

Abstract
Atrial fibrillation (AF), the most common progressive and age-related cardiac arrhythmia, affects millions of people worldwide. AF is associated with common risk factors, including hypertension, diabetes, and obesity, and serious complications such as stroke and heart failure. Notably, AF is progressive in nature and because current treatment options are mainly symptomatic, they have only a moderate effect on prevention of arrhythmia progression. Hereto, there is an urgent unmet need to develop mechanistic treatments directed at root causes of AF. Recent research findings indicate a key role for inflammasomes and derailed proteostasis as root causes of AF. Here, we elaborate on the molecular mechanisms of these two emerging key pathways to drive the pathogenesis of AF. First the role of ‘NACHT, LRR and PYD domains-containing protein 3’ (NLRP3) inflammasome on AF pathogenesis and cardiomyocyte remodeling is discussed. Then we highlight pathways of proteostasis derailment, including exhaustion of cardioprotective heat shock proteins, disruption of cytoskeletal proteins via histone deacetylases, and the recently discovered DNA damage-induced NAD+ depletion to underlie AF. Moreover, potential interactions between the inflammasomes and proteostasis pathways are discussed and possible therapeutic targets within these pathways indicated.
Keywords: Atrial Fibrillation, inflammation, arrhythmia
Introduction
Atrial fibrillation (AF), the most common cardiac rhythm disorder, affects 2–3% of the Western population and is associated with serious complications such as stroke, heart failure and increased mortality.1 Well-known risk factors of AF are associated with the environmentally-induced ‘wear-and-tear’, including ageing and cardiovascular diseases. Whereas in 20% of the patient population, AF is familial suggesting a genetic predisposition.2, 3 AF is progressive in nature, and therefore, most patients progress from paroxysmal AF (pAF) to persistent and longstanding persistent AF (perAF).1, 4 Importantly, therapy of perAF has high failure rates, with 20–60% of patients showing recurrence of AF within three months after ablation or electrical cardioversion.5–7 Therapy failure in AF is related to the presence of structural remodeling of the myocardium (fibrosis and/or myolysis), which, in turn, impairs electrical conduction and contractile function of atrial cardiomyocytes (“electropathology”).8–11 The development of fibrosis can form the substrate which disrupts conduction and facilitates the maintenance and progression of AF. Therefore, current research is increasingly directed at revealing the molecular root causes underlying AF-induced structural, electrical, and contractile remodeling of cardiomyocytes in order to develop mechanism-based therapies which may ultimately result in effective therapeutic strategies to treat AF.
Growing evidence suggests that derailed inflammatory signaling and protein homeostasis (proteostasis) in atrial cardiomyocytes drive structural remodeling and electrical conduction impairment in AF. The innate inflammatory signaling is continuously surveilling the extracellular milieu. Inflammasomes are important components of the innate inflammatory signaling and act as a first line of defense against danger signals such as pathogens (viruses, bacteria) or molecules (DNA, RNA, ATP, nuclear/cytosolic proteins) released from the damaged or dying cells.12, 13 Activation of inflammasomes can lead to the release of cytokines interleukin-1β (IL-1β) and IL-18, which can mediate inflammatory response in the event of infection or tissue injury.12, 13 Recent research findings indicate that inflammasomes are enhanced in atrial cardiomyocytes of experimental and clinical AF. Activation of inflammasomes in atrial cardiomyocytes promote both the ectopic-firing and AF-maintaining substrate that ultimately leads to AF. In addition, derailment of protein homeostasis (proteostasis) has been identified to drive AF. Proteostasis is the balance in protein maturation, transport and ultimately breakdown, and is considered as a first line of defense against intracellular protein stress and therefore crucial for cellular and organismal health.14 With ageing, a gradual derailment in proteostasis has been observed, resulting in proteotoxic stress and compromised defense against reactive oxygen species (ROS)15, 16, a process that seems accelerated in various age-related diseases including cardiac diseases.17–21 The concept that derailed proteostasis drives cardiac diseases, including AF, has been applied, and dissection of important components in the molecular mechanisms revealed exciting and innovative treatment strategies. Key components involve derailment of protein quality control (PQC) system, histone deacetylase 6 (HDAC6)-mediated disruption of the microtubule network, and depletion of mitochondrial nicotinamide adenine dinucleotide (NAD+) levels by excessive poly [ADP-ribose] polymerase 1 (PARP-1) activation due to DNA damage.
In this article, we first review the current understanding of inflammasome and proteostasis derailment in the pathogenesis of AF separately, followed by description of potential interactions between both pathways and identification of druggable targets within the inflammatory signaling and proteostasis derailment pathways. Finally, we discuss therapeutic strategies to treat AF.
Derailment of inflammasome signaling as root cause of AF
Inflammation is a vital biological process mediating tissue healing in response to infection or tissue damage.22, 23 Emerging evidence point to the involvement of inflammatory signaling in the pathogenesis of AF. Inflammasome is a well-established canonical inflammatory signaling pathway. Recent studies have established a causative role of ‘NACHT, LRR and PYD domains-containing protein 3’ (NLRP3) inflammasome in AF.
Inflammation and AF
During infection or tissue damage, immune cells such as macrophages and T-cells are the major players involved in the inflammatory process by releasing inflammatory cytokines into the blood. A plethora of evidence showed that AF is associated with inflammatory process as circulating levels of various inflammatory markers correlate with the incidence of clinical AF and the progression from pAF to the more sustained perAF or long-standing perAF.24 Inflammatory markers like C-reactive protein (CRP), IL-6, IL-1β, myeloperoxidase (MPO), and tumor necrosis factor-α (TNF-α) not only correlate with the progression of AF, but also can predict the outcome of AF ablation.25–29 Table 1 highlights several clinical studies that have demonstrated the positive correlation between inflammatory biomarkers in serum and AF incidence in patients. In addition to results from non-operative AF cohorts, a meta-analysis based on 42 studies and total 8398 patients further confirmed that levels of perioperative inflammatory cytokines, such as CPR, IL-6, IL-8, and IL-10, are significantly associated with post-operative AF (POAF).30 Apart from the cytokines, chemokine monocyte chemoattractant protein-1 (MCP1) level is also associated with the increased risk of AF and atrial fibrosis – a substrate for AF.29, 31 Moreover, immune cells can also be recruited from peripheral tissue to the myocardium and induce a local inflammation response. In line, macrophages and T-cells have been have been found in atrial myocardium of AF patients.32–34 As immune cells can modulate the electrophysiological property of cardiomyocytes through direct cell-cell coupling, or through stimulation of regulatory pathways such as inflammasome or cytokine receptor, these actions may drive AF.35–37
Table 1.
Clinical studies that revealed the association between inflammatory and proteostasis biomarkers and the development of atrial fibrillation
| Biomarkers | Study Design | Conclusion |
|---|---|---|
| CRP | cross-sectional & longitudinal | CRP is not only associated with the presence of AF but also associated with the increased risk for future development of AF.151 |
| prospective | CRP levels were higher in pAF group. CRP levels were significantly associated with successful cardioversion to sinus rhythm.152 | |
| prospective | High-sensitive (hs)-CRP predicts the risk of occurrence of new-onset AF in patients with CAD.153 | |
| prospective | A rise in the CRP level is associated with the incidence of AF.154 | |
| case-control | Serum level of hsCRP was significantly higher in AF patients compared to control patients, and the increase is more prominent in perAF patients than pAF patients.155 | |
| retrospective | CRP levels were increased in AF patients, and CRP levels were higher in perAF than pAF.156 | |
| IL-6 | case control | Serum level of IL-6 was significantly higher in AF patients than in non-AF controls, and the increase is more prominent in perAF than in pAF patients.157 |
| prospective | −174G/C polymorphism of the IL-6 promoter modulates the inflammatory response and the GG genotype is an independent predictor of postoperative atrial AF.158 | |
| prospective | Circulating IL-6 level positively correlates with the prolongation and dispersion of P-waves, and negatively correlates with the circulation connexin protein. Incubation of the atrial myocyte-like cell line (HL-1 cells) with IL-6 reduced the expression of connexins, which is also reversible by removing IL-6.159 | |
| prospective | Dataset from the Chronic Renal Insufficiency Cohort (CRIC) study revealed that plasma IL-6 level was not only associated with the presence of AF at baseline, but also new onset of AF in patients with chronic kidney disease.160 | |
| prospective | The decreased serum level of IL-6 is associated with the free of recurrent AF within 6-months follow-up post ablation.161 | |
| IL-1β | case-control | Microvesicle-bound IL-1β was significantly increased in perAF patients compared to controls.162 |
| IL-2 | prospective | Higher serum level of IL-2 is an independent predictor of AF recurrence in pAF patients post ablation.163 |
| IL-10 | case-control | The plasma level of IL-17A was significantly increased in AF patients. IL-17A level was positively correlated with left atrial diameter.164 |
| case-control | IL-10 promoter polymorphism (−592A/C) have an impact on POAF development in north Indian patients. AA genotype increases AF risk in post-surgery.165 | |
| IL-18 | case-control | The serum level of IL-18 was significantly higher in AF patients than in controls. Among AF subgroups, IL-18 was significantly higher in perAF patients than in pAF patients. IL-18 also correlates with the left atrial diameter, a risk factor of AF.27 |
| IL-17A | case-control | Plasma level of IL-17A was significantly increased in patients with AF compared to controls. There was no significant difference in the level of IL-17A between different types of AF. 164, 166 |
| IL-37 | case-control | The serum level of the anti-inflammatory cytokine IL-37 and the expression of IL-37 mRNA in the peripheral blood mononuclear cells were increased in AF patients compared to non-AF controls.167 |
| TNFα | case-control | The serum level of TNFα was significantly higher in AF patients (including both pAF and perAF patients) compared to control patients.155 |
| case-control | The serum level of TNFα positively correlates with the severity of AF.29 | |
| MPO | case-control | Serum level of MPO was increased in AF patients regardless of the stage.168 |
| prospective | Increased serum level of MPO correlates with the risk of AF recurrence post ablation.169 | |
| HSPB1 (HSP27) | case-control | Serum HSPB1 levels predicted AF recurrence after catheter ablation. HSP27 also correlated with left atrial diameter, left atrial voltage and fractionated intervals.170 |
| case-control | Elevated HSPB1 levels in atrial tissue from patients with pAF compared to perAF or sinus rhythm controls.66, 171, Also, an inverse correlation was observed between HSPB1 levels and the duration of AF and the extent of structural damage (myolysis) was observed.66 | |
| HSPA1A (HSP70) | case-control | Higher HSPA1A levels in atrial tissue of patients in sinus rhythm undergoing cardiac bypass surgery are correlated with a lower incidence of POAF.172, 173 |
| prospective | Mutation in HSPA1A-Hom gene (Met439Thr substitution) is associated with higher levels of serum HSPA1A and incidence of POAF after cardiac bypass surgery.174 | |
| HSPD1/E1 (HSP60/10) | prospective | HSPD1 levels were lower in both atrial tissue and plasma of AF patients undergoing mitral valve replacement and spontaneously restored to sinus rhythm. HSPD1 and was positively correlated with apoptosis, interstitial fibrosis, and inflammation.175 |
| prospective | Pre- and post- operative circulating anti-HSPD1 antibodies are associated with POAF in patients undergoing cardiac bypass surgery.176 | |
| case-control | Higher levels of mitochondrial HSPD1 and HSPE1 in are found in right atrial tissue of patients with peAF compared to controls.177 | |
| cohort study | However, the Multi-Ethnic Study of Atherosclerosis (MESA) reveals no association between HSPD1 levels in blood and occurrence of AF was found.178 | |
AF, atrial fibrillation; CAD, coronary artery disease; CRP, C-reactive protein; IL-1β, interleukin-1β; IL-18, interleukin-18; pAF, paroxysmal atrial fibrillation; perAF, persistent atrial fibrillation; POAF, post-operative atrial fibrillation.
On the other hand, research has revealed that inflammatory cytokines can cause adverse remodeling in cardiomyocytes and enhance AF susceptibility. A study showed that the increased AF incidence, as observed in endurance exercise mouse models (swimming or treadmill-running) is associated with increased inflammation, slowed electrical conduction, enhanced Ca2+ release via ryanodine receptor 2 (RyR2), impaired function of L-type Ca2+ channel, and TNFα-dependent activation of nuclear factor NFκB in atrial cardiomocytes.38 In contrast, inhibition of TNFα by an inhibitor etanercept or genetic ablation (TNFα−/−) mitigated atrial remodeling and AF susceptibility.38 Similarly, in a pressure-overload induced heart failure model, the increased AF inducibility can be prevented by genetic ablation of IL-1β (IL-1β−/−).39 The actions of TNFα and IL-1β are mediated through their respective receptors TNFR1/2 and IL-1R located on immune and non-immune cells. Activation of TNFR1/2 and IL-1R enhanced NFκB- and activating protein 1 (AP1)-mediated transcription of genes involved in inflammatory pathway such as Nlrp3 and Il1b, thereby resulting in further perpetuation of inflammation.40, 41 Consistently, the levels of total and phosphorylated (active) NFκB are increased in the atrial tissue of AF patients.42 Together the findings collectively point to the close connection between inflammation and AF pathogenesis.
NLRP3 inflammasome
Inflammasomes are multimeric protein complexes that act as an intracellular sensor in response to pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs).43 PAMPs initiate and perpetuate the infectious pathogen-induced inflammatory response triggered predominantly by microbe-released lipopolysaccharides (LPS), lipoteichoic acid, or peptidoglycan.40 DAMPs are host-released biomolecules that can initiate and perpetuate a noninfectious inflammatory response as a result of tissue damage.43 DAMPs include intracellular proteins like heat-shock proteins (HSPs)44 and high-mobility group box 1 (HMGB1), as well as non-protein molecules such as ATP, uric acid, heparin sulfate and double strand DNA.45 In most of the innate immune cells, inflammasomes are the canonical pathway that can assist in defending the host against infection or injury by producing both pro-inflammatory and anti-inflammatory cytokines.43
To date, nearly ten inflammasomes have been described, including NLRP1-, NLRP2-, NLRP3-, NLRP6-, NLRC4-, NLRP12-, Pyrin-, AIM2-, and IFI16.40, 46 They are centered around Nod-like receptor (NLR), or Absent-in-melanoma-2 (AIM2)-like receptor proteins. Among these inflammasomes, NLRP3 inflammasome has gained an increasing amount of attention in recent years.47 The NLRP3 inflammasome can be activated by a variety of stimuli, which are involved in two basic steps i.e. ‘priming’ and ‘triggering’ processes (Figure 1). ‘Priming’ refers to the activation of toll-like receptors (TLRs) by PAMPs/DAMPs that leads to the NFκB-mediated transcription of NLRP3, and pro-IL-1β/pro-IL-18. ‘Triggering’ is the process that promotes the inter-domain interactions among NLRP3, ‘apoptosis-associated speck-like protein containing a CARD’ (ASC), and pro-caspase-1.48 The oligomerization of the multiprotein platform facilitates the autocleavage of pro-caspase-1 into the mature caspase-1. Mature caspase-1 is a cysteine protease that can further cleave pro-IL-1β and pro-IL-18 into the active forms IL-1β and IL-18. Mature caspase-1 can also cleave gasdermin-D (GSDMD), and the N-terminus fragment of GSDMD creates pores in the sarcolemma, which facilitate the release of mature IL-1β and IL-18 from secretory immune cells and cardiac fibroblasts.40, 43
Figure 1. Mechanisms responsible for the activation of NLRP3 inflammasome in immune cells via the two basic steps: ‘priming’ and ‘triggering’ processes.

Pharmacological treatments directed at 1) ‘Priming’ stimuli (with IL-1R inhibitor anakinra and TNFα receptor inhibitor etanercept), 2) ‘Triggering’ stimuli (with P2X7R inhibitor AZD9056), 3) NLRP3 (with MCC950 and colchicine), and 4) the effectors capase-1 (with VX-765) and IL-1β (with canakinumab). ASC, apoptosis-associated speck-like protein containing a CARD; DAMPs, damage-associated molecular patterns; GSDMD, gesdermin-D; IL-1R, interleukin-1 receptor; NFκB, nuclear factor kappa-light-chain-enhancer of activated B cells; NLRP3, NACHT, LRR and PYD domain containing protein 3; Nt-GSDMD, N-terminus fragment of gesdermin-D; mtROS, mitochondrial ROS; PAMPs, pathogen-associated molecular patterns; P2X7R, purinergic receptor P2X7R; TLR, toll-like receptor.
In immune cells, the best described signals that contribute to the ‘triggering’ process include 1) increased K+ efflux due to the activation of purinergic receptor P2X7R, 2) increased Ca2+ influx via the Ca2+ sensing receptor (CaSR), 3) increased reactive oxygen species (ROS), 4) release of mitochondrial DNA into cytosol, and 5) cathepsin-B released from lysosome. However, the exact mechanism by which the ‘triggering’ stimuli promotes the inter-domain interaction and oligomerization of inflammasome components remains unclear.
It has been recognized that NLRP3 inflammasome activity is a key modulator in a wide range of pathology involved in immunological and cardiovascular disorders.49–51 It has been proposed that the activation of NLRP3 inflammasome facilitates the development of cardiomyopathy post myocardium infarction via two major mechanisms. First, the maturation of caspase-1 in cardiomyocytes can promote a type of cell death called ‘pyroptosis’, which cause the loss of working cardiomyocytes. Second, active caspase-1 in cardiac fibroblasts can promote the release of IL-1β, which in turn activates fibroblasts to transdifferentiate into myofibroblasts. Myofibroblasts exert a variety of detrimental effects to myocardium by releasing cytokines and collagens, which ultimately lead to structural remodeling.52–56 Myofibroblasts can also alter the electrophysiological property of cardiomyocytes by direct interaction.57 A recent study has shown that human induced pluripotent stem cells (hiPSCs)-derived cardiomyocytes from patient with arrhythmogenic cardiomyopathy can directly secrete IL-1β and several pro-inflammatory cytokines and inflammation-related factors.58 This work suggests that cardiomyocytes represent a source for the release of inflammatory biomarkers associated with the AF development.
NLRP3 inflammasome & AF
The direct involvement of NLRP3 inflammasome in AF pathogenesis was established recently.59 It was observed that the activity of NLRP3 inflammasome increased in the atrial tissue samples of pAF and perAF patients, and animal models of AF including the atrial tachypacing-induced AF dog model and the transgenic CREM-IbΔC-X mouse model that spontaneously develops AF.59 By examining the levels of NLRP3 inflammasome components in separated cardiomyocytes and cardiac fibroblasts respectively, it was found that cardiomyocyte NLRP3 inflammasome activation was the major contributor of the overall increased NLRP3 activity in atrial tissues of pAF patients. To demonstrate the causal association between the cardiomyocyte NLRP3 activity and AF pathology, a mouse model with cardiomyocyte-restricted expression of a constitutively activated NLRP3 was established (αMHC:NLRP3A350V/+). The αMHC:NLRP3A350V/+ mice exhibited more frequent premature atrial contractions (PACs) compared to control animals. In addition, systematic evaluation revealed that this mouse strain exhibits atrial ectopic activity, shortening of atrial effective refractory period (AERP), and enlarged atria, all of which are considered proarrhythmic for AF development (Figure 2). Despite the lack of spontaneous AF, αMHC:NLRP3A350V/+ mice were very susceptible for rapid pacing-induced AF, which was successfully prevented by either the NLRP3 inhibitor MCC950 or a NLRP3-targeting shRNA delivered by adeno-associated virus 9 (AAV9). Consistently, the genetic ablation of NLRP3 in a mouse model of spontaneous AF60, 61 – CREM transgenic mice (CREM:NLRP3−/−) prevented the development of spontaneous AF59, further supporting a causative role for NLRP3 in AF development. Because the enhanced AF inducibility in αMHC:NLRP3A350V/+ mice is proceeding the increased protein level of IL-1β in the atria of αMHC:NLRP3A350V/+ mice, this finding suggests that hyperactive NLRP3-induced AF arrhythmogenesis is independent from IL-1β or caspase-1 activity. Coincidently, an independent study has demonstrated that ablation of ASC in mice (ASC−/−) lacks the protection against AF inducibility in a pressure overload-related heart failure model. 39 These results imply that in a constitutive active NLRP3 model, AF development results from an alternative function of NLRP3. One interesting observation is that mRNA levels of several cardiac specific genes (Ryr2, Kcna5, and Mef2c) were upregulated in the atria of αMHC:NLRP3A350V/+ mice. Since it has been suggested that NLRP3 may function as a transcriptional co-activator in T cells,62 NLRP3 may exert transcriptional activity and/or affect epigenetic regulation in cardiomyocytes. This mode of action of NLRP3 certainly deserves more in-depth investigation.
Figure 2. Mechanisms underlying AF development associated with the enhanced activity of NLRP3 inflammasome.
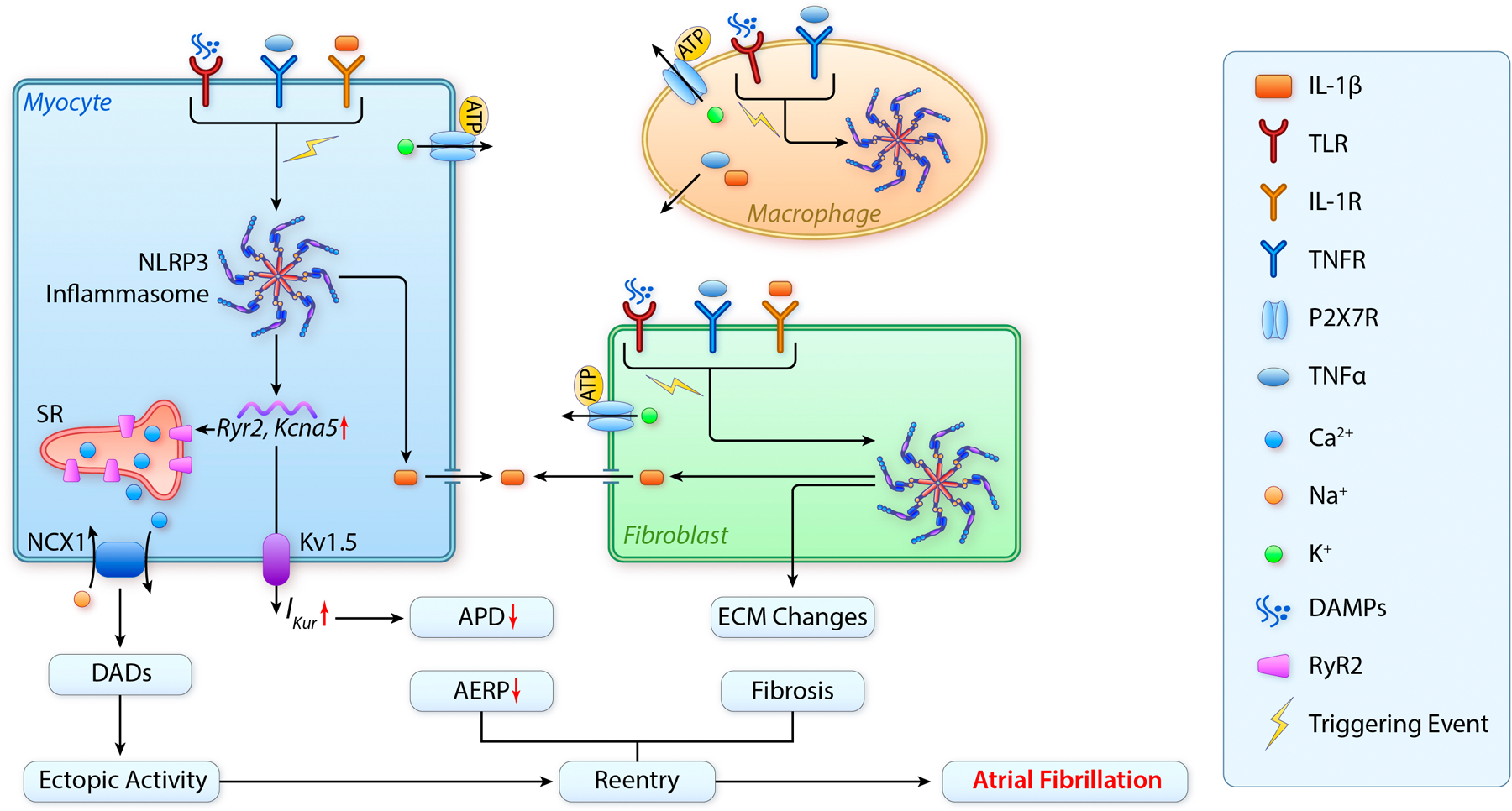
1) The active NLRP3 promotes ectopic activity and reentry by enhancing RyR2-mediated SR Ca2+ release and outward IKur current in cardiomyocytes. 2) The release of IL-1β from cardiomyocytes due to the activation of NLRP3 inflammasome can activate cardiac myofibroblasts and macrophages, promoting the release of inflammatory cytokines like IL-1β and TNFα active fibroblast can also transdifferentiate into myofibroblasts that can promote fibrosis by releasing collagen and altering ECM. 3) The release of TNFα and IL-1β can further activate TNFR and IL-1R and ultimately enhance the transcription (‘priming’) of NLRP3 inflammasome components (‘priming’); whilst the increase in the extracellular ATP level may further activate P2X7R, which subsequently promotes the oligomerization (‘triggering’) of NLRP3 inflammasome in cardiomyocytes and non-myocytes. The feedforward loop causing enhancement of NLRP3 activity can be established because of the interaction between cardiomyocytes and non-myocytes through autocrine and paracrine actions. AERP, atrial effective refractory period; APD, action potential duration; DADs, delayed after-depolorizations; ECM, extracellular matrix; Mac, macrophage; NCX1, sodium-calcium exchanger 1; IKur, ultrarapid outward current; SR, sarcoplasmic reticulum.
Derailment of proteostasis as root cause of AF promotion
In addition to the role of inflammasome on atrial cardiomyocyte remodeling and AF, accumulating evidence indicates that in atrial cardiomyocytes, derailment in proteostasis also represents a root cause for structural damage and AF promotion. As far, three mechanisms contributing to the proteostasis derailment have been identified to play a role in atrial remodeling and AF promotion. These include impaired PQC system, HDAC6-induced disruption of the microtubule network, and DNA damage-induced PARP-1 activation with subsequent depletion of NAD+ levels in mitochondria.
Impaired Protein Quality Control
Proteins are versatile and complex macromolecules involved in the proper function of cardiomyocytes. The balance in protein synthesis, maturation, transport and breakdown (i.e. proteostasis) is closely monitored by the PQC system. Main players within the PQC involve chaperones, especially heat shock proteins (HSPs) 63 and pathways that clear damaged proteins including the ubiquitin-proteasome system and autophagy system.64 While mild stress-induced derailment of proteostasis has beneficial effect on the cardiomyocyte (referred to as hormesis)15, 65, excessive stress is accompanied with loss of PQC. Research findings reveal that AF is causing excessive derailment of the PQC system.66–70 During mild stress, the heat shock response (HSR) is stimulated by activation of heat shock transcription factors, of which HSF1 is the major regulator of HSP transcription in eukaryotes.71 HSPs consist of five HSP families, i.e. HSPA (HSP70), HSPB (small HSPs), HSPC (HSP90), HSPD (HSP60), and DnaJB (HSP40), each with several family members and (specific) co-factors in various cellular localizations, with distinct and overlapping functions.72–74 Under physiological conditions, cardiomyocytes express high levels of HSPBs which localize at contractile and microtubule proteins thereby stabilizing the cardiomyocyte structure, and conserving the contractile and electrophysiological function of the atrial cardiomyocytes.66–70 Although HSPB1 levels are induced in atrial tissue samples of patient with pAF, HSPB1 levels get exhausted in patients with (longstanding) perAF (Table 1).66 In addition, in these patients, HSPB1 levels in atrial tissue correlate inversely with the amount of structural remodeling, suggesting that the HSR becomes exhausted in time resulting in derailment of proteostasis, structural remodeling and AF progression.64, 66 In contrast, boosting of the endogenous HSR with drugs or by genetic overexpression of HSPB1 limited the expansion of the AF substrate and therefore attenuated AF promotion in various experimental AF model systems.66, 75–78 These findings indicate that loss of PQC by exhaustion of the HSR is a prominent contributor to AF promotion.
Recent evidence also indicates that AF results in endoplasmic reticulum (ER) stress and subsequent activation of macroautophagy (hereafter ‘autophagy’).79–81 Autophagy is an evolutionarily-conserved protein-degradation pathway that removes damaged or expired proteins and organelles by sequestration them into autophagosomes for subsequent lysosomal degradation (Figure 3).80 Recent research findings show that the ER stress response can activate the autophagy–lysosome pathway via the unfolded protein response (UPR). The UPR induces the phosphorylation of eIF2α at Serine31, which inhibits protein translation and initiates selective expression of stress-responsive transcripts, including activating transcription factor 4 and 6 (ATF4 and ATF6, respectively). In turn, ATF4 and ATF6 signaling induces CHOP, autophagy genes ATG12, LC3B and HSPA5 expression and as such activates autophagic protein degradation by stimulating elongation of autophagosomes.79, 82 Autophagy is widely recognized as a cell-stress pathway, of which the excessive activation triggers cardiac remodeling in response to degradation of sarcomere proteins.80 In AF, the ER stress-induced activation of autophagy constitutes an important mechanism of atrial remodeling in experimental model systems of AF and in atrial biopsies of patients with AF.81 In line, blocking ER stress, by the chemical chaperone 4-phenyl butyrate, overexpression of the ER chaperone HSPA5 or mutated eIF2α, inhibits activation of autophagy and thereby precludes electrical and contractile dysfunction in both in vitro and in vivo AF models.81 These studies indicate that, as a prominent modulator in proteostasis derailment, ER stress may represent a novel druggable target to attenuate AF promotion.
Figure 3. Mechanisms underlying AF development associated with proteostasis derailment.

1) AF results in impaired of PQC by downregulation of HSP gene expression, so the first line of cell defense against stress is lost and ER stress response is observed with downstream activation of autophagy gene expression. 2) AF also induces the activation of HDAC6, which results in de-acetylation of microtubules, alteration in Ca2+ handling, and loss of contractile function. 3) AF results in DNA-damage, PARP1 activation and consequently NAD+ depletion which underlies electrophysiolocigal and contractile impairment. Pharmacological treatments directed at induction of HSP transcription (with geranylgeranylacetone; GGA), prevention of ER stress (with 4-phenylbutyrate; 4PBA), inhibition of HDAC6 activation (with tubastatin A), and PARP1 inhibition (with ABT888 or olaparib) or NAD+ supplementation protect against proteostasis derailment and AF progression.
Histone deacetylase activation-induced microtubule network disruption
It has been recognized that proper cardiomyocyte function is related to a balanced communication between the components of the proteostasis network and the cytoskeleton.64 In cardiomyocytes, the cytoskeleton provides communication between the organelles and contractile properties. Therefore, the cytoskeleton is highly specialized and consists of actin filaments, intermediate filament proteins and microtubules. Microtubules interact with membrane-associated proteins, sarcomeric proteins and the nuclear envelope and, as such, regulate transmission of signals between organelles including sarcoplasmic and endoplasmic reticulum, mitochondria and the nucleus, and transport of ubiquitinated proteins in the proteostasis network. Research in experimental and clinical AF revealed a role for microtubule disruption in AF promotion.10 HDAC6 was identified to confer tachypacing-induced post-translational modifications of cytoskeleton proteins, both in experimental model systems and clinical AF. Rather than targeting histones, the increased HDAC6 activation specifically deacetylates α-tubulin, resulting in the disruption and subsequent degradation of the microtubule network through the protease calpain, which becomes activated in AF.10 Interestingly, mutated HDAC6, bearing a dominant negative mutation in the α-tubulin catalytic domain of HDAC6, causes suppression of α-tubulin deacetylation and thereby rescues cardiomyocytes from microtubule disruption and contractile dysfunction.10 On the other hand, several reports show that the inhibitors of HDAC class IIa exert protective benefits against AF.83–85 These findings underscore the central role of HDAC6 activation in AF-related cardiomyocyte remodeling and offer a novel therapeutic target in AF.
DNA damage-induced PARP1 activation and NAD+ depletion
Recent published findings reveal important evidence for AF promotion due to dysmorphic nuclei-associated DNA damage, and subsequent PARP-1 activation. In turn, PARP1 activation results in the depletion of NAD+ levels in mitochondria, which causes additional DNA damage, energy depletion and AF progression (Figure 3).86 PARPs constitute a family of 6 nuclear enzymes which are activated by single and double stranded DNA breaks, serving to recruit the DNA repair machinery by synthesis of poly(ADP-ribose) chains (PAR).87 During the synthesis of PAR chains, NAD+ is consumed by PARP up to an extent that it depletes cellular NAD+, thereby triggering a progressive decline in ATP levels, energy loss and cell death in case of excessive PARP activation.88 It has been reported that tachypacing-induced functional loss of atrial cardiomyocytes is precipitated by excessive PARP1 activation in response to oxidative DNA damage.86 Interestingly, replenishment of NAD+ and pharmacological or genetic depletion of PARP1 precludes tachypacing-induced functional loss. Moreover, inhibition of PARP1 protects against tachypacing-induced NAD+ depletion, oxidative stress, DNA damage and contractile dysfunction in HL-1 and rat atrial cardiomyocytes and in Drosophila model systems for AF. Consistent with these findings, cardiomyocytes of patients with perAF also show significant DNA damage, which correlates with PARP1 activity. In addition, DNA damage was associated with electrophysiological deterioration, including prolongation of APD (possibly via the reduction in potassium channel expression)89–92, reduction in cardiomyocyte excitability and increased ADP dispersion, thereby creating a molecular and structural substrate for further arrhythmogenesis.93–96 Although APD shorting was previously recorded in models for tachypacing-induced AF, APD prolongation was also observed in patients with lone pAF, in atrial tissue of patients predisposed to AF and in various patient and animal studies for AF with underlying heart failure and structural changes in the atria91, 97–101. These studies provide compelling evidence that the structural and associated conduction abnormalities due to DNA damage may represent a predominant contributor to the AF substrate. Moreover, the studies may explain why current drug treatment directed at modulation of refractoriness shows limited efficacy, while its usage is further limited by a pro-arrhythmic action and non-cardiovascular toxicity.102 Together, the findings not only indicate a novel mechanism by which tachypacing impairs cardiomyocyte function, but also suggest PARP1-inhibition and/or NAD+ supplementation as a possible therapeutic target that may preserve cardiomyocyte function in clinical AF.
Crosstalk between proteostasis derailment and inflammasome activation
Various studies indicate a potential crosstalk between modulators of proteostasis derailment and inflammation-induced stimuli (Figure 4). Emerging evidence support that derailed proteostasis can enhance the activation of NLRP3 inflammasome by the increased protein misfolding and ER stress pathway in autoinflammatory diseases.103, 104 Misfolded proteins are known to induce the UPR, a common event causing ER stress by activation of sensors like ATF6, inositol-requiring transmembrane kinase/endonuclease 1 (IRE1), and pancreatic ER kinase (PERK). ER stress also affects Ca2+ metabolism and ROS production, both of which modulate the activation of NLRP3 inflammasome.81, 105
Figure 4. Crosstalk between proteostasis derailment and inflammasome activation.

Solid lines indicate established modulations, dashed line indicates the postulated feedforward modulation of NLRP3 inflammasome on NAD+ levels, which requires further investigation. ER, endoplasmic reticulum; IRE1, inositol-requiring transmembrane kinase/endonuclease 1; ROS, reactive oxygen species; XBP1, x-box binding protein 1.
On the other hand, autophagy contributes to the degradation of NLRP3, thereby inhibiting its activity. Ubiquitination of NLRP3 is critically involved in inflammasome inactivation as ubiquitinated NLRP3 is cleared by autophagy.106–108 It has been shown that the macrophage-specific inhibition of autophagy gene 7 (ATG7) in LysMCre+;ATG7flox/flox mice leads to hyperactivation of NLRP3 inflammasome in macrophages. Importantly, induction of protein misfolding by puromycin, thapsigargin, or geldanamycin causes inflammasome activation in macrophages, which can be further exacerbated by the inhibition of ATG7.109 Interestingly, accumulation of misfolded proteins can induce inflammasome activation, which is mediated by increased ROS production and the release of protease cathepsin B as a result of lysosomal damage.109
Furthermore, another target for crosstalk between proteostasis derailment and inflammasome activation is via DNA damage and AIM2-inflammasome activation.110 AIM2 senses DNA damage in the nucleus and mediates inflammasome activation and subsequent cell death.110 Interestingly, genetic ablation of AIM2 in mice prevented the irradiation-induced damage in gastrointestinal tract and bone marrow. In line, recent work from the Brundel lab has shown that both, AF and irradiation of atrial cardiomyocytes result in DNA damage, and subsequent electrical and contractile impairment via PARP1 activation and NAD+ depletion.86 A closer look into whether some of the AF-induced detrimental effects on cardiomyocytes are mediated by AIM2 or NLRP3 inflammasomes is compelling.
On the flip side, NLRP3 may affect the proteostasis by modulating the synthesis of NAD+. A study in cardiac aging reveals that genetic ablation of NLRP3 in mice (NLRP3−/−) improves lifespan, preserves cardiac integrity, and prevents age-associated metabolic changes.111 NLRP3−/− mice are resistant to the age-dependent prolongation of PR-intervals, a known risk factor of AF. The mechanisms underlying the improved cardiac aging in NLRP3−/− mice include the attenuation on telomere shortening and improved autophagic degradation via the inhibition of PI3K/AKT/mTOR pathway. An interesting finding in this study is that the mRNA level of nicotinamide phosphoribosyltransferase (Nampt), a rate-limiting enzyme in NAD+ biosynthesis, and the overall NAD+ content are increased in cardiac tissue of NLRP3−/− mice compared to control mice.111 These results imply that NLRP3 can negatively regulate NAD+ synthesis. It is of interest to evaluate whether hyperactive NLRP3 promotes proteostasis derailment by causing NAD+ depletion, which may further support the notion that NLRP3 exhibit alternative functions independent of IL-1β or caspase-1. Also, it is worth mentioning, that DNA damage-induced NAD+ depletion, which has been identified as one major mechanism underlying proteostasis derailment86, increases mitochondrial ROS production and oxidative stress86, 112, and subsequently can promote the activation of NLRP3 inflammasome. As such, this feedforward modulation (Figure 4) can perpetuate the activation of NLRP3 inflammasome and proteostasis derailment.
Therapeutic potential of pharmacological restoration of inflammasome activity and proteostasis derailment
There has been a remarkable interest in developing effective anti-inflammatory agents by targeting the inflammasome pathway.113, 114 Current strategies to reduce inflammasome pathway activity are mainly achieved by targeting either the effectors (e.g. caspase-1, IL-1β, and IL-1R) or the activating stimuli (e.g. P2X7R, NFκB, and TLR) of inflammasome activation (Table 2, part I). Apart from the chemically synthesized compounds, a number of “biologicals” such as neutralizing antibody and recombined decoy proteins have been developed to reduce the activity of IL-1β or IL-1R. Many of these FDA-approved orphan drugs are indicated for autoinflammatory and autoimmune diseases including rheumatoid arthritis (RA). Surprisingly, in a recent clinical trial (CANTOS), the anti-IL-Iβ antibody canakinumab (150mg every 3-months) can significantly reduce the recurrent of major cardiac events in patients with atherosclerosis,115 suggesting that inhibition of NLRP3 inflammasome may be cardioprotective. Although many of these anti-inflammatory therapies are not indicated for cardiac disease, it would be interesting to evaluate the potential effect of these therapies on prevention of AF development in this particular cohort of patients, especially since RA is a known independent risk factor of AF.
Table 2.
Drugs targeting NLRP3 inflammasome or proteostasis pathway with potential benefit in AF.
| Key modulators | Drug | Action | Clinical Phase | Indication | Ref/identifier (clinicaltrails.gov) |
|---|---|---|---|---|---|
| Targeting NLRP3 inflammasome | |||||
| IL-1β | Canakinumab (Ilaris®) | Neutralizing anti-IL-1β antibody | Phase III Phase III Phase III Phase III Phase III Phase III Phase II |
CAPS Atherosclerosis & MI115 schizophrenia Gout Systemic Juvenile Idiopathic Arthritis Non-Small-Cell Lung Cancer Atrial fibrillation |
NCT01576367 NCT01327846 NCT01390350 NCT01431638 NCT02396212 NCT03626545 NCT01805960* |
| IL-1R | Rilonacept (Arcalyst®) | Soluble IL-1 decoy receptor | Phase II Phase III Phase II Phase IV |
CAPS Gout Recurrent Pericarditis Vascular dysfunction in CKD |
NCT00288704 NCT00855920 NCT03737110 NCT01663103 |
| IL-1R | Anakinra (Kineret®) | IL-1R antagonist | Phase III Phase II Phase II/III Phase II Phase III |
Rheumatoid arthritis Multiple Myeloma Myocarditis Heart failure, acute MI ST segment elevation during acute MI |
NCT00117091 NCT03233776 NCT03018834 NCT01175018 NCT00789724 |
| Caspase-1 | VX-765 (Belnacasan) | Caspase-1 inhibitor | Phase II Phase II |
Epilepsy Psoriasis |
NCT01048255 NCT00205465 |
| NLRP3 | MCC950 (CRID3, CP456773)116 |
Specific NLRP3 inhibitor upstream of ASC oligomerization | Preclinical | AF Atherosclerotic lesion Type-2 diabetes |
59 179 180 |
| KATP | Glibenclamide (Glyburide) |
Prevent intracellular K+ depletion | Phase IV | Type-2 diabetes | 181 |
| Microtubule polymerization | Colchicine | Prevent microtubule polymerization | Phase IV Phase II Phase III Phase IV |
Gout Behcet’s disease POAF Arrhythmia recurrence after acute pericardial effusion |
NCT02500641 NCT03888846 NCT03015831 NCT02260206 |
| TNFα | Etanercept (Enbrel®) | Decoy receptor can bind to TNFα, thereby preventing its binding with the endogenous TNFR. | Phase III Phase IV |
Rheumatoid arthritis Plaque psoriasis |
NCT04079374 NCT02274792 |
| P2X7R | AZD9056 182 | P2X7 receptor antagonist | Phase II | Rheumatoid Arthritis | NCT00520572 |
| P2X7R | 16673-34-0 | Prevent the P2X7R mediated K+-efflux | Preclinical | Ischemic cardiomyopathy | 118 |
| II. Targeting proteostasis derailment | |||||
| ER stess | 4PBA (Buphenyl® & Ammonaps®) |
Chemical chaperone | Phase II Phase II Phase II Phase II Phase III Phase IV Phase III Preclinical |
Cystic fibrosis Amyotrophic lateral sclerosis Huntington’s disease Pulmonary tuberculosis Maple syrup urine disease Diabetes Urea cycle disorder AF |
NCT00590538 NCT00107770 NCT00212316 NCT01580007 NCT01529060 NCT00533559 NCT00947544 81 |
| HDAC6 | tubastatin A | HDAC6 inhibitor | Preclinical | AF | 183 |
| HDAC6 | ACY-1215 (ricolinostat) | HDAC6 inhibitor | Phase II | Lymphoma | NCT02091063 |
| DNA damage | ABT888 | PARP inhibitor | Phase II Phase II Phase I Phase II Phase II Preclinical |
Metastatic breast cancer Hepatocellular carcinoma Adult solid neoplasm Ovarian cancer Colorectal cancer AF |
NCT01009788 NCT01205828 NCT01154426 NCT01113957 NCT01051596 86 |
| Mitochondrial dysfunction | Nicotinamide (niagen®) |
PARP inhibitor Sirt inhibitor NAD-precursor |
Phase III Phase III Phase II Phase II Preclinical |
Lung carcinoma Chronic kidney disease Neurodegenerative disease Alzheimer’s disease AF |
NCT02416739 NCT02258074 NCT01589809 NCT00580931 183 |
| HSP | GGA (teprenone) | HSP induction | Phase IV Preclinical Phase II |
Gastric ulcers Gastritis Gastric lesion AF Cardiac bypass surgery |
NCT01190657 NCT015475559 NCT01397448 75 77 |
| HSP | GGA derivatives | HSP induction | Preclinical | AF | 76 |
AF, atrial fibrillation; CAPS, cryopyrin-associated periodic syndromes; CKD, chronic kidney disease; MI, myocardial infarction.
prematurely ended
In a pre-clinical study,59 shRNA-mediated knockdown of Nlrp3 and the selective NLRP3-inhibitor MCC950116 are both sufficient to reduce AF inducibility in the NLRP3 gain-of-function mice, which provides a proof-of-concept for NLRP3-inhibitor in AF prevention. Several compounds that can target the ‘triggering’ stimuli for inflammasome activation also have shown inhibitory effect on NLRP3 activity. For example, the anti-diabetic glibenclamide, an inhibitor of the ATP-sensitive K+ channels (KATP), was found to suppress activation of NLRP3 inflammasome via prevention of intracellular K+ depletion.117 However, since global inhibition of KATP by glibenclamide can lead to hypoglycemia in non-diabetic patients, a compound 16673-34-0 that lacks the KATP-inhibitory effect has been developed.118 This pre-clinical compound 16673-34-0 is an intermediate substrate of glibenclamide synthesis, without the cycloxyurea moiety that mediates the KATP inhibition. Administration of 16673-34-0 in mice improved cardiac function following ischemia-reperfusion without altering glucose metabolism. 118 Moreover, recent studies have shown that the anti-gout medication colchicine also exhibit NLRP3-inhibitory effects, although colchicine is a well-known inhibitor for microtubule polymerization, the exact mechanism in gout disease remains to be determined. 119, 120 It is known that microtubules mediate assembly of the NLRP3 inflammasome by providing optimal spatial arrangement for signal transduction.121 Thus, it has been proposed that colchicine can suppress the NLRP3 inflammasome activation by inhibiting tubulin polymerization, thereby limiting the necessary spatial requirement for NLRP3 inflammasome activation in immune cells.120, 122 Though, it is worth mentioning that loss of polymerized tubulin leads to proteostasis derailment, remodeling of atrial cardiomyocytes and AF progression as discussed above. Thus, it remains to be determined whether the suppression of NLRP3 inflammasome by colchicine counterbalances its potential detrimental effect on tubulin polymerization and proteostasis.10
As for restoring the proteostasis, a number of compounds have been described to target the three major mechanisms abovementioned (Table 2, part II). First, securing HSP levels at an adequate level may limit the expansion of the AF substrate and as such may protect against AF induction and progression from pAF to perAF.66, 76 A well-known HSP-inducing compound is geranylgeranylacetone (GGA). GGA is a nontoxic acyclic isoprenoid compound with a retinoid skeleton that is originally used as an antiulcer drug in Asian countries.123, 124 GGA induces HSPs in various tissues, including gastric mucosa, intestine, liver, myocardium, retina, and central nervous system.125, 126 The protective effect of GGA-induced HSP expression on tachycardia-induced cardiomyocyte remodeling has been observed in atrial cardiomyocytes, and a Drosophila model for AF, suggesting that the induction of HSPs by GGA might have potential value for clinical AF.66, 75, 127, 128 Furthermore, GGA treatment protected against cardiomyocyte remodeling and tachypacing-induced AF in a dog model of (acute) atrial ischemia and in a rabbit heart failure model.75, 129 A recent study also indicates that a derivative of GGA, GGA*−59, and recombinant HSPB1 can accelerate recovery from tachypacing-induced structural remodeling and contractile dysfunction in HL-1 cardiomyocytes.76 In this study it was shown that post-treatment with GGA*−59 increases HSPB1 levels, represses HDAC6 activity and restores contractile protein and microtubule levels after tachypacing, indicating that HSP-induction may represent an effective approach to combat AF-induced cardiomyocyte damage.76 In addition, compounds directed at prevention of proteotoxic ER stress and subsequent protein degradation may also represent interesting candidates to treat AF. Among the available compounds, the chemical chaperone (sodium)4-phenylbutyric acid (4PBA) is seemingly promising because it has been approved for clinical use to treat urea cycle disorders (Table 2). 4PBA can alleviate ER stress and it has been shown that 4PBA protects against AF promotion in tachypaced atrial cardiomyocytes, Drosophila, and dog model of AF.81 As the ER is the cellular compartment where most proteins (at least one-third) are synthesized and folded.130 This compartment is highly susceptible for proteotoxic stress induced by AF. Data from a phase I clinical study indicates that 4PBA is safe and displays minor adverse effects.131 Because of its protective effect on ER integrity, 4PBA is currently tested in several clinical trials for misfolded protein diseases including cystic fibrosis, amyotrophic lateral sclerosis and Huntington’s disease (Table 2). Results of these studies may inform us on the effectiveness of 4PBA in respect to AF.
Second, specific inhibition of HDACs that have been implicated in AF may be beneficial. Among various HDACs, HDAC6 emerges as a key regulator in AF progression by inducing α-tubulin deacetylation and microtubule disruption and therefore represents as an interesting target for AF (Table 2).10, 132 Two potent HDAC6 inhibitors include tubastatin A and ricolinostat (ACY-1215) and both have shown beneficial effects in mouse models of neurodegenerative diseases and cancer.133–135 In addition, tubastatin A protected against electrical and contractile remodeling and subsequent AF promotion in the dog model of AF.10 Because the specific inhibition of HDAC6 has not been associated with any serious toxicity so far,136, 137 clinical trials may be properly designed to evaluate the anti-AF efficacy of HDAC6 inhibitors.
The third aspect in normalizing proteostasis is to prevent excessive NAD+ depletion and the subsequent cardiomyocyte dysfunction. PARP1 inhibitors are therefore interesting compounds. The first PARP1 inhibitors, such as 3-AB, compete with NAD+ for the enzyme and consequently inhibit PARP1 and other members of the PARP family, as well as mono-ADP-ribosyl-transferases and sirtuins, which are cardiac protective enzymes13. Therefore, the early PARP1 inhibitors probably are less suited to treat AF. However, recently developed PARP inhibitors, including ABT-888 and olaparib, exhibit increased potency and specificity relative to earlier inhibitors. ABT-888 directly inhibits PARP1 and PARP2 without an action on sirtuins.138 ABT-888 is currently in phase I and II clinical studies in cancer.139 Olaparib is used in phase III clinical trials for the treatment of metastatic breast cancers and has no effect on QT/QTc interval and therefore likely safe to treat cardiac diseases.140, 141 Another therapeutic option to protect against AF-induced remodeling is to replenish the NAD+ pool by supplementation with NAD+ or its precursors, such as nicotinamide and nicotinamide riboside. Interestingly, nicotinamide is not only a PARP1 inhibitor, but also a NAD+ precursor. Nicotinamide can be converted into NAD+ via the salvage pathway. In experimental heart failure and dilated cardiomyopathy model systems, nicotinamide displayed a protective effect demonstrating a clear benefit of normalizing NAD+ levels in failing hearts.142–144 An open-label pharmacokinetics study with nicotinamide riboside (Niagen®, Chromadex) in healthy volunteers, showed that nicotinamide riboside stably induced circulating NAD+ levels in the blood and was well tolerated (even up to 2× 1000 mg/day).145 Therefore, nicotinamide riboside represents a potential therapy AF by replenishing the NAD+ levels in atrial cardiomyocytes.
Limitations and future perspectives
It is worth noting that anti-inflammation can be achieved through steroids and nonsteroidal anti-inflammatory drugs (NSAIDs). However, in a randomized, double-blind, and prospective clinical trial, patients treated with dexamethasone before coronary artery bypass graft or valvular surgery still experience POAF, despite the reduction of inflammatory mediators.146 Similarly, oral administration of the corticosteroid prednisone significantly reduces certain cytokines, but fails to improve the outcomes of AF ablation.147 These results suggest that non-specific and non-selective anti-inflammatory therapies yield minimum or have no impact on prevention of AF onset and progression. A specific and selective NLRP3 inhibitor may reveal more desirable effects for AF intervention.
The mechanisms underlying the activation of NLRP3 in the context of AF are currently missing. Although the activation of NLRP3 inflammasomes has been observed in non-cardiomyocytes in the setting of obesity148, diabetes149, and hypertension150, all well known risk factors of AF, evaluation of NLRP3 inflammasome as an intermediator between these environment-induced risk factors and AF pathogenesis is certainly appealing. Moreover, apart from the NLRP3 inflammasome, whether other forms of inflammasome play a role in AF pathogenesis remains elusive. Knowledge on these pathways will open up a new and exciting research avenue.
Finally, the involvement of hyperactive NLRP3-inflammasome in AF pathogenesis is largely based on small animal studies. In order to develop effective therapies for AF, appropriate pharmacodynamic and pharmacokinetic studies of compounds or biologicals targeting this pathway should be performed in large animal models of AF prior to the clinical studies in patients. Since several (marketed) drugs against proteostasis derailment have been successfully tested in dog models for AF10, 75, 81, 129, clinical studies directed at proteostasis derailment in patients with AF is within reach.
Conclusion
NLRP3 inflammasome and proteostasis derailment can promote an array of proarrhythmic events, including ectopic activity, reentry substrate, electrical and contractile remodeling, thereby enhancing AF arrhythmogenesis. As such, both pathways represent two novel causative mechanisms for AF development. The modulators driving NLRP3 inflammasome and proteostasis derailment are diverse, which offers several plausible therapeutic targets. Because of the reciprocal modulation between proteostasis derailment and inflammasome activation, establishing mechanistic evidence for a crosstalk in AF is required to provide a holistic understanding of molecular mechanisms of AF.
Acknowledgments
Source of Funding
This work was supported by grants from the National Institutes of Health (R01HL136389 and R01HL147108 to Dr Li) and CVON-STW2016-14728 AFFIP and Medical Delta (to Dr Brundel).
Abbreviations
- AAV9
adeno-associated virus 9
- AERP
atrial effective refractory period
- AF
atrial fibrillation
- AIM2
absent in melanoma 2
- APD
action potential duration
- ATF4
activating transcription factor 4
- ATF6
activating transcription factor 6
- ATG 7
autophagy gene 7
- ATG12
autophagy gene 12
- ASC
apoptosis-associated speck-like protein containing a CARD
- CAD
coronary artery disease
- CHOP
C/EBP homologous protein
- CRP
C-reactive protein
- DADs
delayed after-depolorizations
- DAMPs
damage-associated molecular patterns
- ECM
extracellular matrix
- eIF2α
α-subunit of eukaryotic initiation factor 2
- ER
endoplasmic reticulum
- GSDMD
gesdermin-D
- hiPSCs
human induced pluripotent stem cells
- HMGB1
high mobility group box 1
- HSP
heat shock protein
- IL-1β
interleukin-1β
- IL-18
interleukin-18
- IL-1R
interleukin-1 receptor
- IRE1
inositol-requiring transmembrane kinase/endonuclease 1
- LC3B
microtubule-associated proteins 1A/1B light chain 3B
- Nampt
nicotinamide phosphoribosyltransferase
- NCX1
sodium-calcium exchanger 1
- NFκB
nuclear factor kappa-light-chain-enhancer of activated B cells
- NLRP3
NACHT, LRR and PYD domain containing protein 3
- Nt-GSDMD
N-terminus fragment of gesdermin-D
- mtROS
mitochondrial ROS
- P2X7R
purinergic receptor P2X7R
- PACs
premature atrial contractions
- pAF
paroxysmal atrial fibrillation
- perAF
persistent atrial fibrillation
- POAF
post-operative atrial fibrillation
- PERK
pancreatic ER kinase
- PAMPs
pathogen-associated molecular patterns
- PQC
protein quality control
- ROS
reactive oxygen species
- RyR2
ryanodine receptor 2
- SR
sarcoplasmic reticulum
- TLR
toll-like receptor
- TNFR
TNFα receptor
- UPR
unfolded protein response
Footnotes
Disclosure
None.
References
- 1.Heijman J, Guichard JB, Dobrev D, Nattel S. Translational challenges in atrial fibrillation. Circ Res. 2018;122:752–773 [DOI] [PubMed] [Google Scholar]
- 2.Palatinus JA, Das S. Your father and grandfather’s atrial fibrillation: A review of the genetics of the most common pathologic cardiac dysrhythmia. Curr Genomics. 2015;16:75–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tucker NR, Clauss S, Ellinor PT. Common variation in atrial fibrillation: Navigating the path from genetic association to mechanism. Cardiovasc Res. 2016;109:493–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jalife J Mechanisms of persistent atrial fibrillation. Current opinion in cardiology. 2014;29:20–27 [DOI] [PubMed] [Google Scholar]
- 5.Brooks S, Metzner A, Wohlmuth P, Lin T, Wissner E, Tilz R, Rillig A, Mathew S, Saguner A, Heeger C, Sohns C, Kuck KH, Ouyang F. Insights into ablation of persistent atrial fibrillation: Lessons from 6-year clinical outcomes. J Cardiovasc Electrophysiol. 2018;29:257–263 [DOI] [PubMed] [Google Scholar]
- 6.Buch E, Share M, Tung R, Benharash P, Sharma P, Koneru J, Mandapati R, Ellenbogen KA, Shivkumar K. Long-term clinical outcomes of focal impulse and rotor modulation for treatment of atrial fibrillation: A multicenter experience. Heart Rhythm. 2016;13:636–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jaakkola S, Lip GY, Biancari F, Nuotio I, Hartikainen JE, Ylitalo A, Airaksinen KE. Predicting unsuccessful electrical cardioversion for acute atrial fibrillation (from the af-cvs score). Am J Cardiol. 2017;119:749–752 [DOI] [PubMed] [Google Scholar]
- 8.de Groot NMS, Allessie MA. Pathophysiology of atrial fibrillation: Focal patterns of activation. Pacing Clin Electrophysiol. 2019;42:1312–1319 [DOI] [PubMed] [Google Scholar]
- 9.Brundel BJ, Henning RH, Kampinga HH, Van Gelder IC, Crijns HJ. Molecular mechanisms of remodeling in human atrial fibrillation. Cardiovasc Res. 2002;54:315–324 [DOI] [PubMed] [Google Scholar]
- 10.Zhang D, Wu CT, Qi X, Meijering RA, Hoogstra-Berends F, Tadevosyan A, Cubukcuoglu Deniz G, Durdu S, Akar AR, Sibon OC, Nattel S, Henning RH, Brundel BJ. Activation of histone deacetylase-6 induces contractile dysfunction through derailment of alpha-tubulin proteostasis in experimental and human atrial fibrillation. Circulation. 2014;129:346–358 [DOI] [PubMed] [Google Scholar]
- 11.Ausma J, Wijffels M, Thone F, Wouters L, Allessie M, Borgers M. Structural changes of atrial myocardium due to sustained atrial fibrillation in the goat. Circulation. 1997;96:3157–3163 [DOI] [PubMed] [Google Scholar]
- 12.Swanson KV, Deng M, Ting JP. The nlrp3 inflammasome: Molecular activation and regulation to therapeutics. Nat Rev Immunol. 2019;19:477–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen G, Chelu MG, Dobrev D, Li N. Cardiomyocyte inflammasome signaling in cardiomyopathies and atrial fibrillation: Mechanisms and potential therapeutic implications. Front Physiol. 2018;9:1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diaz-Villanueva JF, Diaz-Molina R, Garcia-Gonzalez V. Protein folding and mechanisms of proteostasis. Int J Mol Sci. 2015;16:17193–17230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taylor RC, Dillin A. Aging as an event of proteostasis collapse. Cold Spring Harb Perspect Biol. 2011;3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vilchez D, Saez I, Dillin A. The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat Commun. 2014;5:5659. [DOI] [PubMed] [Google Scholar]
- 17.Castillero E, Akashi H, Pendrak K, Yerebakan H, Najjar M, Wang C, Naka Y, Mancini D, Sweeney HL, J DA, Ali ZA, Schulze PC, George I. Attenuation of the unfolded protein response and endoplasmic reticulum stress after mechanical unloading in dilated cardiomyopathy. Am J Physiol Heart Circ Physiol. 2015;309:H459–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ortega A, Rosello-Lleti E, Tarazon E, Molina-Navarro MM, Martinez-Dolz L, Gonzalez-Juanatey JR, Lago F, Montoro-Mateos JD, Salvador A, Rivera M, Portoles M. Endoplasmic reticulum stress induces different molecular structural alterations in human dilated and ischemic cardiomyopathy. PLoS One. 2014;9:e107635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rosello-Lleti E, Tarazon E, Barderas MG, Ortega A, Otero M, Molina-Navarro MM, Lago F, Gonzalez-Juanatey JR, Salvador A, Portoles M, Rivera M. Heart mitochondrial proteome study elucidates changes in cardiac energy metabolism and antioxidant prdx3 in human dilated cardiomyopathy. PLoS One. 2014;9:e112971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dai DF, Hsieh EJ, Liu Y, Chen T, Beyer RP, Chin MT, MacCoss MJ, Rabinovitch PS. Mitochondrial proteome remodelling in pressure overload-induced heart failure: The role of mitochondrial oxidative stress. Cardiovasc Res. 2012;93:79–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Y, Wang J, Qi SY, Ru LS, Ding C, Wang HJ, Zhao JS, Li JJ, Li AY, Wang DM. Reduced endoplasmic reticulum stress might alter the course of heart failure via caspase-12 and jnk pathways. Can J Cardiol. 2014;30:368–375 [DOI] [PubMed] [Google Scholar]
- 22.Buckley CD, Gilroy DW, Serhan CN, Stockinger B, Tak PP. The resolution of inflammation. Nat Rev Immunol. 2013;13:59–66 [DOI] [PubMed] [Google Scholar]
- 23.Origin Medzhitov R. and physiological roles of inflammation. Nature. 2008;454:428–435 [DOI] [PubMed] [Google Scholar]
- 24.Issac TT, Dokainish H, Lakkis NM. Role of inflammation in initiation and perpetuation of atrial fibrillation: A systematic review of the published data. J Am Coll Cardiol. 2007;50:2021–2028 [DOI] [PubMed] [Google Scholar]
- 25.Qu YC, Du YM, Wu SL, Chen QX, Wu HL, Zhou SF. Activated nuclear factor-kappab and increased tumor necrosis factor-alpha in atrial tissue of atrial fibrillation. Scand Cardiovasc J. 2009;43:292–297 [DOI] [PubMed] [Google Scholar]
- 26.Wu N, Xu B, Xiang Y, Wu L, Zhang Y, Ma X, Tong S, Shu M, Song Z, Li Y, Zhong L. Association of inflammatory factors with occurrence and recurrence of atrial fibrillation: A meta-analysis. Int J Cardiol. 2013;169:62–72 [DOI] [PubMed] [Google Scholar]
- 27.Luan Y, Guo Y, Li S, Yu B, Zhu S, Li S, Li N, Tian Z, Peng C, Cheng J, Li Q, Cui J, Tian Y. Interleukin-18 among atrial fibrillation patients in the absence of structural heart disease. Europace. 2010;12:1713–1718 [DOI] [PubMed] [Google Scholar]
- 28.Rudolph V, Andrie RP, Rudolph TK, et al. Myeloperoxidase acts as a profibrotic mediator of atrial fibrillation. Nat Med. 2010;16:470–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li J, Solus J, Chen Q, Rho YH, Milne G, Stein CM, Darbar D. Role of inflammation and oxidative stress in atrial fibrillation. Heart Rhythm. 2010;7:438–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weymann A, Popov AF, Sabashnikov A, et al. Baseline and postoperative levels of c-reactive protein and interleukins as inflammatory predictors of atrial fibrillation following cardiac surgery: A systematic review and meta-analysis. Kardiol Pol. 2018;76:440–451 [DOI] [PubMed] [Google Scholar]
- 31.Abe I, Teshima Y, Kondo H, et al. Association of fibrotic remodeling and cytokines/chemokines content in epicardial adipose tissue with atrial myocardial fibrosis in patients with atrial fibrillation. Heart Rhythm. 2018;15:1717–1727 [DOI] [PubMed] [Google Scholar]
- 32.Fontes ML, Mathew JP, Rinder HM, Zelterman D, Smith BR, Rinder CS, Multicenter Study of Perioperative Ischemia Research G. Atrial fibrillation after cardiac surgery/cardiopulmonary bypass is associated with monocyte activation. Anesth Analg. 2005;101:17–23, table of contents [DOI] [PubMed] [Google Scholar]
- 33.Chen MC, Chang JP, Liu WH, Yang CH, Chen YL, Tsai TH, Wang YH, Pan KL. Increased inflammatory cell infiltration in the atrial myocardium of patients with atrial fibrillation. Am J Cardiol. 2008;102:861–865 [DOI] [PubMed] [Google Scholar]
- 34.Yamashita T, Sekiguchi A, Iwasaki YK, Date T, Sagara K, Tanabe H, Suma H, Sawada H, Aizawa T. Recruitment of immune cells across atrial endocardium in human atrial fibrillation. Circ J. 2010;74:262–270 [DOI] [PubMed] [Google Scholar]
- 35.Hulsmans M, Clauss S, Xiao L, et al. Macrophages facilitate electrical conduction in the heart. Cell. 2017;169:510–522 e520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leuschner F, Nahrendorf M. Novel functions of macrophages in the heart: Insights into electrical conduction, stress, and diastolic dysfunction. Eur Heart J. 2019 [DOI] [PMC free article] [PubMed]
- 37.Dewachter L, Dewachter C. Inflammation in right ventricular failure: Does it matter? Front Physiol. 2018;9:1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aschar-Sobbi R, Izaddoustdar F, Korogyi AS, et al. Increased atrial arrhythmia susceptibility induced by intense endurance exercise in mice requires tnfalpha. Nat Commun. 2015;6:6018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matsushita N, Ishida N, Ibi M, Saito M, Takahashi M, Taniguchi S, Iwakura Y, Morino Y, Taira E, Sawa Y, Hirose M. Il-1beta plays an important role in pressure overload-induced atrial fibrillation in mice. Biol Pharm Bull. 2019;42:543–546 [DOI] [PubMed] [Google Scholar]
- 40.Scott L Jr., Li N, Dobrev D Role of inflammatory signaling in atrial fibrillation. Int J Cardiol. 2019;287:195–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Verstrepen L, Bekaert T, Chau TL, Tavernier J, Chariot A, Beyaert R. Tlr-4, il-1r and tnf-r signaling to nf-kappab: Variations on a common theme. Cell Mol Life Sci. 2008;65:2964–2978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu Q, Bo L, Hu J, Geng J, Chen Y, Li X, Chen F, Song J. High mobility group box 1 was associated with thrombosis in patients with atrial fibrillation. Medicine (Baltimore). 2018;97:e0132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martinon F, Burns K, Tschopp J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proil-beta. Mol Cell. 2002;10:417–426 [DOI] [PubMed] [Google Scholar]
- 44.Panayi GS, Corrigall VM, Henderson B. Stress cytokines: Pivotal proteins in immune regulatory networks; opinion. Curr Opin Immunol. 2004;16:531–534 [DOI] [PubMed] [Google Scholar]
- 45.Kim EJ, Park SY, Baek SE, Jang MA, Lee WS, Bae SS, Kim K, Kim CD. Hmgb1 increases il-1beta production in vascular smooth muscle cells via nlrp3 inflammasome. Front Physiol. 2018;9:313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature. 2012;481:278–286 [DOI] [PubMed] [Google Scholar]
- 47.He Y, Hara H and Núñez G Mechanism and regulation of nlrp3 inflammasome activation. Trends Biochem Sci. 2016;41:1012–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kanneganti TD. The inflammasome: Firing up innate immunity. Immunol rev. Immunol Rev. 2015;265:1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ozaki E, Campbell M, Doyle SL. Targeting the nlrp3 inflammasome in chronic inflammatory diseases: Current perspectives. J Inflamm Res. 2015;8:15–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sutterwala FS, Haasken S, Cassel SL. Mechanism of nlrp3 inflammasome activation. Ann N Y Acad Sci. 2014;1319:82–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Elliott EI, Sutterwala FS. Initiation and perpetuation of nlrp3 inflammasome activation and assembly. Immunol Rev. 2015;265:35–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bracey NA, Beck PL, Muruve DA, Hirota SA, Guo J, Jabagi H, Wright JR Jr., MacDonald JA, Lees-Miller JP, Roach D, Semeniuk LM and Duff HJ The nlrp3 inflammasome promotes myocardial dysfunction in structural cardiomyopathy through interleukin-1β. Exp Physiol. 2013;98:462–473 [DOI] [PubMed] [Google Scholar]
- 53.Mezzaroma E, Toldo S, Farkas D, Seropian IM, Van Tassell BW, Salloum FN, Kannan HR, Menna AC, Voelkel NF and Abbate A The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc Natl Acad Sci USA. 2011;108:19725–19730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Valle Raleigh J, Mauro AG, Devarakonda T, Marchetti C, He J, Kim E, Filippone S, Das A, Toldo S, Abbate A, Salloum FN. Reperfusion therapy with recombinant human relaxin-2 (serelaxin) attenuates myocardial infarct size and nlrp3 inflammasome following ischemia/reperfusion injury via enos-dependent mechanism. Cardiovasc Res. 2017;113:609–619 [DOI] [PubMed] [Google Scholar]
- 55.Toldo S, Marchetti C, Mauro AG, Chojnacki J, Mezzaroma E, Carbone S, Zhang S, Van Tassell B, Salloum FN, Abbate A. Inhibition of the nlrp3 inflammasome limits the inflammatory injury following myocardial ischemia-reperfusion in the mouse. Int J Cardiol. 2016;209:215–220 [DOI] [PubMed] [Google Scholar]
- 56.Baum J, Duffy HS. Fibroblasts and myofibroblasts: What are we talking about? J Cardiovasc Pharmacol. 2011;57:376–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Martin ML, Blaxall BC. Cardiac intercellular communication: Are myocytes and fibroblasts fair-weather friends? J Cardiovasc Transl Res. 2012;5:768–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chelko SP, Asimaki A, Lowenthal J, Bueno-Beti C, Bedja D, Scalco A, Amat-Alarcon N, Andersen P, Judge DP, Tung L, Saffitz JE. Therapeutic modulation of the immune response in arrhythmogenic cardiomyopathy. Circulation. 2019;140:1491–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yao C, Veleva T, Scott L Jr., et al. Enhanced cardiomyocyte nlrp3 inflammasome signaling promotes atrial fibrillation. Circulation. 2018;138:2227–2242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kirchhof P, Marijon E, Fabritz L, et al. Overexpression of camp-response element modulator causes abnormal growth and development of the atrial myocardium resulting in a substrate for sustained atrial fibrillation in mice. Int J Cardiol. 2013;166:366–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li N, Chiang DY, Wang S, et al. Ryanodine receptor-mediated calcium leak drives progressive development of an atrial fibrillation substrate in a transgenic mouse model. Circulation. 2014;129:1276–1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bruchard M, Rebe C, Derangere V, et al. The receptor nlrp3 is a transcriptional regulator of th2 differentiation. Nat Immunol. 2015;16:859–870 [DOI] [PubMed] [Google Scholar]
- 63.Golenhofen N, Perng MD, Quinlan RA, Drenckhahn D. Comparison of the small heat shock proteins alphab-crystallin, mkbp, hsp25, hsp20, and cvhsp in heart and skeletal muscle. Histochem Cell Biol. 2004;122:415–425 [DOI] [PubMed] [Google Scholar]
- 64.Henning RH, Brundel B. Proteostasis in cardiac health and disease. Nat Rev Cardiol. 2017;14:637–653 [DOI] [PubMed] [Google Scholar]
- 65.Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319:916–919 [DOI] [PubMed] [Google Scholar]
- 66.Brundel BJ, Henning RH, Ke L, van Gelder IC, Crijns HJ, Kampinga HH. Heat shock protein upregulation protects against pacing-induced myolysis in hl-1 atrial myocytes and in human atrial fibrillation. J Mol Cell Cardiol. 2006;41:555–562 [DOI] [PubMed] [Google Scholar]
- 67.Kotter S, Unger A, Hamdani N, Lang P, Vorgerd M, Nagel-Steger L, Linke WA. Human myocytes are protected from titin aggregation-induced stiffening by small heat shock proteins. J Cell Biol. 2014;204:187–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bullard B, Ferguson C, Minajeva A, Leake MC, Gautel M, Labeit D, Ding L, Labeit S, Horwitz J, Leonard KR, Linke WA. Association of the chaperone alphab-crystallin with titin in heart muscle. J Biol Chem. 2004;279:7917–7924 [DOI] [PubMed] [Google Scholar]
- 69.Ghosh JG, Houck SA, Clark JI. Interactive domains in the molecular chaperone human alphab crystallin modulate microtubule assembly and disassembly. PLoS One. 2007;2:e498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bryantsev AL, Loktionova SA, Ilyinskaya OP, Tararak EM, Kampinga HH, Kabakov AE. Distribution, phosphorylation, and activities of hsp25 in heat-stressed h9c2 myoblasts: A functional link to cytoprotection. Cell Stress Chaperones. 2002;7:146–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Baler R, Dahl G, Voellmy R. Activation of human heat shock genes is accompanied by oligomerization, modification, and rapid translocation of heat shock transcription factor hsf1. Mol Cell Biol. 1993;13:2486–2496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Haslbeck M, Franzmann T, Weinfurtner D, Buchner J. Some like it hot: The structure and function of small heat-shock proteins. Nat Struct Mol Biol. 2005;12:842–846 [DOI] [PubMed] [Google Scholar]
- 73.Kim HJ, Hwang NR, Lee KJ. Heat shock responses for understanding diseases of protein denaturation. Mol Cells. 2007;23:123–131 [PubMed] [Google Scholar]
- 74.Hu X, Van Marion DMS, Wiersma M, Zhang D, Brundel B. The protective role of small heat shock proteins in cardiac diseases: Key role in atrial fibrillation. Cell Stress Chaperones. 2017;22:665–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brundel BJ, Shiroshita-Takeshita A, Qi X, Yeh YH, Chartier D, van Gelder IC, Henning RH, Kampinga HH, Nattel S. Induction of heat shock response protects the heart against atrial fibrillation. Circ Res. 2006;99:1394–1402 [DOI] [PubMed] [Google Scholar]
- 76.Hu X, Li J, van Marion DMS, Zhang D, Brundel B. Heat shock protein inducer gga*−59 reverses contractile and structural remodeling via restoration of the microtubule network in experimental atrial fibrillation. J Mol Cell Cardiol. 2019;134:86–97 [DOI] [PubMed] [Google Scholar]
- 77.van Marion DMS, Dorsch L, Hoogstra-Berends F, Kakuchaya T, Bockeria L, de Groot NMS, Brundel B. Oral geranylgeranylacetone treatment increases heat shock protein expression in human atrial tissue. Heart Rhythm. 2019 [DOI] [PubMed]
- 78.van Marion DM, Hu X, Zhang D, Hoogstra-Berends F, Seerden JG, Loen L, Heeres A, Steen H, Henning RH, Brundel BJ. Screening of novel hsp-inducing compounds to conserve cardiomyocyte function in experimental atrial fibrillation. Drug Des Devel Ther. 2019;13:345–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li J, Zhang D, Wiersma M, Brundel B. Role of autophagy in proteostasis: Friend and foe in cardiac diseases. Cells. 2018;7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wiersma M, Meijering RAM, Qi XY, Zhang D, Liu T, Hoogstra-Berends F, Sibon OCM, Henning RH, Nattel S, Brundel B. Endoplasmic reticulum stress is associated with autophagy and cardiomyocyte remodeling in experimental and human atrial fibrillation. J Am Heart Assoc. 2017;6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, Nishida K, Hori M, Mizushima N, Otsu K. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13:619–624 [DOI] [PubMed] [Google Scholar]
- 83.Lkhagva B, Chang SL, Chen YC, Kao YH, Lin YK, Chiu CT, Chen SA, Chen YJ. Histone deacetylase inhibition reduces pulmonary vein arrhythmogenesis through calcium regulation. Int J Cardiol. 2014;177:982–989 [DOI] [PubMed] [Google Scholar]
- 84.Lugenbiel P, Govorov K, Rahm AK, Wieder T, Gramlich D, Syren P, Weiberg N, Seyler C, Katus HA, Thomas D. Inhibition of histone deacetylases induces k+ channel remodeling and action potential prolongation in hl-1 atrial cardiomyocytes. Cell Physiol Biochem. 2018;49:65–77 [DOI] [PubMed] [Google Scholar]
- 85.Scholz B, Schulte JS, Hamer S, Himmler K, Pluteanu F, Seidl MD, Stein J, Wardelmann E, Hammer E, Volker U, Muller FU. Hdac (histone deacetylase) inhibitor valproic acid attenuates atrial remodeling and delays the onset of atrial fibrillation in mice. Circ Arrhythm Electrophysiol. 2019;12:e007071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhang D, Hu X, Li J, et al. DNA damage-induced parp1 activation confers cardiomyocyte dysfunction through nad(+) depletion in experimental atrial fibrillation. Nat Commun. 2019;10:1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hassa PO, Hottiger MO. The diverse biological roles of mammalian parps, a small but powerful family of poly-adp-ribose polymerases. Front Biosci. 2008;13:3046–3082 [DOI] [PubMed] [Google Scholar]
- 88.Pillai JB, Isbatan A, Imai S, Gupta MP. Poly(adp-ribose) polymerase-1-dependent cardiac myocyte cell death during heart failure is mediated by nad+ depletion and reduced sir2alpha deacetylase activity. J Biol Chem. 2005;280:43121–43130 [DOI] [PubMed] [Google Scholar]
- 89.Andrade J, Khairy P, Dobrev D, Nattel S. The clinical profile and pathophysiology of atrial fibrillation: Relationships among clinical features, epidemiology, and mechanisms. Circ Res. 2014;114:1453–1468 [DOI] [PubMed] [Google Scholar]
- 90.Bingen BO, Neshati Z, Askar SF, Kazbanov IV, Ypey DL, Panfilov AV, Schalij MJ, de Vries AA, Pijnappels DA. Atrium-specific kir3.X determines inducibility, dynamics, and termination of fibrillation by regulating restitution-driven alternans. Circulation. 2013;128:2732–2744 [DOI] [PubMed] [Google Scholar]
- 91.Brundel BJ, Van Gelder IC, Henning RH, Tieleman RG, Tuinenburg AE, Wietses M, Grandjean JG, Van Gilst WH, Crijns HJ. Ion channel remodeling is related to intraoperative atrial effective refractory periods in patients with paroxysmal and persistent atrial fibrillation. Circulation. 2001;103:684–690 [DOI] [PubMed] [Google Scholar]
- 92.Brundel BJ, Van Gelder IC, Henning RH, Tuinenburg AE, Wietses M, Grandjean JG, Wilde AA, Van Gilst WH, Crijns HJ. Alterations in potassium channel gene expression in atria of patients with persistent and paroxysmal atrial fibrillation: Differential regulation of protein and mrna levels for k+ channels. J Am Coll Cardiol. 2001;37:926–932 [DOI] [PubMed] [Google Scholar]
- 93.Pacher P, Liaudet L, Mabley J, Komjati K, Szabo C. Pharmacologic inhibition of poly(adenosine diphosphate-ribose) polymerase may represent a novel therapeutic approach in chronic heart failure. J Am Coll Cardiol. 2002;40:1006–1016 [DOI] [PubMed] [Google Scholar]
- 94.Pillai JB, Russell HM, Raman J, Jeevanandam V, Gupta MP. Increased expression of poly(adp-ribose) polymerase-1 contributes to caspase-independent myocyte cell death during heart failure. Am J Physiol Heart Circ Physiol. 2005;288:H486–496 [DOI] [PubMed] [Google Scholar]
- 95.Schionning JD, Poulsen EH, Moller-Madsen B, Danscher G. Autometallographic detection of gold in dorsal root ganglia of rats treated with sodium aurothiomalate. Exp Mol Pathol. 1992;56:239–247 [DOI] [PubMed] [Google Scholar]
- 96.Szabo C Roles of poly(adp-ribose) polymerase activation in the pathogenesis of diabetes mellitus and its complications. Pharmacol Res. 2005;52:60–71 [DOI] [PubMed] [Google Scholar]
- 97.Cha YM, Dzeja PP, Shen WK, Jahangir A, Hart CY, Terzic A, Redfield MM. Failing atrial myocardium: Energetic deficits accompany structural remodeling and electrical instability. Am J Physiol Heart Circ Physiol. 2003;284:H1313–1320 [DOI] [PubMed] [Google Scholar]
- 98.Li D, Fareh S, Leung TK, Nattel S. Promotion of atrial fibrillation by heart failure in dogs: Atrial remodeling of a different sort. Circulation. 1999;100:87–95 [DOI] [PubMed] [Google Scholar]
- 99.Allessie M The “second factor”: A first step toward diagnosing the substrate of atrial fibrillation? J Am Coll Cardiol. 2009;53:1192–1193 [DOI] [PubMed] [Google Scholar]
- 100.Stiles MK, John B, Wong CX, Kuklik P, Brooks AG, Lau DH, Dimitri H, Roberts-Thomson KC, Wilson L, De Sciscio P, Young GD, Sanders P. Paroxysmal lone atrial fibrillation is associated with an abnormal atrial substrate: Characterizing the “second factor”. J Am Coll Cardiol. 2009;53:1182–1191 [DOI] [PubMed] [Google Scholar]
- 101.Verheule S, Wilson E, Everett Tt, Shanbhag S, Golden C, Olgin J. Alterations in atrial electrophysiology and tissue structure in a canine model of chronic atrial dilatation due to mitral regurgitation. Circulation. 2003;107:2615–2622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dobrev D, Carlsson L, Nattel S. Novel molecular targets for atrial fibrillation therapy. Nat Rev Drug Discov. 2012;11:275–291 [DOI] [PubMed] [Google Scholar]
- 103.Martinon F, Aksentijevich I. New players driving inflammation in monogenic autoinflammatory diseases. Nat Rev Rheumatol. 2015;11:11–20 [DOI] [PubMed] [Google Scholar]
- 104.Agyemang AF, Harrison SR, Siegel RM, McDermott MF. Protein misfolding and dysregulated protein homeostasis in autoinflammatory diseases and beyond. Semin Immunopathol. 2015;37:335–347 [DOI] [PubMed] [Google Scholar]
- 105.Wiersma M, van Marion DMS, Wust RCI, Houtkooper RH, Zhang D, Groot NMS, Henning RH, Brundel B. Mitochondrial dysfunction underlies cardiomyocyte remodeling in experimental and clinical atrial fibrillation. Cells. 2019;8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Han X, Sun S, Sun Y, Song Q, Zhu J, Song N, Chen M, Sun T, Xia M, Ding J, Lu M, Yao H, Hu G. Small molecule-driven nlrp3 inflammation inhibition via interplay between ubiquitination and autophagy: Implications for parkinson disease. Autophagy. 2019;15:1860–1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Guo C, Xie S, Chi Z, Zhang J, Liu Y, Zhang L, Zheng M, Zhang X, Xia D, Ke Y, Lu L, Wang D. Bile acids control inflammation and metabolic disorder through inhibition of nlrp3 inflammasome. Immunity. 2016;45:802–816 [DOI] [PubMed] [Google Scholar]
- 108.Yan Y, Jiang W, Liu L, Wang X, Ding C, Tian Z, Zhou R. Dopamine controls systemic inflammation through inhibition of nlrp3 inflammasome. Cell. 2015;160:62–73 [DOI] [PubMed] [Google Scholar]
- 109.Shin JN, Fattah EA, Bhattacharya A, Ko S, Eissa NT. Inflammasome activation by altered proteostasis. J Biol Chem. 2013;288:35886–35895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hu B, Jin C, Li HB, et al. The DNA-sensing aim2 inflammasome controls radiation-induced cell death and tissue injury. Science. 2016;354:765–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Marin-Aguilar F, Lechuga-Vieco AV, Alcocer-Gomez E, et al. Nlrp3 inflammasome suppression improves longevity and prevents cardiac aging in male mice. Aging Cell. 2019:e13050. [DOI] [PMC free article] [PubMed]
- 112.Venkataraman K, Khurana S, Tai TC. Oxidative stress in aging--matters of the heart and mind. Int J Mol Sci. 2013;14:17897–17925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Dinarello CA. Anti-inflammatory agents: Present and future. Cell. 2010;140:935–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Dinarello CA, Simon A, van der Meer JW. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov. 2012;11:633–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119–1131 [DOI] [PubMed] [Google Scholar]
- 116.Coll RC, Robertson AA, Chae JJ, et al. A small-molecule inhibitor of the nlrp3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21:248–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Tamura K, Ishikawa G, Yoshie M, Ohneda W, Nakai A, Takeshita T, Tachikawa E. Glibenclamide inhibits nlrp3 inflammasome-mediated il-1beta secretion in human trophoblasts. J Pharmacol Sci. 2017;135:89–95 [DOI] [PubMed] [Google Scholar]
- 118.Marchetti C, Chojnacki J, Toldo S, Mezzaroma E, Tranchida N, Rose SW, Federici M, Van Tassell BW, Zhang S, Abbate A. A novel pharmacologic inhibitor of the nlrp3 inflammasome limits myocardial injury after ischemia-reperfusion in the mouse. J Cardiovasc Pharmacol. 2014;63:316–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hoss F, Latz E. Inhibitory effects of colchicine on inflammasomes. Atherosclerosis. 2018;273:153–154 [DOI] [PubMed] [Google Scholar]
- 120.Demidowich AP, Davis AI, Dedhia N, Yanovski JA. Colchicine to decrease nlrp3-activated inflammation and improve obesity-related metabolic dysregulation. Med Hypotheses. 2016;92:67–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Misawa T, Takahama M, Kozaki T, Lee H, Zou J, Saitoh T, Akira S. Microtubule-driven spatial arrangement of mitochondria promotes activation of the nlrp3 inflammasome. Nat Immunol. 2013;14:454–460 [DOI] [PubMed] [Google Scholar]
- 122.Leung YY, Yao Hui LL, Kraus VB. Colchicine--update on mechanisms of action and therapeutic uses. Semin Arthritis Rheum. 2015;45:341–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Murakami M, Oketani K, Fujisaki H, Wakabayashi T, Ohgo T. Antiulcer effect of geranylgeranylacetone, a new acyclic polyisoprenoid on experimentally induced gastric and duodenal ulcers in rats. Arzneimittelforschung. 1981;31:799–804 [PubMed] [Google Scholar]
- 124.Yanaka A, Zhang S, Sato D, Tauchi M, Suzuki H, Shibahara T, Matsui H, Nakahara A, Hyodo I. Geranylgeranylacetone protects the human gastric mucosa from diclofenac-induced injury via induction of heat shock protein 70. Digestion. 2007;75:148–155 [DOI] [PubMed] [Google Scholar]
- 125.Katsuno M, Sang C, Adachi H, Minamiyama M, Waza M, Tanaka F, Doyu M, Sobue G. Pharmacological induction of heat-shock proteins alleviates polyglutamine-mediated motor neuron disease. Proc Natl Acad Sci U S A. 2005;102:16801–16806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hirakawa T, Rokutan K, Nikawa T, Kishi K. Geranylgeranylacetone induces heat shock proteins in cultured guinea pig gastric mucosal cells and rat gastric mucosa. Gastroenterology. 1996;111:345–357 [DOI] [PubMed] [Google Scholar]
- 127.Brundel BJ, Ke L, Dijkhuis AJ, Qi X, Shiroshita-Takeshita A, Nattel S, Henning RH, Kampinga HH. Heat shock proteins as molecular targets for intervention in atrial fibrillation. Cardiovasc Res. 2008;78:422–428 [DOI] [PubMed] [Google Scholar]
- 128.Zhang D, Ke L, Mackovicova K, Van Der Want JJ, Sibon OC, Tanguay RM, Morrow G, Henning RH, Kampinga HH, Brundel BJ. Effects of different small hspb members on contractile dysfunction and structural changes in a drosophila melanogaster model for atrial fibrillation. J Mol Cell Cardiol. 2011;51:381–389 [DOI] [PubMed] [Google Scholar]
- 129.Sakabe M, Shiroshita-Takeshita A, Maguy A, Brundel BJ, Fujiki A, Inoue H, Nattel S. Effects of a heat shock protein inducer on the atrial fibrillation substrate caused by acute atrial ischaemia. Cardiovasc Res. 2008;78:63–70 [DOI] [PubMed] [Google Scholar]
- 130.Li J, Zhang D, Brundel BJ, Wiersma M. Imbalance of er and mitochondria interactions: Prelude to cardiac ageing and disease? Cells. 2019;8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Carducci MA, Gilbert J, Bowling MK, Noe D, Eisenberger MA, Sinibaldi V, Zabelina Y, Chen TL, Grochow LB, Donehower RC. A phase i clinical and pharmacological evaluation of sodium phenylbutyrate on an 120-h infusion schedule. Clin Cancer Res. 2001;7:3047–3055 [PubMed] [Google Scholar]
- 132.Zhang D, Hu X, Li J, Hoogstra-Berends F, Zhuang Q, Esteban MA, de Groot N, Henning RH, Brundel B. Converse role of class i and class iia hdacs in the progression of atrial fibrillation. J Mol Cell Cardiol. 2018;125:39–49 [DOI] [PubMed] [Google Scholar]
- 133.Butler KV, Kalin J, Brochier C, Vistoli G, Langley B, Kozikowski AP. Rational design and simple chemistry yield a superior, neuroprotective hdac6 inhibitor, tubastatin a. J Am Chem Soc. 2010;132:10842–10846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.d’Ydewalle C, Krishnan J, Chiheb DM, Van Damme P, Irobi J, Kozikowski AP, Vanden Berghe P, Timmerman V, Robberecht W, Van Den Bosch L. Hdac6 inhibitors reverse axonal loss in a mouse model of mutant hspb1-induced charcot-marie-tooth disease. Nat Med. 2011;17:968–974 [DOI] [PubMed] [Google Scholar]
- 135.Santo L, Hideshima T, Kung AL, et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective hdac6 inhibitor, acy-1215, in combination with bortezomib in multiple myeloma. Blood. 2012;119:2579–2589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Vogl DT, Raje N, Jagannath S, Richardson P, Hari P, Orlowski R, Supko JG, Tamang D, Yang M, Jones SS, Wheeler C, Markelewicz RJ, Lonial S. Ricolinostat, the first selective histone deacetylase 6 inhibitor, in combination with bortezomib and dexamethasone for relapsed or refractory multiple myeloma. Clin Cancer Res. 2017;23:3307–3315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Witt O, Lindemann R. Hdac inhibitors: Magic bullets, dirty drugs or just another targeted therapy. Cancer Lett. 2009;280:123–124 [DOI] [PubMed] [Google Scholar]
- 138.Donawho CK, Luo Y, Luo Y, et al. Abt-888, an orally active poly(adp-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin Cancer Res. 2007;13:2728–2737 [DOI] [PubMed] [Google Scholar]
- 139.Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. Parp inhibition: Parp1 and beyond. Nat Rev Cancer. 2010;10:293–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Swaisland H, Plummer R, So K, Garnett S, Bannister W, Fabre MA, Dota C, Fielding A. Olaparib does not cause clinically relevant qt/qtc interval prolongation in patients with advanced solid tumours: Results from two phase i studies. Cancer Chemother Pharmacol. 2016;78:775–784 [DOI] [PubMed] [Google Scholar]
- 141.Olaparib for metastatic breast cancer in patients with a germline brca mutation. N Engl J Med. 2017;377:1700. [DOI] [PubMed] [Google Scholar]
- 142.Diguet N, Trammell SAJ, Tannous C, et al. Nicotinamide riboside preserves cardiac function in a mouse model of dilated cardiomyopathy. Circulation. 2018;137:2256–2273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Lee CF, Chavez JD, Garcia-Menendez L, Choi Y, Roe ND, Chiao YA, Edgar JS, Goo YA, Goodlett DR, Bruce JE, Tian R. Normalization of nad+ redox balance as a therapy for heart failure. Circulation. 2016;134:883–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Walker MA, Tian R. Raising nad in heart failure: Time to translate? Circulation. 2018;137:2274–2277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Airhart SE, Shireman LM, Risler LJ, Anderson GD, Nagana Gowda GA, Raftery D, Tian R, Shen DD, O’Brien KD. An open-label, non-randomized study of the pharmacokinetics of the nutritional supplement nicotinamide riboside (nr) and its effects on blood nad+ levels in healthy volunteers. PLoS One. 2017;12:e0186459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Yared JP, Bakri MH, Erzurum SC, Moravec CS, Laskowski DM, Van Wagoner DR, Mascha E, Thornton J. Effect of dexamethasone on atrial fibrillation after cardiac surgery: Prospective, randomized, double-blind, placebo-controlled trial. J Cardiothorac Vasc Anesth. 2007;21:68–75 [DOI] [PubMed] [Google Scholar]
- 147.Iskandar S, Reddy M, Afzal MR, et al. Use of oral steroid and its effects on atrial fibrillation recurrence and inflammatory cytokines post ablation - the steroid af study. J Atr Fibrillation. 2017;9:1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Rheinheimer J, de Souza BM, Cardoso NS, Bauer AC, Crispim D. Current role of the nlrp3 inflammasome on obesity and insulin resistance: A systematic review. Metabolism. 2017;74:1–9 [DOI] [PubMed] [Google Scholar]
- 149.Yu ZW, Zhang J, Li X, Wang Y, Fu YH, Gao XY. A new research hot spot: The role of nlrp3 inflammasome activation, a key step in pyroptosis, in diabetes and diabetic complications. Life Sci. 2020;240:117138. [DOI] [PubMed] [Google Scholar]
- 150.Pasqua T, Pagliaro P, Rocca C, Angelone T, Penna C. Role of nlrp-3 inflammasome in hypertension: A potential therapeutic target. Curr Pharm Biotechnol. 2018;19:708–714 [DOI] [PubMed] [Google Scholar]
- 151.Aviles RJ, Martin DO, Apperson-Hansen C, Houghtaling PL, Rautaharju P, Kronmal RA, Tracy RP, Van Wagoner DR, Psaty BM, Lauer MS, Chung MK. Inflammation as a risk factor for atrial fibrillation. Circulation. 2003;108:3006–3010 [DOI] [PubMed] [Google Scholar]
- 152.Dernellis J, Panaretou M. C-reactive protein and paroxysmal atrial fibrillation: Evidence of the implication of an inflammatory process in paroxysmal atrial fibrillation. Acta Cardiol. 2001;56:375–380 [DOI] [PubMed] [Google Scholar]
- 153.Nortamo S, Ukkola O, Lepojarvi S, Kentta T, Kiviniemi A, Junttila J, Huikuri H, Perkiomaki J. Association of sst2 and hs-crp levels with new-onset atrial fibrillation in coronary artery disease. Int J Cardiol. 2017;248:173–178 [DOI] [PubMed] [Google Scholar]
- 154.Dastan F, Talasaz AH, Mojtahedzadeh M, Karimi A, Salehiomran A, Bina P, Jalali A. Randomized trial of carnitine for the prevention of perioperative atrial fibrillation. Semin Thorac Cardiovasc Surg. 2018;30:7–13 [DOI] [PubMed] [Google Scholar]
- 155.Cheng T, Wang XF, Hou YT, Zhang L. Correlation between atrial fibrillation, serum amyloid protein a and other inflammatory cytokines. Mol Med Rep. 2012;6:581–584 [DOI] [PubMed] [Google Scholar]
- 156.Chung MK, Martin DO, Sprecher D, Wazni O, Kanderian A, Carnes CA, Bauer JA, Tchou PJ, Niebauer MJ, Natale A, Van Wagoner DR. C-reactive protein elevation in patients with atrial arrhythmias: Inflammatory mechanisms and persistence of atrial fibrillation. Circulation. 2001;104:2886–2891 [DOI] [PubMed] [Google Scholar]
- 157.Stanciu AE, Vatasescu RG, Stanciu MM, Serdarevic N, Dorobantu M. The role of profibrotic biomarkers in paroxysmal and persistent atrial fibrillation. Cytokine. 2018;103:63–68 [DOI] [PubMed] [Google Scholar]
- 158.Gaudino M, Andreotti F, Zamparelli R, Di Castelnuovo A, Nasso G, Burzotta F, Iacoviello L, Donati MB, Schiavello R, Maseri A, Possati G. The −174g/c interleukin-6 polymorphism influences postoperative interleukin-6 levels and postoperative atrial fibrillation. Is atrial fibrillation an inflammatory complication? Circulation. 2003;108 Suppl 1:II195–199 [DOI] [PubMed] [Google Scholar]
- 159.Lazzerini PE, Laghi-Pasini F, Acampa M, et al. Systemic inflammation rapidly induces reversible atrial electrical remodeling: The role of interleukin-6-mediated changes in connexin expression. J Am Heart Assoc. 2019;8:e011006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Amdur RL, Mukherjee M, Go A, et al. Interleukin-6 is a risk factor for atrial fibrillation in chronic kidney disease: Findings from the cric study. PLoS One. 2016;11:e0148189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Osmancik P, Peroutka Z, Budera P, Herman D, Stros P, Straka Z. Changes in cytokine concentrations following successful ablation of atrial fibrillation. Eur Cytokine Netw. 2010;21:278–284 [DOI] [PubMed] [Google Scholar]
- 162.Wang H, Yan HM, Tang MX, Wang ZH, Zhong M, Zhang Y, Deng JT, Zhang W. Increased serum levels of microvesicles in nonvalvular atrial fibrillation determinated by elisa using a specific monoclonal antibody ad-1. Clin Chim Acta. 2010;411:1700–1704 [DOI] [PubMed] [Google Scholar]
- 163.Cabrera-Bueno F, Medina-Palomo C, Ruiz-Salas A, Flores A, Rodriguez-Losada N, Barrera A, Jimenez-Navarro M, Alzueta J. Serum levels of interleukin-2 predict the recurrence of atrial fibrillation after pulmonary vein ablation. Cytokine. 2015;73:74–78 [DOI] [PubMed] [Google Scholar]
- 164.Wu N, Xu B, Liu Y, Chen X, Tang H, Wu L, Xiang Y, Zhang M, Shu M, Song Z, Li Y, Zhong L. Elevated plasma levels of th17-related cytokines are associated with increased risk of atrial fibrillation. Sci Rep. 2016;6:26543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Yadav S, Akhtar S, Agarwal SK, Majumdar G, Vimal S, Sharma M. Il-10(−592a/c) gene variant a predictor of postoperative atrial fibrillation in the north indian population. J Arrhythm. 2018;34:281–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Nikoo MH, Taghavian SR, Golmoghaddam H, Arandi N, Abdi Ardakani A, Doroudchi M. Increased il-17a in atrial fibrillation correlates with neutrophil to lymphocyte ratio. Iran J Immunol. 2014;11:246–258 [PubMed] [Google Scholar]
- 167.Li W, Li S, Li X, Jiang S, Han B. Interleukin-37 elevation in patients with atrial fibrillation. Clin Cardiol. 2017;40:66–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Holzwirth E, Kornej J, Erbs S, Obradovic D, Bollmann A, Hindricks G, Thiele H, Buttner P. Myeloperoxidase in atrial fibrillation: Association with progression, origin and influence of renin-angiotensin system antagonists. Clin Res Cardiol. 2019 [DOI] [PubMed]
- 169.Li SB, Yang F, Jing L, Ma J, Jia YD, Dong SY, Zheng WF, Zhao LS. Myeloperoxidase and risk of recurrence of atrial fibrillation after catheter ablation. J Investig Med. 2013;61:722–727 [DOI] [PubMed] [Google Scholar]
- 170.Hu YF, Yeh HI, Tsao HM, Tai CT, Lin YJ, Chang SL, Lo LW, Tuan TC, Suenari K, Li CH, Chao TF, Chen SA. Electrophysiological correlation and prognostic impact of heat shock protein 27 in atrial fibrillation. Circ Arrhythm Electrophysiol. 2012;5:334–340 [DOI] [PubMed] [Google Scholar]
- 171.Yang M, Tan H, Cheng L, He M, Wei Q, Tanguay RM, Wu T. Expression of heat shock proteins in myocardium of patients with atrial fibrillation. Cell Stress Chaperones. 2007;12:142–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Mandal K, Torsney E, Poloniecki J, Camm AJ, Xu Q, Jahangiri M. Association of high intracellular, but not serum, heat shock protein 70 with postoperative atrial fibrillation. Ann Thorac Surg. 2005;79:865–871 [DOI] [PubMed] [Google Scholar]
- 173.St Rammos K, Koullias GJ, Hassan MO, Argyrakis NP, Voucharas CG, Scarupa SJ, Cowte TG. Low preoperative hsp70 atrial myocardial levels correlate significantly with high incidence of postoperative atrial fibrillation after cardiac surgery. Cardiovasc Surg. 2002;10:228–232 [DOI] [PubMed] [Google Scholar]
- 174.Afzal AR, Mandal K, Nyamweya S, Foteinos G, Poloniecki J, Camm AJ, Jahangiri M, Xu Q. Association of met439thr substitution in heat shock protein 70 gene with postoperative atrial fibrillation and serum hsp70 protein levels. Cardiology. 2008;110:45–52 [DOI] [PubMed] [Google Scholar]
- 175.Cao H, Xue L, Xu X, Wu Y, Zhu J, Chen L, Chen D, Chen Y. Heat shock proteins in stabilization of spontaneously restored sinus rhythm in permanent atrial fibrillation patients after mitral valve surgery. Cell Stress Chaperones. 2011;16:517–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Oc M, Ucar HI, Pinar A, Akbulut B, Oc B, Akinci SB, Akyon Y, Kanbak M, Boke E, Dogan R. Heat shock protein 60 antibody. A new marker for subsequent atrial fibrillation development. Saudi Med J. 2007;28:844–847 [PubMed] [Google Scholar]
- 177.Schafler AE, Kirmanoglou K, Pecher P, Hannekum A, Schumacher B. Overexpression of heat shock protein 60/10 in myocardium of patients with chronic atrial fibrillation. Ann Thorac Surg. 2002;74:767–770 [DOI] [PubMed] [Google Scholar]
- 178.Maan A, Jorgensen NW, Mansour M, Dudley S Jr., Jenny NS, Defilippi C, Szklo M, Alonso A, Refaat MM, Ruskin J, Heckbert SR, Heist EK. Association between heat shock protein-60 and development of atrial fibrillation: Results from the multi-ethnic study of atherosclerosis (mesa). Pacing Clin Electrophysiol. 2016;39:1373–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.van der Heijden T, Kritikou E, Venema W, van Duijn J, van Santbrink PJ, Slutter B, Foks AC, Bot I, Kuiper J. Nlrp3 inflammasome inhibition by mcc950 reduces atherosclerotic lesion development in apolipoprotein e-deficient mice-brief report. Arterioscler Thromb Vasc Biol. 2017;37:1457–1461 [DOI] [PubMed] [Google Scholar]
- 180.Ferreira NS, Bruder-Nascimento T, Pereira CA, Zanotto CZ, Prado DS, Silva JF, Rassi DM, Foss-Freitas MC, Alves-Filho JC, Carlos D, Tostes RC. Nlrp3 inflammasome and mineralocorticoid receptors are associated with vascular dysfunction in type 2 diabetes mellitus. Cells. 2019;8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Damm P, Mathiesen ER. Diabetes: Therapy for gestational diabetes mellitus--time for a change? Nat Rev Endocrinol. 2015;11:327–328 [DOI] [PubMed] [Google Scholar]
- 182.Eser A, Colombel JF, Rutgeerts P, Vermeire S, Vogelsang H, Braddock M, Persson T, Reinisch W. Safety and efficacy of an oral inhibitor of the purinergic receptor p2×7 in adult patients with moderately to severely active crohn’s disease: A randomized placebo-controlled, double-blind, phase iia study. Inflamm Bowel Dis. 2015;21:2247–2253 [DOI] [PubMed] [Google Scholar]
- 183.Zhang D, Wu CT, Qi XY, Meijering RAM, Hoogstra-Berends F, Tadevosyan A, Deniz GC, Durdu S, Akar R, Sibon OCM, Nattel S, Henning RH, Brundel B. Activation of histone deacetylase-6 (hdac6) induces contractile dysfunction through derailment of α-tubulin proteostasis in experimental and human atrial fibrillation. Circulation. 2014;129:346–358 [DOI] [PubMed] [Google Scholar]