% CI 0.06 to 15.92) (one trial,133 participants, very low quality evidence) (Analysis 2.2; Table 4).
2.2. Analysis.

Comparison 2: Hydroxyurea and phlebotomy versus standard treatment (transfusions/chelation), Outcome 2: Mortality
3. Transfusion related complications
Alloimunisation
None of the trials reported this outcome.
Infection from blood component
None of the trials reported this outcome.
Procedural complications
None of the trials reported this outcome.
Transfusion reactions
None of the trials reported this outcome.
Reduced immunocompetence
None of the trials reported this outcome.
Iron overload
Primary prevention
Switching to hydroxyurea and phlebotomy may reduce serum ferritin levels compared to continuing to receive red cell transfusions and chelation in children who have a previous history of abnormal TCD velocities and at least 12 months of transfusions, MD ‐1398.00 μg/L (95% CI ‐1929.23 to ‐866.77) (one trial, 121 participants, low quality evidence) (Analysis 2.3). Quality of the evidence was low due to risk of bias and indirectness.
2.3. Analysis.

Comparison 2: Hydroxyurea and phlebotomy versus standard treatment (transfusions/chelation), Outcome 3: Transfusion‐related complications ‐ Serum ferritin; Primary prevention
Switching to hydroxyurea and phlebotomy may have little or no effect on liver iron concentrations compared to continuing to receive red cell transfusions and chelation in children who have a previous history of abnormal TCD velocities and at least 12 months of transfusions, MD ‐1.80 mg Fe/g dry weight liver (95% CI ‐5.16 to 1.56) (one trial, 121 participants, low quality evidence) (Analysis 2.4; Table 3).
2.4. Analysis.

Comparison 2: Hydroxyurea and phlebotomy versus standard treatment (transfusions/chelation), Outcome 4: Transfusion related complications ‐ Liver iron concentration ‐ Primary prevention
Secondary prevention
Switching to hydroxyurea and phlebotomy may reduce serum ferritin levels compared to continuing to receive red cell transfusions and chelation in children and adolescents with a history of stroke (at the final assessment, median serum ferritin was 1994 μg/L, interquartile range (IQR) 998 to 3475, in the hydroxyurea arm and 4064 μg/L, IQR 2330 to 7126, in the transfusion arm; one trial, 133 participants; P < 0.001 analysis performed by the trial authors) (SWiTCH 2012). Quality of the evidence was low due to risk of bias and indirectness.
Switching to hydroxyurea and phlebotomy may have little or no effect on liver iron concentrations compared to continuing to receive red cell transfusions and chelation in children and adolescents with a history of stroke (at the final assessment, median liver iron concentration was 17.3 mg Fe/g dry weight iron, IQR 10.0 to 30.6 in the hydroxyurea arm and 17.3 mg Fe/g dry weight iron, IQR 8.8 to 30.7 in the transfusion arm (one trial,133 participants, low quality evidence, P = 0.79 analysis performed by the trial authors) (SWiTCH 2012; Table 4).
Secondary outcomes
1. Incidence of TIA
Primary prevention
We are very uncertain whether switching to hydroxyurea and phlebotomy has any effect on the incidence of TIA compared to continuing to receive red cell transfusions and chelation in children who have a previous history of abnormal TCD velocities and at least 12 months of transfusions, RR 1.02 (95% CI 0.21 to 4.84) (one trial, 121 participants, very low quality evidence) (Analysis 2.5; Table 3).
2.5. Analysis.

Comparison 2: Hydroxyurea and phlebotomy versus standard treatment (transfusions/chelation), Outcome 5: Other neurological event
Secondary prevention
We are very uncertain whether switching to hydroxyurea and phlebotomy has any effect on the incidence of TIA compared to continuing to receive red cell transfusions and chelation in children and adolescents who have a previous history of stroke, RR 0.66 (95% CI 0.25 to 1.74) (one trial, 133 participants, very low quality evidence) (Analysis 2.5; Table 4).
2. Measures of neurological impairment
Primary prevention
Neurological status is not yet reported in the TWiTCH trial (TWiTCH 2016).
Secondary prevention
Measures of neurological function are not reported in the SWiTCH trial (SWiTCH 2012).
3. Incidence of other sickle cell complications
Total SCD‐related serious adverse events
The SWiTCH trial reported the number of non‐neurological SCD‐related adverse events (SWiTCH 2012).
Secondary prevention
Switching to hydroxyurea and phlebotomy may increase the risk of SCD‐related serious adverse events compared to continuing to receive red cell transfusions and chelation in children and adolescents with a history of stroke, RR 3.10 (95% CI 1.42 to 6.75) (one trial, 133 participants, low quality evidence) (Analysis 2.6). Quality of the evidence was low due to risk of bias and indirectness.
2.6. Analysis.

Comparison 2: Hydroxyurea and phlebotomy versus standard treatment (transfusions/chelation), Outcome 6: Other sickle cell related complications
Acute chest syndrome (ACS)
Primary prevention
We are very uncertain whether switching to hydroxyurea and phlebotomy has any effect on the risk of ACS compared to continuing to receive red cell transfusions and chelation in children who have a previous history of abnormal TCD velocities and at least 12 months of transfusions, RR 2.03 (95% CI 0.39 to 10.69) (one trial, 121 participants, very low quality evidence) (Analysis 2.6; Table 3).
Secondary prevention
We are very uncertain whether switching to hydroxyurea and phlebotomy has any effect on the risk of ACS compared to continuing to receive red cell transfusions and chelation in children and adolescents who have a previous history of stroke, RR 0.33 (95% CI 0.04 to 3.08) (one trial, 133 participants, very low quality evidence) (Analysis 2.6; Table 4).
Painful crisis
Primary prevention
We are very uncertain whether switching to hydroxyurea and phlebotomy has any effect on the risk of painful crisis compared to continuing to receive red cell transfusions and chelation in children who have a previous history of abnormal TCD velocities and at least 12 months of transfusions, RR 5.08 (95% CI 0.61 to 42.23) (one trial, 121 participants, very low quality evidence) (Analysis 2.6). Quality of the evidence was very low due to imprecision of the effect estimate, risk of bias, and indirectness.
Secondary prevention
Switching to hydroxyurea and phlebotomy may increase the risk of painful crisis compared to continuing to receive red cell transfusions and chelation in children and adolescents who have a previous history of stroke, RR 3.15 (95% CI 1.23 to 8.11), one trial, 133 participants, low quality evidence (Analysis 2.6). Quality of the evidence was low due to risk of bias, and indirectness.
Splenic sequestration
Secondary prevention
No cases of splenic sequestration occurred in the SWiTCH trial (SWiTCH 2012).
4. Quality of life, inpatient stay, immobility and disability
Quality of life has not yet been reported in either trial.
5. Measures of organ damage
Not reported in either trial.
6. Haemoglobin level and haemoglobin S percentage (mean, pre and post transfusion and at time of the event)
Haemoglobin level
Primary prevention
Switching to hydroxyurea and phlebotomy may have little or no effect on haemoglobin levels compared to continuing to receive red cell transfusions and chelation in children who have a previous history of abnormal TCD velocities and at least 12 months of transfusions, MD ‐1.00 g/L (95% CI ‐4.29 to 2.29) (one trial, 121 participants, low‐quality evidence) (Analysis 2.7). Quality of the evidence was low due to imprecision, and indirectness.
2.7. Analysis.

Comparison 2: Hydroxyurea and phlebotomy versus standard treatment (transfusions/chelation), Outcome 7: Haemoglobin levels
Secondary prevention
Switching to hydroxyurea and phlebotomy may have little or no effect on haemoglobin levels compared to continuing to receive red cell transfusions and chelation in children and adolescents with a history of stroke (at the final assessment median Hb level was 90 g/L, IQR 84 to 96 for the hydroxyurea arm and 90 g/L, IQR 87 to 96 for the transfusion arm) (one trial, 133 participants, P = 0.93 analysis performed by the trial authors) (SWiTCH 2012).
None of the trials reported the haemoglobin level pre‐transfusion, post‐transfusion or at the time of the event.
Haemoglobin S level
Primary prevention
As expected, switching to hydroxyurea and phlebotomy increased the haemoglobin S levels compared to continuing to receive red cell transfusions and chelation in children who have a previous history of abnormal TCD velocities and at least 12 months of transfusions, MD 43.10% (95% CI 39.59 to 46.61) (one trial, 121 participants) (Analysis 2.8).
2.8. Analysis.

Comparison 2: Hydroxyurea and phlebotomy versus standard treatment (transfusions/chelation), Outcome 8: Haemoglobin S levels
Secondary prevention
As expected, switching to hydroxyurea and phlebotomy increased the haemoglobin S levels compared to continuing to receive red cell transfusions and chelation in children and adolescents with a history of stroke (At the final assessment median Hb S level was 64.1%, IQR 52.6 to 76.4 for the hydroxyurea arm and 32.3%, IQR 25.0 to 38.3 for the transfusion arm (one trial, 133 participants, P < 0.001 analysis performed by the trial authors) (SWiTCH 2012).
None of the trials reported the haemoglobin S level pre transfusion, post transfusion or at the time of the event.
Discussion
Summary of main results
Stroke is a potentially devastating event which affects up to 10% of children with sickle cell anaemia. Recurrences are common after the first stroke, and result in progressively more severe neurological dysfunction. The use of TCD screening has made it possible to identify a subset of children who are at higher risk for primary stroke (Adams 1992).
This Cochrane Review aimed to evaluate the literature on the effectiveness and safety of red cell transfusions for primary and secondary prevention of stroke.
We identified five randomised controlled trials that met our inclusion criteria and included a total of 660 participants. The trials were published between 1998 and 2016. Three trials compared red cell transfusions to standard care and two trials compared hydroxyurea with phlebotomy to red cell transfusions with chelation. The majority of participants had HbSS SCD, all trials included children and two trials also included adolescents. Four trials were for primary stroke prevention and one trial dealt with secondary stroke prevention. No trials included adults for primary or secondary prevention of stroke.
Red cell transfusions versus standard care
Primary prevention
Three randomised trials compared red cell transfusions to standard care for primary prevention of stroke. Two of these trials included children with no previous long‐term transfusions; one of these included children with abnormal TCD velocities, and the other included children with silent cerebral infarcts on magnetic resonance imaging (MRI) but normal TCD velocities. The third trial included children and adolescents on long‐term transfusion whose TCD velocities had normalised. Two of the three trials were terminated early due to safety concerns.
The findings of the review led to the following main conclusions regarding red cell transfusions versus standard care.
Children with no previous long‐term red cell transfusions
Long‐term transfusions probably reduce the incidence of clinical stroke in children with a higher risk of stroke (abnormal TCD velocities or previous history of SCIs).
Long‐term transfusions may: reduce the incidence of other SCD‐related complications (ACS and painful crisis); make little or no difference to IQ scores in children with SCIs; and may increase quality of life.
We are very uncertain whether long‐term transfusions: reduce all‐cause mortality (no deaths in either trial); reduce the risk of TIAs; or increase the risk for developing alloimmunisation.
Children and adolescents with previous long‐term red cell transfusions and normalised TCD velocities
We are very uncertain whether continuing red cell transfusions reduces the incidence of clinical stroke or all‐cause mortality.
Several review outcomes were only reported in one of the trial arms (SCD‐related complications, alloimmunisation, incidence of TIAs), and the trial did not report neurological impairment or quality of life.
Hydroxyurea and phlebotomy versus red cell transfusions and chelation
Primary prevention in children
There were no deaths or clinical strokes in either arm of the trial
Switching to hydroxyurea and phlebotomy may have little or no effect on liver iron concentrations, but may reduce serum ferritin levels.
We are very uncertain whether switching to hydroxyurea and phlebotomy has any effect on the incidence of TIA, or the risk of other SCD‐related complications (ACS or painful crisis)
Secondary prevention in children and adolescents
Switching to hydroxyurea and phlebotomy may increase the risk of SCD‐related serious adverse events
Switching to hydroxyurea and phlebotomy may have little or no effect on liver iron concentrations, but may reduce serum ferritin levels.
We are very uncertain whether switching to hydroxyurea and phlebotomy increases the risk of stroke.
We are very uncertain whether switching to hydroxyurea and phlebotomy has any effect on: all‐cause mortality, or the risk of a TIA.
Neither trial reported on neurological impairment, alloimmunisation, or quality of life.
Overall completeness and applicability of evidence
This review provides the most up‐to‐date assessment of the effectiveness and safety of red cell transfusions for the prevention of primary and secondary stroke. In this 2016 updated review we include two new trials and we have not identified any ongoing trials.
The results of this review can only be interpreted in consideration of the following factors.
The findings in this review can only be generalised to children with HbSS disease. Two trials also included adolescents (STOP 2 2005; SWiTCH 2012).
Only one of the trials assessed secondary prevention of stroke (SWiTCH 2012).
All of the trials were conducted in high‐income countries (USA, Canada, France and the UK). The potential risks associated with transfusion therapy are increased in low‐income countries due to a lack of trained staff, modern equipment, sanitary conditions and clean, infection‐free blood products (Ansong 2013; Ohene‐Frempong 1999). Therefore, the risk‐benefit ratio will be different in low‐income countries to those in the included trials, and the results discussed in this review may not be generalisable to that setting.
Co‐inheritance of alpha thalassaemia may reduce the frequency of complications in individuals with HbSS (Rumaney 2014; Steinberg 2012). Only one trial reported co‐existence of alpha thalassaemia (STOP 1998). In the Stop trial, alpha thalassaemia (at least one alpha globin gene missing) occurred in 22% in the transfusion arm and 9% in the standard treatment arm (STOP 1998).
The chances of having a stroke must be weighed carefully against the burden of regular blood transfusion. In the STOP trial, of the 192 children eligible to take part in the trial, 52 declined to undergo randomisation due to either reluctance of the child or their parents, or due to concern of the physician about compliance with the regimen (STOP 1998). In the SIT trial, of the 99 randomised to transfusion, 15 crossed over to observation, nine of which declined transfusion immediately after randomisation (SIT 2014). These figures may illustrate a relatively poor level of acceptance of this therapy.
The trials did not answer the question of how long red cell transfusion needs to be continued in this population, once commenced. The STOP 2 trial attempted to determine a safe age, or period, for discontinuation of chronic transfusion therapy for primary stroke prevention (STOP 2 2005). However, because of the high proportion (39%) of participants who reverted to abnormal TCD high stroke risk or had a stroke after transfusion was ceased, the trial was terminated two years early with the implication that transfusion therapy should be continued indefinitely in indicated participants. Although eight participants (20%) of those who discontinued transfusion had no abnormal TCD over 25 months of observation, there is currently no way of predicting which individuals will require continuance of transfusion and which will not. Extended follow‐up analysis of the STOP trial also failed to identify predictors for lower‐risk groups (Lee 2006).
The optimal level of sensitivity and specificity for TCD screening remains unclear. The STOP trial included high‐risk children with abnormal TCD velocities, the rate of stroke in the standard care group was approximately 10% per year (STOP 1998). The number of participants needed to treat (NNT) was approximately 11 participants per year to prevent one stroke. In a lower risk group this NNT would be higher. However, if the specificity of the screening, or the 'cut‐off point' for a high‐risk classification, was increased, only the very highest risk children would be treated and children at risk of stroke may be missed. It must therefore be judged at what level of risk it is justifiable to enrol a child to a potentially life‐long regimen of transfusion. Despite these barriers, the routine use of TCD screening and appropriate prophylactic transfusion has greatly lowered the risk of stroke in people who are managed in contemporary sickle cell programmes (Enninful‐Eghan 2010; Fullerton 2004; McCarville 2008; Telfer 2007). At present, the majority of transfusions provided by large sickle cell centres are for the provision of primary rather than secondary stroke prevention.
Four of the five trials were stopped early. The STOP trial was terminated 16 months before the planned end date because of the high rate of stroke in the standard care arm (STOP 1998). The STOP 2 trial was terminated early over safety concerns because high‐risk TCD results developed in 14 participants and two participants had a stroke in the transfusion halted arm (STOP 2 2005). The SWiTCH trial was stopped early due to futility when hydroxyurea and phlebotomy showed no advantage in iron removal compared with long‐term transfusion and iron chelation (SWiTCH 2012). The TWiTCH trial was stopped early because after the first interim analysis, when 33% of participants had completed the trial, non‐inferiority was found, and non‐inferiority was confirmed again after 50% of participants had exited the trial (TWiTCH 2016).
Children with severe vasculopathy were excluded from the TWiTCH trial, and only individuals with severe vasculopathy had a stroke in the SWiTCH trial (SWiTCH 2012; TWiTCH 2016). So individuals with severe vasculopathy may not be candidates for hydroxyurea. As well, it is not known if the effectiveness of hydroxyurea with phlebotomy is sustained over the long term.
In the SIT trial there was a 20% cross‐over rate to either transfusion or hydroxyurea therapy due to increasing disease severity in children receiving standard care (SIT 2014). Six per cent of children receiving standard care crossed over to regular monthly transfusions and 14% began receiving hydroxyurea treatment. We do not know how this may have influenced outcomes over the long term, but this increases uncertainty as to the efficacy of standard care compared to transfusions in these children.
Quality of the evidence
Overall the quality of the evidence was rated moderate to very low across different outcomes according to GRADE methodology (Table 1; Table 2; Table 3; Table 4). This was due to trials being at serious risk of bias; outcome estimates being imprecise (wide confidence intervals); and a serious risk of indirectness (no direct evidence for adults or children without HbSS) since the trials only included children and adolescents primarily with HbSS disease. The HbSS and HbSβº thalassaemia genotypes included within the trials account for approximately 70% of people with SCD. Individuals with these genotypes have a higher prevalence of stroke that presents earlier than individuals with HbSC and other milder forms of SCD. Nevertheless, the participant inclusion criteria for this review is broad and although the prevalence of stroke is estimated to be rare in children with milder forms of SCD a recent trial in a cohort of 96 children with HbSC disease, estimated the prevalence for SCIs to be 13.5% (Guilliams 2015). The outcome for clinical stroke in primary prevention in children with no previous long‐term transfusions and at higher risk of stroke (abnormal TCD velocities and presence of SCIs) was the only outcome rated as moderate quality evidence.
Red cell transfusions versus standard care
No previous long‐term red cell transfusions
We considered one outcome, clinical stroke in children with higher risk of stroke (abnormal TCD velocities and SCIs), as moderate quality evidence due to indirectness.
We considered three outcomes as low quality evidence due to serious risk of bias and indirectness. These were:
SCD‐related adverse events (acute chest syndrome and painful crises);
neurological impairment (IQ);
quality of life.
We considered three outcomes to be very low quality evidence due to serious risk of bias, indirectness and imprecision. These were:
all‐cause mortality;
TIAs;
transfusion‐related adverse events (alloimmunisation).
Previous long‐term red cell transfusions
We considered two outcomes as very low quality evidence due to serious risk of bias, indirectness and imprecision. These were:
clinical stroke;
all‐cause mortality.
Several review outcomes were only reported in one of the trial arms (SCD‐related complications, alloimmunisation, incidence of TIAs).
The trial did not report neurological impairment or quality of life.
Hydroxyurea and phlebotomy versus red cell transfusions and iron chelation
Primary prevention
We considered one outcome (transfusion‐related adverse events ‐ iron overload) as low quality evidence due to serious risk of bias and indirectness
We considered four outcomes as very low quality evidence due to serious risk of bias, indirectness and imprecision. These were:
clinical stroke;
all‐cause mortality;
TIAs;
SCD‐related adverse events (ACS, painful crises).
The trial did not report neurological impairment or quality of life.
Secondary prevention
We considered one outcome as low quality evidence (transfusion‐related adverse events ‐ iron overload) as low quality evidence due to serious risk of bias and indirectness.
We considered four outcomes to be very low quality evidence due to serious risk of bias, indirectness, and imprecision. These were:
clinical stroke;
all‐cause mortality;
TIAs;
SCD‐related adverse events.
The trial did not report neurological impairment or quality of life.
Potential biases in the review process
To our knowledge, our review process is free from bias. We conducted a comprehensive search; searching data sources (including multiple databases, and clinical trial registries) to ensure that all relevant trials would be captured. There were no restrictions for the language in which the paper was originally published. The relevance of each paper was carefully assessed and all screening and data extractions were performed in duplicate. We pre‐specified all outcomes and subgroups prior to analysis. There were insufficient numbers of included trials within the meta‐analyses for us to use a funnel plot to examine the risk of publication bias.
Agreements and disagreements with other studies or reviews
With regard to the conclusions of the STOP and STOP 2 trials, multiple reports have supported the efficacy of chronic transfusion in preventing primary stroke in those at high risk based on abnormal TCD velocities (Enninful‐Eghan 2010; Fullerton 2004; McCarville 2008); this has become standard of care practice in almost all sickle cell centres and has led to more subjects being transfused for primary than for secondary stroke prevention in those centres. The findings of the SWiTCH trial were somewhat unexpected because of previously published smaller single institution studies which suggested greater efficacy from phlebotomy in reducing iron overload and not as great a degree of protection from chronic transfusion in secondary stroke prophylaxis (SWiTCH 2012).
Several observational studies for secondary stroke prevention have also attempted to define whether transfusion may be safely stopped. One review suggested that transfusion may be safely stopped at age 18 years (Powars 2000), and no recurrences occurred in a small series of seven adults with SCD followed up for three years to 18 years (Rana 1997). However, a study of 10 participants who discontinued transfusion after one to two years showed a 70% recurrence rate after only 11 months (Wilimas 1980). In a subsequent study, five out of 10 people with SCD who had been receiving regular transfusions for five to 12 years experienced an ischaemic event in the 12‐month period after transfusions were stopped (Wang 1991). These findings suggest that the risk of secondary stroke may be almost as high after discontinuing transfusion therapy 'prematurely' as before starting it. Evaluation of risk factors related to the initial stroke might be an important consideration in the design of any further studies. For example, in a retrospective analysis of 137 stroke participants, those who had an identified medical or concurrent event associated with their initial stroke did not have a recurrent stroke two or more years after the initial event (Scothorn 2002).
This systematic review noted a lack of prospective randomised controlled trials of blood transfusion for prevention of secondary stroke (following an initial stroke) in SCD. However, the practice of long‐term transfusion therapy is supported by evidence from several observational studies (Sarnaik 1979; Styles 1994). It was demonstrated in a small study of 15 participants that a target HbS level of 30% to 50% retains a high protective value against stroke, but reduces transfusion requirements and the associated risks (Cohen 1992). However, in a study which observed 60 persons with SCD for 192 person years, HbS was greater than 30% in seven of the 16 reported transient ischaemic events and five of six recurrent infarctions (Pegelow 1995). A recent survey has indicated that an acceptable "community standard" level of pre‐transfusion HbS is less than 45% rather than less than 30% (Aygun 2009).
Authors' conclusions
Implications for practice.
In children with no previous long‐term transfusions, red cell transfusions probably reduce the risk of stroke and may confer some additional advantage by reducing the rates of acute chest syndrome (ACS) and painful crisis. This must be balanced against the adverse effects and costs of a chronic transfusion regimen. We are uncertain whether transfusions can be stopped in children and adolescents at high risk of stroke who have had their transcranial Doppler (TCD) velocities normalised, nor has any treatment duration threshold been established for stopping transfusions.
We are uncertain if switching to hydroxyurea with phlebotomy is non‐inferior to red cell transfusions in a select population of children in primary prevention; we are also uncertain as to how long the drug effects may be maintained. We are uncertain if switching to hydroxyurea with phlebotomy results in an increased risk of stroke in secondary prevention and switching may increase the risk of adverse events in children and adolescents. Hydroxyurea with phlebotomy did not demonstrate superiority in iron removal as measured by liver iron concentration.
Due to lack of evidence this review cannot comment on management for adults with HbSS disease or children and adults with HbSβº, HbSC or HbSβ⁺ disease.
Implications for research.
Information from a well‐designed, prospective, randomised controlled trial of chronic blood transfusion regimens in persons with sickle cell disease (SCD) who have had a previous stroke is desirable in order to make recommendations for the optimal use of this therapy in secondary stroke prevention. Recent improvements in methods of detecting high‐risk individuals are improving clinical outcome, but further research is needed to assess the relative risks and benefits of hydroxyurea in comparison with long‐term transfusion therapy for primary and secondary prevention of cerebral infarcts. Randomised trials are needed comparing blood transfusions with both standard care and hydroxyurea for both primary and secondary prevention in adults with SCD.
What's new
| Date | Event | Description |
|---|---|---|
| 3 February 2020 | New search has been performed | The searches have been updated. No new completed or ongoing eligible trials were identified. We identified 33 additional citations to the already included trials. No new data were added to the review from these citations. We excluded five new trials (10 citations) (Bernaudin 2016; EXTEND 2016; Gwam 2016; SACRED 2018; SPIN 2017) and identified a further five citations to an already excluded trial. |
| 23 January 2020 | New citation required but conclusions have not changed | For this update, minor changes have been made throughout the review. |
History
Protocol first published: Issue 3, 2001 Review first published: Issue 1, 2002
| Date | Event | Description |
|---|---|---|
| 12 January 2017 | New search has been performed | Two new trials have been added to the review (SIT 2014; TWiTCH 2016). One trial, previously listed in 'Ongoing studies' has been moved to 'Excluded studies' (SCATE 2015). |
| 12 January 2017 | New citation required but conclusions have not changed | This update has included GRADE assessment of the quality of the evidence. |
| 13 November 2014 | New search has been performed | A search of the Cystic Fibrosis and Genetic Disorders Group's Haemoglobinopathies Trials Register identified: three additional references to the SWiTCH study (SWiTCH 2012); one additional reference to the STOP trial; two references reporting on both the STOP and the STOP II trials (STOP 1998; STOP 2 2005); and one additional reference to the ongoing SIT trial (SIT 2014). |
| 6 November 2013 | New citation required but conclusions have not changed | The inclusion of additional trials resulted in only minor amendments to the conclusion section of the review. |
| 6 November 2013 | New search has been performed | A Search of the Cochrane Cystic Fibrosis and Genetic Disorders Group's Haemoglobinopathies Trials Register identified six additional references to two already included trials (STOP 1998; STOP 2 2005) and 11 additional references to two trials previously listed in Ongoing studies (SIT 2014; SWiTCH 2012). The SWiTCH 2012 trial has now been listed as an included study. Two further studies have been included in the ongoing studies section of the review (SCATE 2015; TWiTCH 2016). The title has been amended to include the terms 'primary and secondary'. |
| 22 May 2009 | New search has been performed | A search of the Group's Trials Register did not identify any new references which may have been eligible for inclusion in this review. |
| 31 July 2008 | Amended | Converted to new review format. |
| 31 July 2008 | New search has been performed | A search of the Group's Haemoglobinopathies Trials Register identified no new trials potentially eligible for inclusion in the review. |
| 1 August 2007 | New search has been performed | A search of the Trials Register identified a total of 10 new references. Eight were of the STOP trials (two of which refer to both the STOP 1998 and STOP 2 2005 trials and so appear in both lists (Adamkiewicz 2006; Kwiatkowski 2002)): six new references to the STOP trial (STOP 1998) as follows (Adamkiewicz 2006; Kwiatkowski 2002; Kwiatkowski 2006; Lee 2006; Lezcano 2006; Wang 2005). The search also identified a further two abstracts (DeBaun 2005; Ware 2006) which have been listed in the 'Ongoing studies' section of the review. |
| 1 February 2006 | New search has been performed | A search of the Trials Register identified seven new references to the included trial (STOP 1998) as follows: Abboud 2004; Adams 2004; Bulas 2000; Gates 2002; Hsu 2003; Kwiatkowski 2002; Kwiatkowski 2003. It has also been reported that the STOP 2 2005 trial has been terminated early, although published findings are not yet available. |
| 31 August 2004 | New search has been performed | A search of the trials register identified no new trials eligible for inclusion in the review. |
| 31 August 2003 | New search has been performed | Additional references to the already included STOP 1998 study have been added. However, no new data have been found. The references added to the STOP 1998 study ID were: Adams 1998 Wang 1998 Adams 2002 Files 2002 |
| 30 September 2002 | New search has been performed | Additional references to the already included STOP 1998 study have been added. Therefore, further results are presented on the following outcomes ‐ 1. Transfusion related complications 2. Incidence of transient ischaemic attack or silent infarction 3. Incidence of other sickle cell complications The references added to the STOP 1998 study ID were: Abbound 1999 Adams 1996 Adams 1997 Adams 1999 ‐ two references Adams 2001 Duncan 1997 Kutlar 2000 Lee 2002 Nichols 2001 Miller 2000 ‐ two references Miller 2001 Pegelow 1999 Pegelow 2001 Vichinsky 1999 Vichinsky 2001 |
Acknowledgements
We acknowledge the substantial contribution by Dr Ceri Hirst in undertaking the original version of this review (Hirst 2002), and Dr Kerry Kirkham (née Dwan) in the update of the review (Wang 2013).
We thank the editorial base of the Cochrane Cystic Fibrosis and Genetic Disorders Group for their help and support during the course of this review.
We thank NHS Blood and Transplant (NHSBT).
We thank the National Institute for Health Research (NIHR). This review is part of a series of reviews that have been partly funded by the NIHR Cochrane Programme Grant ‐ Safe and Appropriate Use of Blood Components. This research was also supported by the National Institute for Health Research (NIHR) Oxford Biomedical Research Centre Programme.The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health.
Appendices
Appendix 1. Search Strategies
CENTRAL (CENTRAL & DARE) (issue 4, 2016; issue 2, 2015 respectively)
#1 MeSH descriptor: [Anemia, Sickle Cell] explode all trees
#2 MeSH descriptor: [Hemoglobin, Sickle] explode all trees
#3 ("hemoglobin S" or "haemoglobin S" or "hemoglobin SC" or "haemoglobin SC" or "hemoglobin SE" or "haemoglobin SE" or "hemoglobin SS" or "haemoglobin SS" or "hemoglobin C disease" or "hemoglobin D disease" or "hemoglobin E disease" or "haemoglobin C disease" or "haemoglobin D disease" or "haemoglobin E disease" or "Hb SC" or HbSC or HbAS or HbSS or HbAC or "Hb SE" or "Hb SS" or "Hb C disease" or "Hb D disease" or "Hb E disease" or "SC disease" or "SC diseases")
#4 (sickle cell* or sicklemia or sickled or sickling or meniscocyt* or drepanocyt*)
#5 (sickle and SCD)
#6 ((Hb S or HbS or sickle) near/3 (disease* or thalassemi* or thalassaemi*))
#7 #1 or #2 or #3 or #4 or #5 or #6
#8 MeSH descriptor: [Cerebrovascular Disorders] explode all trees
#9 ((brain* or cerebr* or vertebrobasil* or hemispher* or intracran* or intracerebral or infratentorial or supratentorial or MCA* or anterior circulation or basilar artery or vertebral artery or subcortical or cortical or choroidal) near/5 (ischemi* or ischaemi* or infarct* or thrombo* or phlebothrombo* or stenosis or emboli* or occlus* or vaso‐occlus* or vasocclus* or vasooclus* or obstruction* or hypoxi* or accident* or abnormalit* or vasculopath* or vasospasm* or disorder* or damage* or disease* or lesion* or insufficien*))
#10 ((ischemi* or ischaemi*) near/5 (apoplex* or attack* or accident* or seizure* or arrest* or injur* or failure* or lesion* or insult*))
#11 (stroke* or CVA or silent infarct* or incomplete infarct*)
#12 MeSH descriptor: [Cerebrovascular Circulation] explode all trees
#13 MeSH descriptor: [Ultrasonography, Doppler, Transcranial] this term only
#14 (transcranial* near/3 (doppler or ultrasound or sonograph* or ultrasonograph*))
#15 ((brain* or cerebr* or vertebrobasil* or hemispher* or intracran* or intracerebral or infratentorial or supratentorial or MCA* or anterior circulation or basilar artery or vertebral artery or subcortical or cortical or choroidal) near/3 (hemodynamics or haemodynamics or blood flow or blood velocit* or volume))
#16 MeSH descriptor: [Cerebral Arteries] explode all trees
#17 MeSH descriptor: [Blood Volume Determination] this term only
#18 MeSH descriptor: [Blood Flow Velocity] this term only
#19 (TCD near/3 (flow* or velocit*))
#20 #8 or #9 or #10 or #11 or #12 or #13 or #14 or #15 or #16 or #17 or #18 or #19
#21 #7 and #20
MEDLINE (OvidSP, Epub Ahead of Print, In‐Process & Other Non‐Indexed Citations, 1946 to 4 April 2016)
1. exp Anemia, Sickle Cell/
2. Hemoglobin, Sickle/
3. (h?emoglobin S or h?emoglobin SC or h?emoglobin SE or h?emoglobin SS or h?emoglobin C disease or h?emoglobin D disease or h?emoglobin E disease or Hb SC or HbSC or HbAS or HbSS or HbAC or Hb SE or Hb SS or Hb C disease or Hb D disease or Hb E disease or SC disease*).tw,kf.
4. (sickle cell* or sicklemia or sickled or sickling or meniscocyt* or drepanocyt*).tw,kf.
5. (sickle and SCD).tw,kf.
6. ((Hb S or HbS or sickle) adj3 (disease* or thalass?emi*)).tw,kf.
7. or/1‐6
8. exp Cerebrovascular Disorders/
9. ((brain* or cerebr* or vertebrobasil* or hemispher* or intracran* or intracerebral or infratentorial or supratentorial or MCA* or anterior circulation or basilar artery or vertebral artery or subcortical or cortical or choroidal) adj5 (isch?emi* or infarct* or thrombo* or phlebothrombo* or stenosis or emboli* or occlus* or vaso‐occlus* or vasocclus* or vasooclus* or obstruction* or hypoxi* or accident* or abnormalit* or vasculopath* or vasospasm* or disorder* or damage* or disease* or lesion* or insufficien*)).tw,kf.
10. (isch?emi* adj5 (apoplex* or attack* or accident* or seizure* or arrest* or injur* or failure* or lesion* or insult*)).tw,kf.
11. (stroke* or CVA or silent infarct* or incomplete infarct*).tw,kf.
12. or/8‐11
13. exp Cerebrovascular Circulation/
14. Ultrasonography, Doppler, Transcranial/
15. (transcranial adj3 (doppler or ultrasound or sonograph* or ultrasonograph*)).tw,kf.
16. ((brain* or cerebr* or vertebrobasil* or hemispher* or intracran* or intracerebral or infratentorial or supratentorial or MCA* or anterior circulation or basilar artery or vertebral artery or subcortical or cortical or choroidal) adj3 (h?emodynamics or blood flow or blood velocit* or volume)).tw,kf.
17. exp Cerebral Arteries/us
18. Blood Volume Determination/
19. Blood Flow Velocity/
20. (TCD adj3 (flow* or velocit*)).tw,kf.
21. or/13‐20
22. 12 or 21
23. 7 and 22
Embase (OvidSP, 1974 to 4 April 2016)
1. exp Sickle Cell Anemia/
2. Hemoglobin S/
3. (h?emoglobin S or h?emoglobin SC or h?emoglobin SE or h?emoglobin SS or h?emoglobin C disease or h?emoglobin D disease or h?emoglobin E disease or Hb SC or HbSC or HbAS or HbSS or HbAC or Hb SE or Hb SS or Hb C disease or Hb D disease or Hb E disease or SC disease*).tw.
4. (sickle cell* or sicklemia or sickled or sickling or meniscocyt* or drepanocyt*).tw.
5. (sickle and SCD).tw.
6. ((Hb S or HbS or sickl*) adj3 (disease* or thalass?emi*)).tw.
7. or/1‐6
8. exp Cerebrovascular Disease/
9. (isch?emi* adj5 (apoplex* or attack* or accident* or seizure* or arrest* or injur* or failure* or lesion* or insult*)).tw.
10. ((brain* or cerebr* or vertebrobasil* or hemispher* or intracran* or intracerebral or infratentorial or supratentorial or MCA* or anterior circulation or basilar artery or vertebral artery or subcortical or cortical or choroidal) adj5 (isch?emi* or infarct* or thrombo* or phlebothrombo* or stenosis or emboli* or occlus* or vaso‐occlus* or vasocclus* or vasooclus* or obstruction* or hypoxi* or accident* or abnormalit* or vasculopath* or vasospasm* or disorder* or damage* or disease* or lesion* or insufficien*)).tw.
11. (stroke* or CVA or silent infarct* or incomplete infarct*).tw.
12. or/8‐11
13. exp Brain Circulation/
14. Doppler Echography/
15. exp Brain Artery/
16. Brain Blood Volume/
17. Blood Flow Velocity/
18. (TCD adj3 (flow* or velocit*)).tw.
19. ((brain* or cerebr* or vertebrobasil* or hemispher* or intracran* or intracerebral or infratentorial or supratentorial or MCA* or anterior circulation or basilar artery or vertebral artery or subcortical or cortical or choroidal) adj3 (h?emodynamics or blood flow or blood velocit* or volume)).tw.
20. (transcranial adj3 (doppler or ultrasound or sonograph* or ultrasonograph*)).tw.
21. or/13‐20
22. 12 or 21
23. 7 and 22
CINAHL (EBSCOHost, 1937 to 4 April 2016)
S1 (MH "Anemia, Sickle Cell+")
S2 TX ("hemoglobin S" or "haemoglobin S" or "hemoglobin SC" or "haemoglobin SC" or "hemoglobin SE" or "haemoglobin SE" or "hemoglobin SS" or "haemoglobin SS" or "hemoglobin C disease" or "hemoglobin D disease" or "hemoglobin E disease" or "haemoglobin C disease" or "haemoglobin D disease" or "haemoglobin E disease" or "Hb SC" or HbSC or HbSS or HbAC or "Hb SE" or "Hb SS" or "Hb C disease" or "Hb D disease" or "Hb E disease" or "SC disease" or "SC diseases" OR sickle cell* or sicklemia or sickled or sickling or meniscocyt* or drepanocyt*)
S3 TX ((Hb S or HbS or sickle) N3 (disease* or thalass?emi*))
S4 S1 OR S2 OR S3
S5 (MH "Cerebrovascular Disorders+") S6 TX ((brain* or cerebr* or vertebrobasil* or hemispher* or intracran* or intracerebral or infratentorial or supratentorial or MCA* or anterior circulation or basilar artery or vertebral artery or subcortical or cortical or choroidal) N5 (ischemi* or ischaemi* or infarct* or thrombo* or phlebothrombo* or stenosis or emboli* or occlus* or vaso‐occlus* or vasocclus* or vasooclus* or obstruction* or hypoxi* or accident* or abnormalit* or vasculopath* or vasospasm* or disorder* or damage* or disease* or lesion* or insufficien*))
S7 TX ((ischemi* or ischaemi*) N5 (apoplex* or attack* or accident* or seizure* or arrest* or injur* or failure* or lesion* or insult*))
S8 TX (stroke* or CVA or silent infarct* or incomplete infarct*)
S9 (MH "Cerebrovascular Circulation+")
S10 (MH "Ultrasonography, Doppler, Transcranial")
S11 TX (transcranial N3 (doppler or ultrasound or sonograph* or ultrasonograph*))
S12 TX ((brain* or cerebr* or vertebrobasil* or hemispher* or intracran* or intracerebral or infratentorial or supratentorial or MCA* or anterior circulation or basilar artery or vertebral artery or subcortical or cortical or choroidal) N3 (hemodynamics or haemodynamics or blood flow or blood velocit* or volume))
S13 (MH "Cerebral Arteries+/US")
S14 (MH "Blood Volume Determination") S15 (MH "Blood Flow Velocity") S16 (TCD N3 (flow* or velocit*))
S17 S5 OR S6 OR S7 OR S8 OR S9 OR S10 OR S11 OR S12 OR S13 OR S14 OR S15 OR S16
S18 S4 AND S17
S19 (MH Clinical Trials+)
S20 PT Clinical Trial
S21 TI ((controlled trial*) or (clinical trial*)) OR AB ((controlled trial*) or (clinical trial*))
S22 TI ((singl* blind*) OR (doubl* blind*) OR (trebl* blind*) OR (tripl* blind*) OR (singl* mask*) OR (doubl* mask*) OR (tripl* mask*)) OR AB ((singl* blind*) OR (doubl* blind*) OR (trebl* blind*) OR (tripl* blind*) OR (singl* mask*) OR (doubl* mask*) OR (tripl* mask*))
S23 TI randomi* OR AB randomi*
S24 MH RANDOM ASSIGNMENT
S25 TI ((phase three) or (phase III) or (phase three)) or AB ((phase three) or (phase III) or (phase three))
S26 ( TI (random* N2 (assign* or allocat*)) ) OR ( AB (random* N2 (assign* or allocat*)) )
S27 MH PLACEBOS
S28 MH META ANALYSIS
S29 MH SYSTEMATIC REVIEW
S30 TI ("meta analys*" OR metaanalys* OR "systematic review" OR "systematic overview" OR "systematic search*") OR AB ("meta analys*" OR metaanalys* OR "systematic review" OR "systematic overview" OR "systematic search*")
S31 TI ("literature review" OR "literature overview" OR "literature search*") OR AB ("literature review" OR "literature overview" OR "literature search*")
S32 TI (cochrane OR embase OR cinahl OR cinhal OR lilacs OR BIDS OR science AND citation AND index OR cancerlit) OR AB (cochrane OR embase OR cinahl OR cinhal OR lilacs OR BIDS OR science AND citation AND index OR cancerlit)
S33 TI placebo* OR AB placebo*
S34 MH QUANTITATIVE STUDIES
S35 S19 or S20 or S21 or S22 or S23 or S24 or S25 or S26 or S27 or S28 or S29 or S30 or S31 or S32 or S33 or S34
S36 S18 AND S35
PubMed (for epublications ahead of print, in‐process & other non‐indexed citations only on 4 April 2016)
#1 ("hemoglobin S" OR "haemoglobin S" OR "hemoglobin SC" OR "haemoglobin SC" OR "hemoglobin SE" OR "haemoglobin SE" OR "hemoglobin SS" OR "haemoglobin SS" OR "hemoglobin C disease" OR "hemoglobin D disease" OR "hemoglobin E disease" OR "haemoglobin C disease" OR "haemoglobin D disease" OR "haemoglobin E disease" OR "Hb SC" OR HbSC OR HbAS OR HbSS OR HbAC OR "Hb SE" OR "Hb SS" OR "Hb C disease" OR "Hb D disease" OR "Hb E disease" OR "SC disease" OR "SC diseases" OR sickle* OR sickled OR sickling OR meniscocyt* OR drepanocyt*)
#2 (("Hb S" OR HbS) AND (disease* OR thalassemi* OR thalassaemi*))
#3 #1 OR #2
#4 ((ischemi* OR ischaemi*) AND (apoplex* OR attack* OR accident* OR seizure* OR arrest* OR injur* OR failure* OR lesion* OR insult*))
#5 (stroke* OR CVA OR silent infarct* OR incomplete infarct*)
#6 (transcranial* AND (doppler OR ultrasound OR sonograph* OR ultrasonograph*))
#7 ((brain OR brains OR cerebral OR cerebrovascular OR cerebro‐vascular OR vertebrobasil* OR hemispher* OR intracran* OR intracerebral OR infratentorial OR supratentorial OR MCA OR "anterior circulation" OR "basilar artery" OR "vertebral artery" OR subcortical OR cortical OR choroidal) AND (hemodynamics OR haemodynamics OR "blood flow" OR blood velocit* OR volume))
#8 (TCD AND (flow* OR velocit*))
#9 #4 OR #5 OR #6 OR #7 OR #8
#10 #3 AND #9
TRANSFUSION EVIDENCE LIBRARY (1950 to April 4 2016)
sickle AND (ischemic OR ischaemic OR ischemia OR ischaemia OR stroke OR infarct OR doppler OR transcranial OR ultrasound OR sonography OR ultrasonography)
LILACS (1982 to April 4 2016)
tw:(sickle) AND (instance:"regional") AND ( db:("LILACS") AND type_of_study:("clinical_trials"))
IndMed (1986 to April 4 2016)
(sickle OR sickled OR sickling OR SC disease) AND (ischemic OR ischaemic OR ischemia OR ischaemia OR stroke OR strokes OR subcortical OR cortical OR choroidal OR infarct OR infarcts OR doppler OR transcranial OR ultrasound OR sonography OR ultrasonography) AND (random OR randomly OR randomised OR randomized OR blind OR blinded OR control group OR placebo OR controlled study OR groups OR trial OR trials OR systematic review OR meta‐analysis OR metaanalysis OR literature search OR medline OR pubmed OR cochrane OR embase)
KoreaMed (1997 to April 4 2016)
sickle [TI] OR sickle [AB] "Randomized Controlled Trial" [PT]
Web of Science CPCI‐S (Conference Proceedings Citation Index‐ Science (CPCI‐S) ‐ 1990 to to April 4 2016)
TOPIC: (sickle OR sickled OR sickling) AND
TOPIC: (ischemic OR ischaemic OR ischemia OR ischaemia OR stroke OR strokes OR infarct OR infarcts OR brain OR hemisphere* OR intracranial OR intracerebral OR cerebral OR cerebrovascular OR subcortical OR cortical OR choroidal OR doppler OR transcranial OR ultrasound OR sonography OR ultrasonography) AND
TOPIC: (randomly OR randomised OR randomized OR blind OR blinded OR control group OR placebo OR controlled study OR groups OR trial OR trials OR systematic review OR meta‐analysis OR metaanalysis OR literature search OR medline OR pubmed OR cochrane OR embase)
ClinicalTrials.gov
Search Terms: ischemia OR stroke OR infarct OR brain OR hemisphere OR intracranial OR intracerebral OR cerebral OR cerebrovascular OR subcortical OR cortical OR choroidal OR doppler OR transcranial OR ultrasound OR sonography OR ultrasonography
Condition: sickle cell anemia
Study Type: Interventional Studies
WHO ICTRP
Title/Intervention: ischemia OR stroke OR infarct OR brain OR hemisphere OR intracranial OR intracerebral OR cerebral OR cerebrovascular OR subcortical OR cortical OR choroidal OR doppler OR transcranial OR ultrasound OR sonography OR ultrasonography
Condition: sickle cell anemia
Recruitment Status: ALL
Data and analyses
Comparison 1. Blood transfusion versus standard care.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 Clinical stroke | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1.1 No previous long‐term red cell transfusions | 2 | 326 | Risk Ratio (M‐H, Random, 95% CI) | 0.12 [0.03, 0.49] |
| 1.1.2 Previous long‐term red cell transfusions | 1 | 79 | Risk Ratio (M‐H, Random, 95% CI) | 0.22 [0.01, 4.35] |
| 1.2 Clinical stroke ‐ velocity | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.2.1 Normal TCD velocities and no previous long‐term red cell transfusions | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.2.2 Normalised TCD velocities and previous long‐term red cell transfusions | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.2.3 Abnormal TCD velocities | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.3 Clinical stroke ‐ SCI | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.3.1 Presence of previous SCI on MRI | 2 | 243 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.11 [0.02, 0.59] |
| 1.3.2 Absence of previous SCI on MRI | 1 | 79 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.27 [0.03, 2.31] |
| 1.4 Mortality | 1 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Totals not selected | |
| 1.5 Transfusion‐related adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.5.1 Alloimmunisation ‐ No previous long‐term red cell transfusions | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.5.2 Transfusion reactions ‐ No previous long‐term red cell transfusion | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.6 TIA | 2 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Subtotals only | |
| 1.6.1 No previous long‐term red cell transfusions | 2 | 323 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.13 [0.01, 2.11] |
| 1.7 Other sickle cell related complications | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.7.1 Acute chest syndrome | 2 | 326 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.24 [0.12, 0.48] |
| 1.7.2 Painful crises | 2 | 326 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.62 [0.46, 0.84] |
| 1.7.3 Priapism | 1 | 111 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.13 [0.02, 0.99] |
| 1.7.4 Avascular necrosis of the hip | 1 | 196 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.16 [0.02, 1.33] |
Comparison 2. Hydroxyurea and phlebotomy versus standard treatment (transfusions/chelation).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 2.1 Clinical stroke ‐ Secondary prevention | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2.2 Mortality | 2 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Totals not selected | |
| 2.2.1 Mortality ‐ Primary prevention | 1 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Totals not selected | |
| 2.2.2 Mortality ‐ Secondary prevention | 1 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Totals not selected | |
| 2.3 Transfusion‐related complications ‐ Serum ferritin; Primary prevention | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 2.4 Transfusion related complications ‐ Liver iron concentration ‐ Primary prevention | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 2.5 Other neurological event | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2.5.1 TIA ‐ Primary prevention | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2.5.2 TIA ‐ Secondary prevention | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2.6 Other sickle cell related complications | 2 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.6.1 Total SCD‐related SAEs ‐ Secondary prevention | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.6.2 Acute chest syndrome ‐ Primary prevention | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.6.3 Acute chest syndrome ‐ Secondary prevention | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.6.4 Painful crisis ‐ Primary prevention | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.6.5 Painful crisis ‐ Secondary prevention | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.6.6 Infections and infestations SAEs ‐ Primary prevention | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.6.7 Infections and infestations SAEs ‐ Secondary prevention | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.7 Haemoglobin levels | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 2.8 Haemoglobin S levels | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
SIT 2014.
| Study characteristics | ||
| Methods | Multicentre randomised trial in 29 clinical centres in the USA, Canada, France and the UK. Recruitment: December 2004 to May 2010. The last participant enrolled completed the exit visit on July 29, 2013. |
|
| Participants |
Inclusion criteria: children aged 5 to 15 years, confirmed diagnosis of haemoglobin SS or haemoglobin Sβ0 thalassaemia, and at least one infarct‐like lesion on the screening MRI scan defined as an MRI signal abnormality that was at least 3 mm in one dimension and that was visible in two planes on fluid‐attenuated inversion recovery (FLAIR) T2‐weighted images, as determined by agreement of two of the three trial neuroradiologists. Exclusion criteria: history of focal neurologic deficit associated with an infarct on brain MRI, a seizure disorder, treatment with hydroxyurea in the previous 3 months, a history of regular transfusion therapy, or imaging or non‐imaging TCD measurement that was above the trial‐defined thresholds. Participant flow: 1210 registered for screening; 1074 had screening MRI evaluated by neuroradiology committee; 675 had normal, 20 indeterminate MRI; 379 had infarct‐like lesions on screening MRI; 291 had infarct‐like lesions adjudicated by neurology committee; 220 had pre‐randomisation MRIs adjudicated by neuroradiology committee; 196 underwent randomisation. Transfusion arm: N = 99 (15 crossed over to observation) Sex: male: 59 (60%); F: 40 (40%) Age: 5 to 7: 26 (26%): 8 to 10: 35 (35%); 11 to 13: 32 (32%); 14 to 15: 6 (6%) TCD velocity: median (IQR) cm/sec: 147 (123‐168) (N = 98) Lesions on initial MRI: 99 (100%) Parental report of recurring headaches: yes: 37 (37%); No: 62 (63%) Steady state haemoglobin: median (IQR): g/L: 77 (72 to 84) Phenotypes: not stated (Included only HbSS or HbSβº) Hb F% median (IQR): 9.0 (4.0 to 14.0) Alpha thalassaemia: not reported Observation arm: N = 97 (6 crossed over to transfusion) Sex: male: 52 (54%); F: 45 (46%) Age: 5 to 7: 28 (29%): 8 to 10: 32 (33%); 11 to 13: 29 (30%); 14 to 15: 8 (8%) TCD velocity: median (IQR) cm/sec: 143 (131 to 163) Lesions on initial MRI: 97 (100%) Parental report of recurring headaches: yes: 43 (44%); No: 54 (56%) Steady state haemoglobin: median (IQR): g/L: 79 (74 to 89) Phenotypes: not stated trial included only HbSS or HbSβº Hb F% median (IQR): 10.0 (5.0 to 15.0) Alpha thalassaemia: not reported |
|
| Interventions |
Transfusion: transfusion arm received a transfusion approximately monthly to maintain a target haemoglobin concentration greater than 90 g/L and a target haemoglobin S concentration of 30% or less. Red cell component: leucocyte‐depleted, negative for haemoglobin S. Red cell matching: ABO, Rh and Kell antigens. Iron chelation: ferritin levels were monitored before each transfusion. Site investigators were advised to initiate chelation therapy for participants who had ferritin levels greater than 1500 ng per millilitre for 2 or more consecutive months. Observation: observation arm received standard care with no treatment for silent infarcts and no hydroxyurea therapy and were evaluated quarterly |
|
| Outcomes |
Primary outcome: the recurrence of infarct or haemorrhage as determined by neuroimaging, clinical evidence of permanent neurologic injury, or both. A new infarct had to meet the criteria for a SCI; an enlarged SCI was defined as a previously identified silent cerebral infarct that increased by at least 3 mm along any linear dimension in any plane on MRI; TIA, included in secondary analyses of neurologic outcomes, defined as an event that resulted in focal neurologic deficits that lasted less than 24 hours, did not result in abnormalities on T2‐weighted or FLAIR images that were indicative of an acute infarct, and had no other reasonable medical explanation. Secondary outcomes: changes in cognition, assessed by measurement of IQ scores with the Wechsler Abbreviated Scale of Intelligence12 or the Wechsler Preschool and Primary Scale of Intelligence III; also assessed scores on the Behavior Rating Inventory of Executive Function (BRIEF). |
|
| Notes |
Funding: supported by grants from the National Institute of Neurological Disorders and Stroke (5U01NS042804, 3U01NS042804 [American Recovery Reinvestment ACT supplementary grant] to Dr DeBaun); the Institute of Clinical and Translational Sciences, National Center for Research Resources, and the National Center for Advancing Translational Sciences, Clinical and Translational Research; NIH Roadmap for Medical Research (UL1TR000448, to Washington University; UL1TR001079, to Johns Hopkins University; and UL1TR000003, to the Children’s Hospital of Philadelphia); and Research and Development in the National Health Service, UK. Declarations of interest: Dr McKinstry reports receiving honoraria and lecture fees from Siemens Healthcare and consulting fees from Guerbet; Dr Woods, receiving fees for serving on a data and safety monitoring board from Mast Therapeutics and grant support from ClinDatrix and Novartis; Dr Kwiatkowski, receiving fees for serving on an advisory board from Shire Pharmaceuticals, consulting fees from Shire Pharmaceuticals and Sideris Pharmaceuticals, and grant support from Resonance Health; Dr. Heiny, receiving lecture fees from Novartis; Dr. Redding‐Lallinger, receiving grant support from Eli Lilly and Mast Therapeutics; and Dr. Casella, receiving honoraria, travel support, and consulting fees through his institution from Mast Therapeutics and being an inventor and a named party on a patent and licensing agreement for an assay panel of brain biomarkers for the detection of brain injury (PCT US2011/056338), licensed to ImmunArray with pending royalties only. No other potential conflict of interest relevant to this article was reported. Trial registration: NCT00072761 & ISRCTN52713285. Mean length of follow‐up: children were followed for a median of 3 years. Power calculation: a sample size of 204 participants (102 in each group) would give the trial 85% power to detect a decrease of at least 86% in the prevalence of the primary end point, assuming a 10% dropout rate and a crossover rate of 16% from transfusion to observation and 3% from observation to transfusion, at a two‐tailed nominal alpha level of 0.05. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation assignments were provided by the statistical data coordinating centre with the use of a permuted block design, with stratification according to site, age, and sex. Participants were assigned in a 1:1 ratio to the observation group or the transfusion group and were followed until the occurrence of a trial end‐point event or until exit from the trial. |
| Allocation concealment (selection bias) | Low risk | Assignments were provided by the statistical data coordinating centre. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | By the nature of the trial treatments used (blood transfusions vs observation), it is impractical to make SIT trial blinded (masked). |
| Blinding of outcome assessment (detection bias) Stroke and TIA | Low risk | Members of neuroradiology and neurology committees, who were unaware of the trial‐group assignments, adjudicated neurologic and MRI findings. |
| Blinding of outcome assessment (detection bias) All outcomes apart from stroke or all‐cause mortality | High risk | Unblinded trial. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All outcomes reported. All participants accounted for, conducted an intention‐to‐treat and per protocol analysis for the primary outcomes The primary end point was ascertained for 185 of the 196 participants (94%). Of the 99 participants randomly assigned to the transfusion group, 90 started receiving transfusions within 4 weeks after assignment. The cross‐over rate from transfusion to observation was 15% (15 of 99 participants); 9 participants declined blood transfusion, and 6 crossed over to observation at a median time of 34 days. |
| Selective reporting (reporting bias) | Low risk | Protocol available and all planned outcomes reported. |
| Other bias | Unclear risk | Among participants in the observation group, 32% received transfusions (a median of 3 transfusions each), including 6 participants who crossed over to regular monthly transfusions at a median of 1.7 years. During the course of the trial, hydroxyurea was started in 14 of 97 participants (14%) in the observation group and in 3 of 99 (3%) in the transfusion group because of disease severity. Exclusion criteria included treatment with hydroxyurea. Not clear how long or when treatment began ‐ possible contamination and unknown effect on outcomes. 6 also crossed over to regular transfusion giving 20% cross‐over rate to either hydroxyurea or transfusion. |
STOP 1998.
| Study characteristics | ||
| Methods | Multicentre randomised controlled trial conducted in 12 centres in the USA and Canada in children 2 ‐ 16 years of age with HbSS or HbSβº thalassaemia. Screening began in January 1995 and ended in November 1996. The trial was to run to December 1998 but was stopped in September 1997. |
|
| Participants |
Inclusion criteria: children 2 to 16 years of age and who had been given a diagnosis of sickle cell anaemia or sickle β0 thalassaemia at high risk of stroke with a blood flow velocity of at least 200 cm per second on 2 TCD trials. Exclusion criteria: history of stroke, had an indication for or contraindication to long‐term transfusion, were receiving other treatments that affected the risk of stroke, were infected with the human immunodeficiency virus (HIV), had been treated for seizures, were pregnant, or had a serum ferritin concentration above 500 ng per millilitre. Participant flow: screened: N = 1934; eligible: N = 206 randomised: N = 130 Transfusion: N = 63 Sex: male: 31 (49%) Age: mean (SD): 8.2 (3.2) years HbS% mean (SD): 87 (10) HbF% mean (SD): 8.0 (5.2) Alpha thalassaemia: 14 (22%) TCD velocity: mean (SD): 223 (27) cm/sec Lesions on initial MRI: N (%) participants: 19 (31%) Standard care: N = 67 Sex: male: 29 (43%) Age: mean (SD): 8.4 (3.3) years HbS% mean (SD): 87 (7) HbF% mean (SD): 9.4 (5.0) Alpha thalassaemia: 7 (9%) TCD velocity: mean (SD) : 223 (28) cm/sec Lesions on initial MRI N (%): participants 25 (38%) Phenotypes: not reported (trial included only HbSS or HbSβº) |
|
| Interventions |
Transfusion: N = 63 In the transfusion arm the goal was to reach an HbS concentration < 30 per cent of total haemoglobin within 21 days without exceeding a haemoglobin concentration of 120 g/L and a hematocrit of 36%. Exchange or simple transfusion were allowed: 63% were simple transfusions, 12% were exchange; 25% a combination of simple and exchange. Red cells were delivered in a volume of approximately 10 to 15 mL per kg of packed cells per transfusion. Red cell component: leucocyte‐depleted, negative for haemoglobin S. Red cell matching: ABO, Rh and Kell antigens. Iron chelation: none. Potential participants with a ferritin level above 500 ng/mL were excluded from the trial. The intention was to exclude any child with a significant iron burden before initiation of treatment, thus avoiding clinically significant iron overload during the trial. Standard care: N = 67 |
|
| Outcomes |
Primary outcome: cerebral infarction and intracranial haemorrhage. Secondary outcomes: death, transfusion‐related adverse events. |
|
| Notes |
Funding: supported by Cooperative Agreements (U10 HL 52193 and U10 HL 52016) with the National Heart, Lung, and Blood Institute. Declarations of interest: none published. Trial registration: no registration found. Mean (SD) length of follow‐up: transfusion arm: 21.0 (5.7) months; Standard care: 18.3 (7.0) months. Power calculation: "Estimates of stroke risk for patients randomized to standard care were obtained by fitting an exponential model to the follow‐up of TCD (1) patients follow‐up, it was estimated that 47% of patients in this group should develop stroke on study. Assuming transfusion prevents 70% of these strokes, 14% of the patients randomized to transfusion should have strokes on study. Taken together, these values imply that a sample size of 46 per treatment arm should provide the desired statistical power of 90% to detect a 70% reduction in stroke incidence at a type I error rate of 0.05 for a two‐sided test". Analysis: 4 interim analyses and one final analysis were planned. the date of the first analysis changed from 20 months to 14 months after recruitment began. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "The DCC developed permuted blocks within which treatment allocations were randomly and evenly assigned. The blocks themselves were randomly assigned to each of the 12 Centers." |
| Allocation concealment (selection bias) | Low risk | "After telephone verification that the patient was eligible and acquisition of written parental consent, the DCC ran a short randomization program and provided the Investigator with the trial group assignment." " Permuted blocks are used to blind Investigators to the potential treatment assignment of each patient while preserving approximate balance within and across Centers. The DCC provided the Clinical Center Investigator and patient with the treatment assignment." |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Clinicians and participants were unblinded. |
| Blinding of outcome assessment (detection bias) Stroke and TIA | Low risk | A panel of physicians with no knowledge of the children’s treatment assignments who were not affiliated with the trial centres determined whether an event was a stroke. The primary end points were cerebral infarction and intracranial haemorrhage. "The protocol was intended to identify all neurologic events. A panel of physicians with no knowledge of the children’s treatment assignments who were not affiliated with the trial centres determined whether an event was a stroke.". |
| Blinding of outcome assessment (detection bias) All outcomes apart from stroke or all‐cause mortality | High risk | Unblinded trial. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | An ITT analysis was used, despite 12 participants crossing over between groups (2) or withdrawing from the trial (10). Reasons were provided. 10 participants from the transfusion group withdrew from the trial because of problems with compliance (n = 4), multiple alloantibodies (n = 1), ineligibility (n = 1) or other unspecified reasons (n = 4). Two participants from the standard care group crossed over to the transfusion group, one on the second day due to diagnosis of subacute intracerebral hematoma and the other after 12 months for treatment of leg ulcers. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available and no prospective trial registration. |
| Other bias | Unclear risk | "Four interim analyses and one final analysis were planned, with the Lan–DeMets approximation of the O’Brien–Fleming stopping boundary. The date of the first analysis was changed from 20 months to 14 months after recruitment began." "Because of the high rate of stroke in the standard‐care group and the significant effect of transfusion found at the second interim analysis, the data safety and monitoring board recommended that the trial be stopped 16 months before the planned date of December 1998 so that transfusion could be offered to children in the standard‐care group." There was an imbalance between the number of participants with alpha thalassaemia trait between treatment arms (22% transfusion arm versus 9% standard treatment arm). |
STOP 2 2005.
| Study characteristics | ||
| Methods | Multicentre RCT, extension to the STOP trial conducted in 23 centres (including the 12 centres in STOP) in the U.S. and Canada to determine whether regular blood transfusions for the prevention of stroke could be stopped in children and youth 5 ‐ 20 years of age with SCD. Children were monitored by transcranial Doppler examinations after transfusions were halted and by resuming transfusions if the examination indicated a high risk of stroke. This trial was an extension of the previous STOP trial, in which children with abnormal velocities on TCD ultrasonographic examination were administered transfusions to prevent a first stroke. The trial was meant to be a 54‐month trial involving 50 participants in each group, with 60 of the participants enrolled during the first 12 months and 40 during the next 24 months; after recruitment ended, there were 18 months of follow‐up. Four interim analyses and one final analysis were planned for the composite end point. The trial was stopped on the advice of the data safety and monitoring committee because of concern about safety at the fourth interim analysis with 79 participants enrolled. |
|
| Participants |
Inclusion criteria: children whose Doppler studies normalized after 30 or more months of transfusion were eligible for the present trial. In addition, children who had not participated in the previous STOP trial whose condition met the criteria for eligibility and treatment were also eligible for the present trial. Adequate participation in a transfusion program (≥ 24 transfusions in 30 months and Hb S < 30% in at least 20 of the 30 months); 2 Normal TCD examinations at least 2 weeks apart while receiving transfusions within 4 months of randomisation; age, 5 to 20 years; consent to participate in trial. Exclusion criteria: prior stroke; Indication for chronic transfusion; contraindication for chronic transfusion; moderate‐to‐severe intracranial arterial disease on MRA. Participant flow: screened: not reported; eligible: not reported; randomised: N = 79. Transfusion continued: N = 38 Sex: male: 20 (53%) Age: mean (SD): 12.5 (3.3) HbS % mean (SD): 21.0 (8.6) HbF% mean (SD): 2.4 (1.8) Alpha thalassaemia: not reported TCD velocity: mean (SD): 139 (16) cm/sec Lesions on initial MRI: 10 (26%) Phenotypes:Not reported (trial included only HbSS or HbSβº) Transfusion halted: N = 41 Sex: male: 13 (32%) Age: mean (SD): 12.05 (3.1) HbS% mean (SD): 19.0 (11) HbF% mean (SD): 2.3 (1.5) Alpha thalassaemia: not reported TCD velocity: mean (SD): 143 (18) cm/sec Lesions on initial MRI: 11 (27%) Phenotypes: not reported (trial included only HbSS or HbSβº) |
|
| Interventions |
Transfusion continued: n = 38. Transfusion could be simple, manual exchange or automated exchange. antigens. Red cell component: leucocyte‐depleted, negative for haemoglobin S. Red cell matching: ABO, Rh and Kell antigens. Iron chelation: chelation therapy with the use of deferoxamine was recommended if serum ferritin levels exceeded 2500 ng per millilitre. Transfusion halted: n = 41. Participants in the transfusion‐halted group could receive transfusions to treat complications of sickle cell disease. Initiation of hydroxyurea therapy or regular transfusion was designated as a cross‐over and data was censored on the patient as of the date of treatment. |
|
| Outcomes |
Primary outcome: composite end point was a stroke (cerebral infarction or intracranial haemorrhage) or reversion to abnormal velocity on transcranial Doppler ultrasonography, defined as 2 consecutive studies with abnormal velocities, 3 consecutive studies with an average velocity of 200 cm per second or more, or 3 consecutive inadequate studies plus evidence of severe stenosis on MRA. Secondary outcomes: also reports deaths, acute chest syndrome and transfusion adverse events. |
|
| Notes |
Funding: supported by grants (U01 HL 052193 and U01 HL 052016) from the National Heart, Lung, and Blood Institute. Declarations of interest: no potential conflict of interest relevant to this article was reported. Trial registration: no registration found. Mean length of follow‐up: the median time from randomisation to an end‐point event was 3.2 months (range, 2.1 to 10.1), and the mean (SD) was 4.5 (2.6) months. Power calculation: for a 54‐month trial involving 50 participants in each group, with 60 of the participants enrolled during the first 12 months and 40 during the next 24 months; after recruitment ended, there were 18 months of follow‐up. Analysis: the trial was stopped by the National Heart, Lung, and Blood Institute on the advice of the data safety and monitoring committee because of concern about safety at the fourth interim analysis. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were stratified at randomisation according to the presence or absence of ischaemic lesions on MRI; random, permuted blocks of 4 or 6 participants were used within each group as defined by MRI. Institutional balancing with a tolerance of two participants per site was imposed to maintain an approximate balance in treatment assignments at each site. Eligible participants underwent randomisation with equal probability of continuing or halting transfusion. |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment not reported. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Participants and personnel were unblinded. |
| Blinding of outcome assessment (detection bias) Stroke and TIA | Low risk | "Suspected strokes were adjudicated by experts unaware of the treatment assignment using clinical data and all available imaging data. Stroke was defined as persistent neurologic abnormalities or transient symptoms accompanied by a new cerebral lesion appropriate to the patient’s clinical presentation." "The Doppler studies were transmitted to central readers who were unaware of the treatment assignments. All results were recorded as the time‐averaged mean of the maximum velocity in the middle cerebral or internal carotid artery and were classified as normal (all mean velocities of <170 cm per second), conditional (at least one mean velocity of 170 to 199 cm per second but none ≥200 cm per second), abnormal (at least one mean velocity of at least 200 cm per second), or inadequate (no information available on one or both middle cerebral arteries)". |
| Blinding of outcome assessment (detection bias) All outcomes apart from stroke or all‐cause mortality | High risk | Unblinded trial. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | It was not stated whether intent‐to‐treat analysis was used. Data on 9 participants assigned to no continued transfusion who did not have a primary end‐point event were censored: 5 of these participants resumed chronic transfusion and four started treatment with hydroxyurea. Of 38 participants assigned to continued transfusion, 5 discontinued participation in the trial. N = 14 (17%) of participants discontinued or censored. |
| Selective reporting (reporting bias) | Unclear risk | Primary outcomes reported. Did not state secondary outcomes. Not clear if all adverse events reported and also censored data not contributing to outcome reporting. |
| Other bias | Unclear risk | The trial was stopped by the National Heart, Lung, and Blood Institute on the advice of the data safety and monitoring committee because of concern about safety at the fourth interim analysis. |
SWiTCH 2012.
| Study characteristics | ||
| Methods | Multicentre randomised controlled non‐inferiority trial conducted in 26 paediatric sickle cell centres in the USA. Total duration of trial treatment was 30 months after randomisation, with a final trial visit scheduled 6 months after discontinuation of trial treatments. |
|
| Participants |
Inclusion criteria: paediatric participants with severe forms of SCA (HbSS, HbS/βº‐thalassemia, HbS/OArab); age range of 5.0 to 18.9 years, inclusive, at the time of enrolment; initial (primary) completed overt clinical stroke after the age 12 months with documented infarction on brain CT or MRI; at least 18 months of chronic monthly erythrocyte transfusions since primary stroke; transfusional iron overload, defined as a previously documented liver iron concentration ≥ 5.0 mg Fe per g of dry weight liver or serum ferritin ≥ 500 ng/mL on 2 independent measurements; adequate monthly erythrocyte transfusions with average HbS ≤ 45% (the upper limit of the established academic community standard) for the past 6 months before enrolment; parent or guardian willing and able to provide informed consent with verbal or written assent from the child (< 18 years of age), and subject willing and able to provide informed consent (≥ 18 years of age); ability to comply with trial related treatments, evaluations, and follow‐up. Exclusion criteria: inability to receive or tolerate chronic RBC transfusion therapy; inability to take or tolerate daily oral hydroxyurea; clinical and laboratory evidence of hypersplenism (temporary); abnormal laboratory values at initial evaluation (temporary); current participation in other therapeutic clinical trials; current use of other therapeutic agents for sickle cell disease (e.g. arginine, decitabine, magnesium); any condition or chronic illness, such as a positive tuberculin (PPD) test, which in the opinion of the investigator makes participation ill‐advised; inability or unwillingness to complete required screening studies, including blood tests, brain MRI/MRA, and liver biopsy; a sibling enrolled in SWiTCH. Participant flow: screened: N = 202; enrolled: N = 161; randomised: N = 134 Transfusion + chelation: N = 66 (one moved before starting trial treatment) Sex: Male: 31 (47%) Age: Mean (SD): 13.3 (3.8) Phenotype: HbSS: 66 (100%) Previous recurrent stroke: 4 (6%) History of TIA: 11 (17%) Infarction: 65 (98%) Vasculopathy: 54 (82%) Moya‐moya: 5 (8%) Liver iron content (LIC), mg Fe/g dw liver median (IQR): 14.5 (9.5 to 23.3) Serum ferritin, ng/mL median (IQR): 3282.0 (2321.0 to 4306.0) HbS% median (IQR): 27.0 (21.2 to 38.6) HbF% median (IQR): 1.7 (1.0 to 2.5) Alpha thalassaemia: not reported Hydroxyurea + phlebotomy: N = 67 Sex: male: 41 (61%) Age: Mean (SD): 13.0 (4.0) Phenotype: HbSS: 66 (99%) Previous recurrent stroke: 10 (15%) History of TIA: 10 (15%) Infarction: 65 (98%) Vasculopathy: 53 (79%) Moya‐moya: 11 (16%) Liver iron content (LIC), mg Fe/g dw liver median (IQR): 13.9 (8.7 to 22.9) Serum ferritin, ng/mL median (IQR): 3346.0 (2202.0 to 4682.0) HbS% median (IQR): 30.3 (23.8 to 39.6) HbF% median (IQR): 1.4 (0.8 to 2.2) Alpha thalassaemia: not reported |
|
| Interventions |
Standard treatment (transfusion + chelation): N = 66 For standard treatment (blood transfusion + iron chelation) participants received monthly blood transfusions designed to maintain 30% HbS, with local discretion regarding transfusion type (e.g., simple or erythrocytapheresis). Red cell component: not reported. Red cell matching: not reported. Iron chelation: daily iron chelation. Hydroxyurea + phlebotomy: N = 67 Participants randomised to hydroxyurea + phlebotomy commenced hydroxyurea at 20 mg/kg/d with stepwise escalation to MTD. Transfusions continued for 4 to 9 months during an overlap phase designed to protect against recurrent stroke during hydroxyurea dose escalation. Once MTD was reached and transfusions were discontinued, phlebotomy commenced with a target of 10 mL/kg (maximum volume, 500 mL) blood removed monthly to reduce iron burden. |
|
| Outcomes |
Primary outcome: composite primary endpoint of secondary stroke recurrence rate and quantitative liver iron concentration. Secondary outcomes: non‐stroke neurological events, non‐neurological sickle cell clinical events, quality of life evaluation, and measures of organ function. |
|
| Notes |
Funding: supported by National Heart, Lung, and Blood Institute grants U01‐HL078787 (R.E.W.) and U01‐HL078987 (R.W.H.). Declarations of interest: the authors declare no competing financial interests. Trial registration: ClinicalTrials.gov NCT00122980. Mean length of follow‐up: 6‐month follow‐up. Power calculation: not stated. Analysis: because reduction in LIC was not superior on hydroxyurea/phlebotomy, the DSMB concluded that the composite primary trial end point would not be met and recommended trial closure. NHLBI closed SWiTCH N = 40 did not complete treatment phase in transfusion/iron chelation arm and N = 43 did not complete treatment phase in hydroxyurea/phlebotomy arm. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of sequence generation not reported. |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment not reported. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | By the nature of the trial treatments used to prevent recurrent stroke (blood transfusions vs hydroxyurea), it is impractical to make SWiTCH a blinded (masked) trial. |
| Blinding of outcome assessment (detection bias) Stroke and TIA | Low risk | The inclusive independent stroke adjudication process for all suspected new neurological events is a novel feature of the trial. Stroke recurrence is a primary trial endpoint but also is a critical safety endpoint for the SWiTCH trial. Accordingly, it was necessary to develop an inclusive process by which all potential stroke events were recognized and systematically adjudicated using a standardized protocol and masked consultants. Participants who develop any acute neurological change are promptly evaluated for possible stroke. In addition, site personnel are provided with a written script to use at each interval clinic visit, to ensure that subjects and families are asked each month about any signs and symptoms of stroke. After a new neurological event is suspected, the stroke adjudication process begins. The clinical history and neurological exam are reviewed by 3 independent neurologists without knowledge of the imaging findings. Simultaneously, the radiological evaluation is reviewed by 3 independent masked neuroradiologists without knowledge of the clinical history or neurological examination. Only after their independent consensus opinions are formed are these two opinions reconciled into a final stroke adjudication decision; a diagnosis of stroke requires new neurological findings with corresponding radiological changes. |
| Blinding of outcome assessment (detection bias) All outcomes apart from stroke or all‐cause mortality | High risk | The SWiTCH principal investigator was masked to all treatment‐specific results, including laboratory tests and clinical events. In addition, all investigators at the peripheral clinical sites are masked to trial treatment results outside of their own clinical centre. Unblinded trial. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The primary statistical analyses of efficacy and safety will be performed on the ITT population, which consists of all participants who were randomised to a trial treatment and for whom outcome data are available. |
| Selective reporting (reporting bias) | High risk | Several secondary outcomes not reported (i.e. quality of life, growth and development, organ damage, transfusion‐related, chelation‐related and phlebotomy related complications) |
| Other bias | Unclear risk | the DSMB concluded that the composite primary trial end point would not be met and recommended trial closure. NHLBI closed SWiTCH ‐ N = 40 did not complete treatment phase in transfusion/iron chelation arm and N = 43 did not complete treatment phase in hydroxyurea/phlebotomy arm. More participants had moya‐moya in the hydroxyurea arm (11 participants) than the transfusion arm (5 participants), it was not known if there was a difference between treatment arms in the number of participants with other types of severe vasculopathy. |
TWiTCH 2016.
| Study characteristics | ||
| Methods | Multicentre randomised open‐label non‐inferiority trial conducted at 26 paediatric hospital and health centres in the USA and Canada in children with abnormal TCD velocities greater or equal to 200 cm/s but no vasculopathy Duration: 24 months after randomisation with a 6‐month visit after completing exit studies. |
|
| Participants | Screened: N = 159; excluded: N = 38; randomised: N = 121 Inclusion criteria
Exclusion criteria
Transfusion (N = 61): (mean (SD) unless otherwise stated) Sex: male: 19 (31%) Age: mean (SD): 9.5 ± 2.68 Phenotype: HbSS: 59 (97%) Age at index abnormal TCD: mean (SD): 5.7 (2.0) Average index TCD value (cm/s): mean (SD): 226 (25) Average entry TCD value (cm/s): mean (SD): 145 (21) Silent cerebral infarct: 25 (41%) Mild to moderate vasculopathy: 6 (10%) Transusion duration (years): 3.8 (1.8) Simple transfusions: 36 (59%) RBC alloantibodies: 9 (15%) RBC autoantibodies 12 (20%) Liver iron (mg Fe per g dry weight liver) Mean (SD): 8.5 (7.5) Serum ferritin (ng/mL) Mean (SD): 2713 (2207) Current chelation usage: 51 (84%) Haemoglobin (g/L) Mean (SD): 93 (8) HbS% Mean (SD): 26.5 (10.3) HbF% Mean (SD): 10·3 (6·5) Alpha thalassaemia: not reported Hydroxyurea (n = 60): (mean (SD) unless otherwise stated) Sex: male: 29 (48%) Age: mean (SD): 9.7 (3.2) Phenotype: HbSS: 60 (100%) Age at index abnormal TCD: mean (SD): 5.0 (1.8) Average index TCD value (cm/s): mean (SD): 220 (17) Average entry TCD value (cm/s): mean (SD): 145 (26) SCI: 22 (37%) Mild to moderate vasculopathy: 4 (7%) Transfusion duration (years): 4.5 (2.8) Simple transfusions: 39 (65%) RBC alloantibodies: 11 (18%) RBC autoantibodies: 10 (17%) Liver iron (mg Fe per g dry weight liver): Mean (SD): 11.3 (9.5) Serum ferritin (ng/mL): mean (SD): 3080 (2347) Current chelation usage: 48 (80%) Haemoglobin (g/L): mean (SD): 93 (8) HbS% Mean (SD): 27.6 (9.9) HbF% Mean (SD): 8·8 (5·5) Alpha thalassaemia: not reported |
|
| Interventions |
Transfusions N = 61: Participants continued to receive transfusions once per month to maintain HbS at 30% or lower with local discretion with respect to transfusion type (simple, partial exchange, or erythrocytapheresis). Red cell component: not reported. Red cell matching: not reported. Iron chelation: deferasirox was recommended to manage iron overload; children already receiving chelation therapy maintained their current doses; those starting chelation therapy received deferasirox at 10 ‐ 40 mg/kg per day with the dose dependent on liver iron concentration at screening. Hydroxyurea N = 60: Participants initiated treatment of hydroxyurea at 20 mg/kg per day (capsules or liquid formulation) with escalation to MTD defined as the dose at which moderate marrow suppression of neutrophils and reticulocytes was achieved. Transfusions were slowly weaned in accordance with a standard protocol over 4 ‐ 9 months. |
|
| Outcomes |
Primary outcome: TCD time‐averaged mean velocity on the index side defined as the cerebral hemisphere with the higher mean arterial velocity at baseline assessment. Secondary outcomes: TCD velocity on the non‐index side, new stroke or non‐stroke neurological events, new brain MRI/MRA lesions, hepatic iron overload, sickle‐related events, neuropsychological status, quality of life, growth, and treatment‐related complications. |
|
| Notes |
Funding: supported by the National Heart, Lung, and Blood Institute through grants R01 HL‐095647 (REW) and R01 HL‐095511 (BRD). Declarations of interest: all authors declared. Trial registration: ClinicalTrials.gov, number NCT01425307. Mean length of follow‐up: 24 months after randomisation,with a 6‐month visit after completing exit studies Power calculation: 100 participants (50 per treatment arm) who complete the 24‐month post‐randomisation follow‐up period will provide at least 90% power to test the non‐inferiority hypothesis under reasonable scenarios. Analysis: two planned interim analyses after 33% and 67% of participants had completed exit studies. At the first scheduled interim analysis, non‐inferiority was shown and the sponsor terminated the trial after 50% had exited and repeat analysis confirmed the first interim analysis. We did analyses in the ITT population, except for a planned per‐protocol analysis of TCD velocities, which excluded participants who exited the trial early. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants will be randomised to treatment arm by means of an adaptive randomisation algorithm to maintain balance between treatment groups with respect to site, age, and the mean of 2 screening maximum TCD velocities. Participants are randomised to either the Standard or Alternative treatment arm at an approximately 1 to 1 ratio. Randomisation is stratified within site to achieve approximate balance with respect to two baseline factors: (1) the mean of the two screening TCD velocity values and (2) participant age at enrolment. Block sizes will be fixed at 4 to maintain equal sample sizes in the two arms within a given site every fourth randomisation. Random block size was not employed due to the small number of participants excepted at each clinical site. Assignment within a given block of four will be randomly ordered and will vary from block to block. The first 8 participants to be randomised will be assigned to a treatment arm based on the site‐specific unconstrained random allocation scheme. |
| Allocation concealment (selection bias) | Low risk | From appendix: this process assures that the differences in mean baseline age and mean baseline TCD velocity between the two arms, after the participant’s randomisation, do not exceed a critical value determined by a statistical probability distribution. Adaptive randomisation and trial‐wide imbalance constraints and other constraints appear to ensure that allocation is statistically determined. Randomisation was done centrally, stratified by site with a block size of four, and we used an adaptive randomisation scheme to balance the covariates of baseline age and TCD velocity. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Participants and personnel are unblinded. |
| Blinding of outcome assessment (detection bias) Stroke and TIA | Low risk | TCD examinations were done just before transfusions or phlebotomy, and all were read centrally by observers masked to treatment assignment and previous TCD results. All new potential stroke events were assessed with careful neurological evaluation and brain MRI or MRA examinations, then adjudicated centrally by a panel of expert reviewers. Independent and then consensus opinions were obtained from neurologists and neuroradiologists masked to trial treatment. Brain MRI/MRA examinations at trial exit allowed us to confirm that no strokes had been missed by the adjudication process. |
| Blinding of outcome assessment (detection bias) All outcomes apart from stroke or all‐cause mortality | High risk | Unblinded trial. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All randomised participants included in safety and efficacy analysis Transfusion arm: 0 lost to follow‐up, 8 discontinued intervention, 3 adjudicated TIA, 1 TCD velocity > 240 cm/s 2 non‐adherence 1 difficulty finding matched blood 1 chose to withdraw hydroxyurea arm: 6 discontinued intervention 3 adjudicated transient Ischaemic attack 3 non‐adherence. |
| Selective reporting (reporting bias) | Unclear risk | Some outcomes will be reported in future papers. |
| Other bias | Unclear risk | Children with severe vasculopathy were excluded from TWiTCH trial during screening, so these children might not be suitable candidates for hydroxyurea. Mean age of the trial participants was slightly older than the peak age for primary stroke (about 5 ‐ 6 years), yet still within the published range. The duration of hydroxyurea therapy without transfusions was relatively short; longer follow‐up is clearly needed to establish whether these findings are maintained over time. Trial stopped early based on TCD velocities, an accepted surrogate for primary stroke risk in children with sickle cell anaemia, still be some uncertainty with regards to effectiveness for stroke prevention in certain populations and over time. After full enrolment and when 37% of the participants had exited the trial, the first scheduled interim analysis showed that the stopping boundary had been passed and non‐inferiority was shown. After 50% of participants had exited, repeat analyses supported these findings and the trial was terminated by NHLBI. Remaining participants then completed all exit studies before discontinuing protocol‐directed trial treatment. In total, the standard group included 42 participants who completed trial treatment, 11 who had truncated treatment, and eight who exited early; the alternative group included 41 participants who completed treatment, 13 who had truncated treatment, and 6 who exited early. |
DSMB: Data and Safety Monitoring Board HbS: sickle haemoglobin HbSβºThal: sickle beta‐zero thalassaemia HbSS: homozygous sickle cell disease ITT: intention‐to‐treat MRA: magnetic resonance angiography MRI: magnetic resonance imaging MTD: maximum tolerated dose RBC: red blood cells RCT: randomised controlled trial SCI: silent cerebral infarct SD: standard deviation TCD: transcranial Doppler ultrasonography TIA: transient Ischaemic attack
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Bernaudin 2016 | Review. |
| DREPAGREFFE 2017 | Not a randomised trial. |
| EXTEND 2016 | Not a randomised trial. |
| Gwam 2016 | Review. |
| SACRED 2018 | Not a randomised trial. |
| SCATE 2015 | No red blood cell transfusion arm; hydroxyurea versus observation. |
| SPIN 2017 | Not a randomised trial. |
Differences between protocol and review
The title has been amended to include the terms 'primary and secondary'.
Excluded quasi‐randomised trials.
New subgroups TCD and silent cerebral infarcts.
Excluded outcome of SCI as it is being addressed within another review (Estcourt 2016).
Includes cluster‐randomised trials.
Included Summary of Findings tables.
Contributions of authors
The original review was conceived by the Cochrane Cystic Fibrosis and Genetic Disorders Group and designed by Dr Hirst (née Riddington).
In this most recent update all listed authors contributed to the review.
Lise Estcourt: searching; selection of trials; eligibility assessment; content expert, and review content update and development. Patricia Fortin: searching; selection of trials; eligibility assessment; and review content update and development. Sally Hopewell: methodological expert and review development. Marialena Trivella: statistical and methodological expert and review development Winfred Wang: protocol development, review development and content expert.
Sources of support
Internal sources
No sources of support supplied
External sources
-
National Institute for Health Research, UK
This systematic review was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to the Cochrane Cystic Fibrosis and Genetic Disorders Group.
Declarations of interest
Lise Estcourt: partly funded by the NIHR Cochrane Programme Grant ‐ Safe and Appropriate Use of Blood Components. Ruchitka Kohli: none known. Sally Hopewell: partly funded by the NIHR Cochrane Programme Grant ‐ Safe and Appropriate Use of Blood Components. Marialena Trivella: partly funded by the NIHR Cochrane Programme Grant ‐ Safe and Appropriate Use of Blood Components. Winfred Wang: was a PI on several of the included trials (STOP 1998; STOP 2 2005; SWiTCH 2012). He has been a consultant for Novartis and Agios and he has received payment fro lectures and educational presentations from Global Blood Therapeutics. These consultancies did not involve treatment that would be used in the management of stroke in people with SCD.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
SIT 2014 {published data only}
- Bean CJ, Boulet SL, Ellingsen D, Pyle ME, Barron-Casella EA, Casella JF, et al. Heme oxygenase-1 gene promoter polymorphism is associated with reduced incidence of acute chest syndrome among children with sickle cell disease. Blood 2012;120(18):3822-8. [CENTRAL: 899251] [EMBASE: 2012654658] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beverung LM, Strouse JJ, Hulbert ML, Neville K, Liem RI, Inusa B, et al. Health-related quality of life in children with sickle cell anemia: impact of blood transfusion therapy. American Journal of Hematology 2015;90(2):139-43. [CFGD REGISTER: SC184k] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatnagar P, Purvis S, Barron-Casella E, DeBaun MR, Casella JF, Arking DE, et al. Genome-wide association study identifies genetic variants influencing F-cell levels in sickle cell patients. Journal of Human Genetics 2011;56(4):316-23. [CFGD REGISTER: SC184e] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatnagar P, Purvis S, Barron-Casella E, DeBaun MR, Casella JF, Arking DE, et al. Genome-wide association study identifies genetic variants influencing F-cell levels in sickle-cell patients. Journal of Human Genetics 2011;56(4):316-23. Supplementary information. www.nature.com/jhg/journal/v56/n4/suppinfo/jhg201112s1.html?url=/jhg/journal/v56/n4/full/jhg201112a.html. [CFGD REGISTER: SC184f] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casella JF, King AA, Barton B, White DA, Noetzel MJ, Ichord RN, et al. Design of the silent cerebral infarct transfusion (SIT) trial. Pediatric Hematology and Oncology 2010;27(2):69-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury NA, DeBaun MR, Ponisio MR, Jordan LC, Rodeghier M, Pruthi S, et al. Intracranial vasculopathy and infarct recurrence in children with sickle cell anaemia, silent cerebral infarcts and normal transcranial Doppler velocities. British Journal of Haematology 2018;183(2):324-6. [CENTRAL: CN-01952385] [PMID: ] [DOI] [PMC free article] [PubMed]
- Darbari DS, Eigbire-Molen O, Ponisio MR, Milchenko MV, Rodeghier MJ, Casella JF, et al. Progressive loss of brain volume in children with sickle cell anemia and silent cerebral infarct: A report from the silent cerebral infarct transfusion trial. American Journal of Hematology 2018;12:E406-8. [DOI] [PubMed] [Google Scholar]
- Day ME, Rodeghier M, DeBaun MR. Children with HbSbeta(0) thalassemia have higher hemoglobin levels and lower incidence rate of acute chest syndrome compared to children with HbSS. Pediatric Blood & Cancer 2018;65(11):e27352. [CENTRAL: CN-01645299] [EMBASE: 623777045] [PMID: ] [DOI] [PubMed] [Google Scholar]
- DeBaun M. Epidemiology and treatment of silent strokes in sickle cell anemia. In: 28th Annual Meeting of the National Sickle Cell Disease Program; 2005 Apr 9-13; Cincinnati, Ohio. 2005:Plenary.
- *.DeBaun MR, Gordon M, McKinstry RC, Noetzel MJ, White DA, Sarnaik SA, et al. Controlled trial of transfusions for silent cerebral infarcts in sickle cell anemia. New England Journal of Medicine 2014;371:399-10. [NCT00072761: Silent Cerebral Infarct Transfusion Multi-Center Clinical Trial (SIT)] [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBaun MR, Sarnaik SA, Rodeghier MJ, Minniti CP, Howard TH, Iyer RV, et al. Associated risk factors for silent cerebral infarcts in sickle cell anemia: low baseline hemoglobin, sex, and relative high systolic blood pressure. Blood 2012;119(16):3684-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling MM, Noetzel MJ, Rodeghier MJ, Quinn CT, Hirtz DG, Ichord RN, et al. Headache and migraine in children with sickle cell disease are associated with lower hemoglobin and higher pain event rates but not silent cerebral infarction. Journal of Pediatrics 2014;164(5):1175-80. [CFGD REGISTER: SC184h] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eigbire-Molen O, Darbari DS, Ponisio MR, Milchenko MV, Rodeghier MJ, Casella JF, et al. Progressive loss of brain volume in children with sickle cell anemia: a report from the silent cerebral infarct transfusion trial cohort. Blood 2015;126(23):546. [CFGD REGISTER: SC184n] [DOI] [PubMed] [Google Scholar]
- Ford AL, Ragan DK, Fellah S, Binkley MM, Fields ME, Guilliams KP, et al. Silent infarcts in sickle cell disease occur in the border zone region and are associated with low cerebral blood flow. Blood 2018;132(16):1714-23. [CENTRAL: CN-01951923] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gay JC, McCavit T, Bundy D, King A, Lehman HP, Strouse JJ, et al. Cost-effectiveness of blood transfusions versus observation for silent cerebral infarcts from the silent cerebral infarct trial. Blood 2016;128(22). [CFGD REGISTER: SC184q] [3655] [Google Scholar]
- ISRCTN52713285. Silent cerebral infarct multi-center clinical trial. www.isrctn.com/ISRCTN52713285 First submitted 24 August 2004.
- Jordan LC, McKinstry RC 3rd, Kraut MA, Ball WS, Vendt BA, Casella JF, et al. Incidental findings on brain magnetic resonance imaging of children with sickle cell disease. Pediatrics 2010;126(1):53-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawadler JM, Clark CA, McKinstry RC, Kirkham FJ. Brain atrophy in paediatric sickle cell anaemia: findings from the silent infarct transfusion (SIT) trial. British Journal of Haematology 2017;177(1):151-3. [DOI] [PubMed] [Google Scholar]
- King AA, Rodeghier MJ, Panepinto JA, Strouse JJ, Casella JF, Quinn CT, et al. Silent cerebral infarction, income, and grade retention among students with sickle cell anemia. American Journal of Hematology 2014;89(10):E188-92. [CFGD REGISTER: SC184p] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT00072761. Silent cerebral infarct transfusion multi-center clinical trial (SIT). www.clinicaltrials.gov/ct2/show/NCT00072761 First submitted 10 November 2003.
- Ogunsile FJ, DeBaun MR, Currie K, Rodeghier MJ. Parvovirus B19 infection associated with silent cerebral infarcts: a secondary analysis of the silent cerebral infarct trial cohort. Blood 2015;126(23):3410. [CFGD REGISTER: SC184o] [DOI] [PubMed] [Google Scholar]
- Quinn CT, McKinstry RC, Dowling MM, Ball WS, Kraut MA, Casella JF, et al. Acute silent cerebral ischemia occurs more frequently than silent cerebral infarction in children with sickle cell anemia. Blood 2010;116(21):268. [Google Scholar]
- Roberts DO, Covert B, Rodeghier MJ, Parmar N, DeBaun MR, Thompson AA, et al. Randomization is not associated with socio-economic and demographic factors in a multi-center clinical trial of children with sickle cell anemia. Pediatric Blood & Cancer 2014;61(9):1529-35. [CFGD REGISTER: SC184m] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vendt BA, McKinstry RC, Ball WS, Kraut MA, Prior FW, Barton B, et al. Silent Cerebral Infarct Transfusion (SIT) trial imaging core: application of novel imaging information technology for rapid and central review of MRI of the brain. Journal of Digital Imaging 2009;22(3):326-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf RB, Saville BR, Roberts DO, Fissell RB, Kassim AA, Airewele G, et al. Factors associated with growth and blood pressure patterns in children with sickle cell anemia: Silent Cerebral Infarct Multi-Center Clinical Trial cohort. American Journal of Hematology 2015;90(1):2-7. [CFGD REGISTER: SC184l] [DOI] [PubMed] [Google Scholar]
- Land V, Heijboer H, Fijnvandraat K, DeBaun MR, Casella JF. Transfusions for silent cerebral infarcts in sickle cell anemia. New England Journal of Medicine 2014;371(19):1841-2. [CFGD REGISTER: SC184j] [DOI] [PubMed] [Google Scholar]
STOP 1998 {published data only}
- Abboud M, Cure J, Gallagher D, Berman B, Hsu L, Wang W, et al. Magnetic resonance angiography (MRA) in children with abnormal transcranial doppler (TCD) velocities in the STOP study. Blood 1999;94(10 Suppl 1):645a. [DOI] [PubMed] [Google Scholar]
- Abboud MR, Cure J, Gallagher D, Berman B, Hsu L, Wang W, et al. Magnetic resonance angiography in children with abnormal TCD in the STOP study. In: 23rd Annual Meeting of the National Sickle Cell Disease Program. 1999:49.
- Abboud MR, Cure J, Granger S, Gallagher D, Hsu L, Wang W, et al. Magnetic resonance angiography in children with sickle cell disease and abnormal transcranial Doppler ultrasonography findings enrolled in the STOP study. Blood 2004;103(7):2822-6. [DOI] [PubMed] [Google Scholar]
- Adamkiewicz T, Abboud M, Barredo J, Cavalier ME, Peterson J, Rackoff B, et al. Serum Ferritin in children with SCD on chronic transfusion: correlation with serum alanine aminotransferase and liver iron (STOP/STOP II liver iron ancillary study). In: 35th Anniversary Convention of the National Sickle Cell Disease Program and the Sickle Cell Disease Association of America; 2007 Sep 17-22; Washington DC, USA. 2007:316.
- Adamkiewicz T, Abboud MR, Barredo JC, Kirby-Allen M, Alvarez OA, Casella JF, et al. Serum ferritin in children with sickle cell disease on chronic transfusion: measure of iron overload or end organ injury? STOP/ STOP II liver iron ancillary study. In: Blood. Vol. 108. 2006. [ABSTRACT NO: 791]
- Adamkiewicz TV, Abboud MR, Paley C, Olivieri N, Kirby-Allen M, Vichinsky E, et al. Serum ferritin level changes in children with sickle cell disease on chronic blood transfusion are nonlinear and are associated with iron load and liver injury. Blood 2009;114(21):4632-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams R, Brambilla D. Clinical alert: periodic transfusions lower stroke risk in children with sickle cell anemia. www.nlm.nih.gov/databases/alerts/sickle97.html 2000;Accessed 20 January 2020. [CFGD REGISTER: SC4iii]
- Adams R, Fullerton H, Kwiatkowski J, Voeks J. Development of high risk TCD in sickle cell disease. Stroke 2015;46(Suppl 1). [ABSTRACT NO.: 17] [CFGD REGISTER: SC4ccc] [Google Scholar]
- Adams RJ, Brambilla DJ, Granger S, Gallagher D, Vichinsky E, Abboud MR, et al. Stroke and conversion to high risk in children screened with transcranial Doppler ultrasound during the STOP study. Blood 2004;103(10):3689-94. [DOI] [PubMed] [Google Scholar]
- Adams RJ, Brambilla DJ, McKie V, Files B, Vichinsky E, Abboud M, et al. Risk of stroke in children with sickle cell disease and abnormal transcranial doppler ultrasound (TCD). Blood 1999;94(10 Suppl 1):419a. [Google Scholar]
- Adams RJ, Brambilla DJ, McKie V, Files B, Vichinsky E, Abboud M, et al. Stroke prevention in sickle cell disease (STOP): Baseline characteristics of trial patients. In: 22nd Annual Meeting of the National Sickle Cell Disease Program. 1997:41.
- Adams RJ, Brambilla DJ, McKie V, Files B, Vichinsky E, Abboud M, et al. Stroke prevention in sickle cell disease (STOP): final results. Blood 2000;96(11 Pt 1):10a. [Google Scholar]
- Adams RJ, Brambilla DJ, McKie V, Files B, Vichinsky E, Abboud M, et al. Stroke prevention in sickle cell disease (STOP): final results. In: 25th Annual Meeting of the National Sickle Cell Disease Program. 2001. [ABSTRACT NO: #8]
- Adams RJ, Brambilla DJ, McKie V, Files B, Vichinsky E, Abboud M, et al. Stroke prevention in sickle cell disease (STOP): follow-up after closure of the trial. Blood 1998;92(10 Suppl 1):527a. [Google Scholar]
- Adams RJ, Brambilla DJ, McKie VC, Files B, Vichinsky E, Abboud M, et al. Stroke prevention in sickle cell disease (STOP): baseline characteristics of trial patients. Blood 1997;90(10 Suppl 1):123a. [Google Scholar]
- Adams RJ, Brambilla DJ, Vichinsky E, Abboud M, Pegelow C, Carl EM, et al. Risk of stroke and conversion to abnormal TCD in children screened with transcranial doppler (TCD) during the STOP study. In: National Sickle Cell Disease Program 30th Annual Meeting; 2002 Sept 17-21. 2002:3.
- Adams RJ, Brambilla DJ, Vichinsky E, Abboud M, Pegelow C, Carl EM, et al. Stroke prevention trial in sickle cell anemia (STOP study): Risk of stroke in 1933 children screened with transcranial doppler (TCD). In: 23rd Annual Meeting of the National Sickle Cell Disease Program. 1999:54.
- Adams RJ, Brambilla DJ. Stroke prevention trial in sickle cell anemia: STOP. In: 21st Annual Meeting of the National Sickle Cell Disease Program. 1996:19.
- Adams RJ, Carl EM, McKie VC, Odo NA, Kutlar A, Phillips M, et al. A pilot trial of hydroxyurea to prevent strokes in children with sickle cell anemia. In: 23rd Annual Meeting of the National Sickle Cell Disease Program. 1999:53.
- Adams RJ, Lackland DT, Brown L, Brown D, Voeks J, Fullerton HJ, et al. Transcranial Doppler Re-screening of Subjects who Participated in STOP and STOP. American Journal of Hematology 2016;91(12):1191-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams RJ, McKie V, Files B, Vichinsky E, Abboud M, Hsu L, et al. Stroke prevention in sickle cell disease (STOP): Results of transcranial doppler ultrasound screening stroke risk. Stroke 1998;29(1):308. [Google Scholar]
- Adams RJ, McKie VC, Brambilla DJ, Carl E, Gallagher D, Nichols FT, et al. Stroke prevention trial in sickle cell anemia. Controlled Clinical Trials 1998;19(1):110-29. [DOI] [PubMed] [Google Scholar]
- *.Adams RJ, McKie VC, Hsu L, Files B, Vichinsky E, Pegelow C, et al. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial doppler ultrasonography. New England Journal of Medicine 1998;339(1):5-11. [NCT00000592: Stroke Prevention in Sickle Cell Anemia (STOP 1)] [DOI] [PubMed] [Google Scholar]
- Adams RJ, McKie VC, Hsu L. Periodic blood transfusions reduced the risk for stroke in children with sickle-cell anaemia. Evidence-based Medicine 1999;4(2):54. [CENTRAL: CN-01762879] [EMBASE: 29159472]
- Adams RJ, Pavlakis S, Roach ES. Sickle cell disease and stroke: primary prevention and transcranial Doppler. Annals of Neurology 2003;54(5):559-63. [CENTRAL: CN-01715357] [EMBASE: 37346061] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Adams RJ. Lessons from the stroke prevention trial in sickle cell anemia (STOP) study. Journal of Child Neurology 2000;15(5):344-9. [NCT00000592: Stroke Prevention in Sickle Cell Anemia (STOP 1)] [DOI] [PubMed] [Google Scholar]
- Anonymous. Transfusion to prevent first stroke in children with sickle cell anemia. Journal of the American Ostoepathic Association 1998;98(7):363-4. [Google Scholar]
- Bulas DI, Jones A, Seibert JJ, Driscoll C, O'Donnell R, Adams RJ. Transcranial Doppler (TCD) screening for stroke prevention in sickle cell anemia: pitfalls in technique variation. Pediatric Radiology 2000;30(11):733-8. [DOI] [PubMed] [Google Scholar]
- Cohen AR. Sickle cell disease - new treatments, new questions. New England Journal of Medicine 1998;339(1):42-4. [DOI] [PubMed] [Google Scholar]
- Duncan GW. Ischemic stroke in sickle cell disease: a review. Tennessee Medicine 1997;90(12):498-9. [PubMed] [Google Scholar]
- Files B, Brambilla D, Kutlar A, Miller S, Pegelow C, Vichinsky E, et al. A randomized trial of chronic transfusion to prevent stroke in sickle cell anemia: changes in ferritin with chronic transfusion. Blood 1998;92(10 Suppl 1):528a. [Google Scholar]
- Files B, Brambilla D, Kutlar A, Miller S, Vichinsky E, Wang W, et al. Longitudinal changes in ferritin during chronic transfusion: a report from the Stroke Prevention Trial in Sickle Cell Anemia (STOP). Journal of Pediatric Hematology/Oncology 2002;24(4):284-90. [DOI] [PubMed] [Google Scholar]
- Gates A, Rogers MA, Puczynski M. Stroke prevention trial in sickle cell anemia: comments on effects of chronic transfusion on pain. Journal of Pediatrics 2002;141(5):742-3. [DOI] [PubMed] [Google Scholar]
- Hsu LL, Miller ST, Wright E, Kutlar A, McKie V, Wang W, et al. Alpha thalassemia is associated with decreased risk of abnormal transcranial doppler ultrasonography in children with sickle cell anemia. Journal of Pediatric Hematology/Oncology 2003;25(8):622-8. [DOI] [PubMed] [Google Scholar]
- Hyacinth HI, Adams RJ, Greenberg CS, Voeks JH, Hill A, Hibbert JM, et al. Effect of chronic blood transfusion on biomarkers of coagulation activation and thrombin generation in sickle cell patients at risk for stroke. Plos One 2015;10(8):e0134193. [CENTRAL: CN-01170147] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyacinth HI, Adams RJ, Voeks JH, Hibbert JM, Gee BE. Frequent red cell transfusions reduced vascular endothelial activation and thrombogenicity in children with sickle cell anemia and high stroke risk. American Journal of Hematology 2014;89(1):47-51. [CENTRAL: CN-00979157] [EMBASE: 2014063339] [PMID: ] [DOI] [PMC free article] [PubMed]
- Hyacinth HI, Gee BE, Voeks JH, Adams RJ, Hibbert J. High frequency of RBC transfusions in the STOP study was associated with reduction in serum biomarkers of neurodegeneration, vascular remodeling and inflammation. Blood 2012;120(21). [ABSTRACT NO: 244] [CENTRAL: 999896] [Google Scholar]
- Kanter J, Kwiatkowski J, Fullerton HJ, Voeks J, Debenham E, Brown LJ, et al. Impact of TCD screening protocol on the incidence of hemorrhagic stroke in children and young adults with sickle cell disease. Blood 2015;126(23):3402. [CFGD REGISTER: SC4eee] [Google Scholar]
- Kutlar A, Brambilla D, Clair B, Haghighat A, Bakanay S, Adams G, et al. Candidate gene polymorphisms and their association with TCD velocities in children with sickle cell disease. Blood 2007;110(11). [ABSTRACT NO: 429] [Google Scholar]
- Kutlar A, Harbin J, Jackson B, Holley L, Gallagher D, Clair B, et al. Laboratory parameters in patients randomized in the STOP study and their modification by transfusion. Blood 2000;96(11 Pt 2):18b-9b. [Google Scholar]
- Kutlar A, Harbin J, Jackson BB, Holley LGD, Clair B, Brambilla D, et al. Baseline laboratory parameters in patients randomized to the STOP study and their modification by transfusion. In: 24th Annual Meeting of the National Sickle Cell Disease Program. 2000:82a.
- Kwiatkowski JL, Brambilla DJ, Granger S, Adams RJ. Elevated blood flow velocity in the anterior cerebral artery and stroke risk in sickle cell disease. Blood 2003;102(11). [ABSTRACT NO: 408] [DOI] [PubMed] [Google Scholar]
- Kwiatkowski JL, Granger S, Brambilla DJ, Brown RC, Miller ST, Adams RJ. Elevated blood flow velocity in the anterior cerebral artery and stroke risk in sickle cell disease: extended analysis from the STOP trial. British Journal of Haematology 2006;134(3):333-9. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski JL, Kanter J, Fullerton HJ, Voeks J, Debenham E, Brown DG, et al. Ischemic stroke in children and young adults with sickle cell disease (SCD) in the post-stop era. Blood 2015;126(23):68. [CFGD REGISTER: SC4fff] [DOI] [PubMed] [Google Scholar]
- Kwiatkowski JL, Morales K, Brambilla DJ, Files B, Adamkiewicz T, Adams RJ. Long-term follow-up of transcranial doppler ultrasonography in children with sickle cell disease: Results of the STOP and STOP II patient cohorts. Blood 2002;100(11 Pt 1):663a. [Google Scholar]
- Lee MT, Piomelli S, Granger S, Miller ST, Harkness S, Brambilla DJ, et al. Stroke Prevention Trial in Sickle Cell Anemia (STOP): extended follow-up and final results. Blood 2006;108(3):847-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SB, Ramsingh D, Kutlar A, Holley L, McVie VC, Adams RJ. C-reactive protein in children with sickle cell disease at risk for stroke in the STOP study. Stroke 2002;33(1):373. [Google Scholar]
- Lezcano NE, Odo N, Kutlar A, Brambilla D, Adams RJ. Regular transfusion lowers plasma free hemoglobin in children with sickle-cell disease at risk for stroke. Stroke 2006;37(6):1424-6. [DOI] [PubMed] [Google Scholar]
- Lezcano NE, Odo N, Kutlar A, Brambilla D, Adams RJ. Transfusion lowers plasma-free hemoglobin in children with sickle cell disease at risk for stroke. Stroke 2006;37(2):695-6. [CFGD REGISTER: SC4ggg] [DOI] [PubMed] [Google Scholar]
- Miller ST, Wright E, Abboud M, Berman B, Files B, Scher C, et al. Impact of chronic transfusion on non-neurological events during the Stroke Prevention Trial in sickle cell anemia (STOP). Pediatric Research 2000;47(4 Pt 2):251A. [DOI] [PubMed] [Google Scholar]
- Miller ST, Wright E, Abboud M, Berman B, Files B, Scher C, et al. Impact of chronic transfusion on non-neurological events during the stroke prevention trial in sickle cell anemia (TOP). In: 24th Annual Meeting of the National Sickle Cell Disease Program. 2000:61a.
- Miller ST, Wright E, Abboud M, Berman B, Files B, Scher CD, et al. Impact of chronic transfusion on incidence of pain and acute chest syndrome during the Stroke Prevention Trial (STOP) in sickle-cell anemia. Journal of Pediatrics 2001;139(6):785-9. [DOI] [PubMed] [Google Scholar]
- NCT00000592. Stroke Prevention in Sickle Cell Anemia (STOP 1). www.clinicaltrials.gov/show/nct00000592 (first received 1999 October 28). [CENTRAL: CN-01975776]
- Nichols FT, Jones AM, Adams RJ. Stroke prevention in sickle cell disease (STOP) study guidelines for transcranial Doppler testing. Journal of Neuroimaging 2001;11(4):354-62. [DOI] [PubMed] [Google Scholar]
- Olivieri N, Brambilla D, McKie V, Piomelli S, Kutlar A, Files B, et al. Changes in cerebral blood flow velocities during chronic transfusion therapy to prevent stroke in sickle cell disease. Blood 2000;96(11 Pt 1):486a. [Google Scholar]
- Pegelow CH, Adams R, Hsu L, McKie V, Wang W, Zimmerman R, et al. Children with silent infarct and elevated transcranial doppler ultrasonography velocity are at increased risk of subsequent infarctive events. In: 23rd Annual Meeting of the National Sickle Cell Disease Program. 1999:137.
- Pegelow CH, Wang W, Granger S, Hsu LL, Vichinsky E, Moser FG, et al. Silent infarcts in children with sickle cell anemia and abnormal cerebral artery velocity. Archives of Neurology 2001;58(12):2017-21. [DOI] [PubMed] [Google Scholar]
- Sarnaik SA. Prevention of stroke by transfusions in children with sickle cell anemia. New England Journal of Medicine 1998;339:1477-8. [CFGD REGISTER: SC4hhh] [PubMed] [Google Scholar]
- Sayer G, Bowman L, Clair B, Cail A, Blanchard B, Natrajan K, et al. Long term outcome of patients enrolled into STOP and STOP II trials: a single center experience. Blood 2012;120(21). [ABSTRACT NO: 3219] [CENTRAL: 999895] [Google Scholar]
- Styles L, De Jong K, Vichinsky E, Lubin B, Adams R, Kuypers F. Increased RBC phosphatidylserine exposure in sickle cell disease patients at risk for stroke by transcranial Doppler screening. Blood 1997;90(10 Suppl 1):604a-5a. [Google Scholar]
- Vichinsky E, Luban E, Wright E, Olivieri N, Driscoll C, Pegelow C, et al. Prospective red cell phenotype matching in STOP - a multi-centre transfusion trial. In: 23rd Annual Meeting of the National Sickle Cell Disease Program. 1999:163.
- Vichinsky E, Luban N, Wright E, Olivieri N, Driscoll C, Pegelow C, et al. Prospective red cell phenotype matching in STOP - a multi-center transfusion trial. Blood 1998;92(10 Suppl 1):528a. [Google Scholar]
- Vichinsky EP, Luban NL, Wright E, Olivieri N, Driscoll C, Pegelow CH, et al. Prospective RBC phenotype matching in a stroke-prevention trial in sickle cell anemia: a multicenter transfusion trial. Transfusion 2001;41(9):1086-92. [DOI] [PubMed] [Google Scholar]
- Wang W, Morales K, Olivieri N, Styles L, Scher C, Adams R, et al. Effect of chronic transfusion on growth in children with sickle cell anemia: results of the STOP trial. In: National Sickle Cell Disease Program 30th Annual Meeting Conference; 2002 Sept 17-21. 2002:100.
- Wang WC, Morales KH, Scher CD, Styles L, Olivieri N, Adams R, et al. Effect of long-term transfusion on growth in children with sickle cell anemia: results of the STOP trial. Journal of Pediatrics 2005;147(2):244-7. [DOI] [PubMed] [Google Scholar]
STOP 2 2005 {published and unpublished data}
- ASH News Daily 2004. NHLBI stops the STOP II trial early. www.hematology.org/publications/newsdaily/2004/issue_3/NHLBI.cfm (accessed 22 November 2005).
- Abboud MR, Yim E, Adams RJ. The progression and development of silent infarcts in children with sickle cell anemia is prevented by chronic transfusions and is unrelated to level of hemolysis. Blood 2008;12. [ABSTRACT NO: 712] [Google Scholar]
- Abboud MR, Yim E, Musallam KM, Adams, RJ. Discontinuing prophylactic transfusions increases the risk of silent brain infarction in children with sickle cell disease: data from STOP II. Blood 2011;118(4):894-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamkiewicz T, Abboud M, Barredo J, Cavalier ME, Peterson J, Rackoff B, et al. Serum Ferritin in children with SCD on chronic transfusion: correlation with serum alanine aminotransferase and liver iron (STOP/STOP II liver iron ancillary study). In: 35th Anniversary Convention of the National Sickle Celll Disesae Program and the Sickle Cell Disease Association of America; 2007 Sep 17-22; Washington DC, USA. 2007:316.
- Adamkiewicz T, Abboud MR, Barredo JC, Kirby-Allen M, Alvarez OA, Casella JF, et al. Serum ferritin in children with sickle cell disease on chronic transfusion: measure of iron overload or end organ injury? STOP/ STOP II liver iron ancillary study. Blood 2006;108(11). [ABSTRACT NO: 791] [Google Scholar]
- Adamkiewicz T. Transcranial doppler measures in patients with sickle cell disease at high risk for stroke and receiving hydroxyurea: the HyRetro ancillary study. In: 52nd ASH Annual Meeting and Exposition; 2010 Dec 4-7; Orlando, Florida. 2010. [ABSTRACT NO: 1620]
- Adamkiewicz TV, Abboud MR, Paley C, Olivieri N, Kirby-Allen M, Vichinsky E, et al. Serum ferritin level changes in children with sickle cell disease on chronic blood transfusion are nonlinear and are associated with iron load and liver injury. Blood 2009;114(21):4632-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams RJ, Brambilla D, Miller ST. Optimizing primary stroke prevention in children with sickle cell. In: 28th Annual Meeting of the National Sickle Cell Disease Program; 2005 April 9-13; Cincinnati, Ohio. 2005:3.
- *.Adams RJ, Brambilla D. Discontinuing prophylactic transfusions used to prevent stroke in sickle cell disease. New England Journal of Medicine 2005;353(26):2769-78. [NCT00006182: Stroke Prevention in Sickle Cell Anemia (STOP 2)] [DOI] [PubMed] [Google Scholar]
- Adams RJ, Brambilla DJ, STOP II investigators. The study design of optimizing primary stroke prevention in children with sickle cell anemia. In: 27th Annual Meeting of the National Sickle Cell Disease Program; 2004 April 18-21; Los Angeles, California. 2004:174.
- Alvarez O, Miller S, Berman B, Brown C, Casella J, Coates T, et al. Evaluation of chronic transfusion practices in children with sickle cell disease: a survey of STOP II investigators. Blood 2004;104(11). [ABSTRACT NO: 3732] [3732] [Google Scholar]
- Brambilla DJ, Adams RJ. Stroke prevention in sickle cell anemia (STOP 2). National Institutes of Health, Clinical Trials Database Started in 06/2000.
- Brown C, Miller S, Kwiatkowski J, Brambilla D, Adams R. Optimizing primary stroke prevention in sickle cell anemia (STOP 2): an argument for prolonged transfusion? In: 29th Annual Meeting of the National Sickle Cell Disease Program; 2006 April 8-12; Memphis, USA. 2006:78.
- Kanter J, Kwiatkowski J, Fullerton HJ, Voeks J, Debenham E, Brown LJ, et al. Impact of TCD screening protocol on the incidence of hemorrhagic stroke in children and young adults with sickle cell disease. Blood 2015;126(23):3402. [CFGD REGISTER: SC4eee] [Google Scholar]
- Kutlar A, Brambilla D, Clair B, Haghighat A, Bakanay S, Adams G, et al. Candidate gene polymorphisms and their association with TCD velocities in children with sickle cell disease. Blood 2007;110(11). [ABSTRACT NO: 429] [ABSTRACT NO.: 429] [Google Scholar]
- Kwiatkowski JL, Kanter J, Fullerton HJ, Voeks J, Debenham E, Brown DG, et al. Ischemic stroke in children and young adults with sickle cell disease (SCD) in the post-stop era. Blood 2015;126(23):68. [CFGD REGISTER: SC4fff] [DOI] [PubMed] [Google Scholar]
- Kwiatkowski JL, Morales K, Brambilla DJ, Files B, Adamkiewicz T, Adams RJ, et al. Long-term follow-up of transcranial doppler ultrasonography in children with sickle cell disease: results of the STOP and STOP II patient cohorts. Blood 2002;100(11 Pt 1):663a. [Google Scholar]
- Medical College of Georgia. STOP II Questions and Answers. www.mcg.edu/neurology/Research/sicklecell/STOPIIQandA.htm (accessed 22 November 2005).
- Medical News Today. NHLBI stops sickle cell anemia transfusion study. www.medicalnewstoday.com/printerfriendlynews.php?newsid=17363 (accessed 22 November 2005).
- Sayer G, Bowman L, Clair B, Cail A, Blanchard B, Natrajan K, et al. Long term outcome of patients enrolled into STOP and STOP II trials: a single center experience. Blood 2012;120(21). [ABSTRACT NO: 3219] [CENTRAL: 999895] [Google Scholar]
SWiTCH 2012 {published data only}
- Alvarez O, Yovetich NA, Scott JP, Owen W, Miller ST, Schultz W, et al. Pain and other non-neurological adverse events in children with sickle cell anemia and previous stroke who received hydroxyurea and phlebotomy or chronic transfusions and chelation: results from the SWiTCH clinical trial. American Journal of Hematology 2013;88(11):932-8. [CENTRAL: 963136] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anonymous. Stroke prevention study in children with sickle cell anemia, iron overload stopped early. www.nih.gov/news/health/jun2010/nhlbi-03.htm 2010;Accessed 20 January 2020. [CFGD REGISTER: SC220l]
- Aygun B, Mortier NA, Kesler K, Lockhart A, Schultz WH, Cohen AR, et al. Therapeutic phlebotomy is safe in children with sickle cell anaemia and can be effective treatment for transfusional iron overload. British Journal of Haematology 2015;169(2):262-6. [CFGD REGISTER: SC220k] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aygun B, Mortier NA, Kesler K, Schultz WH, Alvarez OA, Rogers ZR, et al. Therapeutice phlebotomy in children with sickle cell anemia, stroke, and iron overload: the SWiTCH experience. In: 53rd ASH Annual Meeting and Exposition; 2011 Dec 10-13; San Diego, California. 2011. [ABSTRACT NO: 1044]
- Helton KJ, Adam RJ, Kesler KL, Lockhart A, Aygun B, Driscoll C, et al. Magnetic resonance imaging/angiography and transcranial Doppler velocities in sickle cell anemia: results from the SWiTCH trial. Blood 2014;124(6):891-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski JL, Cohen AR, Garro J, Alvarez O, Nagasubramanian R, Sarnaik S, et al. Transfusional iron overload in children with sickle cell anemia on chronic transfusion therapy for secondary stroke prevention. American Journal of Hematology 2012;87(2):221-3. [CENTRAL: 864015] [PMID: ] [DOI] [PubMed] [Google Scholar]
- NCT00122980. Stroke with transfusions changing to hydroxyurea (SWiTCH). www.clinicaltrials.gov/show/NCT00122980 First submitted 20 July 2005.
- Sheehan VA, Howard TA, Sabo A, Nagasaswamy U, Crosby JR, Davis B, et al. Genetic predictors of hemoglobin F response to hydroxyurea in sickle cell anemia. Blood 2012;120(21). [ABSTRACT NO: 241] [CENTRAL: 977454] [Google Scholar]
- *.Ware RE, Helms RW, SWiTCH Investigators. Stroke with transfusions changing to hydroxyurea (SWiTCH). Blood 2012;119(17):3925-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware RE, Helms RW. Stroke with transfusions changing to hydroxyurea (SWiTCH): a phase 3 randomised clinical trial for treatment of children with sickle cell anemia. In: 52nd ASH Meeting and Exposition; 2010 Dec 4-7; Orlando, Florida. 2010. [ABSTRACT NO: 844]
- Ware RE, McMurray MA, Schultz WH, Alvarez OA, Aygun B, Cavalier ME, et al. Academic community standards for chronic transfusion therapy in children with sickle cell anemia and stroke. Blood 2006;108(11). [ABSTRACT NO: 1213] [Google Scholar]
- Ware RE, Schultz WH, Yovetich N, Mortier NA, Alvarez O, Hilliard L, et al. Stroke with transfusions changing to hydroxyurea (SWiTCH): A phase III randomized clinical trial for treatment of children with sickle cell anemia, stroke, and iron overload. Pediatric Blood & Cancer 2011;57(6):1011-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
TWiTCH 2016 {published data only}
- Aygun B, Mortier N, Rogers ZR, Owen W, Fuh B, George A, et al. Iron unloading by therapeutic phlebotomy in previously transfused children with sickle cell anemia: the twitch experience. Blood 2016;128(22). [CFGD REGISTER: SC276i] [1018] [Google Scholar]
- Aygun B, Wruck LM, Schultz WH, Mueller BU, Brown C, Luchtman-Jones L, et al. Chronic transfusion practices for prevention of primary stroke in children with sickle cell anemia and abnormal TCD velocities. American Journal of Hematology 2012;87(4):428-30. [CENTRAL: 1139772] [PMID: ] [DOI] [PubMed] [Google Scholar]
- De Montalembert M, Benkerrou M, Grosse R, Kordes U, Brousse V, Pondarre C, et al. Assessment of hematological data in a cohort of european children with sickle cell anemia treated with hydroxyurea: can European centers apply today the lessons from the twitch study? Blood 2016;128(22). [CFGD REGISTER: SC276k] [2494] [Google Scholar]
- Helton KJ, Roberts D, Schultz WH, Davis BR, Kalfa TA, Pressel SL, et al. Effects of chronic transfusion therapy on MRI and MRA in children with sickle cell anemia at risk for primary stroke: baseline imaging from the Twitch Trial. Blood 2014;124(21):4052. [Google Scholar]
- Imran H, Aygun B, Davis BR, Pressel SL, Schultz WH, Jackson S, et al. Effects of chronic transfusion therapy on transcranial doppler ultrasonography velocities in children with sickle cell anemia at risk for primary stroke: Baseline findings from the twitch trial. Blood 2014;124(21):87. [Google Scholar]
- NCT01425307. Transcranial doppler (TCD) with transfusions changing to hydroxyurea (TWiTCH). www.clinicaltrials.gov/ct2/show/NCT01425307 First submitted 19 August 2011.
- Ware RE, Davis BR, Schultz WH, Brown C, Aygun B, Sarnaik SA, et al. TCD with transfusions changing to hydroxyurea (TWITCH): Hydroxyurea therapy as an alternative to transfusions for primary stroke prevention in children with sickle cell anemia. Blood 2015;126(23):3. [Google Scholar]
- *.Ware RE, Davis BR, Schultz WH, Brown RC, Aygun B, Sarnaik S, et al. Hydroxycarbamide versus chronic transfusion for maintenance of transcranial doppler flow velocities in children with sickle cell anaemia - TCD with Transfusions Changing to Hydroxyurea (TWiTCH): A multicentre, open-label, phase 3, non-inferiority trial. Lancet 2016;387(10019):661-70. [CENTRAL: 1138971] [EMBASE: 20160126001] [NCT01425307: TCD With Transfusions Changing to Hydroxyurea (TWiTCH): A Phase III Randomized Trial to Compare Standard Therapy (Erythrocyte Transfusions) With Alternative Therapy (Hydroxyurea) for the Maintenance of Lowered TCD Velocities in Pediatric Subjects With Sickle Cell Anemia and Abnormal Pre-treatment TCD Velocities] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware RE, Lane A, Schultz WH, Imran H, Lee MT, Heeney MM, et al. Variation in serial TCD velocity measurements in the TCD with transfusions changing to hydroxyurea (TWiTCH) trial. Blood 2016;128(22):1019. [CFGD REGISTER: SC276n] [Google Scholar]
- Wood JC, Cohen A, Aygun B, Imran H, Luchtman-Jones L, Thompson AA, et al. Extrahepatic iron deposition in chronically transfused children with sickle cell anemia - Baseline findings from the twitch trial. Blood 2013;122(21):2238. [Google Scholar]
- Wood JC, Cohen AR, Pressel SL, Aygun B, Imran H, Luchtman-Jones L, et al. Organ iron accumulation in chronically transfused children with sickle cell anaemia: Baseline results from the TWiTCH trial. British Journal of Haematology 2016;172(1):122-30. [CENTRAL: 1133502] [EMBASE: 20151011308] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JC, Ofelia A, Heeney MM, George A, Gauger C, Fuh B, et al. Changes in extrahepatic iron load in response to iron chelation versus phlebotomy: observations from the twitch trial. Blood 2016;128(22). [ABSTRACT NO.: 202] [CFGD REGISTER: SC276j] [Google Scholar]
- Wood JC, Pressel S, Rogers ZR, Odame I, Kwiatkowski JL, Lee MT, et al. Liver iron concentration measurements by MRI in chronically transfused children with sickle cell anemia: Baseline results from the TWiTCH trial. American Journal of Hematology 2015;90(9):806-10. [CENTRAL: 1090263] [EMBASE: 2015311270] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JC, St Pierre T, Aygun B, Mortier N, Schultz WH, Piccone CM, et al. Agreement between R2 and R2* liver iron estimates is independent of the type of iron removal therapy: results from the twitch trial. Blood 2016;128(22). [ABSTRACT NO.: 1274] [Google Scholar]
References to studies excluded from this review
Bernaudin 2016 {published data only}
- Bernaudin F, Verlhac S, Chevret S. Treating sickle cell anaemia: the TWiTCH trial. Lancet 2016;388(10048):960. [DOI] [PubMed] [Google Scholar]
DREPAGREFFE 2017 {published data only}
- Bernaudin F, Drain E, Dalle JH, Leveille E, Peffault De Latour R, Brousse V, et al. Health-related quality of life at 1 year in the drepagreffe trial comparing chronic transfusion to transplantation in children with sickle cell anemia and a history of abnormal cerebral velocities. Blood 2017;130(S1):617. [Google Scholar]
- Bernaudin F, Verlhac S, Ducros Miralles E, Peffault Delatour R, Dalle JH, Paillard C, et al. Trial comparing HSCT vs transfusions in sickle cell anemia (SCA)-patients with abnormal cerebral velocities: cerebral vasculopathy outcome at 1 year. Bone Marrow Transplantation 2017;51(1):S49-50. [Google Scholar]
- Bernaudin F, Verlhac S, Ducros-Miralles E, Brousse V, Thuret I, Paillard C, et al. Children with sickle cell anemia on chronic transfusion for abnormal transcranial doppler velocities. Patient/ sibling comparison of cognitive performances and association with neuroimaging data. Haematologica 2016;101:168. [Google Scholar]
- Bernaudin FV, Ducros-Miralles S, Ducros-Miralles E, Delatour RP, Dalle J-H, Petras E, et al. French national drepagreffe trial: Cognitive performances and neuroimaging at enrollment and after 12 months on transfusion program or transplantation (AP-HP: NCT 01340404). Blood 2015;126(23):544. [Google Scholar]
- Bernaudin M, Khelif Y, Chadebech P, Bodivit G, Jouard A, Verlhac S, et al. Probability of TCD-Normalization in the "Drepagreffe" Trial Comparing Transplantation to Chronic Transfusion in Sickle Cell Anemia Children with Abnormal-Transcranial Doppler Is Associated with Lower Ang-2 and BDNF Plasma Levels. Blood 2018;132(S1):506. [Google Scholar]
- *.Chevret S, Verlhac S, Ducros-Miralles E, Dalle JH, Latour RP, Montalembert M, et al. Design of the DREPAGREFFE trial: A prospective controlled multicenter study evaluating the benefit of genoidentical hematopoietic stem cell transplantation over chronic transfusion in sickle cell anemia children detected to be at risk of stroke by transcranial Doppler (NCT 01340404). Contemporary Clinical Trials 2017;62:91-104. [DOI] [PubMed] [Google Scholar]
EXTEND 2016 {published data only}
- Rankine-Mullings A, Badaloo A, Soares D, Little C, Schultz W, Taylor-Bryan C, et al. Transcranial doppler ultrasonography (TCD), organ function, metabolic and nutritional status in children with sickle cell anemia: baseline data from the EXTEND trial. Pediatric Blood & Cancer 2017;64:S24. [Google Scholar]
- *.Rankine-Mullings AE, Little CR, Reid ME, Soares DP, Taylor-Bryan C, Knight-Madden JM, et al. EXpanding Treatment for Existing Neurological Disease (EXTEND): An Open-Label Phase II Clinical Trial of Hydroxyurea Treatment in Sickle Cell Anemia. JMIR Research Protocols 2016;5(3):e185. [DOI] [PMC free article] [PubMed] [Google Scholar]
Gwam 2016 {published data only}
- Gwam M, Nwosu C. Treating sickle cell anaemia: the TWiTCH trial. Lancet 2016;388(10048):960-1. [DOI] [PubMed] [Google Scholar]
SACRED 2018 {published data only}
- Jeste ND, Sanchez LM, Urcuyo GS, Berges ME, Luden JP, Stuber SE, et al. Stroke Avoidance for Children in REpublica Dominicana (SACRED): Protocol for a Prospective Study of Stroke Risk and Hydroxyurea Treatment in Sickle Cell Anemia. JMIR Research Protocols 2017;6(6):e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez LM, Nieves RM, Latham T, Stuber S, Luden JR, Urcuyo GS, et al. Building capacity to reduce stroke in children with sickle cell anemia in the Dominican Republic: the SACRED trial. Blood Advances 2018;2(Suppl 1):50-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
SCATE 2015 {published data only}
- Hankins JS, McCarville MB, Rankine-Mullings A, Reid ME, Lobo CL, Moura PG, et al. Prevention of conversion to abnormal transcranial Doppler with hydroxyurea in sickle cell anemia: A Phase III international randomized clinical trial. American Journal of Hematology 2015;90(12):1099-105. [CFGD REGISTER: SC279b] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT01531387. Sparing conversion to abnormal TCD (transcranial doppler) elevation (SCATE). www.clinicaltrials.gov/show/NCT01531387 First submitted 6 February 2012. [CFGD REGISTER: SC279a]
SPIN 2017 {published data only}
- Galadanci NA, Abdullahi SU, Vance LD, Tabari M, Abubakar S, Raymond B, et al. Feasibility trial for primary stroke prevention in children with sickle cell anemia in Nigeria (SPIN trial). Blood 2016;128(22):122. [Google Scholar]
- *.Galadanci NA, Umar Abdullahi S, Vance LD, Musa Tabari A, Ali S, Belonwu R, et al. Feasibility trial for primary stroke prevention in children with sickle cell anemia in Nigeria (SPIN trial). American Journal of Hematology 2017;92(8):780-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galadanci NA, Umar Abdullahi S, Vance LD, Musa Tabari A, Ali S, Belonwu R, et al. Feasibility trial for primary stroke prevention in children with sickle cell anemia in Nigeria (SPIN trial). American Journal of Hematology 2018;93(3):E83. [DOI] [PubMed] [Google Scholar]
Additional references
Adams 1992
- Adams RJ, Nichols FT, Figueroa R, McKie V, Lott T. Transcranial Doppler correlation with cerebral angiography in sickle cell disease. Stroke 1992;23(8):1073-7. [DOI] [PubMed] [Google Scholar]
Adams 1998a
- Adams RJ, McKie VC, Brambilla D, Carl E, Gallagher D, Nichols FT, et al. Stroke prevention trial in sickle cell anemia. Controlled Clinical Trials 1998;19(1):110-29. [DOI] [PubMed] [Google Scholar]
Adams 1998b
- Adams RJ, McKie VC, Hsu L, Files B, Vichinsky E, Pegelow C, et al. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial doppler ultrasonography. New England Journal of Medicine 1998;339(1):5-11. [DOI] [PubMed] [Google Scholar]
Ansong 2013
- Ansong D, Osei Akoto A, Ocloo D, Ohene-Frempong K. Sickle cell disease: management options and challenges in developing countries. Mediterranean Journal of Hematology and Infectious Diseases 2013;5(1):e2013062. [DOI] [PMC free article] [PubMed] [Google Scholar]
Aygun 2009
- Aygun B, McMurray MA, Schultz WH, Kwiatkowski JL, Hilliard L, Alvarez O, et al. Chronic transfusion practice for children with sickle cell anaemia and stroke. British Journal of Haematology 2009;145(4):524-8. [DOI] [PubMed] [Google Scholar]
Bernaudin 2011
- Bernaudin F, Verlhac S, Arnaud C, Kamdem A, Chevret S, Hau I, et al. Impact of early transcranial Doppler screening and intensive therapy on cerebral vasculopathy outcome in a newborn sickle cell anemia cohort. Blood 2011;117(4):1130-40. [DOI] [PubMed] [Google Scholar]
Chou 2013
- Chou ST, Jackson T, Vege S, Smith-Whitley K, Friedman DF, Westhoff CM. High prevalence of red blood cell alloimmunization in sickle cell disease despite transfusion from Rh-matched minority donors. Blood 2013;122(6):1062-1071. [DOI] [PubMed] [Google Scholar]
Cohen 1992
- Cohen AR, Martin MB, Silber JH, Kim HC, Ohene-Frempong K, Schwarz E. A modified program for prevention of stroke in sickle cell disease. Blood 1992;79(7):1657-61. [PubMed] [Google Scholar]
Cohen 1996
- Cohen AR, Norris CF, Smith-Whitley K. Transfusion therapy for sickle cell disease. In: Capon SM, Chambers LA, editors(s). New Directions in Pediatric Hematology. Bethesda MD: American Association of Blood Banks, 1996:39-85. [Google Scholar]
Covidence 2015 [Computer program]
- Covidence systematic review software. Version accessed prior to 28 November 2016. Melbourne, Australia: Veritas Health Innovation, 2015. Available at www.covidence.org.
DeBaun 2006
- DeBaun MR, Derdeyn CP, McKinstry RC. Etiology of strokes in children with sickle cell anemia. Mental Retardation and Developmental Disabilities Research Reviews 2006;12(3):192-9. [DOI] [PubMed] [Google Scholar]
Deeks 2011
- Deeks JJ, Higgins JP, Altman DG. Chapter 9: Analysing data and undertaking meta-analyses. In: Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Enninful‐Eghan 2010
- Enninful-Eghan H, Moore RH, Ichord R, Smith-Whitley K, Kwiatkowski JL. Transcranial doppler ultrasonography and prophylactic transfusion program is effective in preventing overt stroke in children with sickle cell disease. Journal of Pediatrics 2010;157(3):479-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
Estcourt 2016
- Estcourt LJ, Fortin PM, Hopewell S, Trivella M, Doree C, Abboud MR. Interventions for preventing silent cerebral infarcts in people with sickle cell disease. Cochrane Database of Systematic Reviews 2016, Issue 10. Art. No: CD012389. [DOI: 10.1002/14651858.CD012389] [DOI] [PMC free article] [PubMed] [Google Scholar]
Fullerton 2004
- Fullerton HJ, Adams RJ, Zhao S, Johnston SC. Declining stroke rates in Californian children with sickle cell disease. Blood 2004;104(2):336-9. [DOI] [PubMed] [Google Scholar]
Gravitz 2014
- Gravitz L, Pincock S. Sickle-cell disease. Nature 2014;515:S1. [DOI] [PubMed] [Google Scholar]
Grosse 2011
- Grosse SD, Odame I, Atrash HK, Amendah DD, Piel FB, PhD, et al. Sickle cell disease in Africa. A neglected cause of early childhood mortality. American Journal of Preventive Medicine 2011;41:S398-405. [DOI] [PMC free article] [PubMed] [Google Scholar]
Guilliams 2015
- Guilliams KP, Fields ME, Hulbert ML. Higher-than-expected prevalence of silent cerebral infarcts in children with hemoglobin SC disease. Blood 2015;125:416-7. [DOI] [PubMed] [Google Scholar]
Higgins 2011a
- Higgins JP, Deeks JJ. Chapter 7: Selecting studies and collecting data. In: Higgins JP, Green S, editors(s).Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Higgins 2011b
- Higgins JP, Altman DG, Sterne JA. Chapter 8: Assessing risk of bias in included studies. In: Higgins JP, Green S, editor(s).Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Higgins 2011c
- Higgins JP, Deeks JJ, Altman DG. Chapter 16: Special topics in statistics. In: Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Inati 2011
- Inati A, Khoriaty E, Musallam KM. Iron in sickle-cell disease: what have we learned over the years? Pediatric Blood & Cancer 2011;56(2):182-90. [DOI] [PubMed] [Google Scholar]
Kinney 1999
- Kinney TR, Sleeper LA, Wang WC, Zimmerman RA, Pegelow CH, Ohene-Frempong K, et al. Silent cerebral infarcts in sickle cell anemia: a risk factor analysis. Pediatrics 1999;103(3):640-5. [DOI] [PubMed] [Google Scholar]
Kwiatkowski 2009
- Kwiatkowski JL, Zimmerman RA, Pollock AN, Seto W, Smith-Whitley K, Shults J, et al. Silent infarcts in young children with sickle cell disease. British Journal of Haemtology 2009;146(3):300-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lee 2006
- Lee MT, Piomelli S, Granger S, Miller ST, Harkness S, Brambilla DJ, et al. Stroke Prevention Trial in Sickle Cell Anemia (STOP): extended follow-up and final results. Blood 2006;108(3):847-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
McCarville 2008
- McCarville MB, Goodin GS, Fortner G, Li CS, Smeltzer MP, Adams R, et al. Evaluation of a comprehensive transcranial doppler screening program for children with sickle cell anemia. Pediatric Blood & Cancer 2008;50(4):818-21. [DOI] [PubMed] [Google Scholar]
Nagel 2003
- Nagel RL, Fabry ME, Steinberg MH. The paradox of hemoglobin SC disease. Blood Reviews 2003;17(3):167-78. [DOI] [PubMed] [Google Scholar]
NICE 2010
- (NICE) National Institute for Health and Care Excellence. Sickle cell disease 2010. cks.nice.org.uk/sickle-cell-disease#!backgroundsub:3 (accessed April 2016).
Ohene‐Frempong 1999
- Ohene-Frempong K. Sickle cell disease in the United States of America and Africa. In: Hematology (American Society of Hematology Education Program). American Society of Hematology, 1999:64-72.
Pegelow 1995
- Pegelow CH, Adams RJ, McKie V, Abboud M, Berman B, Miller ST, et al. Risk of recurrent stroke in patients with sickle cell disease treated with erythrocyte transfusions. Journal of Pediatrics 1995;126(6):896-9. [DOI] [PubMed] [Google Scholar]
Piel 2012
- Piel FB, Patil AP, Howes RE, Nyangiri OA, Gething PW, Dewi M, et al. Global epidemiology of sickle haemoglobin in neonates: a contemporary geostatistical model-based map and population estimates. Lancet 2012;381(9861):142-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
Pleasants 2014
- Pleasants S. Epidemiology: a moving target. Nature 2014;515(7526):S2-3. [DOI] [PubMed] [Google Scholar]
Powars 2000
- Powars DR. Management of cerebral vasculopathy in children with sickle cell anaemia. British Journal of Hematology 2000;108(4):666-78. [DOI] [PubMed] [Google Scholar]
Rana 1997
- Rana S, Houston PE, Surana N, Shalaby-Rana EI, Castro OL. Discontinuation of long-term transfusion therapy in patients with sickle cell disease and stroke. Journal of Pediatrics 1997;131(5):757-60. [DOI] [PubMed] [Google Scholar]
Rees 2010
- Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet 2010;376(9757):2018-31. [DOI] [PubMed] [Google Scholar]
Review Manager 5.3 [Computer program]
- Review Manager (RevMan). Version 5.3. Copenhagen: Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Rumaney 2014
- Rumaney MB, Ngo Bitoungui VJ, Vorster AA, Ramesar R, Kengne AP, Ngogang J, et al. The co-inheritance of alpha-thalassemia and sickle cell anemia Is associated with better hematological indices and lower consultations rate in Cameroonian patients and could improve their survival. PLoS ONE 2014;9:e100516. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sarnaik 1979
- Sarnaik S, Soorya D, Kim J, Ravindranath Y, Lusher J. Periodic transfusions for sickle cell anemia and CNS infarction. American Journal of Diseases of Children 1979;133(12):1254-7. [DOI] [PubMed] [Google Scholar]
Schünemann 2011a
- Schünemann HJ, Oxman AD, Higgins JP, Vist GE, Glasziou P, Guyatt GH. Chapter 11: Presenting results and 'Summary of findings' tables. In: Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011.. Available from handbook.cochrane.org.
Schünemann 2011b
- Schünemann HJ, Oxman AD, Vist GE, Higgins JP, Deeks JJ, Glasziou P, et al. Chapter 12: Interpreting results and drawing conclusions. In: Higgins JP, Green S, editors(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Scothorn 2002
- Scothorn DJ, Price C, Schwartz D, Terrill C, Buchanan GR, Shurney W, et al. Risk of recurrent stroke in children with sickle cell disease receiving blood transfusion therapy for at least five years after initial stroke. Journal of Pediatrics 2002;140(3):48-54. [DOI] [PubMed] [Google Scholar]
Serjeant 1992
- Serjeant GR. Surgery and anaesthesia. In: Sickle Cell Disease. 2nd edition. Oxford: Oxford University Press, 1992:455-8. [Google Scholar]
Smith‐Whitley 2012
- Smith-Whitley K, Thompson AA. Indications and complications of transfusions in sickle cell disease. Pediatric Blood & Cancer 2012;59(2):358-64. [DOI] [PubMed] [Google Scholar]
Steinberg 1999
- Steinberg MH. Management of sickle cell disease. New England Journal of Medicine 1999;340(13):1021-30. [DOI] [PubMed] [Google Scholar]
Steinberg 2012
- Steinberg MH, Sebastiani P. Genetic modifiers of sickle cell disease. American Journal of Hematology 2012;87(8):795-803. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sterne 2011
- Sterne JA, Egger M, Moher D. Chapter 10: Addressing reporting biases. In: Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Styles 1994
- Styles LA, Vichinsky E. Effects of a long-term transfusion regimen on sickle cell-related illnesses. Journal of Pediatrics 1994;125(6 Pt 1):909-11. [DOI] [PubMed] [Google Scholar]
Telfer 2007
- Telfer P, Coen P, Chakravorty S, Wilkey O, Evans J, Newell H, et al. Clinical outcomes in children with sickle cell disease living in England: a neonatal cohort in East London. Haematologica 2007;92(7):905-12. [DOI] [PubMed] [Google Scholar]
Venkatesubramanian 1994
- Venketasubramanian N, Prohovnik I, Hurlet A, Mohr JP, Piomelli S. Middle cerebral artery velocity changes during transfusion in sickle cell anemia. Stroke 1994;25(11):2153-8. [DOI] [PubMed] [Google Scholar]
Wang 1991
- Wang W, Kovnar EH, Tonkin IL, Mulhern RK, Langston JW, Day S, et al. High risk of recurrent stroke after discontinuance of five to twelve years of transfusion therapy in patients with sickle cell disease. Journal of Pediatrics 1991;118(3):377-82. [DOI] [PubMed] [Google Scholar]
Wang 2005
- Wang WC, Morales KH, Scher CD, Styles L, Olivieri N, Adams R, et al. Effect of long-term transfusion on growth in children with sickle cell anemia: results of the STOP trial. Journal of Pediatrics 2005;147(2):244-7. [DOI] [PubMed] [Google Scholar]
Ware 2004
- Ware RE, Zimmermann SA, Sylvestre PB, Mortier NA, Davis JS, Treem WR, et al. Prevention of secondary stroke and resolution of transfusional iron overload in children with sickle cell anemia using hydroxyurea and phlebotomy. Journal of Pediatrics 2004;145(3):346-52. [DOI] [PubMed] [Google Scholar]
Ware 2010
- Ware RE. How I use hydroxyurea to treat young patients with sickle cell anemia. Blood 2010;115(26):5300-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wilimas 1980
- Wilimas J, Goff JR, Anderson HR, Langston JW, Thomson E. Efficacy of transfusion therapy for one to two years in patients with sickle cell disease and cerebrovascular accidents. Journal of Pediatrics 1980;96(2):205-8. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Estcourt 2017
- Estcourt LJ, Fortin PM, Hopewell S, Trivella M, Wang WC. Blood transfusion for preventing primary and secondary stroke in people with sickle cell disease. Cochrane Database of Systematic Reviews 2017, Issue 1. Art. No: CD003146. [DOI: 10.1002/14651858.CD003146.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hirst 2002
- Hirst C, Wang WC. Blood transfusion for preventing stroke in people with sickle cell disease. Cochrane Database of Systematic Reviews 2002, Issue 1. Art. No: CD003146. [DOI: 10.1002/14651858.CD003146] [DOI] [PubMed] [Google Scholar]
Wang 2013
- Wang WC, Dwan K. Blood transfusion for preventing primary and secondary stroke in people with sickle cell disease. Cochrane Database of Systematic Reviews 2013, Issue 11. Art. No: CD003146. [DOI: 10.1002/14651858.CD003146.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]